
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScaled-up microwave-assisted batch and flow synthesis and life cycle assessment of a silica mesoporous material: UVM-7†
Miriam
Benítez
a,
Cristina
Rodríguez-Carrillo
a,
Sheila
Sánchez-Artero
 a,
Jamal
El Haskouri
b,
Pedro
Amorós
b and
Jose Vicente
Ros-Lis
*a
a,
Jamal
El Haskouri
b,
Pedro
Amorós
b and
Jose Vicente
Ros-Lis
*a
aREDOLí Research Group, Instituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universitat Politècnica de València, Universitat de València, Doctor Moliner 50, Burjassot, Valencia 46100, Spain. E-mail: J.Vicente.Ros@uv.es; Tel: +34 963870000
bInstitut de Ciència dels Materials (ICMUV), c/Catedrático José Beltrán 2, Paterna, 46980 Valencia, Spain
First published on 31st October 2023
Abstract
The development of scaled-up processes for the synthesis and functionalization of silica materials is a key issue for its adoption in various applications. This report introduces innovative batch and flow methodologies for the synthesis and functionalization of substantial quantities of UVM-7, a silica mesoporous material exhibiting a bimodal pore system. The synthesis benefits from the combination of the atrane route with a reduction in reaction time induced by microwaves generated with a solid-state source. To our knowledge, this is the first example of a flow microwave-assisted synthesis of silica mesoporous materials. The materials were characterized by thermogravimetric analysis, X-ray diffraction, transmission electron microscopy and N2 absorption/desorption isotherms. The scaled-up procedures, encompassing both batch and flow, are capable of synthesizing as-made materials in less than 15 minutes, yielding materials with a topology characteristic of UVM-7 akin to those synthesized via conventional methodologies. The functionalization process can be executed in less than five minutes with loadings of organic moieties of 3.2 mmol of APTES (g of silica)−1. Within an hour, the as-made material for more than 150 g of calcined UVM-7 could be synthesized, and up to 60 g could be functionalized. Life Cycle Assessment (LCA) using the ReCiPe method indicates that the most significant impacts are in the categories of freshwater ecotoxicity, marine ecotoxicity, and human carcinogenic toxicity. The scaled-up synthesis offers a substantial reduction in environmental impacts in these categories, a 5-fold reduction in CO2 equivalent emissions compared to the non-scaled synthesis and up to half compared to conventional synthesis. The main contributors are solvents in the functionalization and energy consumption during the calcination.
Introduction
Silica mesoporous materials have been widely explored at the research level in recent decades. We can cite as the first example the MCM-41 published in 1992.1 These materials are characterized by an ordered mesopore system with a pore diameter of around 2 nm. The presence of a pore system with a uniform size distribution and the ease of functionalization with both organic molecules and inorganic oxides or nanoparticles have allowed the development of a wide range of materials with the most diverse applications.2 The pore size of MCM-41 limited its application, so other materials with larger pore sizes quickly appeared, such as SBA-15 or SBA-16.3 Among the fields in which silica mesoporous materials have been used, we can mention catalysis, biomedicine, sensors, controlled release, etc.4 In contrast to MCM-41, which shows a unimodal mesopore system, nanoparticulated silicas can also offer hierarchical bimodal porosity, and this feature can improve accessibility and mass transfer (in both directions, between the pores and the outside). For example, UVM-7, prepared through a one-pot procedure by using a simple template agent and starting from silicon atrane complexes as inorganic precursors, presents a very open architecture based on a continuous network constructed from covalently bonded pseudo-spherical mesoporous primary nanoparticles whose aggregation defines a nonordered secondary system of large textural pores.5 This organization favours the mass-transfer processes inside UVM-7 silicas, which improve their applicability.6The preparation of mesoporous silica materials is mainly based on the sol–gel method in a hydroalcoholic medium in the presence of a surfactant or polymer. These act as structure directing agents and generate the final material, the mesoporous structure. The typical synthesis process for this class of materials comprises synthesis (at room temperature or heating, generally including an aging process), emptying of the pores by calcination or extraction, and a subsequent functionalization process. This method, which we could describe as “conventional”, has been complemented with other strategies aimed at obtaining materials more quickly, with higher yields or novel morphologies. Some examples of alternative methodologies use ultrasounds, microwaves, or mechanochemistry.7 Among them, microwaves are especially suitable for this purpose since they considerably increase the reaction rate, which means less processing time. In this sense, the authors of the present work reported a microwave-assisted UVM-7 synthesis method that allows the use of solid-state microwave generation technology to prepare, calcinate, and functionalize a mesoporous silica material in only 4 hours.8 In part, the success of the result is due to the use of solid-state (SS) microwave generators that allow the generation of radiation with a controlled and stable power, even for small powers, and with a narrow range of frequencies.9
Compared to other silica materials such as zeolites or fumed silica that are usually prepared in large quantities, mesoporous materials are generally prepared only in batches and at the gram level, limiting their practical application. Thus, the development of synthesis procedures that allow one to obtain larger quantities of materials is a topic of interest. The reported scaled-up synthesis methods tend to depart from organosilanes or a low-cost source of silica such as sodium silicate or rice husk following the common sol–gel procedures.10 In other cases, more scalable alternative techniques such as spray drying are applied.11 However, they are limited in general to a few grams, even only 1 gram. Conversely, the gradual injection of reactants has been revealed as an interesting technique for the preparation of dozens of grams of monodisperse microporous silica particles in 3 hours, but unfortunately, the resulting material had a pore diameter narrower than 1 nm.12 An attempt to use the same technique in the presence of a surfactant to obtain an ordered large-sized pore structure produced only disordered materials with an area of approximately 500 m2 g−1.13 Remarkably, Zhang et al. were able to obtain up to 1 kg of spherical silica nanoparticles with a stellate, worm-like or raspberry-like morphology departing from TEOS upon heating the reaction mixture for 2 hours.14 Going a step further, 1 kilogram of mesoporous silica nanospheres with a surface area of 500 m2 g−1 were prepared in the absence of organic solvents in 3 hours using a combination of cationic and anionic surfactants.15 However, none of these examples take advantage of the possibilities that microwaves offer to speed up synthesis.
