
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGinsenoside Rk1 improves endothelial function in diabetes through activating peroxisome proliferator-activated receptors†
Lingchao
Miao‡
a,
Yan
Zhou‡
a,
Dechao
Tan
ab,
Chunxiu
Zhou
a,
Cheng-Chao
Ruan
c,
Shengpeng
Wang
 ab,
Yitao
Wang
ab,
Chi Teng
Vong
*ab and
Wai San
Cheang
*a
ab,
Yitao
Wang
ab,
Chi Teng
Vong
*ab and
Wai San
Cheang
*a
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau SAR, China. E-mail: annacheang@um.edu.mo; gigictvong@um.edu.mo; Fax: +8532884 1358; Tel: +853 8822 4914
bMacau Centre for Research and Development in Chinese Medicine, University of Macau, Macau SAR, China
cDepartment of Physiology and Pathophysiology, School of Basic Medical Sciences, Shanghai Key Laboratory of Bioactive Small Molecules, Fudan University, Shanghai, China
First published on 17th April 2024
Abstract
Ginsenoside Rk1, one kind of ginsenoside, is a minor ginsenoside found in Panax ginseng and used as traditional Chinese medicine for centuries. It exhibits anti-tumor and anti-aggregation effects. However, little research has been done on its effect on endothelial function. This study investigated whether ginsenoside Rk1 improved endothelial dysfunction in diabetes and the underlying mechanisms in vivo and in vitro. Male C57BL/6 mice were fed with a 12 week high-fat diet (60% kcal % fat), whereas treatment groups were orally administered with ginsenoside Rk1 (10 and 20 mg per kg per day) in the last 4 weeks. Aortas isolated from C57BL/6 mice were induced by high glucose (HG; 30 mM) and co-treated with or without ginsenoside Rk1 (1 and 10 μM) for 48 h ex vivo. Moreover, primary rat aortic endothelial cells (RAECs) were cultured and stimulated by HG (44 mM) to mimic hyperglycemia, with or without the co-treatment of ginsenoside Rk1 (10 μM) for 48 h. Endothelium-dependent relaxations of mouse aortas were damaged with elevated oxidative stress and downregulation of three isoforms of peroxisome proliferator-activated receptors (PPARs), PPAR-α, PPAR-β/δ, and PPAR-γ, as well as endothelial nitric oxide synthase (eNOS) phosphorylation due to HG or high-fat diet stimulation, which also existed in RAECs. However, after the treatment with ginsenoside Rk1, these impairments were all ameliorated significantly. Moreover, the vaso-protective and anti-oxidative effects of ginsenoside Rk1 were abolished by PPAR antagonists (GSK0660, GW9662 or GW6471). In conclusion, this study reveals that ginsenoside Rk1 ameliorates endothelial dysfunction and suppresses oxidative stress in diabetic vasculature through activating the PPAR/eNOS pathway.
1. Introduction
Diabetes is the most common metabolic disease in the world with high prevalence and mortality rates due to its complications such as coronary artery disease. In hyperglycemia, elevated blood glucose levels can cause endothelial damage, thus leading to endothelial dysfunction during the progression of diabetes and obesity.1 The disruption of the balance between nitric oxide (NO) availability and reactive oxygen species (ROS) generation leads to endothelial dysfunction and thereby the development of cardiovascular diseases (CVD).2 Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that act as ligand-inducing transcription factors. They are composed of three subtypes: PPAR-α, PPAR-δ, and PPAR-γ.3 PPARs control metabolic homeostasis, lipid and glucose metabolism, as well as inflammation, which play an important role in CVD, especially in diabetes and obesity.4,5 Evidence has indicated that several transcription factors are related to serine–threonine kinases. The activation of PPARs promotes NO production through protein kinase B (Akt)/endothelial nitric oxide synthase (eNOS) signaling.6 Goetze et al. discovered that both PPAR-α and PPAR-γ activators significantly inhibited the phosphorylation of Akt induced by the vascular endothelial growth factor, thus protecting the vascular endothelium from metabolic disorders.7 Besides, Akt/nuclear factor erythroid-2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathways play an important role in regulating oxidative stress, protecting the cells from various toxins.8,9 The phosphorylation of Akt increases Nrf2 protein levels, thus activating HO-1 expression.10In recent years, natural products have received great attention in cardiovascular benefits. Ginseng is a plant of the Panax genus and exhibits a wide range of pharmacological properties, including anti-tumor, anti-inflammatory, anti-oxidative and anti-apoptotic effects.11 Ginsenosides, as the major active components of Panax ginseng, have been demonstrated to exhibit numerous pharmacological bioactivities, especially in treating cardiovascular and cerebrovascular diseases.12–14 Although varied biological effects of some kinds of ginsenosides, such as Rg1, Rb1, Rb3 and Rh2, have been illustrated in the previous studies,15–18 there is relatively less research on Rk1, which might be due to its low bioavailability or low content in Panax ginseng. Ginsenoside Rk1 is a monomeric compound extracted from ginseng that has been used for the treatment of cancer, inflammation and diabetes.19–21 Most of the previous studies have focused on investigating the anti-tumor effects of ginsenoside Rk1. Although the function of ginsenoside Rk1 in the vasculature is less studied, existing evidence indicates that ginsenoside Rk1 inhibits platelet aggregation. Ginsenoside Rk1 inhibits thrombin-, collagen- and U46619-stimulated human platelet aggregation in a dose-dependent manner.22 However, research on the effects and underlying mechanisms of ginsenoside Rk1 for endothelial dysfunction associated with diabetes and obesity is limited. Therefore, this study aimed to investigate whether ginsenoside Rk1 could ameliorate endothelial dysfunction in diabetes and obesity and whether or not the activation of the three subtypes of PPAR contributes to the vascular benefits of ginsenoside Rk1.
