
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeanut supplementation affects compositions and functions of gut microbiome in Ugandan children†
Jia-Sheng
Wang
 *a,
Kathy
Xue
a,
Zilin
Li
a,
John
Ssempebwa
b,
Gakenia
Wamuyu-Maina
b,
Geofrey
Musinguzi
c,
Jamie
Rhoads
c,
Dave
Hoisington
c and
Lili
Tang
a
*a,
Kathy
Xue
a,
Zilin
Li
a,
John
Ssempebwa
b,
Gakenia
Wamuyu-Maina
b,
Geofrey
Musinguzi
c,
Jamie
Rhoads
c,
Dave
Hoisington
c and
Lili
Tang
a
aDepartment of Environmental Health Science, College of Public Health, University of Georgia, Athens, Georgia 30602, USA. E-mail: jswang@uga.edu
bSchool of Public Health, Makerere University, Kampala, Uganda
cFeed the Future Innovation Lab for Peanut, University of Georgia, Athens, Georgia 30602, USA
First published on 15th March 2024
Abstract
Childhood malnutrition remains a serious global health concern, particularly in low-income nations like Uganda. This study investigated the impact of peanut supplementation on the compositions and functions of gut microbiome with nutritional improvement. School children aged 6–9 years from four rural communities were recruited, with half receiving roasted peanut snacks while the other half served as controls. Fecal samples were collected at the baseline (day 0), day 60, and day 90. Microbial DNA was extracted, and 16S rRNA sequencing was performed, followed by the measurement of SCFA concentration in fecal samples using UHPLC. Alpha and beta diversity analyses revealed significant differences between the control and supplemented groups after 90 days of supplementation. Leuconostoc lactis, Lactococcus lactis, Lactococcus garvieae, Eubacterium ventriosum, and Bacteroides thetaiotaomicron, associated with the production of beneficial metabolites, increased significantly in the supplemented group. Acetic acid concentration also increased significantly. Notably, pathogenic bacteria, including Clostridium perfringens and Leuconostoc mesenteroides, were decreased in the supplemented group. The study indicates the potential of peanut supplementation to modulate the gut metabolome, enrich beneficial bacteria, and inhibit pathogens, suggesting a novel approach to mitigating child malnutrition and improving health status.
1. Introduction
Regular nut consumption has been associated with reduced risks of cardiovascular diseases and diabetes.1 This practice has shown positive effects on lipid profiles, inflammation markers, and endothelial function.2 Peanuts share nutritional similarities with nuts due to their nutrient density, high content of unsaturated fatty acids, plant-based proteins, minerals, and vitamins.3 Additionally, the skin of peanuts contains a significant amount of polyphenols, which are antioxidant substances.4 Furthermore, peanuts are a source of dietary fiber that undergoes fermentation in the gut, producing short-chain fatty acids (SCFAs). These SCFAs, including acetate, propionate, and butyrate, are important mediators of gut health, stemming from their production during the fermentation of indigestible carbohydrates by gut microbes.5 Studies conducted by Griel et al.6 and Moreno et al.7 have demonstrated the positive effects of peanut supplementation on body weight control, dietary quality, and micronutrient status in children. Moreover, peanut paste-based ready-to-use school meals were proven to improve cognition functions.8 A recent study9 showed that regular peanut and peanut butter consumption may enhance memory function and stress response in healthy young populations, which were associated with increased levels of fecal SCFAs. These findings support the potential benefits of incorporating peanuts into the diet to improve nutrition and promote healthy outcomes. However, there is limited understanding about mechanisms of peanut supplementation in the regulation and modification of gut microbiome.Dietary changes can significantly influence the composition and activity of gut microbes, either leading to health or disease.10 As widely known, the Western diet, which contains high-fat and high-sugar intake, may be associated with suspicious microbial changes and lead to various chronic diseases such as obesity11 and inflammatory bowel disease.12 On the contrary, the Mediterranean diet, which emphasizes plant-based foods and healthy fats, could remodel the intestinal microbiome towards a state that promotes metabolism and cardiovascular health.13 The gut houses trillions of microbial cells, mainly bacteria, spanning 500–1000 species and over 70 genera. This collective microbial genome far exceeds the human genome in gene count.14 The dominant phyla of bacteria in the gut microbiome are Firmicutes and Bacteroidetes, followed by Actinobacteria, Proteobacteria, and Verrucomicrobia. These various phyla collectively play a vital role in human health and contribute to the normal functioning of the body. Specifically, the gut microbiome plays a critical role in child development by influencing nutrient absorption and the development of the immune system and function,15 as laid out by Korpela et al.16 on the gut microbiota's influence on child growth patterns and nutritional status. Therefore, understanding the connection between peanut supplementation, gut microbiome modulation, and child development is paramount.
Nuts, such as almonds17 and walnuts18 have been studied for their effects on the gut microbiome. However, there are limited publications about the effects of peanuts on the gut microbiome, especially in school children. This study aimed to bridge the gap by investigating the effects of peanut supplementation on the gut microbiome and microbial metabolism of SCFAs in this vulnerable population. The study's outcomes hold promise for advancing our understanding in this critical domain.
2. Experimental
2.1. Subjects and study design
Fig. 1 illustrates the study design, involving the enrolment of 120 school children residing in four villages (Kituba, Naalya, Nama I, and Nama II), with parental consent obtained for their participation. Children from villages Kituba and Nama II constituted the supplemented group, while children from villages Naalya and Nama I comprised the control group. All 4 villages are within the Mukono District. Following parasite infection screening, which was excluded from the study, a total of 100 children were enrolled in this randomized controlled trial. All participants were provided with the standard local diet. Within the supplemented group, roasted peanuts with skin were administered as snacks, amounting to 1 oz (28.3 g), twice daily for a period of 90 days. The peanut snack was uniformly provided by growers and manufacturers from Uganda via a USAID supported Program and was detected as aflatoxin-free.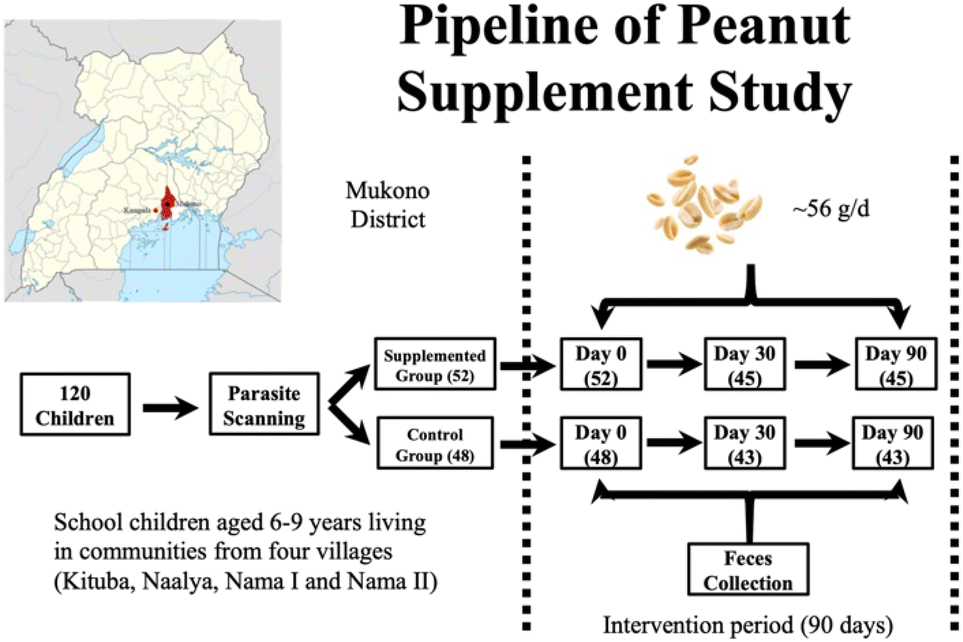 | ||
| Fig. 1 Pipeline of study: the workflow of this study encompassed several stages, commencing with parasite screening, followed by a 90-day intervention period. During this phase, two packages (1 oz or 28.35 g per pack) of peanut snacks were administered daily to the children in the supplemented group. Fecal samples were collected at three time points: baseline (day 0), day 60, and day 90. | ||
Fecal samples were collected at three time points: baseline, day 60, and day 90. Participants were instructed to gather the first complete bowel movement of the day, which was subsequently chilled on ice and stored in a cool box. The assistance of parents was enlisted to facilitate the collection process by collaborating with field-study research assistants. The collected samples were then stored at a temperature of −80 °C until they were ready for further analysis. To ensure a comprehensive grasp of the population-wide impact, samples were pooled from children who shared similar characteristics such as age, sex, and village of residence. To facilitate analysis, samples from 12 subjects were selected for evaluation, evenly divided between the control group (6 pooled batches of samples) and the supplemented group (6 pooled batches of samples). At three time points, the collected samples from these 12 children (0.5 g frozen each) were pooled and pulverized in liquid nitrogen to mix well before DNA extraction. It is noteworthy that there were no statistically significant disparities in the age and sex distributions between the two groups.
