
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeak-coordination-auxiliary aminocatalysis enables directed [3 + 2] cyclization for 2-acylindolizines†
Kui
Zeng‡
 a,
Neeraj Kumar
Pandit
b,
João C. A.
Oliveira
b,
Sebastian
Dechert
c,
Lutz
Ackermann
*bde and
Kai
Zhang
*a
a,
Neeraj Kumar
Pandit
b,
João C. A.
Oliveira
b,
Sebastian
Dechert
c,
Lutz
Ackermann
*bde and
Kai
Zhang
*a
aSustainable Materials and Chemistry, Dept. Wood Technology and Wood-Based Composites, University of Göttingen, Göttingen, Germany. E-mail: kai.zhang@uni-goettingen.de
bInstitute of Organic and Biomolecular Chemistry, University of Göttingen, Göttingen, Germany
cInstitute of Inorganic Chemistry, University of Göttingen, Göttingen, Germany
dWöhler Research Institute for Sustainable Chemistry (WISCh), University of Göttingen, Göttingen, Germany
eDZHK (German Centre for Cardiovascular Research), Potsdamer Straße 58, 10785 Berlin, Germany
First published on 13th December 2023
Abstract
The synthesis of 2-acylindolizines, possessing a readily modifiable ketone group, is of significant importance as it provides versatile precursors for the preparation of various indolizines. However, due to the electronically less active and more sterically demanding nature of α,β-unsaturated ketones toward iminium formation with an aminocatalyst, the efficient one-pot transformation of α,β-unsaturated ketones for distinct 2-acylindolizines bearing sensitive groups represents a challenge for synthetic chemists. Herein, we report a weak-coordination-auxiliary amino-catalyzed approach that enables directed [3 + 2] cyclization of α,β-unsaturated ketones and N-heteroaryl ketones for the desired 2-acylindolizines via an iminium ion/enamine tandem sequence. A highly broad range of commercially available α,β-unsaturated ketones (internal, terminal, and cyclic enones) can act as coupling partners for readily accessible 2-acylindolizines relative to the existing state-of-the-art methods. Control experiments and in-depth DFT calculations highlight the importance of weakly coordinated glycine's carboxylic group in promoting the intramolecular cyclization and 1,5-proton transfer processes.
Introduction
Indolizines are an important group of N-heterocyclic motifs that are widely used for pharmaceutics (Scheme 1a),1–3 materials science4 and chemical feedstocks.5–7 Four representative traditional strategies, including Scholtz reaction,8,9 the use of pyridinium N-methylides,10,11 Tschitschibabin reaction12,13 and cyclization of alkynes with heteroaromatic feedstocks,14–16 are known for the preparation of indolizines. Efficient transformation methods that utilize readily available substrates for the one-pot synthesis of diverse indolizines, particularly those containing an electron-withdrawing group at the C-2 position, are in high demand.17,18 2-Acylindolizines, with a readily modifiable ketone group at the C-2 position, provide versatile precursors for the preparation of various indolizines. In 2012, 2-acylindolizines were prepared via a palladium-catalyzed three component carbonylative cyclization/arylation cascade.19 