
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStarch esterification using deep eutectic solvents as chaotropic agents and reaction promoters†
Guillermo A.
Portillo-Perez
 ,
Kasper B.
Skov
and
Mario M.
Martinez
*
,
Kasper B.
Skov
and
Mario M.
Martinez
*
Center for Innovative Food (CiFOOD), Department of Food Science, Aarhus University, AgroFood Park 48, Aarhus N, 8200, Denmark. E-mail: mm@food.au.dk
First published on 19th January 2024
Abstract
Starch derivatizations require harsh chemicals as solvents or catalysts, which can negate the green nature of starch-based materials. Often, these reaction systems require several hours to achieve moderate degrees of substitution (DS), drastically reduce molar mass and can result in undesired crosslinking compromising solubility. Here, we report an efficient and sustainable starch esterification with acetic anhydride (AA) and the optimization of the process avoiding initiators in terms of molar ratios, time, and temperature. Furthermore, chromatographic (HPSEC-MALS-dRI) and spectroscopic (FTIR, 1H NMR, 2D HSQC and HMBC, Solid State CP/MAS 13C NMR, TG-IR) tools were implemented to elucidate the chemical composition and structure of the resulting starch acetates and potential intermediate side reaction products over the course of the reaction. Different combinations of choline chloride (ChCl) with several hydrogen bond acceptors (urea, tartaric, malonic, and malic acids) were used as both chaotropic solvents and reaction promoters for starch acetylation. The reaction system comprising 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ChCl:urea and AA showed good miscibility at 100 °C after 30 min, representing a seemingly homogeneous reaction system while better preserving starch molar mass. Side products emerging from solvent-reagent interactions, such as starch carbamate and acetylurea, were identified. Reaction optimization resulted in no side products, fast reaction rates (36 min), high DS (2.87) and starch loads (20 wt%), and increased reaction throughput and atom economy. Catalyst-free starch acetylation reduced hygroscopicity and increased glass transition and degradation temperatures (162 °C and 397 °C, respectively), while generally keeping a relatively higher molar mass (1.5–2.9 × 106 Da) than traditional starch acetates.
:1 molar ChCl:urea and AA showed good miscibility at 100 °C after 30 min, representing a seemingly homogeneous reaction system while better preserving starch molar mass. Side products emerging from solvent-reagent interactions, such as starch carbamate and acetylurea, were identified. Reaction optimization resulted in no side products, fast reaction rates (36 min), high DS (2.87) and starch loads (20 wt%), and increased reaction throughput and atom economy. Catalyst-free starch acetylation reduced hygroscopicity and increased glass transition and degradation temperatures (162 °C and 397 °C, respectively), while generally keeping a relatively higher molar mass (1.5–2.9 × 106 Da) than traditional starch acetates.
Introduction
Starch is among the most abundant and largest naturally engineered macromolecules present in terrestrial biomass. It is a major part of our diets and a prominent renewable resource for the development of biobased and biodegradable materials, such as bioplastics. The use of starch as a packaging material could particularly gain attention after the introduction and implementation of different regulations such as the Single-Use Plastic Directive issued by the European Commission.1 Starch is readily available from a large variety of crops, and it is commercialized at low-cost either in pure form or as non-purified starch-rich edible particles, commonly referred to as flours used as commodity food applications.2 On the one hand, starch ingredients can provide bulk, body, and improved texture and mouthfeel to foods, but cooked starch pastes cannot withstand the heat, shear, and acid associated with processing conditions. Native cooked starch also has a strong tendency for retrogradation and syneresis and, in many food systems,3 it can be a major contributing factor in the development of type-2 diabetes.4 On the other hand, native starch-based biomaterials are strongly polar and hydrophilic, which results in poor moisture and thermal resistivity,5,6 and a relatively high residual moisture that can potentiate hydrolysis and molar mass decrease upon processing. The properties of native starch are therefore less than ideal for its direct application as food or as biomaterial.The modification of polysaccharides to produce esters using anhydrides, particularly acetic anhydride, is well documented in the literature.7–10 Starch esters with monofunctional reagents, such as acetic acid, are more stable against retrogradation (critical for the shelf-life of starchy food systems) and more resistant to moisture (key aspect for e.g. packaging applications). Besides the reduction or elimination of the negative attributes of native starch, esterification could also introduce specific functionalities. For instance, starch esters have the potential to enhance colonic health through the delivery of short chain fatty acids (SCFA), such as acetic acid, to the colon.11 During esterification, starch is left to react in a solution consisting primarily of the chosen anhydride and a basic catalyst such as NaOH, or pyridine. This reaction involves the splitting of the anhydride generating acetic acid as a byproduct, which in turn causes the neutralization of the basic catalyst. This creates additional steps, such as catalyst recovery and regeneration, bringing about environmental pollution and human safety problems related to the large volume of wastewater and sodium acetate generated.12,13 Progress has recently been made in this aspect by substituting the alkaline catalysts with organic acids, such as citric and tartaric acid.5,14 Albeit the extent of esterification was a success, these reaction systems still required several hours to achieve moderate degrees of substitution. Furthermore, the acidic medium led to an important starch degradation, reducing the molar mass of the resulting material by several orders of magnitude. Moreover, at long reaction times, the dihydroxy dicarboxylic organocatalyst also participated in the esterification reaction, resulting in covalently crosslinked di-starch tartrate, which notably decreased its solubility in organic solvents.5 The use of ionic liquids (IL) as solvents or catalysts has been another approach explored. IL 1-butyl-3-methyl-imidazolium chloride ([BMIM][Cl]) has commonly been used as solvent/catalyst to produce substituted starch acetates, although several others have been tested with different degrees of success, seemingly related to the ionic combination.15–17 Ren et al. recently observed that shorter alkyl cations paired with an acetate anion resulted in better crystalline disruption of the starch matrix, leading to more substituted starches.18 While ILs are considered safe due to their low flammability and volatility, concerns have been raised over their toxicity, impact on the environment, and high cost of production.19,20
Deep eutectic solvents (DES) are mixtures of salts representing at least a proton donor and a proton acceptor. When mixed at a specific molar ratio, the mixture undergoes a large depression of the melting point of both of its components resulting in a solvent with low vapor pressure and volatility.21,22 This class of solvents have found extensive use in the field of biomass processing, such as pretreatment, fractionation, and purification of lignocellulosic material, proteins, or chitin.23–28 ChCl:urea, commonly dubbed “reline”, is a natural DES which has gained notoriety in the field of biomass valorization due to its non-toxic nature, low melting point (≈12 °C) and for its capability to solubilize large biomacromolecules and polymeric systems up to 10 wt%.21,29–31 Carboxylic acid DESs have been commonly used in the dissolution and modification of cellulose, such as carboxymethylation and cellulose nanofiber production.32–36 Moreover, DES have been shown to work as plasticizers for biobased thermoplastics, as well as to provide moderate fire-retardant properties.37
With the intend of addressing the main principles of green chemistry as posited by Anastas and Warner (Fig. 1), this study approaches starch esterification utilizing fewer hazardous chemicals and synthesis processes, reducing energy consumption by means of diminishing reaction time or temperature, and relying on fully renewable feedstock.38 For the first time, we report the systematic investigation of DES for efficient starch esterification and the optimization of the process avoiding initiators in terms of molar ratios, time and temperature. Furthermore, a combination of chromatographic and spectroscopic tools was implemented to elucidate the chemical composition and structure of the resulting starch acetates and quantify and elucidate potential intermediate side reaction products over the course of the reaction. Thus, the reaction system presented here foregoes the use of any additional catalysts or auxiliaries, relying instead solely on starch solvation and the slightly basic environment provided by the eutectic mixture. Compared to organocatalytic esterification, results showed more favorable conditions for our developed esterification route in terms of preserving the macromolecular integrity of starch molecules and avoiding side-reactions affecting starch performance, such as crosslinking.
 | ||
| Fig. 1 Listed common 12 principles of green chemistry with highlights representing those considered in this study and met in the proposed starch esterification reaction. | ||
Experimental
Chemicals
Choline chloride (≥98%) (ChCl), deuterated dimethyl sulfoxide (DMSO-D6) 99.9% atom D, acetic anhydride for synthesis ≥98% (AA), L-(+)-tartaric acid (99.7% FG), DL-malic acid (99%), malonic acid (99%) and urea 99%, ACS reagent, were purchased from Sigma-Aldrich (Søborg, Denmark). DMSO ≥99.5% for HPLC was purchased from VWR (Søborg, Denmark). Native wheat starch was kindly provided by Roquette Frères (Roquette wheat starch, Lestrem, France). Ethanol 96% v/v was purchased from VWR (Søborg, Denmark).Deep eutectic solvent (DES) preparation
1 M hydrogen bond donor (HBD) and acceptor (HBA) solutions were prepared by dissolving appropriate amounts of either of these into ethanol. Choline chloride (ChCl) was selected as the acceptor, whereas urea, tartaric (TA), malonic (MO), and malic (MA) acids were selected as donors. Solutions were then mixed in different molar ratios ranging from 2:1 to 1:2 HBA:HBD. Ethanol was then evaporated via rotary evaporation (Büchi, Switzerland) resulting in a DES.
Starch acetylation
To avoid the interference of moisture in the acetylation process, starch was oven-dried before chemical modification at 65 °C for 48 h until <3 wt% water, as determined by a halogen moisture analyzer (HR73, Mettler Toledo, Naverland, Denmark). 5 g of DES were heated up in an oil bath to each selected temperature, where given amounts of starch and acetic anhydride (AA) were added and left to react for different times. All reactions were carried out in fume hood and under reflux to avoid any hazards created from acetic acid vapors and the loss of acetic anhydride. The experimental conditions were selected using a rotatable central composite design with five center point replicates, and four variable factors: time, temperature, AA:starch molar ratio, and starch load. A full table of the experimental conditions is shown in Table S1.†
Acetylated starch (AcS) was recovered via precipitation by adding 10 mL ethanol to the solution, which was then transferred to 15 mL falcon tubes followed by vigorous mixing using a vortex mixer (Yellowline TTS2, IKA-Werke, Germany) at 2500 rpms for 30 s, and centrifuging for 5 min at 12000 rpm and 4 °C (SIGMA Centrifuges, UK). The recovered solid material was washed seven times using a 50% ethanol:water mixture, followed by oven drying at 65 °C for 24 h. For samples presenting side products, an additional purification step was performed for further spectroscopy confirmation of the nature of the side products. Specifically, after the ethanol:water washing, starch was solubilized in DMSO and reprecipitated using ethanol to separate starch from other compounds. All samples were kept under controlled relative humidity in a desiccator at room temperature until further analysis.
