
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChallenges and opportunities for the photo-(thermal) synthesis of ammonia
Diego
Mateo
*,
Angel
Sousa
,
Maksim
Zakharzhevskii
and
Jorge
Gascon
 *
*
Advanced Catalytic Materials, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: Jorge.gascon@kaust.edu.sa; Diego.mateo@kaust.edu.sa
First published on 7th December 2023
Abstract
For more than one century, the synthesis of ammonia (NH3) through the Haber–Bosch route has allowed the industrial-scale production of fertilizers and other nitrogen-containing compounds. Despite its success, the development of alternative NH3 synthesis methods with a lower CO2 footprint is highly desirable. In this context, the use of sunlight as source of energy to perform the photo-catalytic nitrogen (N2) reduction reaction represents a promising approach, although the reported efficiencies remain below those required to compete with the traditional HB route. To overcome these limitations, photo-thermal catalysis offers the opportunity to synergistically combine the effect of photo-induced charge carriers and localized heat to perform the synthesis of ammonia. In this review, we first provide an overview of the most recent advances in thermo- and photo-catalytic NH3 production. Next, we describe the merits of photo-thermal catalysis to enhance the catalytic activity towards NH3 synthesis. To conclude, we share our viewpoint on the future prospects and challenges ahead for photo-thermal catalysis to become a reality in the production of NH3 and other industrially relevant chemicals.
Introduction
Ammonia (NH3) synthesis is considered one of the most relevant processes in the chemical industry. Together at BASF, German chemists Fritz Haber (1868–1934) and Carl Bosch (1874–1940) developed in 1913 the so-called Haber–Bosch (HB) process for the industrial-scale manufacturing of ammonia.1,2 This breakthrough contributed to the extensive production of NH3 and its application to the large-scale fabrication of fertilizers, explosives and other nitrogen-containing chemicals of industrial interest. After more than one century, the actual ammonia synthesis has barely changed, and indeed circa 95% of the total ammonia production is still obtained through the Haber–Bosch route.Despite the success of the traditional thermo-catalytic ammonia synthesis, the pursuit of higher efficiency, cost reduction, and environmental sustainability remains ongoing. In this regard, the synthesis of ammonia consumes 3–5% of the world's natural gas production, representing 1–2% of the annual energy supply and approximately 1% of all greenhouse gas generation.3,4 In view of this, the current focus of research aims to further optimize the process and explore novel approaches to meet the rising global demand for ammonia while minimizing energy consumption and greenhouse gas emissions. One promising strategy in this vein is the use of blue hydrogen (H2) for ammonia production.5 This method leverages existing fossil fuel infrastructure for exploitation and distribution while minimizing carbon dioxide (CO2) emissions through carbon capture and storage (CCS). Recent reports indicate, indeed, that the use of blue ammonia can be a present-day convenient solution to mitigate the climate impacts associated with the conventional ammonia production route, at least until green ammonia becomes economically profitable.6 In fact, in September 2020 ARAMCO and the Institute of Energy Economics (Japan), in partnership with SABIC, successfully achieved the production and shipment of blue ammonia from Saudi Arabia to Japan, thus representing the world's first blue ammonia supply-chain demonstration.7 Unfortunately, this approach still relies on fossil gas, leading to a high concentration of HB facilities near the gas supply points and increasing transportation costs to remote or underdeveloped locations.8
To achieve decentralized ammonia production and move towards a carbon-free economy, alternative methods for the reduction of nitrogen (N2) have been proposed. Among these methods, photo-catalysis represents an intelligent approach to drive chemical reactions by taking advantage of the solar energy. Assuming water or renewable H2 as source of protons, photo-catalysis enables a CO2-free NH3 synthesis using sunlight as driving force. Besides, this strategy offers the additional advantage of allowing small on-site NH3 synthesis plants powered by renewable energy, which essentially favors local production of ammonia close to the final consumption points. Nevertheless, and despite all these potential benefits, the reported efficiencies of most of the photo-catalytic systems for N2 fixation described to date are still insufficient to meet industrial-scale requirements.9
In this context, the integration of both thermo-catalysis and photo-catalysis (from now on, “photo-thermal catalysis”) has lately appeared as an excellent opportunity to both reduce the energy requirements and increase the catalytic efficiency of many chemical processes, including CO2 conversion, H2 generation or the degradation of pollutants, among others.10–15 Thanks to the combination of photo-induced localized heating and photo-excited charge carriers, many photo-thermal catalysts have exhibited comparable or even higher catalytic rates than those displayed under pure thermal conditions. For these reasons, it is quite surprising that, to date, only a few works have drawn attention to the synergistic combination of light and heat to perform the NH3 synthesis reaction, thus remaining as an almost unexplored field of research. Given the emerging prospects of photo-thermal catalysis and the extraordinary relevance of ammonia in our modern societies, we herein present a review of the newest advances in thermo-catalytic, photo-catalytic and synergistic photo-thermo-catalytic NH3 synthesis during the last five years. The goal is to give an overview of the up-to-date strategies of traditional thermo- and photo-catalytic NH3 production and, at the same time, to highlight the potential of photo-thermal catalysis as a hybrid tool to break the limitations of its individual thermochemical and photochemical counterparts. In this review, we will first briefly introduce the most well-accepted reaction pathways for NH3 synthesis in thermo- and photo-catalysis. Then, we will describe recent advancements in thermo-catalytic and photo-catalytic NH3 synthesis, paying special attention to novel strategies to moderate reaction conditions and/or improve the catalytic performance. After that, we will outline a summary of the merits of photo-thermal catalysis to boost the catalytic activity towards NH3 production, giving specific examples found in the literature. Lastly, we will share our opinion on the future directions and challenges ahead for photo-thermal catalysis to become a viable technology for the production of NH3 and other commodities of industrial interest.
Reaction pathways for NH3 synthesis
The reaction of N2 and H2 to produce NH3 (eqn (1)) is an exothermic process thermodynamically favored by high pressures and low temperatures.| N2 + 3H2 ↔ 2NH3 ΔH° = −92.2 kJ mol−1 | (1) |
Mechanistically, the catalytic N2 reduction reaction is a multi-step process that involves: (i) N2 adsorption, (ii) N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond activation and/or cleavage, (iii) N2(ads) or N(ads) hydrogenation and (iv) NH3 desorption.16,17 Traditionally, two broadly accepted mechanisms have been proposed for the N2 reduction reaction, namely the dissociative pathway and the associative pathway (Fig. 1).18
N bond activation and/or cleavage, (iii) N2(ads) or N(ads) hydrogenation and (iv) NH3 desorption.16,17 Traditionally, two broadly accepted mechanisms have been proposed for the N2 reduction reaction, namely the dissociative pathway and the associative pathway (Fig. 1).18
 | ||
| Fig. 1 Reaction pathways for the N2 reduction reaction to ammonia. Adapted with permission from ref. 18. Copyright (2017) Elsevier. | ||
In the dissociative pathway, the NN bond cleavage takes place before the hydrogenation of the nitrogen atom. In view of the high dissociation energy of the first N–N bond (410 kJ mol−1), this route is extremely energy-intensive, and, indeed, traditional thermochemical HB systems are believed to follow this mechanism for the production of ammonia.19 On the contrary, the associative pathway is based on the sequential hydrogenation of nitrogen atoms while bonded to each other. Two different sub-types of associative pathways are possible depending on the hydrogenation site.
In the alternating associative pathway, nitrogen atoms of adsorbed N2 are successively hydrogenated and, eventually, NH3 is desorbed after the cleavage of the N–N bond. In the distal associative pathway, however, the hydrogenation occurs preferably on the distal nitrogen atom.9 Overall, the associative pathway does not require the cleavage of the first bond in N2 molecule so in these cases the energy requisites are considerably lower. In consequence, the associative pathway is expected to be the preferential mechanism for electro- or photo-catalytic NH3 production systems.
Thermo-catalytic NH3 synthesis
As previously mentioned, NH3 synthesis reaction is thermodynamically enhanced at high pressures and low temperatures. From a kinetic point of view, however, temperatures above 200 °C are necessary in order to achieve significant conversion values. For these reasons, as a compromise between kinetics and thermodynamics, thermo-catalytic NH3 synthesis is generally conducted under medium temperatures and high pressures (400–450 °C and 150–250 atm), achieving conversion rates of 10–15% per pass.20 These operating conditions represent the main drawback for the thermo-catalytic NH3 synthesis, not only for the elevated consumption of energy but also for the limited stability of the catalysts. For these reasons, one of the main challenges in the field is the development of systems which enable milder reaction conditions, especially in terms of operating pressure, as compression cycles represent the major capital cost of the whole HB process.Fe-based catalysts
Iron-based catalysts represent, undoubtedly, the most investigated and applied materials in the NH3 synthesis reaction. Particularly, iron-fused catalyst derived from hematite (Fe2O3), magnetite (Fe3O4) and/or wüstite (Fe1−xO) are the most commonly used in modern industries. Nevertheless, it is also true that early studies conducted with iron catalysts at BASF facilities showed quite disappointing activities and, in fact, this served as a spur for the study of the effect of additives on the catalytic performance. A comprehensive investigation was developed in between 1909 and 1911 by Carl Bosch's assistant Alwin Mittasch, who conducted a systematic testing of 2500 different materials in order to find an efficient and stable catalyst for the NH3 synthesis.21 The scrutiny was prolonged until 1922 including about 5000 different candidates and eventually the best catalyst resulted to be a multi-component mixture consisting of magnetite-based iron catalyst with small amounts of alumina, calcium oxide, and potassium alkali.21 This exhaustive screening demonstrated the importance of the addition of promoters to the formulation of catalysts in order to provide beneficial structural and electronic effects, as we will later elaborate in this review.Supported iron catalysts are also an appealing subfamily of ammonia synthesis catalysts. In these cases, carbon or metal oxides usually act as support materials for active iron species. When it comes to carbon-based catalysts, the one-step pyrolysis of metal–organic compounds has lately emerged as a straightforward method to obtain well-dispersed metal nanoparticles (NPs) encapsulated into a carbon matrix. For instance, Morlanés et al. developed a novel catalyst based on iron NPs embedded on carbon through the controlled pyrolysis at 735 °C of iron phthalocyanines.22 Upon promotion with Cs, the as-prepared catalyst (10%Cs-FePc) exhibited a stable production of NH3 in the order of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μmol g−1 h−1 at 400 °C and 3 MPa, even surpassing an iron benchmark catalyst (Fe_KM1) under similar reaction conditions (Fig. 2A–C). Long-term stability tests also demonstrated an outstanding stability of the catalyst after more than 100 hours of reaction (Fig. 2D).
000 μmol g−1 h−1 at 400 °C and 3 MPa, even surpassing an iron benchmark catalyst (Fe_KM1) under similar reaction conditions (Fig. 2A–C). Long-term stability tests also demonstrated an outstanding stability of the catalyst after more than 100 hours of reaction (Fig. 2D).
 | ||
| Fig. 2 Catalytic performance of the phthalocyanine-derived catalysts for the ammonia synthesis reaction. (A) Pressure effect on the NH3 synthesis rate at 400 °C. (B) Temperature effect on the NH3 synthesis rate at 1 MPa. (C) Specific activity in μmol NH3 h−1 gmetal−1 and TOF (s−1) (diagonal stripes) at 400 °C and 3 MPa. (D) Stability tests in the range of 400–520 °C and 3–7 MPa. Reproduced with permission from ref. 22. Copyright (2020) Royal Society of Chemistry. | ||
The promotion with Cs was crucial to boost the catalytic activity and authors speculated that the presence of alkali both decreased the energetic barrier for nitrogen dissociation and increased the number of surface active sites by destabilization of adsorbed NHx species. Interestingly, homologous materials prepared by conventional impregnation of carbon with iron/alkali displayed much lower activity. The authors concluded, therefore, that the remarkable stability and catalytic activity of 10%Cs-FePc directly stemmed from the synthetic approach based on the pyrolysis of phthalocyanine precursor. A follow-up work from the same group demonstrated that it was also possible to obtain highly efficient bimetallic Fe–Co catalysts through the physical mixture of Fe and Co phthalocyanines followed by the pyrolysis treatment.23 Under the optimal Fe:Co ratio, the bimetallic catalyst outperformed the monometallic Fe and Co counterparts, exhibiting a NH3 production of 53666 μmol g−1 h−1 at 400 °C and 7 MPa. In a related precedent, the groups of Zou and Ma reported a K-promoted Fe/C catalyst derived from the pyrolysis under He of Fe-based metal–organic-framework (MOF) xerogels.24 Upon addition of 1% K to the MOF-derived Fe catalyst, the catalytic activity reached 30400 μmol g−1 h−1 at 400 °C and 3 MPa, approximately 3 times higher compared to the sample without potassium. X-ray photoelectron spectroscopy (XPS) measurements revealed a shift to lower binding energies for the Fe 2p peak along with the increase of potassium concentration in the samples, thus suggesting a charge transfer from K ions to the surface of iron. Eventually, this electron-rich environment on the Fe surface favors nitrogen activation and improves the catalytic activity of promoted samples. Curiously, potassium not only enhanced the catalytic performance by modulating the electronic properties of the Fe active sites but also by increasing the thermal stability of the catalyst. Authors observed that unpromoted samples suffered from a severe deactivation after 24 h of reaction under high temperature conditions (475 °C). In situ temperature-programed reaction-mass spectrometry (TPR-MS) measurements demonstrated that the deactivation of the samples stemmed from the methanation of the catalyst under reaction conditions, which at the end led to carbon loss and agglomeration of Fe nanoparticles. However, under the same reaction conditions K-promoted samples retained more than 90% of their activity due to the inhibition of the methanation reaction by potassium.
Apart from carbon, oxyhydrides have been recently explored as potential supports in the formulation of new NH3 synthesis catalysts. In these cases, a low content of iron in the order of 1–2% in weight is deposited on the inorganic support, in sharp contrast with the high iron loading of up to 50% that can be achieved through the controlled pyrolysis of Fe-containing carbon-based precursors. For example, the groups of Kobayashi and Kageyama reported the preparation of 1% Fe/BaTiO2.35H0.65 catalyst with a remarkable catalytic activity of 14000 μmol g−1 h−1 at 400 °C and 5 MPa.25 Authors found that the oxyhydride support could successfully inhibit H2 poisoning by removing the excess of adsorbed hydrogen through an effective spillover pathway. Furthermore, the oxyhydride served as electron donation source to the Fe metal sites, thus reducing the strength of the triple bond of adsorbed N2 and, consequently, improving the catalytic activity. Further research from Kitano and co-workers demonstrated that the presence of other heteroanions like nitride (N3−) could enhance even more the performance of oxyhydrides as NH3 catalysts.26 In this regard, authors developed a new perovskite oxynitride–hydride BaCeO3−xNyHz loaded with small amounts of Fe (1.2 wt%). Using this catalyst, the NH3 synthesis rate at 400 °C and 0.9 MPa was 6800 μmol g−1 h−1. In comparison, Fe-supported BaCeO3 displayed a catalytic rate in the range of 30 μmol g−1 h−1. Authors performed additional isotopic experiments in order to disentangle the promotion effect of Fe-BaCeO3−xNyHz and found that, unexpectedly, the rate-determining step shifted from N2 dissociation to N–H bond formation due to the presence of N3− and H− anions. Ultimately, this change in the reaction mechanism translated into a lower activation energy for the ammonia synthesis (46–62 kJ mol−1). In the case of Fe-BaCeO3, however, the NH3 formation followed the conventional mechanism characterized by high-energy barriers (85–121 kJ mol−1).
