
Manufacture of olefins by the selective hydrodeoxygenation of lignocellulosic ketones over a cobalt molybdate catalyst†
Fengan
Han
abc,
Guangyi
Li
a,
Yanting
Liu
a,
Aiqin
Wang
 ad,
Feng
Wang
d,
Tao
Zhang
abd and
Ning
Li
*a
ad,
Feng
Wang
d,
Tao
Zhang
abd and
Ning
Li
*a
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: lining@dicp.ac.cn
bDepartment of Chemical Physics, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, China
cUniversity of Chinese Academy of Sciences, 19 A Yuquan Road, Shijingshan District, Beijing 100049, China
dState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 12th December 2023
Abstract
Olefins are important feedstocks in the production of fuels and many useful chemicals. Herein, cobalt molybdate (CoMoO4) was first synthesized by an environmentally friendly evaporation method and it exhibited excellent catalytic performance for the production of olefins by the selective hydrodeoxygenation (HDO) of ketones that can be derived from lignocellulose. On the basis of the characterization results, the excellent catalytic performance of CoMoO4 was attributed to its larger specific surface area, better pore structure, higher oxygen vacancy (or Mo5+ species) concentration, higher acid strength and higher concentration of acid sites. Under the optimized reaction conditions, 4-heptanone was almost completely converted to heptene, and a high carbon yield of heptene (96%) was achieved over a CoMoO4 catalyst with a Co/Mo atomic ratio of 0.8 (denoted as CoMoO4-0.8). The CoMoO4 catalyst is also active for the selective HDO of other lignocellulosic ketones (such as acetone, butanone, 2-pentanone, 3-pentanone, cyclopentanone, cyclohexanone, 5-nonanone and acetophenone) to their corresponding olefins.
Introduction
Due to the great social concerns about the environmental and sustainable development issues, the utilization of renewable energy has emerged as a hot research topic. As the main component of agricultural and forestry waste, lignocellulose is a cheap and abundant carbon resource.1,2 In recent years, the catalytic conversion of lignocellulose to hydrogen, methane, liquid biofuels and value-added chemicals has aroused widespread concern.3,4Olefins are basic feedstocks in the production of fuels, lubricants, drugs, cosmetics, polymers, coatings, surfactants, detergents, etc.5 Traditionally, olefins are mainly obtained by dehydrogenation of alkanes, metathesis, catalytic cracking and pyrolysis from nonrenewable petrochemical resources.6 The manufacturing procedure of olefins is generally energy-intensive and leads to a significant amount of CO2 emission.7 From a long-term perspective, it is still imperative to develop a highly integrated method for the manufacture of olefins with lignocellulose-derived feedstocks.
Ketones are a class of oxygenated hydrocarbons that can be obtained from lignocellulose by fermentation or catalytic conversion. For instance, acetone, as a useful chemical, can be obtained from the fermentation of lignocellulose by an acetone–butanol–ethanol (ABE) method.8 Apart from this, the ketonization reaction of acetic acid obtained from the production of furfural as a by-product can be used to manufacture acetone.9 Butanone can be synthesized by the decarboxylation reaction of levulinic acid.10 Using a bi-metallic Cu–Ni/SBA-15 catalyst, 2-pentanone can be derived from the hydrogenolysis of furfural.11 Meanwhile, the catalytic conversion of ABE fermentation products is another method to produce 2-pentanone.2 3-Pentanone can be obtained from the ketonization of propionic acid, which is the partial HDO product of lactic acid12 obtained from the chemical or biological degradation of cellulose.13 Cyclopentanone can be obtained from the aqueous-phase selective hydrogenation of furfural.14 Cyclohexanone is the product of hydrogenation of phenol15 which can be obtained from lignin. The ABE fermentation product can be converted to 4-heptanone by tin-doped ceria catalysts.16 As the partial HDO product of levulinic acid, valeric acid can be converted into 5-nonanone by ketonization.17 Acetophenone, as a representative of aromatic ketones, can be derived from lignin.18 The selective hydrodeoxygenation (HDO) of these ketones to obtain the corresponding olefins by a one-step reaction has great significance.
Cobalt molybdate (CoMoO4) is a low cost, non-toxic and abundant molybdate that has a wide range of applications in electrocatalysis,19 energy storage materials20 and catalytic oxidation.21 Generally, CoMoO4 is obtained by a co-precipitation method,22 a hydrothermal method19,20 and a sol–gel method.21 These methods are relatively complicated and environmentally unfriendly. In this work, a CoMoO4 catalyst was first manufactured by a simple and environmentally friendly evaporation method and it demonstrated excellent activity for the selective HDO of lignocellulose-derived ketones to olefins. Under the optimal reaction conditions (673 K, 0.1 MPa H2, WHSV = 10 h−1, H2/4-heptanone molar ratio = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the conversion of 4-heptanone and the carbon yield of heptene could reach 98% and 96%, respectively. According to the characterization results, it was found that the excellent catalytic activity of CoMoO4 was attributed to its larger specific surface area, good pore structure, higher oxygen vacancy (or Mo5+ species) concentration, stronger acidity and higher acid site concentration. Furthermore, cobalt molybdate catalysts also exhibited excellent catalytic activity for the selective HDO of other lignocellulosic ketones (such as acetone, butanone, 2-pentanone, 3-pentanone, cyclopentanone, cyclohexanone, 5-nonanone and acetophenone) to their corresponding olefins (propylene, butene, pentene, cyclopentene, cyclopentadiene, cyclohexene, styrene and nonene). The strategy of our work is shown in Fig. 1.