2. Materials and methods
2.1. Animals
All animal experiments were performed in accordance with the National Institutes of Health guidelines for the Care Use of Laboratory Animals. The animal research protocol was approved by the Animal Research Ethics Committee, University of Macau. In this study, adult male C57BL/6 mice at 6 weeks were supplied from the Faculty of the Healthy Science Animal Centre of the University of Macau, bred in pathogen-free rooms with controlled temperature (21–23 °C) and housed with regular 12/12 h light/dark cycles. C57BL/6 mice were fed with normal chow diet that served as the control group, or high-fat diet (45% kcal % fat) for 8 weeks to induce type 2 diabetes mellitus, which was followed by oral administration of a vehicle (water) or ginsenoside Rk1 (MedChemExpress, HY-N2515, Shanghai, China) at 10 and 20 mg per kg per day for another consecutive 4 weeks. At the end of the treatment period, the mice were sacrificed by CO2 anesthesia to dissect arteries for further studies.2.2. Blood pressure measurement
Systolic (SBP) and diastolic (DBP) blood pressures were detected in the warmed, conscious, restrained state by the tail-cuff method using a CODA noninvasive blood pressure system (Kent Scientific Corporation, Torrington, CT, USA) in mice.2.3. Blood glucose measurement
Fasting blood glucose (FBG) levels were measured in mice post-fasting for 6 h. The FBG levels were measured in venous blood from mouse tails by using a commercial glucometer (Jiangsu Yuyue Medical Equipment and Supply Co. Ltd, Jiangsu, China), and mice with FBG > 11 mM were considered as diabetic mice.2.4. Preparation and incubation of mouse aortas
After 4 weeks of treatment, mice were sacrificed and thoracic aortas with adjacent connective tissues were excised and placed in sterile phosphate-buffered saline (PBS). Next, thoracic aortas were rapidly isolated and carefully dissected from surrounding tissues under a dissecting microscope and subjected for measurement of vascular reactivity by using a wire myograph. The protein expressions were measured by western blotting, while ROS levels were evaluated by fluorescence imaging. For ex vivo incubation experiments, aortas (segment lengths ∼2 mm) were cultured with DMEM low glucose (Gibco, Grand Island, NY, USA) containing 5.5 mM glucose, 10% FBS (Gibco, Paisley, UK), plus 1% P/S (Gibco, Grand Island, NY, USA) as the normal glucose control group (NG). Arteries were incubated with normal medium added with high glucose (HG, 30 mM, Sigma-Aldrich, St Louis, MO, USA), ginsenoside Rk1 (1 or 10 μM), GW6471 (5 μM, MedChemExpress, HY-15372, Shanghai, China), GSK0660 (5 μM, MedChemExpress, HY-12377, Shanghai, China), GW9662 (5 μM, Sigma-Aldrich, M6191, Beijing, China) or a vehicle (mannitol as osmotic control) for 48 h at 37 °C.2.5. Primary culture of rat aortic endothelial cells (RAECs)
Thoracic aortas were removed from Sprague-Dawley (SD) rats and dissected in sterile PBS. The arteries were incubated with sterile PBS containing collagenase type 1A (2 mg mL−1, Sigma-Aldrich, St Louis, MO, USA) at 37 °C for 15 min to detach the endothelial cells. The digestion was halted by adding RPMI-1640 medium with 10% FBS and 1% P/S. The cell suspension was centrifuged at 1500 rpm for 10 min and re-suspended in RPMI-1640 medium. After incubation for 1 h at 37 °C, the medium was refreshed to remove unattached cells and then RAECs were cultured in endothelial cell growth medium until 80–90% confluency. RAECs were treated with mannitol as an osmotic control, HG (44 mM) and co-incubated with ginsenoside Rk1 (10 μM) for 48 h.2.6. Wire myograph
Aortic segments of male C57BL/6 mice (∼2 mm) were transferred to an organ chamber filled with 5 mL oxygenated and ice-cold Krebs–Henseleit solution containing 119 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 25 mM NaHCO3, 1.2 mM KH2PO4, and 11 mM D-glucose. The ring segments were individually mounted onto a Multi Myograph System (Danish Myo Technology, Aarhus, Denmark) for the measurement of isometric tension. Each segment was stretched to optimal baseline tension (3 mN) and was then equilibrated at 37 °C in Krebs solution under continuous oxygenation (95% O2, 5% CO2) for 1 h, followed by the stimulation of contraction using 60 mM KCl. For testing endothelium-dependent relaxations (EDRs), each ring was pre-contracted with phenylephrine (Phe, 3 μM, Sigma-Aldrich, St Louis, MO, USA) in response to cumulative addition of acetylcholine (ACh, 3 nM-10 μM, Sigma-Aldrich, St Louis, MO, USA). Afterwards, endothelium-independent relaxations were measured in response to an endothelium-independent NO donor, sodium nitroprusside (SNP, 1 nM–10 μM, Sigma-Aldrich, St Louis, MO, USA).2.7. Western blotting
