
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic transformation of lignin into mussel-inspired glues: next-generation 2K adhesive for setting corals under saltwater†
Ching-Yi
Choi
 a,
Francisco
Lossada
a,
Keven
Walter
a,
Tom
Fleck-Kunde
a,
Sascha
Behrens
b,
Thomas
Meinelt
b,
Jana
Falkenhagen
c,
Matthias
Hiller
d,
Hartmut
Oschkinat
d,
André
Dallmann
a,
Andreas
Taden
e and
Hans G.
Börner
*a
a,
Francisco
Lossada
a,
Keven
Walter
a,
Tom
Fleck-Kunde
a,
Sascha
Behrens
b,
Thomas
Meinelt
b,
Jana
Falkenhagen
c,
Matthias
Hiller
d,
Hartmut
Oschkinat
d,
André
Dallmann
a,
Andreas
Taden
e and
Hans G.
Börner
*a
aHumboldt-Universität zu Berlin, Department of Chemistry, Laboratory for Organic Synthesis of Functional Systems, Brook-Taylor Straße 2, 12489 Berlin, Germany. E-mail: h.boerner@HU-Berlin.de
bInstitute of Freshwater Ecology and Inland Fisheries, Department of Ecophysiology and Aquaculture, Müggelseedamm 301, 12587 Berlin, Germany
cBundesanstalt für Materialforschung und -prüfung, BAM, Department of Materials Chemistry, Richard-Willstätter-Straße 11, 12489 Berlin, Germany
dLeibniz-Forschungsinstitut für Molekulare Pharmakologie, FMP, Department of NMR assisted structural biology, Robert-Rössle-Straße 10, 13125 Berlin, Germany
eHenkel, Adhesives Technologies, Düsseldorf, Germany
First published on 13th December 2023
Abstract
The 2-methoxyphenol units (G-units) in lignin are modified by demethylation and oxidation to provide the activated lignin as one part of an advanced biobased two-component (2K) adhesive system, which exhibits promising shear strengths in dry and underwater applications. The activation of lignin is straightforward and generates quinones via demethylation and periodate oxidation. These act as Michael acceptors and react smoothly with multi-thiol-star polymers to yield thiol-catechol connectivities (TCCs). The mussel-inspired material platform acts as a very robust and versatile adhesive, combining low-cost and readily available lignin with multi-thiols to achieve outstanding adhesion strengths of up to 15 MPa in dry application. In particular, the 2K system is compatible with the marine biological environment and shows no acute toxicity to sensitive organisms such as fish eggs. Thus, one possible application of this material could be an adhesive for setting temperature-resistant corals in damaged reefs.
Introduction
The need and urgency of feedstock changes from fossil to renewable is a paradigm that has been widely recognized and is accepted in many areas by the chemical industry.1–5 While several strategies are currently being developed and efforts are being made for implementation, the challenges, but more importantly the opportunity spaces, are becoming clear.6–9 Since the dawn of petrochemistry in the 18th century, fossil gas or oil has been the main resource, where the processing of their chemical mixture demands a tremendous amount of energy and water resources.10–12 Given that the structural and functional complexity of the components in fossil feedstocks tends to be rather low, energy-intensive chemical reactions are required to fabricate carbon-based functional materials. Considering this, lignin represents a promising candidate for chemists.13,14 As an abundant biomass, lignin has a high chemical complexity and is a functionally rich compound that has mostly been considered of low value and is, to a large extent, thermally utilized waste material from paper pulp production.15–19 Certainly, lignin has been in focus for chemists for several decades and interesting materials have already been fabricated from it in the 1980s.20,21 Additionally, within the last decade, technical lignin conversion processes overcame both severe batch-to-batch variations and dependence on raw material, resulting in consistent lignin quality at an industrial scale.22 Moreover, advanced lignin structure analysis methods, such as QQ-HSQC, NMR and HR-MS MS coupled with computational evaluation, provide deep insights, setting the stage for the exploitation of lignin as a feedstock platform for materials and fine chemical building blocks.23–27Commonly, lignin is isolated from wood pellets or plant fibers and this process generates low molecular weight polymeric structures that are soluble in alkaline solutions or organic solvents.19 Furthermore, when separated from cellulose and hemicellulose compounds, the chemical network of lignin is reorganized, leading to an artificial lignin structure.23
Depending on the source of lignin, it possesses three types of hydroxycinnamyl alcohol subunits or monolignols, namely p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S), which are connected in varying ratios, forming a biopolymer nanogel. A high amount of phenolic elements that constitute the backbone of lignin structures, in which they are connected via various C–O and C–C bond types, make lignin an interesting candidate for applications in diverse fields, such as medicine, electrochemistry, concrete additives, heavy metal ion absorbers, and adhesive materials.28–34 Among them, the use of lignin as an adhesive has been intensively studied and it has been integrated into several platform chemistries.35 In living vegetation and lignocellulosic biomass, lignin serves as a purpose-adapted crosslinker, having covalent bonds with hemicellulose, which then interface and adhere to cellulose fibers for structural frameworks.36 During industrial lignin isolation processes, these adhesive properties are largely lost in the pure lignin compounds due to their structural reconstitution and the absence of natural binding partners.37–39
In synthetic resin-type adhesives, lignin often acts as a replacement for petrochemical and polyphenolic components, resulting, for example, in the production of greener lignin-phenol-formaldehyde, -epoxy, or -urethane adhesives.40–42 However, although lignin is renewable and reduces the carbon footprint of adhesives, the synthesis of resins often involve the use of critical chemicals such as formaldehyde, epichlorohydrin, and isocyanates, as well as partially environmentally harmful catalysts, which reduce the overall green advances of these material approaches.35,40–42
Process softwood lignins such as kraft or organosolv lignin have been extensively utilized to create innovative materials,43–45 taking advantage of their easy processing and high content of polyphenolic entities that allow further chemistry.19,46–49 Lignin can act as a substitute for environmentally harmful phenol compounds, eventually eliminate bisphenol A as a compound of potential concern in the synthesis of renewable polycarbonates.50–54 The polyvalent phenols present in a fairly compact molecular “nanogel” are beneficial for different adhesive platforms. For example, bisphenol A free epoxy adhesives or glues obtained by crosslinking lignin via the addition of α,ω-vinyl ether functional poly(ethylene glycols) to phenolic hydroxy groups or via polyurethanes using classical diisocyanate polyaddition to oxypropylated lignin or related green chemistry approaches.4,55
While the nanogel structure promises the constitution of effective network points in lignin-hybrid polymer resins, the low abundance of available functional groups for chemical crosslinking reactions and the steric hindrance due to the compact connectivity of the aromatic units within the lignin macromolecule offer room for improvement.50,51 This has triggered the rich field of chemical modification of lignin, which can be categorized into, on the one hand, the introduction of artificial functionalities that usually connect functional entities by consuming lignin hydroxy/phenolic groups forming e.g. esters or aliphatic, allylic or silyl ethers.56–58 On the other hand, the abundance of hydroxyl groups in the lignin structure can be increased artificially by chemical modification of its network. One successful and well-studied pathway employs the demethylation reaction with reagents such as hydroiodic acid (HI), hydrogen bromide (HBr) and iodocyclohexane (ICH), which cleave the methyl ether bonds of the aromatic subunits.
Recently, these demethylated lignins have been successfully used as components of epoxy resins.59,60 However, the introduction of epoxide groups consumes the phenolic/hydroxy functionalities, causing the lignin molecules to lose one of their dominant interaction modes. This leads to a decrease in both inter- and intramolecular H-bonding capabilities, which appears to be one of the vital supramolecular modes for both dynamic network cohesion and adhesion properties.61,62
Hydrogen bonding of hydroxy groups in an inherently hydrophobic environment, such as the aromatic lignin structure, drives interactions through the hydrophobic (entropic) effect, which is well known in water-based self-assembly and becomes particularly important when targeting wet or underwater adhesives.63–69 For instance, the biological adhesive system from blue mussels (Mytilus edulis) utilizes the aromatic hydroxyl groups from the 3,4-dihydroxy-L-phenylalanin (L-DOPA) residues present in various mussel foot proteins (mfps).70,71 It is commonly accepted that L-DOPA residues provide the key components to create cohesive and adhesive properties under the hostile seawater conditions.63,72–79
It should be noted that in biological wet adhesives, non-covalent interactions are often accompanied by covalent crosslinking processes to construct and tune the desired structures from purpose-adapted adhesive proteins, achieving the required cohesive/adhesive performance and realizing remarkable resilience.80–82 This straightforward concept of crosslinking has enabled the industrial application of adhesives using the ill-defined agglomeration of native plant or animal proteins, where chemical modification is not always necessary.83,84
The remarkably versatile and reversible interaction capabilities of L-DOPA residues lead to a rich class of biomimetic catechol-containing peptides and polymers that demonstrate exciting underwater adhesive properties.71,74,85–88 Recently, a new class of mussel glue-inspired adhesives was fabricated by exploiting the biogenic cystenyldopa connectivity, originating from a thiol-quinone Michael-type addition.89 β-Thiols derived from cysteine residues were proven to add rapidly and cleanly to oxidized L-DOPA residues in the quinone form.89–91
The latter could be conveniently generated via enzymatic means using tyrosinase and by oxidizing agents such as periodate and 2-iodoxybenzoic acid (IBX).89,91–93 The reaction pathway is generic and polymerizes AB-type Cys/Tyr-bearing peptides to artificial mussel glue proteins with e.g. implemented cohesion switching mechanisms.91
Moreover, fully synthetic sets of AA/BB-type bisquinones and di-thiols polymerize rapidly, leading to libraries of artificial mussel glue polymers that present the thiol-catechol-connectivities (TCC) as fully synthetic “L-DOPA” derivatives.93,94 The TCC-formation exhibits an effective crosslinking/polymerization mechanism that is not consuming the valuable phenolic functionalities and thus would preserve potentially also H-bonding capabilities of lignin.93 Furthermore, the thioether substituent even stabilizes the catecholic functionality by reducing autooxidation under air and with this unwanted dimerization in analogy to “diDOPA” crosslinking pathways.70 Interestingly, various bisquinones can be accessed from technically available commodity bisphenol monomers, e.g., bisphenol A, via a single-step reaction with IBX, thus offering scale-up possibilities.93,94 However, the compound family based on bisphenol A relies on fossil resources and raises concerns as endocrine-disrupting compounds in animal studies.95 Thus, lignin appears to be an interesting candidate as a sustainable feedstock, which is available on a scale of 50–70 million tons per year from paper pulp production and chemically one demethylation step away from a catechol-bearing scaffold.96
Herein, we present the modification of commercially available lignin by distinct chemical transformations to enable its utilization as a multi-/poly-quinone component in thiol-quinone Michael-type polyaddition reactions. A two-component (2K) strategy was employed to combine the activated lignin with multi-thiol-star polymers containing either hydrophobic poly(ε-caprolactone), PCL4MP, or hydrophilic poly(ethylene oxide), ETTMP, segments, leading to the formation of TCCs (Fig. 1). The application and curing properties of the adhesive platform and its adhesive and fracture properties for gluing technical aluminum substrates were investigated under dry and saltwater conditions. The formation of TCC as one of the primary reaction pathways for building networks ensured that the aromatic hydroxyl groups of lignin remained intact, thus preserving their ability to interact and provide significant structural stability, cohesive strength and adhesive properties even under hostile seawater conditions. The 2K systems established a pure bulk adhesive, proving the modularity and tunability of the material platform. Leaching tests were performed to investigate the stability of the network under seawater conditions and standard 48 h fish egg breeding toxicity tests proved that extracts of the non-cured or fully cured adhesive had no adverse effects. Corals were successfully glued in a saltwater model including underwater glue application and curing to provide a non-toxic alternative to the commonly used epoxy/bisphenol A and cyanoacrylate underwater adhesives.
 | ||
| Fig. 1 Idealized illustration of the bioinspired concept (a) combining toughening mechanisms of wood by lignin (left) with adhesive key functionalities from the bio adhesion apparatus of marine mussels (right) in a lignin-based two-component (2K) adhesive system. Chemical constituents of the 2K adhesive mixture (b) requiring chemical transformation to activate lignin (left and middle) and liquid multi-thiols for crosslinking (right). | ||
Results and discussion
Activation of lignin
The waste material lignin is extensively available as feedstock for the design of renewable adhesive materials. Considering the important role played by catechols in the function of the bio-adhesive apparatus of marine mussels, softwood kraft lignin (SKL) appears to be an ideal feedstock given that it possesses mostly guaiacyl (G)-units with minor traces of p-hydroxyphenyl (H)-units and practically no syringyl (S)-units.23 The G-structural motif offers an ideal starting point for conversion into catechols, and ultimately quinones. In this case, although other lignin sources may be potentially feasible, SKL BioPiva™ 300 lignin was utilized for this study as a readily available source. The technical grade lignin was dissolved in acetone and insoluble parts such as ash, residual saccharides and crosslinked lignin fractions were filtered off. Subsequent drying provided 80% recovery and gel permeation chromatography (GPC) analysis in dimethyl sulfoxide (DMSO) showed only minor fractionation at the high molecular weight flank of the GPC trace (Fig. 2d). This pretreatment increased the processability of the lignin in the following solution-based reactions. | ||
Fig. 2 Chemical transformation steps of technical available softwood kraft lignin (a), involving (i) fractionation, (ii) demethylation of G-type units, and (iii) oxidation, yielding quinone entities that equip lignin ready to react with multi thiols in 2K adhesives. Hydroxy pattern analysis of fractionated and demethylated lignin by 1H NMR (b), 31P NMR analysis comparing the fractionated lignin prior and after demethylation (c), GPC analysis revealing the molecular weight distributions of the different lignin products (d) and FTIR spectra confirming the chemical transformations in the heterogenous lignin network, leading to quinone structures after oxidation (e). (Conditions: 16.6 mM commercial lignin batch in acetone; 11.1 mM fractionated lignin with 0.3 mol ICH, reflux, 6 h; 66.6 mM demethylated lignin in DMF/acetate buffer (0.1 M, pH 5) with 7 mmol NaIO4, r.t., 2 h; 1H NMR: lignin derivatized with acetate anhydride in pyridine, r.t., o.n.; 31P NMR: lignin derivatized with CDP using Cr (acac)3 and Ph3PO as an internal standard in pyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CDCl3-1.6:1 v/v%, r.t., 15 min; GPC: c = 1 g L−1 in DMSO + 0.076 M NaNO3). :CDCl3-1.6:1 v/v%, r.t., 15 min; GPC: c = 1 g L−1 in DMSO + 0.076 M NaNO3). | ||
Regarding TCC-chemistry, catechols are required, which have to be oxidized to quinones as Michael acceptor functionalities. However, in the native state of SKL, catechols are not naturally present, but G-structure elements can be seen as catechol precursors. Thus, to establish catechols in the lignin network, the aromatic methoxy groups were selectively transformed into hydroxyl groups via acidic demethylation by iodocyclohexane (ICH) (Fig. 1). The most common chemical demethylation strategies were carefully studied by Takano and co-workers, reporting that the ICH path yielded the highest demethylation conversion, while keeping the degradation of the lignin as low as possible.60 FTIR analysis suggested the effective ICH demethylation of the lignin, showing an increase in the vibration band at ν = 1200 cm−1, which is characteristic of phenolic-OH, accompanied by a reduction in the intensity of the ether vibration at ν = 1030 cm−1 (Fig. 2e). GPC analysis of SKL before and after demethylation proved that a rather gentle reaction occurred without dramatic degradation of the lignin. Although the apparent peak molecular weights (Mpeak,app.) remained constant at 2.4–2.5 kDa, Mw,app. changed from 6.0 kDa to 3.6 kDa when the fractionated and demethylated samples were compared (Fig. 2d). During the 6 h demethylation reaction, it was anticipated that other ether connectivities in the lignin structural network, including β-O-4′ and β–β linkages will also be partially cleaved (Fig. S5 and S9†).60 Considering the rather similar GPC traces, these side reactions were apparently not dramatic and partial splitting of the above-mentioned bonds would improve the conformational dynamics in the “nanogel”, probably endowing the lignin molecules with better reactivity. The demethylation of lignin was followed conveniently by 1H NMR spectroscopy, using global acetylation of all the hydroxyl groups for quantitative hydroxy-pattern analysis.97 After demethylation, the content of aromatic hydroxy groups per gram lignin increased from 2.8 ± 0.3 to 4.4 ± 0.4 mmol g−1 (Fig. 2b). This indirectly suggests that roughly 1.6 mmol catechols was added per gram lignin, which would ideally be converted to quinone moieties in the subsequent oxidation step.
Consistently, the integral intensity from the characteristic resonance of the aromatic methoxy groups at 3.67 ppm was significantly reduced and the quantitative analysis against an internal standard revealed ∼1.4 mmol demethylated G-units per gram lignin. Considering the heterogeneity of the lignin functionality network, a comparable value was confirmed by quantitative 31P NMR spectroscopy analysis of the lignin after demethylation (Fig. 2c), where an average number of 4.2 ± 0.6 aromatic OHs was determined.98 Both NMR methods confirmed that the lignin primarily consists of structural G-units. Considering the apparent average molecular weight of Mw,app. = 3600 g mol−1 of the demethylated lignin and the ideal oxidation of all the catechol-units, theoretically six quinone entities per average lignin molecule could be expected. Additionally, according to the qualitative HSQC analysis, the disappearance of the resonances corresponding to the β-O-5′ and β–β connectivities in the demethylated lignin was observed (Fig. S9†). The splitting of these ether linkages would even further increase the possible number of quinones.
In the context of green chemistry, it is worth mentioning that enzymatic or water-based demethylation protocols offer perspectives to potentially replace the chemical demethylation step in the future.99,100 Sodium periodate proved to oxidize catechols in solution effectively to quinones and the application of periodate to non-modified lignin has been intensively studied.101,102 Green alternatives, including enzymatic oxidation utilizing fungal lignin peroxidases or laccases have been reported.103–105 Nevertheless, periodate oxidation promises excellent scalability and the electrochemical synthesis route for sodium periodate makes the oxidant an attractive green option.106 The oxidative generation of quinones in raw, non-demethylated lignin commonly suffers from a low abundancy of catechols.102,107,108 Although periodate was proven to be capable of oxidatively cleaving ortho-methoxyphenols to generate quinones without a distinct prior demethylation step, this reaction requires elevated temperatures and is categorized as low-yielding and slow compared to catechol oxidation.109 The demethylated lignin was readily soluble in N,N-dimethylformamide (DMF) and at least 30 wt% of sodium periodate was required to perform the oxidation for 2 h at room temperature. The reaction mixture was quenched by precipitation in water and extensive washing steps ensured that all the soluble byproducts were rinsed off, isolating 81% of the lignin as a salt-free material. The effective oxidation of the catechol moieties in the lignin was suggested by FTIR spectroscopy.
After oxidation, the lignin exhibited a significant increase in the absorption band at ν = 1650 cm−1, which corresponds typically to the vibrations of conjugated ketones (Fig. 2e). As expected, the appearance of quinone vibrations was accompanied by an obvious decrease in the intensity of the band at ν = 1200 cm−1, which is typically assigned to phenolic OH-groups. A direct quantitative determination of the quinone number (QN, quinones per gram lignin) will be technologically very useful but remains analytically challenging. Based on 1H NMR analysis of the demethylated lignin prior to oxidation, up to 1.6 mmol of catechol per gram lignin is present, which will ideally be oxidizable to quinones. However, it should be noted that the quantitative oxidation of catechols to quinones may be limited by (i) accessibility to catechols located in the interior of the compact lignin network, and more importantly (ii) chemical side reactions such as “dimerization” by quinones reacting with catechols, as has been found in DOPA-rich mussel foot proteins forming “diDOPA” crosslinks.70 In addition, sodium periodate may partly cleave the lignin network connectivities, as prominently observed under harsher conditions.110,111 It can be speculated that the opening of the intramolecular network bonds leads to increased accessibility to the interior catechol entities by periodate to also improve the downstream addition of thiols to quinones.
The chemical manipulations may have effects on the thermal stability of the lignin, which determines a constraint for the curing conditions. Thermogravimetric analysis (TGA) of each modification step was performed, showing the network stability of different lignin derivatives (Fig. S18†). Independent of the type of functionalization, the 5% decomposition temperature was found at around 150 °C, showing that neither the ICH ether splitting nor the periodate oxidation dramatically jeopardized the network stability. As the fractionated lignin, both chemical derivatives reached the maximum decomposition rates at temperatures of about 400–450 °C, which was accompanied by a second mode of decomposition occurring above 500 °C. The initial decomposition is consistent with the literature and related to the post-network consolidations, eliminating water and volatile organic components.112 The second stage led to about 40% weight loss regardless of the chemical modification. The maximum is within the range of previous studies and could be assigned to depolymerization and fragmentation, releasing aromatics, carbonyls, alkyls and CO2.112,113 The final degradation mode was related to carbonization and the loss of condensed aromatics.114 The onset degradation temperatures of all the processed lignin samples are well above the anticipated 2K adhesive curing temperatures of 60–90° C. Thus, the oxidized lignin material was investigated for its use in a 2K adhesive system, involving the formation of thiol-catechol connectivities (TCC) as an internal crosslinking method.
To test the viability of lignin as one component in the 2K adhesive system, a model reaction with ethanethiol was conducted in solution according to the previously described method.93N-Methyl-2-pyrrolidon (NMP) readily dissolved the activated lignin, enabling the Michael-type thiol-addition to proceed in solution. Due to the utilization of lignin as a multi-quinone component, the reaction conditions had to be adapted from a previously described polyaddition process, involving low-molecular-weight bisquinone-type monomers.
Lignin is functionally and molecularly highly dispersed in nature, making the determination of its individual functional groups challenging. However, 1H NMR spectroscopy provided indirect evidence of the formation of TCC in the product of the model reaction. Prior to the quantitative analysis, the lignin derivatives were isolated from NMP and residual thiol was carefully removed by multiple precipitation and washing steps. The spectra of the reaction product revealed a distinct resonance at 1.31 ppm, which was assigned to the H3C-group of an ethanethiol derivative. The broadening of the resonance suggested that the thiol added to the lignin polymer, and the use of an internal standard enabled the approximation of 0.6 mmol of thiols per gram lignin (Table S5†). It should be noted that non-activated lignin did not react with ethanethiol under the given conditions.
Analyzing the hydroxyl pattern in the lignin structure after the addition of thiol by 31P NMR revealed the presence of 3.0 ± 0.5 mmol of phenolic OH-groups per gram of lignin. Accounting for the error of the analytical method, the determined value was in the related range of 4.2 ± 0.6 mmol aromatic OH-groups per gram lignin found after demethylation. This suggests that large amounts of phenolic hydroxyl groups were preserved throughout the process-steps of oxidation, desalting, thiol-addition and washing. Consequently, the lignin-derivatives retained the ability to form H-bonds, which potentially serve as one of the important modes of wet-adhesives to achieve adhesion and cohesion. With the activated lignin, exhibiting an increase in Mw,app. to 6000 g mol−1 (GPC), an average number of 20 aromatic OH-groups and 4 TCC linkages per lignin molecule might be estimated. Nevertheless, the authors would like to emphasize that the solution reaction serves as a model and achieving these conversions under bulk conditions as used for the 2K strategy is probably difficult with polymeric multi-thiols.
The 2K adhesive system
The activated lignin presented multiple quinone functionalities per average molecule, and consequentially polymerization with di-thiols lead to the formation of a resin. Considering the application of materials as a structural (underwater) adhesive, a polymer resin will be ideal, providing room for sufficient cohesion. Thus, a 2K strategy was developed, switching from solution polymerization to bulk conditions as a commonly established route in adhesive technologies.However, liquid multi-thiols were required to facilitate the formation of a homogenous mixture under solvent-free conditions with the demethylated, oxidized lignin (DoxL) given that it was a solid powder, where its decomposition occurred before melting. Two different liquid multi-thiols having a three or tetra arm star topology appear to be suitable. Both, the polar poly(ethylene oxide)-based, trivalent thiol ethoxylated trimethylolpropane-tri (3-mercapto-propionate) (ETTMP) with Mn = 1300 g mol−1 and the non-polar poly(ε-caprolactone)-based tetravalent thiol polycaprolactone-pentaerythritol-tetra (3-mercapto propionate) (PCL4MP) having Mn of 1350 g mol−1 could be homogeneously mixed with DoxL. It was anticipated that the hydrophobic PCL4MP may stabilize the resin network, particularly when used for underwater applications. Although the hydrophilic may enhance the wettability of the adhesive, it reduced its water resistance by its unfavorable capability of swelling.
The 2K adhesive mixtures were denoted as DoxLx/ETTMPy and DoxLx/PCL4MPy, where x and y correspond to the weight fraction (wt%) of each component. A remarkably broad mixing window was found, reaching a lignin weight fraction of 60 wt%. Above these compositions, no homogenous binary mixtures were obtained without the addition of further additives and below the 40 wt% threshold, the viscosity of the mixtures was too low to be applicable. In the lignin content range of 40–60 wt%, both multi-thiols lead to the formation of homogeneous and spreadable pastes for every tested composition. Depending on the multi-thiol and DoxL solid content, the paste viscosity changed from a viscous fluid with 40 wt% DoxL to a ductile solid with 60 wt% DoxL (Fig. 3a). After high shear mixing of both components at room temperature with a bladeless SpeedMixer, the paste was easily applied and spread on pre-treated, degreased aluminum substrates, which were then glued together. The two substrates were fixed with office paper clamps, and subsequently curing of the glue was done in a standard dry application setting overnight at 60 °C (Fig. 3b). Curing resulted in obvious changes in physical properties, leading to hardened non-recoverable materials with a rubber-like appearance. Calorimetric curing tests with differential scanning calorimetry (DSC) showed for both multi-thiols, i.e., 40 wt% DoxL, the maximum curing rate occurred at about 105 °C (Fig. S20†). The apparent onset temperature of the exothermic curing reaction was found to be between 30 °C for the ETTMP- and 50 °C for the PCL4MP-based 2K mixtures. Interestingly, only the DoxL/PCL4MP blends showed an obvious 1st-order endothermic phase transition at 30 °C, which also occurred with the same intensity, fully reversibly in the PCL4MP bulk. The SAXS measurements indicated the presence of a liquid crystalline structure due to the broad reflex, which was also observed via polarization microscopy in the form of optical birefringence at 20 °C, which disappeared at 60 °C.
 | ||
| Fig. 3 Two-component (2K) adhesive systems resulting from a bulk reaction of the activated lignin with a set of different multi-thiols. The demethylated oxidized lignin (DoxL) is mixed with multi-thiols at different weight ratios (from 40/60 to 60/40 of DoxL/thiol) using a speed mixer at room temperature to yield a homogeneous viscous adhesive mixture. Representative photograph of DoxL40–60/ETTMP60–40, in which the viscosity of the mixtures increases with an increase in the weight fraction (wt%) of DoxL (a). The 2K adhesive mixtures are easily applied and spread as pastes on aluminium substrates at r.t. and fixed with clamps to be cured overnight at 60 °C. Cured samples are tested in lap shear settings to determine their adhesive strength at failure (b). Representative FTIR spectra showing distinct changes in functionality by comparing activated lignin, with the not cured 2K mixtures (nc) and those after curing for DoxL50/ETTMP50 (c) and DoxL50/PCL4MP50 (d). | ||
However, the most important fact for their applicability is that both homogenized, non-cured glue mixtures preserved their processability at room temperature for several days without showing separation or significant increase in viscosity.
The FTIR spectroscopy analysis enabled a comparison of the pure DoxL with both mixtures of the 2K adhesive systems before and after curing (Fig. 3c, d and Fig. S23†), respectively. The FTIR spectra of representative mixtures with a DoxL content of 50 wt% provided insights into the curing chemistry. The stretching vibration for conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O appeared in the activated lignin at 1650 cm−1 and this characteristic band of quinone structural elements disappeared in both 2K mixtures after curing. Although it remains challenging to directly prove the formation of TCC, the loss of the typical bands of quinones is consistent with the formation of TCC-Michael-addition products. The consumption kinetics of quinones is expected to depend on the local thiol concentration and/or on conformational dynamics of the polymer multi-thiols in the bulk reaction mixtures. However, for both multi-thiols, the quinone bands in DoxL were not reduced or at least not fully reduced prior to curing, which suggests that the reaction is slow in the viscous bulk at room temperature. In addition, the characteristic ETTMP ether and ester νC–O bands at 1090 cm−1 and νCO at 1730 cm−1 as well as the νCO stretching vibration at 1730 cm−1 typical of PCL4MP were observed to not decrease in intensity in the DoxL mixtures during curing, indicating the absence of severe degradation (Fig. 3c).