Another possibility for obtaining large amounts of materials is the development of flow synthesis strategies. This allows the continuous production of materials with homogeneous properties and facilitates automation. However, until now, it has generally been limited to microfluidic systems and the use of this approach to obtain silica materials in large quantities has not been reported. A particular case is the use of a roll-to-roll fabrication technique coupled with evaporation induced self-organization for the preparation of ordered mesoporous materials.16
In the present work, we hypothesize that the use of solid-state microwaves in batch or flow systems may be a good strategy to obtain mesoporous silica, specifically UVM-7, in large quantities while reducing the environmental impact.
Results and discussion
The preparation process of mesoporous silica materials can be divided into two independent stages: (i) the formation of the silica structure, “synthesis” from now on, and (ii) the surface modification with organic silanes, “functionalization” from now on. In the present work we set ourselves the objective of achieving a scaling factor of 100 in both stages. The first approach to obtain large amounts of materials is to continue working in batch while increasing the size of the reactor. This is not immediate since aspects such as the mixture of the reagents, the homogeneity of the reaction, and the transmission of energy can be dependent on the batch size. An alternative approach is the use of flow synthesis, which offers the main advantages of maintaining a smaller reactor size and the possibility of automation.Scaling-up of the microwave-assisted batch synthesis and functionalization
 | ||
| Fig. 1 (a) Scheme of the batch reactor; (b) normalized XRD of the materials BS800 (solid line), BS400 (dashed line) and a conventional synthesis (dotted line); (c) nitrogen adsorption–desorption isotherms of BS800; (d) representative TEM images of BS800. | ||
As expected, the formation of a white solid in the reactor due to condensation and the growth of silica is observed experimentally. The process occurs quickly in about 10 minutes and is accompanied by a decrease in reflected power. This suggests that the gel formed couples better with the incident radiation than the solution or sol before gelation and may be a tool to detect the end point of the reaction. After calcination, 35 g of material is obtained, equivalent to a yield of 95%. Nearly all the silicon from TEOS is incorporated into the final material.
The material obtained under these experimental conditions (BS800) presents the typical characteristics of UVM-7. Low angle XRD shows a peak at around 2° (2θ) corresponding to the (100) reflection of the hexagonal mesoporous structure. In addition, a wide signal of lower intensity is observed at 3.5–4.0° (2θ) due to the overlapped (110) and (200) reflections of a hexagonal lattice (Fig. 1b). TEM shows the structure of this class of materials consisting of small mesoporous particles that aggregate giving rise to the presence of large textural pores between them (Fig. 1d). This type of morphology is confirmed by the N2 absorption/desorption analysis, which includes a high surface area of about 956 m2 g−1 and a bimodal pore system with two well-differentiated adsorption steps (Fig. 1c and Table 1). The first one, occurring at relatively low relative pressures (0.2 < P/P0 < 0.4) is related to capillary condensation in the 2.8 nm mesopores of the particles. Its shape, sharp and without a hysteresis loop, suggests that these mesopores are uniform and cylindrical. The second step, occurring at high relative pressures (P/P0 > 0.8), corresponds to the filling of 37.2 nm inter-particle pores formed between primary nanoparticles. If we compare our material with that obtained by conventional synthesis, it is observed that in general, it has the same properties. The UVM-7 prepared following conventional procedures has larger inter-particle pores, but typically this parameter is one of those that presents greater variation.8 This fact opens the possibility of modulating larger pores in a finer way through a modification of the MW preparative conditions.