Total protein samples were prepared from mouse aorta homogenates or RAECs after treatment. Frozen aortas in liquid nitrogen were lysed in ice-cold RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing a complete protease inhibitor cocktail (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitors (Roche, Basel, Switzerland). RAECs were lysed with RIPA solution, supplemented with 1% phosphatase inhibitor cocktail (Thermo Fisher Scientific, IL, USA) and 1% phenylmethylsulfonyl (PMSF, Thermo Fisher Scientific; IL, USA). The lysates were then centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min at 4 °C. Subsequently, the supernatant was collected and the protein concentration were measured by using a BCA protein assay kit (Beyotime Biotechnology, Shanghai, China). Protein samples were separated by10% SDS-PAGE and transferred to a PVDA membrane (Millipore, Billerica, MA, USA), followed by blocking with 5% non-fat milk for 2 h at room temperature. Then, the membranes were probed with appropriate primary antibodies overnight at 4 °C followed by incubation with secondary antibodies (1:1000 dilution; Beyotime Biotechnology, Shanghai, China) for another 2 h at room temperature. The following primary antibodies were used: phospho (p)-eNOS at Ser1177 (#9570S), eNOS (#32027S), p-Akt at Ser473 (#4060S), Akt (#4685S) [1:1000 dilution; Cell Signaling Technology, Danvers, MA], PPAR-α (1:500 dilution, sc-398394, Santa Cruz Biotechnology, OR, USA), PPAR-γ (1:1000 dilution, ab209350, Abcam, Cambridge, UK), PPAR-δ (1:1000 dilution, ab178866, Abcam, Cambridge, UK), Nrf2 (1:1000 dilution, AF7623, Beyotime Biotechnology, Shanghai, China), HO-1 (1:1000 dilution, 66743-1-Ig, Proteintech Group, Inc., Wuhan, China), and GAPDH (1:5000 dilution, 60004-1-Ig, Proteintech Group, Inc., Wuhan, China). The protein bands were developed with an American ECLTM Advanced Western Blotting Detection Kit (GE Healthcare Life Sciences, Uppsala, Sweden) and quantified by using the ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA).
000 rpm for 30 min at 4 °C. Subsequently, the supernatant was collected and the protein concentration were measured by using a BCA protein assay kit (Beyotime Biotechnology, Shanghai, China). Protein samples were separated by10% SDS-PAGE and transferred to a PVDA membrane (Millipore, Billerica, MA, USA), followed by blocking with 5% non-fat milk for 2 h at room temperature. Then, the membranes were probed with appropriate primary antibodies overnight at 4 °C followed by incubation with secondary antibodies (1:1000 dilution; Beyotime Biotechnology, Shanghai, China) for another 2 h at room temperature. The following primary antibodies were used: phospho (p)-eNOS at Ser1177 (#9570S), eNOS (#32027S), p-Akt at Ser473 (#4060S), Akt (#4685S) [1:1000 dilution; Cell Signaling Technology, Danvers, MA], PPAR-α (1:500 dilution, sc-398394, Santa Cruz Biotechnology, OR, USA), PPAR-γ (1:1000 dilution, ab209350, Abcam, Cambridge, UK), PPAR-δ (1:1000 dilution, ab178866, Abcam, Cambridge, UK), Nrf2 (1:1000 dilution, AF7623, Beyotime Biotechnology, Shanghai, China), HO-1 (1:1000 dilution, 66743-1-Ig, Proteintech Group, Inc., Wuhan, China), and GAPDH (1:5000 dilution, 60004-1-Ig, Proteintech Group, Inc., Wuhan, China). The protein bands were developed with an American ECLTM Advanced Western Blotting Detection Kit (GE Healthcare Life Sciences, Uppsala, Sweden) and quantified by using the ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA).
2.8. Determination of ROS levels
Aortic segments were fixed in Tissue-Tek OCT Compound (Sakura, Nagano, Japan) and frozen in liquid nitrogen as well as sectioned (10 μm) by using a Leica CM 1000 cryostat at −20 °C. Frozen sections of aortas or cultured RAECs were incubated with 5 μM dihydroethidium (DHE, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at 37 °C in the dark. Fluorescence images were observed by using a Leica-DMi8 inverted fluorescence microscope (515 nm excitation/585 nm emission) and the fluorescence intensities were measured by using ImageJ software.2.9. Quantification of NO generation
The amount of NO released from the medium of RAECs was determined by using a Griess reagent (Invitrogen, Oregon, USA) according to the manufacturer's instructions. RAECs were treated with HG (44 mM) and ginsenoside Rk1 (10 μM) in a 24-well plate for 48 h, and the NO levels in the supernatants were determined by using the Griess reagent and evaluated by measuring the absorbance using a SpectraMax M5 microplate reader (Molecular Devices, Silicon Valley, CA, USA) at 548 nm.2.10. Statistical analysis
All data obtained from this study were expressed as mean ± standard error of mean (SEM), and n represents separate experiments. Statistical analysis was determined by using GraphPad Prism software (version 9.0, GraphPad Software Inc., San Diego, CA, USA), and comparison among multiple groups was analyzed using Student's t-test or one-way analysis of variance (ANOVA) by Bonferroni post hoc tests. A p value < 0.05 was considered as statistically significant.3. Results
3.1 Four week treatment with ginsenoside Rk1 ameliorated endothelial dysfunction in diet-induced obese (DIO) mice
For investigating the in vivo impact of ginsenoside Rk1 on arterial function, DIO mice were given a 4 week administration of ginsenoside Rk1 (10 and 20 mg per kg per day) by oral gavage during the experimental process. After 12 weeks of high-fat diet feeding, the body weights of DIO mice obviously increased when compared with those of the control mice fed with a standard chow diet. Yet, a 4 week treatment with two oral concentrations of ginsenoside Rk1 did not significantly decrease the body weights of DIO mice (Fig. 1A). The mean values of FBG in DIO mice were over 15 mM, and were apparently higher than those of the control mice, indicating a successfully established diabetic mouse model. After 4 week treatment with ginsenoside Rk1, the FBG of DIO mice was significantly improved by both the concentrations of Rk1 (Fig. 1B). The elevated mean values of systolic and diastolic blood pressures were normalized markedly by a 4-week treatment with ginsenoside Rk1 in DIO mice (Fig. 1C and D). Additionally, without affecting the SNP-induced endothelium-independent relaxations of mouse aortas, 4 week oral administration of ginsenoside Rk1 at a higher concentration only (20 mg per kg per day) could significantly restore ACh-induced EDRs, which were damaged in DIO mouse aortas (Fig. 1E and F).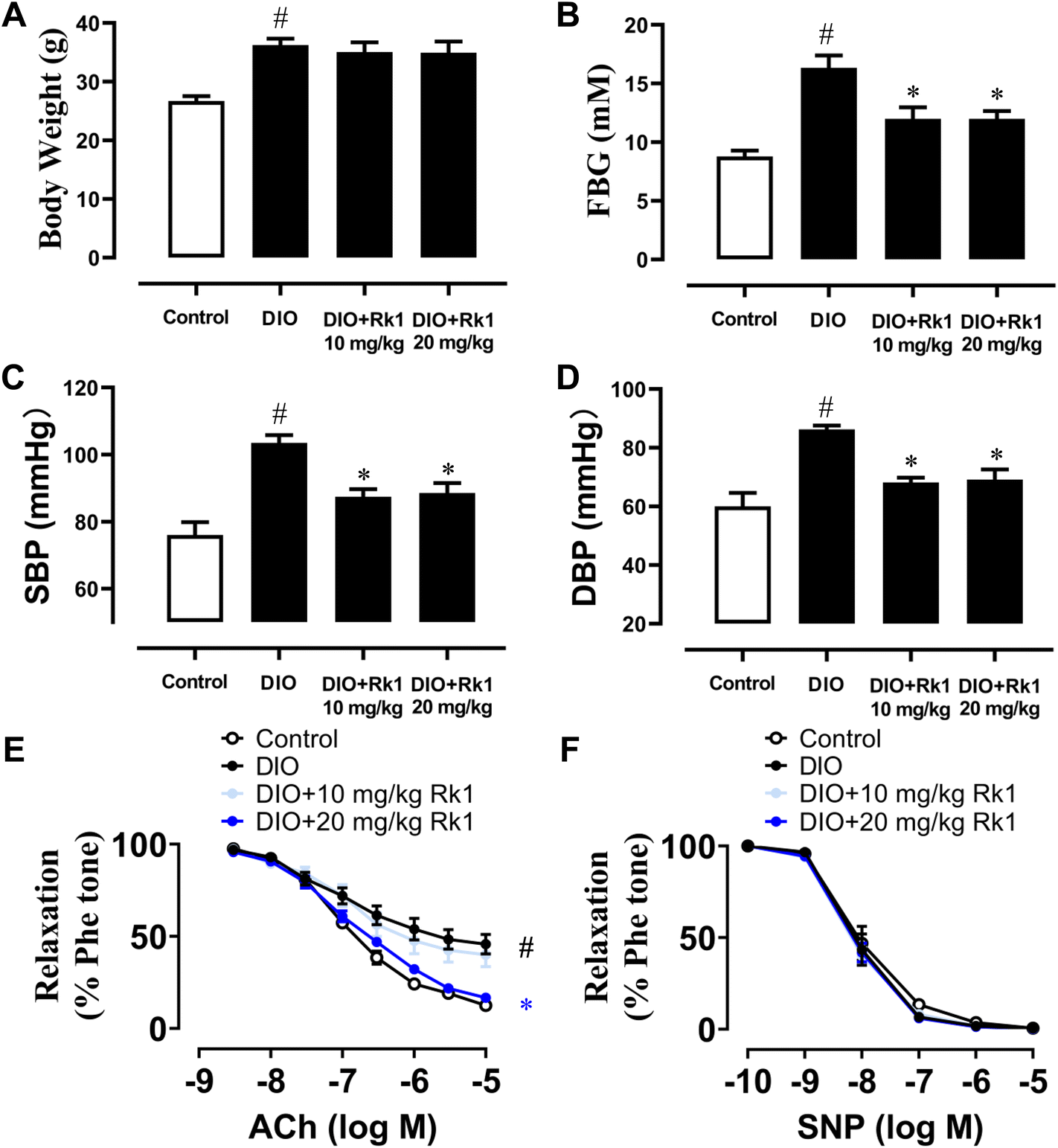 | ||
| Fig. 1 Ginsenoside Rk1 improved endothelial dysfunction in diet-induced obese (DIO) mice. (A) Body weights of all C57 BL/6 mice with a standard chow diet or high-fat diet for a total of 12 weeks, and in ginsenoside Rk1-treated groups. DIO mice were orally administered with ginsenoside Rk1 (10 and 20 mg per kg per day) and other mice with water during the last 4 weeks of a high-fat diet. (B) FBG results upon 6 h fasting in all mice. (C and D) Systolic (SBP) and diastolic (DBP) blood pressure values measured in all mice through the tail-cuff way. (E) Summarized results reflecting the effect of ginsenoside Rk1 treatment on acetylcholine (ACh)-induced endothelium-dependent relaxations (EDRs) in the aortas from DIO mice and (F) sodium nitroprusside (SNP)-induced endothelium-independent relaxations in all mouse aortas. Data are shown as mean ± SEM of 6 mice from each group. #P < 0.05 vs. control; *P < 0.05 vs. DIO. | ||
3.2 Four week treatment with ginsenoside Rk1 improved endothelial function via the activation of eNOS and PPAR signaling pathways in the aortas of DIO mice
For further investigating the underlying mechanisms of the vaso-protective activity of ginsenoside Rk1 in vivo, mouse aortas were isolated to examine the protein expression levels by western blotting. The p-eNOS at Ser1177 and its total expressions were both obviously down-regulated in DIO mouse aortas when compared with those in lean mouse aortas, which were effectively up-regulated by a 4-week treatment with ginsenoside Rk1 at 20 mg per kg per day with significance (Fig. 2A–C). What's more, the expression levels of PPAR three isoforms (γ, δ and α) were all apparently suppressed in DIO mouse aortas, of which the trends were all turned over by ginsenoside Rk1 treatment at 20 mg per kg per day (Fig. 2A and D–F).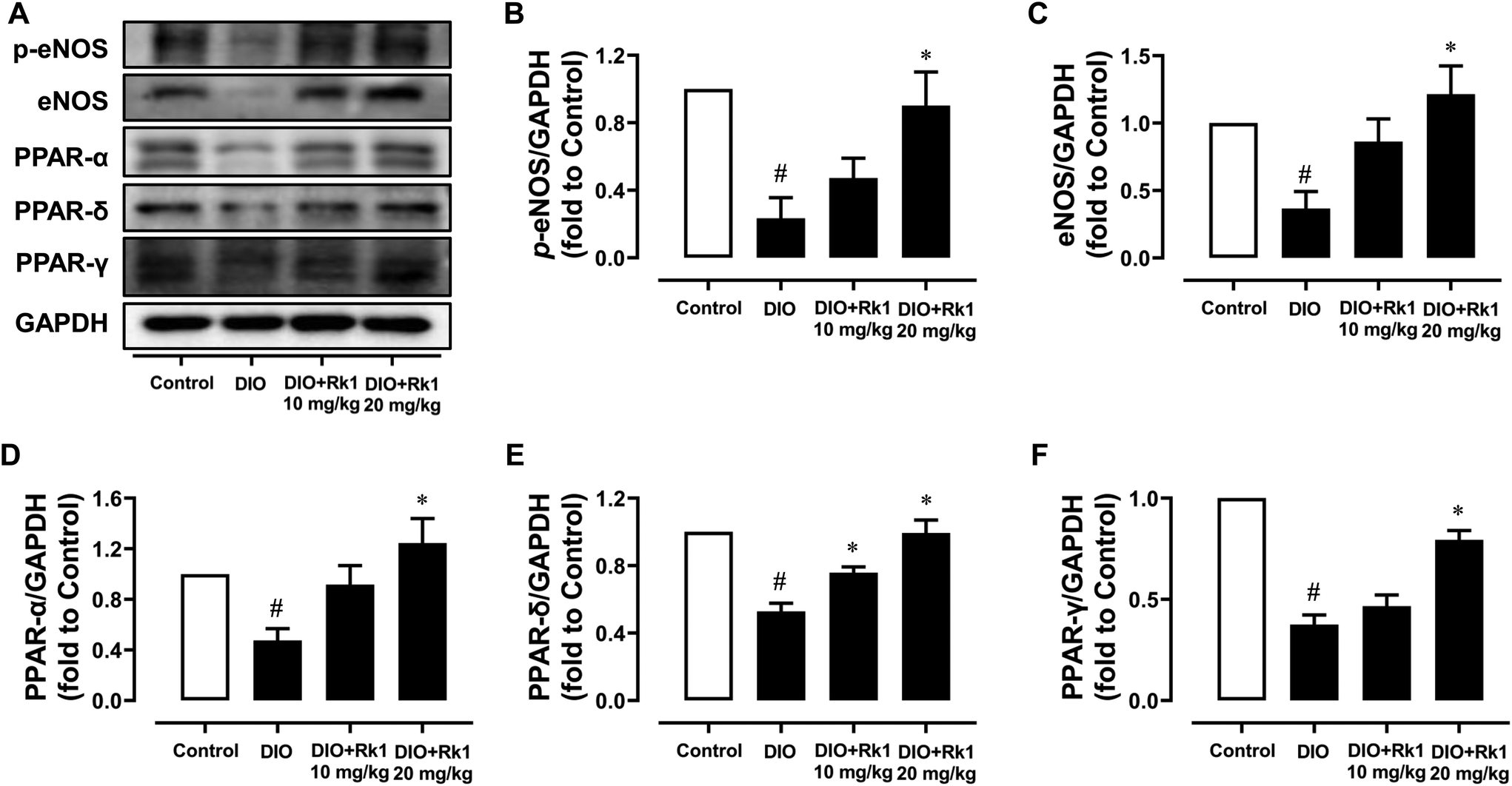 | ||
| Fig. 2 Four-week treatment with ginsenoside Rk1 activated eNOS and PPAR signaling pathways in DIO mouse aortas. (A) Representative western blotting images and summarized graphs showing (B) eNOS phosphorylation at Ser1177, (C) total eNOS, (D) total PPAR-α, and (E) PPAR-δ and (F) PPAR-γ expression levels compared to the corresponding total protein or GAPDH in the aortas of lean control, DIO and ginsenoside Rk1-treated (10 and 20 mg per kg per day, 4 weeks) DIO mice. p-eNOS (Ser1177) and eNOS, 140 kDa; PPAR-α, 55 kDa; PPAR-δ, 50 kDa; PPAR-γ, 57 kDa. Data are shown as mean ± SEM of 4 mice from each group. #P < 0.05 vs. control; *P < 0.05 vs. DIO. | ||
3.3 Ginsenoside Rk1 ameliorated endothelial dysfunction in mouse aortas damaged by HG ex vivo
Aortic rings (about 2 mm per segment) of mice were cultured ex vivo using normal glucose medium (5.5 mM, 48 h) or HG medium (30 mM, 48 h) to mimic the hyperglycemic conditions under diabetes. HG stimulation destroyed ACh-induced EDRs remarkably, while co-treatment with 10 μM but not 1 μM ginsenoside Rk1 could significantly reverse the damage (Fig. 3A and B). Moreover, GW6417 (a PPAR-α antagonist, 5 μM), GSK0660 (a PPAR-β/δ antagonist, 5 μM) and GW9662 (a PPAR-γ antagonist, 5 μM) were applied to examine the role of the PPAR signaling pathway in the vascular protective mechanisms of ginsenoside Rk1. It was found that the vaso-protective activity of ginsenoside Rk1 was totally abolished by all the antagonists of these PPAR sub-types (Fig. 3C). Apart from this, SNP-induced endothelium-independent relaxations were not disturbed among all groups, indicating that the vascular smooth muscle cells could respond to NO release as normal (Fig. 3D and E).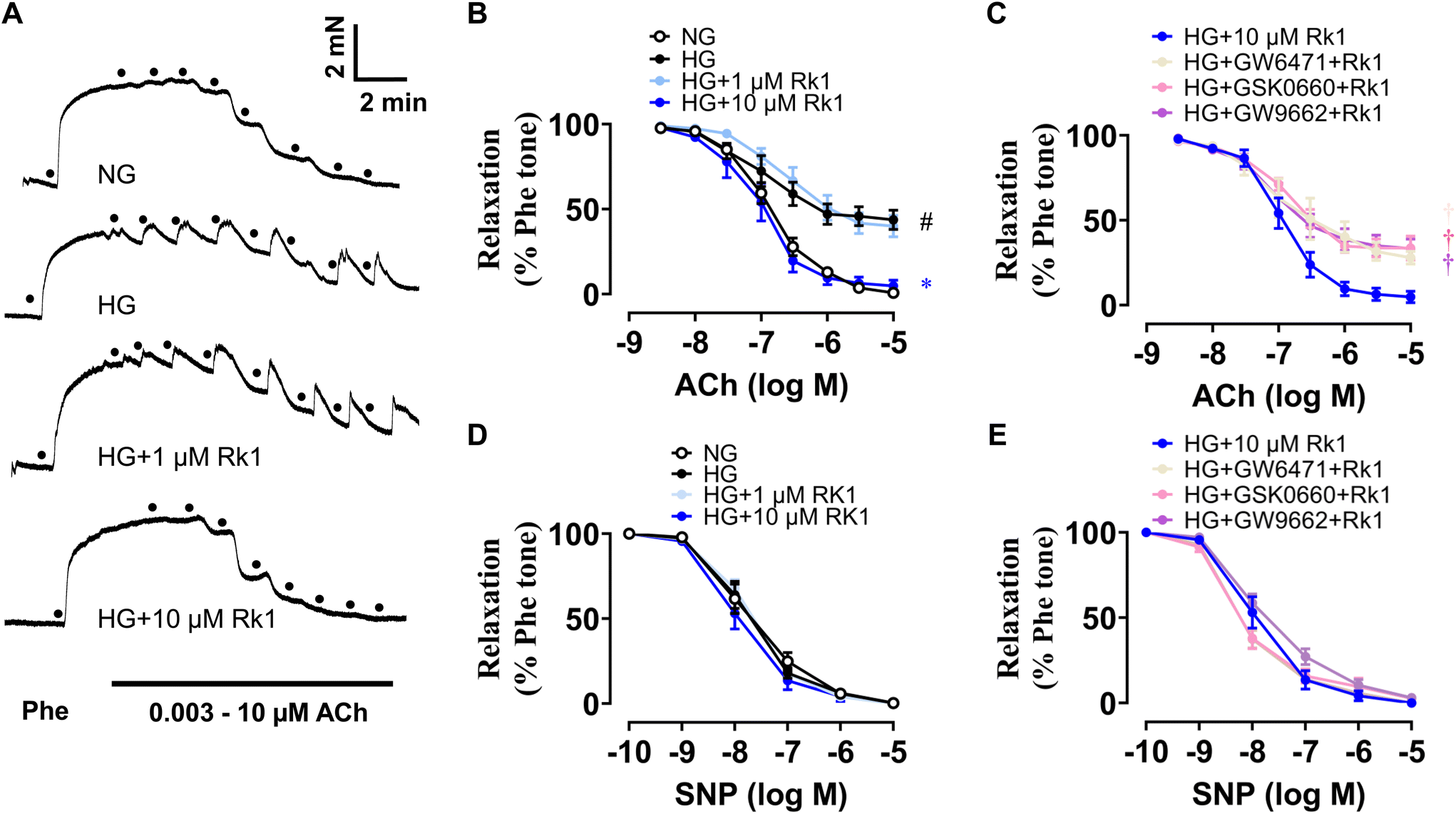 | ||
| Fig. 3 Ginsenoside Rk1 treatment protected against high glucose (HG)-triggered endothelial dysfunction via activating PPAR signaling pathways. (A) Representative traces. (B) Summarized data of the function of ginsenoside Rk1 co-treatment (1 and 10 μM) on ACh-induced endothelium-dependent relaxations (EDRs) of mouse aortas exposed to HG stimulation (30 mM, 48 h). (C) Effects of the PPAR sub-type antagonists (all 5 μM, GW6417: PPAR-α antagonist; GSK0660: PPAR-β/δ antagonist; GW9662: PPAR-γ antagonist) on ACh-induced EDRs in mouse aortas co-treated with 10 μM ginsenoside Rk1 under HG stimulation (30 mM, 48 h). (D and E) SNP-induced relaxations in mouse aortas exposed to HG (30 mM, 48 h). Data are shown as mean ± SEM of 5 independent experiments. #P < 0.05 vs. normal glucose (NG, 5.55 mM glucose); *P < 0.05 vs. HG (30 mM); †P < 0.05 vs. HG + ginsenoside Rk1 (10 μM). | ||
3.4 Ginsenoside Rk1 enhanced Akt/eNOS and PPAR signaling pathways in RAECs when exposed to HG stimulation
RAECs were induced by HG (44 mM, 48 h) and co-treated with ginsenoside Rk1 (10 μM). Exposure to HG inhibited the phosphorylation of eNOS at Ser1177 and phosphorylation of Akt at Ser473 when compared with their total proteins (Fig. 4A–C), and reduced the protein expressions of Nrf2 and HO-1 (Fig. 4A, D and E), as well as suppressing the expression levels of PPAR-α, PPAR-γ and PPAR-δ compared to GAPDH (Fig. 4A and F–H). Besides, co-treatment with ginsenoside Rk1 (10 μM) significantly reversed all these alterations, illustrating that its vaso-protective effects relied on activating Akt/eNOS and PPAR signaling pathways.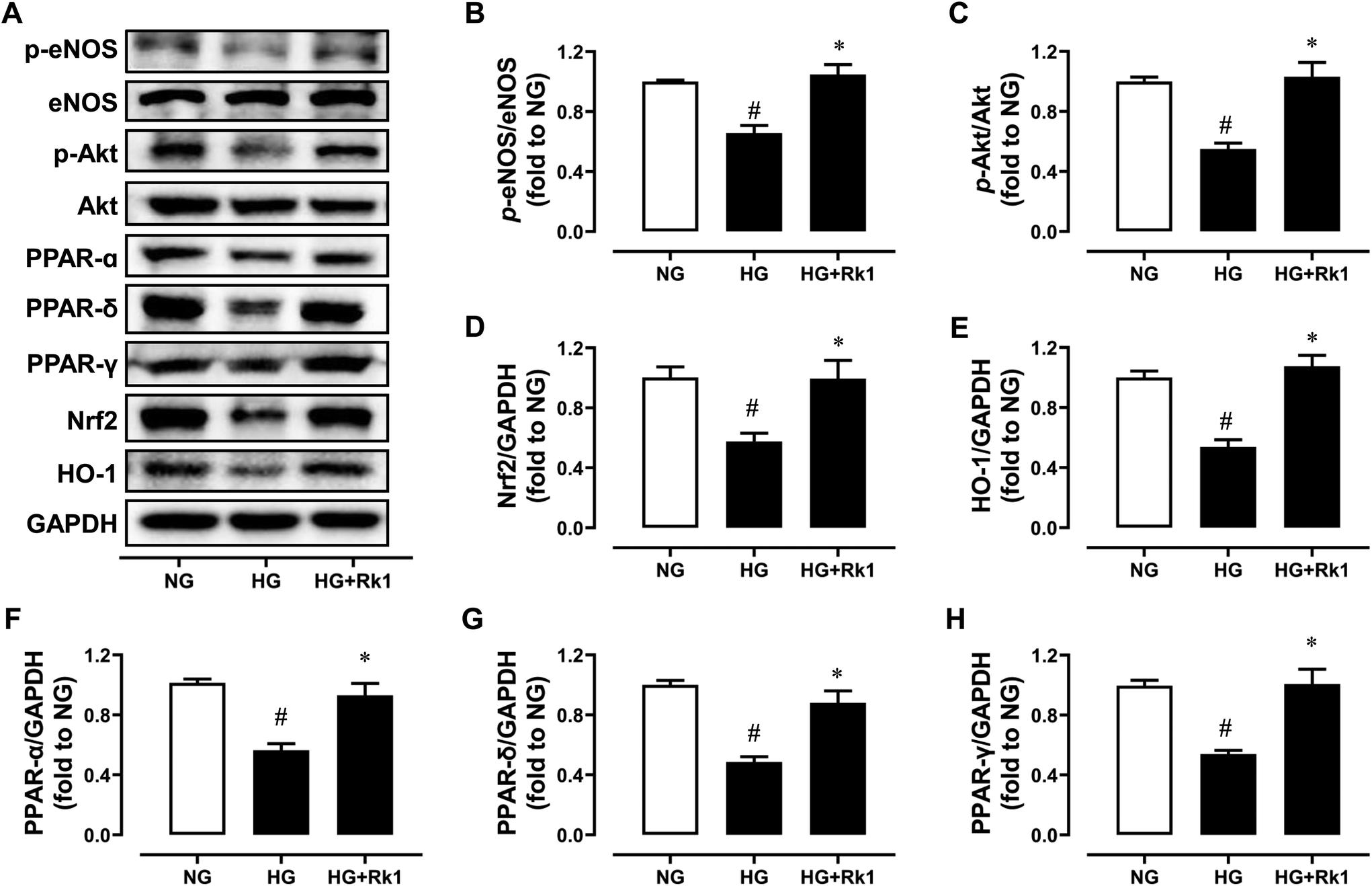 | ||
| Fig. 4 Ginsenoside Rk1 activated Akt/eNOS and PPAR pathways in high glucose (HG)-induced RAECs. (A) Representative western blot images of RAECs treated with normal glucose (NG, 11 mM in RPMI-1640 medium), HG (44 mM) and ginsenoside Rk1 (10 μM) for 48 h. Summarized data showing (B) the phosphorylation of eNOS at Ser1177 compared with total eNOS expression, (C) the phosphorylation of Akt at Ser473 compared to total Akt expression, and (D) Nrf2, (E) HO-1, (F) PPAR-α, (G) PPAR-δ and (H) PPAR-γ expressions compared to GAPDH in RAECs. p-eNOS (Ser1177) and eNOS, 140 kDa; p-Akt (Ser473) and Akt, 60 kDa; PPAR-α, 55 kDa; PPAR-δ, 50 kDa; PPAR-γ, 57 kDa; Nrf2, 100 kDa; HO-1, 28 kDa. Data are shown as mean ± SEM of 6 independent experiments. #P < 0.05 vs. NG (11 mM); *P < 0.05 vs. HG (44 mM). | ||
3.5 Ginsenoside Rk1 suppressed oxidative stress via a PPAR-dependent mechanism
After 4-week oral administration of ginsenoside Rk1 (10 and 20 mg per kg per day), the elevated ROS levels in the aortas of DIO mice were suppressed observably, especially by the higher oral concentration (Fig. 5A and B). In addition, it was also proved in vitro that ginsenoside Rk1 co-treatment (10 μM) could inhibit HG (44 mM, 48 h)-induced ROS elevation in RAECs. However, the anti-oxidative activities of ginsenoside Rk1 on HG-stimulated RAECs were reversed by the antagonists of PPAR sub-types, GW9662, GW6417 and GSK0660. This phenomenon indicated that the anti-oxidative effect of ginsenoside Rk1 relied on the activation of PPAR pathway (Fig. 5C and D). Besides, exposure to HG decreased nitrite production in RAECs, indicating that the NO bioavailability was diminished in RAECs and such diminishment was reversed by co-treatment with ginsenoside Rk1 (Fig. 5E).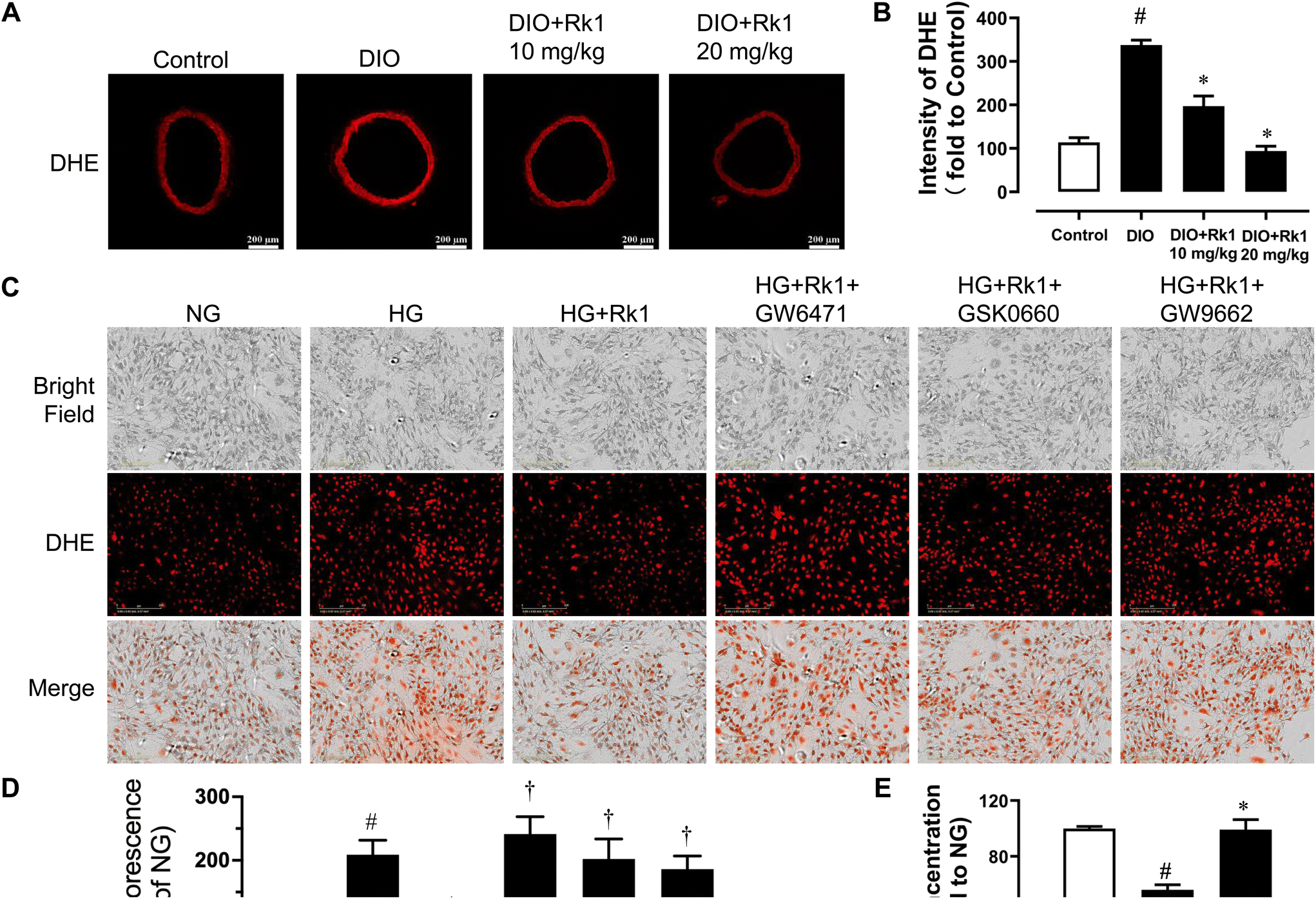 | ||
| Fig. 5 Ginsenoside Rk1 inhibited oxidative stress in RAECs that was dependent via activating the PPAR pathway. (A) Representative mouse aorta section images stained with DHE (oxidative stress indicator) and (B) their summarized intensity graph including control, DIO and DIO mice with 4-week treatment with ginsenoside Rk1. (C) Representative DHE staining images of RAECs treated with normal glucose (NG, 11 mM), high glucose (HG, 44 mM), ginsenoside Rk1 (10 μM) and PPAR sub-type antagonists GW9662, GW6417 and GSK0660 (all 5 μM), which were also shown by using the (D) summarized graph. (E) Summarized graphs showing the NO production levels in RAECs. Data are shown as mean ± SEM of 5 independent experiments. #P < 0.05 vs. control/NG; *P < 0.05 vs. DIO/HG; †P < 0.05 vs. HG + ginsenoside Rk1. | ||
4. Discussion
In this study, by means of varied in vivo, ex vivo and in vitro investigation, the vaso-protective activity of ginsenoside Rk1 was investigated. The main findings of the present study included (i) 4-week oral administration of ginsenoside Rk1 alleviated FBG and high blood pressure in DIO mice; (ii) 4-week treatment with ginsenoside Rk1 improved damaged EDRs of mouse aortas via activating the PPAR/eNOS signaling pathways in DIO mice; (iii) the endothelial dysfunction of mouse aortas was ameliorated by ginsenoside Rk1 treatment that was dependent on PPAR activation under HG condition ex vivo; and (iv) the elevated ROS generation levels in both mouse aortas of DIO mice and in HG-stimulated RAECs were all inhibited by ginsenoside Rk1.By means of comprehensive metabolomic analysis of platelet aggregation induced by collagen and arachidonic acid in the blood samples from male SD rats, Ju et al. have found out some meaningful results, showing that a key element in platelet aggregation, named thromboxane B2 (TXB2), which was downregulated by ginsenoside Rk1 upon refraining activity of cyclooxygenase (COX). In addition, the anti-platelet effect of ginsenoside Rk1 is mediated through decreased 12-hydroxy-5,8,10,14-eicosatetraenoic acid (12-HETE) level, and the cytosol-membrane translocation of 12-lipoxyenase (12-LOX).23 Ginsenoside Rk1 suppresses the permeability of human retinal endothelial cells stimulated by advanced glycation end-products (AGEs), vascular endothelial growth factor (VEGF), histamine, or thrombin; and it ameliorates the vessel leakiness of retina in a diabetic mouse model.24 There is no study investigating the impact of ginsenoside Rk1 on diabetic vasomotor dysfunction. The current study is the first to examine the vaso-protective effect of ginsenoside Rk1 and its potential mechanisms. The present study provides novel evidence supporting that ginsenoside Rk1 can restore endothelial dysfunction in isolated aortas from DIO mice and in aortas subjected to HG challenge through activating PPAR/eNOS pathways and inhibiting oxidative stress in vivo and in vitro. Furthermore, GW9662, GW6417 and GSK0660, as the selective and potent antagonists of PPAR-γ, PPAR-α and PPAR-β/δ, respectively, reversed the endothelial protective effect of ginsenoside Rk1, suggesting that its activity was dependent on PPAR pathway activation. The beneficial effect of ginsenoside Rk1 on endothelial function featured the improved aortic EDRs in HG-stimulated mouse aortas or obese diabetic mouse aortas. The HG concentration used in the current study was 30 mM for the ex vivo culture of mouse aortas but it was 44 mM for the in vitro culture of RAECs to mimic hyperglycemia in diabetes. Indeed, 25 mM or 30 mM HG is more physiological and is widely used for cell culture studies.25–27 A few studies have applied higher concentrations of glucose, 40 mM or 44 mM HG for culturing RAECs and rat aortas.28,29 There is a study limitation that 30 mM HG should be used to stimulate RAECs so that the findings will be more physiologically relevant. Oral administration of ginsenoside Rk1 for 4 weeks improved hyperglycemia and high blood pressure in DIO mice. The abilities of ginsenoside Rk1 in regulating blood glucose levels, inhibiting oxidative stress and increasing NO bioavailability all might partially contribute to ameliorate vascular dysfunction, and other underlying mechanisms are worth paying attention for further study.
The PPARs have been reported to be implicated in regulating cardiovascular diseases.6,30–34 There are drugs already on the market, pioglitazone and rosiglitazone, which are the selective and strong PPAR-γ agonists for treating type-2 diabetes through enhancing insulin sensitivity. They have also been reported to enable rat EDRs to be recovered in resistant arteries and reduce hypertension in angiotensin II–stimulated SD rats through inhibiting angiotensin II type 1 (AT1) receptors, pro-inflammatory mediators and cell cycle proteins.35 In addition, the disruption of endothelial PPAR-γ in endothelial PPAR-γ null mice (ePPAR-γ−/−) produced by crossing the mice expressing Cre recombinase actuated with an endothelial-specific promoter with the mice bearing a floxed PPAR-γ gene results in impaired EDRs, suggesting that endothelial PPAR-γ contributes to vascular NO production.36 Apart from this, the activation of PPAR-δ by two selective PPAR-δ agonists, GW0742 and GW1516, is reported to improve the damaged EDRs of mouse aorta and augment NO bioavailability in mouse aortic endothelial cells (MAECs) through the phosphorylation of eNOS at serine 1177 in an PI3K/Akt-dependent mechanism in db/db and PPAR-δ KO DIO mice.31 Moreover, PPAR-δ transcriptional activity can be increased by resveratrol or sirtuin 1 (SIRT1) activator CAY10602 treatment in endothelial cells through activating SIRT1, thus up-regulating Akt/eNOS signaling pathways.30 As for PPAR-α, its activator docosahexaenoic acid (DHA) is found to attenuate hypertension in angiotensin (Ang) II–infused SD rats and improve endothelial dysfunction, which is closely related to suppressed inflammation and oxidative stress in the vascular wall.32 Fibrate drugs, as the ligands of the fatty acid receptor PPAR-α, are directed by PPAR-α activation to exhibit lipid-lowering effects to have the potential to treat atherosclerosis. In terms of NO production, PPAR-α agonists are reported to increase eNOS expression to elevate NO levels.37 In line with the aforementioned observations that Akt/eNOS is the likely downstream mechanisms for the activation of PPARs to mediate vascular-protective effects, the present results showed that ginsenoside Rk1 upregulated the expression of the three PPAR subtypes and phosphorylation of Akt and eNOS in RAECs exposed to HG and in aortas from DIO mice. Notably, the results on the protein expression of eNOS were different in vitro and in vivo. In RAECs, ginsenoside Rk1 significantly increased eNOS mRNA expression, which was not affected by the antagonists for three PPAR subtypes (data not shown), but it did not affect eNOS protein expression ultimately. The mRNA level might not correlate with protein expression due to translational regulation and different in vivo half-lives.38 In contrast, eNOS protein expression was found to be suppressed in aortas from DIO mice and was upregulated by 4-week ginsenoside Rk1 treatment. The effect of ginsenoside Rk1 on eNOS mRNA expression might not be dependent on PPAR activation, whilst its effect on eNOS phosphorylation might involve both PPAR-dependent and -independent signaling pathways.
It is well known that ROS plays a crucial role in vascular complications in diabetes.39 We herein showed that the ROS level was elevated in aortas from DIO mice and in HG-induced RAECs and ginsenoside Rk1 significantly suppressed ROS production. Ginsenoside Rk1 upregulated the expression of Nrf2 and HO-1, implying the role of Nrf2/HO-1 signaling in its anti-oxidative effect. Noteworthily, the antagonists for the three PPAR subtypes abolished the effect of ginsenoside Rk1 on ROS production in endothelial cells. The interaction between PPARs and Akt activation has been found in previous studies. Furthermore, the phosphorylation of Akt leads to Nrf2/HO-1 activation.10 The present data supported that the activation of PPARs is essential for the beneficial effect of ginsenoside Rk1 on oxidative stress, with Nrf2/HO-1 as one of the most likely downstream mechanisms.
Existing evidence indicates that the low bioavailability of ginsenosides currently limits their clinical application due to their relatively large protopanaxadiol (PPD) or protopanaxatriol (PPT) molecular structures.40 The low content of ginsenoside Rk1 in Panax ginseng, its preparation from heat-processed ginseng and ginseng planting-year requirements all contribute to the high cost of purified ginsenoside Rk1 on the market. More efforts are still needed to increase the production of ginsenoside Rk1 in the future. The analysis of the metabolism of ginsenoside Rk1 and its direct binding protein is also meaningful for clarifying its metabolite in the body and how it works to achieve its vaso-protective effect. Moreover, previous studies have demonstrated that ginsenoside Rk1 suppresses the NF-κB pathway to alleviate inflammation or apoptosis in other circumstances.41–43 Regarding the involvement of NF-κB-mediated inflammatory responses in vascular dysfunction associated with diabetes,44 it is worthwhile to investigate the potential role of NF-kB suppression mediated by ginsenoside Rk1 in the future.
5. Conclusion
In conclusion, we are the first to show that ginsenoside Rk1 treatment relieves endothelial dysfunction, inhibits oxidative stress and increases NO production, through the activation of PPARs, Akt/eNOS and Nrf2/HO-1 signaling pathways in aortas from diabetic obese mice (Fig. 6). The novel findings in the present study suggest the potential of ginsenoside Rk1 to be a vaso-protective drug or dietary supplement for the treatment of endothelial dysfunction.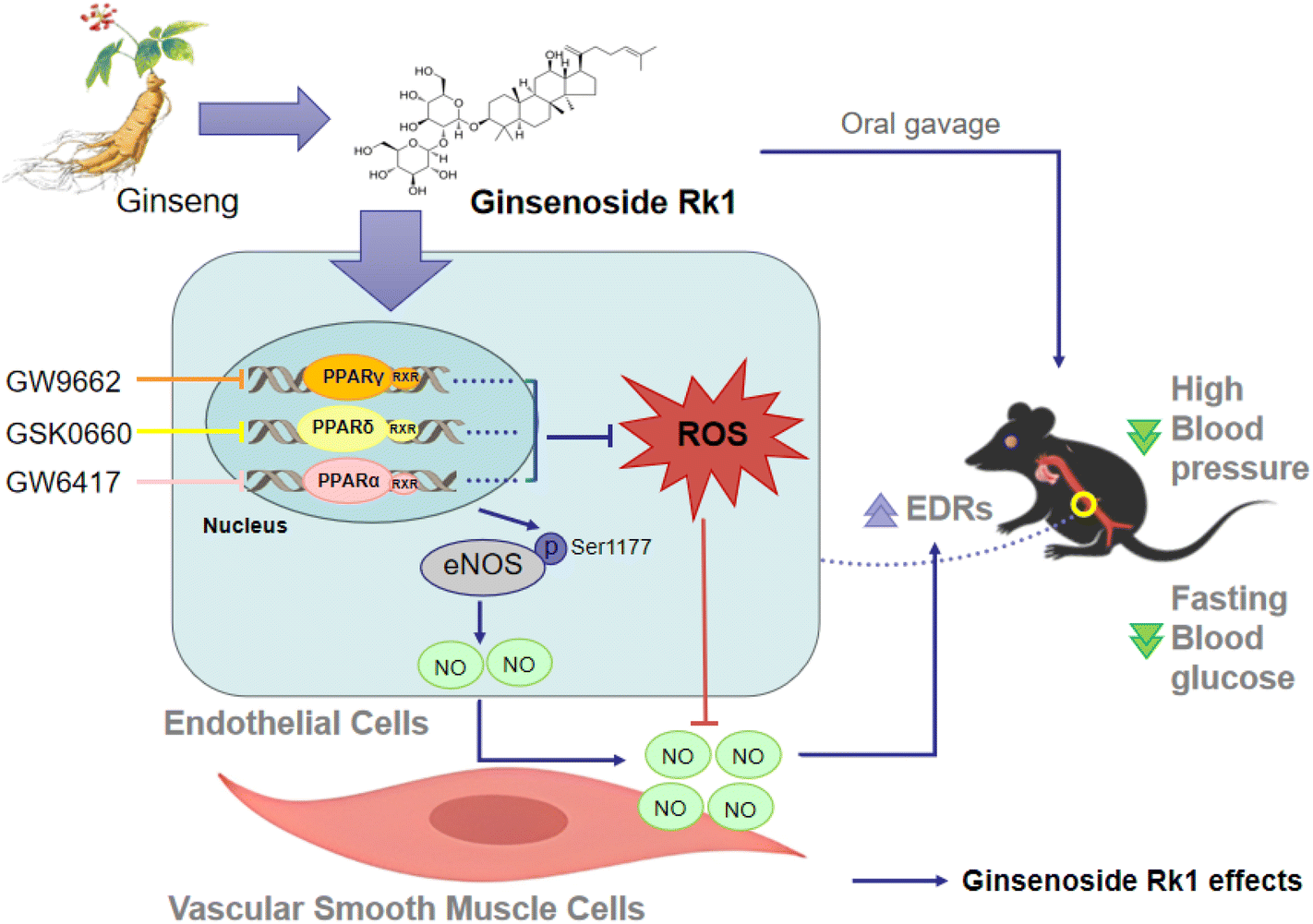 | ||
| Fig. 6 Schematic diagram showing the action of ginsenoside Rk1 to improve endothelial function in diabetes and obesity. Ginsenoside Rk1 treatment leads to the activation of three peroxisome proliferator-activated receptor (PPAR) isoforms to suppress reactive oxygen species (ROS) production and increase nitric oxide (NO) release in endothelial cells, and thereby improves endothelial function in aortas from diabetic obese mice. | ||
Author contributions
Lingchao Miao: investigation, formal analysis, and writing – original draft preparation. Yan Zhou: investigation, formal analysis, and writing – original draft preparation. Dechao Tan: investigation. Chunxiu Zhou: investigation. Cheng-Chao Ruan: writing – reviewing and editing. Shengpeng Wang: writing – reviewing and editing. Yitao Wang: writing – reviewing and editing. Chi Teng Vong: conceptualization, supervision, writing – reviewing and editing, and funding acquisition. Wai San Cheang: conceptualization, supervision, writing – reviewing and editing, and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Science and Technology Development Fund, Macau SAR (FDCT) [005/2023/SKL], University of Macau [MYRG-GRG2023-00211-ICMS-UMDF], International, Hong Kong, Macau and the Taiwan Talent Exchange Project of Guangdong Province, China [KD0120230024], the Major Basic and Applied Basic Research Projects of Guangdong Province of China (2019B030302005; EF013/ICMS-WYT/2019/GPU), and the Young Scientists Fund of the National Natural Science Foundation of China (32200975; EF049/ICMS-CWS/2022/NSFC).References
- W. Bakker, E. C. Eringa, P. Sipkema and V. W. M. van Hinsbergh, Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity, Cell Tissue Res., 2009, 335, 165–189 CrossRef CAS PubMed.
- H. Cai and D. G. Harrison, Endothelial dysfunction in cardiovascular diseases - The role of oxidant stress, Circ. Res., 2000, 87, 840–844 CrossRef CAS PubMed.
- N. Wagner and K. D. Wagner, The Role of PPARs in Disease, Cells, 2020, 9, 2367 CrossRef CAS PubMed.
- L. Han, W. J. Shen, S. Bittner, F. B. Kraemer and S. Azhar, PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-alpha, Future Cardiol., 2017, 13, 259–278 CrossRef CAS PubMed.
- L. Han, W. J. Shen, S. Bittner, F. B. Kraemer and S. Azhar, PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-ss/delta and PPAR-gamma, Future Cardiol., 2017, 13, 279–296 CrossRef CAS PubMed.
- W. S. Cheang, X. Y. Tian, W. T. Wong and Y. Huang, The peroxisome proliferator-activated receptors in cardiovascular diseases: experimental benefits and clinical challenges, Br. J. Pharmacol., 2015, 172, 5512–5522 CrossRef CAS PubMed.
- S. Goetze, F. Eilers, A. Bungenstock, U. Kintscher, P. Stawowy, F. Blaschke, K. Graf, R. E. Law, E. Fleck and M. Grafe, PPAR activators inhibit endothelial cell migration by targeting Akt, Biochem. Biophys. Res. Commun., 2002, 293, 1431–1437 CrossRef CAS PubMed.
- W. Li, F. Ma, L. Zhang, Y. Huang, X. Li, A. Zhang, C. Hou, Y. Zhu and Y. Zhu, S-Propargyl-cysteine Exerts a Novel Protective Effect on Methionine and Choline Deficient Diet-Induced Fatty Liver via Akt/Nrf2/HO-1 Pathway, Oxid. Med. Cell. Longevity, 2016, 2016, 4690857 Search PubMed.
- X. H. Wu, J. M. Wang, L. L. Song, Y. C. Guan, C. Cao, Y. Cui, Y. Y. Zhang and C. Liu, Catalpol Weakens Depressive-like Behavior in Mice with Streptozotocin-induced Hyperglycemia via PI3K/AKT/Nrf2/HO-1 Signaling Pathway, Neuroscience, 2021, 473, 102–118 CrossRef CAS PubMed.
- D. Martin, A. I. Rojo, M. Salinas, R. Diaz, G. Gallardo, J. Alam, C. M. R. de Galarreta and A. Cuadrado, Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol, J. Biol. Chem., 2004, 279, 8919–8929 CrossRef CAS PubMed.
- L. W. Qi, C. Z. Wang and C. S. Yuan, Ginsenosides from American ginseng: Chemical and pharmacological diversity, Phytochemistry, 2011, 72, 689–699 CrossRef CAS PubMed.
- J. H. Kim, Pharmacological and medical applications of Panax ginseng and ginsenosides: a review for use in cardiovascular diseases, J. Ginseng Res., 2018, 42, 264–269 CrossRef PubMed.
- M. Karmazyn, M. Moey and X. T. Gan, Therapeutic Potential of Ginseng in the Management of Cardiovascular Disorders, Drugs, 2011, 71, 1989–2008 CrossRef CAS PubMed.
- H. Liu, X. Lu, Y. Hu and X. Fan, Chemical constituents of Panax ginseng and Panax notoginseng explain why they differ in therapeutic efficacy, Pharmacol. Res., 2020, 161, 105263 CrossRef CAS PubMed.
- Y. Cheng, L.-h. Shen and J.-t. Zhang, Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action, Acta Pharmacol. Sin., 2005, 26, 143–149 CrossRef CAS PubMed.
- P. Zhou, W. Xie, S. He, Y. Sun, X. Meng, G. Sun and X. Sun, Ginsenoside Rb1 as an Anti-Diabetic Agent and Its Underlying Mechanism Analysis, Cells, 2019, 8, 204 CrossRef CAS PubMed.
- J.-j. Xing, J.-g. Hou, Z.-n. Ma, Z. Wang, S. Ren, Y.-p. Wang, W.-c. Liu, C. Chen and W. Li, Ginsenoside Rb3 provides protective effects against cisplatin-induced nephrotoxicity via regulation of AMPK-/mTOR-mediated autophagy and inhibition of apoptosis in vitro and in vivo, Cell Proliferation, 2019, 52, e12627 CrossRef PubMed.
- S. Xiaodan and C. Ying, Role of ginsenoside Rh2 in tumor therapy and tumor microenvironment immunomodulation, Biomed. Pharmacother., 2022, 156, 113912 CrossRef PubMed.
- Y. N. Hong and D. D. Fan, Ginsenoside Rk1 induces cell death through ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway in MCF-7 cells, J. Funct. Foods, 2019, 57, 255–265 CrossRef CAS.
- Q. Yu, K. W. Zeng, X. L. Ma, Y. Jiang, P. F. Tu and X. M. Wang, Ginsenoside Rk1 suppresses pro-inflammatory responses in lipopolysaccharide-stimulated RAW264.7 cells by inhibiting the Jak2/Stat3 pathway, Chin. J. Nat. Med., 2017, 15, 751–757 CrossRef CAS PubMed.
- K. Kim, S. Maharjan, C. Lim, N. J. Kim, V. Agrawal, Y. T. Han, S. Lee, H. An, H. Yun, H. J. Choi, Y. G. Kwon and Y. G. Suh, Glucal-conjugated sterols as novel vascular leakage blocker: Structure-activity relationship focusing on the C-17-side chain, Eur. J. Med. Chem., 2014, 75, 184–194 CrossRef CAS PubMed.
- J. H. Shin, H. W. Kwon, M. Irfan, M. H. Rhee and D. H. Lee, Ginsenoside Rk1 suppresses platelet mediated thrombus formation by downregulation of granule release and alpha(IIb)beta(3) activation, J. Ginseng Res., 2021, 45, 490–497 CrossRef PubMed.
- H. K. Ju, J. G. Lee, M. K. Park, S.-J. Park, C. H. Lee, J. H. Park and S. W. Kwon, Metabolomic Investigation of the Anti-Platelet Aggregation Activity of Ginsenoside Rk1 Reveals Attenuated 12-HETE Production, J. Proteome Res., 2012, 11, 4939–4946 CrossRef CAS PubMed.
- Y.-S. Maeng, S. Maharjan, J.-H. Kim, J.-H. Park, Y. Suk Yu, Y.-M. Kim and Y.-G. Kwon, Rk1, a Ginsenoside, Is a New Blocker of Vascular Leakage Acting through Actin Structure Remodeling, PLoS One, 2013, 8, e68659 CrossRef CAS PubMed.
- Y. S. Lau, X. Y. Tian, Y. Huang, D. Murugan, F. I. Achike and M. R. Mustafa, Boldine protects endothelial function in hyperglycemia-induced oxidative stress through an antioxidant mechanism, Biochem. Pharmacol., 2013, 85, 367–375 CrossRef CAS PubMed.
- J. Su, X. R. An, Q. Li, X. X. Li, X. D. Cong and M. Xu, Improvement of vascular dysfunction by argirein through inhibiting endothelial cell apoptosis associated with ET-1/Nox4 signal pathway in diabetic rats, Sci. Rep., 2018, 8, 12620 CrossRef PubMed.
- X. X. Li, S. K. Ling, M. Y. Hu, Y. Ma, Y. Li and P. L. Huang, Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats, Free Radicals Biol. Med., 2019, 145, 175–186 CrossRef CAS PubMed.
- Y. An, K. Geng, H. Y. Wang, S. R. Wan, X. M. Ma, Y. Long, Y. Xu and Z. Z. Jiang, Hyperglycemia-induced STING signaling activation leads to aortic endothelial injury in diabetes, Cell Commun. Signaling, 2023, 21, 365 CrossRef CAS PubMed.
- M. I. Sonmez, A. Shahzadi, C. Kose, H. Sonmez, S. Ozyazgan and A. G. Akkan, Effect of sulfasalazine on endothelium-dependent vascular response by the activation of Nrf2 signalling pathway, Front. Pharmacol., 2022, 13, 979300 CrossRef CAS PubMed.
- W. S. Cheang, W. T. Wong, L. Wang, C. K. Cheng, C. W. Lau, R. C. W. Ma, A. Xu, N. Wang, Y. Huang and X. Y. Tian, Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor delta, Pharmacol. Res., 2019, 139, 384–394 CrossRef CAS PubMed.
- X. Y. Tian, W. T. Wong, N. Wang, Y. Lu, W. S. Cheang, J. Liu, L. Liu, Y. Liu, S. S.-T. Lee, Z. Y. Chen, J. P. Cooke, X. Yao and Y. Huang, PPARδ Activation Protects Endothelial Function in Diabetic Mice, Diabetes, 2012, 61, 3285–3293 CrossRef CAS PubMed.
- Q. N. Diep, F. Amiri, R. M. Touyz, J. S. Cohn, D. Endemann, M. F. Neves and E. L. Schiffrin, PPARα Activator Effects on Ang II–Induced Vascular Oxidative Stress and Inflammation, Hypertension, 2002, 40, 866–871 CrossRef CAS PubMed.
- D. Bishop-Bailey, Peroxisome proliferator-activated receptors in the cardiovascular system, Br. J. Pharmacol., 2000, 129, 823–834 CrossRef CAS PubMed.
- H. Vosper, G. A. Khoudoli, T. L. Graham and C. N. Palmer, Peroxisome proliferator-activated receptor agonists, hyperlipidaemia, and atherosclerosis, Pharmacol. Ther., 2002, 95, 47–62 CrossRef CAS PubMed.
- Q. N. Diep, M. E. Mabrouk, J. S. Cohn, D. Endemann, F. Amiri, A. Virdis, M. F. Neves and E. L. Schiffrin, Structure, Endothelial Function, Cell Growth, and Inflammation in Blood Vessels of Angiotensin II–Infused Rats, Circulation, 2002, 105, 2296–2302 CrossRef CAS PubMed.
- J. M. Kleinhenz, D. J. Kleinhenz, S. You, J. D. Ritzenthaler, J. M. Hansen, D. R. Archer, R. L. Sutliff and C. M. Hart, Disruption of endothelial peroxisome proliferator-activated receptor-γ reduces vascular nitric oxide production, Am. J. Physiol., 2009, 297, H1647–H1654 CAS.
- F. Zandbergen and J. Plutzky, PPARalpha in atherosclerosis and inflammation, Biochim. Biophys. Acta, 2007, 1771, 972–982 CrossRef CAS PubMed.
- D. Greenbaum, C. Colangelo, K. Williams and M. Gerstein, Comparing protein abundance and mRNA expression levels on a genomic scale, Genome Biol., 2003, 4, 117 CrossRef PubMed.
- Y. Tan, M. S. Cheong and W. S. Cheang, Roles of Reactive Oxygen Species in Vascular Complications of Diabetes: Therapeutic Properties of Medicinal Plants and Food, Oxygen, 2022, 2, 246–268 CrossRef CAS.
- A. Elshafay, N. X. Tinh, S. Salman, Y. S. Shaheen, E. B. Othman, M. T. Elhady, A. R. Kansakar, L. Tran, L. Van, K. Hirayama and N. T. Huy, Ginsenoside Rk1 bioactivity: a systematic review, PeerJ, 2017, 5, e3993 CrossRef PubMed.
- M. Hu, J. Yang, L. Qu, X. Deng, Z. Duan, R. Fu, L. Liang and D. Fan, Ginsenoside Rk1 induces apoptosis and downregulates the expression of PD-L1 by targeting the NF-kappaB pathway in lung adenocarcinoma, Food Funct., 2020, 11, 456–471 RSC.
- Y. Liu, L. Qu, S. Wan, Y. Li and D. Fan, Ginsenoside Rk1 Prevents UVB Irradiation-Mediated Oxidative Stress, Inflammatory Response, and Collagen Degradation via the PI3K/AKT/NF-kappaB Pathway In Vitro and In Vivo, J. Agric. Food Chem., 2022, 70, 15804–15817 CrossRef CAS PubMed.
- Q. Yu, K. W. Zeng, X. L. Ma, Y. Jiang, P. F. Tu and X. M. Wang, Ginsenoside Rk1 suppresses pro-inflammatory responses in lipopolysaccharide-stimulated RAW264.7 cells by inhibiting the Jak2/Stat3 pathway, Chin. J. Nat. Med., 2017, 15, 751–757 CAS.
- S. V. Suryavanshi and Y. A. Kulkarni, NF-kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes, Front. Pharmacol., 2017, 8, 798 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo05222b |
| ‡ Lingchao Miao and Yan Zhou equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |