
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maximization of hydrogen peroxide utilization in a proton exchange membrane H2O2 electrolyzer for efficient power-to-hydrogen conversion†
Jie
Yang
ab,
Ruimin
Ding
*a,
Chang
Liu
 ac,
Lifang
Chen
a,
Qi
Wang
ab,
Shanshan
Liu
a,
Qinchao
Xu
a and
Xi
Yin
*a
ac,
Lifang
Chen
a,
Qi
Wang
ab,
Shanshan
Liu
a,
Qinchao
Xu
a and
Xi
Yin
*a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, Shanxi 030001, China. E-mail: dingrm@sxicc.ac.cn; xiyin@sxicc.ac.cn
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 1st December 2023
Abstract
A hydrogen peroxide electrolyzer (HPEL) is the workhorse for an energy storage system based on the H2O2 electrochemical cycle. The high H2O2 utilization towards power-to-hydrogen conversion in the HPEL is essential to ensure the efficiency and cyclability of the system. Unfortunately, the H2O2 disproportionation at the anode and its crossover to the cathode in a proton exchange membrane (PEM) HPEL is detrimental to H2O2 utilization and must be mitigated. This work investigates the effects of the catalyst type, anode catalyst loading, and PEM thickness on H2O2 utilization in a PEM HPEL. The results show that the Co–N–C catalyst exhibits higher H2O2 utilization than the Fe–N–C and Pt/C catalysts due to its higher selectivity towards the hydrogen peroxide oxidation reaction (HPOR) and the lesser H2O2 disproportionation reaction (HPDR). Increasing the Co–N–C catalyst loading and PEM thickness can effectively inhibit the H2O2 crossover and improve the H2O2 utilization. On the other hand, the portion of the HPDR and the ohmic loss increase with the catalyst loading and PEM thickness, respectively. A maximum H2O2 utilization of over 98% can be achieved by balancing these factors. These results provide valuable guides to the catalyst design and device optimization for efficient energy storage systems based on the electrochemical H2O2–H2 cycle.
1. Introduction
The percentage of electricity generated by intermittent renewable sources (e.g., wind and solar power) is increasing rapidly. Large-scale, efficient, economical, and zero-emission energy storage systems are needed to integrate renewable-sourced electricity with the traditional power grid and store the electricity in the short or long term.1,2 Electrochemical energy storage systems based on the H2–water cycle,3–10 the ammonia–N2 cycle,11–14 the H2O2–water cycle,15–17 metal-ion batteries,18,19 and redox-flow batteries20–22 have been proposed for large-scale energy storage and grid balancing.Recently, we proposed the concept of distributed generation and energy storage systems based on the highly efficient electrochemical cycle of hydrogen peroxide (H2O2).23–25 In this system, H2O2 can be electrolyzed to H2 and O2 to store energy in the short term and then be regenerated via the two-electron oxygen reduction reaction (2e-ORR) in a fuel cell to generate power. Our proof-of-concept studies have demonstrated a highly efficient and stable PEM H2O2 electrolyzer (HPEL) for power-to-hydrogen conversion with a H2 production Faraday efficiency of 96% and a unitized regenerative hydrogen peroxide cycle cell (UR-HPCC) with a round-trip efficiency (RTE) of above 90% for renewable energy storage.23–25 Their unprecedented low system cost and high energy efficiency make the H2O2 electrochemical cycle systems highly promising for short- and long-term energy/hydrogen storage, along with other systems (Table S1†). In addition, it is possible to realize long-term energy/hydrogen storage using H2O2 as a H2 carrier and release the hydrogen using a HPEL for various applications.23–25
Achieving a high H2O2 utilization towards power-to-hydrogen conversion in the HPEL is essential to ensure the efficiency and cyclability of the H2O2–H2 energy storage system. In an ideal PEM HPEL, the H2O2 oxidation reaction (HPOR, H2O2 → 2H+ + 2e− + O2; E0 = 0.695 V vs. RHE) takes place and produces protons in the anode and the coupled hydrogen evolution reaction (HER, 2H+ + 2e− → H2; E0 = 0 V vs. RHE) occurs in the cathode, converting power to H2 gas at voltages of 0.7 V and above. In a practical PEM HPEL, as shown in Scheme 1, H2O2 in the anode can cross over to the cathode through the PEM and be reduced via the hydrogen peroxide reduction reaction (HPRR, H2O2 + 2H+ + 2e− → 2H2O; E0 = 1.78 V vs. RHE) at the cathode. This HPRR competes with the HER, decreasing the H2O2 utilization towards the power-to-H2 conversion (EH2O2-HPOR) and the faradaic efficiency. In addition, the undesired H2O2 disproportionation reaction (HPDR, 2H2O2 → 2H2O + O2) may occur at the anode, decreasing the H2O2 utilization for H2 production (Scheme 1).24,25 Therefore, it is critical to identify the key factors that affect these processes, mitigate the H2O2 crossover, inhibit the competing HPDR, and maximize the H2O2 utilization.