It not only relies on the precious metal palladium and toxic CO as the carbonyl source, the requirement of a pivaloyl (–OPiv) group on 2-propargylpyridine restricts the substitution pattern of the product. Wang et al. developed a cycloaddition reaction between 1-cyanocyclopropane 1-ester and pyridine or benzopyridine for synthesizing 1-cyano,2-acylindolizine.20 Although this protocol is accessible to 2-acylindolizine with a cyano group, multiple steps are required to pre-construct complex 1-cyanocyclopropane 1-ester substrates. Alternatively, the [3 + 2] cyclization of readily available pyridine ketone with α,β-unsaturated ketones provides another viable route to form the 2-acylindolizine core. As an example, 2-acylindolizines can be prepared via one-pot two component Baylis–Hillman reactions (Scheme 1b).21,22 Their α,β-unsaturated ketones can be activated with a Lewis-acid/Brønsted acid. However, an efficient catalyzed approach to a highly expanded range of commercially available versatile internal, terminal and cyclic enone substrates for broader scope of 2-acylindolizine with even higher efficiency via [3 + 2] cyclization is still highly desired.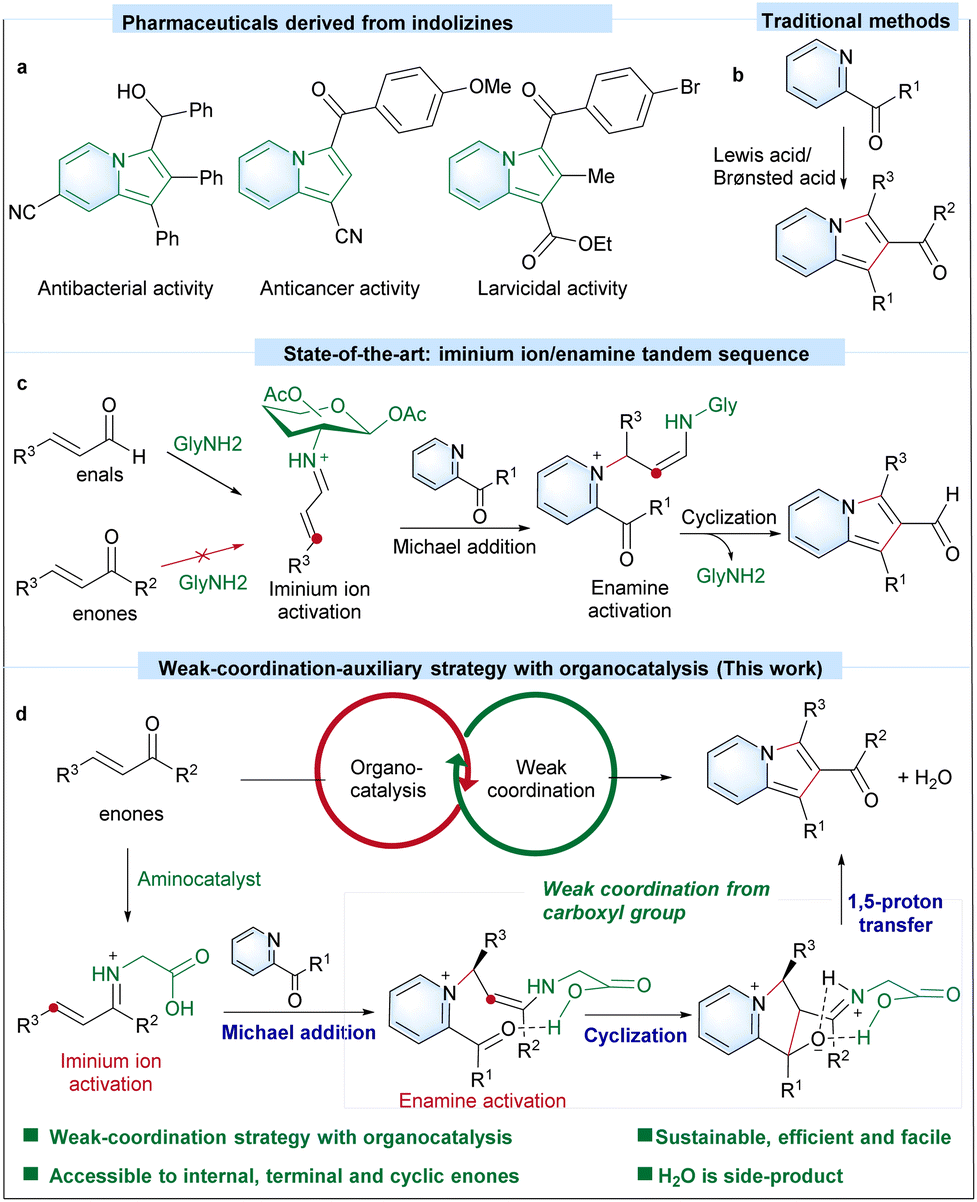 | ||
| Scheme 1 Design of a weak-coordination-auxiliary strategy with a collaborative amino-catalyzed iminium ion/enamine tandem sequence for the preparation of 1,2,3-trisubstituted indolizine-2-ketones via one-pot reaction. | ||
Inspired by the strategies of using an iminium ion and enamine mechanism to activate the carbonyl group with aminocatalysts,23–26 our previous glucosamine-catalyzed strategy via a stereoauxiliary-iminium ion/enamine tandem sequence demonstrated the efficient preparation of a rich library of indolizine-2-aldehydes with one-pot [3 + 2] cyclization of 2-acetylpyridine and α,β-unsaturated aldehydes (Scheme 1c).27 As demonstrated,27 Lewis-acid mediated Baylis–Hillman reaction (Scheme 1b)22 cannot tolerate a group of sensitive functional groups (such as the hydroxyl group, N,N-dimethyl group, heterocyclic group, cyclic enone group etc.), but highlights the ability of the amino-catalyzed method to gain successful access to these α,β-unsaturated aldehydes.27 However, due to less electronic activity and more sterically demanding nature of α,β-unsaturated ketones toward iminium formation with aminocatalysts,28–30 the glucosamine-catalyzed strategy cannot efficiently access α,β-unsaturated ketones for the preparation of 2-acylindolizines. We propose herewith that a weak-coordination-auxiliary strategy involving an aminocatalyst would help overcome the challenges, especially the energy barrier in the intramolecular cyclization step. With our ongoing interest in the carboxylate assisted activation strategy31–34 and the preparation of N-heterocyclic compounds by sustainable organic synthesis methods,27,35,36 herein, we report for the first time glycine-catalyzed [3 + 2] cyclization of α,β-unsaturated ketones and N-heteroaryl ketones for various 2-acylindolizines by a weak-coordination-auxiliary strategy (Scheme 1d).
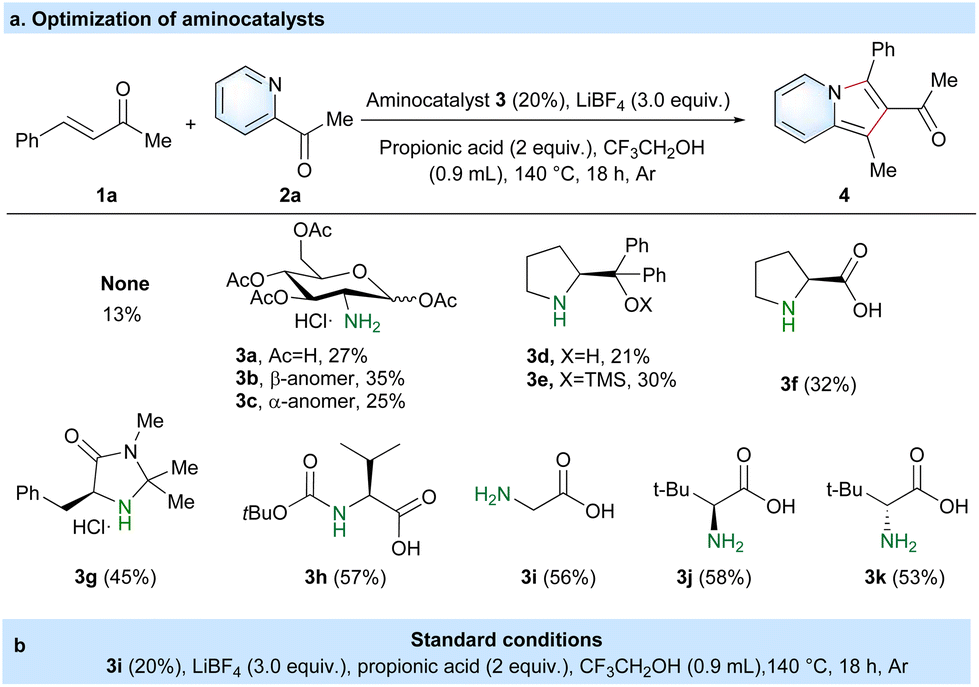