Ru-based catalysts
In spite that iron-based catalysts have enjoyed a historical hegemony in the Haber–Bosch process, it was in the 1970s when a consortium between British Petroleum and M.W. Kellogg companies developed a novel carbon-supported ruthenium catalyst (Ru/C) for the NH3 synthesis reaction.27 Since then, the research on innovative Ru-based catalysts has gained considerable relevance and a vast number of supports such as perovskites, oxides or carbon-derived materials have been investigated.28–33For instance, the use of ceria (CeO2) has been extensively reported due to its abundant oxygen vacancies that increase the electron density of Ru sites to promote N2 activation.34 Furthermore, it has been found that parameters like morphology of CeO2 or metal–support interaction are key to maximize the catalytic activity of the resultant materials.35 In this regard, Ma and co-workers compared the catalytic activity for NH3 synthesis of three different morphologies of ceria (nanocubes, nanorods, and nanoparticles) with a nominal Ru loading of 4%.36 At 400 °C and only 1 MPa, ceria nanorods displayed the highest catalytic activity (3830 μmol g−1 h−1) followed by ceria nanocubes (1289 μmol g−1 h−1) and ceria nanoparticles (529 μmol g−1 h−1). XPS measurements also revealed the presence of CeH2+x hydride on the samples and, curiously, authors identified a positive correlation between the amount of this hydride and the catalytic activity. Basically, the role of the CeH2+x species seemed to be the electron donation to the Ru active sites, thus increasing the catalytic performance towards NH3 synthesis. More recently in 2021, Li and collaborators described how the treatment of Ru-CeO2 catalyst with hydrazine (N2H4) could modulate the electronic metal–support interaction and boost the catalytic activity in the NH3 synthesis reaction.37 Authors found that the mild reduction of Ru–CeO2 with hydrazine followed by H2 treatment at 500 °C caused an increase in (i) the fraction of metallic Ru, (ii) the concentration of Ce3+ and (iii) the number of exposed species of Ru compared to the conventional reduction with only H2. In the absence of N2H4 treatment, oxygen migration from the ceria support to the Ru species led to the lowering of exposed metallic Ru. However, the treatment with N2H4 improved the electronic metal–support interaction to create electron-rich states at the Ru sites and, eventually, increased the activity of Ru-CeO2. Interestingly, a very recent work from Feng and collaborators revealed that ceria could act not only as a support but also as a promoter of Ru-based catalysts.38 In their work, authors deposited small clusters of ceria on a Ru/Al2O3 catalyst and demonstrated that these sub-nanometric ceria domains could prevent the H2 poisoning of Ru active sites. Furthermore, XPS data suggested that ceria clusters acted as electron donors to modulate the electronic structure of Ru, thus facilitating the N2 adsorption and H2 dissociation on its surface. Apart from ceria, other metal oxides such as MgO, La2O3 or Y2O3 have been recently explored as potential supports for Ru-based catalysts for the HB reaction.39–41
Besides metal oxides, other materials like hydrides, nitrides, or amides have lately demonstrated to be excellent hosts for Ru in the preparation of new ammonia synthesis catalysts, even outperforming their Fe-based counterparts.42–45 For instance, Kitano et al. developed a Ru/Ca(NH2)2 catalyst promoted with Ba to create Ru–Ba core–shell nanoparticles supported on the calcium amide.45 The as-prepared catalyst displayed an outstanding activity towards NH3 synthesis of 60400 μmol g−1 h−1 at 360 °C and pressure as low as 0.9 MPa, thus exceeding by 6 times the performance of the industrial benchmark Fe catalyst (Fig. 3). Furthermore, Ru–Ba/Ca(NH2)2 could operate at relatively low temperatures below 300 °C, achieving NH3 production rates of 7500 μmol g−1 h−1 at only 260 °C.
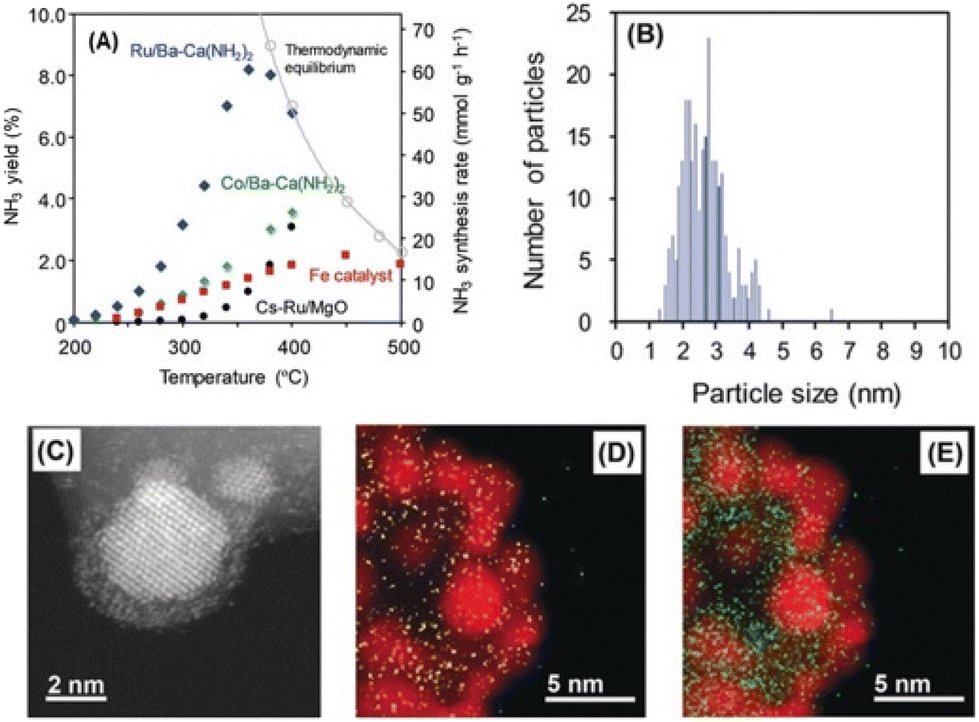 | ||
| Fig. 3 (A) Temperature dependence on the catalytic performance of Ru/Ba-Ca(NH2)2 catalyst for the ammonia synthesis reaction at 0.9 MPa. (B) Particle size distribution and (C) HAADF-STEM image of Ru/Ba-Ca(NH2)2 catalyst. EDX elemental mapping images of (D) Ba (yellow) and (E) Ca (green), where Ru is displayed as red. Reproduced with permission from ref. 45. Copyright (2018) John Wiley and Sons. | ||
In fact, one of the main advantages of Ru catalysts compared to their Fe-based equivalents lies in their improved catalytic activities under mild reaction conditions of temperature and pressure, therefore contributing to a considerable reduction of the capital cost of ammonia plants. Nevertheless, it is still hard to imagine a scenario in which Ru catalysts widely substitute traditional Fe catalysts, especially if one takes into account the high cost and limited availability of Ru. In the specific case of carbonaceous supports, the presence of Ru can also lead to the partial loss of the carbon due to its detrimental methanation, thus reducing the long-term stability of the catalyst.46 All these drawbacks still hamper the broad implementation of Ru catalysts in conventional Haber–Bosch plants and, indeed, by 2010 only sixteen ammonia plants around the globe operated using commercial ruthenium-based catalysts.21
Co-based catalysts
Alternative first-raw transition metals like cobalt or nickel have been explored in recent years as potential catalysts for NH3 synthesis.47,48 Particularly for Co-based catalysts, notable catalytic activities at moderate operating conditions were achieved. For example, in a similar way to Fe catalysts derived from the pyrolysis of carbonaceous precursors, in 2020 Morlanés and collaborators reported the use of a barium-promoted Co/C catalyst obtained from the pyrolysis of Co phthalocyanine at 735 °C.22 The as-prepared materials displayed an ammonia production of 10250 μmol g−1 h−1 at 400 °C and 3 MPa. As in the case of Cs for Fe/C catalyst, the promotional effect of Ba on Co/C catalyst was uniquely ascribed to its electron donation activity but not to a protective effect against methanation of the carbon support. In 2015, the group of Pilecka evaluated the effect of barium as promoter of an activated carbon-supported Co catalyst for ammonia production.49 Under optimal conditions, Ba–Co/C catalyst achieved a NH3 production of 88235 μmol g−1 h−1 at 400 °C and a high pressure of 9 MPa. However, in contradiction with the work of Morlanés et al., in this case authors found that even small amounts of Ba were able to enhance the catalytic activity by suppressing the undesirable methanation of the carbon matrix. All these results suggest that the nature and preparation protocol of the carbonaceous supports are decisive to determine both the activity and stability of the resultant catalysts.
The use of inorganic supports has been also described in the formulation of Co-based catalysts. Kitano and co-workers prepared a perovskite oxynitride-hydride, BaCeO3−xNyHz, which was loaded with a 4.7 wt% of Co.26 The as-prepared catalyst achieved an outstanding NH3 production rate of 10100 μmol g−1 h−1 at 400 °C and a very low pressure of 0.9 MPa. When authors decreased the temperature to 300 °C keeping constant the pressure, the catalytic activity significantly decreased to ∼2000 μmol g−1 h−1. It is worth noticing that, under the same reaction conditions, an analogous catalyst with a 4.3 wt% of Ru displayed a NH3 production rate close to 5000 μmol g−1 h−1, thus highlighting the promising attributes of Co-based catalysts. In the last years, the group of Jiang has performed an extensive research on Co/CeO2-based catalysts for the HB process. As we discussed in the previous section for CeO2-supported Ru catalysts, authors found that the shape of CeO2 (polyhedral, nanorods or hexagonal morphology) was critical to optimize the catalytic performance of a series of Co-based catalysts.50 Authors suggested that the morphology had a strong influence of the reducibility of both Co oxides and CeO2 support, thus modifying the metal–support interaction. Co NPs supported on CeO2 polyhedrons displayed the best catalytic activity achieving 86000 μmol g−1 h−1 at 430 °C and 10 MPa. In a follow-up work, the same authors studied the effect of alkali promoters (Ba and K) on the catalytic activity of Co/CeO2 catalysts.51 Quite surprisingly, Ba showed a positive effect on the performance but the addition of K, even at small quantities, was detrimental to improve the catalytic activity. Later in 2019, authors made a last effort to strengthen the metal–support interaction of Co/CeO2 catalysts.52 To this end, they used dopamine during the synthesis to favor the stability of Co NPs and increase the interaction between Co and CeO2 thanks to a strong interfacial bonding. This improved metal–support interaction was accompanied by a reduction of the particle size of Co, a better reducibility and a lower activation energy for N2 desorption. Under optimal conditions, the Co/CeO2 catalyst exhibited an outstanding NH3 production rate of 19120 μmol g−1 h−1 at 425 °C and a pressure as low as 1 MPa.
In general, most of the productivity values presented in this section are comparable or even outperform those achieved by many Ru-based catalysts. Taking into account that the main advantage of Ru-based catalysts is the possibility to operate under low pressure, Co-based catalysts have exhibited a huge potential to compete with Ru in the development of efficient yet affordable NH3 catalysts operative under mild reaction conditions.
Photo-catalytic NH3 synthesis
As discussed above, the traditional HB synthesis heavily relies on fossil energy sources (particularly the steam methane reformers for the production of H2), thus contributing to estimated CO2 emissions in the order of 1.5–1.6 tons of CO2 per ton of NH3.53 For this reason, the introduction of water or renewable hydrogen (even blue hydrogen in the worst-case scenario) as reducing agents constitutes a key improvement to mitigate the carbon footprint of the NH3 synthesis reaction. In addition to this, the implementation of renewable energy (either wind or solar) to drive the full process would further contribute to alleviate CO2 emissions associated to the HB process.With this in mind, photo-catalysis has emerged as a viable strategy to achieve a low-carbon emitting NH3 synthesis route. On the one hand, photo-catalysis offers the possibility to integrate water as source of protons for the N2 reduction reaction. On the other hand, photo-catalysis uses sunlight as unique driving force and operates under mild reaction conditions of pressure and temperature, therefore decreasing the global energy requirements of the process. Consequently, the photo-catalytic NH3 synthesis represents a promising strategy to overcome the limitations of the conventional thermo-catalytic pathway and, indeed, over the last years it has become a very dynamic field of research.54–57
Despite these encouraging features, the catalytic performance of most of the photo-catalytic NH3 synthesis systems reported to date is still insufficient to meet the requirements for any industrial scale-up of this technology. This is mainly due to the limited utilization of sunlight by many photo-catalysts, the fast recombination rate between photo-generated electrons and holes and the slow reaction dynamics characteristic of photo-catalytic processes. These shortcomings are, obviously, shared by all photo-catalytic reactions but, particularly for the nitrogen reduction reaction, the process is even more complex considering the extremely high reduction potential (−4.2 eV vs. NHE) necessary to activate molecular nitrogen.19 Although it is possible to circumvent this thermodynamic limitation through alternative multiple-electron transfer processes with lower reduction potentials, this entails slower reaction kinetics that, inevitably, decrease the productivity of the photo-catalytic system. Furthermore, even if the photo-catalyst possesses a sufficiently high reduction potential at its conduction band to make the N2 reduction thermodynamically feasible, the selectivity can be compromised due to presence of the undesirable H2 evolution reaction under aqueous media, thus further reducing the overall efficiency of the process.
All the above-mentioned drawbacks have motivated active lines of research devoted to improve the efficiency and the selectivity of photo-induced NH3 synthesis. Therefore, in the following sections of this review we will describe the current advances in the design of novel photo-catalysts, paying special attention to different strategies to overcome the limitations that still impede photo-catalysis from becoming a viable route for NH3 production.