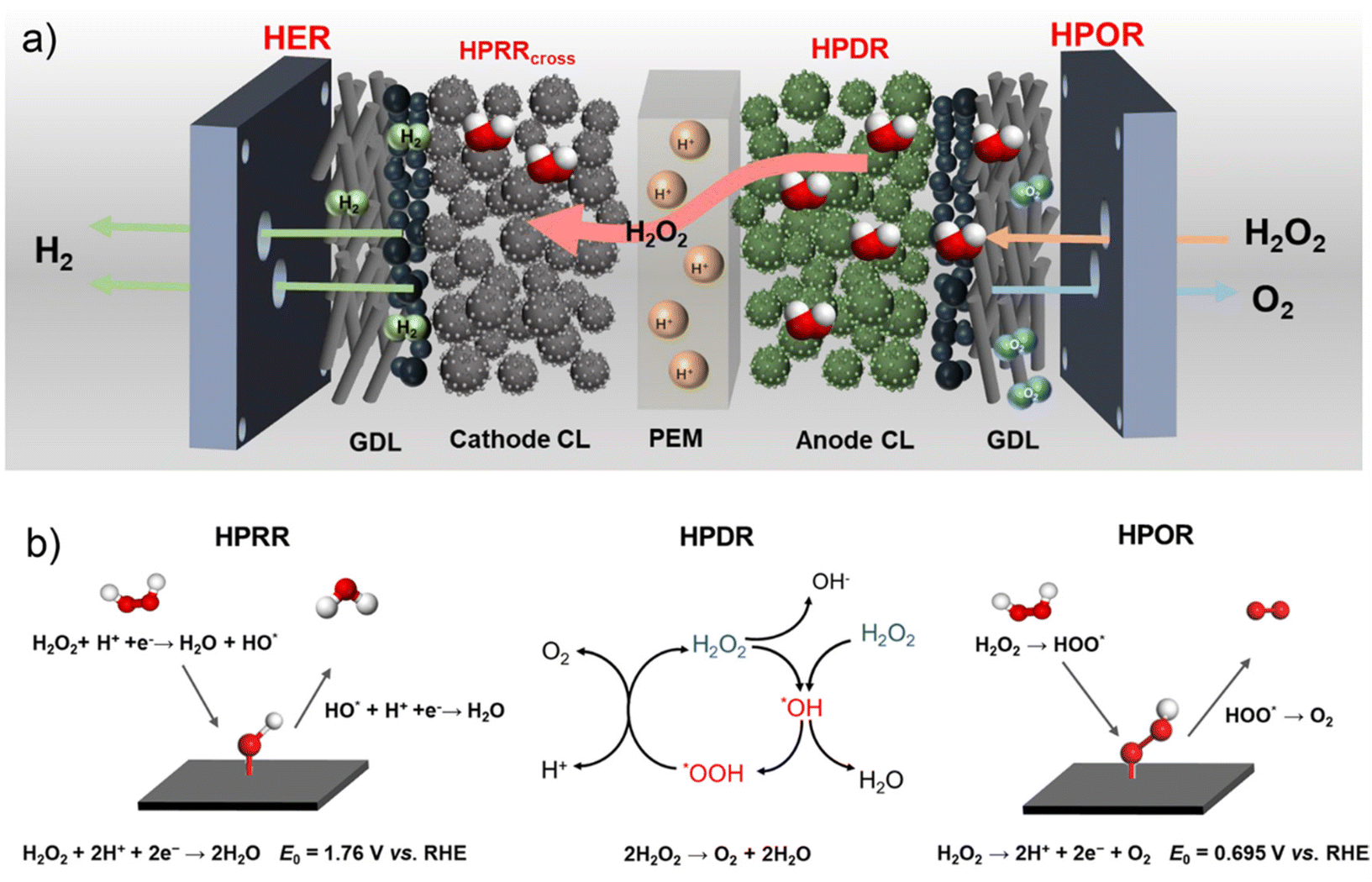 | ||
| Scheme 1 (a) In a PEM HPEL, H2 generation at the cathode stems exclusively from the HER. O2 is generated at the anode via the following three processes: HPOR coupled with the HER, HPOR coupled with the HPRRcross, and HPDR at the anode. (b) Schematic representation of the response mechanisms of the HPOR, HPDR,26,27 and HPRR. | ||
Herein, we focus on the effects of anode catalyst types, anode catalyst loading, and PEM thickness on the H2O2 utilization and device performance of the HPEL. By benchmarking the performance of representative platinum group metal-free (PGM-free) catalysts and Pt/C in the electrochemical cell and PEM HPEL, we identify the cobalt- and nitrogen-doped carbon (Co–N–C) catalyst as a better candidate than the Fe–N–C and Pt/C catalysts for its higher selectivity towards the HPOR and lesser HPDR activity. Using Co–N–C, we reveal that increasing the anode catalyst loading and PEM thickness appropriately can effectively inhibit the crossover and thus improve the H2O2 utilization. On the other hand, the HPDR intensifies as the anode catalyst loading increases and the internal resistance increases with the PEM thickness. Optimizing the above vital factors allows us to achieve a maximum H2O2 utilization of 98% in the PEM HPEL for power-to-hydrogen conversion. Our results elucidate the interdependences of the competing HPOR, HPDR, H2O2 crossover, and H2O2 utilization in the PEM HPEL, providing guides for catalyst design and device optimization of efficient energy storage systems based on electrochemical H2O2–H2 cycles.