:1), the conversion of 4-heptanone and the carbon yield of heptene could reach 98% and 96%, respectively. According to the characterization results, it was found that the excellent catalytic activity of CoMoO4 was attributed to its larger specific surface area, good pore structure, higher oxygen vacancy (or Mo5+ species) concentration, stronger acidity and higher acid site concentration. Furthermore, cobalt molybdate catalysts also exhibited excellent catalytic activity for the selective HDO of other lignocellulosic ketones (such as acetone, butanone, 2-pentanone, 3-pentanone, cyclopentanone, cyclohexanone, 5-nonanone and acetophenone) to their corresponding olefins (propylene, butene, pentene, cyclopentene, cyclopentadiene, cyclohexene, styrene and nonene). The strategy of our work is shown in Fig. 1.
 | ||
| Fig. 1 Strategy for the production of olefins from lignocellulose-derived ketones over the CoMoO4 catalyst. | ||
Experimental
Chemicals
Cobalt oxide (CoO), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, AR), copper nitrate trihydrate (Cu(NO3)2·3H2O, AR), iron nitrate nonahydrate (Fe(NO3)3·9H2O, 99.9%), nickel nitrate hexahydrate (Fe(NO3)2·6H2O, AR) and internal standard substances (such as dodecane, 1,4-dioxane and cyclohexanone) were supplied by Shanghai Aladdin Bio-Chem Technology Co., Ltd. Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, AR) was purchased from Meryer (Shanghai) Chemical Technology Co. Ethanol (AR) and methanol (AR) were supplied by Shanghai Sinopharm Chemical Reagent Co., Ltd.Catalyst preparation
The MoO3 catalyst was manufactured by the calcination of (NH4)6Mo7O24·4H2O at 873 K for 2 h. The MoO3 + CoO catalyst was obtained by physically mixing MoO3 and CoO at a molar ratio of 1:1. The CoMoO4 catalyst was synthesized by a modified evaporation method.23 First, 4 mmol of (NH4)6Mo7O24·4H2O and 28 mmol of Co(NO3)2·6H2O were dissolved in 200 mL of water. The mixture was heated at 373 K to remove the water by evaporation. The product was calcined at 673 K for 2 h. For comparison, we also prepared a MoO3/CoO catalyst (with a Co/Mo atomic ratio of 1:1) by an impregnation method.24 Furthermore, the other molybdate catalysts (Fe2(MoO)3, CuMoO4, and NiMoO4) were synthesized by the same evaporation method.
Characterization
The X-ray diffraction (XRD) spectra of the catalysts were obtained using a PW3040/60X’ Pert PRO (PANAlytical) diffractometer.The scanning electron micrograph (SEM) images of the samples were acquired using a JSM-7800F field-emission SEM device to analyze the morphology. The transmission electron microscopy (TEM) images of the samples were acquired from a JEM-2100F field emission electronic microscope to analyze the morphology and composition.
The N2-physisorption of the catalysts was performed using Micromeritics ASAP 2010 apparatus. Before the tests, the samples were evacuated for 6 h to eliminate the adsorbents.
The H2-temperature programmed reduction (H2-TPR) of the catalysts was conducted using a Micromeritics AutoChem II 2920 Characterization System. Before the tests, the samples were pretreated for 0.5 h under Ar flow at 673 K to remove the impurities that were adsorbed on the surface of the samples. After being cooled down to 333 K under the Ar flow, the catalysts were heated from 333 K to 1073 K at a rate of 10 K min−1 under 10% H2/Ar flow. After the removal of water by a cold trap at the outlet of the reactor, the hydrogen consumed in the measurement was detected using a thermal conductivity detector (TCD).
The NH3-temperature programmed desorption (NH3-TPD) of the catalysts was conducted using a Micromeritics AutoChem II 2920 Characterization System. First, the catalysts were pretreated at 673 K for 1 h under H2 flow (as we did for activity tests) and cooled down to 373 K under He flow. After NH3 adsorption saturation at 373 K, the desorption of NH3 was conducted from 373 K to 1173 K (at a rate of 10 K min−1). The desorbed NH3 was detected using an OminiStar mass spectrometer.
The X-ray photoelectron spectroscopy (XPS) of catalysts was conducted using a Thermo Fisher ESCALAB 250Xi.
The ultraviolet-visible (UV-Vis) diffuse reflectance spectroscopy of catalysts was performed using a PerkinElmer Lambda 950 UV/Vis/NIR spectrometer.
The online-tandem thermogravimetric-mass spectrometry (TG-MS) analysis was conducted using a TA Instruments SDT Q600 connected with an OminiStar mass spectrometer to monitor the formation of gaseous products during the test from 298 K to 1273 K (with a ramp of 10 K min−1) under air.
Activity test
The selective HDO of lignocellulose-derived ketones was conducted using a fixed-bed reactor described in our previous work.24 Prior to the activity test, the catalysts were reduced under H2 flow for 1 h at 673 K. Subsequently, the ketones were pumped into the tubular reactor using a HPLC pump. After passing the reactor, the products were cooled down and separated by a cold trap, the gaseous products were analyzed using an on-line Agilent 6890N GC, and the liquid products were gathered in the cold trap and analyzed using an Agilent 7890A GC after 5 h. The conversions of the substrates and the carbon yields of specific products were calculated using the following equations:

Results and discussion
Characterization
Based on the morphologies of the MoO3, CoO, MoO3/CoO and CoMoO4 catalysts (shown in Fig. 2 and 3), the MoO3 catalyst has an irregular structure, while the CoO catalyst mainly consists of a nanoparticle structure. Different from MoO3 and CoO, the MoO3/CoO obtained by the impregnation method has a nanorod structure. The CoMoO4 manufactured by the evaporation method has a disordered nano-scale particle stacking structure. According to the STEM-EDX results of MoO3/CoO and CoMoO4 (Fig. 4), the Co, Mo and O species were evenly distributed in the surfaces of the MoO3/CoO and CoMoO4 catalysts. The surface Co/Mo atomic ratios of the MoO3/CoO and CoMoO4 catalysts were found to be 0.99 and 0.94, respectively. These values are consistent with the theoretical Co/Mo atomic ratios in the MoO3/CoO and CoMoO4 catalysts (i.e. 1:1).
 | ||
| Fig. 2 SEM images of the (a) MoO3, (b) CoO, (c) MoO3/CoO and (d) CoMoO4 catalysts. | ||
 | ||
| Fig. 3 STEM images of the (a) MoO3, (b) CoO, (c) MoO3/CoO and (d) CoMoO4 catalysts. | ||
 | ||
| Fig. 4 TEM-EDX elemental mapping images of the (a) MoO3/CoO and (b) CoMoO4 catalysts. | ||
The specific BET surface areas (SBET), pore volumes and average pore diameters of the CoO, MoO3, MoO3/CoO and CoMoO4 catalysts were analyzed by N2-physisorption. According to Table 1 and Fig. S1,† the SBET values of CoO (9.9 m2 g−1), MoO3 (2.4 m2 g−1) and MoO3/CoO (7.7 m2 g−1) are smaller than that of CoMoO4 (22.1 m2 g−1). Furthermore, the pore volume and average pore diameter of CoMoO4 are greater than those of MoO3/CoO and CoO.