Inorganic semiconductors
Defect engineering has recently gained considerable relevance as a tool to enhance the catalytic activity in semiconductor photo-catalysts. Thus, for example, the introduction of oxygen vacancies, heteroatom doping or lattice distortions can modify the surface, chemical and/or light absorption properties.63,64 When it comes to light-induced NH3 synthesis, TiO2-based catalysts have also benefited from this strategy to improve their catalytic performance. For instance, Zhang and co-workers developed ultrathin defective TiO2 nanosheets through doping with copper ions.65 Upon doping with Cu, authors increased the oxygen vacancy (VO) defect states and this translated into an enhancement in the light absorption in the range of 400–800 nm (Fig. 4A). These new absorption features derived not only from the presence of oxygen vacancies but also from 2Eg → 2T2g transitions from O to Cu atoms and d–d transitions from copper ions. TiO2 nanosheets samples containing a 6 mol% of Cu displayed the best catalytic results, achieving a NH3 production rate of 78.9 μmol g−1 h−1 and quantum yields in the range of 0.05–0.25% when using different monochromatic lights from 700 to 400 nm (Fig. 4B and C). Additional photocurrent and time-resolved photoluminescence (PL) decay measurements together with DFT calculations demonstrated that doped TiO2 nanosheets exhibited an improved charge separation of electron–hole pairs owing to the presence of VO acting as electron-trapping sites.
 | ||
| Fig. 4 (a) UV-vis diffuse reflectance spectra (DRS) of TiO2 nanosheets with different percentages of Cu doping. (b) NH3 yield of different TiO2 nanosheets samples under UV-vis radiation (c) UV-vis diffuse reflectance spectrum and quantum efficiency for NH3 evolution by 6%-TiO2 sample under monochromatic light of different wavelengths. Reproduced with permission from ref. 65. Copyright (2019) John Wiley and Sons. | ||
In a related precedent, Tang and collaborators reported the synthesis of porous carbon-doped anatase TiOx (C–TiOx) nanosheets obtained from the thermal-oxidation etching of Ti3SiC2.66 As a result of the carbon doping, the obtained C-TiOx material displayed a high concentration of Ti3+ species that could be easily adjusted by modifying the synthetic conditions. According to both steady-state and time-resolved PL analysis, authors found that the Ti3+ sites could act as electron traps to extend the lifetime of charge carriers, compared to the un-doped TiO2. N2 chemisorption measurements also indicated that C–TiOx samples exhibited a high amount of active sites for N2 activation. All these features, together with an extended light absorption in the visible region thanks to the carbon doping, contributed to the enhancement of the catalytic activity of C–TiOx nanosheets towards the photo-catalytic N2 reduction. At the optimal Ti3+ concentration, the as-prepared material showed a remarkable NH3 production of 109.3 μmol g−1 h−1 under visible light and an apparent quantum efficiency of 1.1% at 400 nm. All these results suggest that the introduction of both oxygen vacancies and/or Ti3+ sites is a powerful strategy to improve the efficiency of TiO2-based catalysts for photo-catalytic N2 reduction.
Besides defect engineering, surface functionalization represents an alternative route to enhance the catalytic activity of TiO2-based photo-catalysts. Traditionally, noble metals like platinum, palladium or ruthenium have been proposed as potential co-catalysts in combination with TiO2. However, the rise of plasmonic photo-chemistry has recently shifted the attention to alternative metals like Au or Ag that present intense plasmon bands in the visible-NIR range. Apart from the extended light absorption, the creation of a metal/semiconductor heterojunction favors the formation of a Schottky barrier that facilitates the injection of plasmon-induced energetic electrons from the metal to the semiconductor and, at the same time, prevents the return of those electrons back to the metal, thereby improving the charge separation at the heterojunction interface. An appealing example of this approach can be found in the work of Yang and collaborators, where they describe the decoration of oxygen vacancy-containing TiO2 nanosheets with Au nanocrystals (Au/TiO2-OV) and its application in the N2 photo-reduction reaction.67 Authors claimed a tandem pathway for the N2 reduction by Au/TiO2-OV catalyst, where both oxygen vacancies and Au nanocrystals contributed to improve the catalytic performance. In fact, the catalytic activity increased not only with the number of oxygen vacancies in the TiO2 but also with the loading of Au, achieving a NH3 production rate close to 100 μmol g−1h−1 using a 2.53 wt% Au. It is worth mentioning that authors employed a 420 nm cut-off filter during the experiments, so the catalytic performance entirely derived from the excitation of Au nanocrystals. Indeed, the analysis of the action spectrum revealed a clear match with the absorption spectrum of Au/TiO2-OV, therefore indicating that the photo-induced hot electrons from plasmonic Au crystals were responsible for the N2 reduction. Mechanistic studies including N2 chemisorption, transient absorption spectroscopy (TAS) and photo-voltage measurements suggested that, on the one hand, oxygen vacancies acted as active sites for the activation of N2 molecules while; on the other hand, Au nanoparticles provided plasmon-induced hot electrons to drive the photo-reduction towards NH3.
Feng et al. also explored the functionalization of oxygen vacancy-enriched TiO2−x with plasmonic Ag nanoparticles and PW10V2 to create a ternary composite for N2 photo-reduction.68 In this case, a Z-scheme heterojunction between TiO2 and PW10V2 was responsible of the N2 reduction, where electrons and holes were located at the TiO2 conduction band (CB) and PW10V2 valence band (VB), respectively. Upon plasmon excitation, Ag nanoparticles injected hot electrons to the CB of TiO2, thus improving the charge separation and increasing the number of available electrons for the N2 reduction, as demonstrated by transient photo-current and PL measurements. For a broader analysis on the state-of-the-art for the photo-catalytic N2 reduction reaction by plasmonic structures, readers are referred to an excellent review from Puertolas and collaborators.56
For example, Di et al. prepared Bi3O4Br layers with surface-confined defects based on Bi and O vacancies.71 Authors found a clear positive relationship between the degree of defects of the Bi3O4Br materials and the amount of NH3 generated under light radiation. Both femtosecond transient absorption spectroscopy (fs-TAS) and time-resolved transient PL measurements indicated that the presence of defects was responsible of an enhancement in the charge carrier separation owing to the capacity of defects to act as electron traps. In spite of these encouraging results, a serious weakness of this work relies on the catalytic stability tests, which were limited to only 4 consecutive cycles. Unfortunately, this reduced number of catalytic cycles is insufficient to properly assess the stability of the catalyst under study. Xu and collaborators induced defects on pristine bismuth subcarbonate (Bi2O2CO3) through hydrothermal treatment of NaBiO3 with graphitic carbon nitride.72 Importantly, authors found that the introduction of defect levels improved the light absorption properties of the samples, showing and extended absorption towards the NIR region. Consequently, defective Bi2O2CO3 displayed catalytic activity towards NH3 production under visible light, in sharp contrast with the pristine counterpart. It is worth mentioning that the defect density also determined the position of the defect level, so there was an optimum degree of defects to maximize the catalytic performance.
An alternative approach for the formulation of new Bi-based photo-catalysts is the design of heterojunctions combining two or more elements. The group of Wang recently reported the synthesis of a Bi2MoO6/BiOBr composite containing both bismuth and oxygen vacancies.73 Interestingly, bismuth vacancies not only modulated the band position of the composite but also induced the formation of additional oxygen vacancies. The synergy between bismuth and oxygen vacancies led to an improvement in the light absorption properties of the resulting heterostructure. Furthermore, bismuth vacancies provided adequate adsorption sites for N2 activation. As a result, the composite displayed a ∼4-fold NH3 production compared to the samples with a poor content of defects. Stability tests were performed for 5 consecutive cycles, showing a slightly decrease in the catalytic performance that authors attributed to the partial loss of oxygen vacancies under reaction conditions. The same group expanded this approach through the design of a ternary heterostructure, namely a Bi@BiOBr-Bi2MoO6 composite fabricated by two consecutive solvothermal methods.74 Both semiconductors, BiOBr and Bi2MoO6, created a type-II heterojunction in which, under light radiation, photo-excited electrons migrated to the CB of Bi2MoO6 and holes accumulated at the VB of BiOBr. Owing to the surface plasmon resonance of metallic Bi and the presence of oxygen vacancies derived from the solvothermal treatments, the light absorption properties of the composite extended towards the IR region of the spectrum. All in all, the combination of the three elements had a positive effect on the catalytic performance, achieving a NH3 photo-reduction rate as high as 167.2 μmol g−1 h−1. In this regard, other Bi-based ternary heterojunctions containing plasmonic particles have been proposed, for instance a combination of graphitic carbon nitride, Bi particles and Bi2WO6 to create a g-C3N4@Bi@Bi2WO6 composite or a heterostructure made of TiO2, Au particles and (TiO2/Au/BiO4I).75,76 Furthermore, besides classical semiconductors other potential candidates to fabricate heterojunctions with Bi-based materials have recently included MOFs, MXenes or even graphenes.77–79
Other semiconductor-based catalysts
In addition to TiO2 and Bi-based catalysts, other families of semiconductors have drawn the attention of researchers as promising candidates to drive the photo-catalytic reduction of N2. For instance, niobates are a well-known class of materials that have been extensively explored as catalysts for photo-catalytic H2 evolution and, in the last years, they have shown a great potential for the N2 photo-fixation.80,81 In 2019, the groups of Zhao and He described a Ag/KNbO3 nanocomposite prepared by a hydrothermal method followed by photo-deposition of Ag nanoparticles.82 Authors found that it was possible to modulate the morphology, crystallinity and textural properties of KNbO3 by adjusting both the amount of KOH and the hydrothermal temperature during the material synthesis. Under optimal conditions, KNbO3 nanorods decorated with a 0.5% Ag molar ratio displayed a remarkable NH3 production rate of 385 μmol L−1 gcat−1 h−1 under simulated sunlight using ethanol as hole scavenger. Given the wide bandgap of KNbO3, the photo-activity under visible light was exclusively attributed to Ag nanoparticles, which injected plasmon-induced hot electrons to the CB of KNbO3 to drive the N2 reduction to NH3. In a follow-up work from the same group, authors tried to substitute the precious Ag nanoparticles by a more affordable co-catalyst. Hence, authors photo-deposited NiO nanoparticles on KNbO3 and the as-prepared NiO/KNbO3 composite reached a NH3 productivity as high as 470 μmol L−1 gcat−1 h−1 under simulated sunlight.83 In this case, both NiO and KNbO3 semiconductors formed a type-II heterojunction that favored charge migration across the heterostructure, with electrons and holes concentrating at the CB of KNbO3 and VB of NiO, respectively.Metal sulphides are also a long-established family of materials for photo-catalytic applications. With respect to N2 photo-fixation, a pioneer work from Miyama and collaborators in 1980 proposed for the first time the use of Pt-CdS as photo-catalyst for NH3 production.84 Since then, different examples of sulphide-based photo-catalysts for N2 fixation have been reported.85–88 One of the main advantages of metal sulphides is their low bandgap that allows for the exploitation of low-energy wavelengths in the visible range (λ > 400 nm).89 However, metal sulphides usually suffer from low stability and fast charge carrier recombination, thus hampering their broad applicability.90–92 These drawbacks have motivated intense lines of research to improve the stability and charge separation efficiency of metal sulphides. One of these strategies consists in the fabrication of heterojunctions between metal sulphides and other semiconductors. For instance, in 2020 Sun et al. prepared CdS nanorods functionalized with sulfur vacancy-rich 1T MoS2 nanosheets (SV-1T-MoS2).93 The as-prepared SV-1T-MoS2/CdS composite with optimal MoS2 loading (30%) showed a photo-catalytic NH3 production of 8221 μmol L−1 gcat−1 h−1 with an AQE of 4.4% using methanol as sacrificial agent. This value represented a ∼4-fold enhancement compared to bare CdS, thus evidencing the positive effect of MoS2 nanosheets as co-catalyst. In this case, the introduction of MoS2 containing sulfur vacancies improved the electron mobility and charge carrier separation, as demonstrated by both steady-state and time-resolved PL, transient photo-current and electrochemical impedance spectroscopy (EIS) measurements (Fig. 5).