2. Experimental
2.1 Membrane electrode assembly (MEA) fabrication
Anode catalyst inks were prepared by dispersing 50 mg of the Co–N–C or Fe–N–C catalyst in a mixture of de-ionized water (DI-water, 30.0 mL), isopropanol (IPA, 30.0 mL), and D521 Nafion dispersion (1070 μL, 5 wt%) in an ultrasonic bath for 2 h or by dispersing 16 mg of Pt/C (Hispec 3000, Johnson Matthey Co.) catalyst in a mixture of DI-water (12.0 mL), IPA (3.0 mL), and D521 Nafion dispersion (165 μL, 5 wt%) in an ultrasonic bath for 30 min. Then, the inks were sprayed onto pieces of 4-cm2 hydrophilic carbon cloth (ca. 0.33 mm in thickness) at 80 °C to form anode electrodes. Anodes with different catalyst loadings were obtained by controlling the volume of ink sprayed. The catalyst loadings were further confirmed by analyzing the electrochemical surface area (ECSA) of the anode catalysts. Commercial Pt/C gas diffusion electrodes (GDEs) (0.2 mgPt cm−2, 20 wt% Pt–C on Sigracet 22BB carbon paper, Fuel Cell Store, USA) were used as cathodes. The Pt/C cathode was hot-pressed to Nafion membranes with different thicknesses (NR211, NR212, NR115, and NR117, Chemours) at 120 °C and 5.3 MPa for 5 minutes to form the half-MEAs. Then, the anodes and the half-MEAs were assembled in the HPEL hardware to form the full MEAs. A detailed description of the HPEL hardware and the system setup can be found in our previous publications.24,252.2 PEM HPEL test and H2O2 utilization analysis
The PEM HPEL performance was evaluated at 25 °C and ambient pressure using an established protocol.24,25 During the activation process, the Pt/C cathode was filled with pure H2 gas to form a quasi-reversible hydrogen electrode (RHE). N2-saturated 0.5 M H2SO4 was pumped into the anode and circulated between the anode and a Boro 3.3 glass bottle at a flow rate of 200 mL min−1. The initial cyclic voltammetry (CV) was recorded by polarizing the Co–N–C or Fe–N–C anode from 0 to 1.0 V vs. RHE or the Pt/C anode from 0.05 to 1.2 V vs. RHE at a scan rate of 50 mV s−1 using a CHI 760E bipotentiostat. The double-layer capacitance for the PGM-free catalyst was calculated by integrating the charging current over a potential range of one-volt wide. A capacitance of 30 μF cm−2 was used to calculate the electrochemical surface area (ECSA), which is used to estimate the loading of the Co–N–C and Fe–N–C catalysts.28,29 The loadings of the Pt/C anode catalyst were determined using a Thermo Scientific ARL QUANT'X energy dispersive X-ray fluorescence (EDXRF) spectrometer.During the PEM HPEL operation, a mixture of 0.5 M H2SO4 and 0.5 M H2O2 was pumped into the anode and circulated between the anode and a glass flask at 200 mL min−1. Linear sweep AC voltammetry was performed by increasing the voltage from the OCV to 1.0 V at a scan rate of 5 mV s−1, while a 10 kHz AC wave with 5 mV amplitude was applied to measure the real-time high-frequency resistance (HFR). Electrochemical impedance spectroscopy (EIS) was performed to examine the resistance of MEAs at a constant open cell voltage (OCV) with an amplitude of 10 mV. The frequency ranged from 1 Hz to 100 kHz. The iR-corrected cell voltage (EiR-free) was obtained using the following equation:
| EiR-free = Ecell − Eohm = Ecell − j × HFR | (1) |
The voltage efficiency (VE) was calculated using the following equations:
 | (2) |
The H2 production rate (YH2) was calculated according to the following equation:
 | (3) |
The efficiencies of H2O2 (EH2O2) consumed for the HPOR (EH2O2-HPOR), crossover (EH2O2-HPRRcross), and disproportionation (EH2O2-HPDR) were calculated using the following equation:24,25
 | (4) |
 | (5) |
 | (6) |
3. Results and discussion
3.1 Effects of catalyst selection on the HPEL performance and H2O2 utilization
We first benchmarked the performance and H2O2 utilization of HPELs constructed using Co–N–C, Fe–N–C, and Pt/C anode catalysts under the same operating conditions (see the ESI for details, Fig. S1 and S2†). These catalysts have been used in the prototypes of PEM HPEL and UR-HPCC devices but lack a systematic comparison.24,25Fig. 1a shows the polarization curves of PEM HPELs with Co–N–C, Fe–N–C, and Pt/C anode catalysts at loadings of 1.0 mg cm−2, 1.0 mg cm−2 and 0.06 mgPt cm−2, respectively. The Co–N–C catalyst shows a lower open cell voltage (OCV) of ca. 0.71 V than those of Fe–N–C (0.81 V) and Pt/C (0.80 V) catalysts, closer to the theoretical reversible cell voltage (Erev = 0.704 V). The corresponding 8 mV onset overpotential (ηonset) yields a 98% maximum voltage efficiency (VE), which is higher than those of Fe–N–C (86.4%) and Pt/C catalysts (87.5%). The result is consistent with that in the RDE system (see the ESI for details, Fig. S3†). When the cell voltage (Ecell) increased from the OCV to 1.1 V, the HPOR current density (j) increased monotonically to ca. 380, 397, and 624 mA cm−2 for the Co–N–C, Fe–N–C, and Pt/C catalysts, respectively. Although the current density of the Pt/C catalyst surpasses that of the Co–N–C catalyst above 0.85 V, the Co–N–C catalyst shows higher HPOR activity than the Fe–N–C and Pt/C catalysts in the low voltage range (0.7–0.8 V). Since the HPOR is a two-electron process and the adsorbed hydroperoxyl (HOO*) is the sole intermediate, the binding free energy of HOO* (ΔGHOO*) can be used as the thermodynamic activity descriptor for HPOR catalysts.24,25 According to the Sabatier principle, the ΔGHOO* value for an ideal HPOR catalyst should be 4.225 eV at U = 0 V. The results of the density functional theory (DFT) calculations show that the ΔGHOO* values at the Co–Nx active sites in the Co–N–C catalyst, including Co–N4-pyrrolic (4.16 eV), Co–N2+2-pyridinic (3.66 eV), and Co–N4-pyridinic (3.50 eV), are closer to the ideal value than those of the Fe–N4-pyridinic active site (3.29 eV), Fe–N2+2-pyridinic (3.13 eV), and Fe–N4-pyrrolic (3.72 eV) in the Fe–N–C catalyst and the Pt/C catalyst (including Pt (111) (3.77 eV) and Pt (100) (3.28 eV)). And the corresponding free energy diagram of HPOR on these sites at U = 0.695 V are shown in Fig. 1b (see the ESI for additional details of DFT calculations and results, Fig. S4–S6 and Table S2†). This more ideal ΔGHOO* at Co–Nx sites is likely the origin of the higher HPOR activity and lower ηonset of the Co–N–C catalyst.30–32 Fig. S7a† compares the stability of PEM HPELs with the Fe–N–C, Co–N–C, and Pt–C as anodic catalysts reported elsewhere.24,25 In the 20 h stability tests, the voltage at 50 mA cm−2 changed from 0.852 to 0.840 V, 0.875 to 0.98 V, and 0.86 V to 0.94 V for the Pt–C, Fe–N–C and Co–N–C catalysts, respectively.24,25 These results suggest that Pt/C has the highest stability among these catalysts, and the stability of the Co–N–C catalyst is higher than that of the Fe–N–C catalyst. The results from the XPS analysis of Fe–N–C and Co–N–C anodes after the durability test show a decrease of the M–N percentage in the total amount of N (Fig. S7b–e†).24 These results suggest the loss of Fe–Nx or Co–Nx sites. Overall, the Co–N–C catalyst has higher HPOR performance and durability than the Fe–N–C catalyst, and therefore is more suitable for PEM HPEL systems. | ||
| Fig. 1 (a) Polarization curves of PEM HPELs with Co–N–C, Fe–N–C and Pt/C catalysts. (b) Reaction free energy diagram of the HPOR at Co–N2+2, Co–N4-pyridinic, and Co–N4-pyrrolic sites in the Co–N–C catalyst, Fe–N2+2, Fe–N4-pyridinic, and Fe–N4-pyrrolic sites in the Fe–N–C catalyst, and Pt(111), Pt(100) planes in the Pt/C catalyst. (c) The efficiency of H2O2 consumed for the HPOR (EH2O2-HPOR), disproportionation (EH2O2-HPDR), and crossover (EH2O2-HPRRcross) at different EiR-free values using the Co–N–C, Fe–N–C and Pt/C catalysts. | ||
Furthermore, we analyzed the effect of catalyst type on H2O2 utilization. Fig. 1c shows the efficiencies of H2O2 (EH2O2) consumed for H2 production (EH2O2-HPOR), disproportionation (EH2O2-HPDR), and crossover (EH2O2-HPRRcross) using the Co–N–C, Fe–N–C, and Pt–C anode catalysts, respectively. Among these catalysts, the Co–N–C catalyst shows the highest EH2O2-HPOR and lowest EH2O2-HPDR at all iR-corrected cell voltages (EiR-free) from 0.85 to 1.00 V. For all three catalysts, the EH2O2-HPOR increases with the EiR-free, while the EH2O2-HPDR and the EH2O2-HPRRcross decrease. These three variables are interdependent, and their sum is accountable for the total H2O2 consumption in the HPEL, hence
| EH2O2-HPOR + EH2O2-HPDR + EH2O2-HPRRcross = 100% | (7) |
Their interdependency suggests that by employing an anode catalyst with high activity and selectivity towards the HPOR, the loss of H2O2 to the HPDR and crossover can be reduced significantly. Thus, selecting catalysts with high HPOR activity is the key to enhancing H2O2 utilization.
More specifically, the Co–N–C catalyst's higher HPOR activity and lower HPDR activity resulted in higher EH2O2-HPOR values (consistently above 75%) and lower EH2O2-HPDR value (below 3%) throughout the applied voltage range. However, the excess H2O2 cannot be completely consumed at the anode, especially at a low cell voltage, so the H2O2 crossover can still occur. The corresponding EH2O2-HPRRcross is consistently above 5%, reaching about 18.3% at an EiR-free of 0.85 V for the Co–N–C catalyst. For the Pt/C and Fe–N–C anode catalysts, the higher EH2O2-HPDR is attributed to their high HPDR activity. The intense HPDR and HPOR within the Pt/C and Fe–N–C anode catalyst layers consume most H2O2 and reduce the concentration of H2O2 at the interface between the membrane and the catalyst layer, resulting in a low H2O2 crossover (EH2O2-HPRRcross).
These results show that different catalysts exhibit different behaviors for three competing reactions (HPOR, HPDR, and HPRRcross), directly affecting the HPEL performance and H2O2 utilization. For the Pt/C catalyst, the higher active site density leads to better HPOR activity under high potential operating conditions. However, the high HPDR activity of the Pt/C catalyst significantly reduces the H2O2 utilization. Compared to the Pt/C and Fe–N–C catalysts, the Co–N–C catalyst exhibited higher HPOR activity and weaker HPDR activities, which resulted in higher H2O2 utilization. High activity and selectivity towards the HPOR and low HPDR activities are essential for anode catalyst selection in PEM HPEL systems.
3.2 Effects of anode catalyst loading on HPEL performance and H2O2 utilization
We studied the effects of catalyst loading on the HPEL performance and H2O2 utilization using an anode with different loadings of the Co–N–C catalyst. Fig. 2 shows the cross-sectional scanning electron microscopy (SEM) images of the Co–N–C anode catalyst layer (ACL). As the catalyst loading increases from ca. 1.0 to 4.5 mg cm−2, the thickness of the ACL increases from ca. 6.5 ± 0.5 to 62.5 ± 10.5 μm (Fig. 2f). In the meantime, the coverage of the ACL on the carbon cloth increases with catalyst loading and forms a more continuous ACL, as shown in the top-view SEM micrographs (Fig. S8†). The texture of the carbon cloth, which is still visible at low catalyst loading, gradually becomes buried by the ACL as the loading increases. However, when the catalyst loading reached ca. 3.6 mg cm−2, thin cracks started appearing in the ACL and developed into wider cracks at ca. 4.5 mg cm−2 (Fig. S8†). | ||
| Fig. 2 Cross-sectional SEM micrographs of the Co–N–C ACL with loadings of (a) 1.0, (b) 1.7, (c) 2.8, (d) 3.6 and (e) 4.5 mg cm−2. False colors were applied for clarity. Green: carbon cloth; orange: catalyst layer. (f) Correlation between the catalyst layer thickness and the loading. | ||
Fig. S9† shows the cyclic voltammograms (CV) of anodes with different Co–N–C catalyst loadings. The double layer capacitance and the ECSA increase as the catalyst loading increases from 1.0 to 4.5 mg cm−2. Fig. 3a and Fig. S10a† show the corresponding PEM HPEL polarization curves, which show the close OCV of ca. 0.72–0.73 V. The HPOR current at low voltage (e.g. 0.8 V) increased as the loading increased from 1.0 to 3.6 mg cm−2, and then decreased as the loading reached 4.5 mg cm−2 (Fig. S10b†). The HPOR current density (j) at high voltage above 0.9 V decreased as the catalyst loading and CL thickness increased (e.g. from ca. 342 to 263 mA cm−2 at 1.00 V). Correspondingly, the internal resistance-corrected cell voltage (EiR-free) increased from 0.80 to 1.00 V and the H2 production rate (YH2) increased at each anode catalyst loading (Fig. 3b). However, YH2 increased from 4.54 to 5.6 mmol (cm2 h)−1 at 1.0 V as the catalyst loading increased from 1.0 to 3.6 mg cm−2. However, YH2 decreased to 3.62 mmol (cm2 h)−1 at 1.0 ViR-free as the anode catalyst loading increased to 4.5 mg cm−2. Fig. S11† shows the Nyquist plot of electrochemical impedance spectra (EIS) of the HPELs with different anode catalyst loadings measured at the OCV. The high-frequency resistance (HFR, corresponding to the intercept on the x-axis) increased slightly from 0.031 to 0.042 Ω when the anode catalyst loading increased from 1.0 to 4.5 mg cm−2. We believe that the slightly increased HFR values is the result of cracks in the ACL that lead to an apparent increase of electrical contact resistance.33,34 The anode polarization resistance (Rct,A) fluctuates slightly from 0.8 ± 0.08 to 0.72 ± 0.05 Ohm as the catalyst loading increased, while the cathode polarization resistance (Rct,C) maintained the same low values (ca. 0.01 ± 0.03 Ohm), as shown in Fig. 3c. Clearly, the mass transfer resistance of the anode, as characterized by Rct,A, is the main factor affecting the cell performance compared to Rct,C and RΩ. We suggest that at high voltages (> 0.9 V), oxygen generation in large amounts leads to an increase in mass transfer resistance, which is the main reason for the decrease in current density with anode catalyst loading.33,35,36
 | ||
| Fig. 3 Performance of PEM HPELs with Co–N–C catalyst loadings of 1.0, 1.7, 2.8, 3.6, and 4.5 mg cm−2. (a) Polarization curves, (b) YH2, (c) variation of R at the OCV with varying catalyst loadings. RΩ refers to the ohmic loss resistance; Rct,A and Rct,C refer to the anode and cathode faradaic resistance, respectively. 2D contour plots of (d) EH2O2-HPRRcross, (e) EH2O2-HPDR, and (f) EH2O2-HPOR as a function of the catalysts loading and the iR-free cell voltage. | ||
Fig. 3d–f show the effects of Co–N–C catalyst loading and EiR-free on the H2O2 utilization (EH2O2-HPOR) and the corresponding loss of H2O2 (EH2O2-HPRRcross, EH2O2-HPDR) in the 2D contour plots (see Fig. S12† for the original data). When the catalyst loading increases from 1.0 to 3.6 mg cm−2, the ACL thickness increased and inhibited the crossover of H2O2. As a result, the EH2O2-HPRRcross decreased with the catalyst loading when the loading was below 3.6 mg cm−2, as shown in Fig. 3d and Fig. S12.† However, when the catalyst loading further increased to 4.5 mg cm−2, wide cracks formed in the ACL and assisted the crossover of H2O2 (Fig. S8†). On the other hand, as the anode catalyst loading increased, the HPDR intensified due to the decreased HPOR performance, and the EH2O2-HPDR increased from 3.1% to 6.7% at an EiR-free of 0.85 ViR-free (Fig. 3e and Fig. S12†). Following the interdependent relationship described in eqn (7), the EH2O2-HPOR increases with catalyst loading and peaks at a loading of 3.6 mg cm−2 (ca. 92% at an EiR-free of 1.0 V) (Fig. 3f).
We suggest that the increase of anode catalyst loading leads to the increase of ACL thickness, which effectively inhibits the H2O2 crossover. However, especially under high voltage operating conditions, the ACL thickness increase significantly increases the mass transport resistance, leading to cell performance degradation. An optimal catalyst loading that balances the interdependent HPOR, HPDR and HPRRcross in the PEM HPEL system is required to maximize the H2O2 utilization.
3.3 Effects of PEM thickness on HPEL performance and H2O2 utilization
Furthermore, we investigated the effects of PEM thickness on the PEM HPEL performance and H2O2 utilization using Nafion membranes with different thicknesses, including Nafion NR 211 (25.4 μm), NR 212 (50.8 μm), NR 115 (127.0 μm), and NR 117 (183.0 μm) (Fig. 4 and Fig. S13†). With the same anode Co–N–C catalyst loading of ca. 1.7 mg cm−2, these HPELs show consistent anode CV curves (Fig. S14†). While the performance of HPELs decreases with the PEM thickness (Fig. S15†), their iR-free polarization curves are almost identical (Fig. 4a). When the EiR-free increased from the OCV to 1.0 V, the j of all HPELs increased monotonically to ca. 360 mA cm−2. Correspondingly, the H2 production rate (YH2) monotonically increases with the EiR-free and PEM thickness (Fig. 4b). In particular, YH2 increases from 2.6 to 7.4 mmol (cm2 h)−1 as the EiR-free increases from 0.8 to 1.0 V at a PEM thickness of 183 μm. The HFR obtained from the x-intercept of the EIS Nyquist plots increased from 0.038 to 0.12 Ω as the PEM thickness increased from 25.4 to 183.0 μm following the ohmic law (Fig. 4c, and S15†). The Rct,A and Rct,C maintained the same level as the PEM thickness increased (Fig. S16†). These results suggest that the PEM thickness does not significantly affect the anode polarization behavior within this range but mainly contributes to the ohmic loss of the system. | ||
| Fig. 4 Performance of PEM HPELs using Nafion membranes with different thicknesses. (a) Polarization curves, (b) YH2, and (c) RΩ at the OCV of PEM HPELs with different PEM thicknesses. 2D contour plots of (d) EH2O2-HPRRcross, (e) EH2O2-HPDR, and (f) EH2O2-HPOR as a function of the PEM thicknesses and the iR-free cell voltage. | ||
Fig. 4d–f show the dependence of H2O2 utilization on the PEM thicknesses and EiR-free. When the PEM thickness increases from 25.4 to 183.0 μm, the EH2O2-HPRRcross decreases due to the increased through-plane mass transport resistance for H2O2 crossover within the Nafion membrane (Fig. 4d). The EH2O2-HPDR decreases with the increase of EiR-free due to the accelerated HPOR rate. On the other hand, the EH2O2-HPDR increases with the PEM thickness, which could be attributed to the increased local H2O2 concentration within the CL as the H2O2 crossover is inhibited (Fig. 4e). The EH2O2-HPOR increased with PEM thickness and reached over 90% within the test voltage range (Fig. 4f), following the interdependent relationship described in eqn (6). These results suggest that thicker membranes can effectively inhibit the crossover of H2O2, resulting in lower EH2O2-HPRRcross and higher EH2O2-HPOR, but at the cost of ohmic loss. The tradeoff of EH2O2-HPOR and iR loss via the optimization of the PEM thickness is needed to maximize the H2O2 utilization. Membrane materials with low permeability to H2O2 might also improve the H2O2 utilization in PEM HPELs.
4. Conclusions and outlook
In summary, we investigated the effects of catalyst type, catalyst loading, and PEM thickness on the performance and H2O2 utilization of a PEM HPEL. The results unveiled the interdependency of three fundamental processes, the HPOR, HPDR, and H2O2 crossover, which mutually determine the H2O2 utilization towards the power-to-hydrogen conversion in the HPEL. Different anode catalysts provide various activities and selectivity towards the HPOR and HPDR. The Co–N–C catalyst is identified as the best anode catalyst for its high activity and selectivity towards the HPOR and low degree of the HPDR. The excellent HPOR activity of the Co–N–C catalyst is attributed to the optimal ΔGHOO* values at the Co–N4 sites. In contrast, the Fe–N–C and Pt/C catalysts show higher HPDR activity, lowering H2O2 utilization. Increasing the anode catalyst loading and PEM thickness can effectively inhibit the H2O2 crossover and improve the H2O2 utilization but inevitably lead to higher H2O2 loss due to the HPDR and ohmic loss. These findings imply the need for developing highly selective HPOR catalysts and low-H2O2-permeability membranes to boost the performance and H2O2 utilization of next-generation PEM HPELs for renewable energy storage. In addition, the rational design and fabrication of 2e-ORR electrocatalysts with high activity and stability are still necessary to improve the efficiency of H2–H2O2 cyclic energy storage.Author contributions
Jie Yang: writing – original draft, methodology, investigation, and visualization. Ruimin Ding: methodology, investigation, visualization, and supervision. Chang Liu: methodology, investigation, and visualization. Lifang Chen: investigation and visualization. Qi Wang: investigation and visualization. Shanshan Liu: investigation. Qinchao Xu: investigation. Xi Yin: conceptualization, methodology, writing – review and editing, supervision, and funding acquisition.Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Xi Yin, Ruimin Ding, Chang Liu, Shanshan Liu, and Jie Yang have patent CN 202110721698.6 pending to the Institute of Coal Chemistry, Chinese Academy of Sciences. Xi Yin, Jie Yang, Chang Liu, Ruimin Ding, and Shanshan Liu have patent CN202111123206.X pending to the Institute of Coal Chemistry, Chinese Academy of Sciences.Acknowledgements
This study was financially supported by the Shanxi Province grant (Grant No. 20210302124441 and 20210302123011), the Shanxi Provincial Key Research and Development Projects (202102070301018), the autonomous research project of SKLCC (Grant No. 2021BWZ006), and ICC CAS (Grant No. 2020SC001).References
- S. Chu and A. Majumdar, Opportunities and Challenges for A Sustainable Energy Future, Nature, 2012, 488, 294–303 CAS.
- X. Yang, C. P. Nielsen, S. Song and M. B. McElroy, Breaking the Hard-to-Abate Bottleneck in China's Path to Carbon Neutrality with Clean Hydrogen, Nat. Energy, 2022, 7, 955–965 CAS.
- P. Nikolaidis and A. Poullikkas, A Comparative Overview of Hydrogen Production Processes, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CAS.
- B. Pivovar, N. Rustagi and S. Satyapal, Hydrogen at Scale (H2 @Scale): Key to a Clean, Economic, and Sustainable Energy System, J. Electrochem. Soc., 2018, 27, 47–52 CAS.
- L. Barreto, A. Makihira and K. Riahi, The Hydrogen Economy in the 21st Century: A Sustainable Development Scenario, Int. J. Hydrogen Energy, 2003, 28, 267–284 CAS.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Anion-Exchange Membrane Water Electrolyzers, Chem. Rev., 2022, 122, 11830–11895 CAS.
- H. Jin, B. Ruqia, Y. Park, H. J. Kim, H.-S. Oh, S.-I. Choi and K. Lee, Nanocatalyst Design for Long-Term Operation of Proton/Anion Exchange Membrane Water Electrolysis, Adv. Energy Mater., 2021, 11, 2003188 CAS.
- B. Lee, J. Heo, S. Kim, C. Sung, C. Moon, S. Moon and H. Lim, Economic Feasibility Studies of High Pressure Pem Water Electrolysis for Distributed H2 Refueling Stations, Energy Convers. Manage., 2018, 162, 139–144 CAS.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Green Hydrogen from Anion Exchange Membrane Water Electrolysis: A Review of Recent Developments in Critical Materials and Operating Conditions, Sustainable Energy Fuels, 2020, 4, 2114–2133 CAS.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Durability of Anion Exchange Membrane Water Electrolyzers, Energy Environ. Sci., 2021, 14, 3393–3419 CAS.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Review on Photocatalytic and Electrocatalytic Artificial Nitrogen Fixation for Ammonia Synthesis at Mild Conditions: Advances, Challenges and Perspectives, Nano Res., 2019, 12, 1229–1249 CAS.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, Electrochemical Ammonia Synthesis—The Selectivity Challenge, ACS Catal., 2017, 7, 706–709 CAS.
- X. Cui, C. Tang and Q. Zhang, A Review of Electrocatalytic Reduction of Dinitrogen to Ammonia under Ambient Conditions, Adv. Energy Mater., 2018, 8, 1800369 Search PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, The Evolution and Future of Earth's Nitrogen Cycle, Science, 2010, 330, 192–196 CAS.
- A. E. Sanli and A. Aytaç, Response to Disselkamp: Direct Peroxide/Peroxide Fuel Cell as a Novel Type Fuel Cell, Int. J. Hydrogen Energy, 2011, 36, 869–875 CAS.
- A. E. Sanlı, A Possible Future Fuel Cell: The Peroxide/Peroxide Fuel Cell, Int. J. Energy Res., 2013, 37, 1488–1497 Search PubMed.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Hydrogen Peroxide as a Sustainable Energy Carrier: Electrocatalytic Production of Hydrogen Peroxide and the Fuel Cell, Electrochim. Acta, 2012, 82, 493–511 CAS.
- N. Soltani, A. Bahrami, L. Giebeler, T. Gemming and D. Mikhailova, Progress and Challenges in Using Sustainable Carbon Anodes in Rechargeable Metal-Ion Batteries, Prog. Energy Combust. Sci., 2021, 87, 100929 Search PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Current Status and Future Directions of Multivalent Metal-Ion Batteries, Nat. Energy, 2020, 5, 646–656 CAS.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Redox-Flow Batteries: From Metals to Organic Redox-Active Materials, Angew. Chem., Int. Ed., 2017, 56, 686–711 CAS.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, Redox Flow Batteries: A Review, J. Appl. Electrochem., 2011, 41, 1137–1164 CAS.
- M. Gencten and Y. Sahin, A Critical Review on Progress of the Electrode Materials of Vanadium Redox Flow Battery, Int. J. Energy Res., 2020, 44, 7903–7923 CAS.
- R. Ding, J. Yang, C. Liu, S. Liu, L. Chen, Q. Xu, J. Chen, J. Li and X. Yin, (Digital Presentation) Hydrogen Peroxide Electrolyzer and Reversible Hydrogen Peroxide Cycle Cell for Renewable Energy Storage, Meet. Abstr., 2022, 2495 Search PubMed , MA2022-01.
- J. Yang, R. Ding, C. Liu, S. Liu, Q. Xu, L. Chen, J. Chen, J. Li and X. Yin, Highly Efficient Unitized Regenerative Hydrogen Peroxide Cycle Cell with Ultralow Overpotential for Renewable Energy Storage, J. Power Sources, 2022, 545, 231948 CAS.
- C. Liu, R. Ding, J. Yang, S. Liu, L. Chen, Q. Xu, J. Li and X. Yin, Low-Voltage Hydrogen Peroxide Electrolyzer for Highly Efficient Power-to-Hydrogen Conversion, ACS Sustainable Chem. Eng., 2023, 11, 2599–2606 CAS.
- R. Serra-Maia, M. Bellier, S. Chastka, K. Tranhuu, A. Subowo, J. D. Rimstidt, P. M. Usov, A. J. Morris and F. M. Michel, Mechanism and Kinetics of Hydrogen Peroxide Decomposition on Platinum Nanocatalysts, ACS Appl. Mater. Interfaces, 2018, 10, 21224–21234 CAS.
- N. Kitajima, S. Fukuzumi and Y. Ono, Formation of Superoxide Ion During the Decomposition of Hydrogen Peroxide on Supported Metal Oxides, J. Phys. Chem. C, 1978, 82, 1505–1509 CAS.
- X. Yin, H. T. Chung, U. Martinez, L. Lin, K. Artyushkova and P. Zelenay, PGM-Free ORR Catalysts Designed by Templating PANI-Type Polymers Containing Functional Groups with High Affinity to Iron, J. Electrochem. Soc., 2019, 166, F3240 CAS.
- K. Fic, A. Platek, J. Piwek and E. Frackowiak, Sustainable materials for electrochemical capacitors, Mater. Today, 2018, 21, 437–454 CAS.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. H. Antink, S. Park, K.-S. Lee, S.-P. Cho, J. S. Yoo, Y.-E. Sung and T. Hyeon, Atomic-level tuning of Co-N-C catalyst for high-performance electrochemical H2O2 production, Nat. Mater., 2020, 19, 436–442 CAS.
- J. Gao, H. b. Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Enabling Direct H2O2 Production in Acidic Media through Rational Design of Transition Metal Single Atom Catalyst, Chem, 2020, 6, 658–674 CAS.
- X. X. Wang, D. A. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Nitrogen-Coordinated Single Cobalt Atom Catalysts for Oxygen Reduction in Proton Exchange Membrane Fuel Cells, Adv. Mater., 2018, 30, 1706758 Search PubMed.
- A. M. Damjanović, B. Koyutürk, Y.-S. Li, D. Menga, C. Eickes, H. A. El-Sayed, H. A. Gasteiger, T.-P. Fellinger and M. Piana, Loading Impact of a PGM-Free Catalyst on the Mass Activity in Proton Exchange Membrane Fuel Cells, J. Electrochem. Soc., 2021, 168, 114518 Search PubMed.
- J. Lopata, Z. Kang, J. Young, G. Bender, J. W. Weidner and S. Shimpalee, Effects of the Transport/Catalyst Layer Interface and Catalyst Loading on Mass and Charge Transport Phenomena in Polymer Electrolyte Membrane Water Electrolysis Devices, J. Electrochem. Soc., 2020, 167, 064507 CAS.
- A. Baricci, A. Bisello, A. Serov, M. Odgaard, P. Atanassov and A. Casalegno, Analysis of the Effect of Catalyst Layer Thickness on the Performance and Durability of Platinum Group Metal-Free Catalysts for Polymer Electrolyte Membrane Fuel Cells, Sustainable Energy Fuels, 2019, 3, 3375–3386 CAS.
- J. E. Park, M. Karuppannan, O. J. Kwon, Y.-H. Cho and Y.-E. Sung, Development of High-performance Membrane-electrode Assembly in Unitized Regenerative Fuel Cells, J. Ind. Eng. Chem., 2019, 80, 527–534 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description including the materials, preparation of the catalysts, physical characterization studies, electrochemical measurements, computational details, and supporting figures. See DOI: https://doi.org/10.1039/d3gc03200k |
| This journal is © The Royal Society of Chemistry 2024 |