
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAstaxanthin targets IL-6 and alleviates the LPS-induced adverse inflammatory response of macrophages†
Yahui
Wu
ab,
Mona A.
Bashir
ab,
Changsheng
Shao
c,
Han
Wang
ab,
Jianxia
Zhu
ad and
Qing
Huang
 *ab
*ab
aCAS Key Laboratory of High Magnetic Field and Ion Beam Physical Biology, Anhui Key Laboratory of Environmental Toxicology and Pollution Control Technology, Hefei Institute of Intelligent Agriculture, Institute of Intelligent Machines, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, China. E-mail: huangq@ipp.ac.cn; Fax: +86-551-65595261; Tel: +86-551-65595261
bScience Island Branch of Graduate School, University of Science & Technology of China, Hefei 230026, China
cHigh Magnetic Field Laboratory, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, China
dSchool of Nursing, Anhui Medical University, Hefei, Anhui 230032, China
First published on 7th March 2024
Abstract
Numerous natural compounds are recognized for their anti-inflammatory properties attributed to antioxidant effects and the modulation of key inflammatory factors. Among them, astaxanthin (AST), a potent carotenoid antioxidant, remains relatively underexplored regarding its anti-inflammatory mechanisms and specific molecular targets. In this study, human monocytic leukemia cell-derived macrophages (THP-1) were selected as experimental cells, and lipopolysaccharides (LPS) served as inflammatory stimuli. Upon LPS treatment, the oxidative stress was significantly increased, accompanied by remarkable cellular damage. Moreover, LPSs escalated the expression of inflammation-related molecules. Our results demonstrate that AST intervention could effectively alleviate LPS-induced oxidative stress, facilitate cellular repair, and significantly attenuate inflammation. Further exploration of the anti-inflammatory mechanism revealed AST could substantially inhibit NF-κB translocation and activation, and mitigate inflammatory factor production by hindering NF-κB through the antioxidant mechanism. We further confirmed that AST exhibited protective effects against cell damage and reduced the injury from inflammatory cytokines by activating p53 and inhibiting STAT3. In addition, utilizing network pharmacology and in silico calculations based on molecular docking, molecular dynamics simulation, we identified interleukin-6 (IL-6) as a prominent core target of AST anti-inflammation, which was further validated by the RNA interference experiment. This IL-6 binding capacity actually enabled AST to curb the positive feedback loop of inflammatory factors, averting the onset of possible inflammatory storms. Therefore, this study offers a new possibility for the application and development of astaxanthin as a popular dietary supplement of anti-inflammatory or immunomodulatory function.
1. Introduction
Inflammation is the organism's defense reaction, triggered in response to various injurious stimuli, aimed at achieving self-protection.1 In the early stages of inflammation, inflammatory factors play a crucial role in eliminating harmful agents, promoting wound healing, and generating favorable defense effects on the organism.2 However, when the inflammatory response becomes persistent, these factors can inflict damage on normal tissues and cells, leading to the development of various inflammatory diseases,3 including acute or chronic inflammation, autoimmune diseases, or cancer. Currently, various synthetic and natural immune modulators have been explored to regulate the immune response. However, the majority of synthetic anti-inflammatory drugs are expensive, and their prolonged use can lead to adverse effects on the body, such as gastrointestinal and respiratory irritation, nephrotoxicity, physical dependence, and constipation. Opting for cost-effective natural medicines with low toxicity and better tolerability helps overcome the toxic side-effects and financial burden associated with the long-term use of synthetic anti-inflammatory drugs. Fortunately, there is growing evidence that many bioactive natural products may antagonize inflammatory responses with little or no side effects in humans.4,5 For example, it has been reported that natural carotenoids play an important role in the regulation of immunity and disease etiology.6Astaxanthin (AST) is an oxygen-containing non-vitamin A source of carotenoid, widely found in nature, especially in shrimp, crab, fish, algae (e.g., Haematococcus pluvialis), yeast, etc.7 AST exhibits a broad range of biological activities and has been extensively utilized in the food, nutraceutical, pharmaceutical, cosmetic, and aquaculture industries. The unique long conjugated double bond in the molecular structure of AST, along with the reactive electrons of the terminal ketone and hydroxyl groups, easily provides electrons to free radicals or attracts the unpaired electrons of free radicals, thereby scavenging free radicals and playing an antioxidant role.8 It stands as the most potent natural antioxidant in nature, surpassing vitamin E by more than 500 times in scavenging free radicals and quenching singlet oxygen.9 Its activity is also 10 times higher those of than zeaxanthin, lycopene, lutein, hordenine, and β-carotene, and it is much more effective than anthocyanin—widely used in medicine and the cosmetics industry. It is also claimed that AST is the only carotenoid known to penetrate the blood–brain and blood–retina barriers.10 The conjugated double bond and α-hydroxyl ketone of AST grant it superb antioxidant properties, providing it with high application value and substantial market potential.
While the antioxidant effects of AST have been well recognized, its role in anti-inflammation remains to be demonstrated and investigated. Sustained oxidative stress is a key mechanism leading to chronic inflammation. Over the last two decades, numerous studies have revealed the mechanisms by which sustained oxidative stress leads to chronic inflammation, a mediator of most chronic diseases.11,12 AST may exert anti-inflammatory effects by regulating oxidative stress/antioxidant levels. Kang et al. conducted a review of 45 publications, noting that the antioxidant effect of AST attenuates inflammation and discussing the mechanism by which AST inhibits gastritis induced by Helicobacter pylori, correlating it with the anti-inflammatory effect of AST.13 Actually, reactive oxygen species (ROS) and inflammation are closely related, and ROS may play a dual role: while they kill pathogens and remove mid-end effector molecules from damaged tissues, they also up-regulate the expression of pro-inflammatory factors, triggering pro-inflammatory cytokines and creating a positive feedback loop. Antioxidants and antioxidant enzymes block this positive feedback loop by suppressing the gene expression of inflammatory factors.14In vitro studies have shown that AST as an antioxidant has several times more antioxidant activity than β-carotene and α-tocopherol.15In vivo experiments in mice have also demonstrated that AST inhibits various activities associated with burns, muscle damage, oxidative stress, and inflammation.16 It has been reported that AST enhances macrophage sensitivity to lipopolysaccharides (LPS), thereby increasing the organism's ability to prevent and resist inflammation.17 All these studies collectively suggest that AST has anti-inflammatory effects. However, the specific mechanism of its anti-inflammatory effect remains largely unexplored.
Therefore, in this study, we attempted to explore the mechanism for the anti-inflammatory function of AST, and particularly, the inflammatory factors that AST may target and interact. For this purpose, THP-1 cells were selected for this study, because THP-1 is a cell line established from monocytes in the blood of leukemia patients and typically used to study the immune responsiveness of monocytes and macrophages derived from monocytes.18 Inflammation was induced experimentally using LPS, which is a common predominant pro-inflammatory factor.19 Stimulation of macrophages is often used as a model for the study of related inflammatory responses.20,21 Through cell experiments, we observed that the cells entered a state of oxidative stress after LPS treatment, leading to cell damage. We found that AST significantly reduced the oxidative stress induced by LPS stimulation and facilitated cell repair. Simultaneously, we found that LPS caused a significant elevation in inflammation-related factors at both the transcriptional and translational levels. The addition of AST intervention significantly reduced the expression of inflammation-related molecules. We also revealed that the anti-inflammatory mechanism of AST involved the nuclear activation of NF-κB and the expression of NF-κB in LPS-stimulated macrophages. Additionally, we analyzed the impact of AST on the expressions of p53 and STAT3, which may explain the reduced cell damage with the AST treatment. Furthermore, by virtue of network pharmacology, which applies systems biology techniques and database resources to analyze target molecules,22,23 we successfully screened out interleukin-6 (IL-6) as the most possible core target. This binding of AST with IL-6 was also further verified by in silico calculation through molecular docking and molecular dynamics simulation, as well as by the RNA interference experiment.
The major aim of this study was to examine the impact and clarify the anti-inflammatory mechanism of AST. The purpose of this study is to provide an innovative perspective for the development of dietary immune modulators and the utilization of astaxanthin resources for individuals with inflammation.
2. Materials and methods
2.1. Materials
Astaxanthin was purchased from Aladdin (Shanghai, China). Malondialdehyde (MDA), catalase (CAT), superoxide dismutase (SOD), and glutathione reductase (GR) kits were purchased from Beyotime (Shanghai, China). The ROS fluorescent probe (2′-7′dichlorofluorescin diacetate, DCFH-DA) was purchased from Sigma-Aldrich (St Louis, USA). Phorbol-12-myristate-13-acetate (PMA), the superoxide anion (O2−) fluorescent probe (dihydroethidium, DHE), the NO (nitric oxide) fluorescent probe (3-amino,4-aminomethyl-2′,7′-fluorescein, diacetate, DAF-FM DA), the calcium ion (Ca2+) fluorescent probe (Fluo-3 AM), and the mitochondrial membrane potential (MMP) fluorescent probe (tetramethylrhodamine, ethyl ester, TMRE) were purchased from Beyotime (Shanghai, China). LPS, CCK-8, and bicinchoninic acid assay (BCA) protein quantification kits were purchased from Biosharp (Hefei, Anhui, China). TRIzol, Evo M-MLV reverse transcription reagent premixed solution, and the SYBR® Green Pro Taq HS premixed qPCR kit were purchased from Accurate Biology (Changsha, Hunan, China). γH2AX antibody and NF-κB p65 antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). The BD™ Cytometric Bead Array (CBA) Human Inflammatory Cytokine kit was purchased from BD Pharmingen (San Diego, CA, USA). All primers were synthesized by GENEWIZ (Suzhou, Jiangsu, China).2.2. Cell culture
The human acute monocytic leukemia cell line, THP-1 cell line, was purchased from Procell Life Science & Technology Co., Ltd (Wuhan, China) and cultured in RPMI 1640 medium (Gibco, Shanghai, China) with 10% fetal bovine serum (Gibco, Shanghai, China), and 1% antibiotic–antimycotic (Gibco, Shanghai, China) under 5% CO2 at 37 °C. THP-1 monocytes were incubated in 6-well plates and exposed to 100 ng mL−1 PMA for 24 hours to induce their transformation into adherent macrophages. Morphological observations were recorded, including images. Flow cytometry was also employed to measure the expression level of the THP-1-derived macrophage-specific protein CD11b.2.3. Detection of MDA, CAT, SOD and GR
Cells from each treatment group were collected and then lysed with RIPA lysate on ice to extract the total proteins. The supernatant was collected by centrifugation (4 °C, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min). The protein concentration was determined and normalized using the bicinchoninic acid assay (BCA) method. Subsequently, the MDA level and SOD, CAT and GR activities were evaluated according to the manufacturer's instructions.
000g, 10 min). The protein concentration was determined and normalized using the bicinchoninic acid assay (BCA) method. Subsequently, the MDA level and SOD, CAT and GR activities were evaluated according to the manufacturer's instructions.
2.4. Detection of ROS, O2−, NO, Ca2+ and MMP
Cells from each treatment group were collected and analyzed using various fluorescent probes: the ROS fluorescent probe DCFH-DA, the O2− fluorescent probe DHE, the NO fluorescent probe DAF-FM DA, the Ca2+ fluorescent probe Fluo-3 AM, and the MMP fluorescent probe TMRE. These probes were employed to detect the levels of ROS, O2−, NO, Ca2+, and MMP, respectively. The fluorescence intensities of the corresponding probes were then measured and analyzed using flow cytometry (EXFLOW-206, DAKEWEI, China) according to the manufacturer's instructions. A total of 10000 events were collected for each sample, and the data were further analyzed using FlowJo (version 10; TreeStar, Ashland, USA).
2.5. Apoptosis detection
THP-1 cells were cultured at a density of 1 × 105 cells per mL in 6-well plates for treatment. After washing the cells once with PBS, Annexin V and PI double staining were applied, and the cells were analyzed using flow cytometry (EXFLOW-206, DAKEWEI, China). A total of 10000 events were collected for each sample, and the data were analyzed using FlowJo (version 10; TreeStar, Ashland, USA).