Determination of the chemical structure of starch acetates
Degree of substitution was obtained by calculating the relevant areas present in the sample spectra, namely the starch hydroxyl and the acetyl areas. The areas were normalized based on the protons represented therein, following the formula:
 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (acyl) ester groups, and B represents the area of the peaks between 4.5–5.5 ppm related to the OH– groups and proton belonging to the anomeric carbon C1 of starch.
O (acyl) ester groups, and B represents the area of the peaks between 4.5–5.5 ppm related to the OH– groups and proton belonging to the anomeric carbon C1 of starch.
For samples containing side products, starch-depleted solutions (after semi-purification) were mixed with DMSO-d6 (1:10 w/w), heated at 70 °C and stirred at 750 rpms for 30 min using a thermomixer (Eppendorf, Hamburg, Germany) for further spectroscopic experimentation. Then, they were analyzed via single pulse 13C NMR, with spectra obtained using a relaxation time of 9.3 s, spectral width of 165 ppm, 45° pulse of 15 μs, 1024 scans and at 37 °C. Furthermore, they were also analyzed via 2D 1H–13C heteronuclear single quantum correlation (HSQC) in DMSO (δC/δH = 39.5/2.50 ppm), where spectra were obtained at 37 °C, 8 scans, spectral width of 13.02 and 165 for H and C nuclei, respectively. 2D 1H–13C heteronuclear multiple bond correlation (HMBC) spectra were similarly acquired, with a spectral width of 11.9 ppm and 220 ppm, for H and C, respectively.
Statistical modelling
A statistical model of the acetylation reaction was produced by response surface methodology using JMP Pro ver. 16.0.0 (JMP, Great Britain). Time, temperature, AA:starch ratio, and starch load comprised the analyzed variables, whereas DS was the selected output. The resulting model was used to determine the optimal reaction conditions, namely maximizing starch load and DS, while reducing reaction time and AA:starch. Validation of the model was achieved by replicating the conditions suggested by the statistical software in the laboratory. Graphics were created using MATLAB R2022b (Mathworks, USA).
Efficiency of the reaction was calculated in two scenarios: (1) against a maximum degree of substitution DS = 3 (ya), and; (2) against the actual DS (yr). ya was determined considering a theoretical yield yt of 177% (eqn (1)) based on a maximum degree of substitution (i.e., DS = 3) and the measurement of initial and recovered starch mass at DS = 3 (eqn (2)). In addition, to get complementary information about the efficiency of the process at any DS, where not all the OH– groups are necessarily substituted, the recovery efficiency yr was also calculated considering the actual measured DS, as shown in eqn (3).
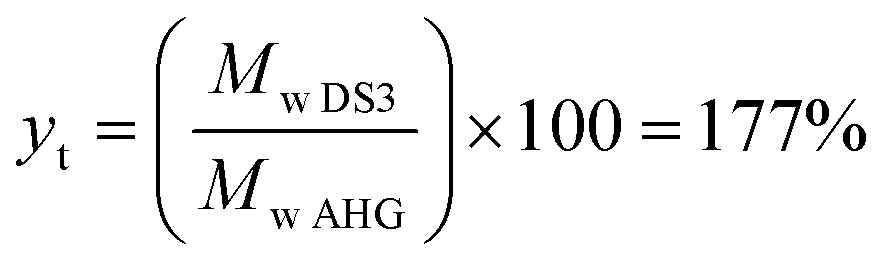 | (1) |
 | (2) |
 | (3) |
Molecular size distribution and molecular weight
Number-average (Mn) and weight-average (Mw) molecular weight of starch and AcS samples were determined using High Performance Size Exclusion Chromatography (HPSEC) as reported by Roman et al.39 A given amount of sample was dissolved in 2 mL of a 0.5% w/w LiBr/DMSO solution at 80 °C and 300 rpms for 24 h in a thermomixer (Eppendorf, Hamburg, Germany), followed by centrifugation at 4000 rpm for 5 min. The supernatant was then transferred to a vial for its analysis. An Agilent 1260 Infinity II (Agilent Technologies, Waldbronn, Germany) with a refractive index (RI) (Shodex RI-501, Munich, Germany) and a Multiangle Laser-Light Scattering (MALS) (Wyatt Technology, Santa Barbara, USA) detectors were used. Columns GRAM 3000 and GRAM 30 (PSS GmbH, Mainz, Germany) were connected in series for size separation. A solution of 0.5% w/w LiBr/DMSO was used as eluent, and the flow rate and temperature were set at 0.3 mL min−1 and 80 °C, respectively. The resulting data was analyzed using ASTRA software v. 8.1, (Wyatt Technology, Santa Barbara, USA). Data was fitted using a second-order Berry plot method. DMSO refractive index increment (dn/dc) was taken as 0.066 ml g−1 as reported in literature for similar starch solutions.40 Second viral coefficient (A2) was assumed to be negligible.Thermal properties
Scanning electron microscopy (SEM)
Starch and AcS samples were observed under a scanning electron microscope (Nova Nano SEM). Images were taken at 5 kV. Before analysis, all samples were coated with 9 nm platinum particles under vacuum, then spread on conductive, double-sided carbon tape (Plano GmbH, Wetzlar, Germany), and attached on an “0.5” aluminum sample stub.Results and discussion
Spectroscopic evaluation of acetylated starch and side reaction products
A series of preliminary experiments were performed to determine the most suitable deep eutectic solvent (DES) for the reaction. The melting point was selected as the first exclusion criterion, with the threshold being <120 °C corresponding to the expected reaction temperature.5 Of all prepared mixtures, only choline chloride (ChCl):tartaric acid (TA) 1:2 and 1:3 were too viscous to be usable at this temperature. Thermal stability at the reaction temperatures, followed by starch solubility, were the next exclusion parameters for DESs. ChCl:Malonic acid (MO) mixtures began to quickly degrade into acetic acid at the range of 110–120 °C. This behavior, together with the formation of esters between ChCl and the carboxylic acid has been previously observed at 125 °C by Rodriguez et al.41 ChCl:Malic acid (MA) mixtures did not solubilize starch, even at high temperatures. While ChCl:TA 1:1 fulfilled the aforementioned criteria, only traces of starch were detected in the recovery, even after only 3 minutes of the reaction at 120 °C (Fig. 2A), likely due to the Brønsted acidity of TA. Interestingly, even at proportions similar to those applied by Vidal et al. (2:1 TA:starch mass ratio) where TA was used as catalysts, acetylation did not occur.5 This would suggest that hydrogen-hydrogen interactions between HBA and HBD inhibit the catalytic effect of the carboxylic acid.
 | ||
| Fig. 2 (A) 1H NMR spectrum of starch acetylation in choline chloride:tartaric acid (ChCl:TA) for 10 min at 135 °C, acetic anhydride:starch ratio of 5, and starch load of 1 g. Starch –OH (reducing): terminal starch –OH groups; TA: tartaric acid protons; ChCl: choline chloride protons. (B) 1H NMR spectrum of starch dissolved in ChCl:urea for 30 min prior to the addiction of acetic anhydride, then acetylated for 5 h at 120 °C, acetic anhydride:starch ratio of 5, and starch load of 1 g. OHn: hydroxyl groups belonging to starch carbon n; Hn: protons belonging to starch carbon n; Hac: protons assigned to acetyl groups. | ||
The reaction system composed of 1:1 molar ChCl:urea and acetic anhydride (AA) showed good miscibility, representing a seemingly homogeneous reaction system (Fig. S1†) at 100 °C after 30 min, while providing a basic environment through the amino groups present in its Hydrogen Bond Donor (HBD) for the acetylation reaction.42,43 While the actual acidity or basicity of DESs, and their determination methods, may still be debated,44,45 our preliminary experiments clearly suggested that ChCl:urea at 1:1 molar ratio much better preserved the structure of starch than ChCl:TA.
As a first approach, starch was allowed to dissolve for 30 min in ChCl:urea prior to AA addition. However, this procedure led to the formation of acetylated starch carbamates, as evidenced by 1H NMR. Precisely, starch samples exhibited peaks related to –NH groups [δ 6.41 (s), 5.40 (s), 5.33 (s), 5.12 (s), 4.51 (s), 3.66 (s), 3.59 (s), 2.04 (m)], featured by the large singlet peak at 6.41 ppm, which can be assigned to the primary amine of the urea, and the characteristic ester peak at 2.04 ppm (Fig. 2B). Simultaneous addition of the reactants shifted the reaction towards acetylation, even at low AA:starch ratios, indicating that this is the preferred pathway for the system. Indeed, simultaneous addition of the reagents resulted in distinct acetylation peaks of starch in the range of 1.9–2.1 ppm (i.e., methyl protons of the acyl groups) as well as a proportional reduction of the hydroxyl groups from the anhydroglucose units between 4.5–5.4 ppm. Moreover, peaks attributed to acetylated starch carbamates were no longer observed (Fig. 3A). This confirms that acetylation was successful, which in turn yielded high degree of substitution (DS) values in short reaction times (0.5 h). Nevertheless, a closer analysis of the 1H NMR spectra exposed the presence of impurities in the solid matrix. Particularly, samples showed atypically large peaks in addition to those from starch at 1.99, 7.07, 7.7 and 10.1 ppm (Fig. 3B). As an example, starch acetylated for 1 h (AcS-1h) displayed the following peaks: δ 10.07 (s), 7.69 (s), 7.10 (s), 5.42 (d, J = 5.9 Hz), 5.34 (s), 5.26 (s), 5.18 (s), 5.11 (s), 4.73 (s), 4.51 (s), 4.47 (s), 4.30 (s), 4.23 (s), 4.16 (s), 3.96–3.92 (m), 3.84 (s), 3.75 (s), 3.66 (s), 3.59 (s), 2.16 (s), 2.10–2.03 (m), 2.05–1.97 (m), 1.99 (s), 1.96 (s) and 1.93 (bs). By all likelihood, these peaks can be attributed to side reactions between acetic acid and urea, namely acetylurea.46 As the acetylation reaction progresses, more acetic acid is generated, and potentially reacts with urea present from ChCl:urea. A scheme of both chemical reactions taking place during ChCl:urea-driven acetylation is shown in Fig. 4.