O appeared in the activated lignin at 1650 cm−1 and this characteristic band of quinone structural elements disappeared in both 2K mixtures after curing. Although it remains challenging to directly prove the formation of TCC, the loss of the typical bands of quinones is consistent with the formation of TCC-Michael-addition products. The consumption kinetics of quinones is expected to depend on the local thiol concentration and/or on conformational dynamics of the polymer multi-thiols in the bulk reaction mixtures. However, for both multi-thiols, the quinone bands in DoxL were not reduced or at least not fully reduced prior to curing, which suggests that the reaction is slow in the viscous bulk at room temperature. In addition, the characteristic ETTMP ether and ester νC–O bands at 1090 cm−1 and νCO at 1730 cm−1 as well as the νCO stretching vibration at 1730 cm−1 typical of PCL4MP were observed to not decrease in intensity in the DoxL mixtures during curing, indicating the absence of severe degradation (Fig. 3c).
The cured adhesive samples were additionally analyzed by solid-state 13C magic-angle-spinning (MAS) NMR using both C–C-insensitive nuclei enhancement by polarization transfer (C–C INEPT) and H–C cross polarization (H–C CP) experiments, which revealed the mobility differences in the carbon-atoms in the adhesive material network (Fig. S24 and S25,† respectively). Although quantification remains difficult, the solid-state NMR spectra analysis revealed several interesting aspects, as follows: (i) the comparison of the resonances of the aliphatic PCL chains in the INEPT spectra of the pure PCL4MP before and after mixing showed an evident decrease in intensity. This suggests the considerable loss of the PCL segment mobilities, which is expected for a product of a crosslinking reaction. Similarly, (ii) in the HC CP spectra of DoxL/PCL4MP with 40 wt% resonances of Cα- and Cβ-atoms neighboring the thiol functionality appeared, confirming the conformational arrest of this prior to curing the flexible PCL chain ends. Indirectly, this is consistent with covalent bond formation with the thiol, leading to a significant decrease in the mobility. The resin-forming crosslinking reactions of two polymeric constituents is not expected to be determined in a quantitative manner. This could be found as (iii) the presence of resonances of the Cα- and Cβ-thiol PCL chain ends in both the HC CP and INEPT spectra. This suggests the presence of two types of chain ends after curing. Although one preserved the conformational dynamics of the unreacted thiol groups, the second species seemed to be involved in crosslinking and had strongly reduced mobility. Given that the 13C MAS NMR spectra are only qualitative, no exact ratio of free vs. bound thiols could be deduced. In addition, the weak carbonyl signal of the conjugated systems disappeared (180 ppm) and a new peak appeared at around 23 ppm, indicating the formation of a thioether bond.
Investigation of adhesive properties under dry conditions
To correlate the composition of both 2K systems with their performance and to elucidate the achievable bond strength, a set of DoxL40–60/ETTMP60–40 and DoxL40–60/PCL4MP60–40 blends were formulated. The resulting material properties were investigated in a dry application by bonding pretreated aluminum substrates (cleaned and degreased). The optimal ratio of the two components in the adhesive systems should primarily depend on the thiol/quinone stoichiometry. However, parameters such as homogeneity and mixability of the powder/liquid mixtures as well as processability of homogenized adhesive pastes may directly influence their application on the substrate surface. Fig. 4a, b and Table 1 presents a summary of the results of the dry adhesion tests. As expected, the feed ratio of DoxL and multi-thiols influenced the obtained adhesive strength and a maximum of about 15 MPa was reached at 55 wt% DoxL, regardless of the use of hydrophilic or hydrophobic multi-thiols. Considering the quinone number from demethylation analysis by 1H NMR, the optimal quinone/thiol (Q/T) ratio was practically fulfilled by DoxL55/PCL4MP45, reaching the ideal stoichiometry within the error of the analysis (Q/T = 1.00/1.52). In contrast, the DoxL55/ETTMP45 adhesive using ETTMP tri-thiol deviated from the ideal stoichiometry with a Q/T ratio of 1.00/1.18. It should be noted that the curing process appears to be remarkably robust with respect to the equivalents of reactive functionalities. This advantage may potentially be rationalized by the character of the lignin multi-quinone. The “nano-gel”-type may be capable of introducing certain buffering capability, where the internal quinone functionalities might not necessarily have to react. In addition, the excess of thiols also appears to be counter balanced. For instance, reducing the DoxL content by 5 wt% resulted in marginally reduced adhesive strengths of 14 MPa for both 2K glues. A more dramatic change in properties was found for the formulations with 60 wt% DoxL, where DoxL60/PCL4MP40 approached the border of homogeneous mixing, and thus exhibited 55% lower adhesive strength. It should be noted that the 2K systems studied are minimal blends of two components and advanced formulation strategies can potentially increase their DoxL content and performance by using viscosity modifiers, plasticizers, primers and wetting agents. | ||
| Fig. 4 Dry adhesive properties of the lignin-based 2K systems gluing aluminum substrates. Lap shear tests shown as a force–distance curve with a representative example for DoxL/ETTMP (a) and DoxL/PCL4MP (b) at varying weight ratios (wt%) of DoxL/thiol. Representative images of sample rupture in the shear adhesive tests using DoxL55/ETTMP45 (c) and DoxL55/PCL4MP45 (d). | ||
| a Quinone number derived from demethylation analysis of 1H NMR. |
|---|
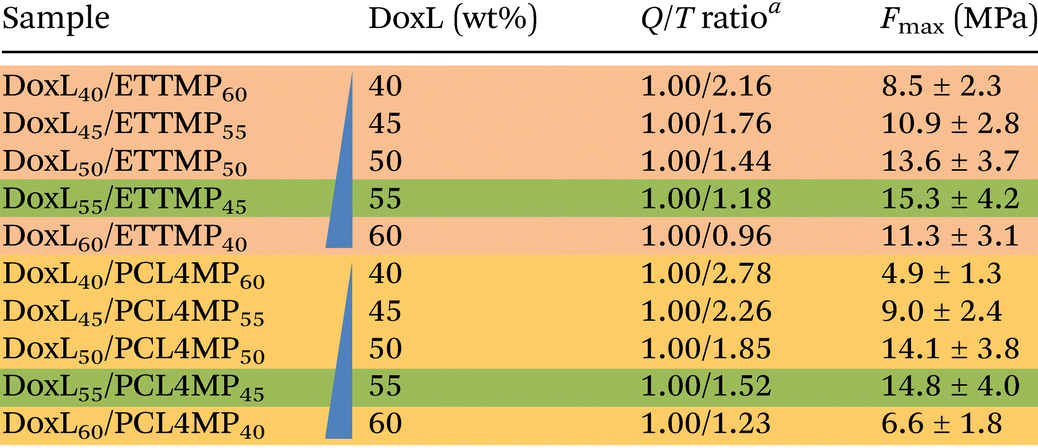
|
The dry performance of both 2K adhesives based on lignin was found to be in the range of structural adhesives. With a strength of 15 MPa, their adhesive performance surpasses some available commodity adhesives and is located at the onset of the regime for professional adhesives (Fig. S27–30†).115 Interestingly, both 2K systems, DoxL55/ETTMP45 and DoxL55/PCL4MP45, showed a mixed mode of adhesive and cohesive failure upon rupture (Fig. 5d, e and Fig. S32†). This highlights both the capability of the adhesive material to offer suitable functionalities, installing a proper adhesive interface in the substrate, and constituting a network with sufficient strength to reach suitable bulk stability. The adhesive strength was determined by lap shear tests, using specimens of 25 mm width but an overlap of 3.0 to 5.0 mm as determined for each sample. It should be noted that the overlap deviated from the DIN EN 1465 experiments116,117 due to the limitations of the measurement apparatus.