| Catalyst | S BET (m2 g−1) | Pore volume (μL g−1) | Average pore size (nm) |
|---|---|---|---|
| CoO | 9.9 | 15.2 | 6.8 |
| MoO3 | 2.4 | 2.3 | 6.1 |
| MoO3/CoO | 7.7 | 22.5 | 10.4 |
| CoMoO4 | 22.1 | 101.6 | 17.8 |
According to the XRD diffraction results illustrated in Fig. 5, the MoO3 manufactured by the calcination method is mainly composed of an orthorhombic MoO3 phase (PDF#05-0508). The CoO supplied by Shanghai Aladdin Bio-Chem Technology is mainly composed of a CoO phase with a cubic crystal structure (PDF#48-1719). The MoO3/CoO synthesized by the impregnation method is mainly composed of a monoclinic CoMoO4 phase (PDF#21-0868). The CoMoO4 obtained by the evaporation method exists in a monoclinic CoMoO4 phase (PDF#21-0868). After reduction under hydrogen flow at 673 K for 1 h, no significant change was observed in the XRD pattern of the MoO3 catalyst (see Fig. 6). In contrast, a metallic Co phase (PDF#05-0727) was generated by the reduction of CoO by H2. The MoO3/CoO and CoMoO4 catalysts were transformed from a monoclinic CoMoO4 phase (PDF#21-0868) to a cubic CoMoO3 phase (PDF#21-0869), a cubic CoO phase (PDF#48-1719) and a Co3O4 phase after reduction. No metallic Co phase was noticed in the XRD of reduced MoO3/CoO and CoMoO4 catalysts. This phenomenon illustrates that Co species in MoO3/CoO and CoMoO4 are difficult to reduce. Moreover, we also found that the crystallinity of CoMoO4 (14.7%) is lower than that of MoO3/CoO (29%). This phenomenon indicates that abundant defect sites may exist on the surface of reduced CoMoO4 catalyst.25
 | ||
| Fig. 5 XRD patterns of the MoO3, CoO, MoO3/CoO and CoMoO4 catalysts. | ||
 | ||
| Fig. 6 XRD patterns of the reduced MoO3, CoO, MoO3/CoO and CoMoO4 catalysts. | ||
The XRD of other molybdate catalysts was analyzed. According to Fig. S2,† the Fe2(MoO4)3 is mainly composed of a monoclinic Fe2(MoO4)3 phase (PDF#35-0183). The NiMoO4 exists in a monoclinic NiMoO4 phase (PDF#33-0948). The CuMoO4 is mainly composed of a triclinic CuMoO4 phase (PDF#22-0242) and an orthorhombic Cu3Mo2O9 phase (PDF#24-0055).
For comparison, we also prepared cobalt molybdate (CoMoO4-X, X = Co/Mo atomic ratio) catalysts with different Co/Mo atomic ratios by the evaporation method. According to the XRD diffraction results illustrated in Fig. S3,† the cobalt molybdate catalysts are mainly composed of a monoclinic CoMoO4 phase (PDF#21-0868).
The reducibility of the CoO, MoO3, MoO3/CoO and CoMoO4 catalysts was analyzed by H2-TPR. According to Fig. 7, a wide peak was found between 883 K and 1070 K in the H2-TPR profile of the MoO3 catalysts. According to the literature,26 this peak was attributed to the partial reduction of MoO3 to MoO2. In the H2-TPR profile of CoO, a wide peak was noticed at about 709 K. This peak can be assigned to the reduction of CoO to metallic Co.27 In the TPR profile of MoO3/CoO or CoMoO4, a peak was observed at about 836 K. Compared with MoO3, the MoO3/CoO and CoMoO4 catalysts are more reducible, which can be observed from the relatively low reduction temperatures in their H2-TPR profiles. These results can be rationalized because the presence of Co species can promote the activation of H2,28 which is beneficial for the reduction of Mo6+ species.
 | ||
| Fig. 7 H2-TPR profiles of the MoO3, CoO, MoO3/CoO and CoMoO4 catalysts. | ||
Surface analysis of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts was carried out using X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of reduced MoO3, MoO3/CoO and CoMoO4 catalysts are illustrated in Fig. S4.† As we can see from Fig. 8, the coexistence of Mo6+, Mo5+ and Mo6+ is evidenced by the Mo 3d XPS spectra of reduced MoO3, MoO3/CoO and CoMoO4 catalysts. According to the literature,4,29 the peaks at about 232.1 eV and 235.3 eV can be assigned to Mo6+ species, the peaks at 230.4 eV and 233.6 eV can be attributed to Mo5+ species, and the peaks at about 229.5 eV and 232.8 eV can be assigned to the Mo4+ species. The O 1s XPS spectra of reduced MoO3, MoO3/CoO and CoMoO4 catalysts have two peaks at about 531.4 eV and 530.5 eV. The peaks at about 531.4 eV can be attributed to the surface oxygen species of catalysts that were adsorbed by the oxygen vacancies (Oads).30,31 The peaks at about 530.5 eV can be assigned to the lattice oxygen (Olatt). The percentages of the various Mo species (Mo6+, Mo5+ and Mo4+) and different O species (Olatt and Oads) in the surface of reduced MoO3, MoO3/CoO and CoMoO4 catalysts are shown in Fig. 9. The surface of reduced MoO3 is mainly composed of Mo6+ (85.3%), accompanied by small amounts of Mo5+ (12.1%) and Mo4+ (2.6%). In contrast, the Mo species in the reduced MoO3/CoO is mainly composed of Mo5+ (47.8%), accompanied by Mo6+ (37.9%) and Mo4+ (14.3%). Compared with the reduced MoO3 and MoO3/CoO, the surface of a reduced CoMoO4 catalyst has a higher proportion of Mo5+ (51.1%) and Mo4+ (18.5%). At the same time, it has a lower proportion of Mo6+ (30.1%). This phenomenon further confirms that the presence of Co species can facilitate the reduction of Mo6+ species to a low oxidation state of Mo species (Mo5+ and Mo4+). According to the literature,32 Mo5+ species can be considered as positively charged oxygen vacancies. Therefore, the reduced CoMoO4 catalyst surface has more oxygen vacancies than MoO3 and MoO3/CoO. Based on the XPS results of O 1s, CoMoO4 has a higher Oads concentration (45.4%) than MoO3 (26.6%) and MoO3/CoO (33.0%). This result further confirms that the surface of the reduced CoMoO4 catalyst has a higher oxygen vacancy concentration. According to Fig. S5,† Mo species mainly exist in the form of Mo6+ in the unreduced MoO3, MoO3/CoO and CoMoO4 catalysts. This phenomenon further indicates that the presence of cobalt species can effectively promote the reduction of molybdenum species.