 | ||
| Fig. 3
1H NMR spectra of native and acetylated (AcS) starches. (A) Full spectra with the starch backbone signals highlighted in the black box. Impurities are observed in the region 7–10 ppm. (B) Enlargement of spectral signals assigned to acetylation. Hac: protons assigned to acetyl groups, either as part of starch or of acetylurea. Reaction conditions: 5 h, 120 °C, acetic anhydride:starch ratio: 15, and starch load: 1.25 g. Reaction was monitored every hour. | ||
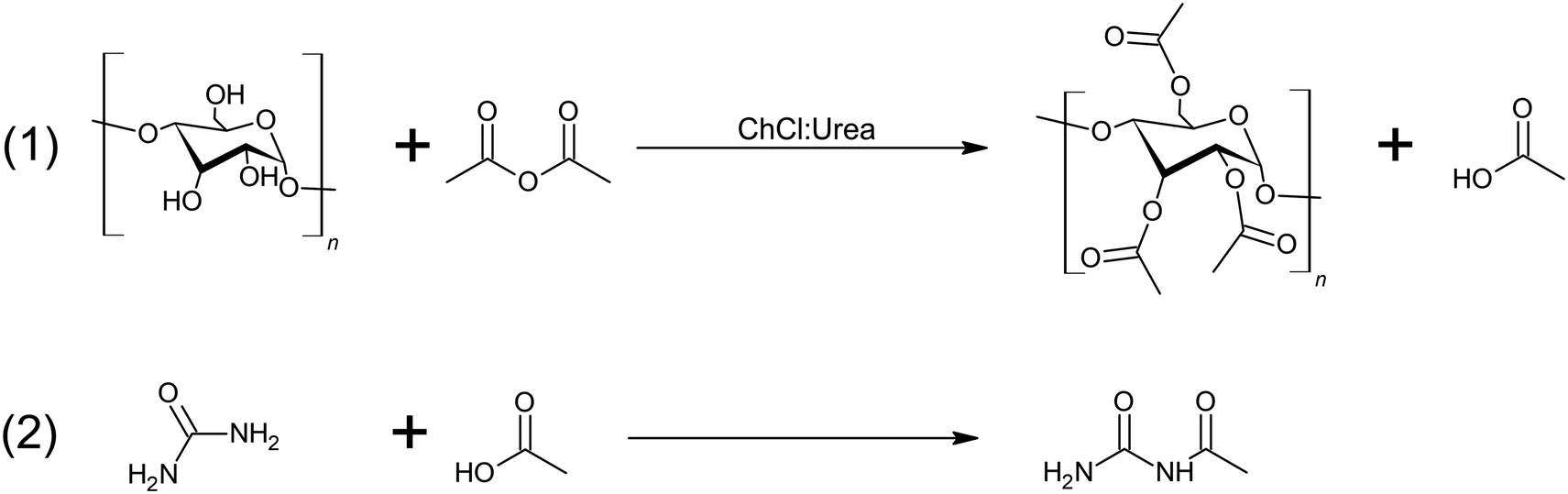 | ||
| Fig. 4 Starch acetylation (1) and formation of acetylurea (2). | ||
The greater intensity of the signals from side reaction products in relation to those from the acetylated material can be attributed to the more soluble nature of acetylurea in comparison to starch. This would result in a drastic underestimation of any signal from undissolved starch protons (removed during centrifugation) together with a much higher concentration of soluble side reaction products. In addition, solubility allows for a much more rapid tumbling in the NMR, thus producing a stronger, clearer signal. Interestingly, the signal corresponding to acetylurea disappeared at longer reaction times, which could be explained by the entrapment of these molecules in the insoluble starch matrix. Samples recovered from shorter reaction times still exhibit a relative integral granular structure, potentially leading to formation and/or entrapment of acetylurea within them, where they are unable to be washed off. As the reaction proceeds, starch loses its semicrystalline and granular structures, allowing for an easier leaching of these impurities upon washing with ethanol:water. To confirm this theory, samples were dissolved in DMSO at 75 °C for 1 hour, reprecipitated using ethanol, then recovered and washed via filtration using a Whatman filter (grade 1). The recovered sample no longer showed any of the related peaks to acetylurea (data not shown), confirming our hypothesis. The recovered acetylurea was analyzed by 13C (151 MHz, DMSO. δ 172.01, 153.80, 39.52, 23.48), as well as 2D 1H–13C HSQC and HMBC. The spectra displayed the expected cross peaks and correlations attributed to acetylurea, i.e., a cross peak at δC/δH 23.5/2.0 representing the acyl group for the HSQC spectra, and a peak at δC/δH 172/1.99, representing the correlation between the carbonyl and methyl groups in HMBC (Fig. 5).
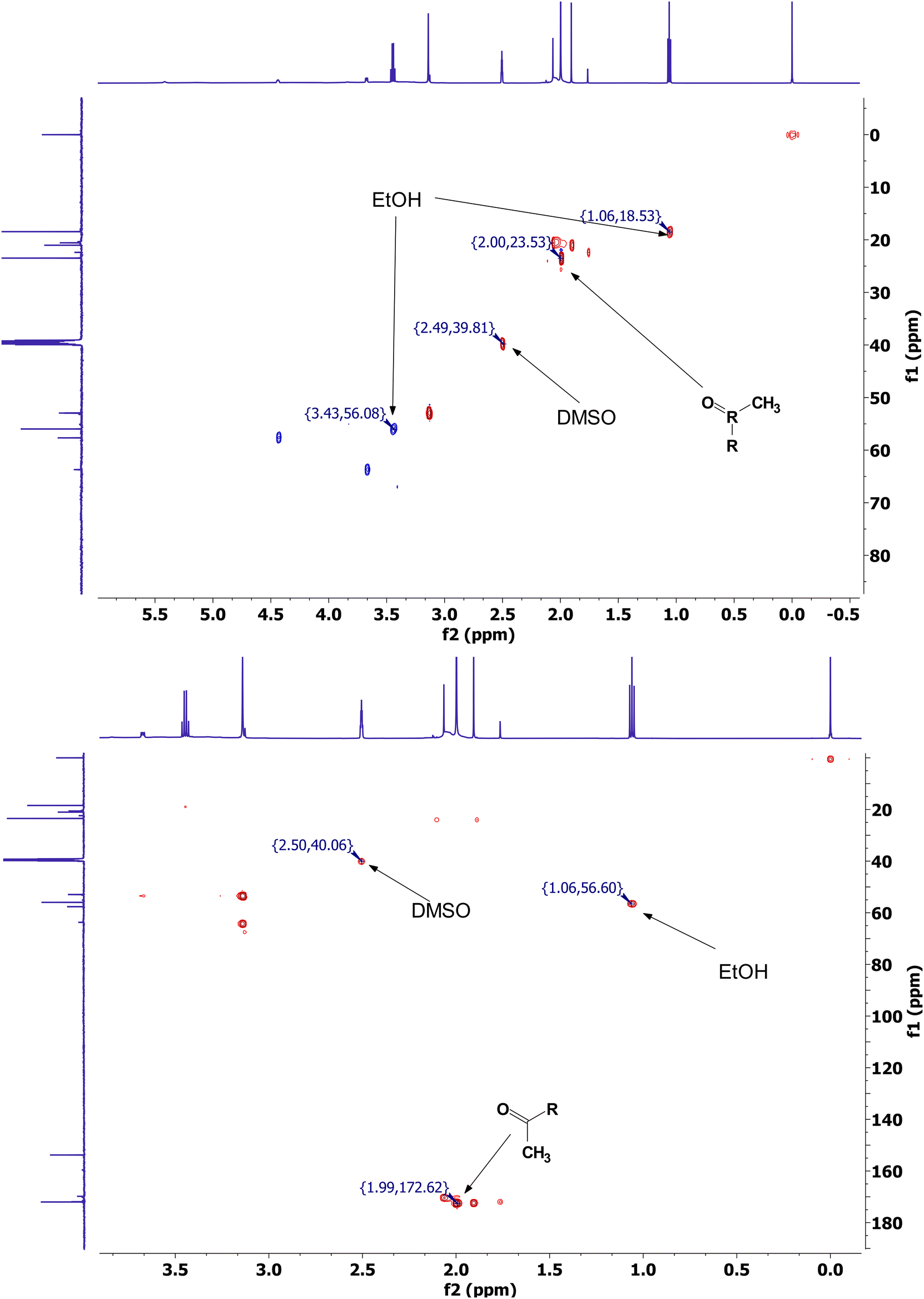 | ||
| Fig. 5 2D 1H–13C heteronuclear single quantum correlation 2D-HSQC (top) and 2D 1H–13C heteronuclear multiple bond correlation HMBC (bottom) spectra of partially purified acetylurea in DMSO-d6 solvent. Spectra also displays traces of ethanol (EtOH), acetic anhydride, acetic acid, and Choline Chloride (ChCl). | ||
Cross Polarization/Magic Angle Spinning (CP/MAS) 13C NMR was performed to corroborate both chemical reactions taking place during the process, to understand the relative proportion of acetylurea in the solid final product, and to study the starch molecular order. All the samples tested exhibited the typical peaks attributed to starch, namely the cluster of peaks C(2), C(3), and C(5) at ∼66 ppm, and C(1) at ∼96 ppm (Fig. 6). Notably, the peak assigned to C(6) disappeared during the reaction, and in turn, two new peaks were observed: the carbonyl group of the acetyl fraction at 166 ppm and its corresponding methyl at 16 ppm, representing the acetyl group attached to starch. The presence of acetylyurea was confirmed by the amide group at 151 ppm and a smaller methyl peak at 19.9 ppm. A liquid NMR analysis of a semipurified sample of acetylurea exhibited similar peaks, more in line with the values observed in spectral databases (Fig. S2†).46 As expected, the relative intensities of these peaks revealed a much lower proportion of acetylurea compared to what 1H NMR peaks showed. Further information regarding the semicrystalline structure of starch could be obtained from the deconvolution of the C(1) peak (Fig. 6B and C), as described before47,48 Three peaks were clearly identified in a triplet arrangement and a smaller shoulder downfield, a common feature of the A-type crystallinity of native starch.49–52 In contrast, the esterified samples no longer presented any type of multiplicity after deconvolution, suggesting that starch became fully amorphous. Such crystallinity loss on acetylated samples using acetic anhydrides as donors, both following the traditional alkaline reaction or by organocatalysis, has been previously reported, with significant changes in morphology and structure even at low DS values (DS = 0.5).7,53,54 Substitution of hydroxyl by acetyl groups leads to the unraveling double helices and the concomitant loss of double helical crystalline packing.