2.2. Chemicals and reagents
Analytical grade of acetic acid, propionic acid, butyric acid, valeric acid, hexanoic acid, 2-ethylbutyric acid, 2nitrophenylhydrazine (2-NPH), N-(3-dimethylaminopropyl)-N′ethylcarbodiimide hydrochloride (EDC), and pyridine were procured from Sigma-Aldrich Inc. (St Louis, MO, USA). All remaining reagents and analytical solvents, including methanol, acetonitrile, and water, were obtained at the highest commercially available grade from Honeywell (Morris Plains, NJ, USA).2.3. DNA extraction
Roughly 150 mg of pooled frozen fecal sample was subjected to homogenization utilizing the MP FastPrep-24 5G (MP Biomedicals, Santa Ana, CA, USA) operating at a velocity of 6.5 m s−1 for a duration of 1 minute. This homogenization procedure involved 5 cycles, each separated by a 5-minute pause on ice to maintain optimal conditions. Subsequent to homogenization, DNA extraction was executed using the Zymo Quick-DNA Fecal/Soil Microbe Miniprep Kit (Zymo Research, Irvine, CA, USA), following the manufacturer's stipulated protocols.2.4. Library preparation and 16s rRNA sequencing
As outlined previously,19 a two-step Quadruple-index PCR methodology was employed to construct the 16S rRNA gene libraries. The primer set targeting the V3–V4 region of the 16S rRNA gene was selected for amplification: S-D-Bact-0341-b-S-17, 5′-CCTACGGGNGGCWGCAG-3′ (341F) and S-D-Bact-0785-a-A-2, 5′-GACTACHVGGGTATCTAATCC-3′ (805R).20 The DNA samples were diluted to a concentration of approximately 20 ng μl−1 and underwent PCR amplification using Kapa HiFi HotStart PCR kits (Kapa Biosystems, Inc, Boston, MA, USA). For the first-round PCR, 5 μl of DNA samples, 1.5 μl of forward primer fused with Read1 sequencing primer, and 1.5 μl of reverse primer fused with Read2 sequencing primer were mixed with reagents to attain a total volume of 25 μl. The thermal cycling involved an initial predenaturation step at 98 °C for 3 minutes, followed by 20–25 cycles of denaturation at 98 °C for 30 seconds, annealing at 55 °C for 30 seconds, elongation at 72 °C for 30 seconds, and a final post-elongation step at 72 °C for 5 minutes. For the second-round PCR, 10 μl of the previous PCR product, 5 μl of iTru5 index, and 5 μl of iTru7 index were mixed with reagents to reach a total volume of 50 μl. The thermal cycling comprised an initial pre-denaturation step at 98 °C for 2 minutes, followed by 5–10 cycles of denaturation at 98 °C for 20 seconds, annealing at 60 °C for 15 seconds, elongation at 72 °C for 30 seconds, and a final post-elongation step at 72 °C for 5 minutes.21 The resulting products were purified using Speedbeads (Thermo-Scientific, Waltham, MA, USA) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, subsequently resuspended in 100 μl of TLE (10 mM Tris-HCl and 0.1 mM EDTA), and stored at −20 °C before sequencing.22 The 16S rRNA sequencing procedures were carried out at the Georgia Genomics and Bioinformatics Core (GGBC, University of Georgia, Athens, GA, USA) utilizing Illumina Miseq with v2 500 cycle chemistry. This led to paired-end 250 base reads, yielding an approximate count greater than 10000 reads per sample. The raw sequence data were deposited into the Sequence Read Archive database with the accession number PRJNA1001060.
:1 ratio, subsequently resuspended in 100 μl of TLE (10 mM Tris-HCl and 0.1 mM EDTA), and stored at −20 °C before sequencing.22 The 16S rRNA sequencing procedures were carried out at the Georgia Genomics and Bioinformatics Core (GGBC, University of Georgia, Athens, GA, USA) utilizing Illumina Miseq with v2 500 cycle chemistry. This led to paired-end 250 base reads, yielding an approximate count greater than 10000 reads per sample. The raw sequence data were deposited into the Sequence Read Archive database with the accession number PRJNA1001060.
2.5. SCFAs extraction and preparation
SCFAs extraction and preparation were performed according to the previous study with some modifications.23 In essence, around 110 mg of fecal samples were precisely measured and placed into MP lysing matrix H tubes (2 mL, MP Biomedicals, Santa Ana, CA, USA). Subsequently, 1 mL of methanol at −80 °C was swiftly introduced into the tube, followed by homogenization on the MP FastPrep-24 5G at a speed of 6.5 m s−1 for 1.5 minutes. This homogenization process comprised 5 cycles, each with a 2-minute interval on ice between runs. After homogenization, the tubes underwent centrifugation at 12000 rpm for 10 minutes. Following centrifugation, 100 μL of supernatant was transferred to a 1.5 mL Eppendorf tube, while the remaining supernatant was stored in a separate tube for future use. For internal standardization, a 10 μL stock solution of internal standard (2-ethylbutyric acid) was introduced into the 100 μL supernatant, achieving a final concentration of 0.1 μg μL−1.