| Material | 2θa (°) | Areab (m2 g−1) | Mesopore volumec (cm3 g−1) | Mesopore diameterc (nm) | Textural diameterc (nm) | Textural volumec (cm3 g−1) |
|---|---|---|---|---|---|---|
| a Angle of incidence for the reflection plane. b BET specific surface calculated from N2 adsorption–desorption isotherms. c Pore volumes and pore sizes (diameter) calculated from N2 adsorption–desorption isotherms. d Average and SD of the fractions FS-2 to FS-4. e Average and SD of the fractions collected during the flow functionalization. | ||||||
| BS800 | 2.11 | 956 | 0.82 | 2.8 | 37.2 | 2.05 |
| BS800c | 2.23 | 801 | 0.85 | 2.6 | 59.9 | 1.62 |
| BS800s | 2.01 | 1024 | 0.67 | 2.6 | 29.9 | 0.58 |
| BS400 | 2.17 | 937 | 0.51 | 2.5 | 30.7 | 1.37 |
| BF-4 | 2.15 | 372 | 0.15 | 2.3 | 33.6 | 0.46 |
| FS-1 | 2.29 | 928 | 0.78 | 2.8 | 47.8 | 2.30 |
| FSd | 2.04 | 980 ± 50 | 0.84 ± 0.04 | 2.82 ± 0.04 | 34.0 ± 1.8 | 0.77 ± 0.13 |
| FFe | 2.01 | 220 ± 160 | 0.08 ± 0.04 | 2.7 ± 0.2 | 54 ± 4 | 1.02 ± 0.10 |
| Conven. Synth | 2.13 | 969 | 0.78 | 2.7 | 49.9 | 1.44 |
| Conven. Func. | 2.14 | 251 | 0.15 | 2.6 | 39.6 | 0.96 |
Since the reactor has a diameter of 14 cm, one of the aspects that worried us was that inhomogeneities could be generated in the reaction medium. The combination of a short reaction time with the limited penetration capacity of the microwaves could lead to hot and cold spots, with a consequent decrease in the homogeneity of the final product. Thus, a new batch irradiated at 800 W for 12.5 minutes was prepared, and samples were taken from the centre (BS800c) and the side (BS800s) of the reaction flask. Although in all cases the material can be qualified as UVM-7 (see Table 1 and Fig. S1 in the ESI†), there is indeed a certain difference between the properties of both samples. The mesopore size in both cases is equal and the BET area, despite its variation, falls into the range of a mesoporous UVM-7 (typically between 800 and 1100 cm3 g−1) (Table 1). Again, the main difference lies in the process of aggregation of the particles to form the large-pore architecture. In any case, it is appropriate to limit the width of the reactor to ensure the homogeneity of UVM-7. Additionally, a longer reaction time was applied (400 W and 25 minutes) maintaining the energy input (BS400). Although BS400 is a bimodal mesoporous material, it presents a lower volume of mesopores and a slight reduction in the pore diameter (see Table 1 and Fig. S1 in the ESI†). Furthermore, only 25 g of material before calcination was obtained, which corresponds to a yield of 68%. It seems that microwave power is not an innocent element and that it has a considerable influence on the kinetics of the hydrolysis and condensation reactions.
The microwave-assisted UVM-7 synthesis process is a widely scalable process. Extrapolating our results and considering our equipment, the proposed synthesis method would allow one to prepare the “as-made” material to obtain more than 150 g of calcined material in just one hour, which opens the way to its industrial application.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :UVM-7 ratio remained constant. The power selected was 200 W and different times of irradiation (4, 8, 12 and 16 minutes) were employed. In only 4 minutes, TGA revealed that a high degree of functionalization was achieved. 3.2 mmol of APTES (g of silica)−1 were added to the surface of UVM-7, similar to the 3.6 mmol of APTES (g of silica)−1 that can be obtained by conventional synthesis. An extension of the reaction time to 8, 12 and 16 minutes increases the organic content to 3.3, 3.7 and 4.0 mmol APTES (g of silica)−1 respectively. However, we believe that for most applications, the slight increase in load does not justify the need for higher energy and time consumption in this synthesis process. This degree of functionalization is significantly higher than that previously found for microwave functionalization (1.8 mmol) for the same time.8 Although we cannot rule out other reasons, we believe that this result could be due to the increase in concentration derived from the reduction in solvent volume. As expected from the functionalization process, chemically mild to silica, the silica structure is not affected (see Table 1 and Fig. S1 in the ESI†). However, the coating of organic matter reduces the size and accessibility of the pores. This effect is more pronounced in the mesopores due to their smaller size, leading to a reduction in the total surface area of the material to 372 m2 g−1 (Table 1). In view of the results, we can conclude that microwave-assisted batch functionalization is also a very scalable process and that it can help considerably reduce reaction times. If we consider that the reaction time is 4 minutes, in 1 hour, about 40 grams of the mesoporous material UVM-7 could be functionalized.