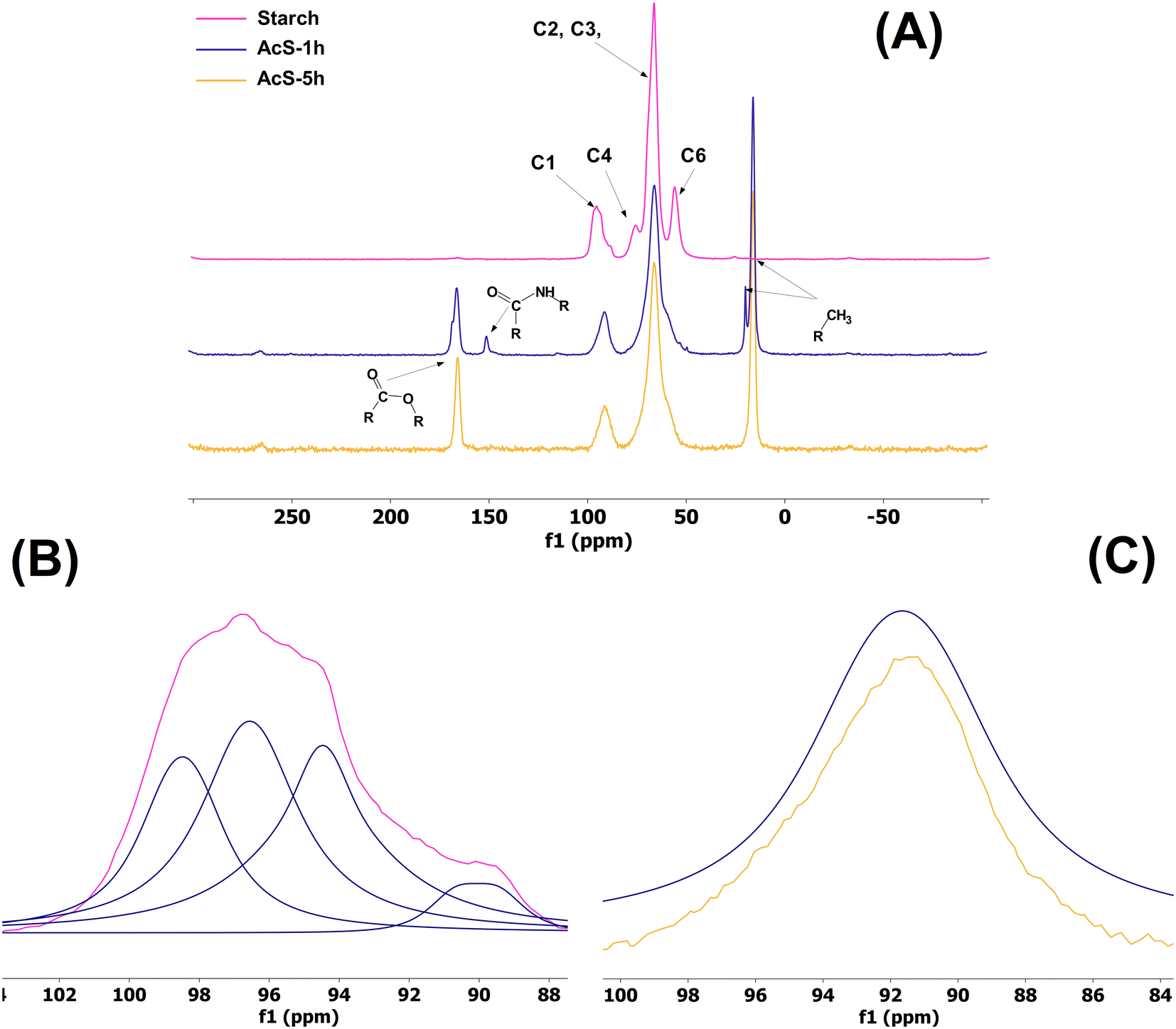 | ||
| Fig. 6 (A) CP/MAS 13C NMR spectra of native starch, and acetylated starches (AcS) that were left to react for 1 (AcS-1h) and 5 h (AcS-5h) at 120 °C, acetic anhydride:starch ratio of 15 and starch load of 1.25 g. (B) Enlargement of the C(1) region of native starch including peak deconvolution of the ordered subspectra into individual peaks. (C) Enlargement of the C(1) region of AcS-1h and AcS-5h. | ||
The results of the Fourier Transform Infrared analysis (FTIR) agreed with those from 1H and solid-state CP/MAS 13C NMR (Fig. 7). Pure wheat starch showed the typical associated peaks, namely a sharp acetal peak related to the C–O–C 1,4/1,6- acetal groups at 1025–994 cm−1, a smaller peak at 2920 cm−1 from the –CH structure, as well as the hydroxyl broad peak of –OH groups at 3275 cm−1. The samples presenting acetylurea side product displayed a set of characteristic peaks, namely a double peak at 3374–3327 cm−1 representing a primary amine, a peak at 3213 cm−1 representing secondary amines, and a series of overlapping peaks in the range of 1640–1629 cm−1 that can be attributed to the CO stretch of amides, both primary and secondary. Furthermore, the typical ester peak at 1733 cm−1 is convoluted due to the presence of amide CO groups. As the reaction progressed and the samples became more unravelled, these peaks disappeared completely, and the ester peak at 1733 cm−1 and that of the methyl group at 1367 cm−1, corresponding to the acetyl group, became clearer. Of note the complete disappearance of the hydroxyl groups at 3275 cm−1, confirming the complete acetylation of starch. Detailed assignment of peaks in IR spectra can be found in Table S2.†
 | ||
| Fig. 7 FTIR spectra of native and acetylated (AcS) starch. Reaction conditions: 120 °C, acetic anhydride:starch ratio of 15, and starch load of 1.25 g. Reaction was monitored every hour. | ||
Although the production of acetylurea is not the main goal of the reaction system, it is by no means an undesirable side product. Acetylurea is a useful source of non-protein nitrogen for animal feed, and an interesting amide precursor for the pharmaceutical industry.55–57 Moreover, the facile, catalyst-free formation of this side product presents an opportunity for synthetic routes for the cogeneration of starch acetates and acetylurea derivatives, which can be applied in the synthesis of antiepileptic drugs.58–60
Optimization of catalyst-free acetylation via statistical modelling
The catalyst-free reaction was studied using a central composite experimental design (CCD) with four variables whose ranges are shown in Table 1. Ranges were determined by the distance from the center point (coded “0”), at distances of +1 to −1 for factorial points and +α to -α for axial points; α was calculated as α = (2k)0.25, where k = number of factors. DS up to DS = 3.1 were obtained within the experimental area of the study, indicating the full substitution of -OH groups. It is noteworthy that the DS values above 3 can be explained by the depolymerization of starch chains, forming smaller starch segments and hence increasing the average number of terminal –OH groups. Though it should also be noted that, as starch fragmentation continues, the substrate becomes more soluble, leaving little precipitable material. Indeed, for experiments with harsher reaction conditions, i.e., high temperature and long reaction times, very little product could be recovered (≈2 wt% of initial starch load).| Variable | −α | −1 | 0 | 1 | α |
|---|---|---|---|---|---|
| Time (h) | 1 | 3 | 5 | 7 | 9 |
| Temperature (°C) | 100 | 110 | 120 | 130 | 140 |
| AA:starch (mol:mol) |
5 | 10 | 15 | 20 | 25 |
| Starch load (g) | 0.25 | 0.50 | 0.75 | 1.00 | 1.25 |
As expected, a direct correlation between reaction time and DS (Fig. 8) was observed. Nevertheless, the relevance of time as a factor appeared to diminish as the other variables increased (AA:starch, starch load, and their interaction). At higher AA ratios and substrate loads, DS > 2 could be obtained only within 1 h. It is also of note that no particular hydroxyl signal presented a more pronounced reduction when compared to others, suggesting that the probability of substitution is equal for all, i.e., no regioselectivity could be observed.
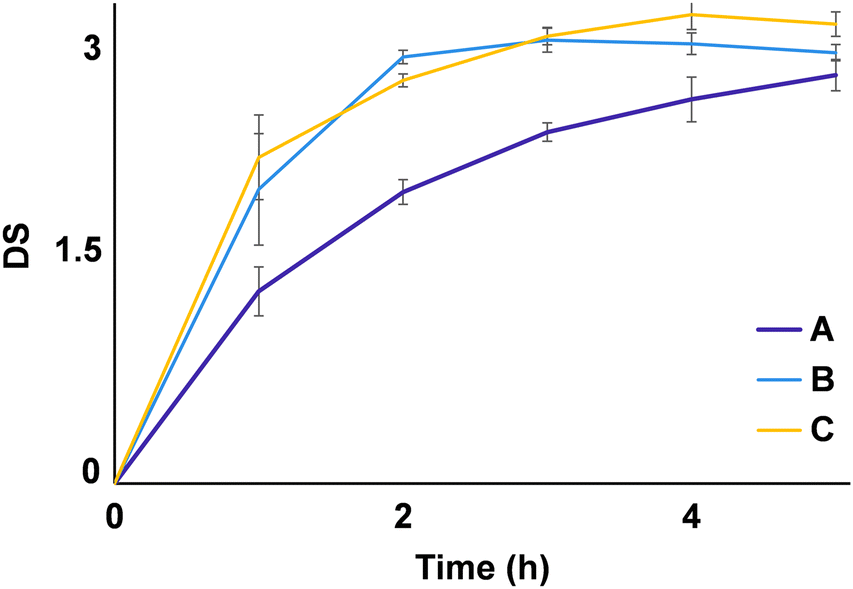 | ||
| Fig. 8 Degree of substitution (DS) during starch acetylation reaction at fixed conditions. (A) Temperature: 120 °C, ratio of acetic anhydride to starch (AA:starch): 15, starch load: 0.75 g. (B) Temperature: 120 °C, AA:starch: 25, starch load:0.75 g. (C) Temperature: 120°, AA:starch: 15, starch load: 1.25 g. | ||
The model obtained from the experimental data fit very accurately to a non-hierarchical quadratic model (R2 = 0.949, P > F = 0.0001, lack of fit P > F = 0.2953). In general, linear, and quadratic terms were found to be significant, while factor interactions were negligible. In both cases, there were two exceptions: factor time was non-significant, albeit marginally (P = 0.0605), whereas the interaction term AA:starch*starch load was well within range with P < 0.0001. The response surface plot of the model is shown in Fig. 9. The complete statistical report can be found in Tables S3–S5.† Importantly, the limited effect of time on the reaction opens the door for optimization conditions with very short processing times, which would in turn reduce the overall energy consumption and increase the total throughput of the reaction.