We initiated our studies using benzalacetone (1a) and 2-acetylpyridine (2a) for the preparation of 2-acylindolizine (4) via the aminocatalyzed [3 + 2] cyclization reaction (Table 1 and Table S1†). In the absence of catalyst 3, only a low yield of 4 (13%) was obtained using LiBF4 and propionic acid in CF3CH2OH solvent at 140 °C for 18 h under an Ar gas atmosphere (Table 1a). To promote the reaction activity with a higher yield of 4, diverse aminocatalysts (3a–3k) were tested under the same conditions. The results revealed that amino acids, e.g. glycine 3i (56%)37 and (tert-butoxycarbonyl)-L-valine 3h (57%),38 were noticeably superior to D-glucosamine 3a (27%),36 1,3,4,6-tetra-O-acetyl-2-amino-2-deoxy-β-D-glucosamine 3b (35%),27 1,3,4,6-tetra-O-acetyl-2-amino-2-deoxy-α-D-glucosamine 3c (25%),27 (S)-diphenyl(pyrrolidin-2-yl)methanol 3d (21%),39 (S)-2-(diphenyl((trimethylsilyl)oxy)methyl)pyrrolidine 3e (30%),39L-proline 3f (32%),23 and (5S)-(−)-5-benzyl-2,2,3-trimethylimidazolidin-4-one 3g (45%).24 Besides, leucine with D/L-tert-butyl bulk steric hindrance did not obviously affect the reaction activity, such as that of 3j (58%) and 3k (53%). Based on the results presented in Table 1a, glycine was chosen as the optimal aminocatalyst, given the similar catalytic activity among these amino acid catalysts and its low price of 0.0837 £ per gram. After considerable experimentation (Table 1b and Table S1†), benzalacetone (1a, 0.2 mmol), 2-acetylpyridine (2a, 0.5 mmol), glycine (3g, 0.04 mmol), LiBF4 (0.6 mmol) and propionic acid (0.4 mmol) in CF3CH2OH solvent (0.9 mL) at 140 °C for 18 h under an Ar atmosphere were taken as optimal conditions.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 4 (%) |
a ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1a (0.2 mmol), 2a (2.5 equiv.), aminocatalyst (20 mol%), LiBF4 (3.0 equiv.), propionic acid (2.0 equiv.), CF3CH2OH (0.9 mL), Ar, 18 h, and 140 °C. Yields were determined by 1H-NMR analysis with CH2Br2 as an internal standard. 1a (0.2 mmol), 2a (2.5 equiv.), aminocatalyst (20 mol%), LiBF4 (3.0 equiv.), propionic acid (2.0 equiv.), CF3CH2OH (0.9 mL), Ar, 18 h, and 140 °C. Yields were determined by 1H-NMR analysis with CH2Br2 as an internal standard.
|
||
| 1 | None | 56 |
| 2 | AcOH/CF3COOH replacing propionic acid | 42/16 |
| 3 | LiSO3CF3/LiCl replacing LiBF4 | 25/18 |
| 4 | HFIP/CH3CN/toluene replacing CF3CH2OH | 31/13/0 |
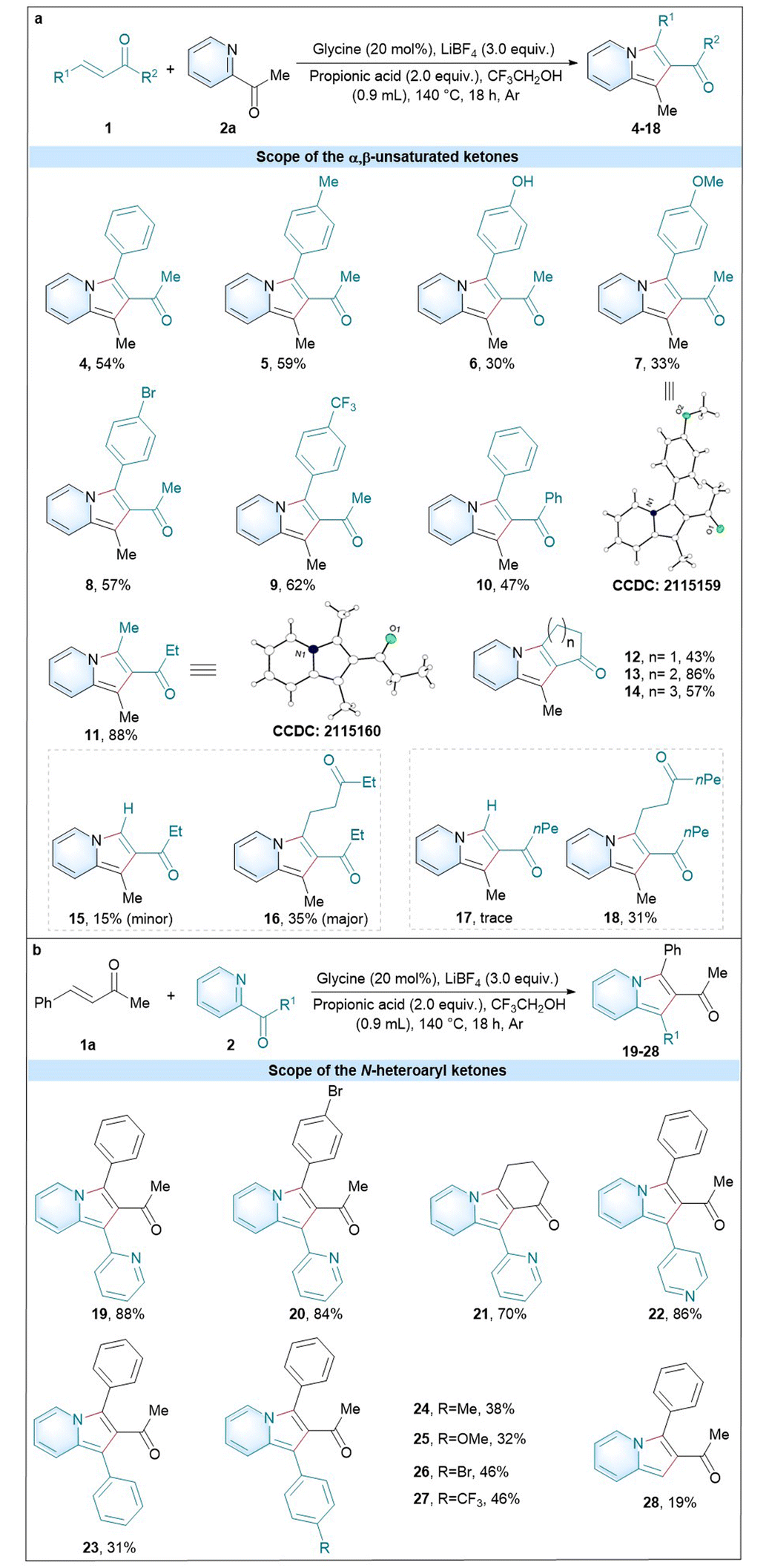
With the established optimal glycine-catalyzed conditions in hand, we proceeded towards examining the scope with a variety of α,β-unsaturated ketones 1 and N-heteroaryl ketones 2 under the standard conditions (Table 2). A group of internal aromatic α,β-unsaturated ketones with electron-donating and -withdrawing groups were compatible with the reaction, such as 4 (54%), 5 (59%), 6 (30%), 7 (33%), 8 (57%), 9 (62%), and 10 (47%). It is worth noting that the internal aromatic α,β-unsaturated ketone (10) with a bulk steric hindrance at the ketone position was also tolerant to the reaction conditions. Besides, the internal aliphatic α,β-unsaturated ketone