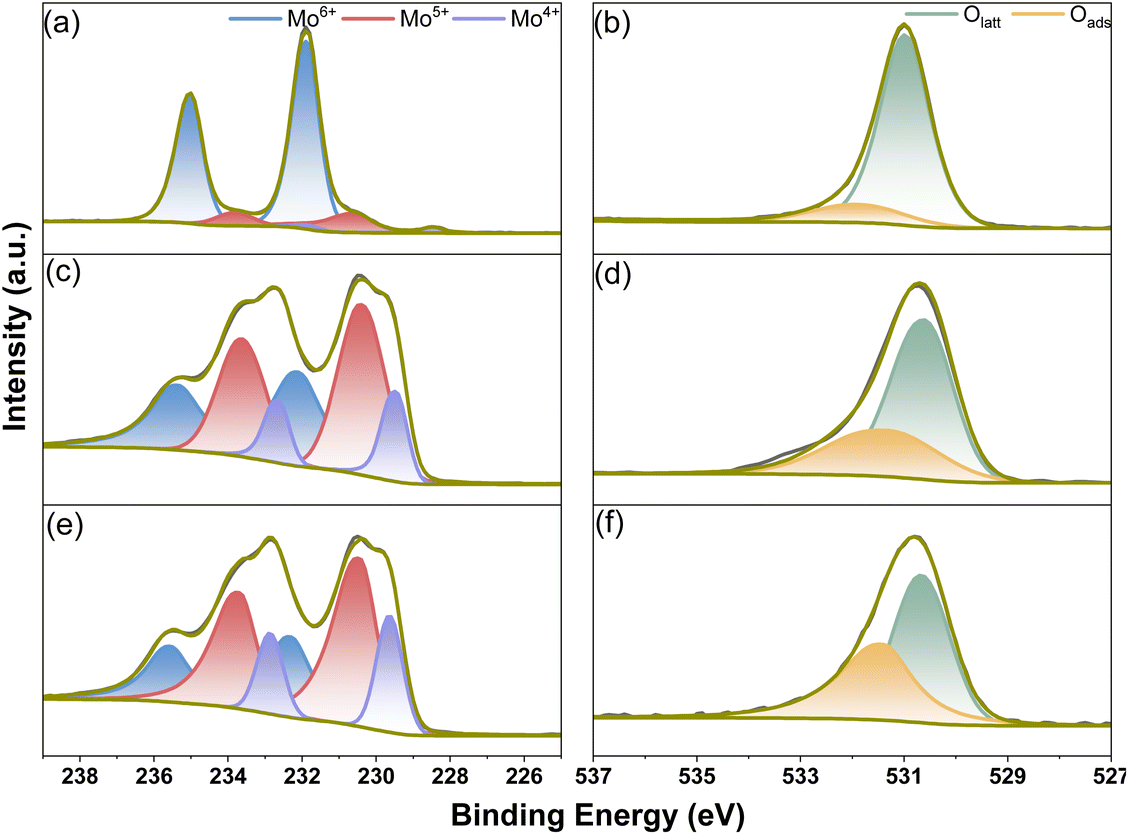 | ||
| Fig. 8 XPS Mo 3d and O 1s core level spectra of the reduced (a and b) MoO3, (c and d) MoO3/CoO and (e and f) CoMoO4 catalysts. | ||
 | ||
| Fig. 9 The percentages of the various Mo species and O species on the reduced Mo-based catalysts. | ||
The oxygen vacancy concentrations of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts were measured by ultraviolet-visible (UV-Vis) diffuse reflectance spectroscopy (see Fig. 10).33,34 Compared to reduced MoO3, the reduced MoO3/CoO and CoMoO4 catalysts exhibit stronger absorption in the wavelength range of 400–800 nm. This result indicates that the surfaces of MoO3/CoO and CoMoO4 catalysts contain more oxygen vacancies than that of MoO3.33 According to the literature,35 there is some relationship between the edge energy (Eg) and the oxygen vacancy concentration of a catalyst. Generally, a catalyst with a lower Eg has a higher oxygen vacancy concentration. To compare the oxygen vacancy concentrations on the surfaces of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts, we also analyzed the Eg values of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts (see Fig. 11). Based on the Eg values of the reduced MoO3 (3.01 eV), MoO3/CoO (1.01 eV) and CoMoO4 (0.82 eV) catalysts, the reduced CoMoO4 has a higher oxygen vacancy concentration than the reduced MoO3 and MoO3/CoO catalysts.
 | ||
| Fig. 10 UV–Vis spectra of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts. | ||
 | ||
| Fig. 11 Band gaps of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts. | ||
The acidity (acid strength and the concentration of acid sites) of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts was analyzed using NH3-TPD (see Fig. 12). The NH3-TPD profile of the reduced MoO3 catalyst shows no significant NH3 desorption peak between 373 K and 1173 K. There is a broad NH3 desorption peak around 518 K for the reduced MoO3/CoO catalyst. The reduced CoMoO4 catalyst exhibits three NH3 desorption peaks at around 518 K, 820 K and 973 K, respectively. Compared to the reduced MoO3 and MoO3/CoO catalysts, the reduced CoMoO4 exhibits NH3 desorption peaks at higher temperatures, which means that the reduced CoMoO4 catalyst has higher acid strength. From Table 2, we can see that the reduced CoMoO4 catalyst also has higher acid concentration than the reduced MoO3 and MoO3/CoO. Based on the results of XPS and UV-Vis spectra, the reduced CoMoO4 catalyst has higher oxygen vacancy concentration than the reduced MoO3 and MoO3/CoO catalysts. In some previous literature,4,30,36 it has been suggested that the oxygen vacancies generated by partially reduced metal oxide catalysts can serve as Lewis acid sites. This may be the reason why the reduced CoMoO4 catalyst has higher concentration of acid sites.