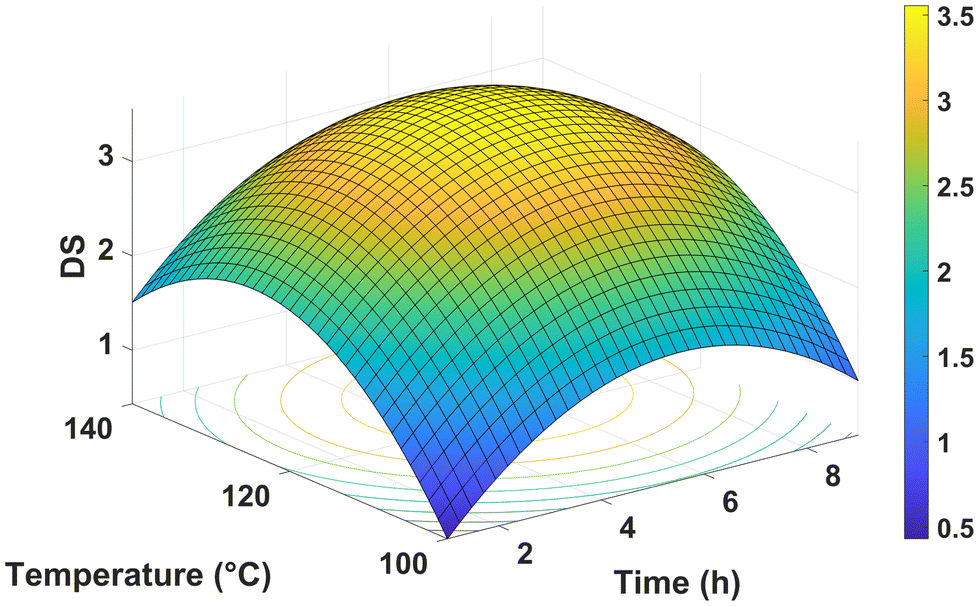 | ||
| Fig. 9 Surface plot of the non-hierarchical quadratic model of catalyst-free starch acetylation at a fixed ratio of acetic anhydride to starch (AA:starch) of 10 and starch load of 1 g. | ||
The statistical model was further validated by replicating the calculated conditions experimentally. An experimental setting of 128 °C, 0.6 h, AA:starch of 20, and 1.25 g of starch was selected. Such conditions would result in DS = 3 as indicated by the model. Indeed, the selected values returned DS = 2.87 ± 0.22, demonstrating that the model can be used confidently to estimate the DS of the catalytic system. Additionally, it proved that high acetylation values can be achieved in short reaction times. Interestingly, samples with higher DS also resulted in higher yr than samples with DS = 1.5. In other words, after esterification, each gram of starch was expected to yield a maximum theoretical yt = 1.77 g against a desired DS = 3. At DS = 1.5, each gram of starch in the reaction resulted in 1.12 g of pure AcS, representing only ya = 63.5 wt% ± 1.45. When ya is calculated against the actual DS of 1.5, a yr = 77.7 wt% ± 1.78 was obtained. At DS > 2.8, 1 g of starch yielded 1.66 g of pure AcS, representing ya = 93.7% ± 1.21 and yr = 95.4 wt% ± 1.23. This is likely due to the recovery method, where smaller, more soluble starch particles were not fully precipitated during centrifugation, and thus lost in the washing solution. While similarly sized particles could be present in more substituted starches, their less water-soluble nature prevented them from being washed away. As expected, lower DS values also translated into low absolute yields (ya > 70 wt%). It should be mentioned that, although the fit of the model to the data was high, its prediction capabilities are more limited at extreme conditions, where DS values can vary by up to 50%.
While expected from high AA:starch, it was surprising that higher starch loads also led to better acetylation. However, once a deeper statistical analysis on the molar proportions was performed, it was observed that it was indeed the ratio between solvent, AA, and starch that led to the improved reaction rates. A larger amount of both AA and starch effectively pushes the reaction equilibrium towards acetylation. This seems to point to the fact that the improved efficiency is due to the chaotropic environment provided by the solvent and the resulting higher solubility and miscibility of the reagents, rather than a possible catalytic effect from urea. Previous evidence notwithstanding, catalytic effects from the DES cannot be completely excluded, given their known catalytic activity attributed to their capacity to form hydrogen bonds with the reactants.61,62 The results imply that ChCl:urea can be used in very small amounts, yet high enough to keep good solubility. Such conditions may find application under certain mechanochemical techniques, such as melt-mixing or extrusion, where starch solubility is not required.
Among all the conditions tested, those developed at 120 °C, AA:starch molar ratio of 15, and starch load of 1.25 g, resulted in acetylated starch with yr >80%, low formation of side reaction products and a broad range of DS depending on the reaction time. Thus, acetylated starches produced at these conditions were collected at different reaction times up to 5 h for further characterization via HPSEC, DSC, TGA and SEM.
Effect of acetylation on molecular size distribution and average molecular weight
Size exclusion Chromatographic (SEC) analysis showed clear changes in molecular size distribution and weight-average molecular weight (Mw) upon acetylation. The SEC-RI elution profile of native wheat starch displayed two distinct peaks for amylose and amylopectin molecules separated at ∼43 min elution volume, both represented by a single peak in the multiangle laser-light scattering (MALS) detector that corresponded to a weight average molar mass for amylopectin molecules of 2.67 × 108 g mol−1 (Fig. 10). This Mw value was very similar to that reported before for wheat starch.63 Acetylation generally resulted in the merging of both amylopectin and amylose peaks into a single broad peak at higher elution times, confirming hydrolytic degradation and a consequent molar mass decrease as well as the coelution of amylose and amylopectin fragments. Similar alkaline catalytic64–67 and organocatalytic5 systems reported this same distribution shift. It is important to highlight that in this research, the concentration and volume of the sample tested, along with the fact that amylose molecules have a wide range of sizes, resulted in the absence of observable signals from laser-light scattering, as reported before.68 As occurred with organocatalytic esterification reactions,5,14 acetylation also resulted in a molar mass reduction of about 2–3 orders of magnitude, in this case from 2.67 × 108 g mol−1 to 3.96 × 105 g mol−1 for the most degraded samples (data not shown). As discussed before, this effect could be explained by the effect of high temperatures in acidic environments. It is difficult to conclude that our reaction system better preserves molas mass than organocatalytic esterification. However, it is noted that the reaction conditions shown in Fig. 9, representing yr >80%, low formation of side reaction products and DS from 1.8 to 3.1 resulted in starches with molar mass above 1 × 106 g mol−1. In any case, traditional alkaline systems still preserved the integrity of the starch molar mass to a higher degree (reduction of 108–107).65,69,70 As opposed to organocatalytic systems, and due to the absence of a dihydroxy dicarboxylic acid as catalyst, no significant regain in molar mass was observed at long reaction times, indicating no crosslinking.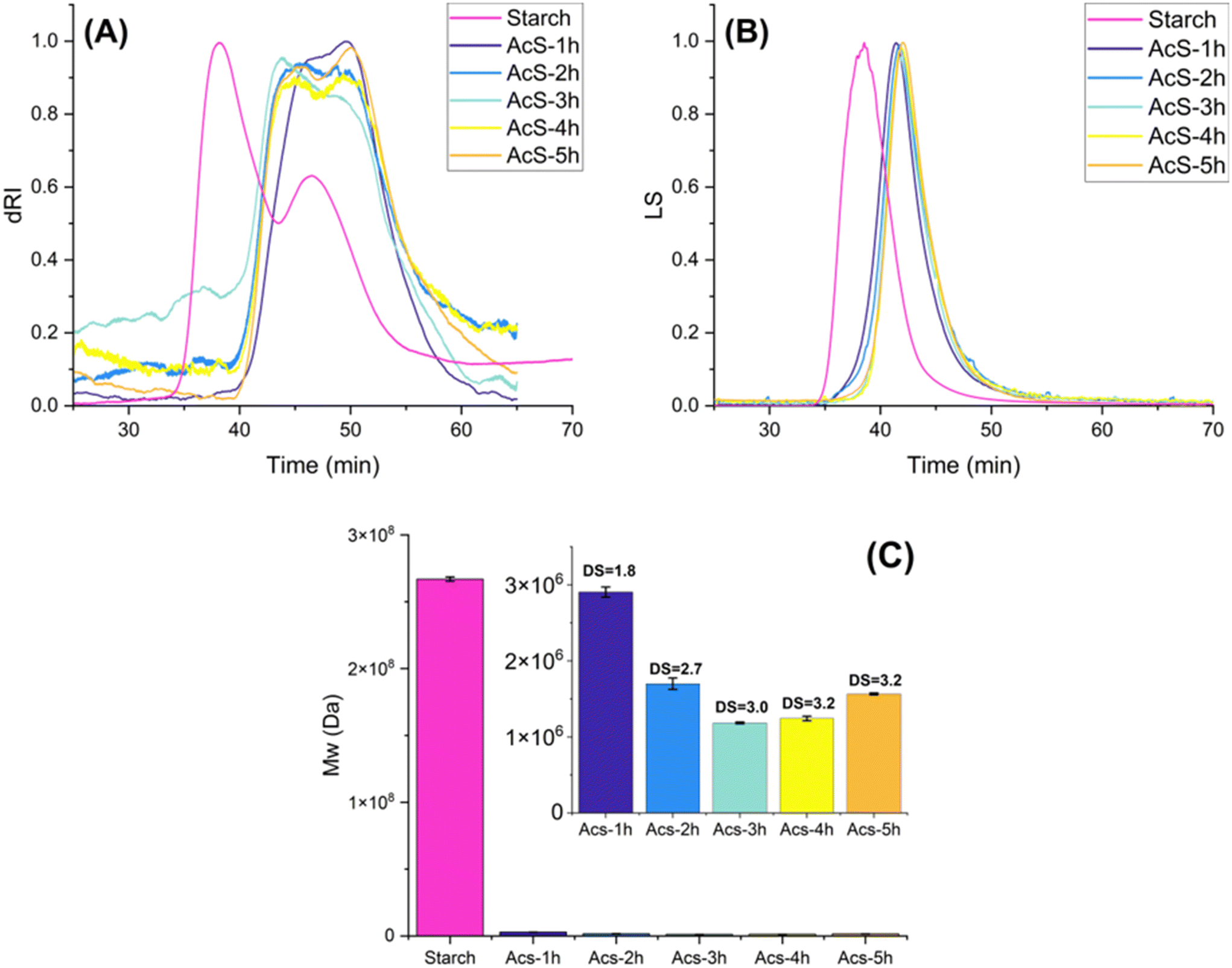 | ||
| Fig. 10 Multiangle laser light scattering (A) and refractive index (B) chromatograms, as well as bar chart displaying weight-average average molecular weight (C). Acetylated starches (AcS) were developed at reaction conditions of 120 °C, acetic anhydride:starch molar ratio of 15, and starch load of 1.25 g. Reaction was monitored every hour, for 5 hours. | ||
Acetylation resulted in Mw ranging between 9.34 × 107 to 3.96 × 105 g mol−1 for the most degraded samples (Fig. 10). Although some tendencies could be observed between DS, temperature, and Mw, no statistically significant model could be derived from the data. Nevertheless, a bivariate analysis of the individual factors revealed that both temperature and DS explain the variability of Mw, albeit to a limited degree (ρ = −0.40, R2 = 0.16, P = 0.036 for DS; ρ = −0.40, R2 = 0.22, P = 0.013 for temperature). This suggests that other factors are at play in the starch fragmentation process. The chain scission of starch macromolecules is likely related to the concentration of acetic acid present in the reaction at any given time, which is in turn generated from the acetylation reaction, and consumed to form acetylurea. On the other hand, –OH esterification leads to an increase in Mw due to their substitution with acetyl groups. Both acetylation and hydrolysis reaction rates are potentiated by temperature increases, leading to a multidimensional tug of war between Mw-increasing and -decreasing factors.