was also compatible with the standard conditions (11, 88%). In particular, a group of aliphatic cyclic α,β-unsaturated ketones gave good to excellent yields (12, 43%; 13, 86%; and 14, 57%). The ring size of cyclic enones could affect the reaction steps like Michael addition and cyclization.27 In addition to the internal α,β-unsaturated ketones, the terminal α,β-unsaturated ketone (pent-1-en-3-one) was also compatible with the standard conditions to give the two products 15:16 (15%:35%). Even the terminal α,β-unsaturated ketone (oct-1-en-3-one) could afford the corresponding product 18 (31%). The structures of 7 and 11 were determined by X-ray crystallographic analysis, and those of the other products in Table 2 were assigned by analogy.
|
a 1 (0.2 mmol), 2 (2.5 equiv.), glycine (20 mol%), LiBF4 (3.0 equiv.), propionic acid (2.0 equiv.), CF3CH2OH (0.9 mL), Ar, 18 h, and 140 °C. Yields were determined by 1H-NMR analysis with CH2Br2 as an internal standard.
|
|---|
|
The scope of the N-heteroaryl ketones was next studied using 4-phenylbut-3-en-2-one (1a) as a model substrate (Scheme 2b). As a result, di(pyridin-2-yl)methanone can well tolerate the optimal conditions with substrates 4-phenylbut-3-en-2-one, (E)-4-(4-bromophenyl)but-3-en-2-one and cyclohex-2-en-1-one to give the corresponding products 19 (88%), 20 (84%) and 21 (70%). Also the substrate pyridin-2-yl(pyridin-4-yl)methanone was also compatible with the conditions to yield 22 (86%). Moreover, a group of aromatic N-heteroaryl ketones with electron-donating and -withdrawing groups were found to give the corresponding products 23–27. Beyond the substrates N-heteroaryl ketones, we also successfully expanded the glycine-catalysed strategy to different substrates, such as picolinaldehyde (28, 19%).
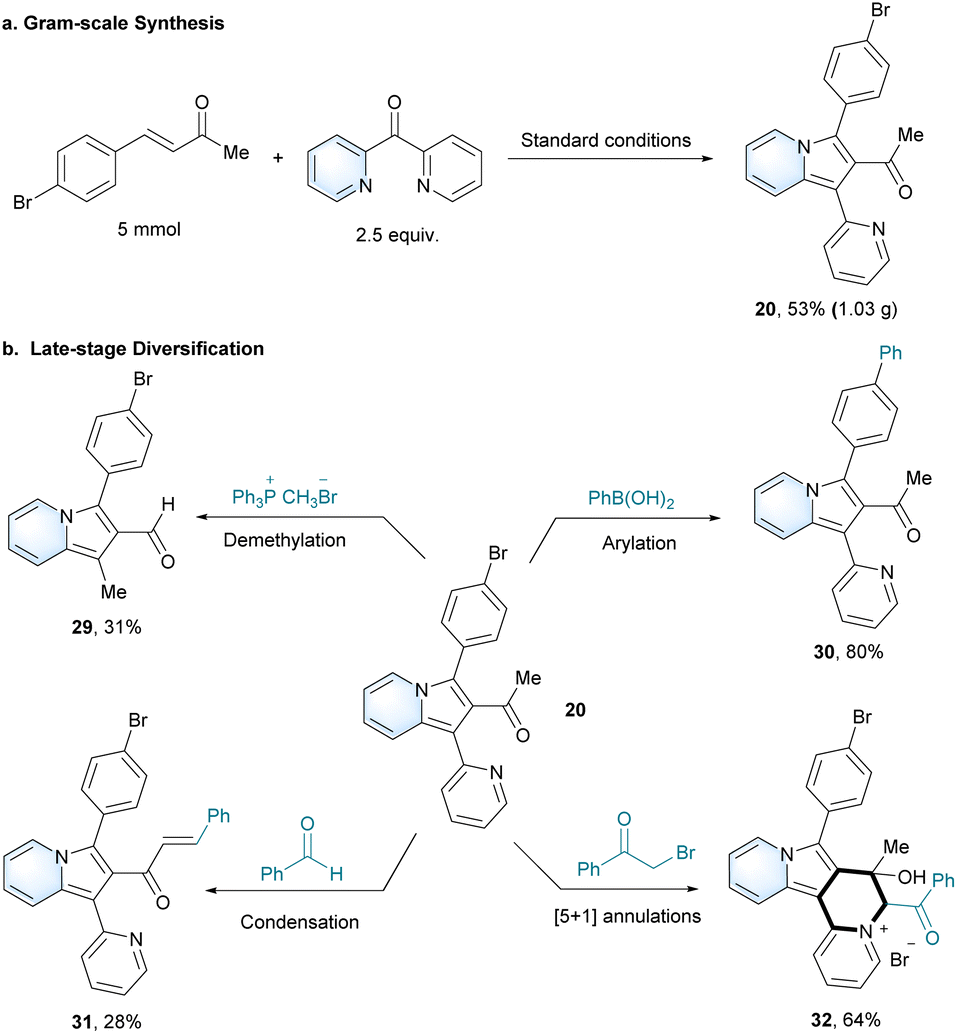 | ||
| Scheme 2 Large-scale synthesis (a) and late-stage diversifications (b). | ||
Large-scale synthesis of 1-(3-(4-bromophenyl)-1-(pyridin-2-yl)indolizin-2-yl)ethan-1-one (20) was achieved with 53% yield under optimal conditions (Scheme 2a). Azaheterocycles are the basic skeletons of many natural compounds and synthetic drugs with important biological activities.40 The ability to prepare and achieve late-stage diversification of azaheterocycles is highly desired.41–43 A group of late-stage transformations was applicable to prepare various 2-acylindolizines, in order to expand their potential applications. As an example, 1-(3-(4-bromophenyl)-1-(pyridin-2-yl)indolizin-2-yl)ethan-1-one (20) was chosen as the substrate for further modifications to synthesize distinct 2-acylindolizine derivatives (Scheme 2b). Starting from 20, 3-(4-bromophenyl)-1-methylindolizine-2-carbaldehyde (29) was unexpectedly synthesized with the Ph3P+CH3Br− reagent. 1-(3-([1,1′-Biphenyl]-4-yl)-1-(pyridin-2-yl)indolizin-2-yl)ethan-1-one (30) was synthesized by the Suzuki–Miyaura coupling reaction. Notably, 3-(5-bromo-[1,1′-biphenyl]-2-yl)-1-(pyridin-2-yl)indolizine-2-carbaldehyde (31) was successfully synthesized via the dehydration coupling of 20 and benzaldehyde. Moreover, a one-pot synthesis of (6S)-6-benzoyl-8-(4-bromophenyl)-7-hydroxy-7-methyl-6,7-dihydroindolizino [1,2-a]quinolizin-5-ium bromide (32) was also realized by the catalyst-free [5 + 1] annulation.