To enhance sensitivity, a 2-NPH derivatization approach was employed.24 Essentially, 110 μL of the sample extract (with the added internal standard) was mixed with 45 μL of the derivatization solution. This solution was freshly prepared by combining 15 μL of EDC solution (0.05 g mL−1 in H2O), 15 μL of 2-NPH solution (12.5 mg ml−1 in methanol), and 15 μL of 3% pyridine in methanol (v/v). After gently vortexing, the tubes were placed in a water bath rack at 60 °C for 60 minutes. Following the water bath, the tubes were allowed to equilibrate at room temperature for 5 minutes and then centrifuged at 12000 rpm for 1 minute to recover any condensate adhering to the tube wall. All prepared sample vials were analysed within a 24-hour timeframe.
2.6. UHPLC analysis for SCFAs
An ultra-high-performance liquid chromatographic (UHPLC) system, the Thermo Scientific™ UltiMate™ 3000, in combination with an ultraviolet detector (UV) from Thermo-Scientific (Waltham, MA, USA), was employed for the quantification of SCFAs concentration. The separation of SCFAs was conducted using Acclaim™ 120 C18 Columns (2.1 × 100 mm, 120 Å, 5 μm) from Thermo-Scientific (Waltham, MA, USA). Each specific SCFA was quantified via an internal standard calibration curve approach. For each analysis, a 10 μl injection volume was utilized, with a consistent flow rate maintained at 0.4 mL min−1. The temperature of the oven was set at 45 °C. The mobile phase configuration consisted of 0.2% formic acid–water as mobile phase A and acetonitrile as mobile phase B. The eluent gradient concentration progression was as follows: initially, the system was held at 95% A for 2 minutes; then transitioned from 95% A to 45% A over 10 minutes, remaining at 45% A for 5 minutes; subsequently, the concentration was shifted from 45% A to 0% A in 3 minutes, sustaining 0% A for 5 minutes; finally, a transition from 0% A back to 95% A was executed over 2 minutes to restore the equilibrium. The UV detector operated at a detection channel of 400 ± 5.0 nm, with the reference wavelength set at 510 ± 5.0 nm. Quantification of all SCFAs was conducted using a standard curve in conjunction with an internal standard (IS). The standards encompass acetic acid, propionic acid, butyric acid, valeric acid, and hexanoic acid. 2-Ethylbutyric acid was utilized as the internal standard (IS) to establish the standard curves and mitigate potential technical variations due to its structural similarity with SCFAs. The standard peaks and analytical parameters are shown in Fig. 2 and Table 1. | ||
| Fig. 2 Peaks of SCFAs and IS on UHPLC. From left to right, the peaks with retention time (min) are acetic acid (8.573), propionic acid (10.023), butyric acid (11.573), valeric acid (13.187), 2-ethylbutyric acid (13.887), and hexanoic acid (14.643). | ||
| SCFAs | Standard curvea | R 2 |
|---|---|---|
| a y: AUC of interested SCFAs/AUC of IS; x: concentration of interested SCFAs/concentration of IS. | ||
| Acetic acid | y = 5.0402x + 0.0589 | 0.9999 |
| Propionic acid | y = 4.3835x − 0.4875 | 0.9999 |
| Butyric acid | y = 3.4867x − 0.3453 | 0.9999 |
| Valeric acid | y = 3.656x − 0.0231 | 0.9999 |
| Hexanoic acid | y = 5.3059x − 1.5994 | 0.9975 |
2.7. Sequencing data preparation
The initial processing of raw paired-end reads from 16S rRNA sequencing was accomplished using QIIME2.25 Briefly, the nonbiological sequences (e.g., primers) were trimmed through Cutadapt 4.4.26 Subsequently, the process encompassed demultiplexing and quality filtering, followed by denoising through DADA2.27 The amplicon sequence variants (ASVs) generated by DADA2 underwent alignment using mafft,28 and a phylogeny was constructed using fasttree2.29 An alpha rarefaction curve was employed to determine the sequence depth based on the observed features. Taxonomy annotation was assigned to the ASVs using the q2-feature-classifier30 to match the Silva ribosomal RNA gene database v138.31 Subsequently, the qza file was exported to R Statistical Software v4.3.132via qiime2R v0.99.6 for subsequent analyses.2.8. Statistical analysis
All data analysis was conducted within the R Statistical Software environment. To explore the impact on gut microbiota composition, α- and β-diversity analyses, as well as Linear discriminant analysis Effect Size (LEfSe) analysis, were employed. To mitigate sequence depth bias, all samples were randomly rarefied to 8000 sequences per sample. α-Diversity indices, including the Fischer index,33 observed features, rarity of log modulo skewness34 and rarity of low abundance relative proportion were calculated using microbiome package.35 The Bray–Curtis distance, which examines shared microbes in each sample, was utilized to construct the distance matrix. Principle Coordinate Analysis (PCoA) was then performed to ascertain clustering based on the Bray–Curtis distance matrix. To ensure robustness in rarefaction, all samples were randomly rarefied to 8000 sequences. For compositional dissimilarity analysis, Analysis of Similarities (ANOSIM) and Permutational Multivariate Analysis of Variance (PERMANOVA) were carried out using the vegan package v2.65.36 Specifically, ANOSIM was used to compare the distances between groups with distances within groups37 and PERMANOVA was used to test whether centroids of distance matrices differed.38 LEfSe analysis was executed through the Galaxy module from Dr Huttenhower's lab to identify potential biomarker bacteria.39 This approach combines standard significance tests with additional tests that encode biological consistency and effect relevance. A significance threshold of 0.05 was set for the factorial Kruskal–Wallis test among classes, and the threshold for discriminative features was set at a logarithmic LDA score of 3.5.Normality was assessed using the Shapiro–Wilk test, and homogeneity was evaluated through Levene's test. For normally distributed and homogenous data, Student's t-test (for two groups) and one-way analysis of variance (ANOVA, for more than two groups) were employed to compare differences. For data that did not meet normality and homogeneity assumptions, the MannWhitney test (for two groups) and Kruskal–Wallis H test (for more than two groups) were used. Post hoc analysis in multi-group tests involved Tukey's test in ANOVA and the Games–Howell test in the Kruskal–Wallis H test. In all statistical analyses, a significance level of P < 0.05 was considered statistically significant.
3. Results
3.1. Overall information about sequencing data
A total of 36 pooled batches of samples were selected at three different time points. Through 16S rRNA sequencing at the Georgia Genomics and Bioinformatics Core (GGBC), an average of 54000 paired-reads (ranging from 10769 to 93850 reads) were successfully generated using the 2-step Quadruple-index PCR method for library preparation. Details of sequencing, including initial reads, filtered reads, merged reads, and reads without chimera, are provided in ESI Table 1.† It is noteworthy that a significant majority of samples exhibited over 80% of sequences post-filtering and merging.
Furthermore, the sequencing depth trend demonstrated in Fig. 3 showed a leveling off, indicating that the most common species had been effectively captured under this sequencing depth. This observation shows the efficiency of our sequencing technology in extracting comprehensive microbial information from the fecal samples.