Morphology and thermal properties thermal properties
The degradation temperature and glass transition of starches were studied to identify their suitability as a component for packaging materials. Thermal degradation profile of the esterified materials was obtained via thermogravimetric analysis (TGA) and further thermogravimetric infrared spectroscopic analysis (TG-IR) (Fig. 11 and 12). The thermal degradation of starch showed two distinct degradation steps. The first one exhibited a maximum degradation rate at 125 °C with a weight loss of 7%, which can be attributed to water, both free and bound. The second, more pronounced step occurred at a maximum degradation rate of 316 °C, with a loss of 73 wt%, which is related to the depolymerization of starch chains and their following pyrolysis to 1,6-anhydroglucose (levoglucosan), and furaldehydes.71,72 Acetylated samples presented a much lower water loss (4–0.7 wt%), starting at 96 °C and finishing at 103 °C. The low water content present in AcS samples suggests a reduced hygroscopicity from the starting material. These results are similar to those observed by Vidal et al.5 The thermal stability of the acetylated samples was also improved, showing a second degradation step at 397 °C. For the samples containing acetylurea, an earlier degradation step can be observed at 258 °C, corroborating their small proportion related to starch as reported by CP/MAS 13C NMR (Fig. 6). A small proportion of non-modified starch could also be identified in sample AcS-1h by a small peak visible in its derivative mass loss curve. | ||
| Fig. 11 Thermal stability represented as weight loss (%) as a function of temperature (A), as well as the derivative mass loss (B), determined by TGA. Reaction conditions: 5 h, 120 °C, acetic anhydride:starch molar ratio: 15, and starch load of 1.25 g. Reaction was monitored every hour. | ||
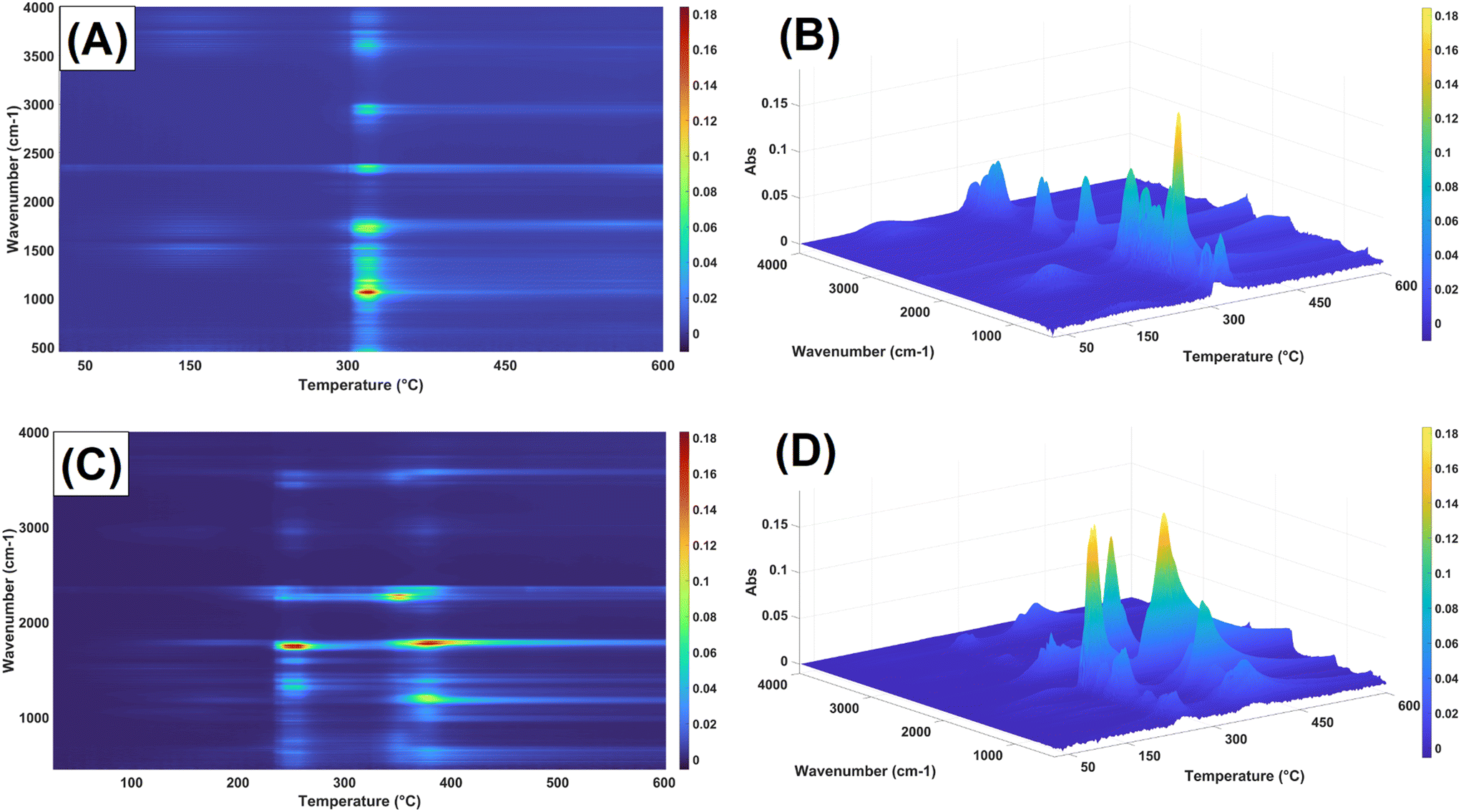 | ||
| Fig. 12 Real-time FT-IR sampling of the evolved gases from the thermogravimetric analysis. Top: 2D (A) and 3D (B) graph with color gradient intensity of native starch. Bottom: 2D (C) and 3D (D) graph with color gradient intensity of acetylated starch containing acetylurea as a side reaction. | ||
Real-time FTIR sampling of the evolved gases from the esterified samples confirmed the above assignation to each degradation peak (Fig. 12). While no water vapor could be detected during the evaporation period due to the small proportion, the gases obtained from the acetylated samples at 380 °C confirmed the degradation of the esterified starch into acetyl groups (3560 cm−1, 1750 cm−1, 1150–1200 cm−1), and CO2 (2300–2400 cm−1). The gas spectrum of acetylated starch containing encapsulated acetylurea as side reaction product showed their degradation peak at approx. 270 °C, represented by secondary amine peaks, CO2, NO2 (3490, 2355, and 950–800 cm−1), usually associated to acetamide, a fractionation product of acetylurea.73 In comparison, starch showed peaks stretching from 85 °C to 150 °C related to water vapor, followed by a single degradation step at 320 °C composed mainly of H2O, CO2, as well as other signals assigned to CH– (2969 cm−1) CO (1730 cm−1), C–O (1057 cm−1) bonds, likely related to volatile products, such as glycol- and acetaldehyde.74,75 Individual vibration peak assignment of the evolved gases can be found in the ESI (Fig. S3†).