 | ||
| Fig. 5 Adhesive stability testing by solvent leaching. Scheme of leaching test procedure of the 2K system with DoxL40/thiol60 (a). Final non-leached content for each 2K adhesive system after the leaching tests in acetone (green) and THF (red/orange) (b). | ||
Thermal stability and solvent leaching studies
After curing of the different sets of DoxL40–60/ETTMP60–40 and DoxL40–60/PCL4MP60–40, the isolated polymer resin materials were characterized by TGA and DSC. Unexpectedly, both material families showed no dramatic differences in thermal decomposition behavior, despite the fact that two rather different multi-thiol components with polyether or polyester segments were employed (Fig. S16†). The materials approached 5% weight loss at about 250 °C and the clear maximum of the decomposition rates was found around 400 °C. Obviously, the lignin constituents stabilized the polymer matrix against thermal degradation, which was most evident for the cured DoxL/PCL4MP, having PCL segments that usually decompose as homopolymer starting at 300 °C.118 Although the main weight loss in the cured DoxL/PCL4MP materials occurred in a rather broad temperature range between 300–500 °C, the cured DoxL/ETTMP material showed a two-stage decomposition with minor 15% weight loss at 300 °C followed by 40% weight loss at 390 °C.As expected, the DSC thermograms of DoxL40–50/ETTMP60–50 and DoxL40–50/PCL4MP60–50 proved that the cured materials were fully amorphous given that no melting phase transition was observed (Fig. S19†). DoxL/ETTMP show more pronounced glass transition temperatures (Tg) at 5 °C and 10 °C for DoxL contents of 40 wt% and 50 wt%, respectively. In contrast, the cured DoxL/PCL4MP showed reduced heat capacity glass transition temperatures at −30 °C and −20 °C for DoxL40/PCL4MP60 and DoxL50/PCL4MP50, respectively. Apparently, the lignin content consistently affected the Tg, which slightly increased. This reduction in polymer segment relaxation can be explained by the increased crosslinking of the polymer chain ends and/or by non-covalent interactions, where lignin complexed polyethyleneoxide or polycaprolactone segments.119,120
The network constitution of a cured 2K adhesive resin is generally challenging to be assessed analytically. Thus, a set of leaching tests was conducted to quantify the fraction of non-covalently attached polymers that can be extracted from the resin bulk. According to the linear thiol-quinone Michael polyaddition reactions, it was established that disulfide bridges may be formed mechanistically, but occur only as a marginal side-reaction, while TCC-functionalities are dominant in the resulting polymer.121 However, the lignin-based multi-quinone may react differently in the 2K bulk reactions. Indirect insight into the types of crosslinks that constitute the resin network was obtained by incubating the cured 2K adhesive with organic solvents, both with and without the presence of reductive cleavage agents capable of splitting disulfide bonds (Fig. 5). Given that disulfides would form most prominently under the conditions of high thiol excess, the cured DoxL40/thiol60 samples were investigated in organo-leaching assays. After curing overnight at 60 °C, the materials were evenly frozen, ground and immersed for 7 days in either acetone or tetrahydrofuran (THF) with and without additional tributyl phosphine reducing agent.
Interestingly, comparing the cured DoxL40/PCL4MP60 with DoxL40/ETTMP60, rather similar swelling and leaching behavior was observed. This was quantitatively determined by gravimetric analysis of the amount of solubilized polymer relative to the undissolved adhesive residue (Fig. 5b). In principle, all the components of the 2K adhesive family, i.e., lignin and both multi-thiols, are readily soluble in acetone. Therefore, it is interesting to note that in the absence of the reducing agent, only 16 wt% and 22 wt% of DoxL40/ETTMP60 and DoxL40/PCL4MP60, respectively, were leached. Moreover, these dissolvable fractions increased only marginally by 5–9 wt% in the case where the disulfides were actively cleaved. THF swelled both 2K adhesive resins even better than acetone, as reflected in the increased leaching. However, when tributyl phosphine was added to THF, the leaching increased similarly by a marginal 5 wt%. Therefore, disulfide bonds were apparently not present to be cleaved even in a well swollen, accessible state. This highlights the fact that disulfide-bonds were not dramatically dominant in the chemical network and further suggests the importance of TCC-bonds in constituting the cured DoxL40/PCL4MP60 and DoxL40/ETTMP60 adhesive resins.
The corresponding supernatants from the leaching experiments of DoxL40/PCL4MP60 were investigated by UV/vis and 1H NMR spectroscopy. The typical UV/vis absorption bands of the extended aromatic systems of lignin suggest the dissolution of the lignin-containing resin parts in both acetone and THF leaching experiments. No dramatic changes in the absorption band structures were found in the UV/vis spectra when comparing the extracts with or without tributyl phosphine (Fig. S26†). 1H NMR analysis of the leaching extracts with acetone and THF enabled the rough estimation of the composition of DoxL and multi-thiols by comparing the characteristic resonance intensities of the extracts with non-reacted DoxL and PCL4MP mixtures (40/60 wt%). Interestingly, a similar composition ratio of approximately 20/80 wt% (DoxL/PCL4MP) was estimated for both leaching extracts with acetone and THF, which only changed marginally to 15/85 wt% in the case of leaching with solvents, where the reductive disulfide cleavage agent was present. The higher content of PCL4MP in the leaching extracts was expected, considering the Q/T ratio of 1:2.78 present in the DoxL40/PCL4MP60 mixtures. Obviously, even under these non-ideal conditions, the residual non-reacted thiol chain ends may lead to the oxidative formation of disulfide potentially by air or quinone reduction. However, these linkages do not dramatically constitute or dominate the network integrity of the 2K resins, given that reductive cleavage would jeopardize the network stability more significantly.
Underwater application and saltwater tolerance of 2K adhesives
Depending on the type of adhesive the cured resin materials, DoxL40/ETTMP60 and DoxL40/PCL4MP60 showed significant differences when exposed to water. Although both adhesives maintained their integrity even after 7 days of direct water contact, the hydrophilic DoxL40/ETTMP60 adhesive showed severe swelling (Fig. S33†). Considering their underlying components, the ETTMP network in DoxL40/ETTMP60 is expected to enable water uptake. This network remained fully intact within the error of the experiments. Gravimetry in the leaching experiments revealed that with either deionized water or sea water model solution, the material extraction was only 2–3 wt% from the cured DoxL40/ETTMP60 materials (Fig. 7a). However, the swelling of the resin network was accompanied by volume changes, weakening the bulk cohesion and introducing shear-stress at the adhesive interfaces. Therefore, further studies focused on the hydrophobic DoxL/PCL4MP adhesive. As expected, the cured DoxL40/PCL4MP60 showed no obvious swelling, suggesting the very good stability of this adhesive in the environment. During the leaching experiments of the cured material with either water or sea water equivalents, practically no material losses were evident by gravimetry and UV/vis spectroscopy only showed the minor adsorption of the supernatant solution at 210 nm (Fig. S26†).To investigate the applicability and achievable stability under practical underwater conditions, a set of three different application and curing scenarios was studied. When the adhesive was applied to aluminum substrates under dry conditions, followed by curing in an oven at 60 °C for 12 h, and then immersed in salt water (599 mM salt concentration) for 7 days, the lap shear tests showed that the DoxL/PCL4MP ratio of 40/60 provided the highest adhesion strength of 3.7 ± 0.9 MPa (Fig. 6a). Increasing the amount of DoxL in the 2K mixture led to a drastic decrease in adhesion strength. This can be rationalized by the fact that the increase in DoxL decreases the PCL4MP content, consequently reducing the hydrophobicity of the adhesive and making it more susceptible to ion and water uptake, which may act as plasticizers.
 | ||
| Fig. 6 Underwater application of the 2K system composed of DoxL and PCL4MP using different curing protocols to glue aluminium substrates and analysis by lap shear tests. Dry application and dry curing followed by immersion under salt water for 7 days (a), dry application followed by curing under salt water for 3 days at 40 °C (b) and wet application and curing under saltwater for 3 days at 40 °C (c). Images of fracture patterns after lap shear test of DoxL/PCL4MP with 40/60, 50/50 and 60/40 wt% ratio after underwater application & curing (d). (Conditions: aluminium substrates, seawater model with 599 mM NaCl salt concentration). | ||
Following these promising initial results, the dry application to substrates and subsequent curing of the adhesive in a water bath under saline conditions at 40 °C was investigated. Due to the onset temperature of curing under dry conditions of approximately 50 °C, the curing time had to be extended to 3 days. Consistent with the initial study, the DoxL40/PCL4MP60 mixture resulted in the highest adhesion strength, reaching 2.8 ± 0.2 MPa (Fig. 6b). Considering that the transfer of the entire curing chemistry to salt water is a drastic change in conditions and the curing temperature was set much lower than ideal, the achieved adhesive strength in marine environments is in a suitable window and meets the same regions as epoxy putty glue that is optimized for aquaristic reef building applications (3.8 ± 0.4 MPa) (Fig. S31†). The most demanding protocol involved direct application and curing under model saltwater conditions. The premixed DoxL40/PCL4MP60 was applied underwater to submerged aluminum substrates, and finally cured at 40 °C for 3 days. Only a marginal decrease in adhesion strength was observed for the most promising DoxL40/PCL4MP60 system, which reached 2.7 ± 0.5 MPa (Fig. 6c). It should be noted that wet substrates and a salt content of 599 mM present major challenges for some adhesives, given that the adhesive interface is difficult to be installed effectively. However, the presented DoxL40/PCL4MP60 adhesive was less affected by the harsh conditions given that the dry application/water curing reached the same adhesive strength as when both steps were performed under water. The fracture side analysis enabled to categorize the failure of DoxL40/PCL4MP60 bonded aluminum as the cohesive mechanism, regardless of the used saltwater application protocol (Fig. 6d). Interestingly, an increase in lignin content shifted the fracture mechanism toward adhesive failure. The analysis of the aluminum substrates after immersion in saltwater for 7 days revealed significant degradation of the substrate surfaces under the harsh saline conditions. It can be speculated that the lower viscosity of the 2K mixtures with a higher PCL4MP content wetted the substrate surface more homogenously. This may prevent or reduce the formation of micro channels, where saltwater could penetrate to compromise the adhesive interface. Therefore, DoxL40/PCL4MP60 appears to exhibit the best balance between cohesive and adhesive strength due to the improved wettability.
The hydrophobic lignin-based 2K adhesive showed stable and reproducible underwater adhesion as well as resistance to saltwater conditions. Potential applications have to be explored to ultimately extend the use of this bio-friendly adhesive platform to saltwater applications, such as reef shaping or coral reef reforestation through the setting of temperature-resistant corals.122,123
Adhesive applications in aquaristics commonly use cyanoacrylates or epoxy cements to attach corals.124 Professional saltwater adhesives are mainly based on epoxy resins,125 and therefore prone to leaching components of concern, potentially harming the fragile oceanic ecosystem.126
Thus, the direct toxicity of the DoxL40/PCL4MP60-based adhesive was investigated by determining its acute toxicity according to the DIN 15088 fish egg test.127 This method provides a critical concentration, which is a measure of the toxic effect of an effluent on fish eggs within 48 h. Cured and non-cured samples of DoxL40/PCL4MP60 were intensively ground, and then dispersed in standardized water.