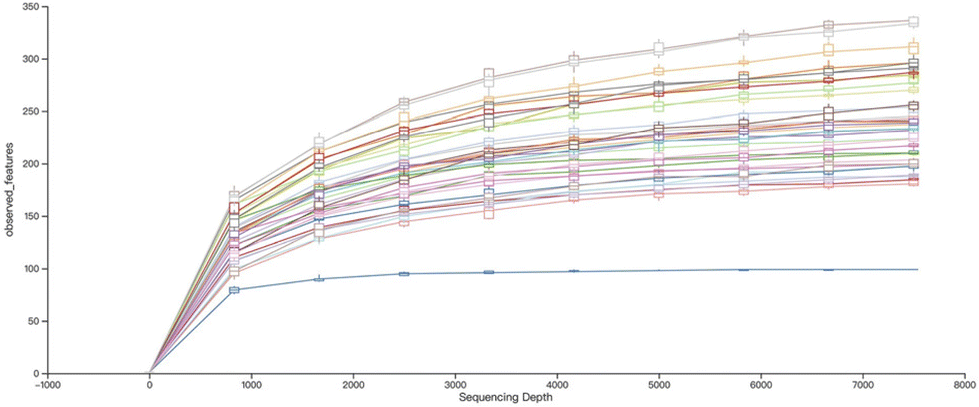 | ||
| Fig. 3 Alpha rarefaction curve based on observed features was presented after the processes of filtering, merging, and elimination of chimeric reads. | ||
Following taxonomy annotation using the Silva ribosomal RNA gene database, the bar plot illustrating the dominant phyla is depicted in Fig. 4. It is notable that both the control and supplemented groups exhibited a consistent pattern of phylum-level composition. The foremost three phyla identified were Firmicutes, Actinobacteria, and Bacteroidetes, which aligns with findings from previous investigations.40 Beyond the bacterial kingdom, archaea also constituted a part of the gut microbial community. This visual representation in the barplot provides valuable insight into the prevailing phylum-level composition within the studied population and indicates the complexity and diversity of the gut microbiota, shedding light on the prominent phyla and their respective abundances within the studied population.
 | ||
| Fig. 4 Bar plot of dominant phyla. Top three were Firmicutes, Actinobacteria, and Bacteroidetes. The most abundant kingdom of microbiome was bacteria, but archaea also existed. | ||
3.2. Alpha diversity
Alpha diversity analysis encompasses various aspects of microbial composition, including richness, evenness, dominance, rare or low abundance species, and coverage. In our study, we examined several indices to capture these characteristics. Initially, we focused on two indices highlighting richness and evenness: the Fischer index (Fig. 5A) and observed features (Fig. 5B). After 90 days of intervention, both indices exhibited significant differences between the control and supplemented groups (P = 0.0186, 0.0177, respectively). These discrepancies suggest that the supplementation may directly influence the quantities of microbial species and their distribution patterns.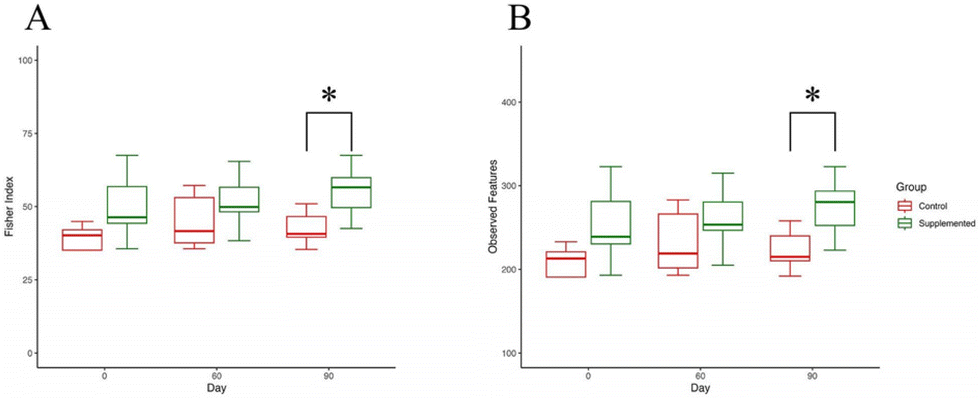 | ||
| Fig. 5 Fischer index and observed features. (A) Fischer index describes the relationship between the number of species and the number of individuals in those species. (B) Observed features counted for richness, which simply reflected the amounts of features found after data preparation. Error bars indicate a 95% confidence interval. *: Student's t-test P < 0.05. | ||
Additionally, we delved into indices addressing the prevalence of rare or low abundance species: rarity of log modulo skewness (Fig. 6A) and rarity of low abundance (Fig. 6B). Remarkably, significant differences were once again identified between the two groups on day 90 (P = 0.0250, 0.0226, respectively). This signifies that the supplementation might impact the microbial composition by modifying the presence of rare species. However, no time-dependent effects were observed across the three time points.
 | ||
| Fig. 6 Rarity of log modulo skewness and rarity of low abundance. (A) Rarity of log modulo skewness used the skewness of the frequency distribution of arithmetic abundance classes and log-modulo transformation to avoid taking the log of occasional negative skews. (B) Rarity of low abundance focused on species whose relative proportion was below the detection level of 0.2%. *: Student's t-test P < 0.05; **: Mann–Whitney test P < 0.05. | ||
3.3. Beta diversity
Unlike alpha diversity, which emphasizes the “amount” of the microbial community, beta diversity centers on the precise “composition” of the community. It enables differentiation between subjects with identical alpha diversity scores, as their beta diversity scores may diverge significantly. A prevalent approach to scrutinizing beta diversity involves constructing a distance matrix for Principle Coordinate Analysis (PCoA). The Bray–Curtis distance, spanning from 0 to 1, signifies the extent of shared species between two samples. Proximity to 1 signifies greater compositional dissimilarity. As illustrated in Fig. 7A and B, during the baseline (day 0) and day 60, both PERMANOVA and ANOSIM analyses exhibited no statistically significant differences in composition (P = 0.68, 0.6, respectively; P = 0.36, 0.33, respectively). However, following the 90-day intervention, both PERMANOVA and ANOSIM indicated a notable dissimilarity between the groups (P = 0.02, 0.01, respectively), visually evident in the PCoA plot where the two groups segregate (Fig. 7C). Notably, PERMANOVA revealed that approximately 15% of the variation between the groups could be attributed to the supplementation. This underlines the significant impact of the supplementation on the microbial composition over time. | ||
| Fig. 7 PCoA of beta diversity. A–C were three time points (day 0, day 60, and day 90). Each dot represented one subject in the group, and two colors represented two groups (green was the supplemented group and red was the control group). The elliptical area around the dots in the same color represented the uncertainty inside the group. Axis.1 and Axis.2 were two mathematical dimensions, which could mostly explain the variation without biological characteristics, and the percentage was the percent of total variation explained by that axis. PERMANOVA and ANOSIM were used to examine the dissimilarity between two groups. | ||
3.4. LEfSe analysis of biomarker bacteria
LEfSe, an algorithm designed for high-dimensional data, serves to identify and explain biomarkers. It identifies genomic features such as genes, pathways, or taxa that characterize distinctions between different biological conditions.39 In our study, we employed LEfSe to explore microbial differences in two contexts: between the control and supplemented groups at day 90 and within the supplemented group between day 0 and day 90.Fig. 8 portrays the outcomes of these comparisons. Notably, three species—Lactococcus garvieae, Lactococcus lactis, and Leuconostoc lactis—associated with fermentation were found to be significantly higher in the supplemented group after 90 days of peanut supplementation. Additionally, two pathogens—Leuconostoc mesenteroides and Clostridium perfringens—were found at lower levels in the supplemented group, hinting that peanut supplementation could potentially contribute to pathogen resistance. Turning attention to the changes within the supplemented group, a similar trend emerged (Fig. 9). Lactococcus garvieae and Lactococcus lactis showed increased abundance after 90 days compared to the baseline. Furthermore, two beneficial species—Eubacterium ventriosum, and Bacteroides thetaiotaomicron—both increased after supplementation.