 | ||
| Fig. 12 NH3-TPD profiles of the reduced MoO3, MoO3/CoO and CoMoO4 catalysts. | ||
| Catalyst | Acid site concentration (mmol g−1) |
|---|---|
| MoO3 | 0.096 |
| MoO3/CoO | 0.543 |
| CoMoO4 | 1.450 |
Activity tests
As a representative of lignocellulosic ketones, 4-heptanone can be directly produced by the reaction of ABE fermentation products over tin-doped ceria catalysts.16 Heptene is a C7 olefin that can be used to manufacture aviation fuel by oligomerization and hydrogenation reactions. As the main purpose of this research work, we investigated the activity of CoO, MoO3 + CoO, MoO3, MoO3/CoO and CoMoO4 catalysts in the selective HDO of 4-heptone to heptene. According to the analysis of GC-MS (Fig. S6 and S7†), heptene was obtained as the only product. No heptane was detected in the products. The reaction pathway is shown in Scheme 1. | ||
| Scheme 1 Reaction pathway for the generation of heptene and heptane from the HDO of 4-heptanone. | ||
From Fig. 13, we can see that the CoO catalyst has low activity for the selective HDO of 4-heptanone to heptene. Different from CoO, the MoO3 catalyst is active for the selective HDO of 4-heptanone to heptene. Moreover, a good 4-heptanone conversion (53%) and heptene carbon yield (51%) were achieved under the investigated reaction conditions. This is consistent with the previous work of Román-Leshkov et al. about similar reaction systems.37 It is very interesting that the activity of MoO3 was further improved after it was loaded on CoO. To understand this phenomenon, we studied the catalytic activity of MoO3 + CoO prepared by physical mixing of MoO3 and CoO at a Mo/Co atomic ratio of 1:1. It was found that the activity of MoO3 + CoO was between those of the CoO and MoO3. Based on these results, we believe that a close interaction of Co and Mo species is necessary for the higher activity of the MoO3/CoO catalyst. To verify this speculation, we studied the activity of the CoMoO4 catalyst prepared by the evaporation method. As we expected, evidently, a higher heptane conversion (95%) and heptene carbon yield (93%) were achieved over this catalyst under the investigated conditions.
 | ||
| Fig. 13 Conversions of 4-heptanone and the carbon yields of heptene over the CoO, MoO3 + CoO, MoO3, MoO3/CoO and CoMoO4 catalysts. Reaction conditions: T = 673 K, PH2 = 0.1 MPa, WHSV = 15 h−1, initial H2/4-heptanone molar ratio = 50:1. | ||
For comparison, we also studied the catalytic performance of Fe2(MoO4)3, CuMoO4 and NiMoO4 catalysts. From Fig. S8,† it could be found that the CoMoO4 catalyst has better catalytic performance than other molybdates for the selective HDO of 4-heptanone to heptene. Moreover, an evident higher 4-heptanone conversion and a heptene carbon yield were achieved under the same reaction conditions.
The effects of the Co/Mo atomic ratio and reaction conditions on the catalytic performance of the CoMoO4 catalyst were investigated (see Fig. 14–16). Under the optimal reaction conditions (CoMoO4-0.8, T = 673 K, PH2 = 0.1 MPa H2, WHSV = 10 h−1, initial H2/4-heptanone molar ratio = 50:1), 98% 4-heptanone conversion and 96% heptene yield were achieved, respectively.
 | ||
| Fig. 14 Conversion of 4-heptanone and the carbon yield of heptene over the cobalt molybdate catalyst as a function of Co/Mo atomic ratio. Reaction conditions: T = 673 K, PH2 = 0.1 MPa, WHSV = 15 h−1, initial H2/4-heptanone molar ratio = 50:1. | ||
 | ||
| Fig. 15 Conversion of 4-heptanone and the carbon yield of heptene over the CoMoO4-0.8 catalyst as a function of reaction temperature. Reaction conditions: PH2 = 0.1 MPa, WHSV = 15 h−1, initial H2/4-heptanone molar ratio = 50:1. | ||
 | ||
| Fig. 16 Conversion of 4-heptanone and the carbon yield of heptene over the CoMoO4-0.8 catalyst as a function of WHSV. Reaction conditions: T = 673 K, PH2 = 0.1 MPa H2, initial H2/4-heptanone molar ratio = 50:1. | ||
According to the characterization results, there are three reasons for the higher activity of the CoMoO4 catalyst. (1) Higher BET specific surface area and good pore structure. According to the N2-physisorption results (see Table 1), CoMoO4 has a higher specific surface area and a larger pore volume and average pore size than CoO, MoO3 and MoO3/CoO. This can be considered as one of the reasons for its higher activity. (2) Higher concentration of oxygen vacancies (Mo5+). According to previous literature reports,37 the oxygen vacancies formed on the surface of a metal oxide by partial reduction can adsorb oxygenated organic compounds and convert oxygenated organic compounds into olefins by a deoxygenation reaction that follows a reversed Mars–van Krevelen mechanism (see Fig. 17). First, the CoMoO4 catalyst is partially reduced by hydrogen and generates CoMoO3 that contains oxygen vacancies (Mo5+). Subsequently, the oxygen vacancies interact with the carbonyl oxygen of 4-heptanone and remove the carbonyl oxygen in the form of electron transfer to generate heptene. Finally, the catalyst is reduced by hydrogen and generates oxygen vacancies and H2O. In the previous reports about a similar reaction system,38 it has been suggested that the Mo5+ species (oxygen vacancies) produced by partially reducing MoO3 is the active center for the selective HDO of biomass platform compounds. Based on the H2-TPR, XPS and UV-Vis spectral results, the presence of Co species promotes the reduction of Mo species. As a result, more surface Mo5+ species (oxygen vacancies) are generated under the investigated conditions, which will lead to the high activity of CoMoO4. To substantiate the significance of Mo5+ species (oxygen vacancy) in the selective HDO of 4-heptanone, a “H2-OFF” experiment was carried out by switching the carrier gas from H2 to N2 during the selective HDO of 4-heptanone over the CoMoO4 catalyst (see Fig. 18). As we expected, the 4-heptanone conversion and heptene carbon yield over the CoMoO4 catalyst plummeted within 60 minutes. After we switched back the carrier gas to H2, the catalytic activity of CoMoO4 was slowly restored. This result indicated that the selective HDO reaction was catalyzed by the Mo5+ species (oxygen vacancy) generated through in situ reduction of H2. (3) Stronger acidity and higher acid site concentration. The NH3-TPD results reveal that the reduced CoMoO4 catalyst exhibits higher acid strength and higher acid site concentration than other catalysts. Based on the literature,39 the Lewis acid site of a metal oxide can interact with a lone pair of oxygen atoms and weaken the energy of C–O bonds. Therefore, the higher acid strength and higher acid concentration of the reduced CoMoO4 catalyst are more conducive to the selective HDO reaction.