After 7 days of leaching, the solids were filtered off, and the filtrates were used as the basis for the acute toxicity tests. During the tests, the fish eggs were incubated separately in the non-diluted (G1) and in 1:1 diluted extracts (G2). These tests indicated that studies at higher dilutions are not required, given that the acute toxicity of the G1 extracts was found to be below the statistical threshold (Fig. 7b). After 48 h, the tests were finalized by determining the number of dead eggs. The tests provide the lowest dilution (Gegg) value at which at least 90% of the eggs survived, and thus the critical G value is correlated with the toxicity of the adhesive extracts under marine conditions. Fig. 7b clearly shows the absence of significant acute toxicity for the tested adhesives. Even without dilution (G1) of the extracts of the cured and non-cured DoxL40/PCL4MP60 materials, no negative effect on the development of the fish embryos was observed. These results highlight that the investigated lignin system exhibits remarkable biocompatibility and fulfills standard ecotoxicity guidelines for use in marine environments.
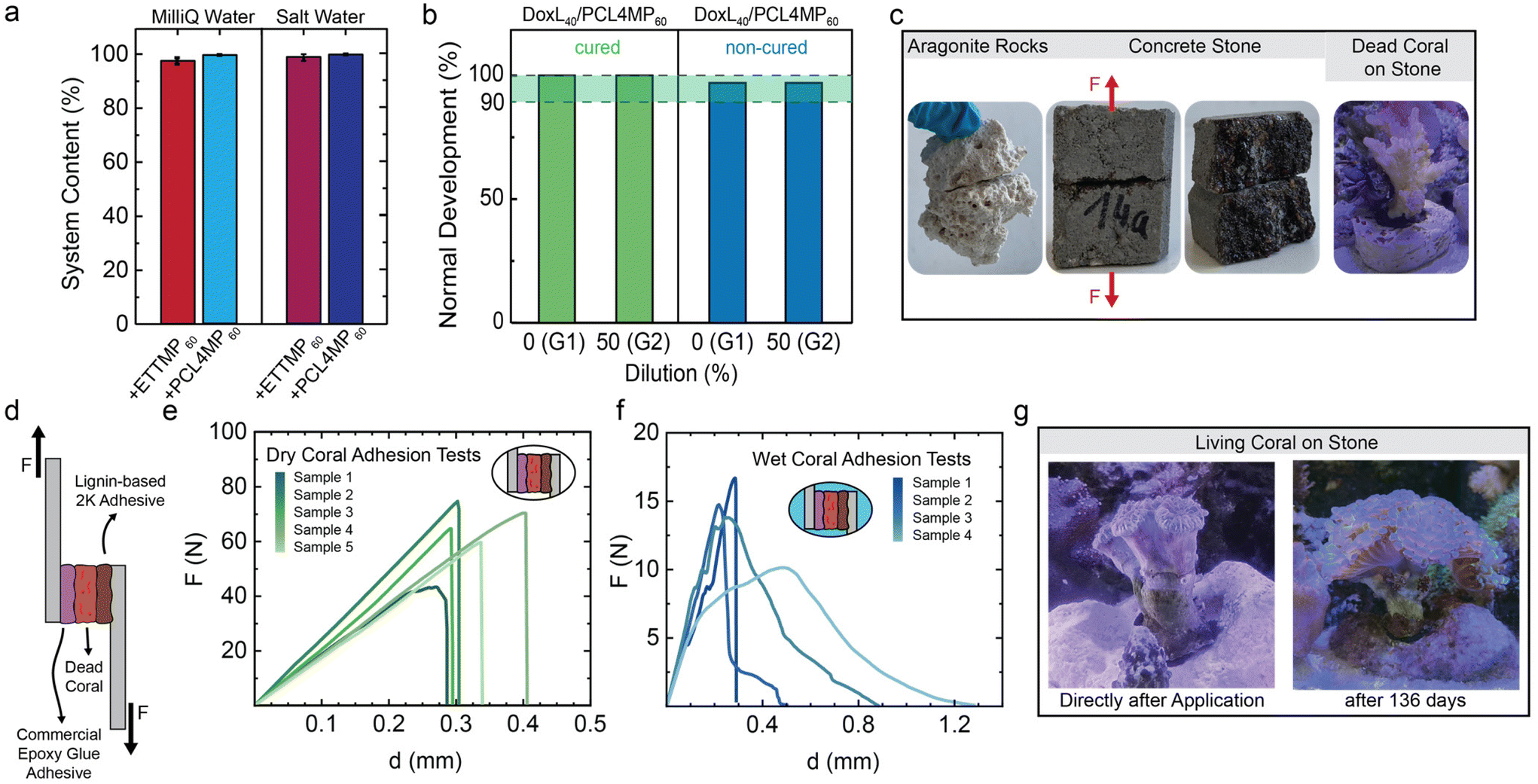 | ||
| Fig. 7 Properties of DoxL/PCL4MP adhesives for gluing under salt water conditions. Recovered solid content of 2K adhesive after leaching in water or salt-water (a). Toxicity tests on breeding fish eggs in DIN water that was mixed with cured (green) and non-cured (blue) adhesives for 7 days. The percentages of surviving eggs correlated to no dilution or 1:1 dilution with clean DIN water (b). Dry application of the DoxL40/PCL4MP60 system on different rough surfaces and underwater curing under saline conditions at 23 °C (c). Schematic representation of lap shear experiments, using dead coral as the substrate for adhesion to two aluminium substrates using a commercial epoxy glue on the left side and DoxL40/PCL4MP60 on the other substrate (d). Lap shear test results from the adhered Al-Coral-Al substrates without water contact (e) or after immersion of the system in saline conditions for 7 days at 40 °C (f). Image of living coral after direct placement and after 136 days (g). | ||
Bonding of natural features is always challenged by rough and irregular surfaces, which require adhesives with pronounced filling properties. This was demonstrated with initial adhesion tests by employing DoxL40/PCL4MP60 to constitute an adhesive joint between rocks, which is useful for reef shaping applications. Fig. 7c, left shows a representative example of aragonite rocks bonded with DoxL40/PCL4MP60 and cured in artificial seawater at 23 °C for 14 days. The construct was capable of maintaining its adhesive integrity for several months, even with direct exposure to harsh saltwater conditions and simulated currents inside a reef demonstration tank as a functional ecosystem.
Further quantitative insight was obtained by gluing fractured non-standardized concrete stones with DoxL40/PCL4MP60 adhesives, using the saltwater curing protocols and water from the demonstration tank. Underwater curing at 70 °C for 1 day established adhesive joints showing rupture adhesion forces of 380 N, whereas curing at 40 °C (4 days) reduced the force slightly to 300 N, and 30 °C (7 days) resulted in 160 N (Fig. 7c, middle and Fig. S34†). Given that the temperature of marine coral ecosystems is usually around 23 °C,128 the curing of the concrete specimens at 23 °C after 14 days gave an adhesive force of 106 N. Remarkably, dry application protocols for gluing concrete specimens led to bond lines that withstood forces of 1000 N, which corresponds to 100 kg load without breaking. It should be noted that the adhesive used is still a non-optimized, pure two-component system, which is expected to be improved by advanced formulation strategies. However, the results highlight its potential, given that the adhesive joint was capable of holding a mass of around 10 kg after curing under harsh saltwater conditions.
Thus, the underwater application of the DoxL40/PCL4MP60 2K adhesive was extended to successfully and firmly bond non-living corals to rocks directly inside a reef demonstration tank (conditions: 23 °C for 14 days, Fig. 7c, right). Apparently, sufficiently strong adhesive joints were formed to hold the corals firmly in place. Fracture during the tensile tests occurred at approximately 70 N and resulted in failure at the body of the coral but not at the adhesive interface between the coral and stone.
The experimental setup was expanded to include bonding of non-living corals in modified lap shear settings. Firstly, a piece of non-living coral was dry-bonded on one side of an aluminum substrate using a commercial 2K epoxy putty adhesive for saltwater applications. After careful curing according to the manufacturer's instructions, the supported coral construct was also bonded to the aluminum substrate on the other side using DoxL40/PCL4MP60 2K (Fig. 7d). Dry curing at 60° C led to lap shear tests that reaching about 63 ± 12 N. Interestingly, failure occurred either in the coral structure or on the epoxy adhesive side, suggesting that the prepared DoxL-based 2K joint was stronger (Fig. 7e). Using the epoxy-bonded non-living corals for the saltwater application of DoxL40/PCL4MP60 and curing at 30 °C for 7 days resulted in the lap shear tests failing in an ill-defined manner. Fracturs were observed by adhesive rupture at each of the two different bond sites. Fig. 7f shows the representative fracture sites and lap test curves that reached about 14 ± 2 N. These results indicate that under the challenging conditions of saltwater application and underwater curing, some of the resins were not fully cured.
For the ultimate test of the DoxL40/PCL4MP60 system as a coral adhesive, a living Fimbriaphyllia ancora coral was subjected to an underwater gluing trial in a reef demonstration tank (Fig. 7g). During the curing process, the coral defensively withdrew its polyps and excreted a secretion that shielded its surface from microbial growth and fouling. At the two-hour mark, the polyps of the coral were observed to expand and were nearly entirely extended 6 h after application (Fig. S36†). Simultaneously, it was clear that the bonding between the coral and the rock remained intact despite the harsh saltwater conditions, currents, and crustaceans climbing over the coral, necessitating extra support. Additionally, it was evident that the lignin adhesive did not compromise the delicate ecosystem of the reef. The coral that was glued remained in place for 136 days and was able to grow naturally.
Conclusions
Herein, we demonstrated that readily available lignin, which in the past has most prominently been used as a material for heat generation, could be converted into a very promising material platform by straightforward and easy modification. By demethylating the G-units in lignin using iodocyclohexane, we not only opened up the internal lignin structure by reducing its intramolecular connectivities, but also introduced around 1.6 mmol g−1 of new catechol motifs. These catechols could then be oxidized using NaIO4 to generate reactive quinone groups inside the macromolecule. By combining these reactive species in an almost 1/1 (ETTMP or PCL4MP) weight ratio with tri- or tetra-functional thiols, we generated a 2K adhesive system, which exhibited extraordinarily high adhesion strengths of up to 15 MPa under dry conditions. By simply varying the lignin/thiol ratio, we developed a 2K adhesive system, which was ultimately used under maritime conditions and could be applied to glue corals onto rocky surfaces. During the development process, we demonstrated that the adhesive system is very robust. Dry and underwater applications as well as curing under saltwater conditions were demonstrated. Furthermore, the compatibility of the 2K system with the maritime biological environment was confirmed by acute toxicological investigations. The material platform developed herein combines activated lignin with multi-thiols, leading to the formation of TCC functionalities to constitute a highly robust and widely usable adhesion system. Further investigations may explore variations of green multi-thiols and optimize the basic 2K system by developing formulation strategies to improve the wettability, maximize the lignin content and broaden the applicability of the present adhesive platform.Author contributions
C-YC, FL, TF-K: investigation, experiments, data analysis, writing, correction; SB: experiments, materials analysis, data curation; KW, JF, MH, AD: materials analysis, data curation, methodology development; HO, TM: analysis resources and supervision; AT: scientific supervision; HGB: conceptualization, supervision, funding, resources, validation, writing, reviewing, correcting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The chair acknowledges the financial support by the German Federal Ministry of Education and Research (BMBF) within the project “LigNovolac – Fusing material concepts from wood and mussels” grant No 13XP5150. We thank A. Thünemann (BAM Federal Institute of Materials Research and Testing) for SAXS measurements, and M. Hübsch (UPM, Leuna, Germany) for valuable support with base materials.References
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656–8664 RSC.
- L. Fertier, H. Koleilat, M. Stemmelen, O. Giani, C. Joly-Duhamel, V. Lapinte and J.-J. Robin, Prog. Polym. Sci., 2013, 38, 932–962 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 5, 7393–7420 RSC.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS PubMed.
- B. Erickson and P. Winters, Biotechnol. J., 2012, 7, 176–185 CrossRef CAS PubMed.
- P. M. Foley, E. S. Beach and J. B. Zimmerman, Green Chem., 2011, 13, 1399–1405 RSC.
- P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- L. E. Manzer, Top. Catal., 2010, 53, 1193–1196 CrossRef CAS.
- M. S. Nazir, A. J. Mahdi, M. Bilal, H. M. Sohail, N. Ali and H. M. N. Iqbal, Sci. Total Environ., 2019, 683, 436–444 CrossRef CAS PubMed.
- L. Yang, X.-C. Wang, M. Dai, B. Chen, Y. Qiao, H. Deng, D. Zhang, Y. Zhang, C. M. V. B. de Almeida and A. S. Chiu, Energy, 2021, 228, 120533 CrossRef.
- T. D. Moshood, G. Nawanir, F. Mahmud, F. Mohamad, M. H. Ahmad and A. AbdulGhani, Curr. R. Green Sustain. Chem., 2022, 100273 CrossRef CAS.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- G. Chatel and R. D. Rogers, ACS Sustainable Chem. Eng., 2014, 2, 322–339 CrossRef CAS.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- O. Yu and K. H. Kim, Appl. Sci., 2020, 10, 4626 CrossRef CAS.
- M. Tuomela, M. Vikman, A. Hatakka and M. Itävaara, Bioresour. Technol., 2000, 72, 169–183 CrossRef CAS.
- G. Brunow, in Biorefineries–Industrial Processes and Products, 2005, pp. 151–163. DOI:10.1002/9783527619849.ch21.
- C. I. Simionescu, V. Rusan, M. M. Macoveanu, G. Cazacu, R. Lipsa, C. Vasile, A. Stoleriu and A. Ioanid, Compos. Sci. Technol., 1993, 48, 317–323 CrossRef CAS.
- M. Rixom and J. Waddicor, Spec. Publ., 1981, 68, 359–380 CAS.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- M. Sette, R. Wechselberger and C. Crestini, Chem. – Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- E. A. Crawford, S. Gerbig, B. Spengler and D. A. Volmer, Anal. Chim. Acta., 2017, 994, 38–48 CrossRef CAS PubMed.
- A. K. Sangha, L. Petridis, J. C. Smith, A. Ziebell and J. M. Parks, Environ. Prog. Sustain., 2012, 31, 47–54 CrossRef CAS.
- E. Terrell, V. Carre, A. Dufour, F. Aubriet, Y. Le Brech and M. Garcia-Perez, ChemSusChem, 2020, 13, 4428–4445 CrossRef CAS PubMed.
- A. Agrawal, N. Kaushik and S. Biswas, Sci. Tech. J., 2014, 1, 30–36 Search PubMed.
- W. Liu, R. Zhou, H. L. Goh, S. Huang and X. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 5810–5817 CrossRef CAS PubMed.
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Prog. Mater. Sci., 2018, 93, 233–269 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- X. Guo, S. Zhang and X. Q. Shan, J. Hazard. Mater., 2008, 151, 134–142 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman Jr. and P. H. Steele, J. Colloid Interface Sci., 2006, 297, 489–504 CrossRef CAS PubMed.
- A. Demirbas, Energy Sources A: Recovery Util. Environ., 2007, 29, 117–123 CrossRef CAS.
- Y. Liu and K. Li, J. Adhes., 2006, 82, 593–605 CrossRef CAS.
- P. E. Marriott, L. D. Gómez and S. J. McQueen-Mason, New Phytol., 2016, 209, 1366–1381 CrossRef CAS PubMed.
- S. Sahay, Handbook of Biofuels, Academic Press, 2021 Search PubMed.
- F. Cherubini and A. H. Strømman, Biofuels, Bioprod. Biorefin., 2011, 5, 548–561 CrossRef CAS.
- X. Ge, C. Chang, L. Zhang, S. Cui, X. Luo, S. Hu, Y. Qin and Y. Li, in Advances in Bioenergy, ed. Y. Li and X. Ge, Elsevier, 2018, vol. 3, pp. 161–213 Search PubMed.
- Y. Zhang, Z. Yuan, N. Mahmood, S. Huang and C. C. Xu, Ind. Crops Prod., 2016, 79, 84–90 CrossRef CAS.
- F. Wang, J. Kuai, H. Pan, N. Wang and X. Zhu, Wood Sci. Technol., 2018, 52, 1343–1357 CrossRef CAS.
- Y. C. Chen, S. Fu and H. Zhang, Colloids Surf. A: Physicochem. Eng., 2020, 585, 124164 CrossRef CAS.
- Ł. Klapiszewski, K. Siwińska-Stefańska and D. Kołodyńska, Chem. Eng. J., 2017, 330, 518–530 CrossRef.
- Ł. Klapiszewski, K. Siwińska-Stefańska and D. Kołodyńska, Chem. Eng. J., 2017, 314, 169–181 CrossRef.
- E. Windeisen and G. Wegener, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 255–266. DOI:10.1016/B978-0-444-53349-4.00263-6.
- R. Vanholme, K. Morreel, J. Ralph and W. Boerjan, Curr. Opin. Plant Biol., 2008, 11, 278–285 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- E. K. Pye, in Biorefineries–Industrial Processes and Products, 2005, pp. 165–200. DOI:10.1002/9783527619849.ch22.
- X. Gong, Y. Meng, J. Lu, Y. Tao, Y. Cheng and H. Wang, Macromol. Chem. Phys., 2022, 2100434 CrossRef CAS.
- S. Yang, Y. Zhang, T. Q. Yuan and R. C. Sun, J. Appl. Polym. Sci., 2015, 132, 41727 CrossRef.
- S. Zhao and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2017, 5, 5059–5066 CrossRef CAS.
- S.-F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde and H. Witters, Green Chem., 2017, 19, 2561–2570 RSC.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J. P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- A. Moreno, M. Morsali and M. H. Sipponen, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS PubMed.
- D. Feldman, in Chemical modification, properties, and usage of lignin, Springer New York, NY, 2002, pp. 81–99 Search PubMed.
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- R. Paul, B. John and S. K. Sahoo, Biomacromolecules, 2022, 23, 816–828 CrossRef CAS PubMed.
- K. Sawamura, Y. Tobimatsu, H. Kamitakahara and T. Takano, ACS Sustainable Chem. Eng., 2017, 5, 5424–5431 CrossRef CAS.
- H. Silau, A. G. Garcia, J. M. Woodley, K. Dam-Johansen and A. E. Daugaard, ACS Appl. Polym. Mater., 2022, 4(1), 444–451 CrossRef.
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS PubMed.
- J. H. Waite, J. Exp. Biol., 2017, 220, 517–530 CrossRef PubMed.
- M. Schauperl, M. Podewitz, B. J. Waldner and K. R. Liedl, J. Chem. Theory Comput., 2016, 12, 4600–4610 CrossRef CAS PubMed.
- C. Camilloni, D. Bonetti, A. Morrone, R. Giri, C. M. Dobson, M. Brunori, S. Gianni and M. Vendruscolo, Sci. Rep., 2016, 6, 28285 CrossRef CAS PubMed.
- H. G. Börner, Macromol. Chem. Phys., 2007, 208, 124–130 CrossRef.
- D. Eckhardt, M. Groenewolt, E. Krause and H. G. Börner, Chem. Commun., 2005, 2814–2816 RSC.
- J. Hentschel, E. Krause and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 7722–7723 CrossRef CAS PubMed.
- E.-K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Börner and P. Bäuerle, Adv. Mater., 2009, 21, 1562–1567 CrossRef CAS.
- J. H. Waite, N. H. Andersen, S. Jewhurst and C. Sun, J. Adhes., 2005, 81, 297–317 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- R. Höfer, in Renewable Resources for Surface Coatings, Inks, and Adhesives, ed. A. S. Matharu and Z. Zhang, RSC Publ., Cambridge, 1 edn., 2023, pp. 65–91 Search PubMed.
- P. Kord Forooshani and B. P. Lee, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 9–33 CrossRef CAS PubMed.
- S. Moulay, Polym. Rev., 2014, 54, 436–513 CrossRef CAS.
- S. Peplau, T. J. Neubert, K. Balasubramanian, J. Polleux and H. G. Börner, Macromol. Rapid Commun., 2023, 2300300 CrossRef CAS PubMed.
- N. L. Venkatareddy, P. Wilke, N. Ernst, J. Horsch, A. Dallmann, M. Weber and H. G. Börner, Adv. Mater. Interfaces, 2019, 6, 1900501 CrossRef.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- P. Wilke and H. G. Börner, ACS Macro Lett., 2012, 1, 871–875 CrossRef CAS PubMed.
- H. G. Silverman and F. F. Roberto, Mar Biotechnol (NY), 2007, 9, 661–681 CrossRef CAS PubMed.
- K. Kamino, in Green Chemistry for Surface Coatings, Inks and Adhesives, Royal Society of Chemistry, 2019, pp. 49–68 Search PubMed.
- J. B. Puthoff, in Green Chemistry for Surface Coatings, Inks and Adhesives, 2019 Search PubMed.
- H. Onusseit, R. Wefringhaus, G. Dreezen, J. Wichelhaus, J. Schall, L. Thiele and A. van Halteren, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 7th edn, 2011 Search PubMed.
- G. Westwood, T. N. Horton and J. J. Wilker, Macromolecules, 2007, 40, 3960–3964 CrossRef CAS.
- C. Cui and W. Liu, Prog. Polym. Sci., 2021, 116, 101388 CrossRef CAS.
- A. Grafl, A. Müller, A. Preuß, B. Röder and H. G. Börner, Adv. Eng. Mater., 2022, 2201279 Search PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- A. A. Putnam and J. J. Wilker, Soft Matter, 2021, 17, 1999–2009 RSC.
- J. Horsch, P. Wilke, M. Pretzler, M. Seuss, I. Melnyk, D. Remmler, A. Fery, A. Rompel and H. G. Börner, Angew. Chem., 2018, 130, 15954–15958 CrossRef.
- S. Arias, S. Amini, J. Horsch, M. Pretzler, A. Rompel, I. Melnyk, D. Sychev, A. Fery and H. G. Börner, Angew. Chem., Int. Ed., 2020, 59, 18495–18499 CrossRef CAS PubMed.
- J. M. Kohn, J. Riedel, J. Horsch, H. Stephanowitz and H. G. Börner, Macromol. Rapid Commun., 2020, 41, 1900431 CrossRef CAS PubMed.
- S. Arias, S. Amini, J. M. Krüger and H. G. Börner, Soft Matter, 2021, 17, 2028–2033 RSC.
- J. M. Kruger and H. G. Borner, Angew. Chem., Int. Ed., 2021, 60, 6408–6413 CrossRef PubMed.
- J. M. Krüger, C.-Y. Choi, F. Lossada, P. Wang, O. Löschke, D. Auhl and H. G. Börner, Macromolecules, 2022, 55, 989–1002 CrossRef.
- S. A. Hafezi and W. M. Abdel-Rahman, Curr. Mol. Pharmacol., 2019, 12, 230–238 CrossRef CAS PubMed.
- I. Haq, P. Mazumder and A. S. Kalamdhad, Bioresour. Technol., 2020, 312, 123636 CrossRef CAS PubMed.
- Z. Li, E. Sutandar, T. Goihl, X. Zhang and X. Pan, Green Chem., 2020, 22, 7989–8001 RSC.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- B. Venkatesagowda and R. F. H. Dekker, Biomass Convers. Biorefin., 2020, 10, 203–225 CrossRef CAS.
- H. Wikberg, T. Ohra-aho, F. Pileidis and M.-M. Titirici, ACS Sustainable Chem. Eng., 2015, 3, 2737–2745 CrossRef CAS.
- L. Burdine, T. G. Gillette, H. J. Lin and T. Kodadek, J. Am. Chem. Soc., 2004, 126, 11442–11443 CrossRef CAS PubMed.
- R. J. Gosselink, J. E. van Dam, E. de Jong, G. Gellerstedt, E. L. Scott and J. P. Sanders, Holzforschung, 2011, 65, 155–162 CAS.
- B. Beer, M. J. Bartolome, L. Berndorfer, G. Bochmann, G. M. Guebitz and G. S. Nyanhongo, Int. J. Biol. Macromol., 2020, 161, 1440–1446 CrossRef CAS PubMed.
- R. Paliwal, A. P. Rawat, M. Rawat and J. Rai, Appl. Biochem. Biotechnol., 2012, 167, 1865–1889 CrossRef CAS PubMed.
- P. Harvey and J. Palmer, J. Biotechnol., 1990, 13, 169–179 CrossRef CAS.
- S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef CAS PubMed.
- B. Zhou, J. Li, W. Liu, H. Jiang, S. Li, L. Tan, L. Dong, L. She and Z. Wei, ChemSusChem, 2020, 13, 2628–2633 CrossRef CAS PubMed.
- A. M. ElSohly, J. I. MacDonald, N. B. Hentzen, I. L. Aanei, K. M. El Muslemany and M. B. Francis, J. Am. Chem. Soc., 2017, 139, 3767–3773 CrossRef CAS PubMed.
- S. Weidman and E. Kaiser, J. Am. Chem. Soc., 1966, 88, 5820–5827 CrossRef CAS.
- A. Y. Kozhevnikov, S. Ul'yanovskaya, M. Semushina, S. Pokryshkin, A. Ladesov, I. Pikovskoi and D. Kosyakov, Russ. J. Appl. Chem., 2017, 90, 516–521 CrossRef CAS.
- S. Y. Lin and C. W. Dence, Methods in lignin chemistry, Springer Science & Business Media, 2012 Search PubMed.
- M. Zhang, F. L. Resende, A. Moutsoglou and D. E. Raynie, J. Anal. Appl. Pyrolysis, 2012, 98, 65–71 CrossRef CAS.
- M. Wądrzyk, R. Janus, M. Lewandowski and A. Magdziarz, Renewable Energy, 2021, 177, 942–952 CrossRef.
- T. Fisher, M. Hajaligol, B. Waymack and D. Kellogg, J. Anal. Appl. Pyrolysis, 2002, 62, 331–349 CrossRef CAS.
- G. Habenicht, Kleben: Grundlagen, Technologie, Anwendungen, Springer, 1997 Search PubMed.
- L. F. Da Silva, A. Öchsner and R. D. Adams, Handbook of adhesion technology, Springer Science & Business Media, 2011 Search PubMed.
- M. Rasche, Adhäsion, 1990, 11, 36–43 Search PubMed.
- J. M. Ferri, O. Fenollar, A. Jorda-Vilaplana, D. García-Sanoguera and R. Balart, Polym. Int., 2016, 65, 453–463 CrossRef CAS.
- B. S. Chiou and P. E. Schoen, J. Appl. Polym. Sci., 2002, 83, 212–223 CrossRef CAS.
- F. Lossada, D. Jiao, J. Guo, D. Hoenders, A. Eckert and A. Walther, ACS Appl. Polym. Mater., 2019, 1, 3334–3342 CrossRef CAS.
- T. W. Thannhauser, Y. Konishi and H. A. Scheraga, in Methods in Enzymology, Academic Press, 1987, vol. 143, pp. 115–119 Search PubMed.
- M. J. van Oppen, J. K. Oliver, H. M. Putnam and R. D. Gates, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2307–2313 CrossRef CAS PubMed.
- J. A. Barton, B. L. Willis and K. S. Hutson, Rev. Aquac., 2017, 9, 238–256 CrossRef.
- I. V. Matus, J. Lino Alves, J. Gois, A. Barata da Rocha, R. Neto and C. Da Silva Mota, Rapid Prototyp. J., 2021, 27, 692–706 CrossRef.
- SELLEYS KNEAD IT AQUA, https://selleys.com.sg/wp-content/uploads/2020/04/SDS-Knead-It-Aqua.pdf, (accessed 23.11., 2021).
- R. M. Dizon, A. J. Edwards and E. D. Gomez, Aquat. Conserv.: Mar. Freshw., 2008, 18, 1140–1148 CrossRef.
- Water quality - Determination of the acute toxicity of waste water to zebrafish eggs (Danio rerio) (ISO 15088:2007); German version EN ISO 15088:2008, https://www.beuth.de/de/norm/din-en-iso-15088/113162875, (accessed 18.07., 2022).
- C. J. Crossland, B. G. Hatcher and S. V. Smith, Coral Reefs, 1991, 10, 55–64 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03680d |
| This journal is © The Royal Society of Chemistry 2024 |