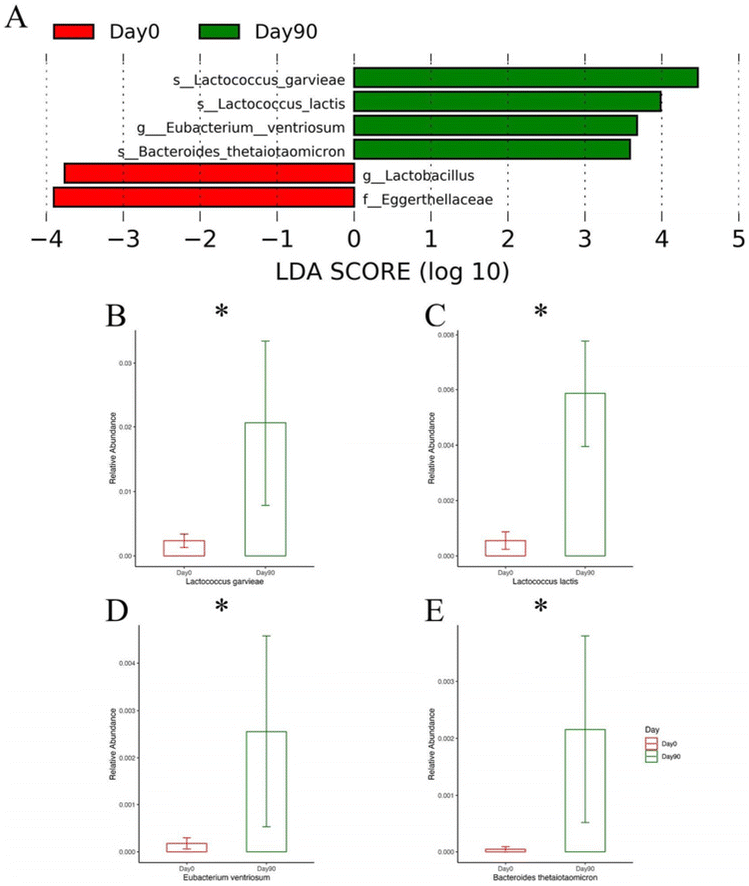 | ||
| Fig. 8 LEfSe analysis compared between day 0 and day 90 in the supplemented group. (A) LEfSe analysis based on LDA score, green color means relatively higher abundance on day 90 and red color means relatively higher abundance on day 0. (B–E) Relative abundance of species and genus higher on day 90 shown on LEfSe analysis plot (Lactococcus garvieae, Lactococcus lactis, Eubacterium ventriosum, Bacteroides thetaiotaomicron, respectively). *: Mann–Whitney test P < 0.05. | ||
 | ||
| Fig. 9 LEfSe analysis compared between the control and supplemented groups at day 90. (A) LEfSe analysis based on LDA score, green color means relatively higher abundance in the supplemented group and red color means relatively higher abundance in the control group. (B–F) Relative abundance of species shown on LEfSe analysis plot (Lactococcus garvieae, Lactococcus lactis, Leuconostoc lactis, Leuconostoc mesenteroides, Clostridium perfringens, respectively); (B–D) species higher in the supplemented group; (E and F) species higher in the control group. *: Mann–Whitney test P < 0.05; **: Mann–Whitney test P < 0.01. | ||
These results collectively suggest that peanut supplementation may enhance the presence of beneficial bacteria and suppress the occurrence of certain pathogens compared to a normal local diet. This provides valuable insights into the potential beneficial effects of the supplementation on the gut microbiota compositions.
3.5. SCFAs concentrations via UHPLC
Following the examination of microbial community composition, our focus shifted to the measurement of short-chain fatty acid (SCFAs) concentrations produced by gut microbes. The concentration of five SCFAs—acetic acid, propionic acid, butyric acid, valeric acid, and hexanoic acid—were quantified using UHPLC. A significant difference in acetic acid concentration was found between different time points in the supplemented group (F2,15 = 3.869, P = 0.044). Moreover, a notable increase in acetic acid levels (12.344 μg per mg fecal samples; 95% CI: 1.256, 23.432) after 90 days were observed compared to day 0 (8.689 μg per mg fecal samples; 95% CI: 1.979, 15.399) (P = 0.036) (Fig. 10A). Furthermore, we noted a significant discrepancy in acetic acid concentration between the control and supplemented groups at day 90 (P = 0.038). It is important to note that no significant differences were observed in the concentrations of other SCFAs. This analysis suggested how peanut supplementation might influence the levels of specific SCFAs and its potential impact on gut microbial metabolism.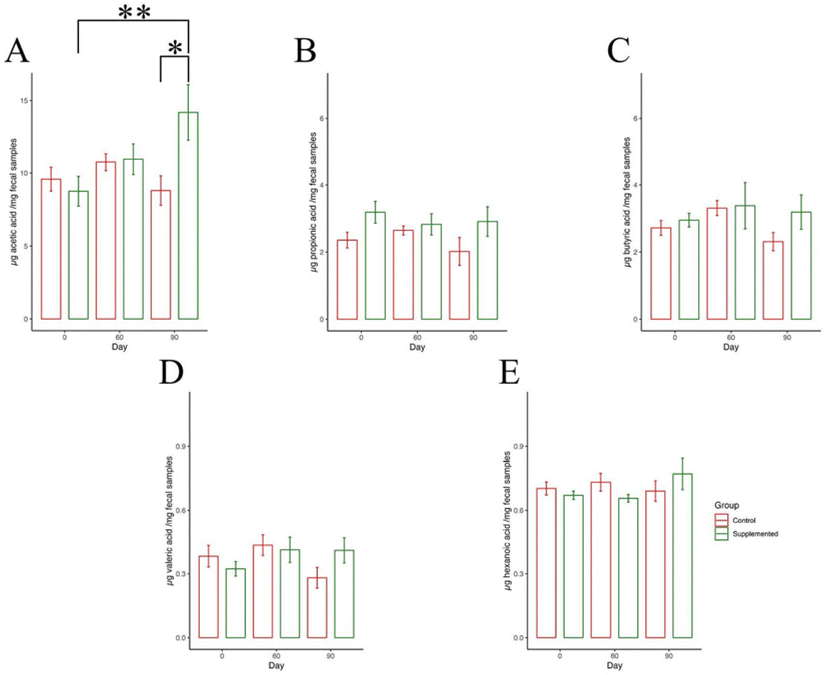 | ||
| Fig. 10 SCFA concentration assessed via UHPLC. (A) Acetic acid (μg mg−1 fecal samples); (B) propionic acid (μg mg−1 fecal samples); (C) butyric acid (μg mg−1 fecal samples); (D) valeric acid (μg mg−1 fecal samples); (E) hexanoic acid (μg mg−1 fecal samples). Student's t-test was used to compare between groups on the same day, and one-way ANOVA was used to compare between three time points within the same group while Tukey's test was used for post hoc analysis; *: Student's t-test P < 0.05; **: Tukey's test P < 0.05. | ||
4. Discussion
Malnutrition, encompassing deficiencies, excesses, or imbalances in nutrient intake, gives rise to under- or overnutrition. In the global context of 2020, an estimated 149 million children experienced stunting, 38.9 million were underweight, and 45.4 million faced wasting.41 Childhood malnutrition continues to loom as a prominent global health issue, particularly in low-income nations such as Uganda.42 Although childhood stunting rates are declining worldwide, Africa remains an exception.41 The aftermath of the COVID-19 pandemic has aggravated the global socioeconomic turmoil, posing a substantial threat to the nutrition and survival of children in low- and middle-income nations.Perturbations in food systems have led to reduced access to nourishing sustenance, restricted healthcare services, and heightened vulnerability to infections. These multifaceted challenges exacerbate the preexisting malnutrition predicament, manifesting the urgency of addressing both immediate and long-term consequences to safeguard children's well-being.43 This study endeavors to probe the effects of peanut supplementation on the gut microbiome and microbial metabolome as an innovative avenue in improving childhood nutrition and health. Such understanding holds promise in shaping strategies to counteract child malnutrition effectively.
The daily peanut snack quantity used in our study, approximately 57 g, is equivalent to about two standard snack packs (1 oz or 28.35 g each) readily available in most grocery stores, making it easily attainable for the average family. These roasted peanuts boast a diverse array of nutrients, including proteins, fats, vitamins, and more, making them a rich source of high-quality protein and essential fatty acids necessary for daily dietary needs. Notably, the presence of polyphenols and dietary fiber within peanuts serves as valuable substrates for fermentation, generating postbiotics like SCFAs, including acetic acid. These SCFAs play a pivotal role in nurturing the growth and maturation of the gut microbial community.44 Importantly, SCFAs such as acetic acid hold significant sway over various aspects of host health, such as fortifying the integrity of the gut epithelial barrier,45 influencing energy metabolism,46 modulating appetite control,47 and even maintaining stable blood pressure levels.48 This complex orchestration involves the participation of a large quantity of microbial species in SCFAs production, albeit in dynamically shifting proportions across different life stages and dietary regimens.49 For instance, infant stages are characterized by the dominance of Bifidobacteria strains, followed by an increase in the relative abundance of Firmicutes as infants transition to a more diverse diet. Our 16s rRNA sequencing results affirm that the supplementation of peanut snacks spurs the significant augmentation of SCFAs-producing species within the Firmicutes phylum. Notably, species like Leuconostoc lactis, Lactococcus garvieae, Lactococcus lactis, and Eubacterium ventriosum exhibit considerable increases, in line with the observed rise in acetic acid concentration as evidenced by UHPLC analysis. This alignment reinforces our contention that peanut supplementation can effectively act as a dietary adjunct, fostering the development of gut microbes that participate in the metabolism of food substrate. Furthermore, the escalation of Bacteroides thetaiotaomicron, a contributor to bile-dependent gut biofilm formation,50 following 90 days of supplementation is noteworthy. This suggests a direct potential benefit to the host in terms of bolstering gut maturation.
On the other hand, gut microbes and their interaction with the gutintestinal system were proven to protect the host from pathogenic infection. For instance, the fermentation activities of lactic acid bacteria (LAB) within the gut ecosystem exhibit the capacity to hinder the proliferation of foodborne pathogens. The ability of LAB to produce organic acids, hydrogen peroxide, and bacteriocins is believed to be responsible for the antimicrobial activity.51 In our study, the reduction of two pathogens, namely Clostridium perfringens and Leuconostoc mesenteroides, when compared to the population consuming a normal local diet, can be attributed to an apparent increase in LAB populations (Leuconostoc lactis and Lactococcus lactis). Clostridium perfringens, is recognized as a common cause of foodborne illnesses in Uganda, potentially leading to severe foodborne necrotic enteritis. Similarly, though possessing low virulence, Leuconostoc mesenteroides has been associated with severe infectious diseases such as bacteremia.52,53 These findings indicate the potential of peanut supplementation in diminishing the risk of pathogen infection, even in scenarios prone to foodborne illnesses. However, further in-depth research is required to illuminate the precise mechanisms underlying this inhibitory effect.
This study has distinct merits. While previous research has studied the effects of other nuts, such as walnuts and almonds, on gut microbiome, our study examined the impact of peanuts on school children. Moreover, our results from both 16s rRNA sequencing and SCFAs measurement via UHPLC exhibit consistency. Whether comparing between groups or time points, the congruence of findings indicates the supplementation's capacity to shape the gut microbial community in terms of compositions, functions and SCFAs metabolism. In summary, the inclusion of peanuts seems to engender a more diverse microbial community, offering protection against pathogen intrusion, promoting the enrichment of beneficial bacteria, and enhancing SCFAs production.
5. Conclusions
This study demonstrates the significant impact of peanut supplementation on the gut microbiome and microbial metabolome in children. The findings highlight the potential of peanuts to promote the growth of beneficial bacteria associated with metabolism, such as the production of SCFAs, while also reducing the presence of pathogenic microbes. These changes are indicative of a healthier gut environment and show the potential of peanut supplementation against childhood malnutrition. This study contributes to our understanding of how dietary interventions can influence the gut microbiome and offers insights into potential strategies for improving childhood nutrition, health and well-being.Author contributions
Jia-Sheng Wang: Study concept, design, manuscript editing, revising, and funding acquisition. Kathy S. Xue: Data analysis, lab supervision, and manuscript revising. Zilin Li: Lab analysis and writing the draft of the manuscript. Lili Tang: Study design and manuscript review. John Ssempebwa: Supplementation study and sample collections. Gakenia W. Maina: Supplementation study and sample collections. Geofrey Musinguzi: Supplementation study and sample collections. Jamie Rhoads: Study design and funding acquisition. Dave Hoisington: Study design and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the research contract (ECG-A-00-1800001-00) from the United States Agency for International Development (USAID) Feed the Future Peanut Innovation Laboratory at the University of Georgia, Athens, Georgia, USA.References
- D. Zhou, H. Yu, F. He, K. H. Reilly, J. Zhang, S. Li, T. Zhang, B. Wang, Y. Ding and B. Xi, Am. J. Clin. Nutr., 2014, 100, 270–277 CrossRef CAS PubMed.
- S. Gulati, A. Misra, R. M. Pandey, S. P. Bhatt and S. Saluja, Nutrition, 2014, 30, 192–197 CrossRef CAS PubMed.
- O. T. Toomer, Crit. Rev. Food Sci. Nutr., 2018, 58, 3042–3053 CrossRef CAS PubMed.
- L. M. Christman, L. L. Dean, C. B. Almeida and J. R. Weissburg, J. Food Sci., 2018, 83, 2571–2577 CrossRef CAS PubMed.
- E. E. Blaak, E. E. Canfora, S. Theis, G. Frost, A. K. Groen, G. Mithieux, A. Nauta, K. Scott, B. Stahl, J. van Harsselaar, R. van Tol, E. E. Vaughan and K. Verbeke, Benefic. Microbes, 2020, 11, 411–455 CAS.
- A. E. Griel, B. Eissenstat, V. Juturu, G. Hsieh and P. M. KrisEtherton, J. Am. Coll. Nutr., 2004, 23, 660–668 CrossRef CAS PubMed.
- J. P. Moreno, C. A. Johnston, A. A. El-Mubasher, M. A. Papaioannou, C. Tyler, M. Gee and J. P. Foreyt, Nutr. Res., 2013, 33, 552–556 CrossRef CAS PubMed.
- K. B. Stephenson, D. R. Wegner, T. G. Hershey, T. Doty, E. Davis, M. Steiner-Asiedu, F. K. Saalia, I. Shani and M. J. Manary, Am. J. Clin. Nutr., 2023, 118, 782–791 CrossRef PubMed.
- I. Parilli-Moser, I. Dominguez-Lopez, M. Trius-Soler, M. Castellvi, B. Bosch, S. Castro-Barquero, R. Estruch, S. HurtadoBarroso and R. M. Lamuela-Raventos, Clin. Nutr., 2021, 40, 5556–5567 CrossRef CAS PubMed.
- L. A. David, C. F. Maurice, R. N. Carmody, D. B. Gootenberg, J. E. Button, B. E. Wolfe, A. V. Ling, A. S. Devlin, Y. Varma, M. A. Fischbach, S. B. Biddinger, R. J. Dutton and P. J. Turnbaugh, Nature, 2014, 505, 559–563 CrossRef CAS PubMed.
- P. J. Turnbaugh, R. E. Ley, M. A. Mahowald, V. Magrini, E. R. Mardis and J. I. Gordon, Nature, 2006, 444, 1027–1031 CrossRef PubMed.
- S. Devkota, Y. Wang, M. W. Musch, V. Leone, H. FehlnerPeach, A. Nadimpalli, D. A. Antonopoulos, B. Jabri and E. B. Chang, Nature, 2012, 487, 104–108 CrossRef CAS PubMed.
- V. Meslier, M. Laiola, H. M. Roager, F. D. Filippis, H. Roume, B. Quinquis, R. Giacco, I. Mennella, R. Ferracane, N. Pons, E. Pasolli, A. Rivellese, L. O. Dragsted, P. Vitaglione, S. D. Ehrlich and D. Ercolini, Gut, 2020, 69, 1258–1268 CrossRef CAS PubMed.
- J. Xu and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10452–10459 CrossRef CAS PubMed.
- L. T. Stiemsma and K. B. Michels, Pediatrics, 2018, 141, e20172437 CrossRef PubMed.
- K. Korpela, P. Costea, L. P. Coelho, S. Kandels-Lewis, G. Willemsen, D. I. Boomsma, N. Segata and P. Bork, Genome Res., 2018, 28, 561–568 CrossRef CAS PubMed.
- J. Dhillon, Z. Li and R. M. Ortiz, Curr. Dev. Nutr., 2019, 3, nzz079 CrossRef PubMed.
- A. M. Tindall, C. J. McLimans, K. S. Petersen, P. M. KrisEtherton and R. Lamendella, J. Nutr., 2020, 150, 806–817 CrossRef PubMed.
- J. Wang, L. Tang, T. C. Glenn and J. S. Wang, Toxicol. Sci., 2016, 150, 54–63 CrossRef CAS PubMed.
- A. Klindworth, E. Pruesse, T. Schweer, J. Peplies, C. Quast, M. Horn and F. O. Glockner, Nucleic Acids Res., 2013, 41, e1 CrossRef CAS PubMed.
- T. C. Glenn, T. W. Pierson, N. J. Bayona-Vasquez, T. J. Kieran, S. L. Hoffberg, J. C. T. Iv, D. E. Lefever, J. W. Finger, B. Gao, X. Bian, S. Louha, R. T. Kolli, K. E. Bentley, J. Rushmore, K. Wong, T. I. Shaw, M. J. Rothrock Jr., A. M. McKee, T. L. Guo, R. Mauricio, M. Molina, B. S. Cummings, L. H. Lash, K. Lu, G. S. Gilbert, S. P. Hubbell and B. C. Faircloth, PeerJ, 2019, 7, e7786 CrossRef PubMed.
- B. Faircloth and T. Glenn, V 2.3 Speedbeads (AKA Serapure): Preparation of an AMPure XP Substitute, University of California, Los Angeles, California (USA), 2016 Search PubMed.
- J. Zhou, L. Tang, J. Wang and J. S. Wang, Toxicol. Sci., 2018, 164, 453–464 CrossRef CAS PubMed.
- R. Peters, J. Hellenbrand, Y. Mengerink and S. V. der Wal, J. Chromatogr. A, 2004, 1031, 35–50 CrossRef CAS PubMed.
- E. Bolyen, J. R. Rideout, M. R. Dillon, N. A. Bokulich, C. C. Abnet, G. A. Al-Ghalith, H. Alexander, E. J. Alm, M. Arumugam, F. Asnicar, Y. Bai, J. E. Bisanz, K. Bittinger, A. Brejnrod, C. J. Brislawn, C. T. Brown, B. J. Callahan, A. M. CaraballoRodriguez, J. Chase, E. K. Cope, R. D. Silva, C. Diener, P. C. Dorrestein, G. M. Douglas, D. M. Durall, C. Duvallet, C. F. Edwardson, M. Ernst, M. Estaki, J. Fouquier, J. M. Gauglitz, S. M. Gibbons, D. L. Gibson, A. Gonzalez, K. Gorlick, J. Guo, B. Hillmann, S. Holmes, H. Holste, C. Huttenhower, G. A. Huttley, S. Janssen, A. K. Jarmusch, L. Jiang, B. D. Kaehler, K. B. Kang, C. R. Keefe, P. Keim, S. T. Kelley, D. Knights, I. Koester, T. Kosciolek, J. Kreps, M. G. I. Langille, J. Lee, R. Ley, Y. X. Liu, E. Loftfield, C. Lozupone, M. Maher, C. Marotz, B. D. Martin, D. McDonald, L. J. McIver, A. V. Melnik, J. L. Metcalf, S. C. Morgan, J. T. Morton, A. T. Naimey, J. A. Navas-Molina, L. F. Nothias, S. B. Orchanian, T. Pearson, S. L. Peoples, D. Petras, M. L. Preuss, E. Pruesse, L. B. Rasmussen, A. Rivers, M. S. R. Robeson 2nd, P. Rosenthal, N. Segata, M. Shaffer, A. Shiffer, R. Sinha, S. J. Song, J. R. Spear, A. D. Swafford, L. R. Thompson, P. J. Torres, P. Trinh, A. Tripathi, P. J. Turnbaugh, S. Ul-Hasan, J. J. J. van der Hooft, F. Vargas, Y. Vazquez-Baeza, E. Vogtmann, M. von Hippel, W. Walters, Y. Wan, M. Wang, J. Warren, K. C. Weber, C. H. D. Williamson, A. D. Willis, Z. Z. Xu, J. R. Zaneveld, Y. Zhang, Q. Zhu, R. Knight and J. G. Caporaso, Nat. Biotechnol., 2019, 37, 852–857 CrossRef CAS PubMed.
- M. Martin, EMBnet. J., 2011, 17, 10–12 CrossRef.
- B. J. Callahan, P. J. McMurdie, M. J. Rosen, A. W. Han, A. J. Johnson and S. P. Holmes, Nat. Methods, 2016, 13, 581–583 CrossRef CAS PubMed.
- K. Katoh, K. Misawa, K. Kuma and T. Miyata, Nucleic Acids Res., 2002, 30, 3059–3066 CrossRef CAS PubMed.
- M. N. Price, P. S. Dehal and A. P. Arkin, PLoS One, 2010, 5, e9490 CrossRef PubMed.
- N. A. Bokulich, B. D. Kaehler, J. R. Rideout, M. Dillon, E. Bolyen, R. Knight, G. A. Huttley and J. G. Caporaso, Microbiome, 2018, 6, 90 CrossRef PubMed.
- C. Quast, E. Pruesse, P. Yilmaz, J. Gerken, T. Schweer, P. Yarza, J. Peplies and F. O. Glockner, Nucleic Acids Res., 2013, 41, D590–D596 CrossRef CAS PubMed.
- R. Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2018 Search PubMed.
- R. A. Fisher, A. S. Corbet and C. B. Williams, J. Anim. Ecol., 1943, 12, 42–58 CrossRef.
- Biological Diversity: Frontiers in Measurement and Assessment, ed. A. E. Magurran and B. J. McGill, Oxford University Press, New York, 2011 Search PubMed.
- S. S. Leo Lahti, et al., Tools for microbiome analysis in R, https://microsud.github.io/Tools-Microbiome-Analysis/, 2017.
- G. J. S. Oksanen, F. Blanchet, R. Kindt, P. Legendre, P. Minchin, R. O'Hara, P. Solymos, M. Stevens, E. Szoecs, H. Wagner, M. Barbour, M. Bedward, B. Bolker, D. Borcard, G. Carvalho, M. Chirico, M. De Caceres, S. Durand, H. Evangelista, R. FitzJohn, M. Friendly, B. Furneaux, G. Hannigan, M. Hill, L. Lahti, D. McGlinn, M. Ouellette, E. R. Cunha, T. Smith, A. Stier, C. T. Braak and J. Weedon, vegan:Community Ecology Package, https://cran.r-project.org/web/packages/vegan/index.html, 2022 Search PubMed.
- K. R. Clarke, Aust. J. Ecol., 1993, 18, 117–143 CrossRef.
- M. J. Anderson, Austral Ecol., 2001, 26, 32–46 Search PubMed.
- N. Segata, J. Izard, L. Waldron, D. Gevers, L. Miropolsky, W. S. Garrett and C. Huttenhower, Genome Biol., 2011, 12, R60 CrossRef PubMed.
- D. Rothschild, O. Weissbrod, E. Barkan, A. Kurilshikov, T. Korem, D. Zeevi, P. I. Costea, A. Godneva, I. N. Kalka, N. Bar, S. Shilo, D. Lador, A. V. Vila, N. Zmora, M. Pevsner-Fischer, D. Israeli, N. Kosower, G. Malka, B. C. Wolf, T. Avnit-Sagi, M. Lotan-Pompan, A. Weinberger, Z. Halpern, S. Carmi, J. Fu, C. Wijmenga, A. Zhernakova, E. Elinav and E. Segal, Nature, 2018, 555, 210–215 CrossRef CAS PubMed.
- Levels and trends in child malnutrition: UNICEF/WHO/The World Bank Group joint child malnutrition estimates: key findings of the 2021 edition, World Health Organization Nutrition and Food Safety Department Technical Report 9789240025257, 2021.
- Uganda Demographic and Health Survey 2016, Uganda Bureau of Statistics and ICF Technical Report FR333, 2018.
- N. Akseer, G. Kandru, E. C. Keats and Z. A. Bhutta, Am. J. Clin. Nutr., 2020, 112, 251–256 CrossRef PubMed.
- Q. Yang, Q. Liang, B. Balakrishnan, D. P. Belobrajdic, Q. J. Feng and W. Zhang, Nutrients, 2020, 12, 381 CrossRef CAS PubMed.
- A. Gonzalez, R. Krieg, H. D. Massey, D. Carl, S. Ghosh, T. W. B. Gehr and S. S. Ghosh, Nephrol., Dial., Transplant., 2019, 34, 783–794 CrossRef CAS PubMed.
- P. Portincasa, L. Bonfrate, M. Vacca, M. D. Angelis, I. Farella, E. Lanza, M. Khalil, D. Q. Wang, M. Sperandio and A. D. Ciaula, Int. J. Mol. Sci., 2022, 23, 1105 CrossRef CAS PubMed.
- Z. Li, C. X. Yi, S. Katiraei, S. Kooijman, E. Zhou, C. K. Chung, Y. Gao, J. K. van den Heuvel, O. C. Meijer, J. F. P. Berbee, M. Heijink, M. Giera, K. W. van Dijk, A. K. Groen, P. C. N. Rensen and Y. Wang, Gut, 2018, 67, 1269–1279 CrossRef CAS PubMed.
- E. S. Chambers, T. Preston, G. Frost and D. J. Morrison, Curr. Nutr. Rep., 2018, 7, 198–206 CrossRef CAS PubMed.
- W. Fusco, M. B. Lorenzo, M. Cintoni, S. Porcari, E. Rinninella, F. Kaitsas, E. Lener, M. C. Mele, A. Gasbarrini, M. C. Collado, G. Cammarota and G. Ianiro, Nutrients, 2023, 15, 2211 CrossRef CAS PubMed.
- N. Bechon, J. Mihajlovic, A. A. Lopes, S. Vendrell-Fernandez, J. Deschamps, R. Briandet, O. Sismeiro, I. Martin-Verstraete, B. Dupuy and J. M. Ghigo, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2111228119 CrossRef PubMed.
- Z. Gao, E. B. Daliri, J. Wang, D. Liu, S. Chen, X. Ye and T. Ding, J. Food Prot., 2019, 82, 441–453 CrossRef CAS PubMed.
- M. G. Menegueti, G. G. Gaspar, A. M. Laus, A. Basile-Filho, F. Bellissimo-Rodrigues and M. Auxiliadora-Martins, BMC Infect. Dis., 2018, 18, 547 CrossRef PubMed.
- G. Bou, J. L. Saleta, J. A. S. Nieto, M. Tomas, S. Valdezate, D. Sousa, F. Lueiro, R. Villanueva, M. J. Pereira and P. Llinares, Emerging Infect. Dis., 2008, 14, 968–971 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo04645a |
| This journal is © The Royal Society of Chemistry 2024 |