Amino acids such as 3h–3k showed significantly better catalytic activity than 3a–3g for amino-catalyzed preparation of 2-acylindolizine by directed [3 + 2] cyclization under the standard conditions (Table 1). These higher catalytic activities of amino acids could be derived from a weak cooperation effect of the carboxyl group of amino acids. Therefore, to investigate the proposed weak-coordination-auxiliary effect, a series of aminocatalysts (3l, 3m, 3i and 3n) with different moieties (e.g. alkyl, hydroxyl, carboxyl and ester groups) at the β position were tested under the standard conditions (Scheme 3a). As a result, aminocatalyst 3l gave only a low yield of 4 (19%), which showed almost the same reactivity as in the case without a catalyst (13%). Compared with catalyst 3l, catalyst 3m with a hydroxyl group could achieve a higher yield (31%). In particular, catalyst 3i (56%) with a carboxyl group and catalyst 3n (52%) with a ester group can obviously generate a better yield than 3l (19%) and 3m (31%). Therefore, the carboxyl group of glycine should have significantly promoted the efficiency of the reaction via a weak-coordination-auxiliary strategy, which results in a lower energy barrier in the intramolecular cyclization step (Scheme 3b and c).
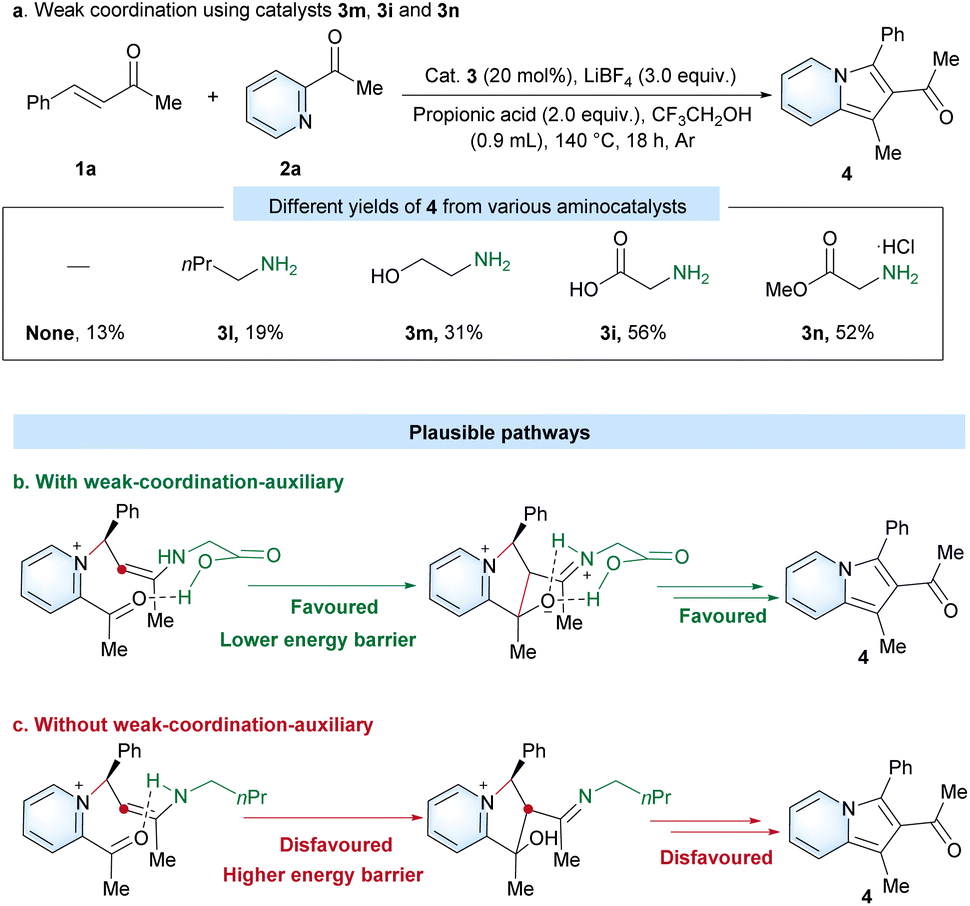 | ||
| Scheme 3 Control experiments for mechanistic study. | ||
To gain further insights into the details of the reaction mechanism, the DFT calculations for Michael addition and cyclization steps were performed at the PW6B95-D4/def2-TZVPP+SMD(2,2,2-trifluoroethanol)//TPSS-D3(BJ)/def2-SVP level of theory (Scheme 4) for three plausible pathways. Pathway A proceeds with a syn-conformed iminium ion, whereas pathways B and C proceed with an anti-conformed iminium ion in the absence and presence of a weak cooperation effect of the carboxyl group of amino acid, respectively. All pathways commence with the attack of 2-acetylpyridine on the iminium ion via Michael addition TS1 to generate the enamine intermediate Int1. The pathways B and C were found to be more energetically favorable than pathway A by approximately 4.1 kcal mol−1 for the transition state TS1. Unfortunately, on proceeding further, the transition state for pathway A could not be located, which may be due to the larger distance between the H and O (4.9 Å) in the intermediate Int2A (Fig. S4†). This provides support for such a pathway not being involved. Calculations indicate that pathway C is kinetically favorable over pathway B, given the negligible and extremely facile energy barriers for the C–C formation and 1,5-proton transfer steps. The weak cooperation effect of the carboxyl group of the amino acid appears to promote the cyclization step and the formation of intermediate Int4C. Thus, DFT calculations highlight the role of the glycine's carboxyl group in the reaction mechanism, strongly supporting the outcome of control experiments (Scheme 3).
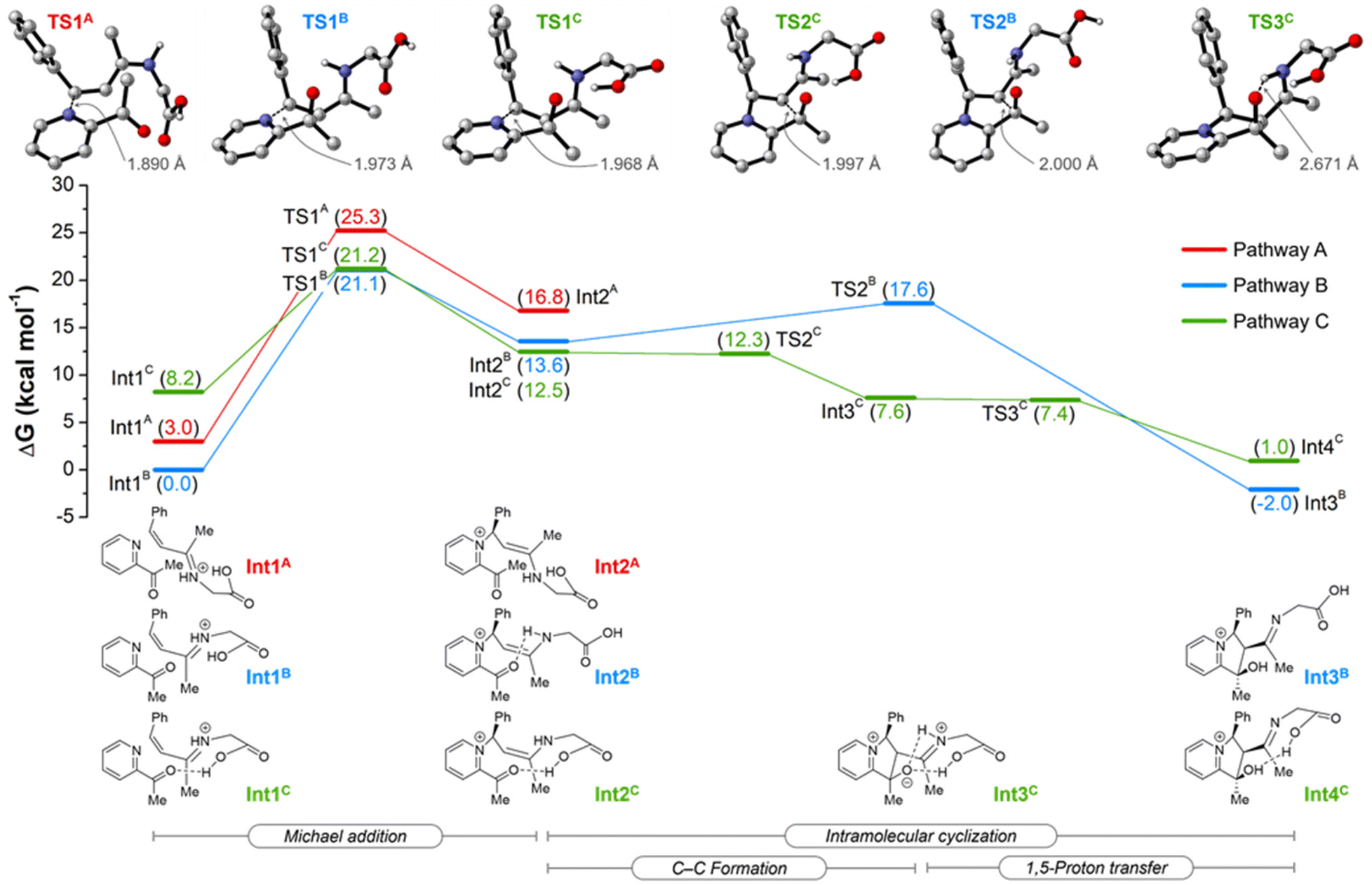 | ||
| Scheme 4 Computed relative Gibbs free energies (ΔG413.15) in kcal mol−1 for the amino-catalyzed 2-acylindolizine formation for the three considered plausible pathways. Calculations were performed at the PW6B95-D4/def2-TZVPP+SMD(2,2,2-trifluoroethanol)//TPSS-D3(BJ)/def2-SVP level of theory. Non-relevant hydrogens in the transition state structures were omitted for clarity. | ||
On the basis of our experimental and computational findings, a plausible catalytic cycle is proposed (Scheme 5). First, a more stable anti-conformed iminium ion C forms following the dehydration step. The carboxyl group of glycine cooperates with 2-acetylpyridine,44 and enamine D is generated via Michael addition. Next, E forms after the intramolecular cyclization due to the lower energy barriers of C–C formation promoted by the weak-coordination-auxiliary effect of the carboxylic group. This was strongly verified by an energy difference of 5.3 kcal mol−1 between TS2C (12.3 kcal mol−1) and TS2B (17.6 kcal mol−1) in Scheme 4. The weak-coordination-auxiliary effect further promotes the 1,5-proton transfer to form F. Then, intermediate I forms after the dehydration of G and deprotonation reaction of H. Finally, product J is generated from hydrolyzed I and the catalyst A regenerates to enter the next catalytic cycle.