 | ||
| Fig. 17 Reaction mechanism for the selective HDO of 4-heptanone to heptene over the CoMoO4 catalyst. | ||
 | ||
| Fig. 18 Conversion of 4-heptanone and the carbon yield of heptene over the CoMoO4-0.8 catalyst as a function of time-on-steam under a H2 (or N2) atmosphere. Reaction conditions: T = 673 K, PH2 = 0.1 MPa H2, WHSV = 14 h−1, initial H2 (or N2)/4-heptanone molar ratio = 50:1. | ||
For the practical application, the stability of CoMoO4-0.8 for the selective HDO of 4-heptanone to heptane was investigated under a relatively high WHSV (20 h−1). As shown in Fig. 19, the 4-heptanone conversion and heptene carbon yield decrease with the reaction time. However, such a problem can be solved by regeneration. After being calcined at 673 K under air flow for 1 h and reduced at 673 K under H2 flow for 1 h, the activity of the CoMoO4-0.8 catalyst was restored to its initial value. Based on this result, we think that the decreased catalyst activity of CoMoO4-0.8 may be caused by a carbon deposit generated on the catalyst surface under high WHSV. To verify this, we characterized the fresh and spent CoMoO4-0.8 catalysts by TG-MS. According to Fig. S9,† about 10% weight increase was observed at 400 K–720 K in the TG-MS profile of the fresh CoMoO4-0.8 catalyst. This phenomenon can be explained by the oxidation of the catalyst. No CO2 was generated at 300 K–900 K. From Fig. S10,† about 4% weight loss was observed at 650 K–800 K in the TG-MS profile of spent CoMoO4-0.8. Meanwhile, CO2 was detected in the gaseous product. These results indicate that carbon deposition has occurred on the surface of the catalyst during stability testing, which may be the reason for catalyst deactivation.
 | ||
| Fig. 19 Conversion of 4-heptanone and the carbon yield of heptene over CoMoO4-0.8 as a function of time-on-stream. Reaction conditions: T = 673 K, PH2 = 0.1 MPa, WHSV = 20 h−1, initial H2/4-heptanone molar ratio = 50:1. | ||
The applicability of the CoMoO4-0.8 catalyst for the selective HDO of other lignocellulosic ketones to olefins was checked as well. As shown in Fig. 20 and Fig. S11–S29 in the ESI,† the selective HDO of acetone, butanone, 2-pentanone, 3-pentanone, cyclopentanone, cyclohexanone, 5-nonanone and acetophenone over the CoMoO4-0.8 catalyst led to high carbon yields of the corresponding olefins (propylene, butene, pentene, cyclopentene, cyclopentadiene, cyclohexene, styrene and nonene) under similar reaction conditions to those we used for 4-heptanone. These olefins can be used as feedstocks in the production of polymers, diesel fuel, aviation fuel, etc. For example, propylene is an important feedstock in the production of fuels, rubber, plastics, synthetic fibers, medicines, etc.40 Butene can not only be used to synthesize rubber and plastics by polymerization reactions, but also be used to manufacture aviation fuel by oligomerization reactions followed by hydrogenation.41 Styrene, as a significant chemical, is widely used to synthesize plastics, resins, coatings, medicines, etc.42 As another potential application, CoMoO4-0.8 also shows good catalytic performance in the selective HDO of lignin-derived aryl ethers (such as anisole and guaiacol)43 to aromatics (Fig. S30–S34†). It is worth mentioning that some alkylated benzenes were also obtained at the same time. These products may be generated by the alkylation of benzene (theoretical HDO product) with methanol over the acid sites on the surface of the CoMoO4-0.8 catalyst. Taking into consideration the eco-friendly and simple preparation method, high activity, good selectivity, regenerability and universality of the CoMoO4-0.8 catalyst, we believe it can be considered as a promising catalyst in future applications.
 | ||
| Fig. 20 Conversions of lignocellulosic ketones and the carbon yields of different products over the CoMoO4-0.8 catalyst. | ||
Conclusions
In summary, the cobalt molybdate (CoMoO4) catalyst manufactured by an eco-friendly and simple evaporation method demonstrated outstanding performance for the selective HDO of lignocellulose-derived ketones to olefins. Moreover, a high 4-heptanone conversion (98%) and a good carbon yield (96%) of heptene were achieved under the optimal conditions. According to the characterization results, the remarkable catalytic performance of CoMoO4 is ascribed to its larger specific surface area, good pore structure, higher oxygen vacancy (Mo5+ species) concentration, higher acid strength and higher acid site concentration compared with the other investigated catalysts. Furthermore, the CoMoO4 catalyst is also applicable for the selective HDO of other lignocellulose-derived ketones (such as acetone, butanone, 2-pentanone, 3-pentanone, cyclopentanone, cyclohexanone, 5-nonanone and acetophenone) to their corresponding olefins (propylene, butene, pentene, cyclopentene, cyclopentadiene, cyclohexene, styrene and nonene). These olefins can be used as feedstocks in the production of fuels, lubricants, drugs, cosmetics, polymers, coatings, surfactants, detergents, etc. Considering its eco-friendly and simple preparation method, high activity and selectivity, good regenerability and applicability, the CoMoO4 catalyst has a good application prospect for the manufacture of olefins with lignocellulose-derived ketones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2022YFB4201802) and the National Natural Science Foundation of China (no. 21721004, 22178335 and 22078318).References
- T. Zhang, Science, 2020, 367, 1305 CrossRef CAS PubMed; T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222 CrossRef PubMed; A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed; K. Sanderson, Nature, 2011, 474, S12 CrossRef PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235 CrossRef CAS PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed; G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef PubMed; G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef PubMed; Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499 RSC; X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198 RSC; H. Zhou, M. Wang and F. Wang, Joule, 2021, 5, 3031 CrossRef; S. Wang, A. Cheng, F. Liu, J. Zhang, T. Xia, X. Zeng, W. Fan and Y. Zhang, Ind. Chem. Mater., 2023, 1, 188 RSC; X. Tong, P. Guo, S. Liao, S. Xue and H. Zhang, Green Chem., 2019, 21, 5828 RSC.