Glass transition temperature (Tg) was analyzed by differential scanning calorimetry (DSC). Native wheat starch exhibited a Tg of 102 °C, with an onset temperature (To) and conclusion temperature (Tc) of 65 °C and 129 °C respectively. After acetylation, Tg was increased significantly to 162 °C, with an average To of 159.5 °C and Tc of 165 °C (Table 2). The increase in Tg and range reduction (To–Te) highlighted the improved thermal stability of the acetylated material, allowing it to keep its structure for longer with no gradual deformation. While it has been posited that acetylation should lead to a reduction of Tg due to the weakening of hydrogen bonding and the related loss of crystallinity, the results show otherwise. This unexpected behavior has been previously reported in literature.5,14,76,77 Given the complex interactions that determine the glass transition, such as crystallinity, moisture, and chemical interactions, it is difficult to establish a source for this increase in Tg. Liu et al. have shown that low moisture content and high amylose content lead to higher Tg.78 The hydrophobicity of the material and fragmentation into a more linear arrangement may explain some of this increase in Tg. The corresponding DSC figure has been added to ESI as Fig. S4.†
| T o | Mid | T c | |
|---|---|---|---|
| T o: onset temperature of glass transition; Mid: middle point of glass transition; Tc: conclusion temperature; AcS: Acetylated starch. | |||
| Starch | 65.0 | 102.0 | 129.0 |
| AcS-1h | 160.6 | 163.7 | 166.7 |
| AcS-2h | 159.6 | 162.1 | 164.4 |
| AcS-3h | 158.4 | 161.4 | 163.7 |
| AcS-4h | 157.2 | 160.6 | 163.9 |
| AcS-5h | 158.7 | 161.2 | 163.7 |
| Average | 159.5 | 162.4 | 165 |
| Std. Dev | 1.1 | 1.2 | 1.6 |
Scanning electron microscopy (SEM) images were obtained at different magnifications to investigate several morphological details. Starch images showed the typical lenticular and spherical shapes of A- and B-type granules, respectively.79 These granules range from 5–30 μm (Fig. 13A). After acetylation, while no intact granules could be identified, evidence of their presence remained in the voids of the particles, with maximum dimension ranging between 30–5 μm (Fig. 13B). Similar reconfiguration of starch macromolecules has been reported by others in literature, even at low DS.7,53,54,64 This phenomena could be explained by the inhomogeneous propensity to gelatinize, solubilize and undergo acetylation among the different granules as observed by Huang et al.80 Starch granules begin reacting on their surface, with the substitution of hydroxyl by acetyl groups disrupting the hydrogen bond interactions between starch chains. Further degradation by breaking glycosidic chains causes the granules to be completely consumed leaving only the void. As time progresses, the material continues to rearrange itself and begins to form molten material, which seems to fuse into longer strands (Fig. 13C). This could be due to the degradation of the amorphous lamella of amylopectin, which is known to be rich in 1,6-glycosidic bonds,81 leaving primarily long 1,4-glycosidic bonds. Indeed, this is in accordance with the data obtained from the dRI chromatograms showing an increase of the amylose fraction. Magnified images of acetylated samples can be found as Fig. S5.†
 | ||
| Fig. 13 Scanning electron microscope (SEM) images of (A) native starch (scale bar = 10 μm); and acetylated starches (AcS) (B) AcS-1h (100 μm); (C) AcS −5h (scale bar = 100 μm). AcS-1h and AcS-5h: 120 °C, acetic anhydride:starch ratio of 15 and starch load of 1.25 g, and 1h and 5h, respectively. | ||
On the subject of recyclability
Recyclability of the reagents involved in the process is an important issue to address when developing new production methods. The reaction system proposed in this paper faces clear challenges when it comes to this point. Mainly, owing to the use of acetic anhydride, which has an inherently low atom economy, due to the formation of acetic acid. This is further complicated by the formation of side products, such as acetylurea, which partially consumes the eutectic solvent. In this unbalanced state, left alone to react without the presence of starch, ChCl began to form acetylcholine. Thus, evaporative techniques to separate acetic acid were ineffective. Nevertheless, further avenues of recovery and recycling can be explored, such as solvent extraction, crystallization, or chromatographic methods.It is noteworthy to mention that both compounds have been successfully used to produce eutectic solvents themselves, and even applied in the valorization of biomass and the capture of emission gases.82–84 Indeed, the recovered samples of acetylcholine:urea 3:1 remained liquid at room temperature. Their potential as green solvent in esterification reactions warrants further experimentation.
Conclusions
The research presented here demonstrates an acetylation process, using environmentally friendly DES ChCl:urea. The results showed that ChCl:urea is an ideal chaotropic solvent for this reaction, since it was not only capable of completely solubilizing starch, but it also provided a favorable environment for the synthesis of esterified starches. While the mechanism by which the improved reaction rates take place is still not clear, chaotropic dissolution, alkalinization and presumably weak interactions affecting the conformation of the reactants could jointly contribute to the success of the reaction, though some catalytic effect from urea cannot be completely ruled out. Combined FTIR and 1H NMR analysis allowed for clear and rapid identification of acetylated starch and side products. The results from the response surface experimental design allowed to reliably calculate the desired degrees of acetylation under several reaction conditions, ranging between DS = 0.2–3.16. Under certain reaction conditions, a potentially interesting side product, acetylurea, could be produced simultaneously from the residual acetic acid, as confirmed by CP/MAS 13C NMR, 2D HSQC and 2D HMBC.
The obtained acetylated starches were thoroughly characterized. Thermal stability was considerably improved for highly substituted samples by up to 81 °C above that of starch. Glass transition was similarly increased, with a higher Tg and a narrower transition range. Moreover, hygroscopicity was reduced, as evidenced by the small amounts of water present in the samples. While Mw was reduced by two orders of magnitude in the most substituted samples, molar mass remained above 106 Da in most cases, with less esterified samples (DS < 1) preserving Mw in the range of 1 × 107 Da.
The process implemented here is not only very energy efficient due to the reduced reaction times, but also a sustainable alternative to the traditional standard catalytic acetylation reactions. ChCl and urea, both non-hazardous, environmentally friendly solids, are safer options to conventional solvents, and their solvation capabilities on macromolecules gives them the additional advantage of creating a highly reactive homogeneous system. While the use of acetic anhydride is less than ideal due to low atom economy and health and recyclability issues, the process may be adapted to less hazardous, cyclic anhydrides, such as maleic or succinic acids, where the reaction does not inherently have low atom economy. Our developed starches here show many promising characteristics ideal for their use in food and packaging applications. Furthermore, the statistical tools developed here open the possibilities for targeted tuning and tailoring (e.g., for low DS starches used as edible food ingredients).
Author contributions
Guillermo A. Portillo Perez: investigation, methodology, formal analysis, visualization, writing – original draft. Kasper Skov: investigation, methodology. Mario M. Martinez: conceptualization, supervision, project administration, funding acquisition, formal analysis, visualization, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support of Independent Research Fund Denmark – Danmarks Frie Forskningsfond (project number 1032-00491B). Some of the data was generated through accessing research infrastructure funded by FOODHAY (Food and Health Open Innovation Laboratory, Danish Roadmap for Research Infrastructure).References
- Directive (EU) 2019/904 of the European Parliament and of the Council of 5 June 2019 on the reduction of the impact of certain plastic products on the environment European Commission, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32019L0904&from=EN, 2019 Search PubMed.
- A. Shafqat, A. Tahir, A. Mahmood, A. B. Tabinda, A. Yasar and A. Pugazhendhi, Biocatal. Agric. Biotechnol., 2020, 27, 101540 CrossRef.
- J. N. BeMiller, Carbohydrate chemistry for food scientists, Elsevier, 2018 Search PubMed.
- M. M. Martinez, Curr. Opin. Food Sci., 2021, 38, 112–121 CrossRef.
- N. P. Vidal, W. Bai, M. Geng and M. M. Martinez, Carbohydr. Polym., 2022, 294, 119780 CrossRef CAS PubMed.
- X. Wang, L. Huang, C. Zhang, Y. Deng, P. Xie, L. Liu and J. Cheng, Carbohydr. Polym., 2020, 240, 116292 CrossRef CAS PubMed.
- P. Cuenca, S. Ferrero and O. Albani, Food Hydrocolloids, 2020, 100, 105430 CrossRef CAS.
- B. F. Bergel, S. Dias Osorio, L. M. da Luz and R. M. C. Santana, Carbohydr. Polym., 2018, 200, 106–114 CrossRef CAS PubMed.
- J. Ye, S. Luo, A. Huang, J. Chen, C. Liu and D. J. McClements, Food Hydrocolloids, 2019, 92, 135–142 CrossRef CAS.
- L. Ren, Q. Wang, X. Yan, J. Tong, J. Zhou and X. Su, Food Res. Int., 2016, 87, 180–188 CrossRef CAS PubMed.
- G. Annison, R. J. Illman and D. L. Topping, J. Nutr., 2003, 133, 3523–3528 CrossRef CAS PubMed.
- K. V. Ragavan, O. Hernandez-Hernandez, M. M. Martinez and T. J. Gutiérrez, Trends Food Sci. Technol., 2022, 119, 45–56 CrossRef CAS.
- Đ. Ačkar, J. Babić, A. Jozinović, B. Miličević, S. Jokić, R. Miličević, M. Rajič and D. Šubarić, Molecules, 2015, 20, 19554–19570 CrossRef PubMed.
- B. Imre and F. Vilaplana, Green Chem., 2020, 22, 5017–5031 RSC.
- W. Xie, L. Shao and Y. Liu, J. Appl. Polym. Sci., 2010, 116, 218–224 CrossRef CAS.
- R. L. Shogren and A. Biswas, Carbohydr. Polym., 2010, 81, 149–151 CrossRef CAS.
- Z. Luo and Z. Zhou, Starch – Stärke, 2012, 64, 37–44 CrossRef CAS.
- F. Ren, J. Wang, J. Yu, C. Zhong, F. Xie and S. Wang, Carbohydr. Polym., 2022, 288, 119353 CrossRef CAS PubMed.
- S. Magina, A. Barros-Timmons, S. P. M. Ventura and D. V. Evtuguin, J. Hazard. Mater., 2021, 412, 125215 CrossRef CAS PubMed.
- G. Cevasco and C. Chiappe, Green Chem., 2014, 16, 2375–2385 RSC.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- L. Y. Ee, Y. K. Tan, J. Miao, H. T. Chu and S. F. Y. Li, Green Chem., 2023, 25, 3137–3151 RSC.
- I. Bodachivskyi, U. Kuzhiumparambil and D. B. G. Williams, ACS Sustainable Chem. Eng., 2020, 8, 678–685 CrossRef CAS.
- M. Bisht, I. P. E. Macário, M. C. Neves, J. L. Pereira, S. Pandey, R. D. Rogers, J. A. P. Coutinho and S. P. M. Ventura, ACS Sustainable Chem. Eng., 2021, 9, 16073–16081 CrossRef CAS.
- M. Pan, G. Zhao, C. Ding, B. Wu, Z. Lian and H. Lian, Carbohydr. Polym., 2017, 176, 307–314 CrossRef CAS PubMed.
- G. Colombo Dugoni, A. Mezzetta, L. Guazzelli, C. Chiappe, M. Ferro and A. Mele, Green Chem., 2020, 22, 8680–8691 RSC.
- Q. Chen, L. Chaihu, X. Yao, X. Cao, W. Bi, J. Lin and D. D. Y. Chen, ACS Sustainable Chem. Eng., 2021, 9, 10083–10092 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC.
- A. Biswas, R. L. Shogren, D. G. Stevenson, J. L. Willett and P. K. Bhowmik, Carbohydr. Polym., 2006, 66, 546–550 CrossRef CAS.
- C. L. Yiin, A. T. Quitain, S. Yusup, M. Sasaki, Y. Uemura and T. Kida, Bioresour. Technol., 2016, 199, 258–264 CrossRef CAS PubMed.
- J. A. Sirviö, E. Isokoski, A. M. Kantola, S. Komulainen and A. Ämmälä, Cellulose, 2021, 28, 6881–6898 CrossRef.
- X. Cao, M. Liu, W. Bi, J. Lin and D. D. Y. Chen, Carbohydr. Polym. Technol. Appl., 2022, 4, 100222 CAS.
- S. Liu, Q. Zhang, S. Gou, L. Zhang and Z. Wang, Carbohydr. Polym., 2021, 251, 117018 CrossRef CAS PubMed.
- W. Yu, C. Wang, Y. Yi, H. Wang, L. Zeng, M. Li, Y. Yang and Z. Tan, ACS Omega, 2020, 5, 5580–5588 CrossRef CAS PubMed.
- X. Li, C. Ning, L. Li, W. Liu, Q. Ren and Q. Hou, Carbohydr. Polym., 2021, 274, 118650 CrossRef CAS PubMed.
- M. Zdanowicz, K. Sałasińska, K. Lewandowski and K. Skórczewska, ACS Sustainable Chem. Eng., 2022, 10, 4579–4587 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: Theory and practice, 1998, pp. 29 Search PubMed.
- L. Roman, O. Campanella and M. M. Martinez, Carbohydr. Polym., 2019, 215, 198–206 CrossRef CAS PubMed.
- L. Roman, M. Gomez, B. R. Hamaker and M. M. Martinez, Food Chem., 2019, 274, 664–671 CrossRef CAS PubMed.
- N. Rodriguez Rodriguez, A. van den Bruinhorst, L. J. B. M. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- C. Cao, B. Nian, Y. Li, S. Wu and Y. Liu, J. Agric. Food Chem., 2019, 67, 12366–12373 CrossRef CAS PubMed.
- F. S. Mjalli and O. U. Ahmed, Korean J. Chem. Eng., 2016, 33, 337–343 CrossRef CAS.
- A. Valvi, J. Dutta and S. Tiwari, J. Phys. Chem. B, 2017, 121, 11356–11366 CrossRef CAS PubMed.
- A. R. R. Teles, E. V. Capela, R. S. Carmo, J. A. P. Coutinho, A. J. D. Silvestre and M. G. Freire, Fluid Phase Equilib., 2017, 448, 15–21 CrossRef CAS PubMed.
- John Wiley & Sons Inc., SpectraBase Compound ID=JxHCsgR4PJ5 SpectraBase Spectrum ID=5DEaDduL8An, https://spectrabase.com/spectrum/5DEaDduL8An, (accessed 02/05/2023).
- M. J. Gidley and S. M. Bociek, J. Am. Chem. Soc., 1985, 107, 7040–7044 CrossRef CAS.
- R. Veregin, C. Fyfe, R. Marchessault and M. Taylor, Macromolecules, 1986, 19, 1030–1034 CrossRef CAS.
- M. Paris, H. Bizot, J. Emery, J. Y. Buzaré and A. Buléon, Carbohydr. Polym., 1999, 39, 327–339 CrossRef CAS.
- M. Paris, H. Bizot, J. Emery, J. Y. Buzaré and A. Buléon, Int. J. Biol. Macromol., 2001, 29, 127–136 CrossRef CAS PubMed.
- C. Chevigny, L. Foucat, A. Rolland-Sabaté, A. Buléon and D. Lourdin, Carbohydr. Polym., 2016, 146, 411–419 CrossRef CAS PubMed.
- M.-Y. Baik, L. C. Dickinson and P. Chinachoti, J. Agric. Food Chem., 2003, 51, 1242–1248 CrossRef CAS PubMed.
- L. Zhang, W. Xie, X. Zhao, Y. Liu and W. Gao, Thermochim. Acta, 2009, 495, 57–62 CrossRef CAS.
- R. Wang, J. Wang, M. Liu, P. Strappe, M. Li, A. Wang, M. Zhuang, J. Liu, C. Blanchard and Z. Zhou, Food Hydrocolloids, 2022, 128, 107556 CrossRef CAS.
- L. A. Sinclair, J. A. Huntington and D. Wilde, Proceedings of the British Society of Animal Science, 2008, 2008, 224–224 Search PubMed.
- H. Bergner, Can. J. Anim. Sci., 1984, 64, 37–38 CrossRef CAS.
- H. Münchow, H. Bergner and J. N. Gupta, Arch. Tierernaehr., 1973, 23, 329–339 CrossRef PubMed.
- E. A. Swinyard, in Antiepileptic Drugs, ed. H.-H. Frey and D. Janz, Springer Berlin Heidelberg, Berlin, Heidelberg, 1985, pp. 601–610. DOI:10.1007/978-3-642-69518-6_22.
- P. Debnath, M. Baeten, N. Lefèvre, S. V. Daele and B. U. W. Maes, Adv. Synth. Catal., 2015, 357, 197–209 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- A. Shaabani and S. E. Hooshmand, Tetrahedron Lett., 2016, 57, 310–313 CrossRef CAS.
- R. Amoroso, F. Hollmann and C. Maccallini, Molecules, 2021, 26(20), 6286 CrossRef CAS PubMed.
- S.-H. Yoo and J.-L. Jane, Carbohydr. Polym., 2002, 49, 307–314 CrossRef CAS.
- P. D. Mbougueng, D. Tenin, J. Scher and C. Tchiégang, J. Food Eng., 2012, 108, 320–326 CrossRef CAS.
- S. Simsek, M. Ovando-Martínez, K. Whitney and L. A. Bello-Pérez, Food Chem., 2012, 134, 1796–1803 CrossRef CAS PubMed.
- S. Garg and A. K. Jana, Carbohydr. Polym., 2011, 83, 1623–1630 CrossRef CAS.
- A. B. Das, G. Singh, S. Singh and C. S. Riar, Carbohydr. Polym., 2010, 80, 725–732 CrossRef CAS.
- L. Roman, O. Campanella and M. M. Martinez, Carbohydr. Polym., 2019, 215, 198–206 CrossRef CAS PubMed.
- L. A. Bello-Pérez, E. Agama-Acevedo, P. B. Zamudio-Flores, G. Mendez-Montealvo and S. L. Rodriguez-Ambriz, LWT – Food Sci. Technol., 2010, 43, 1434–1440 CrossRef.
- W. Berski, A. Ptaszek, P. Ptaszek, R. Ziobro, G. Kowalski, M. Grzesik and B. Achremowicz, Carbohydr. Polym., 2011, 83, 665–671 CrossRef CAS.
- F. Shafizadeh, R. A. Susott and G. D. McGinnis, Carbohydr. Res., 1972, 22, 63–73 CrossRef CAS.
- P. Aggarwal and D. Dollimore, Thermochim. Acta, 1998, 319, 17–25 CrossRef CAS.
- H. F. G. Barbosa, D. S. Francisco, A. P. G. Ferreira and É. T. G. Cavalheiro, Carbohydr. Polym., 2019, 225, 115232 CrossRef PubMed.
- M. Pigłowska, B. Kurc, Ł. Rymaniak, P. Lijewski and P. Fuć, Polymers, 2020, 12, 357 CrossRef PubMed.
- T. J. Johnson, R. L. Sams, L. T. M. Profeta, S. K. Akagi, I. R. Burling, R. J. Yokelson and S. D. Williams, J. Phys. Chem. A, 2013, 117, 4096–4107 CrossRef CAS PubMed.
- C. I. K. Diop, H. L. Li, B. J. Xie and J. Shi, Food Chem., 2011, 126, 1662–1669 CrossRef CAS PubMed.
- C. Fringant, J. Desbrieres and M. Rinaudo, Polymer, 1996, 37, 2663–2673 CrossRef CAS.
- P. Liu, L. Yu, X. Wang, D. Li, L. Chen and X. Li, J. Cereal Sci., 2010, 51, 388–391 CrossRef CAS.
- L. Roman, E. d. l. Cal, M. Gomez and M. M. Martinez, Food Hydrocolloids, 2018, 82, 510–518 CrossRef CAS.
- J. Huang, H. A. Schols, Z. Jin, E. Sulmann and A. G. J. Voragen, Carbohydr. Polym., 2007, 68, 397–406 CrossRef CAS.
- Y. Nakamura and K. Kainuma, Plant Mol. Biol., 2022, 108, 291–306 CrossRef CAS PubMed.
- R. Lu, S. Ma, Y. Zhai, Z. Geng, Z. Jin, Y. Fu, W. Wang and Y. Xu, J. Mol. Liq., 2023, 383, 121937 CrossRef CAS.
- Y. Egi and J.-i. Kadokawa, Tetrahedron Green Chem., 2023, 1, 100012 CrossRef.
- X. Yang, Z. Liu, P. Chen, F. Liu and T. Zhao, J. CO2 Util., 2022, 58, 101936 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02833j |
| This journal is © The Royal Society of Chemistry 2024 |