
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent triazine-based frameworks – switching selectivity in HMF photooxidation†
Daniel
Ditz
a,
Nina M.
Sackers
a,
Felix
Müller
b,
Mirijam
Zobel
 b,
Sebastian
Bergwinkl
c,
Patrick
Nuernberger
c,
Leonie Sophie
Häser
a,
Sarah
Brettschneider
a,
Florian M.
Wisser
d,
Christoph
Bannwarth
e and
Regina
Palkovits
*af
b,
Sebastian
Bergwinkl
c,
Patrick
Nuernberger
c,
Leonie Sophie
Häser
a,
Sarah
Brettschneider
a,
Florian M.
Wisser
d,
Christoph
Bannwarth
e and
Regina
Palkovits
*af
aHeterogeneous Catalysis and Technical Chemistry, Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bInstitute of Crystallography, RWTH Aachen University, Jägerstraße 17-19, 52066 Aachen, Germany
cInstitute of Physical and Theoretical Chemistry, University of Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany
dErlangen Center for Interface Research and Catalysis (ECRC) Friedrich-Alexander-Universität Erlangen-Nürnberg Egerlandstraße 3, 91058 Erlangen, Germany
eTheoretical Physical Chemistry of Large Molecules, Institute of Physical Chemistry, RWTH Aachen University, Melatener Str. 20, 52074 Aachen, Germany
fMax-Planck-Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 12th February 2024
Abstract
Covalent triazine-based frameworks (CTFs) – a class of porous organic polymers – excel as photocatalysts due to their chemical and thermal robustness, their highly conjugated and nitrogen rich nature, facile syntheses, and most important unprecedented structural variability. This allows the design of tailor-made photocatalysts. Here, we present how a controlled modification of the CTF allows us to switch the selectivity of 5-hydroxymethylfurfural (HMF) oxidation into two different valuable biomass-based molecules which can substitute oil-based products in existing value chains. Two reaction mechanisms can be chosen by the choice of the CTF's building block depending on the activation mechanism of molecular oxygen: either oxygen is reduced and HMF is directly oxidized to 2,5-diformylfuran (DFF) or molecular oxygen is photoactivated to singlet oxygen which reacts in a cycloaddition with HMF to yield 5-hydroxy-5-hydroxymethyl-furan-2-one (H2MF). Extensive characterization of the optoelectronic properties combined with quantum chemical calculations and detailed investigation of the reaction mechanism allowed us to elucidate the origin of this switch in selectivity.
Introduction
The latest political decisions like the European Green Deal and increasing socioeconomic pressure push the transformation towards a sustainable economic system.1 For the chemical industry in Europe, this implies carbon neutrality by 2050, establishment of more circular value chains, and increasing independence of fossil resources.2 Combined with the growing electrification of the energy sector, there is a high demand for alternative energy and carbon feedstock usage. Photocatalytic valorization of biomass over metal-free organic photocatalysts is one promising alternative to reach these goals.3–11Amongst organic photocatalysts, CTFs, a subclass of covalent organic frameworks (COFs) and porous polymers, stand out for their high chemical and thermal stability,12 their highly conjugated and nitrogen rich nature combined with facile syntheses, and most important easily variable chemical structure.13–18 In recent years, the huge potential of CTFs in various applications, crucial for achieving the afore mentioned transformation of our economic system, has been demonstrated. This includes their use in gas storage,19–23 separation techniques,24–26 electrode and battery materials,27–31 thermocatalysis12,32–35 and photocatalysis.16,17,36,37 While most photocatalytic studies focus on boosting water splitting,38–40 hydrogen production41–44 or carbon dioxide reduction,45–47 only a few reports consider the potential of tailor-made CTF photocatalysts for metal-free organosynthesis.48–55 Especially, CTF's ability to activate molecular oxygen makes it promising for benign oxidations.35 Zhang et al. demonstrated the successful oxidation of several benzyl alcohols to the corresponding aldehydes or ketones with a thiophene based CTF.53 Some of the authors extended the range of CTF photocatalysts for this reaction in a study on structure–activity relationships, showing that sulfur containing heterocycles improve the light absorption and charge carrier mobility properties as well as the interaction of substrate and catalyst.56 The selective oxidation of aromatic sulfides was shown by Wang et al.57 Ayed et al. were the first to report the selective oxidation of HMF to DFF over a thiophene based CTF.54 Since 2005 HMF continues to stay in the center of research on biomass valorization and by now first commercial production sites are realized.58,59 It provides a platform for many target molecules that can substitute oil-based products in existing value chains, two of which are DFF and H2MF. DFF can be produced by partial oxidation of HMF avoiding 5-hydroxymethyl-2-furancarboxylic acid (HMFA) formation or over oxidation to 5-formyl-2-furancarboxylic acid (FFCA) and 2,5-furan-dicarboxylic acid (FDCA).32 H2MF is the product of a [4 + 2] cycloaddition of singlet oxygen (1O2) and HMF under elimination of formic acid. H2MF quickly undergoes ring opening under elevated temperatures to form 5-hydroxy-4-keto-pentenoic acid (HKPA) which is prone to polymerization (Scheme 1).60
 | ||
| Scheme 1 Oxidation of HMF to various valuable products such as DFF or H2MF. While the top pathway is expected to proceed via direct oxidation of HMF, the bottom pathway proceeds via singlet oxygen. | ||
In this work, we elaborate on a variation of the CTF structure that triggers a switch in selectivity of the photoactivation of molecular oxygen and hence in the valorization of the substrate HMF. Either molecular oxygen is reduced and HMF is directly oxidized to DFF or O2 is activated to singlet oxygen which then undergoes a cycloaddition with HMF. Achieving different selectivity in the photoactivation of O2 over structurally modified COFs has been reported before.61,62 However, it was neither analyzed in great detail for CTFs so far, nor was this phenomenon applied to a highly relevant, benign platform chemical. Combining extensive characterization of the optoelectronic properties with detailed knowledge of the reaction mechanism allowed us to understand the switch in selectivity, adding another tool to the toolbox of CTF photocatalysts.