:UVM-7 ratio remained constant. The power selected was 200 W and different times of irradiation (4, 8, 12 and 16 minutes) were employed. In only 4 minutes, TGA revealed that a high degree of functionalization was achieved. 3.2 mmol of APTES (g of silica)−1 were added to the surface of UVM-7, similar to the 3.6 mmol of APTES (g of silica)−1 that can be obtained by conventional synthesis. An extension of the reaction time to 8, 12 and 16 minutes increases the organic content to 3.3, 3.7 and 4.0 mmol APTES (g of silica)−1 respectively. However, we believe that for most applications, the slight increase in load does not justify the need for higher energy and time consumption in this synthesis process. This degree of functionalization is significantly higher than that previously found for microwave functionalization (1.8 mmol) for the same time.8 Although we cannot rule out other reasons, we believe that this result could be due to the increase in concentration derived from the reduction in solvent volume. As expected from the functionalization process, chemically mild to silica, the silica structure is not affected (see Table 1 and Fig. S1 in the ESI†). However, the coating of organic matter reduces the size and accessibility of the pores. This effect is more pronounced in the mesopores due to their smaller size, leading to a reduction in the total surface area of the material to 372 m2 g−1 (Table 1). In view of the results, we can conclude that microwave-assisted batch functionalization is also a very scalable process and that it can help considerably reduce reaction times. If we consider that the reaction time is 4 minutes, in 1 hour, about 40 grams of the mesoporous material UVM-7 could be functionalized.
Flow microwave-assisted synthesis
Flow synthesis offers an interesting option for the preparation of materials in large quantities due to its versatility and the homogeneity that is conferred on the products. In addition, in the case of microwaves, it allows the avoidance of the formation of hot and cold spots because the liquid is in motion, and the diameter of the conduction can be adjusted to avoid significant power losses. Unlike batch synthesis, as the dissolution mass is lower, the power requirements are lower. Thus, we work with a source of only 200 W and one antenna. | ||
| Fig. 2 (a) Scheme of the batch reactor; representative TEM images of FS-1 (b), FS-2 (c), FS-3 (e) and FS-4 (f); (d) normalized XRD of the materials FS-1 (dashed line), FS-2 to 4 (dotted line), and conventional synthesis (solid line). | ||
:glycerol 2:1 v/v mixtures without obvious improvements. We also tested water, which gave an adequate drag for the material, but we discarded it due to the risk of hydrolysis and condensation reactions between the APTES molecules in parallel to the reaction on the surface of UVM-7. Finally, we evaluated the possibility of using DMSO, which, although offered good transport of materials and functionalization of around 18.0 ± 0.5% by mass, was discarded due to the difficulty of working with this type of solvent in large-scale productions. Therefore, we decided to use acetonitrile as the solvent and focus on modifying the system. Among these adaptations we can highlight the achievement of a tube of adequate inner diameter, small enough to drive the solution homogeneously but large enough to avoid blocking the conduction by the material, and a configuration in which the tube was always advancing in favour of gravity. We opted for a rubber with an internal diameter of 6 mm. The organic load stands at 13.2 ± 1.4% (equivalent to 3.1 mmol APTES (g of silica)−1). The measurements of nitrogen adsorption/desorption show the expected reduction in the total area due to the blockage of the pores by APTES. The variation in the total area between the fractions is higher in the flow functionalization in comparison with the flow synthesis, which could be assigned to the heterogeneous character of the reaction and possible variations in the residence time of the material. Continuous microwave-assisted functionalization allows the production of 59 g of functionalized material in one hour.
Life cycle assessment and reduction of the environmental impact
Life cycle analysis (LCA) is a methodology that allows the identification of those processes that have the greatest environmental impact. It covers not only global warming, but also many other parameters of great importance for the sustainability of human actions. Although at first glance it may seem that one process is more sustainable than another, this methodology allows us to quantify it and identify those reagents or processes with the greatest impact to try to reduce it.In our case, the LCA has been applied to the four scaled-up processes, the non-scaled synthesis and functionalization, and the conventional synthesis. The objective is to determine if, through the application of microwaves and scaling, in addition to a reduction in time, there is a variation in the environmental impact of the synthesis and functionalization of UVM-7. The system boundaries have been defined as a cradle-to-gate system. The use and disposal of the material have been left out of the study, as well as the emissions. The functional unit has been defined as the preparation of 1 kg of calcined UVM-7 or the functionalization of 1 kg of UVM-7. Life cycle inventories can be found in Table 2. For a better data analysis, energy and solvents have been broken down into the different stages of the process and/or functions.