2.6. Immunofluorescence assay
THP-1 cells were uniformly seeded in 24-well plates with a low cell density to prevent intercellular contact affecting the morphological observation. After treating each group of cells accordingly, the culture medium was removed, and the cells were fixed with 4% paraformaldehyde for 15 minutes at 4 °C. Subsequently, the fixative was removed, and the cells were washed three times with pre-cooled PBS buffer at 4 °C. The samples were then completely covered with an immunostaining blocking solution and incubated in a constant temperature and humidity incubator at 37 °C for 30 minutes. Following this, the blocking solution was removed, and the diluted primary antibody working solution was added dropwise to the samples, ensuring complete coverage. The samples were incubated at 4 °C overnight. The next day, the samples were rewarmed at room temperature for 15 minutes, and washed once with PBST and three times with PBS. Subsequently, the fluorescent secondary antibody working solution was added dropwise and incubated at 37 °C for 1 hour. After incubation, the samples were washed once with PBST and three times with PBS. DAPI staining solution with a mass concentration of 1 mg mL−1 was diluted 5000-fold with 1× PBS. The diluted solution was then added dropwise to the cell samples for nuclei labeling. The samples were incubated at room temperature, protected from light for 10 minutes, and washed once with PBST and three times with PBS. Finally, the samples were observed and photographed under a confocal microscope (ZEISS LSM710) (Jena, Germany).2.7. RNA extraction, reverse transcription, and qPCR
Quantitative real-time PCR was employed to detect the relative expression of the mRNA of inflammation-related genes after treating LPS-stimulated THP-1-derived macrophages with AST. The procedure involved collecting cells treated under optimal conditions, followed by cellular RNA extraction according to the RNA TRIzol instructions. Subsequently, cDNA synthesis was performed as per the Evo M-MLV RT Premix for qPCR reagent instructions. Finally, qPCR was conducted using a 20 μL reaction system: SYBR premix (10 μL), 1.0 μL each of upstream and downstream primers, ddH2O (6 μL), and 2.0 μL of the cDNA template. A Roche 480 fluorescence PCR instrument (Basel, Switzerland) was used for qPCR, following this procedure: pre-denaturation at 95 °C for 5 min; denaturation at 95 °C for 10 s, annealing at 60 °C, extension at 60 °C for 30 s, 40 cycles; 95 °C, 15 s, 60 °C, 60 s, 95 °C, 15 s to generate a melting curve. The relative expression of the mRNA of inflammation-related genes was calculated using the comparative CT method (2−ΔΔCt) and normalized to β-actin. The primer sequences are provided in Table S1.†2.8. Cytokine detection
Cytokine expression was detected using the BD™ Cytometric Bead Array (CBA) Human Inflammatory Cytokines kit. 50 μL of the capture microsphere mix was added per 50 μL of the standard or sample, followed by 50 μL of a detection antibody, and incubated (3 h at room temperature away from light). Washing with 1 mL of wash buffer was performed, followed by centrifugation, and the supernatant was discarded. Each tube was resuspended with 300 μL of washing buffer. The samples were obtained and detected by flow cytometry (EXFLOW-206, DAKEWEI, China), and the data were analyzed using FCAP Array™ analysis software (version 3.0.1).2.9. Network pharmacology
Using the PubChem database (https://pubchem.ncbi.nlm.nih.gov/),24 AST was input to find its 3D structure and obtain the SMILE name. The SMILE name of AST was then input into the SWISS Target Prediction database (https://www.swisstargetprediction.ch/) to identify the target genes of AST.25 In most cases of utilizing web-based pharmacological methods to study diseases, the collection of disease-related genes is often confined to a single database. This approach does not guarantee data integrity, as it may result in issues due to untimely updates of databases or discrepancies with similar databases. In this study, recognized disease databases were collectively utilized to search and collect inflammation-related genes. The genes were sourced from the GeneCards (https://www.genecards.org/) and DisGeNET (https://www.disgenet.org/) databases. Duplicate entries were merged to obtain a comprehensive set of inflammation-related targets.26 The obtained AST targets and inflammation targets were organized and counted. Intersecting targets were then screened, and a Venn diagram was created using the online mapping tool VENNY 2.1.27 The shared protein targets of AST and inflammation were uploaded to the STRING database (https://cn.string-db.org/) to establish a protein–protein interaction (PPI) network. The downloaded files were then imported into Cytoscape 3.9.1 software for mapping analysis.28,292.10. Molecular docking
The AST applied in this study had a 3S,3′S configuration, and its structure was retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/compound/) in SDF files and converted to the pdbqt format using the OpenBabel toolbox in the PyRx 0.8 virtual tool. The crystal structure of the IL-6 protein (PDB ID 1ALU) was downloaded from the PDB database (https://www.rcsb.org/structure/) and saved in the PDB format. PDB file was prepared by removing the water molecules and heat atoms, inserting charge, and adding polar hydrogen to the 3-D protein moiety using PyMol and BIOVINA Discovery Studio Visualizer v21.1.0.20298 virtual tool. Molecular docking of AST against IL-6 was performed using the AutoDock Vina 1.1.2 virtual screening tool. The interaction between AST and protein complexes was visualized in 2D and 3D structures using Molecular Operating Environment MOE 2015.10 software and PyMol.302.11. Molecular dynamics simulation
The AST–IL-6 complex file was subjected to molecular dynamics studies using the System Builder module in Schrödinger Desmond with default protocols.31 The system was solvated in a TIP3P water box, in which 0.15 M NaCl was introduced to neutralize the system. Energy minimization was achieved using the NPT (number of atoms, pressure, and temperature) ensemble at a temperature of 310 K with a pressure of 1.013 bar.32 The hydrogen bonds in the system were constrained using the SHAKE algorithm. The production run was performed using the NVT (number of atoms, volume, and temperature) ensemble for 100 ns with a Langevin thermostat and PME (particle mesh Ewald) method for long-range electrostatics.33 The force field parameters were generated using the Force Field Builder module in Schrödinger Desmond, and the OPLS3 (Optimized Potentials for Liquid Simulations) force field was used. The trajectory file generated from the production run was analyzed for RMSD, RMSF, hydrogen bonds, salt bridges, and free energy of binding using Desmond, VMD, and PyMOL.34MM-GBSA analysis was performed utilizing the thermal_MMGBSA.py script from the Prime Desmond module in the Schrödinger suite. The analysis involved calculating the free binding energy by generating 0–1000 frames. For the solvent model, the VSGB 2.0 new energy model was chosen, and the force field selected was 89 OPLS-2005. The remaining settings were kept at their default values. The MM-GBSA calculation of 100 ns MDS data involved processing and analyzing 200 frames. Subsequently, the binding free energies (in kcal mol−1) were computed.35
2.12. Small interfering RNA (siRNA) interference assay
The designs of siRNA sequences that interfere with the IL-6 and TNF-α gene were based on the human IL-6 and TNF-α gene sequences. The interference sequence information was as follows: si-IL-6 (sense), 5′-GCUGUGCAGAUGAGUACAATT-3′; si-IL-6 (antisense), 5′-UUGUACUCAUCUGCACAGCTT-3′; si-TNF-α (sense), 5′-UGGCGUGGAGCUGAGAGAUTT-3′; and si-TNF-α (antisense), 5′-AUCUCUCAGCUCCACGCCATT-3′. The negative control sequence was as follows: NC (sense), 5′-UUCUCCGAACGUGUCACGUTT-3′; and NC (antisense), 5′-ACGUGACACGUUCGGAGAATT-3. The experimental cells were divided into four groups: LPS (NC), AST + LPS (NC), LPS (si-IL-6 or si-TNF-α), and AST + LPS (si-IL-6 or si-TNF-α). These groups were transfected with the above si-IL-6 (or si-TNF-α) and NC. After 24 hours of transfection, the cells in each group were collected with the corresponding LPS and AST treatments. RNA was then extracted to detect the mRNA expression levels of IL-6 (or TNF-α), NF-κB and STAT3.2.13. Statistical analysis
Prism Version 9.0.0 (GraphPad, San Diego, USA) software was used to analyze the statistical data and create graphs. FlowJo (version 10; TreeStar, Ashland, USA) was employed for analyzing the flow cytometry experimental data. ImageJ software was utilized to analyze the average fluorescence intensity. The presented data are expressed as mean ± standard deviation (mean ± SD). Student's t-test and one-way analysis of variance (ANOVA) were employed to test for inter-sample differences. In all trials, a significance level of p < 0.05 was considered statistically significant. Adobe Illustrator (version 2020) software was used for plotting.3. Results
3.1. AST alleviates LPS-stimulated oxidative stress in macrophages
For the macrophage inflammation experiment, we initially induced prompt differentiation into macrophages in THP-1 cells, in which we observed a notable elevation in the expression of the cell surface-specific protein CD11b after induction with PMA, which suggests the successful differentiation of THP-1 monocytes into macrophages (Fig. S1†). LPS is a predominant pro-inflammatory factor, so we used LPS as the effective inflammation stimulus. For the application of AST, an optimal dose of 50 μM was found in the experiment (Fig. S2†), which ensured consistency with the conditions used in previous studies.36It is generally believed that the anti-inflammatory effect of AST can be manifested as an antioxidant effect, and for this reason, we first inspected the antioxidant effect of AST. MDA is a product from membrane lipid peroxidation, and it is generally regarded as an effective indicator of the level of oxidative stress imposed on the cell.21 To investigate whether AST could modulate lipid peroxidation activity in LPS-stimulated THP-1-derived macrophages, we measured the intracellular MDA level. Indeed, we observed a significant increase in the MDA level after LPS treatment compared with the CK group (the control group) and a lower amount of MDA in THP-1-derived macrophages treated with AST compared with the LPS group (Fig. 1A). CAT, SOD and GR are the key enzymes in the antioxidant enzyme system. By detecting the activity of CAT, SOD and GR in anti-oxidative stress, we found that after LPS treatment, the activity levels of CAT, SOD and GR were significantly lower than those in the CK group. In the experimental group pretreated with AST, the activity levels of CAT, SOD and GR in the cells increased, and the activity could be restored to the level of the CK group cells (Fig. 1B–D).
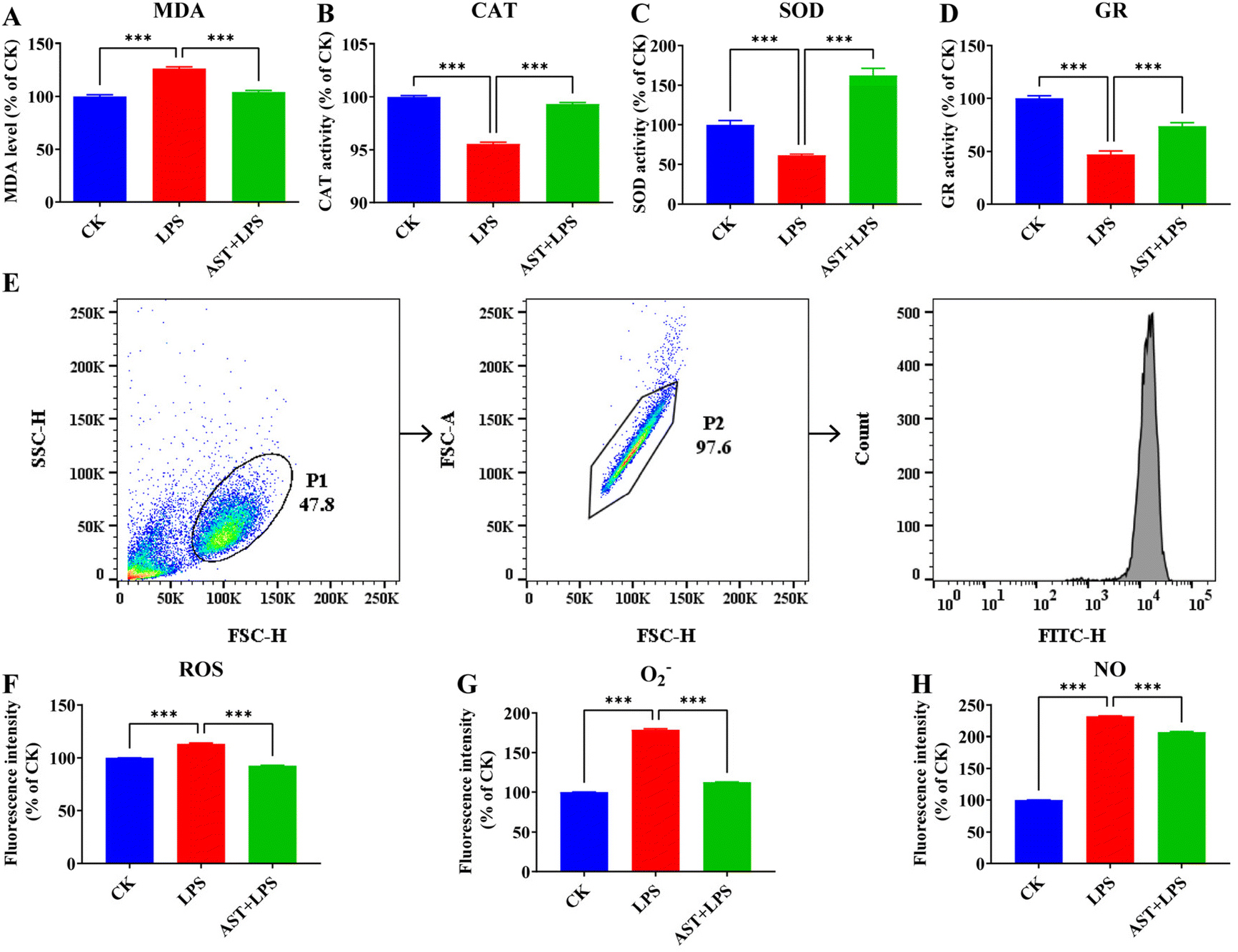 | ||
| Fig. 1 Effects of AST on LPS-stimulated oxidative stress in macrophages. (A) The effect of AST on the MDA level in LPS-stimulated macrophages. (B) The effect of AST on the CAT activity of LPS-stimulated macrophages. (C) The effect of AST on the SOD activity of LPS-stimulated macrophages. (D) The effect of AST on the GR activity of LPS-stimulated macrophages. (E) The gate strategy for flow cytometric analysis. (F) The effect of AST on ROS in LPS-stimulated macrophages. (G) The effect of AST on O2− in LPS-stimulated macrophages. (H) The effect of AST on NO in LPS-stimulated macrophages. (n = 3 per group; mean ± SD; *** represents p < 0.001.) | ||
To further check the antioxidant effect, we also investigated the impact of AST on reactive oxygen/nitrogen species (RONS) in the LPS-stimulated macrophages. Previous studies have indicated that increased ROS is associated with inflammatory activation.37Fig. 1E shows the results measured by flow cytometry. The effect of AST on the production of ROS in LPS-stimulated macrophages is illustrated in Fig. 1F. Compared with the non-stimulated group (CK group), the level of ROS in THP-1 cells significantly increased after 3 h of LPS treatment, while the level of ROS in the experimental group treated with AST for 3 h (AST + LPS) showed a much lower level of ROS. O2− is a kind of ROS, which is the product of single electron reduction of oxygen molecules in cells,38,39 and it participates in many physiological activities such as cell proliferation and apoptosis and plays an important role in the redox metabolism of cells. The effect of AST on O2− in macrophages is shown in Fig. 1G. Compared with the CK group, the level of O2− in THP-1 cells after 3 h of LPS treatment increased significantly. Compared with the LPS group, the level of O2− in the experimental group treated with AST for 3 h decreased significantly. NO is a key inflammatory intermediate.40–42 Excessive NO production can lead to severe inflammatory injury or disease. In the LPS-stimulated THP-1-derived macrophages, NO was significantly increased. After 3 h of LPS treatment, compared with the CK group, the NO production was significantly increased, and the NO production in the experimental group pretreated with AST (AST + LPS group) was significantly lower than that in the LPS group (Fig. 1H). These results confirmed that AST could alleviate lipid peroxidation, enhance antioxidant enzyme activities, and resist the abnormal increase of RONS.