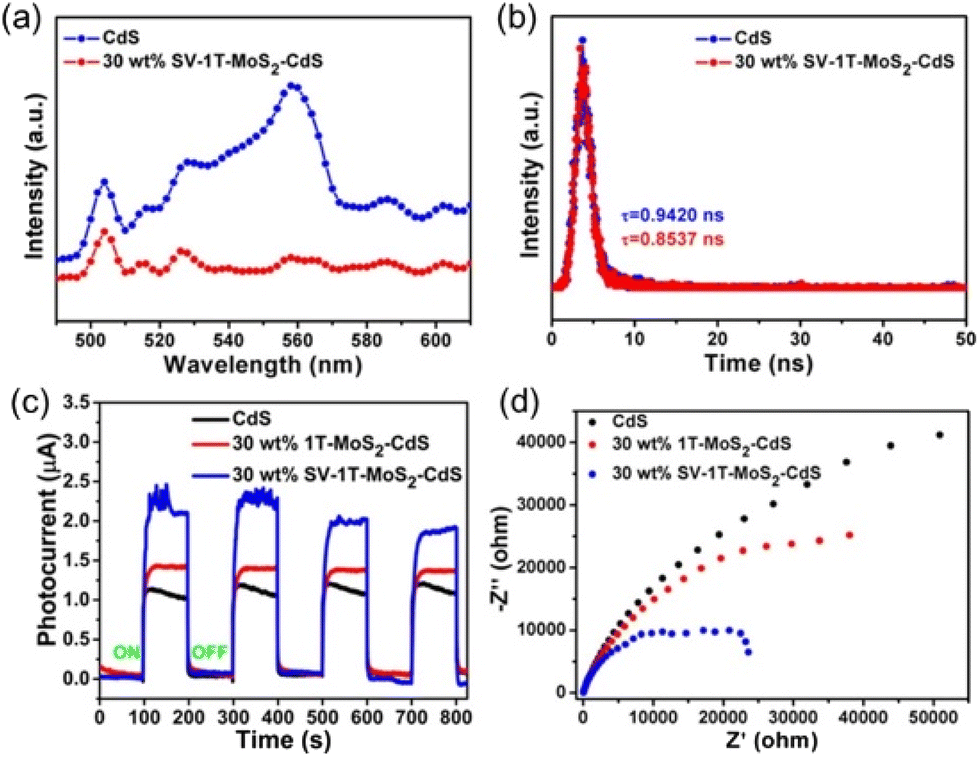 | ||
| Fig. 5 (a) Steady state and (b) time-resolved PL spectra of CdS nanorods and 30 wt% SV-1T-MoS2/CdS composites at λex = 405 nm. (c) Transient photo-current measurements and (d) EIS Nyquist plots of CdS nanorods, 30 wt% SV-1T-MoS2/CdS and 30 wt% 1T-MoS2/CdS composites. Reproduced with permission from ref. 93. Copyright (2020) American Chemical Society. | ||
DFT studies also revealed that the presence of both MoS2 and sulfur vacancies was critical to promote N2 adsorption. In spite of this, the study on the stability of the SV-1T-MoS2/CdS composite was limited to only three consecutive uses, showing a remarkable 20% decrease compared to the fresh sample. This highlights again the importance of performing longer stability tests to demonstrate the reusability of the photo-catalysts under study, especially on those affected by photo-corrosion events. In a related precedent, the group of Yang fabricated nanoporous films of a WS2@TiO2 type II heterojunction.94 Compared to bare TiO2, the presence of WS2 improved the charge carrier separation and transfer, as demonstrated by both the increase in the fluorescence time and the reduced semicircle in the Nyquist plots. Upon optimal WS2 loading, the WS2@TiO2 photo-catalyst rendered a maximum NH3 production rate of 1390 μmol gcat−1 h−1 in the absence of any sign of deactivation after 4 consecutive cycles. The proposed reaction mechanism involved the charge carrier generation at WS2 and subsequent electron transfer to the CB of TiO2, where they were finally injected into anti-bonding orbitals of N2. Photo-generated holes were accumulated at the VB of WS2 and then consumed by the hole scavenger. Other composites including In2O3/In2S3 microspheres, MoS2/n-MgIn2S4 sheets or g-C3N4/ZnMoCdS have been also reported although in all these cases the catalytic performance was far from adequate.95–97
In addition to the above-discussed semiconductor-based photo-catalysts, the use of tungsten oxides has recently sparked the interest of the scientific community. Although pristine WO3 has shown a slight activity towards the N2 photo-reduction, the doping with Fe (0.25%) increased by 4 times the productivity up to 477 μg gcat−1 h−1 thanks to the improved charge mobility and charge separation efficiency.98 As for the case of TiO2-based catalysts, the introduction of oxygen vacancies or dopants to tungsten oxides is a straightforward method to enhance their catalytic activity. In 2022, Yang and collaborators compared the catalytic activity of both 2D WO3−x nanosheets and WO3−x nanoparticles containing a high and low content of oxygen vacancies, respectively.99 The introduction of oxygen vacancies in the tungsten oxide lattice featured a series of advantages including extended light absorption, improved charge carrier separation and enhanced N2 adsorption. Consequently, 2D WO3−x nanosheets with abundant oxygen vacancies displayed a NH3 production of 82.41 μmol gcat−1 h−1 under irradiation with a visible LED (λ = 420 nm), which was almost 4 times higher compared to the sample with a low content of oxygen vacancies. The stability tests, however, were limited to only 5 cycles and authors did not provide any explanation to the observed deactivation of the catalyst upon consecutive uses. Zhang et al. studied the effect of Mo doping on the photo-catalytic activity of W18O49 nanowires and it was found that the Mo–W centers acted as active sites for N2 activation thanks to the polarization of chemisorbed nitrogen molecules.100 Consequently, Mo-doped W18O49 nanowires displayed a 7-fold higher NH3 production compared to pristine W18O49, thus achieving 195 μmol NH3 gcat−1 h−1. In a related precedent, Zhang and co-workers demonstrated that oxygen vacancies displayed a dual effect to both induce the hydrogen spillover and promote N2 adsorption on Ru-decorated W18O49.101 Under irradiation, photo-generated electrons migrated to Ru NPs to activate and dissociate H2O in the form of H* while oxygen vacancies facilitated the migration of the H* species to W18O49. Eventually, H* species reacted with adsorbed N2 to produce NH3. Recycling experiments showed, however, that the catalyst lost its activity upon consecutive reuses. Interestingly, apart from tungsten, other oxides from the same group like molybdenum ones have been investigated as active materials for N2 photo-fixation. Thanks to the presence of oxygen vacancies, defective molybdenum oxides featured plasmonic behavior that provided extended light absorption along the visible and NIR regions, thus exhibiting remarkable catalytic activities for N2 photo-reduction.102,103 More recently in 2023, Fu et al. also described the use of defective ZrO2−x in combination with Ru NPs to create a metal/semiconductor heterojunction.104 In this case, the presence of oxygen vacancies in the zirconia lattice induced a higher concentration of carriers, therefore providing more electrons for the N2 reduction reaction. Furthermore, the formation of a Schottky barrier at the interface between the defective ZrO2−x and the Ru NPs improved the charge separation and the effective injection of electrons from the semiconductor to the metal.
Carbon-based materials
In addition to inorganic semiconductors, metal-free carbon-based materials have attracted a great deal of attention as affordable and sustainable candidates for applications in catalysis, especially when these materials are obtained from biomass or other renewable sources.105 In this regard, graphitic carbon nitride (g-C3N4) is one of the most relevant examples of this family of materials and its applications span from heterogeneous organic synthesis to photo- and electro-catalysis, among others.106,107 When it comes to N2 photo-fixation, a cutting-edge work of 2015 from Dong and co-workers demonstrated for the first time that g-C3N4 containing nitrogen vacancies could perform the photo-reduction of N2 gas under visible light.108 Not surprisingly, the study demonstrated that nitrogen vacancies are crucial to endow g-C3N4 with the capability to photo-reduce N2 thanks to the improved nitrogen adsorption and enhanced charge transfer efficiency.Apart from the introduction of vacancies, doping with heteroatoms is a straightforward approach to expand the light absorption properties and modulate the redox potentials of g-C3N4.109 In 2020, Liang and collaborators explored the possibility to merge the positive effects of both nitrogen vacancies and boron-doping in a graphitic carbon nitride.110 With that purpose, authors first obtained a defective carbon nitride (CNx) through calcination of melamine followed by an annealing treatment under N2 in presence of NaBH4 (Fig. 6A). The as-prepared boron-doped defective carbon nitride (BNUCNx) displayed a remarkable NH3 production rate of 435 μmol g−1 h−1 under visible light. Both steady-state PL and EIS spectra and photo-current measurements revealed that the defective boron-doped carbon nitride featured a good charge separation efficiency and a low charge-transfer resistance that ultimately improved the catalytic activity (Fig. 6B and C). Furthermore, the introduction of N vacancies and B dopants led to a narrowing in the bandgap that enhanced the optical absorption properties of the final material. In the same spirit, other researchers have investigated the effect of the doping of g-C3N4 with diverse heteroatoms such as S or P.111,112
 | ||
| Fig. 6 (a) Schematic illustration of the synthesis protocol for the preparation of boron-doped defective ultrathin carbon nitride (BNUCNx). (b) Steady state PL and (c) EIS spectra of different samples. Reproduced with permission from ref. 110. Copyright (2020) Elsevier. | ||
As in the case of inorganic semiconductors, the construction of heterojunctions containing g-C3N4 can be a useful tool to refine its electronic and catalytic properties. Among all of the different types of heterojunctions, type-II heterojunctions are one of the most investigated systems. For instance, the groups of Zhang and Wu studied the combination of p-type Cu2O and n-type C3N4 semiconductors to build a composite for N2 photo-fixation.113 The strong electric field at the interface between both elements greatly enhanced the charge separation efficiency, with electrons and holes accumulating at g-C3N4 and Cu2O, respectively. Another example of type-II heterojunction between g-C3N4 and ZrO2 was reported recently by Mou et al.114 Upon light radiation, photo-excited electrons migrated from the CB of g-C3N4 to the CB of ZrO2, remaining the holes at g-C3N4. Thanks to the effective charge separation, the g-C3N4/ZrO2 heterostructure displayed a NH3 production rate as high as 1446 μmol L−1 h−1, compared to the negligible activity of both elements separately. Apart from type-II heterojunctions, Z-scheme heterojunctions are also a very representative exponent of composite materials with improved catalytic efficiencies. In spite that Z-scheme heterostructures feature a distinct design compared to type-II heterojunctions, both architectures provide a spatial separation of electron and holes and the creation of well-defined redox active sites.115 Several examples of Z-scheme heterostructures for N2 photo-reduction include g-C3N4/Fe2O3, g-C3N4/Ga2O3 or B-doped g-C3N4/WO3.116–118 For a more comprehensive revision on g-C3N4-based materials and heterostructures for photo-catalytic N2 reduction, readers are referred to excellent reviews covering this topic.115,119
Although g-C3N4 has been, by far, the most widely explored metal-free material, other carbon-based materials such as doped graphenes, reduced graphene oxides or carbon nanotubes have been investigated, alone or in the form of heterostructures, as potential photo-catalysts for NH3 production.120–124 It is worth reminding that, especially for N containing photo-catalysts such as g-C3N4 or N-doped carbons, the incorporation of a set of control experiments including the use of isotopically labelled 15N2, blank experiments working under N2-free atmosphere and/or in the absence of photo-catalyst is crucial to ensure the reliability of the reported results. Despite it is out of the scope of the present review, readers will find excellent guidelines for the accurate evaluation of the catalytic activity in N2 photo-fixation experiments elsewhere.125–127
Metal–organic frameworks (MOFs)
In addition to commonly studied inorganic semiconductors and carbon-based materials, metal–organic frameworks are a novel class of materials that can be effectively used as photocatalysts due to their tunability, wide light harvesting properties and large surface area. Applications include photocatalytic water splitting, CO2 reduction and organic photosynthesis, among others.128–130 Unfortunately, the study of metal–organic frameworks as photo-catalysts for ammonia synthesis is still in its initial stage.To date, the greatest number of publications is devoted to transition metal-based metal–organic frameworks. Quite remarkable NH3 production rates were achieved using iron-based MOFs.131 In the case of MIL-101(Fe), MIL-88(Fe) and MIL-100 (Fe), the ammonia yield reached 1007.1 μmol g−1 h−1, 800.7 μmol g−1 h−1 and 930.64 μmol g−1 h−1, respectively (Fig. 7). The difference between isoreticular iron-based and chromium-based MIL-101, which exhibited no activity, was explored in the same study. Lower activation energy and higher electron density due to two additional filled d-orbitals were identified as fundamental reasons for such a different behavior.
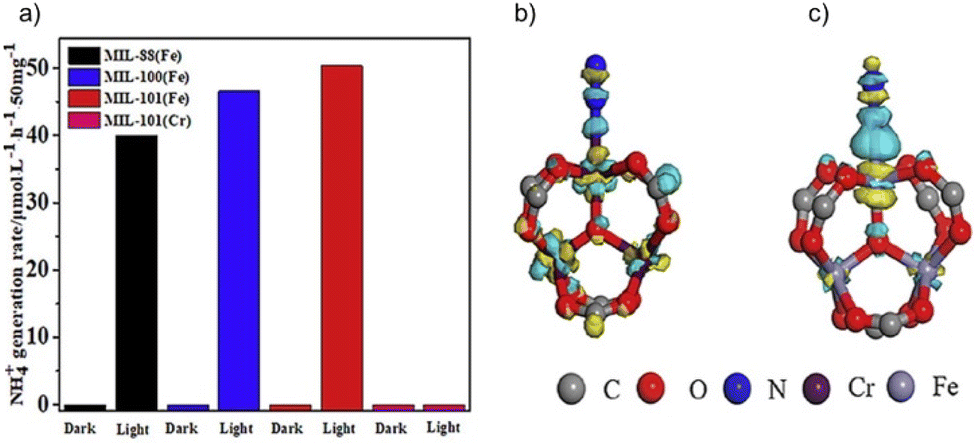 | ||
| Fig. 7 (a) Photo-catalytic nitrogen fixation activity of different MIL MOFs. Charge density difference of a N2 molecule adsorbed at (b) MIL-101(Cr) and (c) MIL-101(Fe). Reproduced with permission from ref. 131. Copyright (2020) Elsevier. | ||
However, Min Luo et al. showed that the incorporation of silver particles into the MIL-101(Cr) cavity allowed reaching an activity of 138.81 μmol NH3 g−1 h−1, which they attributed to the synergy of ligand–metal charge transfer (LMCT) and localized surface plasmon resonance effect (LSPR).132 Inspired by naturally occurring multi-metal cluster nitrogenases, Zhao et al. recently synthesized MIL-53(Fe2+/Fe3+), which displayed high nitrogen fixation activity as high as 306 μmol NH3 g−1 h−1.133 It is important to note that the sample was irradiated only in the visible range above 420 nm, which means that it can potentially be used to convert solar energy. In a similar vein, Li and co-workers adopted a strategy to mimic the functionality of natural enzymes in synthetic materials.134 In this case, the approach involved modifying the MIL-88A iron MOF with polyoxometalates, which acted as charge regulators and simulated the π*-back-donating mechanism of nitrogenase. This modification resulted in a remarkable six-fold increase in the photocatalytic activity of the MOF.
Some Ti-based MOFs have been explored in photocatalytic reactions and have shown good nitrogen sorption capacity, thus standing for potential catalysts in the N2 photo-fixation reaction. Huang et al. showed that a decrease in the optical band gap of NH2-MIL-125(Ti) led to an increase in the photocatalytic performance, as in the case of hydrogen evolution reaction.135 However, this Ti-MOF, which exhibits one of the most impressive H2 evolution activities, reached a very modest NH3 production of 12.3 μmol g−1 h−1. It should be mentioned, however, that the photocatalytic nitrogen reduction tests were tested under visible light and without a sacrificial agent. Interestingly, hafnium and zirconium-containing MOFs (UiO-66-Vis and U(0.5 Hf)) turned out to be ten times more active (256.6 μmol NH3 g−1 h−1 and 351 μmol NH3 g−1 h−1, respectively).136,137 In fact, a recent work indicated that Zr-based MOFs activity could be increased via the incorporation of Ca2+ dopants, resulting in a noteworthy increase in photocatalytic activity from 105 μmol g−1 h−1 in the unmodified MOF to 222.2 μmol g−1 h−1 in the modified counterpart.138 Authors suggested that the introduction of Ca2+ dopants promoted the redistribution of electrons around active Zr sites, thus reducing the activation energy of N2 triple bonds as elucidated through DFT calculations. Furthermore, the utilization of porphyrins and metalloporphyrins as ligands for stable Zr-based MOF represents an alternative approach to formulate new photocatalysts for NH3 production.139 Hence, using porphyrin ligands for MOF construction led to NH3 production rates of 776.24 μmol g−1 h−1, while replacing the ligands with metalloporphyrin resulted in a significant boost in photocatalytic activity, reaching 1502.51 μmol g−1 h−1.