| Inventory | Reagents (kg) or energy (kW h) | |||
|---|---|---|---|---|
| MW escalated batch | MW escalated flow | MW non-escalated | Conventional | |
| TEOS | 3.7 | 3.7 | 3.7 | 3.7 |
| TEAH | 8.4 | 8.4 | 8.4 | 9.2 |
| CTABr | 1.3 | 1.3 | 1.3 | 1.6 |
| DIW synthesis | 28.6 | 28.6 | 28.6 | 28.5 |
| DIW washing | 71.4 | 71.4 | 428.6 | 177.9 |
| Ethanol washing | 2.9 | 2.9 | 57.1 | 8.9 |
| APTES | 1.3 | 1.3 | 1.3 | 2.2 |
| Acetonitrile functionalization | 47.2 | 47.2 | 235.8 | 23.6 |
| Acetonitrile washing | 15.7 | 7.9 | 157.2 | 23.6 |
| Energy synthesis | 3.5 | 3.5 | 22.9 | 201.4 |
| Energy calcination | 360.5 | 360.5 | 970.0 | 1924.0 |
| Energy functionalization | 24.0 | 18.0 | 200.0 | 55.0 |
The main results of the life cycle impact assessment (LCIA) have been collected in Fig. 3–5. First, we considered the sum of the normalized impact values to identify which categories generated the greatest impact (see Fig. 3a). Unlike other studies where the contribution to global warming is highlighted, in our case the greatest impacts are in the categories of freshwater ecotoxicity, marine ecotoxicity, and human carcinogenic toxicity and these are largely dependent on the type of synthesis process. Also, the global warming potential has been calculated and displayed in Fig. 3b. The scaled synthesis developed in this work offers a 5-fold reduction in the kg of CO2 eq. generated per kg of material with respect to the non-scaled synthesis and up to half compared to conventional synthesis.
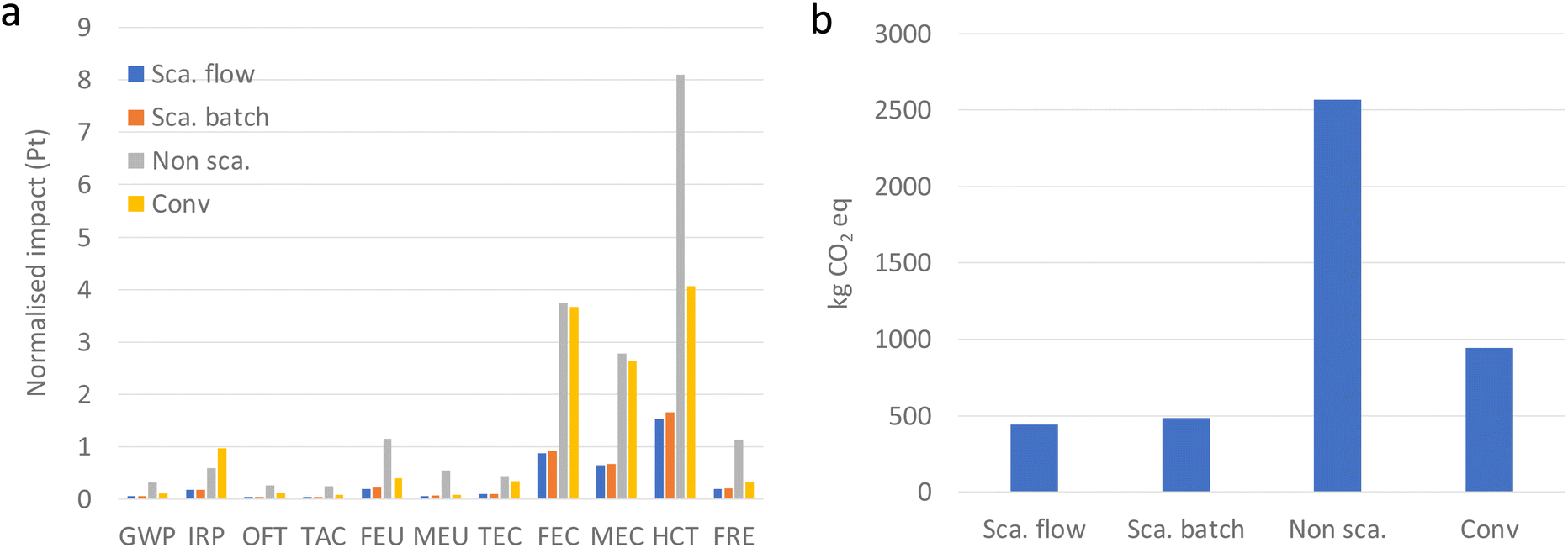 | ||
| Fig. 3 (a) Normalised impact of different synthesis methods and impact categories: global warming (GWP), ionizing radiation (IRP), ozone formation (terrestrial ecosystems) (OFT), terrestrial acidification (TAC), freshwater eutrophication (FEU), marine eutrophication (MEU), terrestrial ecotoxicity (TEC), freshwater ecotoxicity (FEC), marine ecotoxicity (MEC), human carcinogenic toxicity (HCT), and fossil resource scarcity (FRE). (b) Emission of CO2 eq. | ||
 | ||
| Fig. 4 (a) Single score obtained from the synthesis methods [conventional (Conv), non-scaled-up MW assisted synthesis (Non sca.), scaled-up MW assisted batch synthesis (Sca. batch), and scaled-up MW assisted flow synthesis (Sca. flow)] and the steps of reaction; (b) single score obtained from the synthesis methods and the items of the inventory. | ||
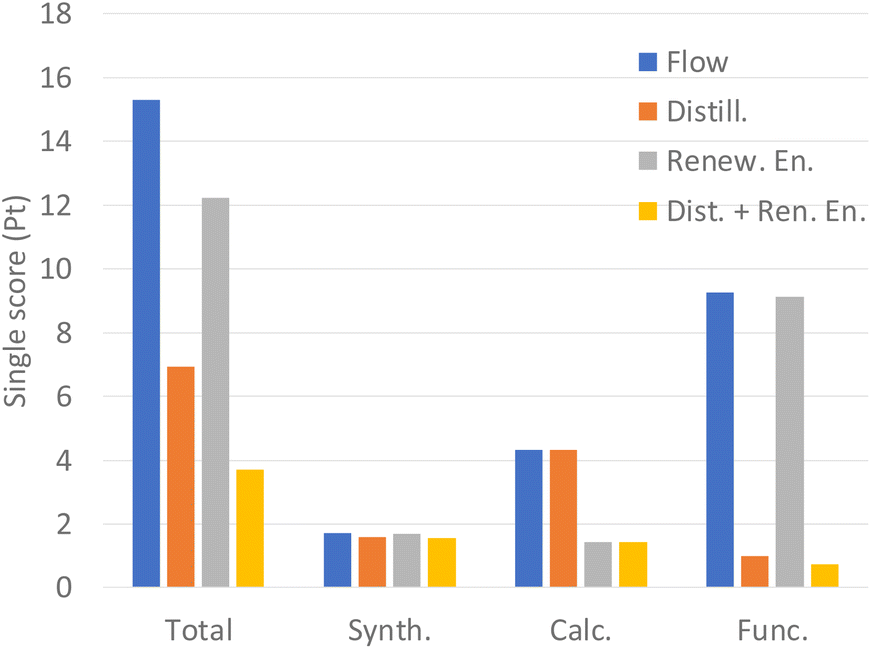 | ||
| Fig. 5 Single score for each scenario and synthesis step. | ||