3.2. AST reduces the LPS-stimulated macrophage damage
One phenomenon associated with cell damage is the overload of intracellular free Ca2+.43 Therefore, to investigate the impact of AST on LPS-induced macrophage damage, we assessed the intracellular calcium levels. The results indicated that the CK group exhibited a certain level of Ca2+. In comparison, macrophages in the LPS group showed a significant increase in Ca2+ levels. The addition of AST and LPS to macrophages effectively inhibited the excessive increase in Ca2+ levels induced by macrophage overactivation (Fig. 2A). Besides, we also observed that the MMP of the LPS group was significantly lower than that of the CK group, while the MMP of the AST-pretreated group showed a significant increase (Fig. 2B).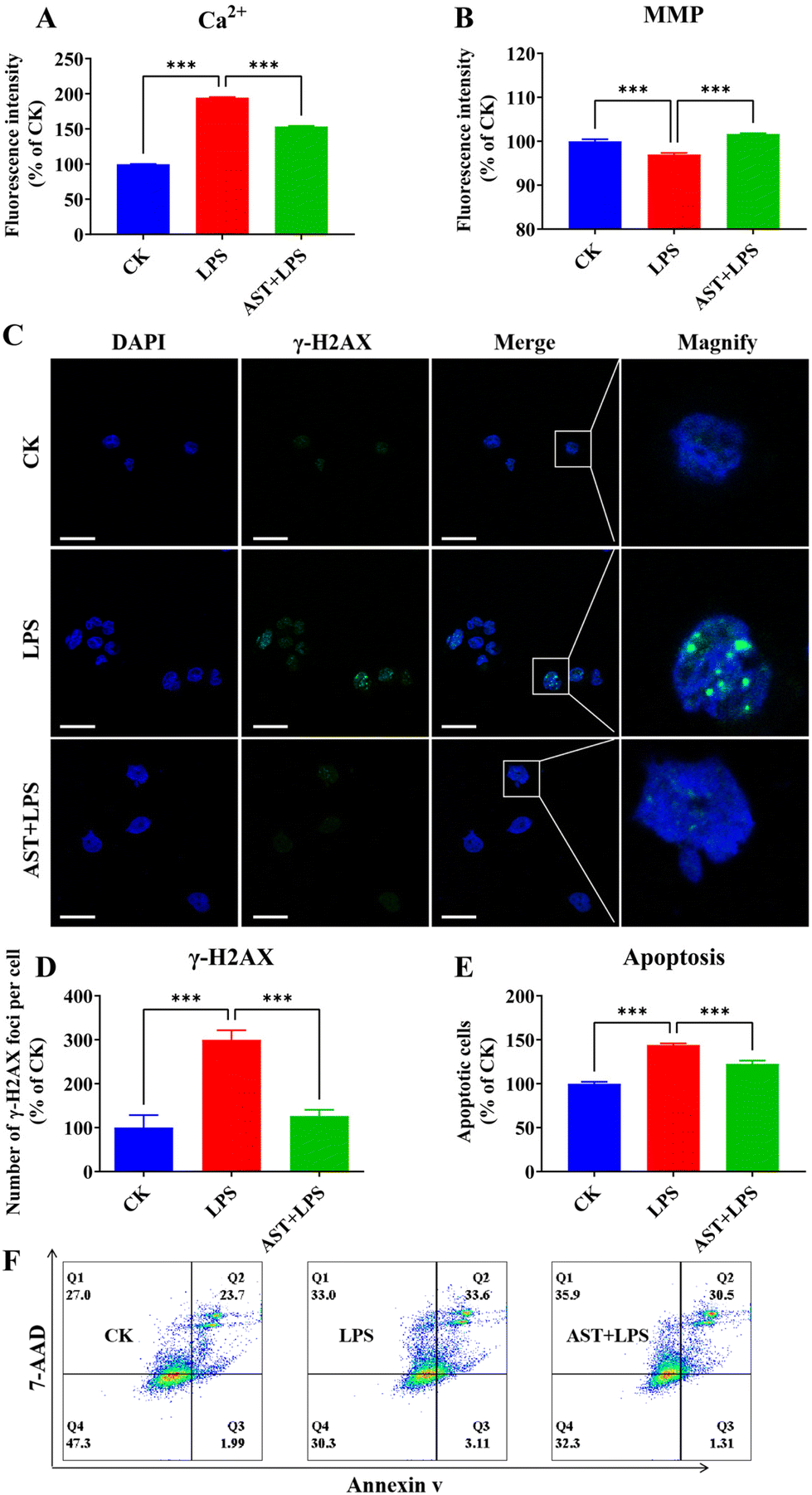 | ||
| Fig. 2 Effects of AST on LPS-stimulated macrophage damage. (A) The effect of AST on intracellular Ca2+ in LPS-stimulated macrophages. (B) The effect of AST on MMP in LPS-stimulated macrophages. (C and D) The effect of AST on DNA damage in LPS-stimulated macrophages. (E and F) The effect of AST on LPS-stimulated macrophage apoptosis. (n = 3 per group; mean ± SD; *** represents p < 0.001; the scale bar is 25 μm.) | ||
Histone γH2AX plays a crucial role in nucleosome formation, chromatin remodeling and DNA repair. γH2AX labels the site of double-strand breaks, and it recruits cell cycle checkpoints and DNA repair factors to the damage site.44 Therefore, quantification of γH2AX expression levels can be used to assess the extent of DNA damage. We found a significant increase in the number of macrophage foci after 3 h of LPS treatment. In contrast, AST reduced the number of macrophage foci significantly (Fig. 2C and D).
Apoptosis occurs when DNA damage cannot be properly repaired.45 To further study the effect of AST on the apoptosis of LPS-stimulated macrophages, the cells were divided into three groups: the CK group, the LPS group, and the AST + LPS group. The effect of AST was evaluated by the sum of early apoptosis and late apoptosis, representing the total number of apoptotic cells. When the normalized control group was set at 100%, the number of apoptotic cells in the LPS group was significantly higher than that in the CK group, indicating that LPS could cause apoptosis of cells. However, when cells were treated with AST, the number of apoptotic cells was significantly reduced, suggesting that AST effectively reduced LPS-induced apoptosis (Fig. 2E and F). This observation was consistent with the results of DNA damage. Therefore, all these results confirmed that AST reduced apoptosis by alleviating Ca2+ overload, MMP reduction and DNA damage.
3.3. AST affects inflammation-related mRNA and protein expression levels in LPS-stimulated macrophages
The main cytokines produced by macrophages include IL-1β, IL-6, IL-8, and TNF-α.46 Quantitative analysis of inflammation-related factor expression is a crucial measure reflecting the severity of inflammation. Therefore, the relative expressions of Pro-IL-1β, IL-1β, IL-6, IL-8, TNF-α, COX-2, NLRP3, and caspase-1 mRNA were assessed through qPCR to determine whether AST can inhibit the release of inflammation-related factors in LPS-stimulated THP-1-derived macrophages. As shown in Fig. 3A–H, the relative expressions of Pro-IL-1β, IL-1β, IL-6, IL-8, TNF-α, COX-2, NLRP3, and caspase-1 mRNA in the LPS group were the highest, significantly differing from those in the control group. In comparison with the LPS group, the relative expression of the inflammation-related factor mRNA in the AST + LPS group was significantly decreased, demonstrating that AST could effectively inhibit the expression of inflammation-related factors.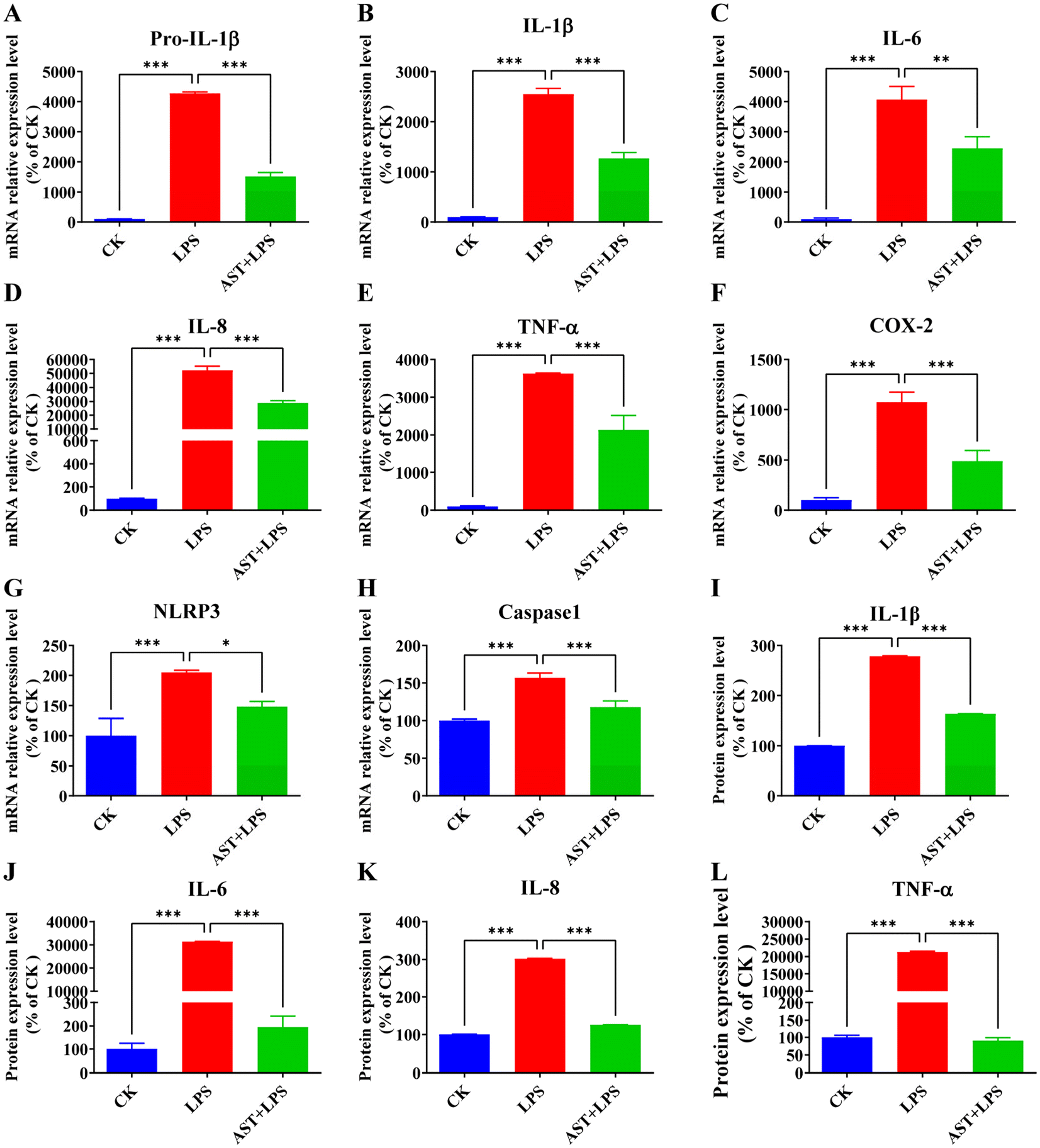 | ||
| Fig. 3 Effects of AST on inflammation-related mRNA and protein expression levels in LPS-stimulated macrophages. (A–H) The effects of AST on inflammation-related mRNA expression levels in LPS-stimulated macrophages. (I–L) The effects of AST on inflammation-related protein expression levels in LPS-stimulated macrophages. (n = 3 per group; mean ± SD; * represents p < 0.05; ** represents p < 0.01; *** represents p < 0.001.) | ||
Therefore, the aforementioned findings have demonstrated that AST effectively inhibits the expression of mRNA levels associated with the inflammation-related factors (Fig. 3A–H). Subsequently, the protein expression levels of inflammation-related factors (IL-1β, IL-6, IL-8 and TNF-α) were further detected. As shown in Fig. 3I–L, compared with the control group, the expression levels of IL-1β, IL-6, IL-8, and TNF-α in the LPS group were significantly elevated, with a notable difference. However, when AST was introduced as an intervention, the expression levels of IL-1β, IL-6, IL-8, and TNF-α were markedly lower than those in the LPS group.
3.4. AST inhibits NF-κB and reduces the production of inflammatory factors through the antioxidant mechanism
NF-κB is a key transcription factor that regulates pro-inflammatory mediators in activated macrophages.47 To detect the inhibitory effect of AST on the NF-κB signaling pathway, we examined the effect of AST on the activation of NF-κB into the nucleus in LPS-stimulated cells. As a result, we found that NF-κB in the CK group was mainly expressed in the cytoplasm, and the blue nucleus boundary was clear. The green fluorescence of the nucleus in the model group gradually increased. After merging the image, the nucleus showed obvious blue-green fluorescence, indicating that NF-κB was activated and translocated into the nucleus. The AST + LPS group showed reduced blue-green fluorescence, and the blue nucleus boundary was clear, indicating that AST could inhibit the translocation of NF-κB from the cytoplasm to the nucleus of THP-1-derived macrophages, that is, AST inhibited the activation of NF-κB, and ultimately reduced the synthesis and release of inflammatory factors, as shown in Fig. 4A and B. The impact of AST on the expression of NF-κB mRNA in LPS-stimulated THP-1 cells was also investigated. As indicated in Fig. 4C, there was a significant increase in the expression level of NF-κB mRNA after LPS treatment. However, in the AST treatment group, these levels were markedly decreased. This result confirmed that AST inhibited NF-κB and reduced the production of inflammatory factors by decreasing lipid peroxidation, enhancing the activity of antioxidant enzymes, and countering the abnormal increase in RONS.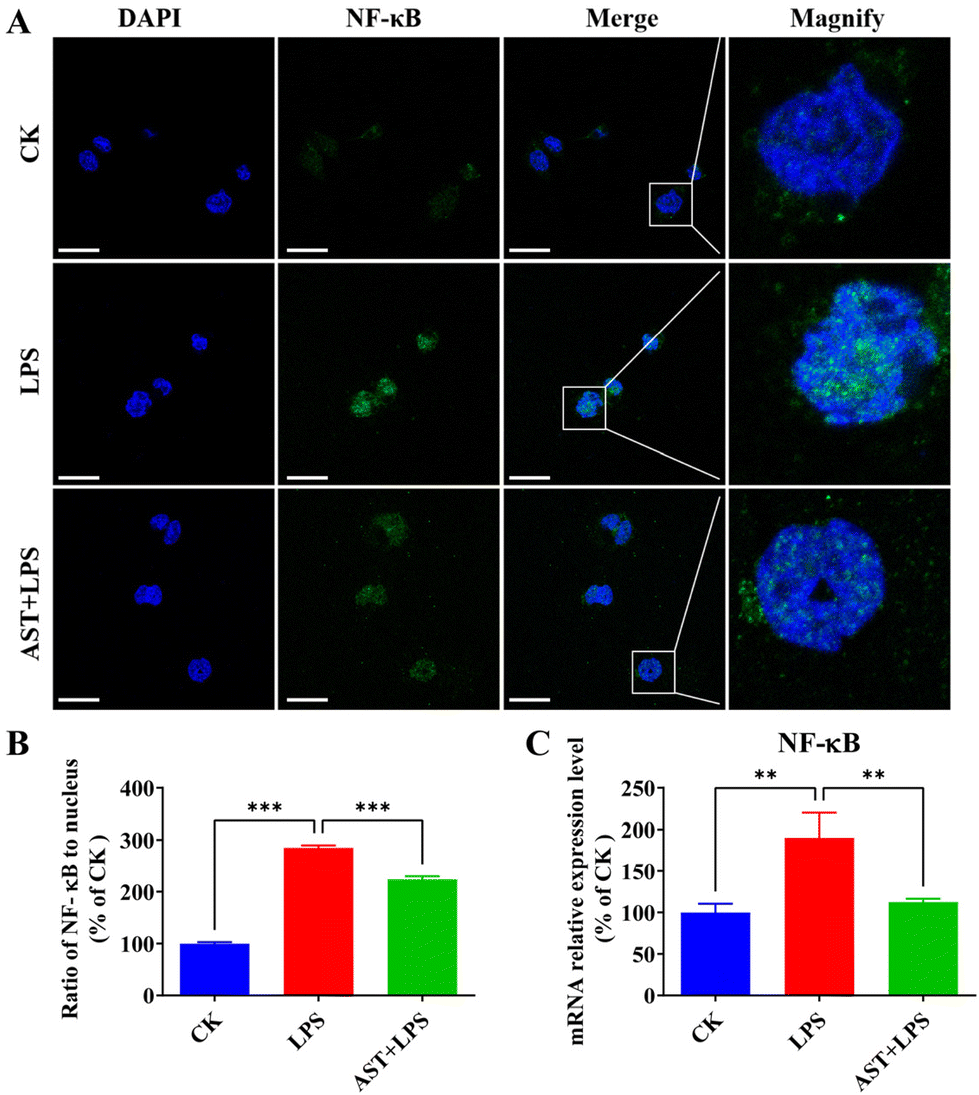 | ||
| Fig. 4 Effects of AST on NF-κB signaling in LPS-stimulated macrophages. (A) The effect of AST on NF-κB p65 nuclear translocation in LPS-stimulated macrophages. (B) Ratio of NF-κB p65 to nucleus. (C) The effect of AST on NF-κB mRNA expression level in LPS-stimulated macrophages. (n = 3 per group; mean ± SD; ** represents p < 0.01; *** represents p < 0.001, the scale bar is 25 μm.) | ||
3.5. AST suppresses inflammatory cytokine-related cell damage by activating p53 and inhibiting STAT3
It is known that up-regulation of NF-κB activity promotes the secretion of IL-6, which can indirectly activate STAT3.48,49 Also, the IL-6 transmembrane signaling pathway can induce STAT3 activation, and activated STAT3 activates the NF-κB signaling pathway, thereby regulating the expression of inflammatory cytokines and exacerbating the organism's inflammatory response.50 The expression of STAT3 could lead to a significant rise in the frequency of DNA damage and gene mutation and reduce the tolerance of cells to oxidative stress and damage.51 Therefore, we detected the expression levels of STAT3 in terms of both mRNA and protein, and investigated whether AST inhibited the activation of the STAT3 signaling pathway in LPS-stimulated THP-1-derived macrophages. As shown in Fig. 5, after LPS stimulation, the expression levels of STAT3 mRNA significantly increased. However, in the AST pretreatment group, the expression levels of STAT3 mRNA were significantly decreased. This indicates that under LPS treatment, the expression of STAT3 increased, leading to enhanced inflammatory damage in cells, while AST inhibited the expression of STAT3, thereby alleviating inflammatory damage in cells (Fig. 5A). In the meantime, we also detected the expression level of p53 mRNA to check whether AST activated p53 to enhance DNA repair in LPS-stimulated macrophages, as p53 is a transcription factor that plays a key role in regulating cell cycle progression, apoptosis in response to different stress signals, and DNA repair.52,53 The results showed a significant elevation in p53 mRNA expression after AST pretreatment in LPS-stimulated macrophages, indicating an enhanced DNA repair capacity by AST (Fig. 5B). Therefore, all these findings confirmed that AST could protect against cell damage and reduce inflammatory cytokines caused by injury by activating p53 and inhibiting STAT3.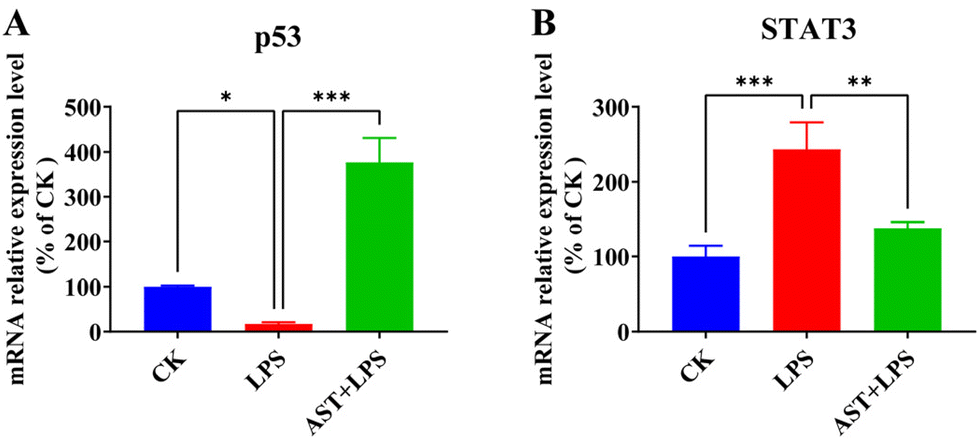 | ||
| Fig. 5 Effects of AST on p53 and STAT3 mRNA expression levels in LPS-stimulated macrophages. (A) The effect of AST on the p53 mRNA expression level in LPS-stimulated macrophages. (B) The effect of AST on the STAT3 mRNA expression level in LPS-stimulated macrophages. (n = 3 per group; mean ± SD; * represents p < 0.05; ** represents p < 0.01; *** represents p < 0.001.) | ||
3.6. Pharmacologic analysis of the AST anti-inflammatory target network
To identify the specific target of AST in the anti-inflammatory function, we initially employed network pharmacology, a very useful system biology tool for analyzing target molecules.22 AST targets and inflammatory targets were obtained through the corresponding websites, and a total of 52 intersection targets were obtained by comparing AST targets with inflammatory targets (Fig. S3†). VENNY 2.1 was used to draw the AST–inflammation intersection gene map PPI network (Fig. 6A). The aforementioned 52 potential targets were entered into the String database to construct (Fig. 6B), and the TSV format file was downloaded and imported into Cytoscape for visualization (Fig. 6C). The color and size of the nodes reflected the degree of the nodes in the network. The PPI network was topologically analyzed based on the ‘Network Analyzer’ function built into the Cytoscape platform. The node connectivity (degree value), node closeness (closeness value), and node betweenness (betweenness value) of all targets were obtained. The larger the degree value, the more important the position in the network. The core target attributes are shown in Table S2,† and it is found that the first target by degree is IL-6 (84), followed by TNF-α (82), MAPK3 (52) and PPARG (42).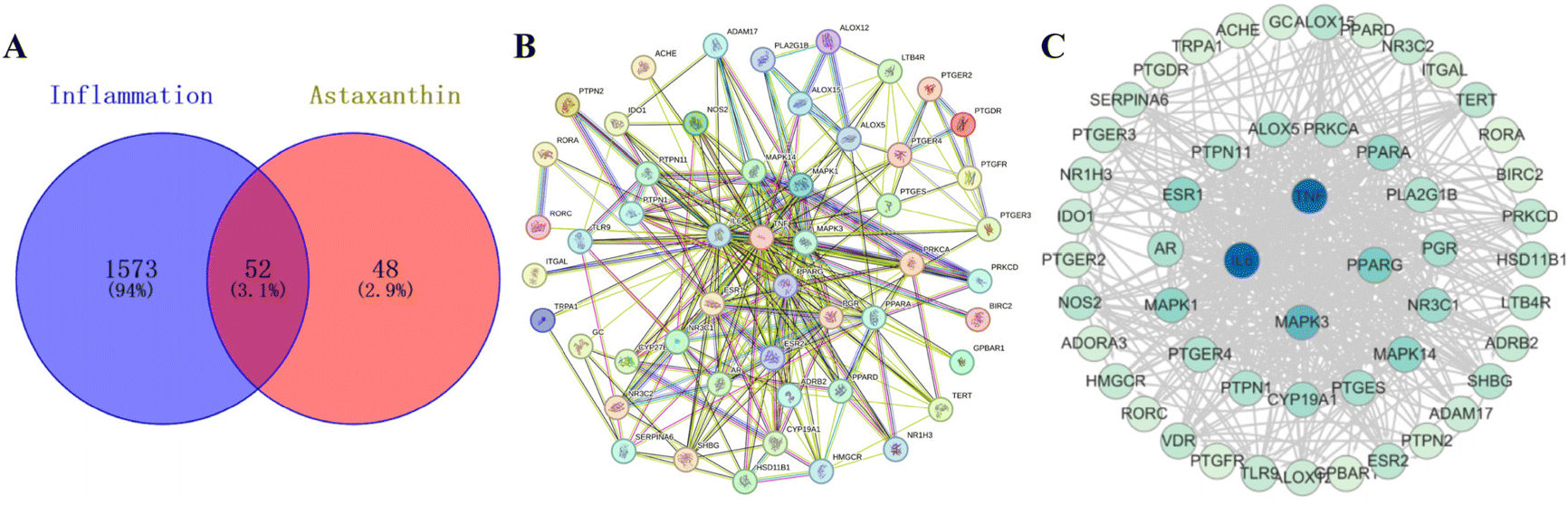 | ||
| Fig. 6 Network interaction analysis of inflammatory targets inhibited by AST. (A) Venn diagram of AST–inflammation intersection genes. (B) PPI network of AST anti-inflammatory related genes constructed using the STRING database. (C) Visualization of the Cytoscape network reconstruction of protein interaction of AST anti-inflammatory related genes. (The color of nodes is from large to small according to the degree value.) | ||
3.7. Validation of AST–IL-6 binding via molecular docking, MMGBSA analysis and molecular dynamics simulation
Molecular docking is a computational chemistry method that has become essential for the rational drug design process.54,55 Therefore, the intermolecular interaction between IL-6 and AST was also investigated using molecular docking analysis. AST interacted with residues in the binding sites of IL-6 protein with binding energies illustrated in Table S3.†Fig. 7A–F show the interaction of IL-6 with AST and the co-crystal. One residue (Phe74) participated in the hydrogen bond interaction between AST and IL-6, while two residues were observed to form the same interaction in the co-crystal. Many other amino acids in the IL-6 participated in several interactions with AST, e.g. polar, greasy acidic, and basic interactions, as shown in Fig. 7E and F.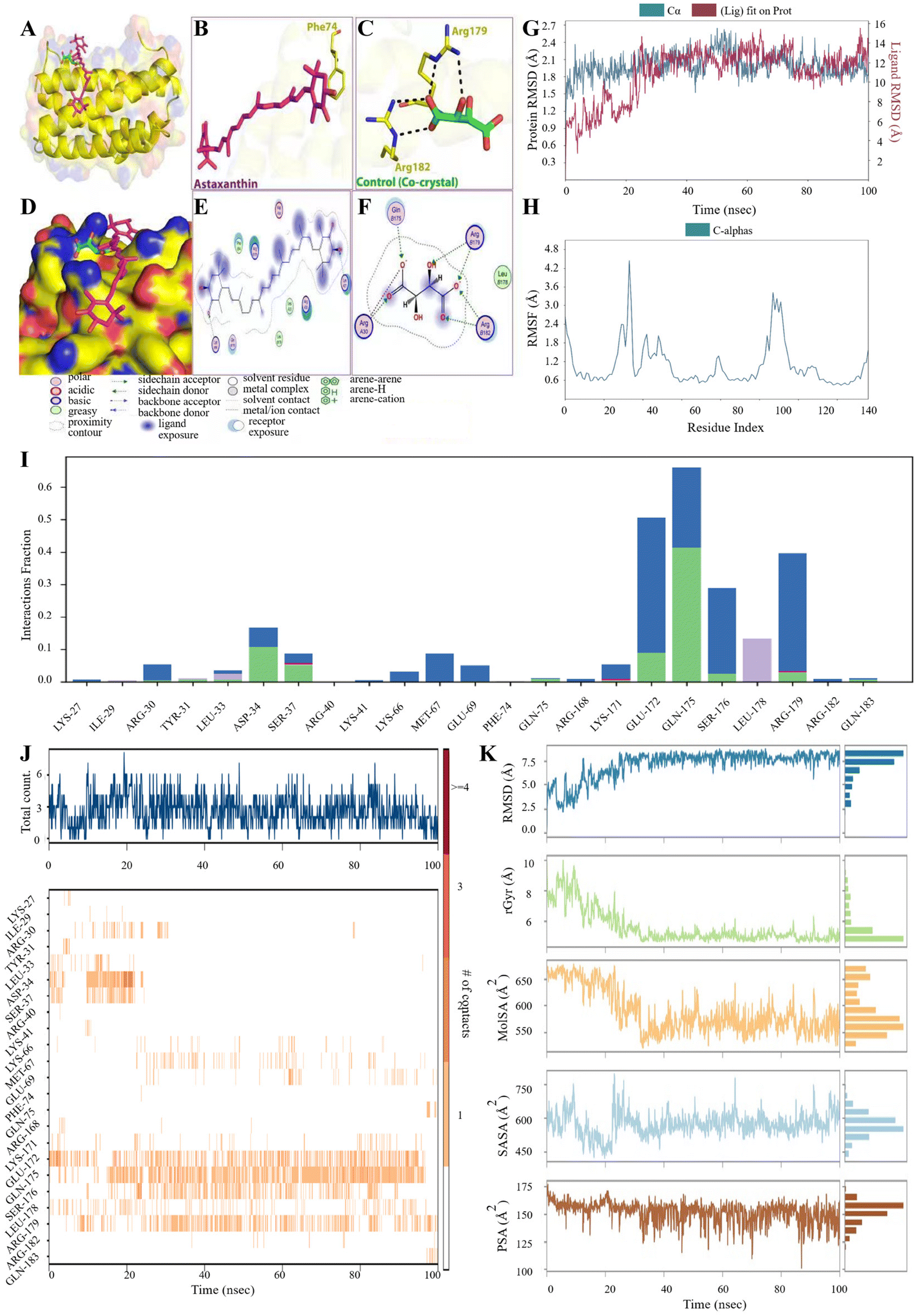 | ||
| Fig. 7 Molecular docking analysis and molecular dynamics simulation of AST–IL-6. (A–F) 2D and 3D visualization of the AST–IL-6 complex. (G and H) RMSD (left) and RMSF (right) of the AST–IL-6 interaction. (I) AST–IL-6 contact mapping shows the hydrogen bonding during the 100 ns simulation time. (J and K) AST–IL-6 contact mapping shows that many amino acids participate in the interaction of AST with IL-6 (left) and the behavior of AST inside the IL-6 pocket during the 100 ns simulation time (right). | ||
Besides, molecular dynamics (MD) simulation was utilized to uncover the stability of the AST–IL-6 complex through RMSD, RMSF, and protein–ligand contacts. Both RMSD and RMSF are important criteria to evaluate the ligand–protein stability, through a 100 ns simulation time. The stability of a protein relative to its conformation can be determined by analyzing trajectory deviations during simulations. Proteins with higher deviations are generally considered less stable.56 As shown in Fig. 7G and H, the RMSD analysis shows a stable trajectory with a fluctuation range between 1.5 and 2.4 Å until the end of the simulation time. The RMSF trajectory shows a decrease in the values, which were below 2.4 Å except in the residues between 20 and 40 and between 100 and 140, which shows a slight increase in the fluctuation. RMSD and RMSF show a stable binding of AST with IL-6. As in Fig. 7I, five residues exhibited interaction with AST. GLU172, GLN175 and Ser176 show hydrogen bonding and water bridge in the AST-IL-6 complex, while Arg179 shows ionic, hydrogen bonding and water bridge. Leu178 participated in the complex formation by hydrophobic interaction. The total number of specific contacts (top panel) and the nature of residues involved in the interactions (bottom panel) in every frame of the 100 ns trajectory are summarized for AST binding with IL-6 in Fig. 7J. The number of specific contacts reached 6 in the AST–IL-6 complex during 100 ns. More interacted residues (darker bands) were observed with IL-6 by GLU172, GLN175, SER176 and ARG179. The behavior of AST inside the binding pocket of IL-6 was observed, as shown in Fig. 7K, which was evaluated by calculating the ligand RMSD, molecular surface area (MolSA), radius of gyration (rGyr), polar surface area (PSA), and solvent accessible surface area (SASA) values. AST RMSD analysis showed a fluctuation range of 2.5–7.5 Å. The obtained rGyr values of AST were below 6 Å after 20 ns until the end of the simulation time. On the other hand, the MolSA values were around 5550–650 Å2, the obtained SASA values showed fluctuations ranging from 450 to 750 Å, while PSA values ranged from 100 to 175 Å2 and for AST in IL-6.
3.8. IL-6 interference abolishes the AST–IL-6 binding and affects NF-κB and STAT3
To verify whether IL-6 is the target of AST in cells, IL-6 interference was conducted on the macrophages. The cells were divided into four groups: the LPS (NC) group, the AST + LPS (NC) group, the LPS (si-IL-6) group, and the AST + LPS (si-IL-6) group. After treating the cells accordingly, cell samples were collected and the expression of the IL-6 mRNA level was assessed. The result, as shown in Fig. 8A, indicates that when the normalized LPS (NC) group was set at 100%, the expression of IL-6 in the AST + LPS (NC) group significantly decreased, demonstrating that AST could inhibit the expression of IL-6. The IL-6 in the LPS (si-IL-6) group also significantly decreased, confirming the success of the interference experiment. In fact, no significant difference in IL-6 expression was observed between the AST + LPS (si-IL-6) group and the LPS (si-IL-6) group, indicating that AST could not reduce IL-6 expression after IL-6 interference, thereby proving that IL-6 is the target of AST.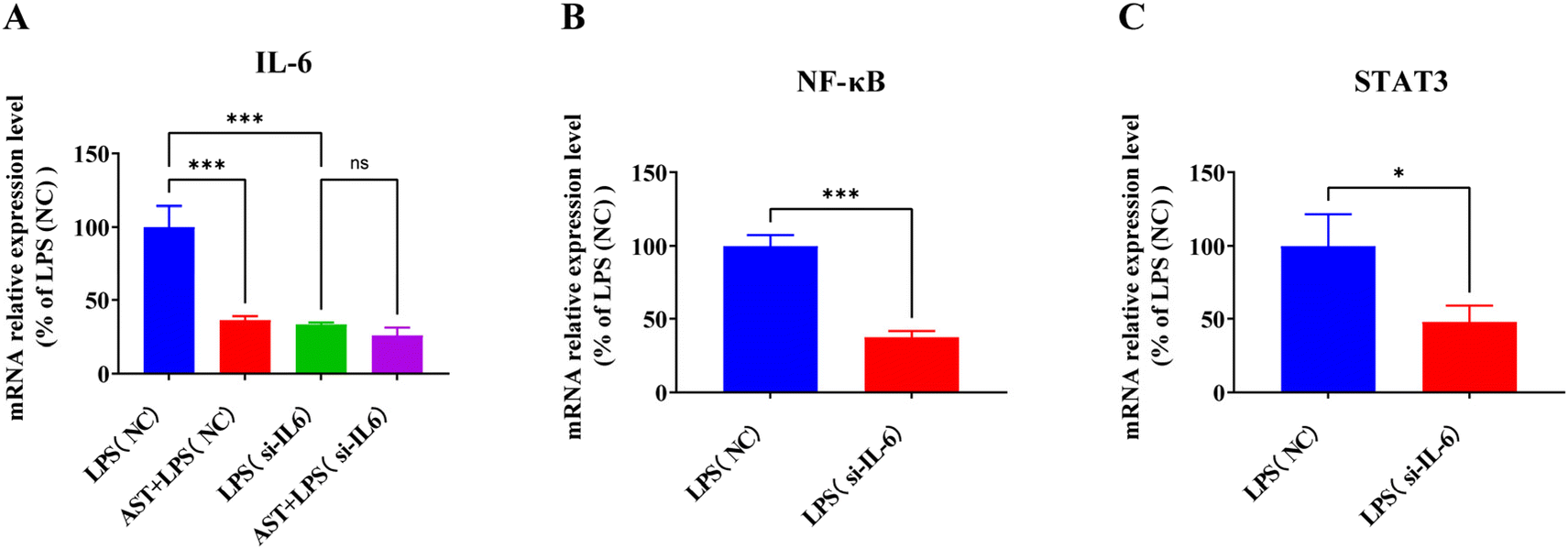 | ||
| Fig. 8 The expression of IL-6, NF-κB and STAT3 mRNA levels after the interference of IL-6 siRNA. (n = 3 per group; mean ± SD; * represents p < 0.05; *** represents p < 0.001; ns represent no significant difference compared with the control group.) | ||
IL-6 is one of the cytokines along with others, e.g. TNF-α, IL-1β and IL-8, and it is regulated by NF-κB and plays a critical role in the immune response.57 As a pro-inflammatory cytokine, IL-6 preferentially activates STAT3-dependent gene expression.58 Activation of STAT3 by IL-6 plays a crucial role in inflammation-induced disease pathogenesis.59–61 To confirm the correlation between IL-6 and NF-κB and STAT3, we also examined the expression levels of NF-κB and STAT3 in macrophages after IL-6 interference. As presented in Fig. 8B and C, the expressions of NF-κB and STAT3 in the LPS (si-IL-6) group were significantly decreased compared with the LPS (NC) group, indicating that interference with IL-6 could affect the expression levels of NF-κB and STAT3 in LPS-stimulated macrophages.
4. Discussion
Many experimental studies have shown that AST has many biological activities,62–65 such as antioxidant activity,66–69 anti-inflammatory activity,70–73 immunity enhancing activity,74,75 neuroprotective activity,76 liver protection activity,77 anti-diabetic activity,78–80 and anti-cancer activity.81,82 Recently, researchers noticed that AST could strongly inhibit LPS-induced immune dysfunction of dendritic cells,83 and even demonstrated the antioxidant and anti-inflammatory effects of AST on dendritic cells and mice.84 Nevertheless, very limited research had been conducted on the anti-inflammatory mechanism of AST. Thus, the major aim of this study was to clarify the anti-inflammatory mechanism of AST.To explore the mechanism, firstly, we examined the anti-oxidative effect of AST, because it is well known that one of the important anti-inflammatory mechanisms is closely connected to anti-oxidative stress. Oxidative stress interferes with the dynamic balance between pro-inflammatory factors and anti-inflammatory factors,85–88 and ultimately produces an enhanced inflammatory response. In particular, immune cells are easily susceptible to oxidative stress due to the prevalence of polyunsaturated fatty acids in the plasma membrane.89 In our experiments, we observed a decrease in antioxidant enzyme activity after LPS treatment, as well as a significant increase when AST was added. Dong et al. also found that pilose antler peptide attenuated the increase of MDA and the decrease of SOD induced by LPS challenge.90 ROS, such as O2− and the hydroxyl radical, are produced in cells due to aerobic oxidation of cellular respiration and energy metabolism in human and animal bodies. When cells are stimulated by the internal and external environment, an increase of ROS may destroy the balance between the organism's oxidation and antioxidant systems, interfere with intracellular redox homeostasis, and ultimately lead to oxidative stress. Obviously, our results clearly showed that AST could remarkably alleviate LPS-stimulated ROS in macrophages.
Secondly, because oxidative stress can cause damage to all types of macromolecules (including DNA molecules), thereby impairing their normal function,91 we then examined whether AST could protect the cell against oxidative damage. For the cell damage, we observed that LPS caused not only cell fates such as cell death including apoptosis, but also damage to proteins and DNA. Oxidative stress can also lead to Ca2+ overload and decreased MMP, which eventually leads to apoptosis.92 Our experimental results confirmed that AST could effectively alleviate the damage from the oxidative stress by LPS.
But how does AST protect the cells against oxidative stress by LPS? It is therefore necessary to explore the protection effect and the involved mechanism. Since the oxidative stress may stem from LPS-stimulated inflammatory response-induced DNA damage, we examined p53 and STAT3. The p53 tumor suppressor plays an important role in regulating cell survival and death.93 When DNA damage occurs, cells undergo normal repair, at which point p53 expression increases. STAT3 is an important transcription factor that regulates a series of genes related to cell proliferation, survival, stress response, and immune function.94,95 When cells undergo abnormal repair, cell deterioration will occur, and in this case, STAT3 expression will be increased. Indeed, we found that AST increased the expression of p53, while decreasing the expression of STAT3, confirming the role of AST in the protection of oxidative stress-induced DNA damage.
On the other hand, oxidative stress induces the elevation of inflammatory factors, and it is necessary to examine the related signal pathways. Oxidative stress plays an important role in the process of inflammation,96,97 which can activate a variety of transcription factors, leading to the activation of some inflammatory pathways, such as the NF-κB signaling pathway. NF-κB is a nuclear factor that regulates gene transcription in the process of inflammation and immunity. The activation of NF-κB triggers the expression of a variety of inflammatory and adhesion factors, which directly leads to the inflammatory response.46 Indeed, we found that AST inhibited the NF-κB signaling pathway through an antioxidant mechanism, confirming its role in exerting an anti-inflammatory effect on oxidative stress.
Nevertheless, it is still necessary to answer the question whether AST plays a role in the inhibition of unfavorable inflammatory factors. Macrophages as the central cells in the inflammatory response fulfill crucial roles in phagocytosis, secretion, and antigen presentation. Upon activation by LPS, they can generate a substantial number of inflammatory cytokines.98 These cytokines establish intricate relationships, either inhibiting or promoting each other, thereby forming a complex cytokine regulatory network. Therefore, when assessing the extent of inflammatory response in the body, it is essential to consider the secretion of inflammatory factors such as TNF-α, IL-1β, IL-8, and IL-6 in cells, as well as the expression of their genes, which serve as important reference indicators for evaluating the degree of inflammatory response injury.99 In fact, as demonstrated in this study, AST notably inhibits the expressions of these inflammatory factors in LPS-stimulated macrophages.
It is therefore important to clarify which critical inflammatory factor AST could directly interact with, so that AST could most efficiently suppress the unfavorable inflammatory response. To address this, we employed multiple tools and methods including network pharmacology, in silico computation, and RNA interference for validation. As an emerging discipline, network pharmacology was first systematically expounded by Hopkins, a pharmacology expert at the University of Dundee in the UK.100 Based on the structural and efficacy similarities between drugs, network pharmacology applies systems biology technology and database resources to analyze target molecules, construct related networks, predict drug targets and guide new drug discovery. Wu et al. applied network pharmacology to identify potential core targets and possible signal pathways of the anti-inflammatory effects of β-carotene, including IL-6,101 but did not perform experimental validation. Guo et al. also found that TNF-α, IL-6 and IL-1β were the core target proteins of Er Miao San (EMS) for the treatment of inflammation, and suggested that EMS mainly exerted its anti-inflammatory effects by inhibiting the multiple pro-inflammatory cytokines.102 In our analysis, strikingly, we found that AST itself could interact with certain inflammatory factors, especially IL-6 (the highest degree is 84) (Table S2†). Furthermore, to improve the accuracy of the prediction results of the study, we also applied molecular docking, molecular dynamics simulation, and RNA interference experiments.
Certainly, AST has other molecular targets, for example, TNF-α. As indicated by the pharmacology network analysis, TNF-α is indeed also a core target of AST, as confirmed also by our in silico calculations based on molecular docking, molecular dynamics simulation (Fig. S4†). However, in this study, we focused on the interaction between AST and IL-6 because we found that IL-6 could cause adverse inflammatory response of macrophages. Different from IL-6, TNF-α did not critically affect the regulation of NF-κB and STAT3, as confirmed by the TNF-α interference experiment (Fig. S5†), in which no significant difference in the expression of NF-κB and STAT3 was observed between the LPS (si-TNF-α) group and the LPS (NC) group.
Moreover, we also confirmed that IL-6 could suppress the LPS-induced adverse inflammatory response in both primary peripheral blood mononuclear cells (PBMCs) and zebrafish (as the animal model) (data not shown). All these results indicate that AST can bind to certain inflammatory factors, particularly IL-6, thereby inhibiting the positive feedback of inflammatory factors and preventing the occurrence of inflammatory storms. The involved mechanisms are summarized and schematically depicted in Fig. 9.
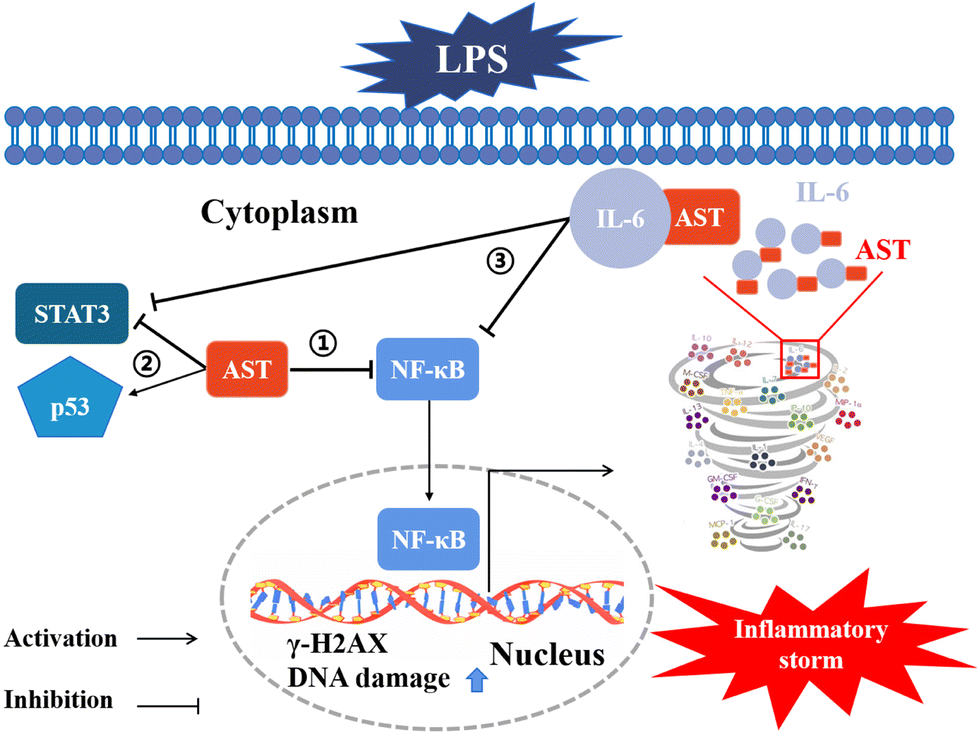 | ||
| Fig. 9 Schematic diagram of the anti-inflammatory effects of AST. ① AST inhibits NF-κB and reduces the production of inflammatory factors through antioxidant mechanisms. ② AST protects against cell damage and reduces inflammatory cytokines caused by injury by activating p53 and inhibiting STAT3. ③ AST binds to IL-6 to inhibit the positive feedback loop of inflammatory factors and prevent the occurrence of inflammatory storms. | ||
5. Conclusions
In summary, our findings indicate that AST exhibits anti-oxidative stress, anti-cell damage, and anti-inflammatory effects on LPS-stimulated macrophages, and AST protects against cell damage, stimulates p53, inhibits STAT3, and mitigates inflammatory factors resulting from damage. We have also confirmed that AST inhibits NF-κB, reducing the production of inflammatory factors through antioxidant mechanisms, and in particular, we have revealed that IL-6 is the key core target for AST's anti-inflammatory action. Therefore, we propose that AST can effectively inhibit the positive feedback loop of inflammatory factors and prevent the occurrence of inflammatory storms.Author contributions
Yahui Wu: data curation, formal analysis, investigation, methodology, and writing – original draft. Mona A. Bashir and Changsheng Shao: data curation and formal analysis. Han Wang and Jianxia Zhu: investigation. Qing Huang: conceptualization, formal analysis, investigation, methodology, supervision, and writing – review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work also was partly supported by the National Nature Science Foundation of China (No. 11635013 and 11775272).References
- M. White, Mediators of inflammation and the inflammatory process, J. Allergy Clin. Immunol., 1999, 103, S378–S381 CrossRef CAS PubMed.
- D. Maslinska and M. Gajewski, Some aspects of the inflammatory process, Folia Neuropathol., 1998, 36, 199–204 CAS.
- D. L. Laskin and K. J. Pendino, Macrophages and inflammatory mediators in tissue-injury, Annu. Rev. Pharmacol., 1995, 35, 655–677 CrossRef CAS PubMed.
- K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas and D. N. Nicolaides, Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities, Curr. Pharm. Des., 2004, 10, 3813–3833 CrossRef CAS PubMed.
- M. Mueller, S. Hobiger and A. Jungbauer, Anti-inflammatory activity of extracts from fruits, herbs and spices, Food Chem., 2010, 122, 987–996 CrossRef CAS.
- J. S. Park, B. P. Chew and T. S. Wong, Dietary lutein from marigold extract inhibits mammary tumor development in BALB/c mice, J. Nutr., 1998, 128, 1650–1656 CrossRef CAS PubMed.
- N. Misawa, Y. Satomi, K. Kondo, A. Yokoyama, S. Kajiwara, T. Saito, T. Ohtani and W. Miki, Structure and functional-analysis of a marine bacterial carotenoid biosynthesis gene-cluster and astaxanthin biosynthetic-pathway proposed at the gene level, J. Bacteriol., 1995, 177, 6575–6584 CrossRef CAS PubMed.
- X. X. Zheng and Q. Huang, Assessment of the antioxidant activities of representative optical and geometric isomers of astaxanthin against singlet oxygen in solution by a spectroscopic approach, Food Chem., 2022, 395, 133584 CrossRef CAS PubMed.
- Y. M. A. Naguib, Antioxidant activities of astaxanthin and related carotenoids, J. Agric. Food Chem., 2000, 48, 1150–1154 CrossRef CAS PubMed.
- H. Jyonouchi, S. Sun, K. Iijima and M. D. Gross, Antitumor activity of astaxanthin and its mode of action, Nutr. Cancer, 2000, 36, 59–65 CrossRef CAS PubMed.
- G. Scapagnini, S. Davinelli, L. Di Renzo, A. De Lorenzo, H. H. Olarte, G. Micali, A. F. Cicero and S. Gonzalez, Cocoa bioactive compounds: Significance and potential for the maintenance of skin health, Nutrients, 2014, 6, 3202–3213 CrossRef CAS PubMed.
- D. Bar-Or, R. Bar-Or, L. T. Rael and E. N. Brody, Oxidative stress in severe acute illness, Redox Biol., 2015, 4, 340–345 CrossRef CAS PubMed.
- H. Kang and H. Kim, Astaxanthin and β-carotene in Helicobacter pylori-induced gastric inflammation: A mini-review on action mechanisms, J. Cancer Prev., 2017, 22, 57–61 CrossRef PubMed.
- T. Z. Liu, K. T. Lee, C. L. Chern, J. T. Cheng, A. Stern and L. Y. Tsai, Free radical-triggered hepatic injury of experimental obstructive jaundice of rats involves overproduction of proinflammatory cytokines and enhanced activation of nuclear factor kappaB, Ann. Clin. Lab. Sci., 2001, 31, 383–390 CAS.
- M. Kurashige, E. Okimasu, M. Inoue and K. Utsumi, Inhibition of oxidative injury of biological membranes by astaxanthin, Physiol. Chem. Phys. Med. NMR, 1990, 22, 27–38 CAS.
- D. R. Brown, L. A. Gough, S. K. Deb, S. A. Sparks and L. R. McNaughton, Astaxanthin in exercise metabolism, performance and recovery: A review, Front. Nutr., 2018, 4, 76 CrossRef PubMed.
- C. Farruggia, M. B. Kim, M. Bae, Y. Lee, T. X. Pham, Y. Yang, M. J. Han, Y. K. Park and J. Y. Lee, Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners, J. Nutr. Biochem., 2018, 62, 202–209 CrossRef CAS PubMed.
- P. R. Augusti, A. Quatrin, S. Somacal, G. M. Conterato, R. Sobieski, A. R. Ruviaro, L. H. Maurer, M. M. Duarte, M. Roehrs and T. Emanuelli, Astaxanthin prevents changes in the activities of thioredoxin reductase and paraoxonase in hypercholesterolemic rabbits, J. Clin. Biochem. Nutr., 2012, 51, 42–49 CrossRef CAS PubMed.
- M. Guha and N. Mackman, LPS induction of gene expression in human monocytes, Cell Signal., 2001, 13, 85–94 CrossRef CAS PubMed.
- G. Moalem and D. J. Tracey, Immune and inflammatory mechanisms in neuropathic pain, Brain Res. Rev., 2006, 51, 240–264 CrossRef CAS PubMed.
- J. Zou, D. Feng, W. H. Ling and R. D. Duan, Lycopene suppresses proinflammatory response in lipopolysaccharide-stimulated macrophages by inhibiting ROS-induced trafficking of TLR4 to lipid raft-like domains, J. Nutr. Biochem., 2013, 24, 1117–1122 CrossRef CAS PubMed.
- T. T. Luo, Y. Lu, S. K. Yan, X. Xiao, X. L. Rong and J. Guo, Network pharmacology in research of Chinese medicine formula: Methodology, application and prospective, Chin. J. Integr. Med., 2020, 26, 72–80 CrossRef CAS PubMed.
- R. Z. Zhang, X. Zhu, H. Bai and K. Ning, Network pharmacology databases for traditional Chinese medicine: Review and assessment, Front. Pharmacol., 2019, 10, 123 CrossRef CAS PubMed.
- Q. L. Li, T. J. Chen, Y. L. Wang and S. H. Bryant, PubChem as a public resource for drug discovery, Drug Discovery Today, 2010, 15, 1052–1057 CrossRef CAS PubMed.
- V. Zoete, A. Daina, C. Bovigny and O. Michielin, SwissSimilarity: A web tool for low to ultra high throughput ligand-based virtual screening, J. Chem. Inf. Model., 2016, 56, 1399–1404 CrossRef CAS PubMed.
- M. Safran, I. Solomon, O. Shmueli, M. Lapidot, S. Shen-Orr, A. Adato, U. Ben-Dor, N. Esterman, N. Rosen, I. Peter, T. Olender, V. Chalifa-Caspi and D. Lancet, GeneCards™ 2002: Towards a complete, object-oriented, human gene compendium, Bioinformatics, 2002, 18, 1542–1543 CrossRef CAS PubMed.
- P. Bardou, J. Mariette, F. Escudié, C. Djemiel and C. Klopp, jvenn: An interactive Venn diagram viewer, BMC Bioinf., 2014, 15, 293 CrossRef PubMed.
- C. J. Zhang, Y. X. Zheng, X. Li, X. Hu, F. Qi and J. Luo, Genome-wide mutation profiling and related risk signature for prognosis of papillary renal cell carcinoma, Ann. Transl. Med., 2019, 7, 427 CrossRef CAS PubMed.
- C. T. Lopes, M. Franz, F. Kazi, S. L. Donaldson, Q. Morris and G. D. Bader, Cytoscape Web: An interactive web-based network browser, Bioinformatics, 2010, 26, 2347–2348 CrossRef CAS PubMed.
- S. Kiliç, M. Andaç and A. Denizli, Binding modes of cibacron blue with albumin in affinity chromatography using docking tools, Int. J. Biol. Macromol., 2021, 183, 110–118 CrossRef PubMed.
- K. J. Bowers, E. Chow., H. Xu., R. O. Dror., M. P. Eastwood., B. A. Gregersen., J. L. Klepeis., I. Kolossvary., M. A. Moraes., F. D. Sacerdoti., J. K. Salmon., Y. Shan and D. E. Shaw, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Association for Computing Machinery, New York, NY, USA, 2006, pp. 84–es, DOI:10.1145/1188455.1188544.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, Molecular-dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. A. Darden, D. M. York and L. G. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. Abdalla, W. A. Eltayb, A. A. El-Arabey, K. Singh and X. Y. Jiang, Molecular dynamic study of SARS-CoV-2 with various S protein mutations and their effect on thermodynamic properties, Comput. Biol. Med., 2022, 141, 105025 CrossRef CAS PubMed.
- M. Rudrapal, W. A. Eltayeb, G. Rakshit, A. A. El-Arabey, J. Khan, S. M. Aldosari, B. Alshehri and M. Abdalla, Dual synergistic inhibition of COX and LOX by potential chemicals from Indian daily spices investigated through detailed computational studies, Sci. Rep., 2023, 13, 8656 CrossRef CAS PubMed.
- W. M. Zhao, L. Ma, C. Cai and X. H. Gong, Caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-kappa B and A2aR signaling in LPS-induced THP-1 macrophages, Int. J. Biol. Sci., 2019, 15, 1571–1581 CrossRef CAS PubMed.
- X. J. Han, T. S. Xu, Q. J. Fang, H. J. Zhang, L. J. Yue, G. Hu and L. Y. Sun, Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy, Redox Biol., 2021, 44, 102010 CrossRef CAS PubMed.
- Z. S. Jie, J. Liu, M. C. Shu, Y. Ying and H. F. Yang, Detection strategies for superoxide anion: A review, Talanta, 2022, 236, 122892 CrossRef CAS PubMed.
- M. Togo, N. Konari, M. Tsukamoto, R. Kimoto, T. Yamaguchi, H. Takeda and I. Kambayashi, Effects of a high-fat diet on superoxide anion generation and membrane fluidity in liver mitochondria in rats, Int. Soc. Sports Nutr., 2018, 15, 13 CrossRef CAS PubMed.
- G. Nagy, J. M. Clark, E. I. Buzas, C. L. Gorman and A. P. Cope, Nitric oxide, chronic inflammation and autoimmunity, Immunol. Lett., 2007, 111, 1–5 CrossRef CAS PubMed.
- K. Maurent, C. Vanucci-Bacque, M. Baltas, A. Negre-Salvayre, N. Auge and F. Bedos-Belval, Synthesis and biological evaluation of diarylheptanoids as potential antioxidant and anti-inflammatory agents, Eur. J. Med. Chem., 2018, 144, 289–299 CrossRef CAS PubMed.
- F. Aktan, iNOS-mediated nitric oxide production and its regulation, Life Sci., 2004, 75, 639–653 CrossRef CAS PubMed.
- C. M. Wang, S. Q. Qi, C. J. Liu, A. X. Yang, W. J. Fu, C. Quan, P. Duan, T. T. Yu and K. D. Yang, Mitochondrial dysfunction and Ca2+ overload in injured Sertoli cells exposed to bisphenol A, Environ. Toxicol., 2017, 32, 823–831 CrossRef CAS PubMed.
- J. Maeda, E. J. Roybal, C. A. Brents, M. Uesaka, Y. Aizawa and T. A. Kato, Natural and glucosyl flavonoids inhibit poly(ADP-ribose) polymerase activity and induce synthetic lethality in BRCA mutant cells, Oncol. Rep., 2014, 31, 551–556 CrossRef CAS PubMed.
- S. J. Sudharshan and M. Dyavaiah, Astaxanthin protects oxidative stress mediated DNA damage and enhances longevity in Saccharomyces cerevisiae, Biogerontology, 2021, 22, 81–100 CrossRef CAS PubMed.
- C. Feng, Y. M. Luo, Y. Y. Nian, D. Liu, X. R. Yin, J. Wu, J. Di, R. Zhang and J. Zhang, Diallyl disulfide suppresses the inflammation and apoptosis resistance induced by DCA through ROS and the NF-κB signaling pathway in human Barrett's epithelial cells, Inflammation, 2017, 40, 818–831 CrossRef CAS PubMed.
- C. Espinosa-Diez, V. Miguel, D. Mennerich, T. Kietzmann, P. Sánchez-Pérez, S. Cadenas and S. Lamas, Antioxidant responses and cellular adjustments to oxidative stress, Redox Biol., 2015, 6, 183–197 CrossRef CAS PubMed.
- M. Sironi, F. Breviario, P. Proserpio, A. Biondi, A. Vecchi, J. Vandamme, E. Dejana and A. Mantovani, Il-1 stimulates Il-6 production in endothelial-cells, J. Immunol., 1989, 142, 549–553 CrossRef CAS.
- G. Kaplanski, N. Teysseire, C. Farnarier, S. Kaplanski, J. C. Lissitzky, J. M. Durand, J. Soubeyrand, C. A. Dinarello and P. Bongrand, IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway, J. Clin. Invest., 1995, 96, 2839–2844 CrossRef CAS PubMed.
- S. L. Kim, M. W. Shin, S. Y. Seo and S. W. Kim, Lipocalin 2 potentially contributes to tumorigenesis from colitis via IL-6/STAT3/NF-kappaB signaling pathway, Biosci. Rep., 2022, 42, BSR20212418 CrossRef CAS PubMed.
- F. Colotta, P. Allavena, A. Sica, C. Garlanda and A. Mantovani, Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability, Carcinogenesis, 2009, 30, 1073–1081 CrossRef CAS PubMed.
- S. E. Kern, K. W. Kinzler, A. Bruskin, D. Jarosz, P. Friedman, C. Prives and B. Vogelstein, Identification of P53 as a sequence-specific DNA-binding protein, Science, 1991, 252, 1708–1711 CrossRef CAS PubMed.
- S. L. Harris and A. J. Levine, The p53 pathway: positive and negative feedback loops, Oncogene, 2005, 24, 2899–2908 CrossRef CAS PubMed.
- D. Ramírez and J. Caballero, Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target?, Int. J. Mol. Sci., 2016, 17, 525 CrossRef PubMed.
- E. Kellenberger, J. Rodrigo, P. Muller and D. Rognan, Comparative evaluation of eight docking tools for docking and virtual screening accuracy, Proteins, 2004, 57, 225–242 CrossRef CAS PubMed.
- Y. Li, Y. Li, C. Ning, J. Yue, C. Zhang, X. He, Y. Wang and Z. Liu, Discovering inhibitors of TEAD palmitate binding pocket through virtual screening and molecular dynamics simulation, Comput. Biol. Chem., 2022, 98, 107648 CrossRef CAS PubMed.
- B. Hoesel and J. A. Schmid, The complexity of NF-kappaB signaling in inflammation and cancer, Mol. Cancer, 2013, 12, 86 CrossRef CAS PubMed.
- N. Somensi, T. K. Rabelo, A. G. Guimaraes, L. J. Quintans-Junior, A. A. de Souza Araujo, J. C. F. Moreira and D. P. Gelain, Carvacrol suppresses LPS-induced pro-inflammatory activation in RAW 264.7 macrophages through ERK1/2 and NF-kB pathway, Int. Immunopharmacol., 2019, 75, 105743 CrossRef CAS PubMed.
- S. Matsumoto, T. Hara, K. Mitsuyama, M. Yamamoto, O. Tsuruta, M. Sata, J. Scheller, S. Rose-John, S. Kado and T. Takada, Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model, J. Immunol., 2010, 184, 1543–1551 CrossRef CAS PubMed.
- S. A. Jones and S. Rose-John, The role of soluble receptors in cytokine biology: The agonistic properties of the sIL-6R/IL-6 complex, Biochim. Biophys. Acta, 2002, 1592, 251–263 CrossRef CAS PubMed.
- S. Rose-John, J. Scheller, G. Elson and S. A. Jones, Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer, J. Leukoc. Biol., 2006, 80, 227–236 CrossRef CAS PubMed.
- W. Chew, B. Mathison, L. Kimble, P. F. Mixter and B. P. Chew, Astaxanthin decreases inflammatory biomarkers associated with cardiovascular disease in human umbilical vein endothelial cells, Am. J. Adv. Food Sci. Technol., 2013, 27, 1–14 Search PubMed.
- J. S. Park, J. H. Chyun, Y. K. Kim, L. L. Line and B. P. Chew, Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans, Nutr. Metab., 2010, 7, 18 CrossRef PubMed.
- M. Ikeuchi, T. Koyama, J. Takahashi and K. Yazawa, Effects of astaxanthin supplementation on exercise-induced fatigue in mice, Biol. Pharm. Bull., 2006, 29, 2106–2110 CrossRef CAS PubMed.
- F. J. Pashkow, D. G. Watumull and C. L. Campbell, Astaxanthin: A novel potential treatment for oxidative stress and inflammation in cardiovascular disease, Am. J. Cardiol., 2008, 101, 58d–68d CrossRef CAS PubMed.
- A. Ranga Rao, R. L. Raghunath Reddy, V. Baskaran, R. Sarada and G. A. Ravishankar, Characterization of microalgal carotenoids by mass spectrometry and their bioavailability and antioxidant properties elucidated in rat model, J. Agric. Food Chem., 2010, 58, 8553–8559 CrossRef CAS PubMed.
- A. R. Rao, H. N. Sindhuja, S. M. Dharmesh, K. U. Sankar, R. Sarada and G. A. Ravishankar, Effective inhibition of skin cancer, tyrosinase, and antioxidative properties by astaxanthin and astaxanthin esters from the green alga Haematococcus pluvialis, J. Agric. Food Chem., 2013, 61, 3842–3851 CrossRef CAS PubMed.
- A. R. Rao, V. Baskaran, R. Sarada and G. A. Ravishankar, In vivo bioavailability and antioxidant activity of carotenoids from microalgal biomass – A repeated dose study, Food Res. Int., 2013, 54, 711–717 CrossRef.
- Y. Yang, J. Min, A. Nguyen, T. X. Pham, H. J. Park, Y. Park, B. Kim, R. S. Bruno and J. Lee, Astaxanthin-rich extract from the green alga lowers plasma lipid concentrations and enhances antioxidant defense in apolipoprotein E knockout mice, J. Nutr., 2011, 141, 1611–1617 CrossRef CAS PubMed.
- S. Bhuvaneswari, B. Yogalakshmi, S. Sreeja and C. V. Anuradha, Astaxanthin reduces hepatic endoplasmic reticulum stress and nuclear factor-kappa B-mediated inflammation in high fructose and high fat diet-fed mice, Cell Stress Chaperones, 2014, 19, 183–191 CrossRef CAS PubMed.
- J. S. Park, B. D. Mathison, M. G. Hayek, J. Zhang, G. A. Reinhart and B. P. Chew, Astaxanthin modulates age-associated mitochondrial dysfunction in healthy dogs, J. Anim. Sci., 2013, 91, 268–275 CrossRef CAS PubMed.
- A. F. Gal, S. Andrei, C. Cernea, M. Taulescu and C. Catoi, Effects of astaxanthin supplementation on chemically induced tumorigenesis in Wistar rats, Acta Vet. Scand., 2012, 54, 50 CrossRef CAS PubMed.
- K. Wibrand, K. Berge, M. Messaoudi, A. Duffaud, D. Panja, C. R. Bramham and L. Burri, Enhanced cognitive function and antidepressant-like effects after krill oil supplementation in rats, Lipids Health Dis., 2013, 12, 6 CrossRef CAS PubMed.
- J. S. Park, J. H. Chyun, Y. K. Kim, L. L. Line and B. P. Chew, Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans, Nutr. Metab., 2010, 7, 18 CrossRef PubMed.
- J. S. Park, B. D. Mathison, M. G. Hayek, S. Massimino, G. A. Reinhart and B. P. Chew, Astaxanthin stimulates cell-mediated and humoral immune responses in cats, Vet. Immunol. Immunop., 2011, 144, 455–461 CrossRef CAS PubMed.
- Y. P. Lu, S. Y. Liu, H. Sun, X. M. Wu, J. J. Li and L. Zhu, Neuroprotective effect of astaxanthin on H2O2-induced neurotoxicity in vitro and on focal cerebral ischemia in vivo, Brain Res., 2010, 1360, 40–48 CrossRef CAS PubMed.
- H. Turkez, F. Geyikoglu and M. I. Yousef, Beneficial effect of astaxanthin on 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced liver injury in rats, Toxicol. Ind. Health, 2013, 29, 591–599 CrossRef CAS PubMed.
- K. C. Chan, P. J. Pen and M. C. Yin, Anticoagulatory and antiinflammatory effects of astaxanthin in diabetic rats, J. Food Sci., 2012, 77, H76–H80 CrossRef CAS PubMed.
- L. Y. Dong, J. Jin, G. Lu and X. L. Kang, Astaxanthin attenuates the apoptosis of retinal ganglion cells in db/db mice by inhibition of oxidative stress, Mar. Drugs, 2013, 11, 960–974 CrossRef CAS PubMed.
- K. Uchiyama, Y. Naito, G. Hasegawa, N. Nakamura, J. Takahashi and T. Yoshikawa, Astaxanthin protects β-cells against glucose toxicity in diabetic db/db mice, Redox Rep., 2002, 7, 290–293 CrossRef CAS PubMed.
- T. Maoka, H. Tokuda, N. Suzuki, H. Kato and H. Etoh, Anti-oxidativeanti-tumor-promoting, and anti-carcinogensis activities of nitroastaxanthin and nitrolutein, the reaction products of astaxanthin and lutein with peroxynitrite, Mar. Drugs, 2012, 10, 1391–1399 CrossRef CAS PubMed.
- J. Huangfu, J. Liu, Z. Sun, M. Wang, Y. Jiang, Z. Y. Chen and F. Chen, Antiaging effects of astaxanthin-rich alga Haematococcus pluvialis on fruit flies under oxidative stress, J. Agric. Food Chem., 2013, 61, 7800–7804 CrossRef CAS PubMed.
- Y. Y. Yin, N. Xu, Y. Shi, B. Y. Zhou, D. R. Sun, B. X. Ma, Z. Z. Xu, J. Yang and C. M. Li, Astaxanthin protects dendritic cells from lipopolysaccharide-induced immune dysfunction, Mar. Drugs, 2021, 19, 346 CrossRef CAS PubMed.
- Y. Y. Yin, N. Xu, T. Qin, B. Y. Zhou, Y. Shi, X. Y. Zhao, B. X. Ma, Z. Z. Xu and C. M. Li, Astaxanthin provides antioxidant protection in LPS-induced dendritic cells for inflammatory control, Mar. Drugs, 2021, 19, 534 CrossRef CAS PubMed.
- M. Fujihara, M. Muroi, K. Tanamoto, T. Suzuki, H. Azuma and H. Ikeda, Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: Roles of the receptor complex, Pharmacol. Ther., 2003, 100, 171–194 CrossRef CAS PubMed.
- Y. C. Lu, W. C. Yeh and P. S. Ohashi, LPS/TLR4 signal transduction pathway, Cytokine, 2008, 42, 145–151 CrossRef CAS PubMed.
- P. Kubes and D. M. McCafferty, Nitric oxide and intestinal inflammation, Am. J. Med., 2000, 109, 150–158 CrossRef CAS PubMed.
- W. H. Watson, Y. Zhao and R. K. Chawla, S-Adenosylmethionine attenuates the lipopolysaccharide-induced expression of the gene for tumour necrosis factor alpha, Biochem. J., 1999, 342(Pt 1), 21–25 CrossRef CAS PubMed.
- J. S. Park, B. P. Chew and T. S. Wong, Dietary lutein from marigold extract inhibits mammary tumor development in BALB/c mice, J. Nutr., 1998, 128, 1650–1656 CrossRef CAS PubMed.
- Y. Dong, L. Liu, X. Shan, J. J. Tang, B. M. Xia, X. L. Cheng, Y. Y. Chen and W. W. Tao, Pilose antler peptide attenuates LPS-induced inflammatory reaction, Int. J. Biol. Macromol., 2018, 108, 272–276 CrossRef CAS PubMed.
- J. R. Cheng and A. Eroglu, The promising effects of astaxanthin on lung diseases, Adv. Nutr., 2021, 12, 850–864 CrossRef PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Regulation of cell death: The calcium–apoptosis link, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS PubMed.
- M. Lacroix, R. Riscal, G. Arena, L. K. Linares and L. Le Cam, Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer, Mol. Metab., 2020, 33, 2–22 CrossRef CAS PubMed.
- G. B. He and M. Karin, NF-κB and STAT3 - key players in liver inflammation and cancer, Cell Res., 2011, 21, 159–168 CrossRef CAS PubMed.
- J. Bollrath and F. R. Greten, IKK/NF-κB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis, EMBO Rep., 2009, 10, 1314–1319 CrossRef CAS PubMed.
- H. S. Park, S. R. Kim and Y. C. Lee, Impact of oxidative stress on lung diseases, Respirology, 2009, 14, 27–38 CrossRef PubMed.
- I. Rahman and I. M. Adcock, Oxidative stress and redox regulation of lung inflammation in COPD, Eur. Respir. J., 2006, 28, 219–242 CrossRef CAS PubMed.
- D. Elia, D. Madhala, E. Ardon, R. Reshef and O. Halevy, Sonic hedgehog promotes proliferation and differentiation of adult muscle cells: Involvement of MAPK/ERK and PI3K/Akt pathways, Biochim. Biophys. Acta, 2007, 1773, 1438–1446 CrossRef CAS PubMed.
- X. F. Qin, C. G. Qiu and L. S. Zhao, Lysophosphatidylcholine perpetuates macrophage polarization toward classically activated phenotype in inflammation, Cell. Immunol., 2014, 289, 185–190 CrossRef CAS PubMed.
- A. L. Hopkins, Network pharmacology, Nat. Biotechnol., 2007, 25, 1110–1111 CrossRef CAS PubMed.
- S. Wu, R. Chen, J. Chen, N. Yang, K. Li, Z. Zhang and R. Zhang, Study of the anti-inflammatory mechanism of β-carotene based on network pharmacology, Molecules, 2023, 28, 7540 CrossRef CAS PubMed.
- B. Guo, C. P. Zhao, C. H. Zhang, Y. Xiao, G. L. Yan, L. Liu and H. D. Pan, Elucidation of the anti-inflammatory mechanism of Er Miao San by integrative approach of network pharmacology and experimental verification, Pharmacol. Res., 2022, 175, 106000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fo00610k |
| This journal is © The Royal Society of Chemistry 2024 |