Besides transition metal-based, attention should be paid to lanthanide-based MOFs since Hu et al. developed two radical-containing gadolinium-based MOFs.140 Purple crystals of Gd-IHEP-7 could be obtained using a solvothermal technique. The procedure for obtaining Gd-IHEP-8 included heating Gd-IHEP-7 to 120 °C in the air when single-crystal-to-single-crystal transformation occurred. The rearrangement of the Gd3+ coordination environment was accompanied by an increase in photocatalytic nitrogen fixation activity (up to 220 μmol NH3 g−1 h−1). Authors suggested that the factor responsible for the higher activity of Gd-IHEP-7 was the calculated change in the stability of the intermediates. Interestingly, Gd-IHEP-7 and Gd-IHEP-8, unlike most other viologen-based compounds, were stable in air and water due to radical–radical interaction. Compared to Gd-based MOFs, deficient activity (34.2 μmol NH3 g−1 h−1) was shown in the case of cerium-based MOF-76(Ce) even using full-spectrum light of Xe-lamp.141
Photo-thermal catalytic NH3 synthesis
As we have earlier discussed in this review, the thermo-catalytic NH3 synthesis route is constrained by its harsh operative conditions whereas the photo-catalytic NH3 production is extremely hampered by its poor catalytic efficiency and low selectivity. In order to overcome these limitations, a smart approach would be the combination of these two strategies into a single process. In this spirit, photo-thermal catalysis arises from the synergistic incorporation of thermal and non-thermal contributions of light and features exceptional advantages compared by its individual counterparts. In fact, this cooperative effect has implications in both reaction thermodynamics and kinetics. For instance, from the thermodynamic point of view, non-thermal effects can reduce the energetic barriers of certain reaction steps. The reduction in the activation energy can derive from charge transfer events to the surface of the catalyst that alter the acidity/basicity of the active sites. Besides, the energy transfer to certain vibrational modes of the transition state can also favour bond elongation, thus decreasing the energy requirements of the process.142 In this regard, Song and collaborators recently demonstrated the potential of non-thermal effects to adjust the thermodynamics of dry reforming reaction.143 Authors demonstrated that photo-induced hot electrons from plasmonic Pt and Au nanoparticles could be injected to adsorbed CO2 and CH4 molecules, thereby weakening the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H bonds and facilitating bond dissociation. In this case, the apparent activation energy under radiation decreased from 241 to 171 kJ mol−1, suggesting a change in the energetics of the reaction. In addition to this, the presence of non-thermal effects opens up the possibility to activate specific chemical bonds that unlock certain reaction pathways that are inaccessible under pure thermal conditions, thus facilitating tailored selectivity patterns or alternative low-energy reaction intermediates. For instance, the groups of Liu and Everitt demonstrated that it was possible to adjust product selectivity in the photo-thermal CO2 hydrogenation by plasmonic Rh nanoparticles.144 The use of blue or UV LEDs to perform the reaction facilitated the CH4 production with selectivities up to 98%, while the reaction under dark conditions displayed comparable amounts of CH4 and CO. Theoretical calculations demonstrated that the injection of photo-induced hot electrons from Rh nanoparticles to anti-bonding orbitals of adsorbed intermediates played a crucial role to determine the selectivity patterns. Curiously, the presence of hot carriers can not only tune the reaction selectivity towards the desired products, but also prevent the formation of detrimental surface species. Indeed, several recent studies have shown the hot carriers’ potential in mitigating the formation of unwanted byproducts, especially in the context of photo-thermal dry reforming of methane.145,146
O and C–H bonds and facilitating bond dissociation. In this case, the apparent activation energy under radiation decreased from 241 to 171 kJ mol−1, suggesting a change in the energetics of the reaction. In addition to this, the presence of non-thermal effects opens up the possibility to activate specific chemical bonds that unlock certain reaction pathways that are inaccessible under pure thermal conditions, thus facilitating tailored selectivity patterns or alternative low-energy reaction intermediates. For instance, the groups of Liu and Everitt demonstrated that it was possible to adjust product selectivity in the photo-thermal CO2 hydrogenation by plasmonic Rh nanoparticles.144 The use of blue or UV LEDs to perform the reaction facilitated the CH4 production with selectivities up to 98%, while the reaction under dark conditions displayed comparable amounts of CH4 and CO. Theoretical calculations demonstrated that the injection of photo-induced hot electrons from Rh nanoparticles to anti-bonding orbitals of adsorbed intermediates played a crucial role to determine the selectivity patterns. Curiously, the presence of hot carriers can not only tune the reaction selectivity towards the desired products, but also prevent the formation of detrimental surface species. Indeed, several recent studies have shown the hot carriers’ potential in mitigating the formation of unwanted byproducts, especially in the context of photo-thermal dry reforming of methane.145,146
When it comes to thermal effects, the photo-induced heat at the nanoscale provides high temperatures at the active sites in a more efficient way compared to traditional thermo-catalysis. These elevated temperatures translate into catalytic activities that are several orders of magnitude above those obtained with conventional photo-catalysis, mainly due to both the improved reaction kinetics and energy-mass transfer. In addition to this, thermal effects favor the desorption of products or poisonous species from the catalyst surface, thereby enhancing the activity and stability of the catalysts.147 Last but not least, while in photo-catalytic systems the use of light is limited by the band gap and the position of both conduction and valence bands of the photocatalysts, photo-thermal catalysis takes advantage of the full solar spectrum, even the low-energy photons from the visible and IR that are typically unable to trigger photo-chemical reactions. A comprehensive discussion on the fundamentals of photo-thermal catalysis and the benefits from the synergies underlying the photo-thermal phenomenon lies beyond the scope of this review, so readers are referred to specific works on this topic.148–151
Photo-thermal catalysis can be considered a relatively recent sub-field of catalysis and, in fact, only a few research groups have explored the combination of light and heat to perform the HB process under mild reaction conditions. For instance, García and co-workers lately reported the use of Cs-promoted ruthenium NPs supported on strontium titanate (CsyRux@ST) as photo-catalyst for the NH3 synthesis reaction under 350 °C and ambient pressure.152 All the tests were performed using a continuous-flow set up equipped with a heating ribbon and a thermocouple to control the temperature. Under optimal Cs and Ru loading, the photo-catalyst achieved an impressive NH3 production rate in the order of 3.6 mmol g−1 h−1, outperforming by a 68% a control experiment in the dark at the same temperature. Authors suggested that the remarkable catalytic activity derived from the combination of both photo-induced hot carrier and local thermal effect mechanisms taking place at the irradiated Ru NPs. Altogether, both contributions facilitated the activation of N2 and its further hydrogenation (Fig. 8A). Interestingly, this work also showed that the choice of the support for Ru NPs was not trivial at all, as authors demonstrated that both the basicity and low thermal conductivity of strontium titanate were key to improve the catalytic performance (Fig. 8B). In 2023, the same group explored the MOF-derived approach to obtain Cs-promoted Ru clusters supported on ZrO2 as photocatalyst for the photo-thermal NH3 synthesis.153 In this case, the authors impregnated the UiO-66 MOF with Ru and Cs precursors and calcined the resulting material in air at 550 °C. Under these conditions, the MOF served as sacrificial template to obtain ZrO2 decorated with Cs and Ru clusters. The as-obtained catalyst exhibited a notable activity under 350 °C and 1 sun radiation, achieving a NH3 production rate of 5.1 mmol g−1 h−1. Analogous materials prepared by impregnation of commercial ZrO2 displayed negligible catalytic activity, thus stressing the potential of the MOF-derived strategy to formulate efficient catalysts that are not achievable by conventional synthetic routes. Mechanistic experiments revealed that both thermal and non-thermal effects operated simultaneously to enhance the catalytic activity. On the one hand, Ru sites absorbed visible and NIR light and locally increased the temperature of the catalyst owing to plasmon excitation. On the other hand, plasmon excitation could also generate hot electrons that could be injected into anti-bonding orbitals of adsorbed N2.
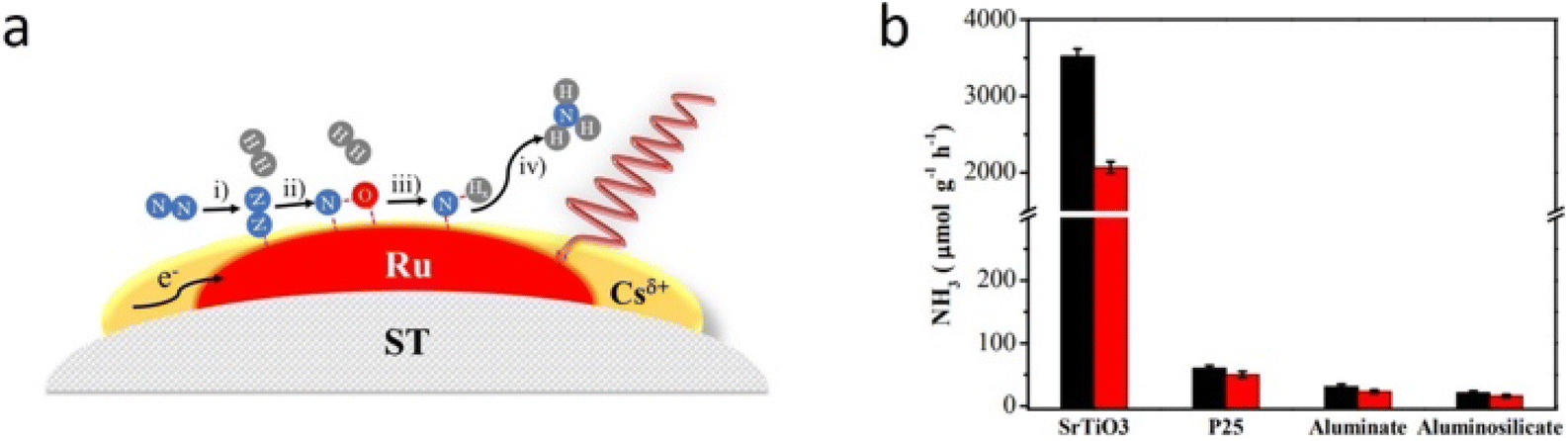 | ||
| Fig. 8 (a) Proposed mechanism for the photo-thermal NH3 synthesis by CsyRux@ST catalyst: (i) N2 adsorption and activation, (ii) N2 dissociation and formation of intermediate species, (iii) hydrogenation of activated nitrogen species, and (iv) NH3 formation and desorption. (b) NH3 production rate using different supports. Dark bars correspond to experiments under light irradiation and red bars correspond to dark conditions. Reproduced with permission from ref. 152. Copyright (2022) American Chemical Society. | ||
Nevertheless, for most of the photo-thermal systems, it is still extremely challenging to provide an accurate evaluation of the specific role of each of the contributions to the overall catalytic activity. Zhang and collaborators recently addressed this issue employing a model catalyst based on Ru on carbon.154 Authors first estimated a local temperature of the catalyst (Treaction) based on the thermodynamic equilibrium conversion. By introducing this Treaction into the Arrhenius equation under thermo-catalytic conditions, authors calculated the actual reaction rate derived from pure thermal effects (Rtemperature). Once the thermal component was isolated, it was possible to extract the non-thermal contribution (Relectron) by subtracting the thermal component to the total photo-thermal rate (RPTC). Interestingly, authors found that Relectron transitioned from a linear to superlinear relationship with the light intensity, thus confirming the presence of non-thermal effects in the system. In fact, under 5.3 W cm−2 light intensity and 350 °C the hot-electron contribution accounted for a 73.6% of the total NH3 production rate. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements also revealed the presence of *NHx species under light radiation at 230 °C, while no intermediates were detected under dark conditions at temperatures below 260 °C. Based on these observations and additional DFT calculations, authors concluded that light could promote the NH3 synthesis reaction at lower temperatures thanks to the injection of photo-induced hot-electrons into unoccupied anti-bonding orbitals of adsorbed N2, thus facilitating the cleavage of the NN triple bond. Overall, this study demonstrated that it is possible to differentiate and quantify both the thermal and non-thermal contributions of photo-thermal systems using Le Chatelier's principle to determine the actual reaction temperature.
As commented before, local heat generation at the nanoscale is considered one of the main advantages of photo-thermal catalysis. In 2019, Mao and co-workers proposed a dual-temperature-zone catalyst to tentatively trick the thermodynamic equilibrium limit while using sunlight to promote the NH3 synthesis.155 The idea here was to locate the N2 dissociation in a “local high-temperature” center, where the nitrogen dissociation takes place, and a local low-temperature center for the N-hydrogenation in order to physically separate these two elemental steps of ammonia synthesis. Thanks to the afore-mentioned capability of photo-thermal catalysis to create hot spots, authors succeeded in designing a catalytic system to provide this non-uniform heat distribution, something otherwise extremely complex to achieve under pure thermal conditions. In this case, plasmonic Fe nanostructures acted as nano-heaters to perform the N2 dissociation step while subsequent N–H hydrogenation took place at the “cooler” surface of defective TiO2−xHy owing to its low thermal conductivity. When irradiated at 102 sun, the catalyst reached a temperature as high as 495 °C, thus achieving a NH3 concentration of 1939 ppm. This concentration was above the thermodynamic equilibrium value of NH3 synthesis at 495 °C and 1 atm (1249 ppm) and even outperformed the value obtained by commercial-wüstite-based Fe catalyst. The presence of the dual-temperature-zone between Fe and TiO2−xHy was key to allocate the elementary steps of NH3 synthesis into two spatially separated catalytic sites, thus surpassing the thermodynamic limit of the reaction (Fig. 9).
 | ||
| Fig. 9 (A) NH3 concentration under combined solar irradiation and external heating by TiO2−xHy/Fe at a constant temperature of 495 °C. (B) NH3 concentration as a function of the local temperature difference (LTD). (C) Comparison of equilibrium limits at the temperature of “cool” TiO2−xHy and “hot” Fe for photo-thermal ammonia synthesis on TiO2−xHy/Fe (1 atm). Reproduced with permission from ref. 155. Copyright (2019) Elsevier. | ||
Despite the potential of the aforementioned dual-temperature-zone catalyst, some authors have called the attention to the existence of important thermal gradients due to the shallow penetration of light, especially in the case of thick catalyst beds. This situation would, inevitably, confine the effect of photo-thermal catalysis to the illuminated surface region. In their work of 2019, Li et al. made the most of this light-induced thermal gradient to boost the NH3 production rate using a conventional cesium-promoted Ru on magnesium oxide (Cs–Ru/MgO) catalyst.156 Authors demonstrated that the non-isothermal gradient between the surface and the bottom of the catalyst bed favored high temperatures on the surface, where the N2 scission took place, while the cooler temperatures from the bottom preserved the NH3 yield. In addition to this, authors found that not only the presence but also the sign of the thermal gradient was critical to enhance the catalytic activity. Under a negative gradient, thermophoretic forces place parallel to the flow of reactants, thus removing NH3 products from the hottest region and circumventing the undesirable reverse reaction of NH3 decomposition. However, under the same absolute value, a positive gradient displayed much lower reaction rates and product yields. Overall, these results evidenced the potential of light-induced thermal gradients to act as thermodynamic pumps to shift the equilibrium and improve the catalytic activity of photo-thermal systems (Fig. 10).
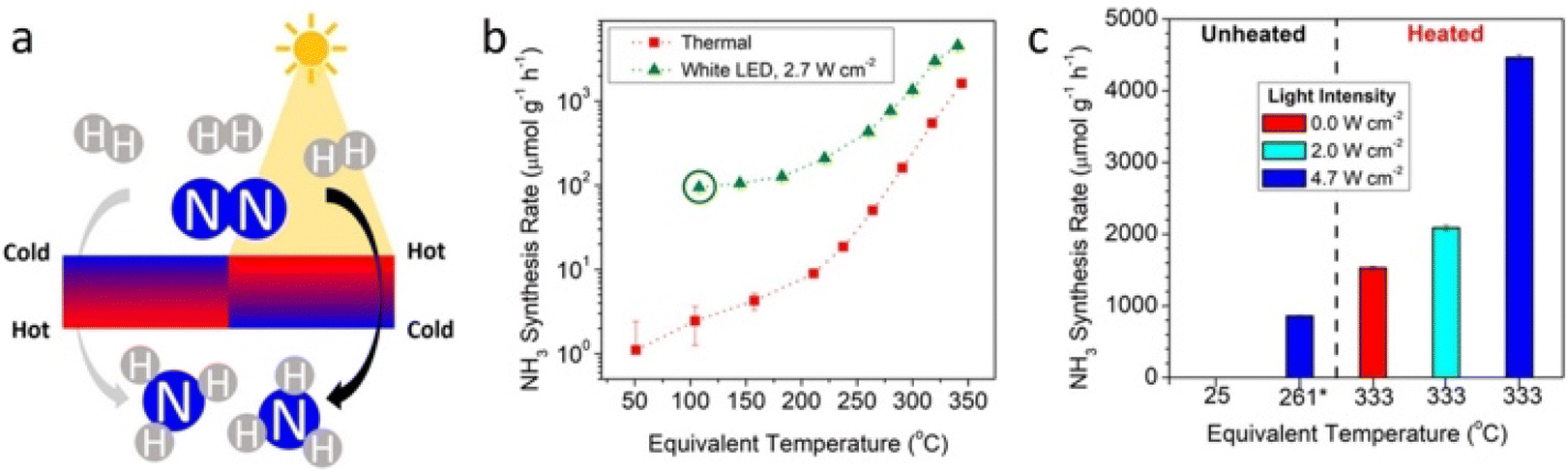 | ||
| Fig. 10 (a) Illustration of the thermal gradient generated by the photo-thermal heating of the irradiated catalyst surface. Effect of light-induced thermal gradients on NH3 synthesis. (b) Measured dark thermal (red squares) and heated white light illumination NH3 synthesis rates as a function of equivalent temperature. Circled data point indicates the light-only condition with no external heating applied. (c) NH3 synthesis rates under dark and additionally illuminated conditions using a blue LED. Reproduced with permission from ref. 156. Copyright (2019) American Chemical Society. | ||
An appealing example of the potential of photo-thermal catalysis to unlock new reaction pathways was reported in 2018 by the group of Zhang. In their work, authors described the use of K-promoted Ru NPs supported on disordered TiO2−xHx (K/Ru/TiO2−xHx) as photo-thermal catalyst for the solar-driven NH3 synthesis.157 The K/Ru/TiO2−xHx catalyst featured a series of advantages that have been described along previous sections of this review such as the introduction of oxygen vacancies to expand the light absorption properties of TiO2 or the use of alkaline promoters to tune the electronic environment of Ru NPs and promote N2 activation. However, the most striking observation in this work was the presence of two simultaneous reaction mechanisms within the K/Ru/TiO2−xHx catalyst. Hence, activated N2 could react not only with H atoms on the Ru surface, but also with the H atoms incorporated in the TiO2−xHx support. Remarkably, this latter pathway was notably different from those reported to date and it could even occur under room temperature, thus suggesting a mechanism with a considerably low energy barrier. In fact, authors calculated the activation energies of both thermal and photo-thermal reactions, achieving 86 and 65 kJ mol−1, respectively. This decrease in the activation energy was explained by the presence of both plasmon-induced confined magnetic fields and heat generation at the vicinity of Ru NPs. In general, this study opened a new venue for the development of alternative NH3 synthesis routes driven by solar light thanks to the combination of low-energy reaction pathways and the confined energy of plasmonic NPs.
Catalyst engineering and optimization
As mentioned before, photo-thermal catalysts benefit from a synergistic contribution of both photo and thermal components of light. To further intensify the photo-thermal effect and enhance production rates, researchers have focused on improving the light-to-heat conversion by optimizing size and shape of active sites, adjusting the porosity and nature of support or through structural engineering, among other strategies.148For plasmonic materials, size and shape of nanoparticles affect not only the capability of charge carrier generation, but also the thermal properties.158 Theoretical calculations indicate that smaller nanoparticles produce electron–hole pairs with higher energy in comparison to larger nanoparticles.159 This characteristic holds significant importance in the context of photo-thermal catalysis, as it enhances the likelihood of populating acceptor states and overcoming Schottky barriers. However, a small particle size may lead to inefficient charge carrier separation due to spatial confinement. In addition, larger particles also show a red-shift on the absorption capability of the catalyst, unlocking the use of visible and IR light ranges.160 Another point is that the plasmon-heating effect in metallic nanoparticles scales quadratically with their radius.161 Therefore, the engineering of the ideal catalyst relies on the perfect balance of the size. As for the morphology, geometries that display strong and inhomogeneous electric fields associated with confinement effects contribute to high-energy hot electrons and holes.162 The opposite is also true, geometries without defects and homogeneous electric fields, like nanospheres, exhibit a less efficient production of hot carriers.
Another possibility is to use secondary light-absorbing elements, whereby a catalytically active metal is coupled with another material able to absorb light in a wider range and generate photo-carriers and/or heat.163–165 Core–shell structures have also received great attention, as the idea is to mimic a nanoreactor, confining the heat in the core, increasing the temperature precisely where the active sites are located.166 Still in this context, MOFs can also be used as sacrificial templates to obtain carbon-based materials featuring broadband light absorption in the visible and IR light ranges.11 The combination of carbonaceous supports with highly dispersed metal nanoparticles makes MOF-derived materials a very attractive family of catalysts for photo-thermal applications.167
Conclusions and outlook
Due to its widespread use as building block for the production of fertilizers, NH3 is considered one of the key chemicals in our society. Despite the technological maturity of the Haber–Bosch process, there is still room for improvement. With this objective in mind, ongoing research is being devoted not only to find catalysts able to operate under milder conditions without losing productivity, but also to the development of alternative synthetic routes. One of the possibilities in this regard is the integration of light into the NH3 synthesis process to achieve photo-catalytic N2 fixation routes powered by sunlight. However, to date the reported efficiencies remain far from those required to compete with the conventional HB approach. In an effort to overcome the limitations of both thermo- and photo-catalytic approaches, photo-thermal catalysis has demonstrated potential to achieve high activities at moderate reaction conditions through the synergistic combination of thermal and non-thermal contributions of sunlight.In this review, we have first provided an overview of the main reaction pathways for NH3 synthesis. Next, we have reviewed recent strategies for the formulation of novel catalysts for the thermo-catalytic NH3 production able to operate at milder conditions. In this regard, catalysts based on Ru or Co are considered promising candidates although their higher costs compared to inexpensive Fe-based catalysts still represent a major pitfall for their wide implementation. Later, we have discussed potential methodologies to improve the catalytic performance of photo-catalytic systems for N2 photo-fixation, mainly the introduction of vacancies, defects or the construction of heterojunctions. In the last section of the review, we highlighted the significance of photo-thermal catalysis to surpass the limitations of its individual counterparts thanks to the synergistic integration of thermal and non-thermal effects. This cooperative effect can improve reaction rates, create positive thermal gradients or even trigger alternative lower-energy reaction pathways.
Although the technological advantages are evident, to date there has been little discussion on the benefits of photo-thermal catalysis in terms of sustainability. A rigorous analysis in this regard would require appropriate metrics to evaluate the environmental impact of photo-thermal catalysis but, unfortunately, these figures of merit have not been perfectly established in the field. Ozin and collaborators recently tried to address this issue and proposed a series of equations to assess the CO2 footprint of thermal and photo-thermal CO2 hydrogenation reactions.168 These equations considered multiple parameters related to the photo-thermal process such as CO2 emissions per kWh of electricity, CO2 produced per mol of H2, total irradiation time or the power consumption of lamps, pumps, etc., among others. Based on their model, authors found that the source of radiation was critical to reduce the CO2 emissions associated with the photo-thermal process. In other words, energy-saving lamps with low power are preferred, as the net CO2 emission rate scales up with the power of the lamp. In the best-case scenario, the direct use of sunlight instead of electricity would further reduce the carbon footprint or even achieve negative CO2 emissions, although this strategy would require additional technological development. Equations also indicated that reactor configuration plays an important role, as flow reactors displayed a higher potential to reduce CO2 emissions compared to their batch counterparts. Assuming a CO2-free source of H2, authors concluded that photo-thermal catalysis has the potential to achieve net CO2 emission rates lower than traditional thermal catalysis, especially in the case of flow reactors coupled with energy-efficient lamps or directly powered with solar radiation. Overall, this pioneer work reinforced the prospects of the photo-thermal approach in terms of its CO2 footprint and set the basis for subsequent studies focused on other chemistries of interest beyond CO2 hydrogenation. In addition to this, assessing the economic feasibility of the entire process is imperative to ensure a realistic scale-up of this technology. While performing an accurate economic analysis of the photo-thermal HB process is highly challenging due to the immaturity of the field and the lack of actual data from large-scale prototypes, we would like to share some considerations for future estimations. Based on available techno-economic analyses, the main contributors to the total energy consumption in blue ammonia production include the ammonia separation refrigerant, cooling water and gas used to provide heat to the reactors.6 In the case of green ammonia, electricity for compressors, water for general cooling and refrigerant for separation purposes are the major contributors to the energy requirements of the operation. With this in mind, the photo-thermal approach could potentially reduce the energy demand (and consequently, the cost) of the process by harnessing natural sunlight instead of relying on electricity or gas to power the HB reactors. Furthermore, considering that photo-thermal processes can operate at lower temperatures than pure thermal systems, the energy costs could be further reduced. The techno-economic analysis should also include the cost of the photo-thermal reactor based on parameters such as the average solar intensity, illumination hours, irradiated area, type of solar collector, amount and lifetime of the photocatalyst, etc. Nevertheless, previous works have revealed that the major capital costs for NH3 synthesis primarily stem from feed costs rather than energy costs. This implies that, ultimately, the hydrogen or gas market prices will play a pivotal role to determine the profitability of any potential route for NH3 production.6
Despite its promising future, if photo-thermal catalysis is to lead a paradigm shift in the field of heterogeneous catalysis and compete with long-established thermo-catalytic processes, a series of challenges need to be addressed. For instance, a deeper understanding of the synergies between hot carriers and thermal effects at the fundamental level would be desirable in order to fully comprehend the mechanisms underlying photo-thermal reactions. To this end, in situ/in operando techniques (X-ray diffraction, electron microscopy, XPS, Fourier-transformed IR, Raman, UV-vis absorption, etc.…) could help not only to assess the contribution of thermal and non-thermal effects on the catalytic performance, but also to provide full information on the catalytic sites and transitional species present under reaction conditions.169–172 In this regard, computational methods can be also interesting tools to disentangle not only the reaction pathways but also the intermediate states involved.
An accurate measurement of the local temperature is crucial in photo-thermal catalysis as imprecise temperature records can lead to misinterpretations of the thermal contribution.173 For this reason, and especially in the case of thick catalyst beds, one or more sets of thermocouples should be utilized to ensure a correct monitoring of the temperature across the catalyst. Ideally, these measurements should be complemented with additional temperature registrations using IR cameras or temperature-responsive photoluminescence probes.
From the point of view of scalability, photo-thermal catalysis requires adequate photo-reactors capable of harvesting sunlight and channeling it directly to the catalyst. Based on lessons learned from a mature technology like the solar thermal energy, solar concentrators or condenser lenses could serve as elements to both harness and concentrate sunlight.174–176 For instance, the groups of Steinfeld and Furler recently developed a solar fuel system that enables the production of syngas from water and CO2 using concentrated solar energy.177 The solar redox unit consisted of a sun-tracking parabolic concentrator coupled with a secondary reflector. Under normal solar radiation conditions, the system could reach a peak flux concentration above 5000 suns and an average flux of approximately 2700 suns. Many reaction systems, however, require less severe solar flux concentrations in the order of 50 to 100 suns. In these cases, the use of parabolic troughs to reflect and concentrate sunlight can be a more affordable and practical option. These systems use sets of concave mirrors to concentrate light into its focal line, achieving temperatures in the range of 500–700 K.178 Alternatively, high-power LEDs or incandescent light bulbs could also act as irradiation sources in conjunction or not with solar light radiation.179 In addition to that, further considerations on heat management should be taken into account when designing new photo-reactors, as thermal management has proved to be critical if one wants to maximize the benefits of the photo-thermal effect. Moreover, the use of batch reactors at the laboratory scale should be progressively shifted to continuous-flow reactors that are better suited for potentially scalable systems at the industrial scale. In any case, it is necessary to perform extensive research in reactor design and engineering to piece together all these considerations and develop suitable photo-reactors for applications in photo-thermal catalysis.
As we have discussed in detail along this review, the HB process is one of the best examples of energy integration in the chemical industry. In this particular case, for photo-thermal catalysis to be successful, catalysts with excellent light absorption and light-to-heat conversion properties are crucial. Examples of this kind of materials include carbon-based solids, defective semiconductors or transition metal carbides and nitrides. These materials, alone or in combination with adequate metallic active sites, can provide exceptional electronic and thermal effects to synergistically boost the production of NH3 from N2 and H2. Along with absorption properties, stability is equally important when assessing the potential application of a catalyst. Conventional stability cycles usually cover only a few hours of activity, however, this limited timespan is insufficient to gain any insight on the real robustness of the system, so long-term stability tests in the order of tens of hours are imperative.
The present review has described alternative NH3 synthesis routes based on the use of solar light as source of energy, namely photo-catalysis and photo-thermal catalysis. However, it is worth reminding that the use of electro-catalysis to drive the N2 reduction reaction has recently gained considerable interest. Compared to the traditional HB process, the possibility to produce NH3 using renewable electricity would allow a decentralized route for N2 fixation with lower CO2 emissions and improved energy efficiency. However, in spite of these promising features, the viability and reliability of most of the available reports on electrocatalytic N2 reduction is still under debate, given the low amount of NH3 produced (in the orders of nmol to μmol) and the presence of multiple impurities that may lead to false positives.180–182 For instance, a recent work from Chen and collaborators demonstrated the presence of nitrogen-containing compounds such as NO3− or NO2− in many commercial catalysts for ammonia electro-synthesis, thus stressing the necessity to develop specific protocols to ensure reliable and reproducible results.183 In any case, electrochemical NH3 synthesis has become a very dynamic field of research and we certainly believe it will play a decisive role in the future NH3 production systems beyond the HB process. For these reasons, and despite being out of the scope of this work, those readers interested in the topic are referred to specific reviews available in the literature.181,184,185
In summary, photo-thermal catalysis has proved to hold great promise as a viable route to drive the N2 hydrogenation to NH3 using solar light as energy source. Apart from its advantages in terms of energy efficiency and operational costs, a potential niche for the photo-thermal NH3 synthesis would be the distributed production of fertilizers. Conventional HB facilities tend to be localized close to the natural gas supply points and require daily productions in the range of thousands of metric tons of NH3 to be economically feasible.186 These facts illustrate some of the constraints of the HB process, particularly the heavy dependence on fossil fuels and a certain inflexibility in the scale.4 On the contrary, photo-thermal NH3 synthesis represents an opportunity for the deployment of small-scale delocalized NH3 plants powered by renewable energy. This on-site production of ammonia would increase the access to fertilizers, especially in remote or isolated areas, promoting their economic and social growth. Interestingly, besides the well-known application of NH3 as precursor for the manufacture of fertilizers, in recent years NH3 has been considered as an excellent H2 carrier owing to its high volumetric and gravimetric energy density and well-established distribution infrastructure.187 Nevertheless, the decomposition of NH3 to obtain N2 and H2 is a highly endothermic reaction that requires elevated temperatures in the range of 500–600 °C to achieve significant conversion rates, thus posing a considerable penalty to the overall efficiency. Given its ability to reach elevated local temperatures, photo-thermal catalysis has the potential to drive the light-induced cracking of NH3 under milder operating conditions, thus lowering the cost and improving the efficiency of the whole process, in a similar way to the NH3 synthesis reaction. Within the context of hydrogen economy, the development of such a light-mediated NH3 synthesis-decomposition cycle would be considered a major breakthrough with multiple implications in the decarbonization of a wide variety of areas including transportation or the industrial sector. All in all, we hope that this review will stimulate the progress of new photo-thermal systems focused not only on the chemistry of NH3 but also on other commodities of industrial interest.
Author contributions
Conceptualization: D.M. and J.G. Writing – original draft: D.M., A.S., M. Z. Writing – review and editing: J.G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from this work was provided by King Abdullah University of Science and Technology (KAUST). Authors would like to thank Mrs Sandra Ramirez for the artwork in this manuscript.References
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- K. H. Rouwenhorst, Y. Engelmann, K. van‘t Veer, R. S. Postma, A. Bogaerts and L. Lefferts, Green Chem., 2020, 22, 6258–6287 RSC.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- C. A. Del Pozo and S. Cloete, Energy Convers. Manage., 2022, 255, 115312 CrossRef.
- P. Mayer, A. Ramirez, G. Pezzella, B. Winter, S. M. Sarathy, J. Gascon and A. Bardow, IScience, 2023, 26, 107389 CrossRef CAS.
- https://www.aramco.com/en/news-media/news/2020/first-blue-ammonia-shipment , (accessed 14/11/2023).
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 CrossRef CAS.
- D. Mateo, P. Maity, G. Shterk, O. F. Mohammed and J. Gascon, ChemSusChem, 2021, 14, 5525–5533 CrossRef CAS PubMed.
- I. S. Khan, D. Mateo, G. Shterk, T. Shoinkhorova, D. Poloneeva, L. Garzón-Tovar and J. Gascon, Angew. Chem., 2021, 133, 26680–26686 CrossRef.
- S. W. L. Ng, M. Gao, W. Lu, M. Hong and G. W. Ho, Adv. Funct. Mater., 2021, 31, 2104750 CrossRef CAS.
- S. Lu, F. Liu, P. Qiu, M. Qiao, Y. Li, Z. Cheng, N. Xue, X. Hou, C. Xu and Y. Xiang, Chem. Eng. J., 2020, 379, 122382 CrossRef CAS.
- J. Liao, Y. Xu, Y. Zhao, C.-C. Wang and C. Ge, ACS Appl. Nano Mater., 2021, 4, 1898–1905 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem. Catal., 2022, 2, 52–83 CrossRef CAS.
- H. Shen, M. Yang, L. Hao, J. Wang, J. Strunk and Z. Sun, Nano Res., 2022, 1–37 Search PubMed.
- G. Zhang, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Adv. Energy Mater., 2021, 11, 2003294 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- M. Li, H. Huang, J. Low, C. Gao, R. Long and Y. Xiong, Small Methods, 2019, 3, 1800388 CrossRef.
- K. H. Rouwenhorst, A. G. Van der Ham and L. Lefferts, Int. J. Hydrogen Energy, 2021, 46, 21566–21579 CrossRef CAS.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- N. Morlanés, W. Almaksoud, R. K. Rai, S. Ould-Chikh, M. M. Ali, B. Vidjayacoumar, B. E. Al-Sabban, K. Albahily and J.-M. Basset, Catal. Sci. Technol., 2020, 10, 844–852 RSC.
- R. K. Rai, W. Al Maksoud, N. Morlanés, M. Harb, R. Ahmad, A. Genovese, M. N. Hedhili, L. Cavallo and J.-M. Basset, ACS Catal., 2021, 12, 587–599 CrossRef.
- P. Yan, W. Guo, Z. Liang, W. Meng, Z. Yin, S. Li, M. Li, M. Zhang, J. Yan, D. Xiao, R. Zou and D. Ma, Nano Res., 2019, 12, 2341–2347 CrossRef CAS.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Loyer, K. Mitsuhara and K. Yamanaka, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- M. Kitano, J. Kujirai, K. Ogasawara, S. Matsuishi, T. Tada, H. Abe, Y. Niwa and H. Hosono, J. Am. Chem. Soc., 2019, 141, 20344–20353 CrossRef CAS PubMed.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202, 420–448 CrossRef CAS.
- Z. Wang, J. Lin, R. Wang and K. Wei, Catal. Commun., 2013, 32, 11–14 CrossRef CAS.
- N. Shimoda, Y. Kimura, Y. Kobayashi, J. Kubota and S. Satokawa, Int. J. Hydrogen Energy, 2017, 42, 29745–29755 CrossRef CAS.
- B. C. McClaine and R. J. Davis, J. Catal., 2002, 210, 387–396 CrossRef CAS.
- X. Zhang, L. Liu, A. Wu, J. Zhu, R. Si, J. Guo, R. Chen, Q. Jiang, X. Ju and J. Feng, ACS Catal., 2022, 12, 2178–2190 CrossRef CAS.
- Y. Ma, G. Lan, W. Fu, Y. Lai, W. Han, H. Tang, H. Liu and Y. Li, J. Energy Chem., 2020, 41, 79–86 CrossRef.
- C. Liang, Z. Wei, Q. Xin and C. Li, Appl. Catal., A, 2001, 208, 193–201 CrossRef CAS.
- Z. Ma, X. Xiong, C. Song, B. Hu and W. Zhang, RSC Adv., 2016, 6, 51106–51110 RSC.
- M. Carltonbird, S. Eaimsumang, S. Pongstabodee, S. Boonyuen, S. M. Smith and A. Luengnaruemitchai, Chem. Eng. J., 2018, 344, 545–555 CrossRef CAS.
- Z. Ma, S. Zhao, X. Pei, X. Xiong and B. Hu, Catal. Sci. Technol., 2017, 7, 191–199 RSC.
- C. Li, Y. Shi, Z. Zhang, J. Ni, X. Wang, J. Lin, B. Lin and L. Jiang, J. Energy Chem., 2021, 60, 403–409 CrossRef CAS.
- J. Feng, L. Liu, X. Ju, Q. Jiang, J. Wang, J. Guo, T. He and P. Chen, Catal. Sci. Technol., 2023, 13, 4156–4167 RSC.
- Y. Baik, M. Kwen, K. Lee, S. Chi, S. Lee, K. Cho, H. Kim and M. Choi, J. Am. Chem. Soc., 2023, 145, 11364–11374 CrossRef CAS.
- X. Zhang, L. Liu, J. Wang, X. Ju, R. Si, J. Feng, J. Guo and P. Chen, J. Catal., 2023, 417, 382–395 CrossRef CAS.
- J. Feng, L. Liu, X. Zhang, J. Wang, X. Ju, R. Li, J. Guo, T. He and P. Chen, Catal. Sci. Technol., 2023, 13, 844–853 RSC.
- L. Li, T. Zhang, J. Cai, H. Cai, J. Ni, B. Lin, J. Lin, X. Wang, L. Zheng and C.-T. Au, J. Catal., 2020, 389, 218–228 CrossRef CAS.
- Y. Zhou, X. Peng, T. Zhang, H. Cai, B. Lin, L. Zheng, X. Wang and L. Jiang, ACS Catal., 2022, 12, 7633–7642 CrossRef CAS.
- M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2016, 7, 4036–4043 RSC.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama and M. Hara, Angew. Chem., 2018, 130, 2678–2682 CrossRef.
- I. Rossetti, N. Pernicone and L. Forni, Catal. Today, 2005, 102, 219–224 CrossRef.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS.
- W. Gao, J. Guo, P. Wang, Q. Wang, F. Chang, Q. Pei, W. Zhang, L. Liu and P. Chen, Nat. Energy, 2018, 3, 1067–1075 CrossRef CAS.
- A. Tarka, M. Zybert, E. Truszkiewicz, B. Mierzwa, L. Kępiński, D. Moszyński and W. Raróg-Pilecka, ChemCatChem, 2015, 7, 2836–2839 CrossRef CAS.
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, Catal. Commun., 2017, 101, 15–19 CrossRef CAS.
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, J. Rare Earths, 2018, 36, 703–707 CrossRef CAS.
- X. Wang, L. Li, T. Zhang, B. Lin, J. Ni, C.-T. Au and L. Jiang, Chem. Commun., 2019, 55, 474–477 RSC.
- Y. Bicer, I. Dincer, C. Zamfirescu, G. Vezina and F. Raso, J. Cleaner Prod., 2016, 135, 1379–1395 CrossRef CAS.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- S. Chen, D. Liu and T. Peng, Sol. RRL, 2021, 5, 2000487 CrossRef CAS.
- B. Puértolas, M. Comesaña-Hermo, L. V. Besteiro, M. Vázquez–González and M. A. Correa–Duarte, Adv. Energy Mater., 2022, 12, 2103909 CrossRef.
- Y. Shi, Z. Zhao, D. Yang, J. Tan, X. Xin, Y. Liu and Z. Jiang, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- G. Schrauzer and T. Guth, J. Am. Chem. Soc., 2002, 99, 7189–7193 CrossRef.
- V. Augugliaro, A. Lauricella, L. Rizzuti, M. Schiavello and A. Sclafani, Int. J. Hydrogen Energy, 1982, 7, 845–849 CrossRef CAS.
- J. Soria, J. Conesa, V. Augugliaro, L. Palmisano, M. Schiavello and A. Sclafani, J. Phys. Chem., 1991, 95, 274–282 CrossRef CAS.
- N. Rao, S. Dube and P. Natarajan, Appl. Catal., B, 1994, 5, 33–42 CrossRef CAS.
- W. Zhao, J. Zhang, X. Zhu, M. Zhang, J. Tang, M. Tan and Y. Wang, Appl. Catal., B, 2014, 144, 468–477 CrossRef CAS.
- D. Maarisetty and S. S. Baral, J. Mater. Chem. A, 2020, 8, 18560–18604 RSC.
- L. Ran, J. Hou, S. Cao, Z. Li, Y. Zhang, Y. Wu, B. Zhang, P. Zhai and L. Sun, Sol. RRL, 2020, 4, 1900487 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, 2008180 CrossRef CAS PubMed.
- J. Yang, Y. Guo, R. Jiang, F. Qin, H. Zhang, W. Lu, J. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS.
- C. Feng, P. Wu, Q. Li, J. Liu, D. Wang, B. Liu, T. Wang, H. Hu and G. Xue, New J. Chem., 2022, 46, 1731–1740 RSC.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- Y. Huang, N. Zhang, Z. Wu and X. Xie, J. Mater. Chem. A, 2020, 8, 4978–4995 RSC.
- J. Di, J. Xia, M. F. Chisholm, J. Zhong, C. Chen, X. Cao, F. Dong, Z. Chi, H. Chen and Y. X. Weng, Adv. Mater., 2019, 31, 1807576 CrossRef.
- C. Xu, P. Qiu, L. Li, H. Chen, F. Jiang and X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 25321–25328 CrossRef CAS PubMed.
- Y. Zhang, S. Gu, X. Zhou, K. Gao, K. Sun, D. Wu, J. Xia and X. Wang, Catal. Sci. Technol., 2021, 11, 4783–4792 RSC.
- M. Lan, N. Zheng, X. Dong, X. Ren, J. Wu, H. Ma and X. Zhang, Sustainable Energy Fuels, 2021, 5, 2927–2933 RSC.
- J. Wang, L. Tang, G. Zeng, Y. Liu, Y. Zhou, Y. Deng, J. Wang and B. Peng, ACS Sustainable Chem. Eng., 2017, 5, 1062–1072 CrossRef CAS.
- X. Yu, H. Qiu, Z. Wang, B. Wang, Q. Meng, S. Sun, Y. Tang and K. Zhao, Appl. Surf. Sci., 2021, 556, 149785 CrossRef CAS.
- J. Liu, R. Li, X. Zu, X. Zhang, Y. Wang, Y. Wang and C. Fan, Chem. Eng. J., 2019, 371, 796–803 CrossRef CAS.
- X. Chen, Y. Li, Z. Wu, X. Xu, W. Zhu and X. Gao, J. Colloid Interface Sci., 2021, 602, 553–562 CrossRef CAS.
- T. Fei, L. Yu, Z. Liu, Y. Song, F. Xu, Z. Mo, C. Liu, J. Deng, H. Ji and M. Cheng, J. Colloid Interface Sci., 2019, 557, 498–505 CrossRef CAS PubMed.
- C. Ladasiu, N. Kulischow and R. Marschall, Catal. Sci. Technol., 2022, 12, 1450–1457 RSC.
- F. Yang, Q. Zhang, L. Zhang, M. Cao, Q. Liu and W.-L. Dai, Appl. Catal., B, 2019, 257, 117901 CrossRef CAS.
- P. Xing, S. Wu, Y. Chen, P. Chen, X. Hu, H. Lin, L. Zhao and Y. He, ACS Sustainable Chem. Eng., 2019, 7, 12408–12418 CAS.
- P. Xing, W. Zhang, L. Chen, X. Dai, J. Zhang, L. Zhao and Y. He, Sustainable Energy Fuels, 2020, 4, 1112–1117 RSC.