We can use the single score calculated from the endpoint analysis to gain a deeper understanding of the factors that influence the overall impact. To facilitate the analysis, we have divided the synthesis into the preparation of the “as-made” material, which has the surfactant inside the pores, and the calcination. If we review the influence of the different stages of preparation of the functionalized UVM-7 (see Fig. 4a), we can observe that the different steps have a diverse level of impact and that this largely depends on the process type. Once again, it can be seen how the scaled processes have a much lower impact than the conventional or non-scaled processes and are similar between them. Regarding the steps, the preparation of the “as-made” material has in all cases a lower impact than the calcination and functionalization phases. Functionalization is especially important in the case of a non-scaled process. A better understanding of these results can be deduced from the impact of the different elements of the inventory (see Fig. 4b). The two main elements contributing to the impact are the use of acetonitrile in the functionalization and the electricity used in the calcination, which account for between 85 and 91% of the impact depending on the type of process.
The cost of synthesis was estimated based on the LCI data and our unitary costs (see ESI Tables S1 and S2†). Although the cost can be overestimated due to the use of laboratory cost data, it can be useful to compare the four synthesis methods and identify opportunities for saving. The scaled-up processes offer a reduction of cost from almost 36€ g−1 to approximately 7.5€ g−1, a 5-fold reduction. The most expensive step is functionalization, due to the use of acetonitrile. Next, the second source of cost is the acquisition of reagents. By contrast, the energy cost is relatively low. If we compare the scaled processes with the conventional ones, the main reduction is due to the reduction in the consumption of electricity.
From the LCA results and cost analysis we can conclude that the next steps to improve the scaled synthesis should be focused on reducing acetonitrile consumption and the impact of energy use. To reduce the consumption of acetonitrile, we can vary the solvent, but a more sustainable option may be to recover the solvent by distillation, using it in successive syntheses. Accordingly, we have proposed a scenario in which the energy cost increases due to distillation and 95% of acetonitrile can be recovered in each cycle. To reduce the impact of energy consumption in calcination, one option is to use electricity from renewable sources such as photovoltaics. Finally, we have proposed a scenario in which the previous two options are combined. The results of the scenarios applied to scaled-up flow synthesis, the one with the lower impact, can be seen in Fig. 5. The use of renewable energy in calcination almost avoids any impact in this phase, while distillation reduces the impact of functionalization by 9 times. When we combine both strategies, the sum of the single score drops from 15.3 to just 3.7. Regarding the cost, savings higher than 60% could be obtained.
Experimental
Chemicals
All the synthesis reagents were analytically pure and were used without further purification. Tetraethyl orthosilicate (TEOS), triethanolamine (TEAH3), cetyl-trimethylammonium bromide (CTABr), and (3-aminopropyl)triethoxysilane (APTES) were purchased from Sigma Aldrich (Madrid, Spain). Acetonitrile and ethanol were purchased from Scharlab S.L.Microwave devices
Two different solid-state (SS) devices, one for batch processes and the other for flow processes, were used. Both devices were provided by Microbiotech S.L. The batch device consists of four microwave sources connected through a coaxial cable to the irradiation cavity. Each source operates at a maximum power of 200 W with selectable potency in 1 W increments rising to a maximum power of 800 W in total. The frequency of each source can be adjusted independently to tune the system and optimise the absorbed power within the 2420 MHz–2480 MHz range in 1 Hz increments. The flow device consists of a SS microwave source with characteristics similar to the batch device (maximum power of 200 W) but with selectable potency in 5 W increments.Microwave-assisted synthesis of UVM-7
:TEAH3:CTABr in a 1:3.4:0.25 mole ratio (263 mL of TEAH3, 137 mL of TEOS, and 45 g of CTABr). The mixture was smoothly shaken until homogeneity, then placed into the microwave oven cavity and then irradiated for 12 minutes at 800 W until the gel was formed. As a result, a white solid was obtained. This solid was isolated by filtration, washed with water and ethanol, dried in an oven at 80 °C overnight and calcined at 550 °C under a static air atmosphere, as an oxidant atmosphere, for 5 h to remove the CTABr surfactant. The resulting material was denoted as BS800 (batch synthesis at 800 W). Samples collected at the centre and the side of the reactor were denoted as BS800c and BS800s respectively. Also, a batch of material irradiated at 400 W for 25 minutes (BS400) and UVM-7 prepared with the conventional method in the absence of MW were synthesized for comparative purposes.5
:water ratio (0.41:1 v/v) and an irradiation time (remaining time inside the cavity) of 1 min. The microwave potency was set at 100 W. The product generated during the first minute was rejected and fractions were collected each 5 minutes. All the fractions were filtered, washed with water and ethanol, dried at 80 °C in an oven overnight, and calcined at 550 °C under a static air atmosphere, as an oxidant atmosphere, for 5 h to remove the CTABr surfactant. The fractions were denoted as FS-1 to FS-4 (flow synthesis).
Characterization of the materials
All solids were characterized by X-ray powder diffraction (XRD) at low angles (Bruker D8 Advance diffractometer) using a monochromatic Cu Kα source operated at 40 kV and 40 mA. Patterns were collected in steps of 0.04° (2θ) over the angular range 1.3–8.3° (2θ) for 1 s per step. Transmission electron microscopy (TEM) images were taken with a JEOL-JEM-1010 instrument operated at 100 kV (equipped with the digital camera MegaView III and “ANALYSIS” software). Samples were gently ground in ethanol, and grains were deposited on a holey carbon film supported on a Cu grid. Nitrogen adsorption–desorption isotherms (−196 °C) were recorded on a Micromeritics ASAP 2020 automated analyser. The samples were degassed in situ at 120 °C and 10–6 Torr for 15 h prior to analysis. Specific surface areas were calculated from the adsorption data within the low-pressure range using the Brunauer–Emmett–Teller (BET) model. Pore sizes and pore volumes were determined following the Barrett–Joyner–Halenda (BJH) model on the adsorption branch. Thermogravimetric analysis (TGA) was performed with a SETARAM SETSYS 16–18 instrument (Caluire-et-Cuire, France) under a dry air flow at a heating rate of 10 °C min−1 up to 1000 °C.Life cycle analysis
Life cycle analysis (ISO 14040:2006) on a cradle-to-gate basis was applied to the scaled processes. This technique allows the identification of the environmental impacts of the processes and determines opportunities to improve their sustainability. The inventory was defined using the synthesis procedures and the energy consumption measured in the laboratory. The impact was calculated using the Ecoinvent 3 database and the ReCiPe 2016 Endpoint (H) V1.08/World (2010) H/A model available in the SimaPro software. Data from Spain or Europe have been used if they were available in the database, otherwise worldwide data were used. The method includes as impact categories global warming, stratospheric ozone depletion, ionizing radiation, ozone formation (human health), fine particulate matter formation, ozone formation (terrestrial ecosystems), terrestrial acidification, freshwater eutrophication, marine eutrophication, terrestrial ecotoxicity, freshwater ecotoxicity, marine ecotoxicity, human carcinogenic toxicity, human non-carcinogenic toxicity, land use, mineral resource scarcity, fossil resource scarcity and water consumption. CTABr and APTES are not available in the database, thus, estequat (a cationic surfactant) and TEOS (a silane) were used for the analysis.Conclusions
We have reported for the first time a scaled-up batch microwave-assisted synthesis of UVM-7, a silica mesoporous material. Also, we have described the first, to our knowledge, flow microwave-assisted synthesis of a silica mesoporous material. The materials obtained following batch and flow scaled procedures have structures and properties similar to those prepared under conventional conditions. The as-made material for the preparation of 168 and 59 g of calcined material per hour can be obtained with the batch and flow methods respectively. The functionalization of UVM-7 can be performed in less than 5 minutes with loadings of organic groups of 3.2 mmol of APTES (g of silica)−1. Up to 59 g of UVM-7 per hour can be functionalized. LCA shows that the scaled-up synthesis offers a strong reduction in the environmental impact, but it could be further reduced by the reutilization of solvents and the use of renewable energy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was carried out under the grant PID2021-126304OB-C43 funded by MCIN/AEI/10.13039/501100011033 and “ERDF A way of making Europe”, the grant INNVA1/2020/44 funded by the Agencia Valenciana de Innovación, Genertalitat Valenciana, and the AGROALNEXT programme supported by MCIN with funding from European Union Next Generation EU (PRTR-C17.I1) and by Generalitat Valenciana grant number EUAGROALNEXT/2022/065.References
- A. Kresge, M. Leonowicz, W. Roth, J. Vartuli and J. Beck, J. Nat., 1992, 359, 710 CrossRef.