Results and discussion
Catalyst preparation and characterization
Two CTFs containing either a 1,4-phenyl (CTF-1) or a 2,6-naphthyl (CTF-2) linker have been synthesized via the polycondensation of the respective amidine hydrochloride and alcohol (Fig. 1a). It is worth noting that naphthalene-2,6-dicarboxamidine dihydrochloride was directly synthesized from the corresponding methyl ester avoiding a nitrile intermediate (see ESI† for further details).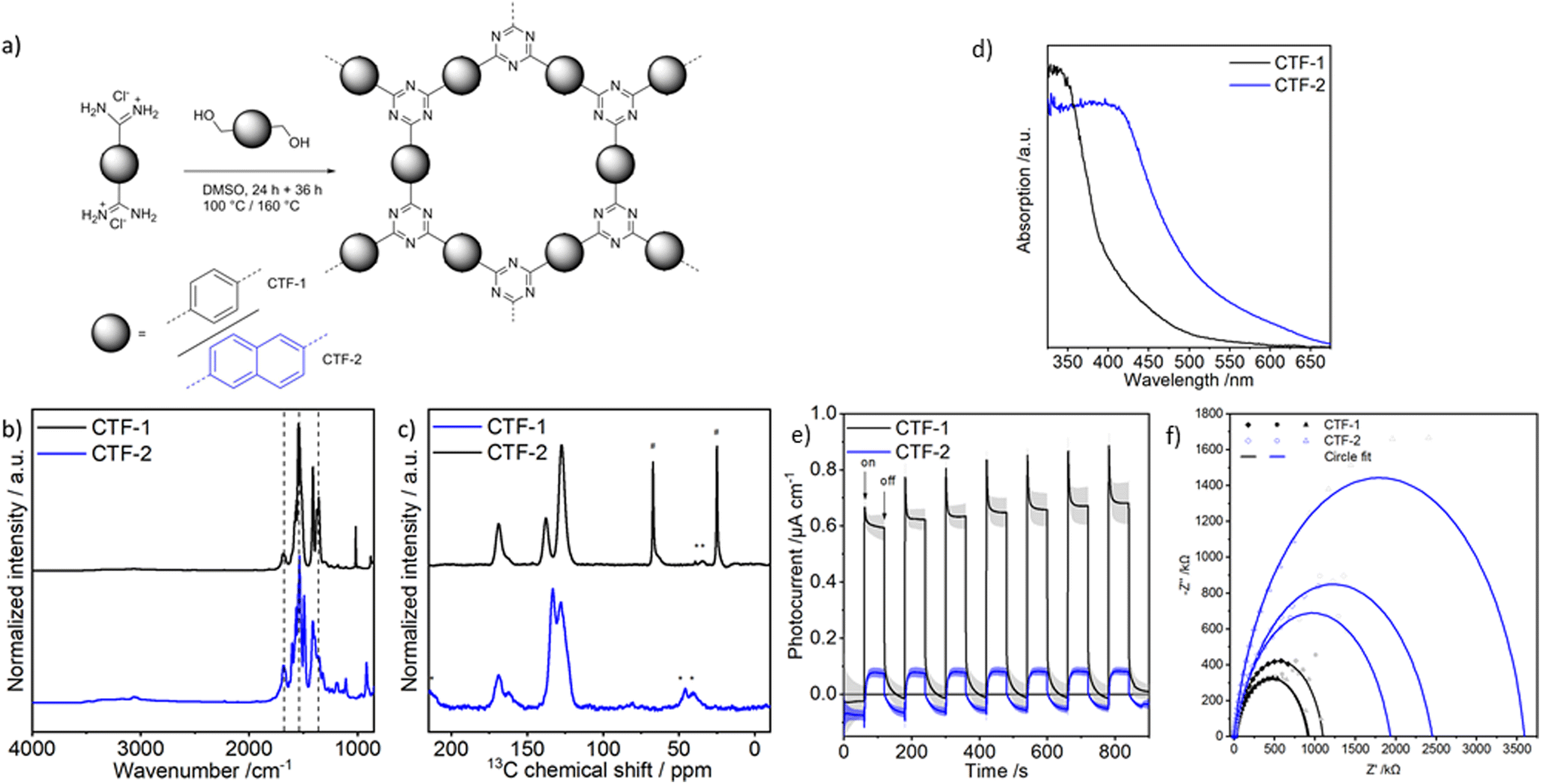 | ||
Fig. 1 (a) CTF synthesis via polycondensation of diamidine hydrochloride and dialcohol. (b) DRIFTS and (c) 13C HPDEC MAS-NMR spectra of CTF-1 (MAS frequency 13 kHz, 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 432 scans) and CTF-2 (MAS frequency 11 kHz 1024 scans). * Spinning sidebands #signals from ethanol. (d) Diffuse reflectance UV-Vis spectra of CTFs. (e) Periodic photocurrent measurements at open circuit potential in aqueous HMF solution. (f) Nyquist plot and simplified Randles circuit fit of EIS data under illumination. 432 scans) and CTF-2 (MAS frequency 11 kHz 1024 scans). * Spinning sidebands #signals from ethanol. (d) Diffuse reflectance UV-Vis spectra of CTFs. (e) Periodic photocurrent measurements at open circuit potential in aqueous HMF solution. (f) Nyquist plot and simplified Randles circuit fit of EIS data under illumination. | ||
The successful formation of CTF-1 and CTF-2 was confirmed by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and solid-state carbon-13 magic angle spinning nuclear magnetic resonance spectroscopy (13C MAS NMR) (Fig. 1b and c). The 13C MAS NMR spectra show the characteristic triazine signal at approx. 170 ppm, which corresponds to the triazine vibration bands in the IR spectra at around 1500 and 1350 cm−1. In both materials a broad shoulder at approx. 163 ppm, integrating to 20–30% of the total triazine signal intensity, can be detected next to the triazine resonance at 170 ppm. An assignment of the 13C NMR signal at 163 ppm to unreacted amidine groups is unlikely as no signal of the characteristic amidine N–H stretching vibration bands between 2500 and 3500 cm−1 can be detected in the IR spectra of both materials (Fig. S18†). Also, CHN analysis does not provide any hint for unreacted amidine groups as the experimental carbon to nitrogen ratios fit well with the expected ratios (see section S2.2† for more details). Thus, the observed 13C shoulders at 163 ppm are most likely due to difference in the stacking of the 2-D layers in the material. Zhao and co-workers recently reported that the chemical shift of the triazine carbon atoms is sensitive to changes in