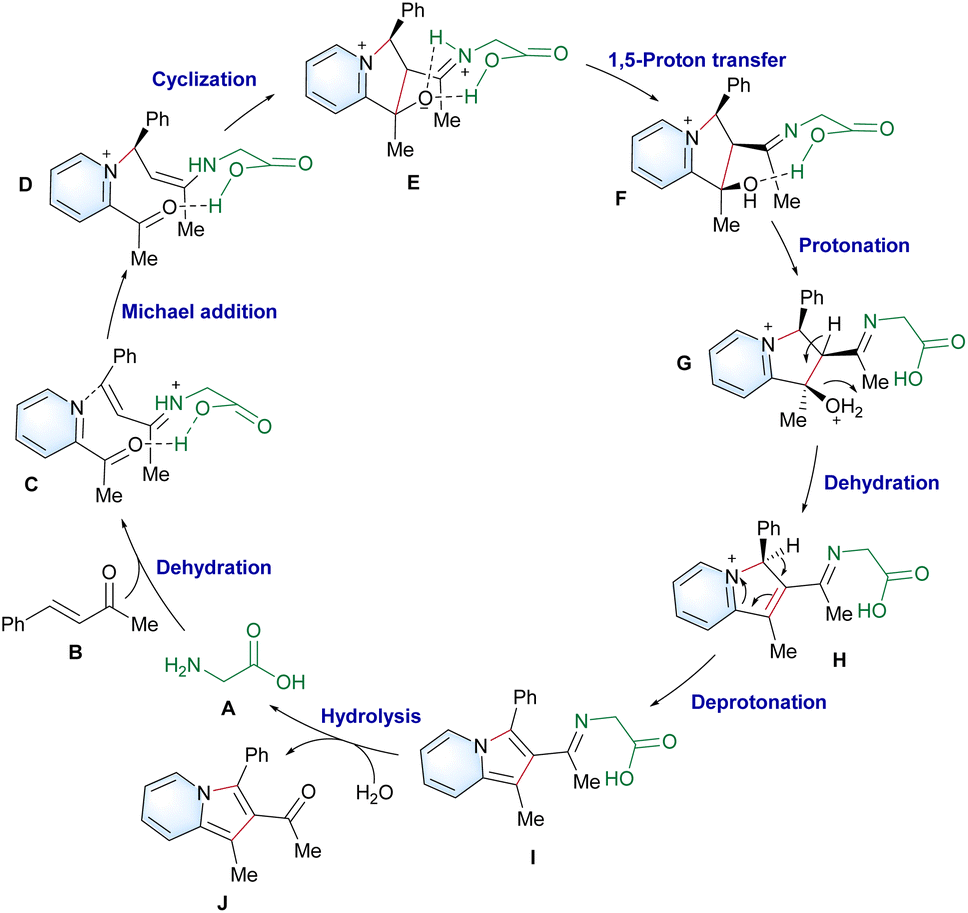 | ||
| Scheme 5 The proposed mechanism. | ||
Process mass intensity (PMI) is a key mass-based metric to evaluate the green credentials of reactions during process and chemical development.45 The PMI of our current work and representative approaches is shown comparatively using product 13 as an example (Table 3). In addition to the highest yield of 13 (83%) achieved in our current work, the PMIRRC (expressed as the amount of reagents, reactants and catalyst) and PMIsolv (solvent relative to the amount of isolated product) of our current approach and the work from the Badsara group22 are obviously lower than those in the state-of-the-art works.21,27 It should be noted that PMIsolv does not take into account any solvent consumed during purification processes since the reference values for the comparative works are not available.
Conclusions
We have shown how a combination of organocatalysis and the weak-coordination-auxiliary strategy can be used to address the problem of activating enones in the one-pot [3 + 2] cyclization of α,β-unsaturated ketones (internal, terminal, and cyclic enones) and N-heteroaryl ketones for the one-pot preparation of valuable 2-acylindolizine molecules. Glycine acts as a representative aminocatalyst to favor the reaction through an iminium ion/enamine tandem sequence, and it also provides a weak-coordination-auxiliary effect from its carboxyl group to overcome the energy barrier in intramolecular cyclization and 1,5-proton transfer processes. These were demonstrated by control experiments and DFT calculations. This novel glycine-catalyzed strategy with a weak-coordination-auxiliary effect is versatile enough to enable the development of other functionalization processes with an iminium ion/enamine tandem sequence.Experimental
Preparation of 2-acylindolizines
A mixture of α,β-unsaturated ketones (0.2 mmol), heteroaryl ketones (2.5 equiv.), catalyst 3g (0.04 mmol), propionic acid (2.0 equiv.) and LiBF4 (3.0 equiv.) in CF3CH2OH (0.9 mL) was stirred at 140 °C under an Ar atmosphere for 18 h. The reactions were conducted in a sealed Schlenk tube and heated with an IKA magnetic heating agitator with a heating block. The reaction temperature was directly read from the temperature detector of the IKA apparatus and was calibrated with a thermometer. After cooling to room temperature, the reaction mixture was basified up to pH 7 using saturated Na2CO3 aqueous solution, before extracting with diether (3 × 3 mL) and drying over anhydrous Na2SO4. After filtration and concentration in a rotary evaporator, the crude product was purified by flash chromatography on a silica gel (ethyl acetate:n-hexane) to give the products. More experimental details and characterization are available in the ESI.†
Author contributions
K. Zh. conceived the concept. L. A. and K. Zh. supervised the project. K. Z. undertook all of the experimental work, analytical characterization, and spectroscopic analysis. N. K. P. and J. C. A. O. performed the DFT analysis. S. D. performed X-ray crystallography. K. Z., L. A., and K. Zh. analyzed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Kai Zhang thanks German Research Foundation (DFG) for the financial support for the project with the grant number ZH546/8-1 and the University of Göttingen for the Department Start-up funding. Kui Zeng thanks China Scholarship Council for the PhD grant. The generous support from the DZHK, the ERC advanced grant no. 101021358, the DFG Gottfried-Wilhelm-Leibniz-Preis and SPP 2363 to Lutz Ackermann is gratefully acknowledged.References
- V. R. Vemula, S. Vurukonda and C. K. Bairi, Int. J. Pharm. Sci. Rev. Res., 2011, 11, 159–163 Search PubMed.