- J.AS. Costa, R.A. de Jesus, D.O. Santos, J.F. Mano, L.PC. Romao and C.M. Paranhos, Microporous Mesoporous Mater., 2020, 291, 109698 CrossRef CAS; C. Liu, Q.X. Li, D. Zhang, Y.J. Li, J.Q. Liu and X.L. Xiao, Prog. Chem., 2021, 33, 2085 Search PubMed; S. Pasricha, P. Gahlot, K. Mittal, D. Rai, N. Avasthi, H. Kaur and S. Rai, ChemistrySelect, 2022, 7, 202103674 CrossRef.
- D.Y. Zhao, Q.S. Huo, J.L. Feng, B.F. Chmelka and G.D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- V.C. Niculescu, Front. Mater., 2020, 7, 36 CrossRef CAS; B. Singh, J. Na, M. Konarova, T. Wakihara, Y. Yamauchi, C. Salomon and M.B. Gawande, Bull. Chem. Soc. Jpn., 2020, 93, 1459 CrossRef.
- J. El Haskouri, D.O. de Zarate, C. Guillem, J. Latorre, M. Caldes, A. Beltran, D. Beltran, A.B. Descalzo, G. Rodriguez-Lopez, R. Martinez-Manez, M.D. Marcos and P. Amoros, Chem. Commun., 1992, 4, 330 Search PubMed.
- S. Muñoz-Pina, J.V. Ros-Lis, A. Arguelles, C. Coll, R. Martinez-Mañez and A. Andres, Food Chem., 2018, 241, 199 CrossRef.
- M.D. Márquez-Medina, S. Mhadmhan, A.M. Balu, A.A. Romero and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 9537 CrossRef CAS; B.D. de Greñu, R. de los Reyes, A.M. Costero, P. Amoros and J.V. Ros-Lis, Nanomaterials, 2020, 10, 1092 CrossRef; S. Glowniak, B. Szczesniak, J. Choma and M. Jaroniec, Molecules, 2023, 28, 2639 CrossRef PubMed.
- B.D. de Greñu, S. Muñoz-Pina, R. de los Reyes, M. Benítez, J. El Haskouri, P. Amoros and J.V Ros-Lis, ChemSusChem, 2023, e202300123 CrossRef.
- X. Zhou, P.D. Pedrow, Z.W. Tang, S. Bohnet, S.S. Sablani and J.M. Tang, Innov. Food Sci. Emerg. Technol., 2023, 83, 103240 CrossRef CAS.
- J.M. Morales, J. Latorre, C. Guillem, A. Beltran-Porter, D. Beltran-Porter and P. Amoros, Solid State Sci., 2005, 7, 415 CrossRef CAS; R.R. Castillo, L. de la Torre, F. Garcia-Ochoa, M. Ladero and M. Vallet-Regi, Int. J. Mol. Sci., 2020, 21, 7899 CrossRef PubMed.
- H.C. Wang, C.S. Lu, H.L. Bai, J.F. Hwang, H.H. Lee, W. Chen, Y.H. Kang, S.T. Chen, F.S. Su, S.C. Kuo and F.C. Hu, Appl. Surf. Sci., 2012, 258, 6943 CrossRef CAS; I. Emmanuelawati, J. Yang, J. Zhang, H.W. Zhang, L. Zhou and C.Z. Yu, Nanoscale, 2013, 5, 6173 RSC.
- T. Shimogaki, H. Tokoro, M. Tabuchi, N. Koike, Y. Yamashina and M. Takahashi, J. Sol-Gel Sci. Technol., 2015, 74, 109 CrossRef CAS.
- T. Shimogaki, H. Tokoro, M. Tabuchi, N. Koike, Y. Yamashina and M. Takahashi, J. Sol-Gel Sci. Technol., 2016, 79, 440 CrossRef CAS; C. Kim, S. Yoon and J.H. Lee, Microporous Mesoporous Mater., 2019, 288, 109595 CrossRef.
- K. Zhang, LL. Xu, JG. Jiang, N. Calin, KF. Lam, SJ. Zhang, HH. Wu, GD. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427 CrossRef CAS.
- P.C. Liu, Y.J. Yu, B. Peng, S.Y. Ma, T.Y. Ning, B.Q. Shan, T.Q. Yang, Q.S. Xue, K. Zhang and P. Wu, Green Chem., 2017, 19, 5575 RSC.
- Z. Qiang, Y.H. Guo, H. Liu, S.ZD. Cheng, M. Cakmak, K.A. Cavicchi and B.D. Vogt, ACS Appl. Mater. Interfaces, 2015, 7(7), 4306–4310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02875e |
| This journal is © The Royal Society of Chemistry 2024 |