- F. Han, Y. Liu, G. Li, L. Yuan, A. Wang, F. Wang, T. Zhang and N. Li, Green Chem., 2023, 25, 1056 RSC.
- B. G. Harvey and R. L. Quintana, Energy Environ. Sci., 2010, 3, 352 RSC; J. A. Martens, W. H. Verrelst, G. M. Mathys, S. H. Brown and P. A. Jacobs, Angew. Chem., Int. Ed., 2005, 44, 5687 CrossRef CAS PubMed; Z. Xu, J. P. Chada, D. Zhao, C. A. Carrero, Y. T. Kim, D. C. Rosenfeld, J. L. Rogers, S. J. Rozeveld, I. Hermans and G. W. Huber, ACS Catal., 2016, 6, 3815 CrossRef; H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297 RSC; M. A. Ershov, V. D. Savelenko, U. A. Makhova, V. M. Kapustin, T. M. M. Abdellatief, N. V. Karpov, E. V. Dutlov and D. V. Borisanov, Fuel, 2022, 321, 124016 CrossRef; Y. Peng, M. Dong, X. Meng, B. Zong and J. Zhang, AIChE J., 2009, 55, 717 CrossRef.
- V. P. Doronin, O. V. Potapenko, T. P. Sorokina, P. V. Lipin, K. I. Dmitriev, K. S. Plekhova, D. O. Kondrashev and A. V. Kleimenov, Catal. Today, 2021, 378, 75 CrossRef CAS; A. Y. Borisevich, S. Wang, S. N. Rashkeev, M. Glazoff, S. J. Pennycook and S. T. Pantelides, Adv. Mater., 2007, 19, 2129 CrossRef; Y. Xu, X. Li and M. Ding, Chem, 2021, 7, 1977 Search PubMed.
- Z. Zhao, J. Jiang and F. Wang, J. Energy Chem., 2021, 56, 193 CrossRef CAS.
- H. Luo, L. Ge, J. Zhang, J. Ding, R. Chen and Z. Shi, Bioresour. Technol., 2016, 200, 111 CrossRef CAS PubMed; M. I. Piñón-Muñiz, V. H. Ramos-Sánchez, N. Gutiérrez-Méndez, S. B. Pérez-Vega, J. C. Sacramento-Rivero, C. I. Vargas-Consuelos, F. M. Martinez, O. A. Graeve, R. E. Orozco-Mena, A. Quintero-Ramos, M. A. Sánchez-Madrigal and I. Salmerón, Renewable Energy, 2023, 212, 632 CrossRef; K. Sandesh, R. K. Shishir and C. Vaman Rao, Fuel, 2020, 262, 116499 CrossRef.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456 CrossRef CAS; R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193 RSC.
- K. Min, S. Kim, T. Yum, Y. Kim, B.-I. Sang and Y. Um, Appl. Microbiol. Biotechnol., 2013, 97, 5627 CrossRef CAS PubMed; C. R. Mehrer, J. M. Rand, M. R. Incha, T. B. Cook, B. Demir, A. H. Motagamwala, D. Kim, J. A. Dumesic and B. F. Pfleger, Metab. Eng., 2019, 55, 92 CrossRef PubMed.
- G. Singh, T. S. Khan, C. Samanta, R. Bal and A. Bordoloi, Biomass Bioenergy, 2022, 156, 106321 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, ChemSusChem, 2009, 2, 581 CrossRef CAS PubMed.
- Y. L. Wang, W. P. Deng, B. J. Wang, Q. H. Zhang, X. Y. Wan, Z. C. Tang, Y. Wang, C. Zhu, Z. X. Cao, G. C. Wang and H. L. Wan, Nat. Commun., 2013, 4, 2141 CrossRef PubMed.
- Z. Hu, A. Xie, C. Chen, Z. Zou, Y. Shen, Z. Fu, Y. Zhang, H. Zhang, H. Zhao and G. Wang, Fuel, 2022, 319, 123815 CrossRef CAS; X. Gao, Y. Ding, L. Peng, D. Yang, X. Wan, C. Zhou, W. Liu, Y. Dai and Y. Yang, Fuel, 2022, 314, 123074 CrossRef; M. Dohade and P. L. Dhepe, Catal. Sci. Technol., 2018, 8, 5259 RSC.
- H. Zhou, B. Han, T. Liu, X. Zhong, G. Zhuang and J. Wang, Green Chem., 2017, 19, 3585 RSC; H. Jiang, Z. Qu, Y. Liu, X. Liu, G. Wang, Y. Wang, L. Xu, K. Ding, W. Xing and R. Chen, Ind. Eng. Chem. Res., 2020, 59, 13848 CrossRef CAS.
- Y. Wang, M. Peng, J. Zhang, Z. Zhang, J. An, S. Du, H. An, F. Fan, X. Liu, P. Zhai, D. Ma and F. Wang, Nat. Commun., 2018, 9, 5183 CrossRef PubMed.
- A. D. Patel, J. C. Serrano-Ruiz, J. A. Dumesic and R. P. Anex, Chem. Eng. J., 2010, 160, 311 CrossRef CAS; J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184 CrossRef.
- H.-M. Yang, W. Zhao, K. Norinaga, J.-J. Fang, Y.-G. Wang, Z.-M. Zong and X.-Y. Wei, Sep. Purif. Technol., 2015, 152, 238 CrossRef CAS; A. Undri, M. Abou-Zaid, C. Briens, F. Berruti, L. Rosi, M. Bartoli, M. Frediani and P. Frediani, J. Anal. Appl. Pyrolysis, 2015, 114, 208 CrossRef; Q. Yu, Z. Song, X. Chen, J. Fan, J. H. Clark, Z. Wang, Y. Sun and Z. Yuan, Green Chem., 2020, 22, 6415 RSC.
- K. Wang, Y. Li, J. Hu, Z. Lu, J. Xie, A. Hao and Y. Cao, Chem. Eng. J., 2022, 447, 137540 CrossRef CAS.
- M. Li, S. Xu, C. Cherry, Y. Zhu, D. Wu, C. Zhang, X. Zhang, R. Huang, R. Qi, L. Wang and P. K. Chu, J. Mater. Chem. A, 2015, 3, 13776 RSC.
- J. Zhou, S. Deng, L. Liu, Y. Lan and C. Chen, Chem. Eng. J., 2023, 451, 138754 CrossRef CAS.
- S. Chen, J. Wu, G. Wang, J. Wang, L. Fan, J. Hao, S. Wang, Y. Liu, H. Wu, Y. Li, J. Gao and M. Yang, Coatings, 2022, 12, 1771 CrossRef CAS.
- I. Kashif, A. A. Soliman and Z. M. El-Bahy, J. Alloys Compd., 2008, 452, 384 CrossRef CAS.
- Y. Liu, R. Wang, H. Qi, X. Y. Liu, G. Li, A. Wang, X. Wang, Y. Cong, T. Zhang and N. Li, Nat. Commun., 2021, 12, 46 CrossRef CAS PubMed.
- X. Liu, J. Meng, J. Zhu, M. Huang, B. Wen, R. Guo and L. Mai, Adv. Mater., 2021, 33, 2007344 CrossRef CAS PubMed; P. Wang, X. Ma, X. Hao, B. Tang, A. Abudula and G. Guan, Catal. Rev., 2022, 1 Search PubMed.
- P. Novotný, S. Yusuf, F. Li and H. H. Lamb, Catal. Today, 2018, 317, 50 CrossRef.
- Y. Ji, Z. Zhao, A. Duan, G. Jiang and J. Liu, J. Phys. Chem. C, 2009, 113, 7186 CrossRef CAS.
- S. Xiang, L. Dong, Z.-Q. Wang, X. Han, L. L. Daemen, J. Li, Y. Cheng, Y. Guo, X. Liu, Y. Hu, A. J. Ramirez-Cuesta, S. Yang, X.-Q. Gong and Y. Wang, Nat. Commun., 2022, 13, 3657 CrossRef CAS PubMed; H. Zhang, X. Zhou, L. Liu, F. Lan, T. Zhao, M. Qiu, Q. Guan and W. Li, Appl. Catal., B, 2023, 338, 123026 CrossRef; H. Wang, S. Bai, Y. Pi, Q. Shao, Y. Tan and X. Huang, ACS Catal., 2019, 9, 154 CrossRef.
- M. Vasilopoulou, A. M. Douvas, D. G. Georgiadou, L. C. Palilis, S. Kennou, L. Sygellou, A. Soultati, I. Kostis, G. Papadimitropoulos, D. Davazoglou and P. Argitis, J. Am. Chem. Soc., 2012, 134, 16178 CrossRef CAS PubMed.
- Q. Deng, R. Gao, X. Li, J. Wang, Z. Zeng, J.-J. Zou and S. Deng, ACS Catal., 2020, 10, 7355 CrossRef CAS.
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586 CrossRef CAS; P. Wang, X. Li, S. Fan, X. Chen, M. Qin, D. Long, M. O. Tadé and S. Liu, Appl. Catal., B, 2020, 279, 119340 CrossRef.
- O. Ambriz-Peláez, S. Durón, A. Olivas, R. Valdez, L. G. Arriaga, L. Álvarez-Contreras, M. Guerra-Balcázar and N. Arjona, Appl. Surf. Sci., 2019, 498, 143842 CrossRef.
- J. Li, M. Zhang, Z. Guan, Q. Li, C. He and J. Yang, Appl. Catal., B, 2017, 206, 300 CrossRef CAS.
- I. M. Szilágyi, B. Fórizs, O. Rosseler, Á. Szegedi, P. Németh, P. Király, G. Tárkányi, B. Vajna, K. Varga-Josepovits, K. László, A. L. Tóth, P. Baranyai and M. Leskelä, J. Catal., 2012, 294, 119 CrossRef.
- S. Huang, R. Bao, J. Wang, J. Yi, Z. Zhang, L. Liu, Y. Han, Z. Li, D. Min, W. Zhang, Z. Ge and X. Zhang, J. Alloys Compd., 2023, 961, 170945 CrossRef CAS; J. Nie, X. Yu, Z. Liu, J. Zhang, Y. Ma, Y. Chen, Q. Ji, N. Zhao and Z. Chang, J. Cleaner Prod., 2022, 363, 132593 CrossRef.
- L. Wen, X. Li, R. Zhang, H. Liang, Q. Zhang, C. Su and Y.-J. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 14181 CrossRef CAS PubMed; X. Wang, L. Lu, B. Wang, Z. Xu, Z. Xin, S. Yan, Z. Geng and Z. Zou, Adv. Funct. Mater., 2018, 28, 1804191 CrossRef; J. Qi, S. Zhou, K. Xie and S. Lin, J. Energy Chem., 2021, 60, 249 CrossRef.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732 RSC.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76 CrossRef CAS PubMed.
- W. Wang, S. Chen, C. Pei, R. Luo, J. Sun, H. Song, G. Sun, X. Wang, Z.-J. Zhao and J. Gong, Science, 2023, 318, 886 CrossRef PubMed.
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397 CrossRef CAS; H. Kim, D. Kim, Y.-K. Park and J.-K. Jeon, Res. Chem. Intermed., 2018, 44, 3823 CrossRef.
- M. Ghadiri, A. Hemmati and M. Rezakazemi, Int. J. Hydrogen Energy, 2021, 46, 28641 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03825d |
| This journal is © The Royal Society of Chemistry 2024 |