the material's stacking.63 Therefore, we assign the signal at lower chemical shifts to triazine moieties in a disordered or non AA stacking. First, the shoulder has a significant broader linewidth than the main triazine signal (Table S6†), indicative for a less ordered system. Second, the high-field shift caused by a higher deshielding of the C-atoms (higher electron density) which can be explained by a positioning of the observed triazine ring on top of a benzene (CTF-1) or a naphthalene moiety (CTF-2) rather than on top of a second triazine in the energetically most favored, slightly shifted AA stacking (for further discussion see ESI section 1.3†). The further 13C NMR signals observed can be attributed to the aromatic linkers: phenyl (13C NMR 138, 128 ppm) and naphthyl (13C NMR 134, 129, 124 ppm). Note that the 13C signals of CTF-1 are characterized by smaller linewidths than the 13C signals of CTF-2 (Table S6†), pointing to a more ordered structure in CTF-1 (see below). Nitrogen physisorption experiments reveal a type-I isotherm for CTF-1, indicative for a microporous material with an apparent surface area of 660 m2 g−1 (Fig. S19†). In contrast CTF-2 is characterized by a much lower apparent surface area of 80 m2 g−1. Powder X-ray diffraction indicates decently ordered frameworks which can be interpreted as layered structures with hexagonal pores (Fig. S21†). Pair distribution function analysis identifies interatomic distances of local structural motifs (Fig. S22†). Scanning electron microscopy shows large aggregates for both materials which seem to be composed of smaller particles for CTF-1 (Fig. S23†). The characterization of the optoelectronic properties reveals a distinct difference between CTF-1 and CTF-2 which stresses again the facile tailoring of CTF photocatalysts. Both materials absorb visible light (Fig. 1d). According to the Tauc plot (Fig. S24†), CTF-1 has a direct band gap transition of 3.37 eV (∼370 nm), although its absorption onset lies at 600 nm. This pronounced Urbach tail indicates certain localized or mid-band gap states caused by the polydispersity and possible defects of the network.64 Defects might be versatile comprising stacking defects or any other deviations from the idealized structure (Fig. S3†). The same applies to CTF-2 with a direct band gap transition of 2.79 eV (470 nm) and an absorption edge at 675 nm. As expected,65 both materials hold smaller indirect transitions resulting in band gaps of 3.20 eV and 2.63 eV, for CTF-1 and CTF-2, respectively (Fig. S24†). Band gaps are reduced by approx. 0.6 eV in CTF-2 compared to CTF-1 due to its larger delocalized system.
The reduction potential is in first approximation equal to the conduction band minimum (CBM) and the flat band potential determined by Mott–Schottky measurements at the point of zero charge (Fig. S11†).66,67 This was further confirmed by, Zou et al. who showed that there is a difference of 0.2 to 0.6 V between the CBM position and the flat band potential determined from Mott–Schottky analysis of CTFs at pH7.68 Here, the naphthalene linker causes a decrease in the reduction potential to −0.81 V vs. NHE compared to −0.97 V vs. NHE for CTF-1. Assuming that the oxidation potential equals the valence band maximum (VBM) which is separated by the band gap from the CBM, the resulting oxidation potentials are 2.23 V and 1.82 V vs. NHE for CTF-1 and CTF-2, respectively (Fig. S24†). Consequently, both materials have the potential to reduce molecular oxygen and oxidize HMF. Further, CTF-1 should be able to oxidize water depending on the overpotential.69 Here we note, that the true surface potential under reaction conditions might deviate from the value determined from Mott–Schottky analysis, hampering predictions for overpotentials and possible reactions.67
Photocurrent measurements at open circuit potential under chopped illumination were performed to get a qualitative comparison of the materials’ photon efficiency. Both CTFs show a positive photocurrent, which is typical for n-type semiconductors. Here, CTF-1 shows a distinctively larger photocurrent than CTF-2 (Fig. 1e). The photocurrent depends on multiple factors such as: absorption quantity, charge transfer resistance, charge carrier diffusion and recombination rates.5 Both materials were illuminated with UV-light (360 nm) to minimize the influence of the different band gaps. Quantum mechanical methods can be used to calculate the electron density of the VBM and CBM which provides an insight of the materials’ charge carrier separation ability. Here, we simplified the excited state with a periodic double layer of either a CTF+ cation or a CTF− anion to avoid resource intensive TD-DFT calculations. The CBM of the anion should mimic the excited electron state while the VBM of the cation should describe the corresponding hole. The CBM shows a clear localization of electron density on the triazine rings for both models (Fig. S4†). In the case of CTF-2, this localization is slightly more pronounced. The hole in the VBM (absence of electron density) is rather delocalized and differences between the two CTFs are negligible. Hence, the electron density does not reveal a charge separation advantage for any of the two materials. Electrochemical impedance spectroscopy (EIS) under illumination allows extraction of the charge transfer resistance (Fig. 1f). CTF-2 has a charge transfer resistance of 2700 ± 700 kΩ which is more than twice as much as the resistance of CTF-1 with 970 ± 90 kΩ. Thus, the higher photocurrent of CTF-1 can be explained by a lower charge transfer resistance and shorter diffusion lengths of the charge carriers due to higher porosity.
Photocatalytic HMF oxidation
Both CTFs were used as photocatalyst for photooxidation of HMF under visible blue LED light (460 nm, Fig. 2). CTF-1 is more than twice as active as CTF-2 since the reaction over CTF-1 reaches nearly full conversion after 4 h, while over CTF-2, the reaction has reached merely 40% conversion. We previously demonstrated that the activity of CTF photocatalysts is the sum of many factors, including light absorption, overpotential, porosity, wettability and interaction of substrate and catalyst.41 Additionally, the CTFs were also tested using simulated solar light, proving the applicability of both materials under these conditions. Noticeably, the yields and selectivities cannot be compared to those at 460 nm due to different irradiation intensities of the LEDs. As former studies have shown, the photocatalytic mechanism and thus photocatalytic activity are influenced by the incident light intensities.70,71 CTF-1 does have a larger band gap than CTF-2, which reduces the spectral range that initiates the photocatalysis. However, the quantity of absorbed photons also depends on the number of accessible chromophores, which in turn varies with morphology and behavior in solution (e.g. wettability, dispersibility and swelling). We utilized different equivalent molar amounts of CTF-1 and CTF-2 to ensure maximum possible light absorption, thus maximum activity for both materials (Fig. S1 and S2d†). The higher porosity of CTF-1 increases the accessible adsorption sites for the substrate, shown by adsorption tests (Table S2†). Combining these factors with lower charge transfer resistance, shorter charge carrier diffusion lengths and higher overpotential, the higher activity of CTF-1 can be explained. | ||
| Fig. 2 Time course of HMF conversion, DFF yield and H2MF yield in the photocatalysis with CTF-1 and -2. Conditions: 2 mg mL−1 catalyst, 0.01 M HMF, H2O, 25 °C, 460 nm (35 mW cm−2) and 0.5 bar overpressure synthetic air. | ||
CTF-1 shows very low selectivity towards DFF, reaching a maximum yield of 4.3% after two hours, before the over oxidation to FFCA and FDCA gets predominant (Fig. S2a†). CTF-2 shows a largely increased maximum DFF yield of 11.0% for the same conversion (approx. 59%), which equals a nearly three times higher selectivity (Fig. S2b†). On first sight, this could be explained by the higher oxidation potential of CTF-1 (Fig. S24†), which in theory is sufficient to split water into hydroxyl radicals (OH˙). Hydroxyl radicals are known to cause fast, unselective oxidation of organic compounds and might even cause their complete mineralization.72 Contrary, CTF-2 has an oxidation potential below 1.98 V, hence it should not be able to split water leading to a more selective oxidation. However, it is a common pitfall in photocatalysis to justify the performance of the catalyst too heavily on the redox potentials.73 In this study, we want to stress the importance of a detailed knowledge of the reaction mechanism to evaluate selectivity in photocatalysis.
After determining all detected main products from our HPLC analysis, H2MF and its joint product formic acid were identified next to the expected direct oxidation products DFF, FFCA and FDCA (Fig. S17†). This revealed two separate valorization pathways of HMF, namely the direct oxidation to DFF and the cycloaddition with singlet oxygen to H2MF. Interestingly, CTF-1 and CTF-2 show opposite selectivity towards these pathways. Comparing both materials at a similar conversion of around 33%, CTF-1 reaches a selectivity towards DFF of 11% and towards H2MF of 51% (Fig. S2b†). CTF-2 shows 30% selectivity to DFF and 35% to H2MF. In total, CTF-1 produces a maximum yield of 79% H2MF after 6 h while CTF-2 produces only 19% after the same reaction time. In order to elucidate the reaction step that decides the difference in selectivity, several control experiments were conducted (Fig. 3a).
 | ||
| Fig. 3 (a) Mechanistic study of HMF photooxidation over CTFs via changed reaction conditions (#) or addition of radical scavengers (*). Conditions if not stated otherwise: 2 mg mL−1 catalyst, 0.01 M HMF, 0.01 M scavenger, H2O, 25 °C, 460 nm (35 mW cm−2) and 0.5 bar overpressure synthetic air. (b and c) In situ EPR spectra of the reaction solution in the range of a possible DMPO–OH adduct and TEMPO radical during light irradiation at 455 nm. | ||
No conversion was detected without light irradiation, ensuring the presence of a photocatalytic reaction. Under argon atmosphere (Fig. S2f†), there was also no yield visible which pinpoints the importance of oxygen for these oxidations. Despite the fact that both CTFs hold frontier orbital positions allowing for hydrogen evolution as reductive half reaction couplet to HMF surface oxidation, hardly any catalytic activity was observed. This is most likely explained by the kinetically unfavored H2 elimination from the surface of an organic photocatalyst.8 To elucidate the role of reactive oxygen species (ROS), radical scavengers were added to the reaction and electron paramagnetic resonance experiments (EPR) were performed.74 The addition of tert-butanol as a hydroxyl radical scavenger decreases the conversion hinting towards an influence of OH˙. However, since EPR experiments with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) show no DMPO–OH adduct (Fig. 3b and c), the decrease in conversion is more likely due to competitive adsorption between HMF and tert-butanol (Table S2†). The presence of singlet oxygen was confirmed by EPR studies with tetramethylpiperidine (TEMP), yielding EPR active TEMPO. In situ EPR studies reveal an approx. five times faster increase of TEMPO formation over CTF-1 as compared to CTF-2, under otherwise identical conditions (Fig. S16†). Adding HMF to the EPR experiment reduces the signal intensity, supporting the assumed reaction of HMF with 1O2. In case of CTF-1 the intensity of TEMPO formed in the presence of HMF is reduced by a factor of 10 (after 20 min) while in case of CTF-2 only a factor of 2 is observed. The in situ EPR data show a higher 1O2 formation capacity and a significantly preferred HMF conversion over TEMP to TEMPO conversion for CTF-1. In order to suppress 1O2 formation under catalytic conditions, sodium azide was added. The presence of this singlet oxygen quencher diminishes the yield of H2MF and slightly reduces the yield of DFF. This confirms the reaction pathway to H2MF via cycloaddition of 1O2 and HMF. The conversion is increased in this case. A control experiment of stirring a mixture of CTF, HMF and NaN3 in the dark showed no conversion, so a possible side reaction of NaN3 with HMF can be excluded. Most likely azide radicals are formed during quenching which consequently react with HMF to a yet unknown product. Ayed et al. also observed the significant role of singlet oxygen without detecting H2MF. The differences in the product distribution might be caused by the different sample preparation techniques (extraction,54 here direct HPLC injection of the reaction mixture, S3.5†). Nonetheless, their postulated mechanism to form DFF out of HMF and 1O2 – although less dominant as they describe – might still explain the decreased DFF yield after the addition of sodium azide. Oxygen can be reduced to superoxide radicals (O2˙−) and consequently hydrogen peroxide. A simple analysis for H2O2 with appropriate test strips was positive, indicating the formation of H2O2 and thus the reduction of oxygen. Most often benzoquinone is used as a superoxide radical scavenger. However, it reacts on its own with HMF which makes the use of an alternative necessary. Therefore, superoxide dismutase (SOD) was added to scavenge potential superoxide radicals. SOD does hardly influence the conversion and yield of the catalysis which indicates no direct interaction of superoxide radicals with HMF. This is further supported by the absence of any DMPO–OOH adduct during in situ EPR experiments (Fig. 3b and c).75,76 We thus reason, that oxygen is needed as a complementary reduction substrate to the HMF oxidation, but we do not observe a connection with the direct oxidation to DFF, as reported previously.54 Hence, to validate if HMF is directly oxidized at the CTF surface, potassium iodide (KI) was added as a hole (h+) scavenger. KI has a very low oxidation potential and should be preferably oxidized to iodine, which subsequently forms triiodide.74 Conversion and yield are significantly decreased in the presence of KI and the formation of triiodide was successfully detected via the iodine-starch test. Consequently, HMF is most likely directly oxidized to DFF and further oxidized to FFCA and FDCA at the CTF surface.
The higher capability of CTF-1 to produce singlet oxygen explains phenomenologically its higher activity towards H2MF. However, this does not yet explain why CTF-1 and CTF-2 show opposite selectivity to these two mechanisms. While the direct oxidation is catalyzed by an excited singlet state (e.g. S1), singlet oxygen is formed via the interaction with a triplet state.77 A potential influence of the irradiation wavelength on the selectivity could be excluded (Fig. S2e†). Therefore, the singlet and triplet states of the two CTF materials are characterized in more detail. Steady state and time resolved emission spectroscopy can provide information about excited states and their lifetimes (Fig. 4a–c). The solid-state emission spectra after excitation at 378 nm of both CTFs are characterized by a roughly Gaussian-shaped emission with maxima around 510 nm and 540 nm for CTF-1 and CTF-2, respectively. However, the emission spectrum of CTF-2 is broader with a pronounced tailing in the red part of the spectrum. Time-correlated single photon counting (TCSPC), performed on the materials in the bulk, allows to separate the emission spectrum into its short- and long-lived emission parts which gives insight about the nature of the excited state. Short-lived emission derives from fluorescing singlet states while long-lived emission occurs either via trapped states or phosphorescing triplet states. From the TCSPC of both materials, two emissive contributions with distinctive different lifetimes can be discerned. An emissive state with a short lifetime of a few ns, which we tentatively assign to a fluorescent state (singlet) and the long-lived state (several 100 ns lifetime) assigned to a phosphorescent state (triplet). In CTF-1, the first state has an average lifetime around 10 ns and at the monitored emission wavelength roughly contributes to about 2/3 of the emission, and the second state has a lifetime of more than 1 μs and contributes to 1/3 of the emission (Table S5†). In contrast, in CTF-2 the relative emission contribution for the short-lived state (∼9 ns) is the minor contribution at the monitored wavelengths, whereas that of the long-lived state (>1.3 ns) is up to 70% (Table S5†). In the solid-state, both materials possess a quantum yield of the emission at room temperature of around 1% (data not shown). The low emissive quantum yield is in line with the indirect bandgap observed in both materials leading to mainly no radiative relaxation pathways. The emission spectra of the phosphorescent states are very similar in both materials, pointing towards the same, most likely triazine based state. This observation is in line with the DFT calculations showing that the CBM is mainly located on the triazine moiety for both CTFs (see above). In both materials, there is thus intersystem crossing from photoexcited singlet to triplet states which can be quenched by O2 forming 1O2. Interestingly, the rate of singlet oxygen formation as deduced from the signal intensity of the radical scavenger adduct TEMPO in EPR spectroscopy is higher in case of CTF-1 as compared to CTF-2 (Fig. S16†), albeit the relative fraction of emissive triplet states did not indicate this. To shine light on the emission under catalytically more relevant conditions, aqueous suspensions of the CTFs were measured by steady state spectroscopy. For CTF-1, a strong hypsochromic shift of about 600 cm−1 (50 nm) is observed for the maximum of the emission and of the excitation spectrum. In contrast, the emission of CTF-2 seems not to be affected by the solvent, e.g., the profile of the emission measured on the dry solid material and of the material suspended are the same (Fig. S15†). A possible explanation for the observed differences of the solvent induced shift in CTF-1 and CTF-2 could be the higher porosity and hydrophilicity on CTF-1 (for further discussion see ESI section 3.9†), increasing the interaction probability between solvent and the exited states.
 | ||
| Fig. 4 (a and b) Normalized steady state excitation and emission spectra of CTF-1 (black) and CTF-2 (blue). (c) Time-resolved emission spectra of CTF-1 (black) and CTF-2 (blue), dissected into short- and long-lived emission. (d and e) Simplified Tamm-Dancoff-approximated TD-DFT38 calculations based on ωB97X/def2-SV(P) calculations using cutouts of CTF layers. No shift was applied to the energies and the density of states was plotted for better visibility by convoluting the discrete singlet and triplet state positions with Gaussian functions with width 0.2 eV. The densities were normalized with respect to the maximum of the first triplet peak of CTF-2. | ||
Simplified TD-DFT calculations on cluster models of the CTFs show that CTF-1 has nine triplet states (located between 4.51–5.25 eV) per CTF-layer beneath the lowest singlet state at 5.25 eV (Fig. 4d). Singlet and triplet states overlap which is favorable for intersystem crossing (ISC). In CTF-2, the triplet states (3.63–3.76 eV) are clearly separated from the lowest singlet state (4.18 eV) making ISC less likely (Fig. 4e). Consequently, CTF-1 should be able to form triplet states more easily during catalysis. If this process is accompanied with energy transfer to oxygen his results in higher selectivity towards H2MF. Hence, the variation of the CTF linker from phenyl to naphthyl reduces the formation of triplet states, pushing the selectivity in the HMF valorization to the direct oxidation pathway.
In summary, we elucidate in detail the reaction mechanism for HMF valorization over CTF photocatalysts (Scheme 2). Two reaction mechanisms are possible depending on the optoelectronic properties. In the first pathway, HMF is directly oxidized at the CTF surface to DFF while molecular oxygen is reduced to O2˙− simultaneously. In the second pathway, oxygen is converted to 1O2 which consequently reacts in a cycloaddition with HMF to H2MF. The selectivity towards any of these two mechanisms can be controlled by the choice of the building blocks used for the catalyst's synthesis. Tailoring the structure of the CTF to form energetically proximate singlet and triplet state manifolds leads to a high selectivity towards 1O2 formation and, thus, towards H2MF. Vice versa, building a network with well-separated singlet and triplet states increases the selectivity towards the direct oxidation.
 | ||
| Scheme 2 Illustration of the two possible reaction mechanisms, 1O2 cyclo-addition or direct oxidation, of HMF over CTF photocatalysts. | ||
Conclusion
We designed two CTF photocatalysts with altered optoelectronic properties that revealed opposing selectivity in HMF valorization. Thus, we report an updated reaction mechanism for HMF photooxidation over CTFs: either HMF is directly oxidized to DFF at the CTF surface or, via photoinduced energy transfer, molecular oxygen is converted to singlet oxygen which then reacts in a cycloaddition with HMF to yield H2MF. Detailed investigation of the reaction mechanism combined with simplified TD-DFT calculations elucidated the decisive property that controls the switch in selectivity. Depending on the probability to form excited triplet states, the cycloaddition or the direct oxidation is favored. Future studies will focus on more tailor-made CTFs to broaden application of our hypothesis and to find structure–property relationships about the affinity towards singlet and triplet states in CTFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. P. acknowledges the Fuel Science Center (EXC 3782186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments. D. D. gratefully thanks the German Federal Environmental Foundation for financial support. N. M. S. thanks RWTH Aachen University for financial support. F. M. W. gratefully acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, grant number WI 4721/3-1) and from CNRS through Momentum 2018 excellence grant. C. B. acknowledges funding by the Ministry of Culture and Science of the German State of North Rhine-Westphalia (MKW) via the NRW Rückkehrprogramm. Felix Müller and Mirijam Zobel acknowledge Florian Puchtler of Institute of Inorganic Chemistry I at University Bayreuth for PDF data collection.References
- Europäischer Grüner Deal, available at: https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal_de, accessed 11 November 2021.
- VCI Online, So gelingt der Green Deal, available at: https://www.vci.de/themen/europa/green-deal/so-gelingt-der-green-deal.jsp, accessed 11 November 2021.
- C. Dai and B. Liu, Environ. Sci. Technol., 2020, 13, 24–52 CAS.
- T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- J. Warnan and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 17344–17354 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS.
- Y. Meng, Y. Jian, J. Li, H. Wu, H. Zhang, S. Saravanamurugan, S. Yang and H. Li, Chem. Eng. J., 2023, 452, 139477 CrossRef CAS https://www.sciencedirect.com/science/article/pii/S1385894722049567 .
- Q. Dou and H. Zeng, Curr. Opin. Green Sustain. Chem., 2023, 40, 100766 CrossRef CAS https://www.sciencedirect.com/science/article/pii/S2452223623000159 .
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed.
- J. Artz, ChemCatChem, 2018, 10, 1753–1771 CrossRef CAS.
- C. Krishnaraj, H. S. Jena, K. Leus and P. van der Voort, Green Chem., 2020, 22, 1038–1071 RSC.
- R. Sun, X. Wang, X. Wang and B. Tan, Angew. Chem., Int. Ed., 2022, e202117668 CAS.
- Z. Qian, Z. J. Wang and K. A. I. Zhang, Chem. Mater., 2021, 33, 1909–1926 CrossRef CAS.
- J. Byun and K. A. I. Zhang, Mater. Horiz., 2020, 7, 15–31 RSC.
- L. Guan, G. Cheng, B. Tan and S. Jin, Chem. Commun., 2021, 57, 5147–5150 RSC.
- G. Tuci, M. Pilaski, H. Ba, A. Rossin, L. Luconi, S. Caporali, C. Pham-Huu, R. Palkovits and G. Giambastiani, Adv. Funct. Mater., 2017, 27, 1605672 CrossRef.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Environ. Sci. Technol., 2013, 6, 3684 CAS.
- S. Hug, M. B. Mesch, H. Oh, N. Popp, M. Hirscher, J. Senker and B. V. Lotsch, J. Mater. Chem. A, 2014, 2, 5928–5936 RSC.
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6263 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., 2008, 120, 3499–3502 CrossRef.
- A. Iemhoff, J. Deischter, S. Jung, G. Tuci, G. Giambastiani and R. Palkovits, J. Mater. Chem. A, 2021, 9, 5390–5403 RSC.
- Y. P. Tang, H. Wang and T. S. Chung, ChemSusChem, 2015, 8, 138–147 CrossRef CAS PubMed.
- S. Dey, S. Bügel, S. Sorribas, A. Nuhnen, A. Bhunia, J. Coronas and C. Janiak, Front. Chem., 2019, 7, 693 CrossRef CAS PubMed.
- O. Buyukcakir, J. Ryu, S. H. Joo, J. Kang, R. Yuksel, J. Lee, Y. Jiang, S. Choi, S. H. Lee, S. K. Kwak, S. Park and R. S. Ruoff, Adv. Funct. Mater., 2020, 30, 2003761 CrossRef CAS.
- S. N. Talapaneni, T. H. Hwang, S. H. Je, O. Buyukcakir, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2016, 55, 3106–3111 CrossRef CAS PubMed.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef PubMed.
- T. Sönmez, K. S. Belthle, A. Iemhoff, J. Uecker, J. Artz, T. Bisswanger, C. Stampfer, H. H. Hamzah, S. A. Nicolae, M.-M. Titirici and R. Palkovits, Catal. Sci. Technol., 2021, 11, 6191–6204 RSC.
- J. Liu, Y. Hu and J. Cao, Catal. Commun., 2015, 66, 91–94 CrossRef CAS.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- M. Pilaski, J. Artz, H.-U. Islam, A. M. Beale and R. Palkovits, Microporous Mesoporous Mater., 2016, 227, 219–227 CrossRef CAS.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- S. Abednatanzi, P. Gohari Derakhshandeh, K. Leus, H. Vrielinck, F. Callens, J. Schmidt, A. Savateev and P. van der Voort, Sci. Adv., 2020, 6, eaaz2310 CrossRef CAS PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, e1904433 CrossRef PubMed.
- Y. Shen, C. Zhu, S. Song, T. Zeng, L. Li and Z. Cai, Environ. Sci. Technol., 2019, 53, 9091–9101 CrossRef CAS PubMed.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- Q. Pan, T. Chen, L. Ma, G. Wang, W.-B. Hu, Z. Zou, K. Wen and H. Yang, Chem. Mater., 2019, 31, 8062–8068 CrossRef CAS.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Angew. Chem., 2020, 132, 6063–6070 CrossRef.
- M. Alves Fávaro, D. Ditz, J. Yang, A. C. Ghosh, C. Lorentz, J. Roeser, E. A. Quadrelli, A. Thomas, R. Palkovits, J. Canivet and F. M. Wisser, ACS Appl. Mater. Interfaces, 2022, 14(12), 14182–14192 CrossRef PubMed.
- L. Guo, Y. Niu, S. Razzaque, B. Tan and S. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS.
- W. Huang, Q. He, Y. Hu and Y. Li, Angew. Chem., Int. Ed., 2019, 58(26), 8676–8680 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- S. Guo, P. Yang, Y. Zhao, X. Yu, Y. Wu, H. Zhang, B. Yu, B. Han, M. W. George and Z. Liu, ChemSusChem, 2020, 13(23), 6278–6283 CrossRef CAS PubMed.
- J. Li, P. Liu, H. Huang, Y. Li, Y. Tang, D. Mei and C. Zhong, ACS Sustainable Chem. Eng., 2020, 8, 5175–5183 CrossRef CAS.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem. – Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- W. Huang, J. Byun, I. Rörich, C. Ramanan, P. W. M. Blom, H. Lu, Di Wang, L. Caire da Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., 2018, 130, 8449–8453 CrossRef.
- Y. Liu, H. Wu and Q. Wang, J. Mater. Chem. A, 2023, 11, 11470–11497, 10.1039/D3TA04472F.
- N. Xu, R.-L. Wang, D.-P. Li, X. Meng, J.-L. Mu, Z.-Y. Zhou and Z.-M. Su, Dalton Trans., 2018, 47, 4191–4197 RSC.
- S.-R. Zhu, Q. Qi, Y. Fang, W.-N. Zhao, M.-K. Wu and L. Han, Cryst. Growth Des., 2018, 18, 883–891 CrossRef CAS.
- W. Huang, Z. J. Wang, B. C. Ma, S. Ghasimi, D. Gehrig, F. Laquai, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2016, 4, 7555–7559 RSC.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- C. Ayed, W. Huang, G. Kizilsavas, K. Landfester and K. A. I. Zhang, ChemPhotoChem, 2020, 4, 571–576 CrossRef CAS.
- X. Liu, R. Qi, S. Li, W. Liu, Y. Yu, J. Wang, S. Wu, K. Ding and Y. Yu, J. Am. Chem. Soc., 2022, 144, 23396–23404 CrossRef CAS PubMed.
- L. Liao, D. Ditz, F. Zeng, M. Alves Fávaro, A. Iemhoff, K. Gupta, H. Hartmann, C. Szczuka, P. Jakes, P. J. C. Hausoul, J. Artz and R. Palkovits, ChemistrySelect, 2020, 5, 14438–14446 CrossRef CAS.
- X. Wang, S. Zhang, X. Li, Z. Zhan, B. Tan, X. Lang and S. Jin, J. Mater. Chem. A, 2021, 9, 16405–16410 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Green Chem., 2021, 23, 3154–3171 RSC.
- T. S. A. Heugebaert, C. V. Stevens and C. O. Kappe, ChemSusChem, 2015, 8, 1648–1651 CrossRef CAS PubMed.
- W. Sun, Y. Xiang, Z. Jiang, S. Wang, N. Yang, S. Jin, L. Sun, H. Teng and H. Chen, Sci. Bull., 2022, 67, 61–70 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- C. Kang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, K. Yu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2022, 144, 3192–3199 CrossRef CAS PubMed.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- R. Gutzler, Phys. Chem. Chem. Phys., 2016, 18, 29092–29100 RSC https://pubs.rsc.org/en/content/articlelanding/2016/CP/C6CP06101J .
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 RSC.
- Photocatalysis. Fundamentals and perspectives, ed. D. Bahnemann, D. D. Dionysiou, G. L. Puma, J. Schneider and J. Ye, Royal Society of Chemistry, Cambridge, 2016, vol. 14 Search PubMed.
- Y. Zou, S. Abednatanzi, P. Gohari Derakhshandeh, S. Mazzanti, C. M. Schüßlbauer, D. Cruz, P. van der Voort, J.-W. Shi, M. Antonietti, D. M. Guldi and A. Savateev, Nat. Commun., 2022, 13, 2171 CrossRef CAS PubMed https://www.nature.com/articles/s41467-022-29781-9 .
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- Z. Zhu, Y. Xuan, X. Liu, K. Zhang, Y. Zhang, Q. Zhu and J. Wang, ChemPhysChem, 2022, 23, e202100851 CrossRef CAS PubMed.
- Z. Wang, W. Qiao, M. Yuan, N. Li and J. Chen, J. Phys. Chem. Lett., 2020, 11, 2369–2373 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- P. Christopher, S. Jin, K. Sivula and P. V. Kamat, ACS Energy Lett., 2021, 6, 707–709 CrossRef CAS.
- J. T. Schneider, D. S. Firak, R. R. Ribeiro and P. Peralta-Zamora, Phys. Chem. Chem. Phys., 2020, 22, 15723–15733 RSC.
- G. R. Buettner, Free Radicals Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- J. Vasquez-Vivar, B. Kalyanaraman and M. C. Kennedy, J. Biol. Chem., 2000, 275, 14064–14069 CrossRef CAS PubMed.
- W. Wu, D. Mao, S. Xu, Kenry, F. Hu, X. Li, D. Kong and B. Liu, Chem, 2018, 4, 1937–1951 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01210g |
| This journal is © The Royal Society of Chemistry 2024 |