- G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237–5257 CrossRef.
- V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 CrossRef.
- E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef PubMed.
- J. T. M. Correia, B. List and F. Coelho, Angew. Chem., Int. Ed., 2017, 56, 7967–7970 CrossRef PubMed.
- D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef PubMed.
- J. Hui, Y. Ma, J. Zhao and H. Cao, Org. Biomol. Chem., 2021, 19, 10245–10258 RSC.
- V. Boekelheide and R. Windgassen Jr., J. Am. Chem. Soc., 1959, 81, 1456–1459 CrossRef.
- M. Scholtz, Ber. Dtsch. Chem. Ges., 1912, 45, 734–746 CrossRef CAS.
- G. Poissonnet, M.-H. Théret-Bettiol and R. H. Dodd, J. Org. Chem., 1996, 61, 2273–2282 CrossRef CAS.
- A. R. Katritzky, G. Qiu, B. Yang and H.-Y. He, J. Org. Chem., 1999, 64, 7618–7621 CrossRef CAS.
- A. Tschitschibabin, Ber. Dtsch. Chem. Ges. A and B, 1927, 60, 1607–1617 CrossRef.
- U. Bora, A. Saikia and R. C. Boruah, Org. Lett., 2003, 5, 435–438 CrossRef CAS PubMed.
- S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757–4759 CrossRef CAS.
- A. V. Kel'in, A. W. Sromek and V. Gevorgyan, J. Am. Chem. Soc., 2001, 123, 2074–2075 CrossRef.
- I. V. Seregin and V. Gevorgyan, J. Am. Chem. Soc., 2006, 128, 12050–12051 CrossRef CAS PubMed.
- D. A. Burnett and J. P. Vacca, International patent No, WO2018/195075A1, 2018 Search PubMed.
- M. Biagetti, A. Accetta, A. M. Capelli, M. Guala and M. Retini, US patent No, US2015/0361100A1, 2015 Search PubMed.
- Z. Li, D. Chernyak and V. Gevorgyan, Org. Lett., 2012, 14, 6056–6059 CrossRef CAS.
- J. Liu, L. Zhou, W. Ye and C. Wang, Chem. Commun., 2014, 50, 9068–9071 RSC.
- Y.-C. Yuan, T.-Z. Liu and B.-X. Zhao, J. Org. Chem., 2021, 86, 12737–12744 CrossRef CAS PubMed.
- D. Basavaiah, G. Veeraraghavaiah and S. S. Badsara, Org. Biomol. Chem., 2014, 12, 1551–1555 RSC.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef.
- P. Chauhan, S. Mahajan and D. Enders, Acc. Chem. Res., 2017, 50, 2809–2821 CrossRef CAS PubMed.
- K. Zeng, R. Mei, S. Dechert, L. Ackermann and K. Zhang, Commun. Chem., 2023, 6, 40 CrossRef CAS PubMed.
- Q. Li, W. Y. Wong, W. H. Chan and A. W. Lee, Adv. Synth. Catal., 2010, 352, 2142–2146 CrossRef CAS.
- C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS.
- X. Lu and L. Deng, Angew. Chem., Int. Ed., 2008, 47, 7710–7713 CrossRef CAS.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- J. Hubrich, T. Himmler, L. Rodefeld and L. Ackermann, ACS Catal., 2015, 5, 4089–4093 CrossRef CAS.
- S. Warratz, C. Kornhaass, A. Cajaraville, B. Niepoetter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS.
- L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS.
- K. Zeng, R. Mei, X. C. Zhang, L. B. Andreas and K. Zhang, Chem. Commun., 2022, 58, 6068–6071 RSC.
- K. Zeng, J. Ye, X. Meng, S. Dechert, M. Simon, S. Gong, R. A. Mata and K. Zhang, Chem. – Eur. J., 2022, 28, e202200648 CrossRef CAS PubMed.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
- M. Wasa, K. M. Engle and J. Q. Yu, Isr. J. Chem., 2010, 50, 605–616 CrossRef CAS.
- L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Chem. Soc. Rev., 2017, 46, 1080–1102 RSC.
- B. Wang, X. Hu, F. Sun, Z. Yang and W. Huang, Interdiscip. Med., 2023, 1, e20220010 CrossRef.
- C.-C. Zhang, H.-L. Wu, X.-C. Yu, L.-T. Wang, Y. Zhou, Y.-B. Sun and W.-T. Wei, Org. Lett., 2023, 25, 5862–5868 CrossRef CAS.
- X.-S. Zhou, Z. Zhang, W.-Y. Qu, X.-P. Liu, W.-J. Xiao, M. Jiang and J.-R. Chen, J. Am. Chem. Soc., 2023, 145, 12233–12243 CrossRef CAS PubMed.
- K. Sun, D. Zhao, Q. Li, S. Ni, G. Zheng and Q. Zhang, Sci. China: Chem., 2023, 66, 2309–2316 CrossRef CAS.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS.
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–135 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2115159 and 2115160. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03552b |
| ‡ Current address: School of Pharmacy, University of Wisconsin–Madison, Madison, US. |
| This journal is © The Royal Society of Chemistry 2024 |