- H. Miyama, N. Fujii and Y. Nagae, Chem. Phys. Lett., 1980, 74, 523–524 CrossRef CAS.
- S. Hu, Y. Li, F. Li, Z. Fan, H. Ma, W. Li and X. Kang, ACS Sustainable Chem. Eng., 2016, 4, 2269–2278 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters and L. C. Seefeldt, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- S. Hu, X. Chen, Q. Li, Y. Zhao and W. Mao, Catal. Sci. Technol., 2016, 6, 5884–5890 RSC.
- Z. He, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, RSC Adv., 2019, 9, 21646–21652 RSC.
- K. Zhang and L. Guo, Catal. Sci. Technol., 2013, 3, 1672–1690 RSC.
- X. Ning, J. Li, B. Yang, W. Zhen, Z. Li, B. Tian and G. Lu, Appl. Catal., B, 2017, 212, 129–139 CrossRef CAS.
- T. Di, Q. Xu, W. Ho, H. Tang, Q. Xiang and J. Yu, ChemCatChem, 2019, 11, 1394–1411 CrossRef CAS.
- M. Lan, Y. Wang, X. Dong, F. Yang, N. Zheng, Y. Wang, H. Ma and X. Zhang, Appl. Surf. Sci., 2022, 591, 153205 CrossRef CAS.
- B. Sun, Z. Liang, Y. Qian, X. Xu, Y. Han and J. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7257–7269 CrossRef CAS PubMed.
- L. Shi, Z. Li, L. Ju, A. Carrasco-Pena, N. Orlovskaya, H. Zhou and Y. Yang, J. Mater. Chem. A, 2020, 8, 1059–1065 RSC.
- H. Xu, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, Appl. Catal., B, 2019, 257, 117932 CrossRef CAS.
- G. Swain, S. Sultana and K. Parida, ACS Sustainable Chem. Eng., 2020, 8, 4848–4862 CrossRef CAS.
- Q. Zhang, S. Hu, Z. Fan, D. Liu, Y. Zhao, H. Ma and F. Li, Dalton Trans., 2016, 45, 3497–3505 RSC.
- Y. Shen, J. Shou, L. Chen, W. Han, L. Zhang, Y. Chen, X. Tu, S. Zhang, Q. Sun and Y. Chang, Appl. Catal., A, 2022, 643, 118739 CrossRef CAS.
- Z. Yang, J. Wang, J. Wang, M. Li, Q. Cheng, Z. Wang, X. Wang, J. Li, Y. Li and G. Zhang, Langmuir, 2022, 38, 1178–1187 CrossRef CAS PubMed.
- N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao, W. Ye, Z. Qi, H. Ju and C. Wang, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- L. Zhang, R. Li, L. Cui, Z. Sun, L. Guo, X. Zhang, Y. Wang, Y. Wang, Z. Yu and T. Lei, Chem. Eng. J., 2023, 461, 141892 CrossRef CAS.
- X. Liu, Y. Luo, C. Ling, Y. Shi, G. Zhan, H. Li, H. Gu, K. Wei, F. Guo and Z. Ai, Appl. Catal., B, 2022, 301, 120766 CrossRef CAS.
- H. Wu, X. Li, Y. Cheng, Y. Xiao, R. Li, Q. Wu, H. Lin, J. Xu, G. Wang and C. Lin, J. Mater. Chem. A, 2020, 8, 2827–2835 RSC.
- R. Fu, G. Wang, Q. Zhan, L. Zhang and L. Liu, Green Chem., 2023, 25, 8531–8538 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- C. Liang, H.-Y. Niu, H. Guo, C.-G. Niu, D.-W. Huang, Y.-Y. Yang, H.-Y. Liu, B.-B. Shao and H.-P. Feng, Chem. Eng. J., 2020, 396, 125395 CrossRef CAS.
- Z. Li, G. Gu, S. Hu, X. Zou and G. Wu, Chin. J. Catal., 2019, 40, 1178–1186 CrossRef CAS.
- Y. Shiraishi, S. Shiota, Y. Kofuji, M. Hashimoto, K. Chishiro, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, ACS Appl. Energy Mater., 2018, 1, 4169–4177 CrossRef CAS.
- K. Zhang, Z. Ai, M. Huang, D. Shi, Y. Shao, X. Hao, B. Zhang and Y. Wu, J. Catal., 2021, 395, 273–281 CrossRef CAS.
- H. Mou, J. Wang, D. Yu, D. Zhang, W. Chen, Y. Wang, D. Wang and T. Mu, ACS Appl. Mater. Interfaces, 2019, 11, 44360–44365 CrossRef CAS PubMed.
- L. Zhang, S. Hou, T. Wang, S. Liu, X. Gao, C. Wang and G. Wang, Small, 2022, 18, 2202252 CrossRef CAS PubMed.
- S. Liu, S. Wang, Y. Jiang, Z. Zhao, G. Jiang and Z. Sun, Chem. Eng. J., 2019, 373, 572–579 CrossRef CAS.
- S. Cao, N. Zhou, F. Gao, H. Chen and F. Jiang, Appl. Catal., B, 2017, 218, 600–610 CrossRef CAS.
- K. Zhang, L. Deng, M. Huang, H. Tu, Z. Kong, Z. Liang, Y. Shao, X. Hao and Y. Wu, Colloids Surf., A, 2022, 633, 127830 CrossRef CAS.
- C. H. Ng, S. H. Teo, N. Mansir, A. Islam, C. G. Joseph, S. Hayase and Y. H. Taufiq-Yap, Sustainable Energy Fuels, 2021, 5, 4457–4511 RSC.
- L. K. Putri, B.-J. Ng, W.-J. Ong, H. W. Lee, W. S. Chang and S.-P. Chai, J. Mater. Chem. A, 2018, 6, 3181–3194 RSC.
- J. Gu, Y. Yu, X. Fu, H. Chen, F. Jiang and X. Wang, Appl. Surf. Sci., 2021, 555, 149733 CrossRef CAS.
- S. Hu, W. Zhang, J. Bai, G. Lu, L. Zhang and G. Wu, RSC Adv., 2016, 6, 25695–25702 RSC.
- X.-H. Li, W.-L. Chen, H.-Q. Tan, F.-R. Li, J.-P. Li, Y.-G. Li and E.-B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 37927–37938 CrossRef CAS PubMed.
- M. Lashgari and P. Zeinalkhani, Nano Energy, 2018, 48, 361–368 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. Waterhouse, L. Z. Wu and C. H. Tung, Adv. Sci., 2019, 6, 1802109 CrossRef.
- Y. Zhao, F. Wu, Y. Miao, C. Zhou, N. Xu, R. Shi, L. Z. Wu, J. Tang and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21728–21731 CrossRef CAS PubMed.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- Y. Xiao, X. Guo, N. Yang and F. Zhang, J. Energy Chem., 2021, 58, 508–522 CrossRef CAS.
- W. Zhan, H. Gao, Y. Yang, X. Li and Q. L. Zhu, Adv. Energy Sustainability Res., 2022, 3, 2200004 CrossRef CAS.
- S. Abednatanzi, M. Najafi, P. G. Derakhshandeh and P. Van Der Voort, Coord. Chem. Rev., 2022, 451, 214259 CrossRef CAS.
- G. Li, F. Li, J. Liu and C. Fan, J. Solid State Chem., 2020, 285, 121245 CrossRef CAS.
- X. Zhang, X. Li, S. Su, M. Tan, G. Liu, Y. Wang and M. Luo, Catal. Sci. Technol., 2023, 13, 705–713 RSC.
- Z. Zhao, D. Yang, H. Ren, K. An, Y. Chen, Z. Zhou, W. Wang and Z. Jiang, Chem. Eng. J., 2020, 400, 125929 CrossRef CAS.
- X.-H. Li, H. Li, S.-L. Jiang, L. Yang, H.-Y. Li, Q.-L. Liu, W. Bai, Q. Zhang, C. Xiao and Y. Xie, ACS Catal., 2023, 13, 7189–7198 CrossRef CAS.
- H. Huang, X.-S. Wang, D. Philo, F. Ichihara, H. Song, Y. Li, D. Li, T. Qiu, S. Wang and J. Ye, Appl. Catal., B, 2020, 267, 118686 CrossRef CAS.
- W. Gao, X. Li, X. Zhang, S. Su, S. Luo, R. Huang, Y. Jing and M. Luo, Nanoscale, 2021, 13, 7801–7809 RSC.
- K. An, H. Ren, D. Yang, Z. Zhao, Y. Gao, Y. Chen, J. Tan, W. Wang and Z. Jiang, Appl. Catal., B, 2021, 292, 120167 CrossRef CAS.
- J. Tan, H. Ren, Z. Zhao, X. Xin, Y. Shi, D. Yang and Z. Jiang, Chem. Eng. J., 2023, 466, 143259 CrossRef CAS.
- B. Bagherpour and S. Dehghanpour, J. Solid State Chem., 2023, 324, 124079 CrossRef CAS.
- K. Q. Hu, P. X. Qiu, L. W. Zeng, S. X. Hu, L. Mei, S. W. An, Z. W. Huang, X. H. Kong, J. H. Lan and J. P. Yu, Angew. Chem., Int. Ed., 2020, 59, 20666–20671 CrossRef CAS PubMed.
- C. Zhang, Y. Xu, C. Lv, X. Zhou, Y. Wang, W. Xing, Q. Meng, Y. Kong and G. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 29917–29923 CrossRef CAS PubMed.
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC.
- H. Song, X. Meng, T. D. Dao, W. Zhou, H. Liu, L. Shi, H. Zhang, T. Nagao, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 408–416 CrossRef CAS PubMed.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 CrossRef CAS PubMed.
- J. Zhang, K. Xie, Y. Jiang, M. Li, X. Tan, Y. Yang, X. Zhao, L. Wang, Y. Wang and X. Wang, ACS Catal., 2023, 13, 10855–10865 CrossRef CAS.
- S. Wu, Y. Li, Q. Hu, J. Wu and Q. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 11635–11651 CrossRef CAS.
- Y. Wu, S. Han, Y. Huang, Y. Shi and B. Zhang, J. Mater. Chem. A, 2018, 6, 18426–18429 RSC.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- N. Keller, J. Ivanez, J. Highfield and A. M. Ruppert, Appl. Catal., B, 2021, 296, 120320 CrossRef CAS.
- Z. Wang, Z. Yang, R. Fang, Y. Yan, J. Ran and L. Zhang, Chem. Eng. J., 2022, 429, 132322 CrossRef CAS.
- J. Hong, C. Xu, B. Deng, Y. Gao, X. Zhu, X. Zhang and Y. Zhang, Adv. Sci., 2022, 9, 2103926 CrossRef CAS.
- Y. Peng, J. Albero, A. Franconetti, P. Concepción and H. García, ACS Catal., 2022, 12, 4938–4946 CrossRef CAS.
- Y. Peng, A. Melillo, R. Shi, A. Forneli, A. Franconetti, J. Albero and H. García, Appl. Catal., B, 2023, 339, 123143 CrossRef CAS.
- X. Bian, Y. Zhao, G. I. Waterhouse, Y. Miao, C. Zhou, L. Z. Wu and T. Zhang, Angew. Chem., 2023, 135, e202304452 CrossRef.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen and J. Li, Chem, 2019, 5, 2702–2717 CAS.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706–1711 CrossRef CAS PubMed.
- C. Mao, L. Yu, J. Li, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 224, 612–620 CrossRef CAS.
- W. He, X. Huang, X. Ma and J. Zhang, J. Opt., 2022, 51, 142–153 CrossRef.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef.
- J. Grand, B. Auguié and E. C. Le Ru, Anal. Chem., 2019, 91, 14639–14648 CrossRef CAS PubMed.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong and L. Henderson, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- Y. Yuan, L. Zhou, H. Robatjazi, J. L. Bao, J. Zhou, A. Bayles, L. Yuan, M. Lou, M. Lou and S. Khatiwada, Science, 2022, 378, 889–893 CrossRef CAS PubMed.
- R. Verma, R. Tyagi, V. K. Voora and V. Polshettiwar, ACS Catal., 2023, 13, 7395–7406 CrossRef CAS.
- J. Shen, R. Tang, Z. Wu, X. Wang, M. Chu, M. Cai, C. Zhang, L. Zhang, K. Yin and L. He, Trans. Tianjin Univ., 2022, 28, 236–244 CrossRef CAS.
- I. S. Khan, L. Garzon-Tovar, D. Mateo and J. Gascon, Eur. J. Inorg. Chem., 2022, 2022, e202200316 CrossRef CAS.
- S. Wang, A. A. Tountas, W. Pan, J. Zhao, L. He, W. Sun, D. Yang and G. A. Ozin, Small, 2021, 17, 2007025 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu and E. E. Storey, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, X. Huang, L. Tao and Y. Bi, Appl. Catal., B, 2021, 283, 119633 CrossRef CAS.
- T. H. Tan, B. Xie, Y. H. Ng, S. F. B. Abdullah, H. Y. M. Tang, N. Bedford, R. A. Taylor, K.-F. Aguey-Zinsou, R. Amal and J. Scott, Nat. Catal., 2020, 3, 1034–1043 CrossRef CAS.
- D. Mateo, J. Albero and H. Garcia, Joule, 2019, 3, 1949–1962 CrossRef CAS.
- L. Mascaretti, A. Schirato, T. Montini, A. Alabastri, A. Naldoni and P. Fornasiero, Joule, 2022, 6, 1727–1732 CrossRef.
- M. Romero and A. Steinfeld, Energy Environ. Sci., 2012, 5, 9234–9245 RSC.
- Y. Tian and C.-Y. Zhao, Appl. Energy, 2013, 104, 538–553 CrossRef CAS.
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS.
- R. Schäppi, D. Rutz, F. Dähler, A. Muroyama, P. Haueter, J. Lilliestam, A. Patt, P. Furler and A. Steinfeld, Nature, 2022, 601, 63–68 CrossRef PubMed.
- J. Zhang, H. Chen, X. Duan, H. Sun and S. Wang, Mater. Today, 2023, 68, 234–253 CrossRef CAS.
- X. E. Cao, Y. Kaminer, T. Hong, P. Schein, T. Liu, T. Hanrath and D. Erickson, Iscience, 2020, 23, 101856 CrossRef PubMed.
- L. F. Greenlee, J. N. Renner and S. L. Foster, Journal, 2018, 8, 7820–7827 CAS.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- J. Choi, B. H. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nat. Catal., 2020, 3, 1055–1061 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- N. Okuzumi, B. Jones and S. Nielsen, Start-up of the world largest ammonia plant, Proceedings of NITROGEN, 2001 Search PubMed.
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |