
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHepatoprotective efficacy and interventional mechanism of the panaxadiol saponin component in high-fat diet-induced NAFLD mice
Ai
Mi†
 ab,
Qinxue
Hu†
ab,
Ying
Liu
ab,
Yanna
Zhao
ab,
Fenglin
Shen
ab,
Jinjian
Lan
ab,
Keren
Lv
ab,
Bolin
Wang
ab,
Ruilan
Gao
*ab and
Xiaoling
Yu
*ab
ab,
Qinxue
Hu†
ab,
Ying
Liu
ab,
Yanna
Zhao
ab,
Fenglin
Shen
ab,
Jinjian
Lan
ab,
Keren
Lv
ab,
Bolin
Wang
ab,
Ruilan
Gao
*ab and
Xiaoling
Yu
*ab
aInstitute of Hematology Research, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China. E-mail: 20093022@zcmu.edu.cn; gaoruilan@126.com
bZhejiang Provincial Hospital of Traditional Chinese Medicine, Hangzhou, Zhejiang, China
First published on 7th December 2023
Abstract
Dietary administration is a promising strategy for intervention in non-alcoholic fatty liver disease (NAFLD). Our research team has identified a biologically active component, the panaxadiol saponin component (PDS-C) isolated from total saponins of panax ginseng, which has various pharmacological and therapeutic functions. However, the efficacy and mechanism of PDS-C in NAFLD were unclear. This study aimed to elucidate the hepatoprotective effects and underlying action mechanism of PDS-C in NAFLD. Mice were fed a high-fat diet (HFD) for 8 weeks to induce NAFLD and treated with PDS-C and metformin as the positive control for 12 weeks. PDS-C significantly alleviated liver function, hepatic steatosis and blood lipid levels, reduced oxidative stress and inflammation in NAFLD mice. In vitro, PDS-C has been shown to reduce lipotoxicity and ROS levels while enhancing the antioxidant and anti-inflammatory capabilities in HepG2 cells induced by palmitic acid. PDS-C induced AMPK phosphorylation, leading to upregulation of the Nrf2/HO1 pathway expression and downregulation of the NFκB protein level. Furthermore, our observations indicate that PDS-C supplementation improves insulin resistance and glucose homeostasis in NAFLD mice, although its efficacy is not as pronounced as metformin. In conclusion, these results demonstrate the hepatoprotective efficacy of PDS-C in NAFLD and provide potential opportunities for developing functional products containing PDS-C.
1. Introduction
Non-alcoholic fatty liver (NAFLD) is a syndrome in which long-term accumulation of triglyceride-dominated lipids in hepatocytes causes a series of pathological changes in the liver.1 According to an epidemiological survey, the prevalence of NAFLD is more than 25% globally, and those affected are getting younger; thus, it has become a world-wide public health problem.2,3 A chronically imbalanced diet and lack of exercise are major causes of NAFLD.4 The pathogenesis of NAFLD is very complicated, and the “second strike” theory is the most widely accepted, i.e., hepatic lipid deposition and insulin resistance (the first strike) lead to disturbances in intracellular triglyceride synthesis and transport, which triggers a cascade of cytotoxic events dominated by oxidative stress and results in hepatic inflammation (the second strike).5 Actually, with the deepening of research, the “multiple-hit” theory has gradually emerged, which emphasizes the role of systemic, multifactorial metabolic disorders in the development of NAFLD.6 The development from the “second strike” theory to the “multiple-hit” theory gradually reflects the complexity and research trends in NAFLD.7AMP-activated protein kinase (AMPK) is a heterotrimeric complex widely found in eukaryotic organisms and plays a key role in cell growth and energy metabolism. Studies have shown that activated AMPK can attenuate lipid accumulation and improve insulin sensitivity.8 Additionally, AMPK is involved not only in metabolic regulation but also in the regulation of oxidative stress and inflammation.9,10 Indeed, AMPK could directly phosphorylate NF-E2-related factor 2 (Nrf2) to enhance cell defence against oxidants.11,12 Besides, AMPK can also indirectly influence NrF2-mediated responses by altering abundance, posttranslational modification, or transactivation capacity of the transcription factor.13 In comparison, AMPKα deficiency significantly attenuated Nrf2-mediated signaling and functional protection.14,15 It has been shown that the specific activation of AMPK could reduce inflammation in the liver.16 The nuclear factor kappa-B (NFκB) intracellular signal pathway, as a key valve in the inflammatory response, plays an important role in the pathogenesis of NAFLD.17 Both AMPK and Nrf2 can regulate the NFκB signaling pathway, which in turn regulates the expression of various proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β.18–20 Numerous studies have demonstrated that activated AMPK can enhance the anti-inflammatory potential through Nrf2; further studies are required to confirm its inhibitory effect on NFκB and pro-inflammatory factors.21,22
Panax ginseng (Chinese ginseng herb) is widely recognized for its high edible and medicinal value. It has been used as a highly esteemed tonic in China for more than a millennium. Our research team has identified a biologically active component, the panaxadiol saponin component (PDS-C) isolated from total saponins of panax ginseng; this component was formulated into capsules and named as Pai-neng-da (PND).23,24 In a series of long-term and acute toxicity tests, oral PDS-C was found to be non-toxic and safe within the recommended dose range. It did not cause any toxic reactions or injuries in the body. We successfully obtained two certificates of new class-five Chinese patent medicine authorized and granted by State Food and Drug Administration (SFDA) of China.25 PDS-C was used subsequently for animal studies and clinical trials.26–28 Our previous research confirmed that PSD-C was effective in reversing hemocytopenia in mice and rats and efficacious and safe in treatment of primary immune thrombocytopenia (ITP),29 aplastic anemia,30,31 myelosuppression & hemocytopenia.32 Our research demonstrated the multiple effects of PDS-C in promoting energy metabolism, modulating immunity, and improving oxidative damage. However, the effectiveness of PDS-C in NAFLD is currently unknown. Therefore, this study comprehensively evaluated the effects of PDS-C on glucolipid metabolism, oxidative stress, and inflammation in NAFLD. Additionally, this study also elucidated the underlying mechanisms of action, thereby establishing a solid experimental foundation for drug development and clinical application.
2. Materials and methods
2.1. Preparation of PDS-C
As mentioned previously,23 PDS-C (purity 88.93%) was isolated from total ginseng saponins using a macroporous resin method. The preparation process was also reasonably modified and optimized to ensure efficacy while enriching the effective component. Further, the composition of PDS-C was analyzed by high performance liquid chromatography-mass spectrometry (HPLC-MS), and its components were identified by HPLC using specific ginsenoside monomers as reference standards.24,26,29,30 The isolation of PDS-C was carried out using scientifically and technologically advanced methods, which ensured its high quality and stability.The dried fine powder of PDS-C was completely dissolved in distilled water before gavage treatment of the animal model, and the working solution of PDS-C was freshly prepared before each gavage. PDS-C was fully solubilized by the culture solution and filtered to remove bacteria with 0.2 μm filter for cell culture. In addition, the positive control drug metformin was purchased from Shanghai Xinyi Tianping Pharmaceutical Co., Ltd (67210914, Shanghai, China).
2.2. Animal models, diet, and sample collection
All experimental protocols involving animals in this study were approved by the Animal Ethics Committee of Zhejiang Chinese Medical University (Ethical approval no. SYXK (Zhe) 2021-0012) and following the National Institute of Animal Health guidelines for the care and use of laboratory animals. Four-week-old SPF-grade C57BL/6 male mice procured from Shanghai Slaughter Laboratory Animal Co. Ltd (Shanghai, China) were housed in the Animal Experimentation Center of Zhejiang Chinese Medical University, under the appropriate environmental conditions (temperature 22 ± 2 °C, humidity 60%, and 12 h/12 h light/dark cycle). All mice were acclimatized for 1 week.Mice were fed with regular chow (RC, 10%calories from fat, D12450J) as the normal control and HF diet (60% calories from fat, D12492) for NAFLD model, which were purchased from Jiangsu Medicience Biopharmaceutical Co., Ltd, China. All mice were randomly divided into 4 groups (n = 10 per group). The normal control group was fed with regular chow, and other mice of three group were fed with high-fat diet (HFD) for 8 weeks to prepare the mouse model with NAFLD. Subsequently, the RC group and HFD group were given a daily oral gavage with distilled water, the HFD + PDS-C group gavaged with PDS-C 80 mg kg−1 day−1 (the optimal dose in pre-experiment), positive control group (HFD + MET) gavaged with metformin 250 mg kg−1 day−1; all mice were treated continuously for 12 weeks. After treatment, the blood was collected from the ophthalmic venous plexus for Elisa and biochemical determinations, and mice were anesthetized with 1% sodium pentobarbital solution and sacrificed by cervical dislocation. The liver tissues were weighed on an analytical balance, then stored at −80 °C. A part of liver tissues was fixed with 4% polyformaldehyde buffer for histological analysis. Whole blood was centrifuged with 4000 rpm for 15 min to separate serum and stored at −80 °C for further analysis.
2.3. Cell culture and treatment
The human hepatocellular carcinoma cell line HepG2 was obtained from the First Affiliated Hospital of Zhejiang Chinese Medical University. Cells were cultured in low glucose DMEM with 10% fetal bovine serum (FBS, Sigma-Aldrich, USA), 1% penicillin–streptomycin solution (Thermo Scientific Inc., VA), and incubated at 37 °C, 5% CO2, in the presence of 25.6 mg of palmitic acid (PA, Solarbio, China), which was dissolved in 1 mL NaOH with a concentration of 0.1 mol L−1 at 70 °C. It was mixed thoroughly with 9 mL 10% fatty acid-free BSA (Solarbio, China) solution without fatty acids at 55 °C for 2 h. After natural cooling, the solution was filtered and sterilized to obtain a 10 mM L−1 solution of PA, which was stored at −20 °C and water-bathed at 55 °C for 15 min before use.33 HepG2 cells were seeded in culture plates including 6, 24, and 96 wells, with or without 0.5 mM PA for 24 h. For the PDS-C group, the cells were treated with PDS-C for 24 h. In addition, cells treated with AICAR (0.5 mM, HY-13417), compound C (10 mM, HY-13418A), ML385 (0.5 mM, HY-100523), and TBHQ (10 mM, HY-100489) were purchased from MEC (New Jersey, USA).2.4. The analysis of serology, lipid, and liver tissue
The expression levels of serum TNF-α and IL-6 were detected with ELISA kits (Doctoral Biologicals, China) according to the manufacturer's instructions. Serum total cholesterol (TC), triglycerides (TG), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) were measured using automated biochemistry analyzer (HITACHI, Tokyo, JPN). According to the manufacturer's instructions of TC (A111-1), TG (A110-1), malondialdehyde (MDA, A001-3), and superoxide dismutase (SOD, A003-1) kits were purchased from Jiancheng Bioengineering Institute (Nanjing, China). Equivalent weights of frozen liver tissues were homogenized in ethanol, and the supernatants were collected for determination of TC, TG, MDA, and SOD.2.5. The staining of HE, Oil Red, IHC, PAS
Fresh liver tissues were fixed with 10% formalin buffer, then dehydrated, embedded, and cut into sections of 5 μm thickness for staining analysis. The sections were stained with hematoxylin and eosin (H&E) stain by a fully automated stainer (Thermo Fisher Scientific, USA). For lipid staining, fresh livers were dehydrated with 30% sucrose, and 8 μm sections were cut by a frozen slicer and prepared for staining with Oil Red O using a commercial kit (G1261, Solarbio). The sections for immunohistochemical staining were dewaxed, dehydrated, and then permeabilized with 0.05% Triton X 100 and heat-induced citrate solution for antigen repair. Subsequently, SP link detection kit (Gene Tech, China) and DAB kit (ZSGB-BIO, China) were used for IHC staining according to the instructions. The glycogen accumulation in the liver was detected by staining deparaffinized liver tissue sections with a commercial kit (PAS, G1281, Solarbio) according to the manufacturer's instructions. The pathological sections were observed using Nikon Ni-E microscope (Nikon, Tokyo, Japan) or Axiolab 5 microscope (Zeiss, Germany) to collect images. H&E staining analyses according to methods by Elizabeth M. Brunt.34 Oil Red O staining and immunohistochemical staining were quantitatively analyzed using the ImageJ software. For the staining analysis, each group provided a minimum of three samples, with each tissue section observed in at least three or more areas. The blind principle was employed to eliminate subjective bias.2.6. Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Glucose tolerance test was measured respectively at the 8th week of HFD as well as after treatment at the 8th and 12th weeks. Mice were fasted for 12–16 h and injected intraperitoneally with glucose (2g kg−1), the blood was obtained by clipping the tail of the mouse, and blood glucose was measured using a glucometer (Roche, Switzerland) at 0, 15, 30, 60, and 120 min after injection.35Insulin tolerance tests were measured respectively at the end of the 8th week with HFD as well as after treatment at the 8th and 12th weekends. Mice were fasted for 4 h and injected with insulin (0.75 U kg−1) via intraperitoneal injection. Mice blood glucose was measured at 0, 15, 30, 45, and 60 min after insulin injection.36 The area under the curve (AUC) of GTT and ITT were calculated as described previously.37
2.7. Cell viability analysis
The cell activity was assessed using the CCK8 assay, according to the manufacturer's instructions. HepG2 cells were seeded in 96-well plates with 103 cell number per well, and the plates were placed in an incubator overnight. Cells were treated by PDS-C at different concentrations for 24 h. Subsequently, CCK8 working solution was added to each well and incubated for another 1 h, and the absorbance was determined using a multimode reader at 450 nm.2.8. Activation analysis of ROS
The levels of reactive oxygen species (ROS) in the cells were detected using the ROS Assay Kit (S0033S, Beyotime). According to the manufacturer's instructions, DCFH-DA was diluted with serum-free culture medium to make a working solution (10 μM), and the cells were incubated with the DCFH-DA working solution, 2 × 105 cell numbers per well for 20 min. The cells were washed three times using serum-free culture medium, and then the ROS activity was detected using a fluorescence microplate at 488 nm excitation wavelength and 525 nm emission wavelength.2.9. Total RNA extraction and gene expression assessment
Total RNAs were extracted from thawed livers using TRIzol reagent (Absin, China). Reverse transcription of RNA to cDNA was done using a reverse transcription kit (Themo Scientific, Lithuania). The relative expression levels of the genes were detected using TransStart Tip Green qPCR SuperMix kit (Applied biosystems, USA) with β-actin and GAPDH genes as an internal reference. The relative gene expression data were analyzed by the 2−ΔΔCT method. The primer sequences are listed in Table 1.| Gene | Forward | Reverse |
|---|---|---|
| Acc1 | TGCCACCACCTTATCACTATGTA | CCTGCCTGTCTCCATCCA |
| Fasn | TCGTCTATACCACTGCTTACTAC | ACACCACCTGAACCTGAG |
| Srebp1c | CTGTTGGTGCTCGTCTCCT | TTGCGATGCCTCCAGAAGTA |
| Fsp27 | ATCATGGCTCACAGCTTGG | ATTGTGCCATCTTCCTCCAG |
| Cpt1 | CTTCCATGACTCGGCTCTTC | AGCTTGAACCTCTGCTCTGC |
| Cpt2 | CAAAAGACTCATCCGCTTTGTTC | CATCACGACTGGGTTTGGGTA |
| Sod | CCACTGCAGGACCTCATTTT | CACCTTTGCCCAAGTCATCT |
| Nrf2 | CTTTAGTCAGCGACAGAAGGAC | AGGCATCTTGTTTGGGAATGTG |
| Ho1 | AGGTACACATCCAAGCCGAGA | CATCACCAGCTTAAAGCCTTCT |
| β-Actin | AAGACCTCTATGCCAACACAGT | AGCCAGAGCAGTAATCTCCTTC |
| GAPDH | GGGCTGGCATTGCTCTCAATG | CATGTAGGCCATGAGGTCCAC |
2.10. Protein extraction and western blotting
For total protein extraction, 100 mg frozen liver tissue was homogenized by 1 mL RIPA lysis buffer (PP1901, Beyotime) with protease and phosphatase inhibitor cocktail for 30 min. The samples were centrifuged at 4 °C and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min, and the total protein in the supernatant was collected. For the extraction of nuclear proteins, equal weight (100 mg) frozen liver tissues were cut into small pieces, homogenized, and lysed with specific reagents using a nuclear protein extraction kit (KGP150, Key GEN Bio TECH). The samples were centrifuged at 4 °C and 3000 rpm for 10 min; the supernatants were collected to obtain the cytoplasmic proteins. The remaining precipitated samples of cell nucleus were lysed using the Nucleoprotein Reagent for 1 h and shaken vigorously according to the instructions. Subsequently, the samples were centrifuged at 4 °C and 14000g for 30 min; the supernatants were collected as nucleoprotein. All protein samples were quantified to a concentration and a volume of 30 μg per 10 μL using the BCA kit (P0011, Beyotime, China). Proteins were separated on 8%–12% SDS-PAGE (Novozymes, Nanjing, China) and transferred to the NC membrane (42 μm), then incubated by the primary antibodies overnight. Then, the membranes of proteins above were incubated with peroxidase conjugated secondary antibodies (Bio-Rad, USA). The special bands from the conjugation reaction of protein antigen and antibody were visualized by Luminol of the ECL kit (Bio-Rad, USA). The experiment above was repeated three times. The following primary antibodies were used: H3 (ABL1070, Abbkine), PPARα (ABP55667, Abbkine), SREBP1C (ABP52497, Abbkine), NLRP3 (ET1610-93, HUABIO), NRF2(R1312-8, HUABIO), AMPK (HA600078, HUABIO), p-AMPK (ET1701-37, HUABIO), NFκB (ET1603-12, HUABIO), IL-6 (EM1701-45, HUABIO), IL-1β (ET170-39, HUABIO), TNF-α (R1203-1, HUABIO), p-NFκB (GB113882, Servicebio), HO1 (GB1214, Servicebio), PPARγ (GB112205, Servicebio), GAPDH (GB15004, Servicebio), β-ACTIN (GB11001, Servicebio).
000 rpm for 15 min, and the total protein in the supernatant was collected. For the extraction of nuclear proteins, equal weight (100 mg) frozen liver tissues were cut into small pieces, homogenized, and lysed with specific reagents using a nuclear protein extraction kit (KGP150, Key GEN Bio TECH). The samples were centrifuged at 4 °C and 3000 rpm for 10 min; the supernatants were collected to obtain the cytoplasmic proteins. The remaining precipitated samples of cell nucleus were lysed using the Nucleoprotein Reagent for 1 h and shaken vigorously according to the instructions. Subsequently, the samples were centrifuged at 4 °C and 14000g for 30 min; the supernatants were collected as nucleoprotein. All protein samples were quantified to a concentration and a volume of 30 μg per 10 μL using the BCA kit (P0011, Beyotime, China). Proteins were separated on 8%–12% SDS-PAGE (Novozymes, Nanjing, China) and transferred to the NC membrane (42 μm), then incubated by the primary antibodies overnight. Then, the membranes of proteins above were incubated with peroxidase conjugated secondary antibodies (Bio-Rad, USA). The special bands from the conjugation reaction of protein antigen and antibody were visualized by Luminol of the ECL kit (Bio-Rad, USA). The experiment above was repeated three times. The following primary antibodies were used: H3 (ABL1070, Abbkine), PPARα (ABP55667, Abbkine), SREBP1C (ABP52497, Abbkine), NLRP3 (ET1610-93, HUABIO), NRF2(R1312-8, HUABIO), AMPK (HA600078, HUABIO), p-AMPK (ET1701-37, HUABIO), NFκB (ET1603-12, HUABIO), IL-6 (EM1701-45, HUABIO), IL-1β (ET170-39, HUABIO), TNF-α (R1203-1, HUABIO), p-NFκB (GB113882, Servicebio), HO1 (GB1214, Servicebio), PPARγ (GB112205, Servicebio), GAPDH (GB15004, Servicebio), β-ACTIN (GB11001, Servicebio).
2.11. Correction systematic pharmacology data and analysis interaction network of protein–protein
The keywords of panaxadiol saponin monomers contained in PDS-C as screening targets were searched from the Traditional Chinese Medicine System Pharmacology (TCMSP) database (https://tcmspw.com/tcmsp.php) and Swiss Target (https://www.swisstargetprediction.ch). Duplicate data were removed to obtain PDS-C potential targets. The potential targets related to NAFLD were screened from Genecards (https://www.genecards.org), DisGeNET (https://www.disgenet.org), OMIM (https://omim.org), and Therapeutic Target Database (TTD, https://bidd.nus.edu.sg/group/ttd/ttd.asp). Potential targets related to NAFLD were obtained after the removal of duplicate data. All target names were standardized through the UniProt database (https://sparql.uniprot.org). The common potential targets between PDS-C and NAFLD were imported into the String database (https://string-db.org) to obtain interaction (PPI) the network data of protein–protein, which were imported into the Cytoscape 3.8 software to obtain key potential target proteins of PDS-C for NAFLD.2.12. Statistical analysis
All data are expressed as the means ± SEM from at least three independent experiments. Student's t-test was used to analyze the experimental data containing two groups, and one-way analysis of variance was used to analyze the experimental data with ≥3 groups. All data was analyzed by GraphPad Prism 9.0 (GraphPad Software Inc., CA, USA). P < 0.05 was considered to indicate a statistically significant difference.3. Results
3.1. PDS-C reduced body weight and alleviated dyslipidemia in NAFLD mice
The body weights of mice were measured weekly during the experimental period. As shown in Fig. 1A–C, mice fed with a high-fat diet exhibited significantly larger weight and body type compared to the RC group (p < 0.05). However, this phenomenon was reversed by both the PDS-C and MET treatments. Moreover, mice supplemented with PDS-C or MET showed significantly lower growth rates of body weight compared to HFD mice (p < 0.05, Fig. 1D). Biochemical assays showed that serum TG, TC, and LDL were significantly increased (p < 0.05), while HDL-C was significantly decreased (p < 0.05) in HFD mice compared with the RC group. However, PDS-C showed its efficacy in reversing these abnormalities (p < 0.05, Fig. 1E–H). Furthermore, no abnormal changes in skin or hair were observed in mice during the experimental period. These results suggested that PDS-C could safely and effectively reduce body weight and dyslipidemia in HFD mice.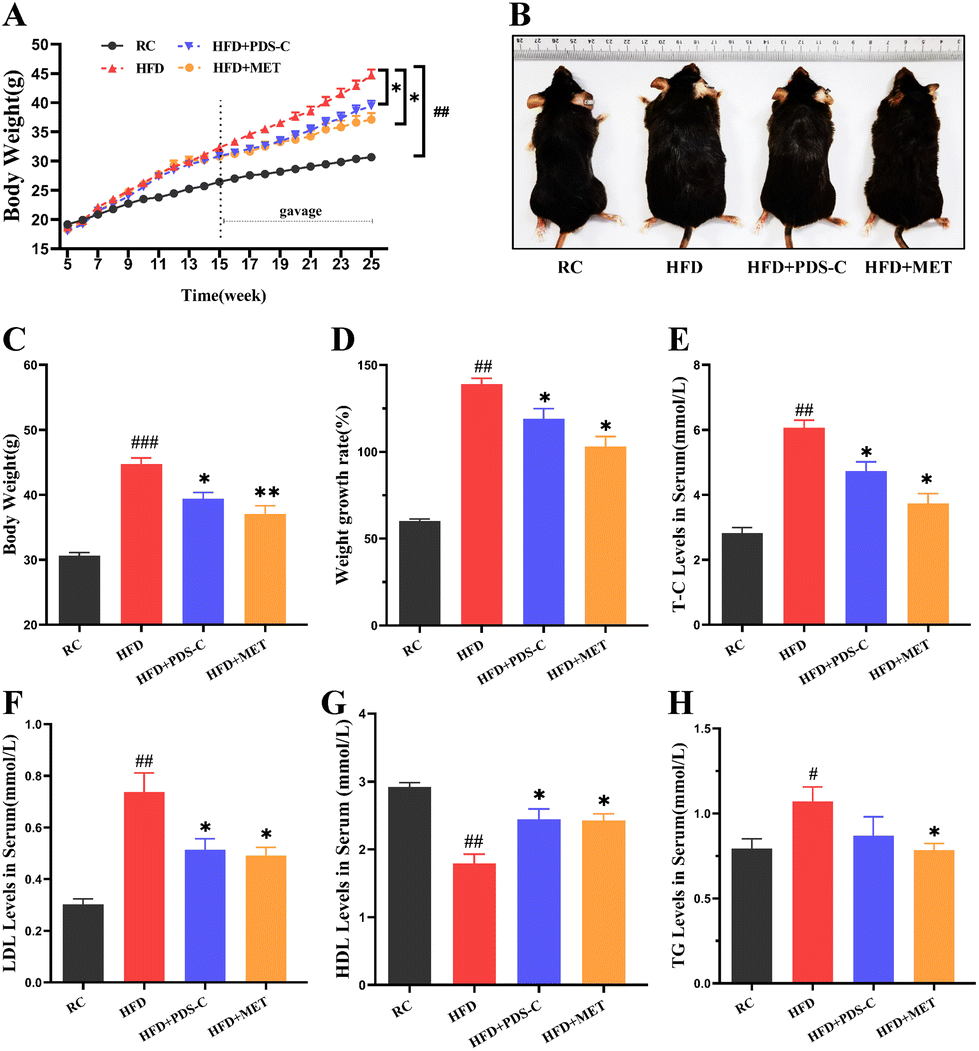 | ||
| Fig. 1 PDS-C reduced body weight and alleviated dyslipidemia in NAFLD mice. (A) Dynamic alteration of body weight during RC or HFD feeding (n = 9). (B) Morphological photographs of the mice. (C) Body weight in mice was measured in the last week. (D) The growth rate of body weight. (E) Serum total cholesterol level (n = 5). (F) Serum triglycerides level. (G) Serum low-density lipoprotein-cholesterol level. (H) Serum high-density lipoprotein-cholesterol level. The data are expressed as mean ± SEM. Comparisons with RC group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
3.2. PDS-C regulated lipid metabolism and improved hepatic steatosis in NAFLD mice
Hepatic steatosis is an important hallmark of NAFLD. In the HFD group, the liver was diffusely enlarged, light, and whitish in color with yellowish-white granules on the surface (Fig. 2A); the liver mass and liver index were significantly increased (p < 0.05, Fig. 2C and D) compared with the RC group. The pathological examination showed that hepatocytes in the HFD group were disorganized, swollen, and also filled with a large number of lipid vacuoles and balloon-like changes (Fig. 2B and E). The Oil Red O (Fig. 2B and F) staining showed that the accumulation of the lipid droplets of liver was significantly increased in the HFD group. Besides, the TG and TC levels of liver tissues and serum ALT and AST were significantly increased in the HFD group (p < 0.05, Fig. 2G–J). However, treatment with PDS-C showed promising results in alleviating the hepatic pathological changes. These results implied that PDS-C treatment could improve hepatic steatosis, revealing relatively normal liver tissue. | ||
| Fig. 2 PDS-C regulated lipid metabolism and improved hepatic steatosis in HFD mice. (A) Morphological photographs of livers. (B) H&E and Oil Red O staining of liver section (400×). (C) Liver weight. (D) Liver index (liver weight/body weight). (E) Steatosis stage of H&E, red arrow: cytoplasmic vacuolation; green arrow: inflammatory cell infiltration. (F) The quantification of the Oil Red positive area. (G) Liver TG content. (H) Liver TC content. (I) Serum ALT level. (J) Serum AST level. (K) The expression levels of genes related to liposynthesis. (L) The expression levels of genes related to fatty acid β-oxidation. The data are expressed as mean ± SEM, n = 5. Comparisons with RC group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
To clarify the role of PDS-C in lipid metabolism, we examined the mRNA expression levels of genes related to lipid synthesis and fatty acid β-oxidation in the liver. As shown in Fig. 2K, the normal heightening genes related to the lipogenesis of liver were induced by HFD, including acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), sterol regulatory element-binding protein 1C (SREBP1C), and fat-specific protein of 27(FSP27). However, treatment with PDS-C effectively downregulated the abnormally high expression of these genes. The expression of fatty acid oxidation-related genes, including carnitine palmitoyl transferase I (CPT1), carnitine palmitoyl transferase II (CPT2), and peroxisome proliferator-activated receptor alpha (PPARα), were significantly upregulated after PDS-C treatment (Fig. 2L). These results indicated that PDS-C could regulate lipid metabolism by inhibiting the expression of liposynthesis genes and promoting the expression of fatty acid β-oxidation genes so as to ameliorate hepatic steatosis.
3.3. PDS-C reduced hyperglycemia, increased insulin sensitivity, and improved glucose homeostasis in NAFLD mice
We monitored important parameters related to the IR in each group of mice throughout the experimental period. After 8 weeks of PDS-C supplementation, the blood glucose of the PDS-C group was lower than the HFD group, but it was still at a high level (Fig. 3A and B). GTT, ITT, and their area under the curve showed that the ability to utilize glucose and the improvement of insulin sensitivity were not significant difference in the PDS-C group (Fig. 3C and D). Interestingly, fasting and random blood glucose (Fig. 3E and F), GTT (Fig. 3G), and ITT (Fig. 3H) were significantly reduced in the PDS-C group with continued administration for 12 weeks (p < 0.05), although PDS-C was not as effective as metformin. Additionally, Periodic Acid-Schiff (PAS) staining showed that PDS-C treatment can partially reverse the excessive glycogen accumulation in the liver of HFD mice (Fig. 3I). These findings indicate that PDS-C treatment for a longer period of time can improve glucose homeostasis by reducing hyperglycemia and increasing insulin sensitivity, which are caused by oversupplied nutrients.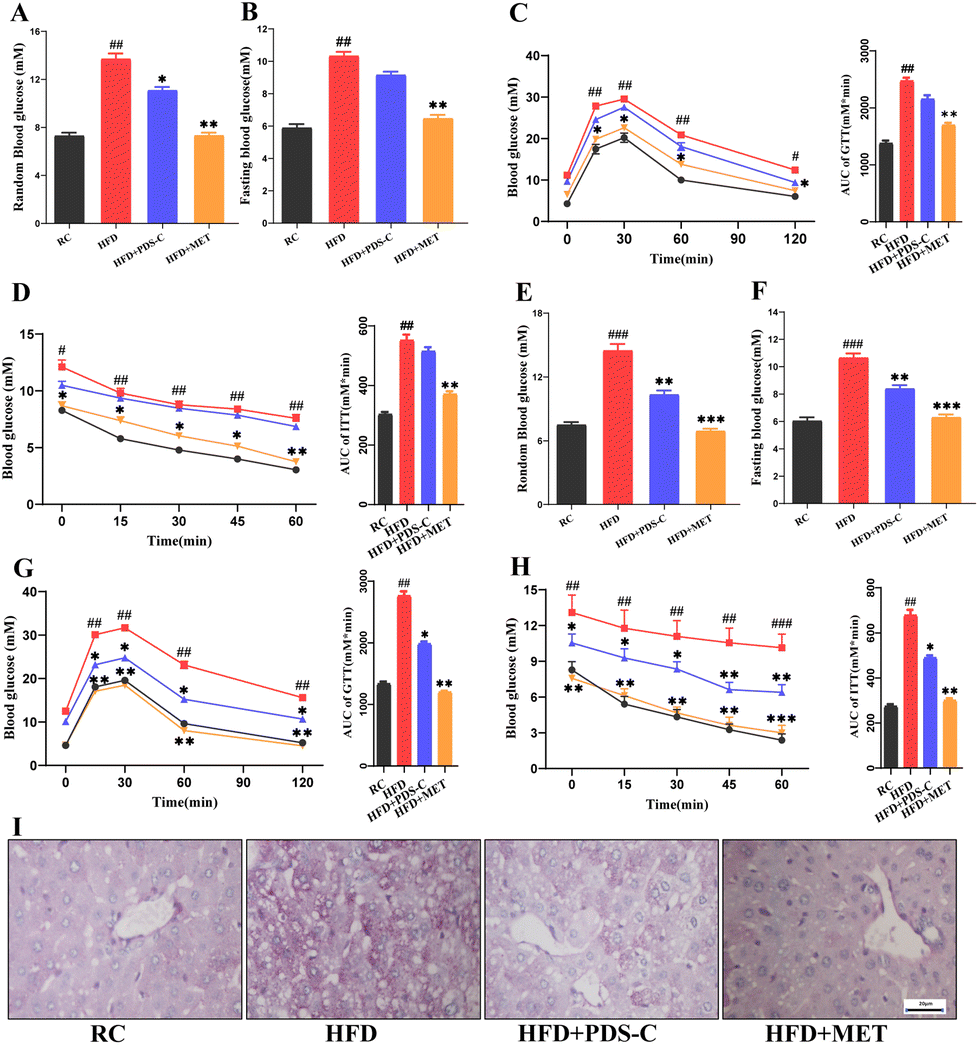 | ||
| Fig. 3 PDS-C reduced hyperglycemia, increased insulin sensitivity in HFD mice. (A) Random blood glucose after treatment for 8 weeks. (B) Fasting blood glucose for 8 weeks of treatment. (C) GTT and its area under the curve after 8 weeks of treatment. (D) ITT and its area under the curve after 8 weeks of treatment. (E) Random blood glucose at 12 weeks of treatment. (F) Fasting blood glucose for 12 weeks of treatment. (G) GTT and its area under the curve after 12 weeks of treatment. (H) ITT and its area under the curve after 12 weeks of treatment. (I) PAS staining of liver section (400×). The data are expressed as mean ± SEM, n = 9. Comparisons with RC group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
3.4. PDS-C exerted antioxidant effects by upregulating the Nrf2/HO1 signaling pathway
The study aimed to evaluate the impact of PDS-C on oxidative stress by measuring the activity of superoxide dismutase (SOD) and the levels of malonic dialdehyde (MDA) in the liver. The results showed that in the HFD group, there was a decrease in the SOD activity and an increase in MDA levels. PDS-C administration reversed these abnormalities (p < 0.05, Fig. 4A and B). Consistently, we found that PDS-C treatment significantly decreased the mRNA levels of various antioxidation-related genes (Fig. 4C), including Heme oxygenase 1 (HO1), NF-E2-related factor 2 (Nrf2), and SOD. Western blot showed that the expression of Nrf2 and HO1 nuclear proteins were significantly decreased in the HFD group compared with the RC group (p < 0.05, Fig. 4D). However, both PDS-C and MET treatment significantly enhanced the nuclear protein expression of Nrf2 and HO1 (p < 0.05). Furthermore, the expression levels of HO1 in total liver protein and immunohistochemical staining exhibited a consistent trend (Fig. 4D and E). It suggested that PDS-C enhanced the antioxidant capacity of hepatocytes by upregulating the levels of Nrf2/HO1 so as to improve HFD-induced oxidative damage, and its effect was similar to MET. | ||
| Fig. 4 PDS-C exerted antioxidant effects by upregulating the Nrf2/HO1 signaling pathway. (A) The activity of SOD in the liver tissues of mice (n = 5). (B) The levels of MDA in liver tissues of mice (n = 5). (C) mRNA expression levels of Ho1, Nqo1, Sod in liver tissue by RT-qPCR (n = 5). (D) Western blot and densitometric analysis the protein levels of total HO1, nuclear Nrf2, HO1 in the liver. (E) Representative images and quantitative analysis of HO1 immunohistochemistry staining in liver. The data are expressed as mean ± SEM. Comparisons with RC group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
3.5. PDS-C exerts anti-inflammatory effects by downregulating the expression of NFκB
In order to clarify the anti-inflammatory effect of PDS-C, the potential drug–disease network was constructed. As mentioned previously, we obtained 151 potential targets mediated by PDS-C active compounds in the database of Traditional Chinese Medicine System Pharmacology (TCMSP). Subsequently, a total of 2406 related targets of NAFLD were screened out from four databases (Fig. 5A). To obtain the intersection of PDS-C target genes and NAFLD target genes, Venn diagram analysis was performed. The results showed that a total of 53 target genes (Fig. 5B) was identified. A protein–protein interaction (PPI) network was constructed using String database and Cytoscape 3.8 software to obtain the pivotal proteins induced by PDS-C in the treatment of NAFLD, which played the role of crucial targets including IL-6, IL-1β, and TNF (Fig. 5C). We discovered that PDS-C has the potential to target NAFLD-associated IL-6 and L-1β and TNF-α from the sub-network (Fig. 5D), which are regulated by NFκB.17,38 The levels of pro-inflammatory cytokines IL-6 and TNF-α in the serum were significantly increased in the HFD group (p < 0.05). However, both PDS-C or MET treatment obviously reduced the levels of IL-6 and TNF-α compared with the HFD group (p < 0.05, Fig. 5E and F). According to these results, we hypothesized that NFκB was probably a key target of PDS-C in NAFLD.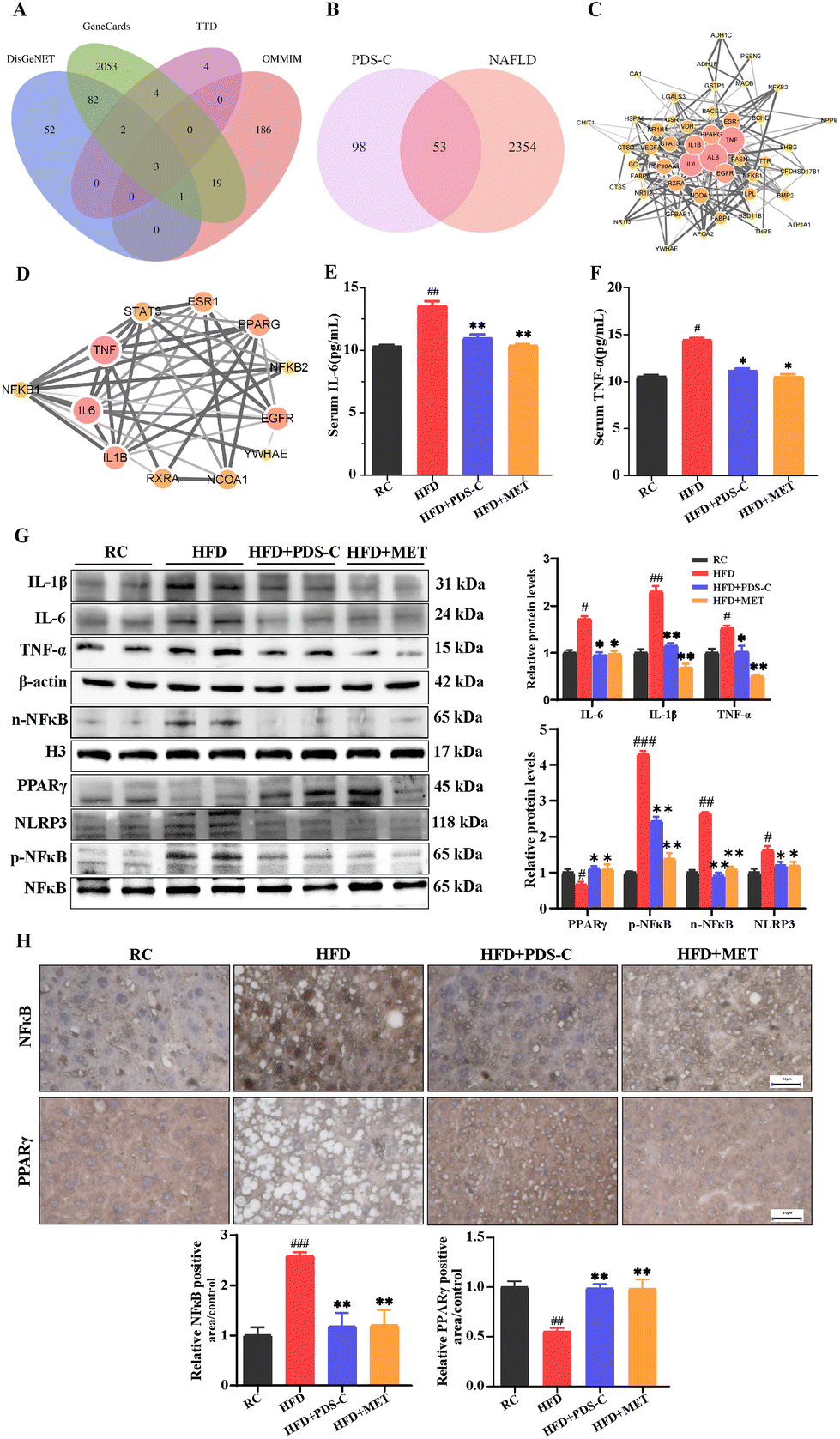 | ||
| Fig. 5 PDS-C exerts anti-inflammatory effects by downregulating the expression of NFκB. (A) Related Targets for NAFLD. (B) Common target genes of PDS-C and NAFLD. (C) Protein–protein interaction (PPI) network. (D) The potential targets of NFκB subnetwork. (E) Serum IL-6 level. (F) Serum TNF-α level. (G) The protein levels of IL-6, IL-1β, TNF-α, p-NFκB, nuclear NFκB, NLRP3, and PPARγ by western blot in liver. (H) Representative images of NFκB and PPARγ detected by immunohistochemistry staining in liver. The data are expressed as mean ± SEM. Comparisons with RC group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
To investigate the effects of PDS-C on anti-inflammatory through NFκB, next, the expression levels of NFκB (p65) and its downstream proteins in the liver were measured by western blotting (Fig. 5G). The protein expression levels of IL-6, IL-1β, and TNF-α in the liver were significantly decreased following PDS-C and MET treatment. HFD feeding led to an increase in nuclear NFκB protein and its phosphorylation status. However, PDS-C treatment effectively reversed the abnormal upregulation of p-NFκB and pyrin domain-containing protein 3 (NLRP3) induced by HFD. Moreover, the protein expression of peroxisome proliferator-activated receptor γ (PPARγ) was significantly decreased in the HFD group compared with the RC group (p < 0.05), while both PDS-C and MET treatment significantly up-regulated the expression of PPARγ in the liver. Consistently, immunohistochemistry analysis was consistent with the trend of NFκB and PPARγ shown by western blot (Fig. 5H). Our results showed that PDS-C inhibited the expression of NFκB and its downstream inflammatory factors, resulting in decreasing inflammatory injury in the NAFLD.
3.6. PDS-C alleviated PA-induced oxidative stress and inflammatory response in HepG2 cells
In vitro, palmitic acid was used to simulate fatty acid influx to construct the NAFLD cell model in HepG2 cells. Subsequently, the cells were treated with different concentrations of PDS-C for 24 h. The CCK8 assay revealed that there was no significant inhibition effect on the growth of HepG2 cells after treatment with PDS-C within 160 mg L−1 (p > 0.05, Fig. 6A). However, the viability of HepG2 cells (PA-HepG2) was significantly inhibited to 60% after incubation with PA (0.5 mM) for 24 h; treatment with PDS-C at a concentration of 80 mg L−1 was able to maintain the cell activity above 80% (p < 0.05, Fig. 6B). In addition, we found that PDS-C (80 mg L−1) significantly decreased ROS in HepG2 cells induced by PA (Fig. 6C). These findings suggested that PDS-C effectively protects HepG2 cells from lipotoxicity. More importantly, PDS-C (80 mg L−1) significantly decreased SREBP1C and elevated PPARa expression in PA-HepG2 cells (Fig. 6D). | ||
| Fig. 6 PDS-C alleviated PA-induced oxidative stress and inflammatory response in HepG2 cells. (A) HepG2 cells were planted and treated with different concentrations of PDS-C for 24 h; the cell viability was detected by the CCK8 assay. (B) PA-treated HepG2 cells were incubated in the presence of PDS-C at 0, 80, 160 mg L−1 for 24 h. (C) The content of ROS in HepG2 cells. (D) Western blot analyzed the protein levels of SREBP1C and PPARα in HepG2 cells. (E) Western blot analyzed the protein levels of HO1, nuclear Nrf2, and HO1 in HepG2 cells. (F) The protein levels of p-NFκB, nuclear NFκB protein (n-NFκB), and cytoplasm NFκB protein (c-NFκB) in HepG2 cells. The data are expressed as mean ± SEM. Comparisons with control group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with PA-HepG2 group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
To further confirm the antioxidant effects of PDS-C, the results showed that the expression of nuclear Nrf2 and HO1 was significantly decreased in PA-HepG2 cells, but PDS-C (80 mg L−1) treatment remarkably upregulated the levels of these proteins (p < 0.05, Fig. 6E). These results indicate that PDS-C can alleviate the abnormal increase in ROS levels and enhance cellular antioxidant capacity by upregulating the Nrf2/HO1 pathway in vitro. Furthermore, we also examined the levels of NFκB induced by PA in HepG2 cells to assess the anti-inflammatory activity of PDS-C. Treatment with PDS-C (80 mg L−1) significantly downregulated phosphorylated NFκB and nuclear NFκB protein, which were abnormally high in PA-induced HepG2 cells (p < 0.05, Fig. 6F). Our results indicated that PDS-C could inhibit the phosphorylation activity of NFκB, reduce the protein abundance of NFκB in the cell nucleus, and alleviated the inflammatory response in NAFLD in vitro.
3.7. PDS-C regulated NAFLD-related metabolic disorders by activating p-AMPK protein
AMPK is a key molecule in bioenergy metabolism and plays an important role in metabolism-related diseases. Western blot was used to detect the phosphorylation status of AMPK in the liver of HFD mice and PA-induced HepG2 cells. In vivo, we observed a significant reduction in the specific band density of p-AMPK in the HFD group compared with the RC group (p < 0.05, Fig. 7A). Consistently, the protein level of p-AMPK was significantly elevated after PDS-C 80 mg L−1 treatment in vitro (p < 0.05, Fig. 7B). All these results suggested that PDS-C could activate the phosphorylation statue of AMPK in vivo and in vitro, thereby participating in the regulation of NAFLD metabolic disorders.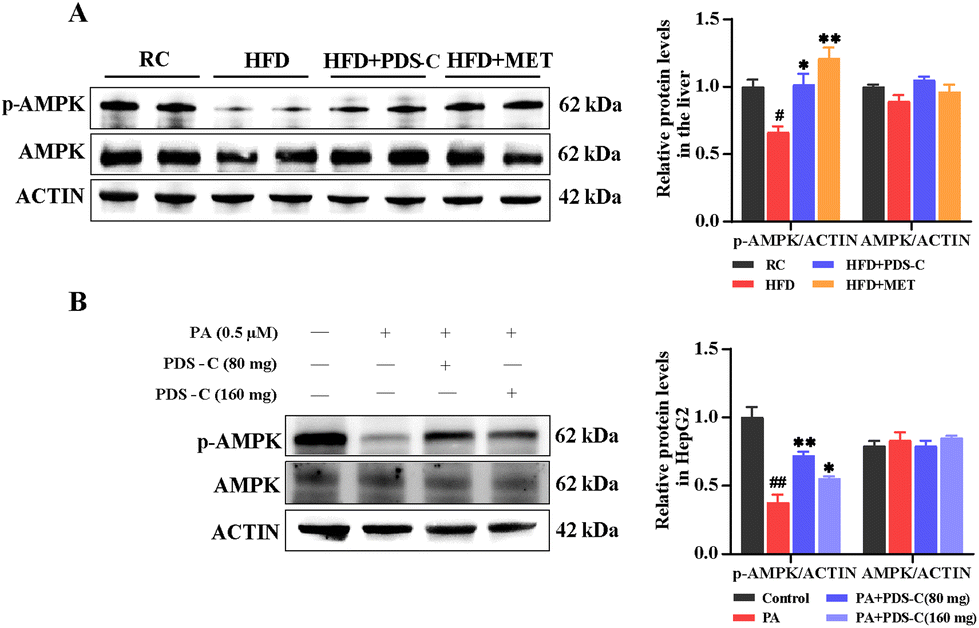 | ||
| Fig. 7 PDS-C regulated NAFLD-related metabolic disorders by activating p-AMPK. (A) Western blot analyzed phosphorylation levels of AMPK in the liver. (B) Western blot analyzed phosphorylation levels of AMPK in the HepG2 cells. The data are expressed as mean ± SEM. Comparisons with RC or control group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with HFD or PA-HepG2 group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
3.8. PDS-C suppressed oxidative stress and inflammatory injury by regulating AMPK/Nrf2/NFκB signaling axis
To further clarify whether PDS-C inhibits oxidative stress and inflammatory injury through AMPK activating Nrf2/HO1 and inhibiting NFκB, PA-HepG2 cells were treated with compound C (CpC) as AMPK inhibitor and AICAR as its agonist, respectively, to observe the regulatory effect of PDS-C on Nrf2/HO1. As shown in Fig. 8A, the inhibitor CpC reduced the expression levels of nuclear Nrf2 and HO1 proteins, located downstream of AMPK, as well as partially counteracted the effect of PDS-C on p-NFκB and nuclear NFκB protein abundance (p < 0.05). However, the efficacy of agonist AICAR was similar to PDS-C in the regulation expression of AMPK. Consistent with previous findings, activated AMPK could decrease oxidative stress and inflammation.12,39 These results suggested that PDS-C could inhibit NFκB by means of enhancing Nrf2/HO1 in PA-HepG2 cells, and the mechanism was related to the activation of AMPK protein. The ML385 as a specific Nrf2 inhibitor and TBHQ as its agonist were used to observe the anti-oxidative effect of PDS-C on inflammatory response. The upregulation of PDS-C on HO1 protein levels in PA-HepG2 cells was partially blocked by the Nrf2 inhibitor ML385. The p-NFκB and nuclear NFκB protein expression levels were significantly increased in PA-HepG2 cells, which were markedly reversed by PDS-C treatment (p < 0.05, Fig. 8B). The Nrf2 inhibitor ML385 revealed partial upregulation of NFκB in PDS-C treated cells. The results suggested that PDS-C exerted beneficial effects on anti-inflammatory by downregulating the expression of p-NFκB and nuclear NFκB protein in PA-HepG2 cells, which was similar to Nrf2 agonist. In addition, the expression levels of cytoplasm NFκB protein in PA-HepG2 cells was lower than that of the control, and PDS-C treatment significantly increased the expression levels of cytoplasm NFκB protein. However, the NFκB protein was downregulated after treatment with the Nrf2 inhibitor ML385. It is suggested that PDS-C was able to obstruct NFκB protein transposition from the cytoplasm into the nucleus. These results indicated that PDS-C could inhibit oxidative stress and inflammatory injury by means of regulating the AMPK/Nrf2/NFκB signaling pathway.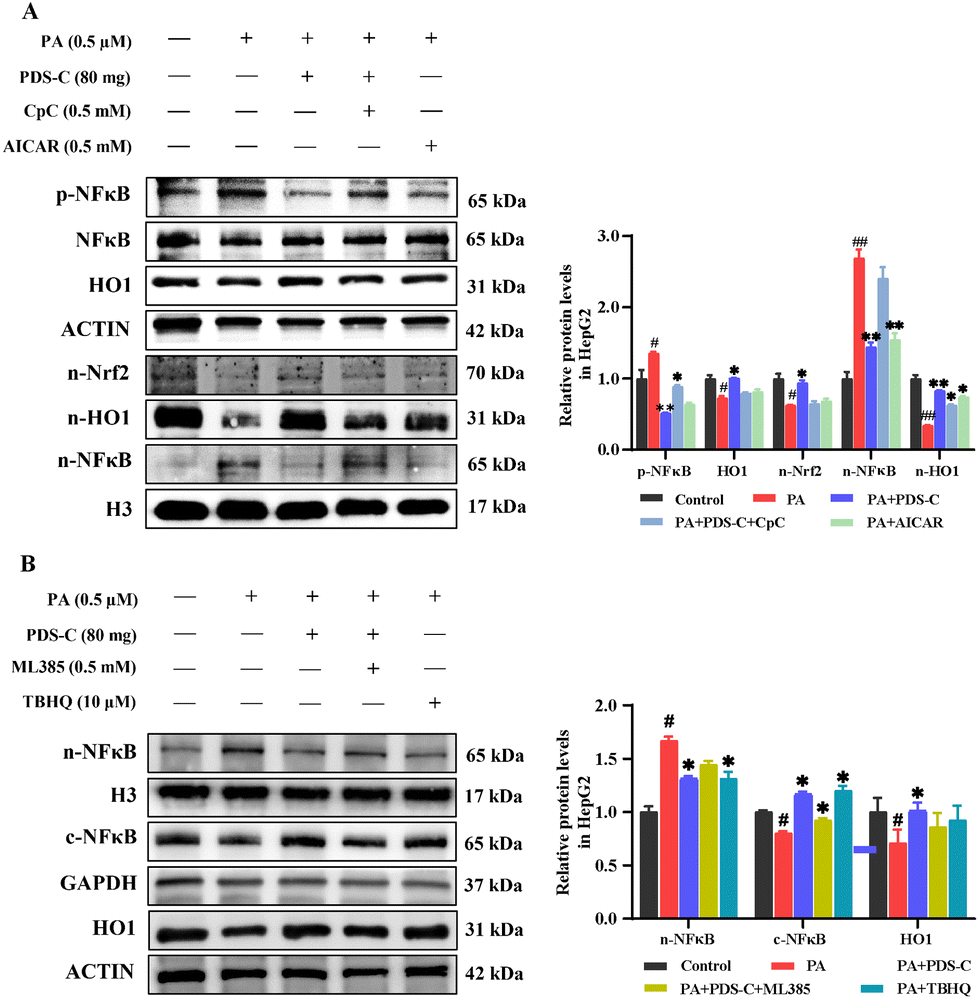 | ||
| Fig. 8 PDS-C suppressed oxidative stress and inflammatory injury by regulating AMPK/Nrf2/NFκB signaling axis. (A) The protein expression levels of p-NFκB, HO1, and nuclear Nrf2, HO1, NFκB in HepG2 cells were observed with the intervention of AMPK inhibitor Compound C and agonist AICAR. (B) The expression levels of HO1, p-NFκB, n-NFκB, c-NFκB protein in HepG2 cells were observed with the intervention of Nrf2 inhibitor ML385 and agonist TBHQ. The data are expressed as mean ± SEM. Comparisons with control group: #p < 0.05, ##p < 0.01, and ###p < 0.001. Comparisons with the PA-HepG2 group: *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
4. Discussion
NAFLD is the main symptoms of metabolic syndrome in the liver; further deterioration would lead to NASH, end-stage liver disease and hepatocellular carcinoma (HCC).40 Lipid accumulation, oxidative stress, and inflammation are the key risk factors for NAFLD.41,42 In recent years, Chinese ginseng herb and its saponins have attracted much attention because of their beneficial effects, including hyperglycemia reduction, fat reduction, antioxidant, and anti-inflammatory.43–45 Previously, we have focused on the study of PDS-C in hematological diseases23,26 and rarely paid attention to its role in protecting the liver and regulating metabolism. In fact, several active monomers of ginsenosides have been shown to have effects on liver injury. Ginsenoside Rb1 could activate AMPK, regulate mitochondrial energy metabolism, improve insulin resistance, and enhance antioxidant capacity.46,47 In addition, Rb1 could reduce inflammatory injury by regulating the NFκB pathway, which was beneficial to the prevention and treatment of metabolic liver disease.48,49 Ginsenoside Rg3 is a star component of panaxadiol saponins, which can effectively improve lipid metabolism disorders and reduce acute and chronic liver injury.50,51 In addition, ginsenoside monomers such as Rb2,52 compound K,53 Rd,54 and Rc55 were also considered to be beneficial for liver protection. In view of the effectiveness of panaxadiol saponin monomers in metabolic liver disease, combined with network pharmacology analysis, we hypothesized that PDS-C may have beneficial effects in reducing blood sugar, serum lipid levels, and exhibiting anti-inflammatory and antioxidant properties. To confirm this hypothesis, we constructed the NAFLD mice model and PA-induced HepG2 cell model to evaluate the effect of PDS-C on NAFLD. Our research was the first to observe that PDS-C ameliorates hepatic steatosis, insulin resistance, reduces oxidative stress, and inflammation by activating AMPK.NAFLD is a metabolic syndrome characterized by lipid accumulation and dyslipidemia, accompanied by liver dysfunction and inflammation production.3,7 The results of our study showed that serum TG, TC, LDL, ALT, AST, inflammatory factors (IL-6, TNF-α) levels, and the content of TC and TG in liver tissue were significantly elevated in NAFLD mice. However, treatment with PDS-C could reverse these abnormalities.
AMPK, a major target of lipid metabolism,56 regulates several key transcription factors of lipid metabolism, including SREBP1C, ACC, and FASN,57 which are key molecules for TG synthesis in the liver, while FSP27 controls lipid droplet synthesis.58 Lipid accumulation disrupts mitochondrial β-oxidation of hepatocytes; however, activated AMPK pathway enhances the expression of PPARα, which can reduce lipid accumulation by promoting the oxidation of mitochondrial fatty acid.59,60 The mRNA expression levels of SREBP1C, ACC, FASN, and FSP27 genes related with lipid synthesis were significantly upregulated in the NAFLD mice. The mRNA expression levels of CPT1, CPT2, and PPARα genes related to fatty acid oxidation, were significantly downregulated; on the other hand, PDS-C treatment could reverse these abnormalities in NAFLD mice, which indicated that the role of PDS-C was similar to metformin as the positive control. In addition, the potential of PDS-C in regulating lipid metabolism is comparable to that of ginsenoside monomers, such as Rb1 and Rb2.46,52 Importantly, we observed that PDS-C significantly increased the phosphorylation level of AMPK in vivo and in vitro, suggesting that the effects of PDS-C on improving metabolic disorders may be attributed to the activation of AMPK as a key factor for NAFLD mice.
Insulin resistance (IR) is closely associated with NAFLD, and studies have shown that individuals with IR tend to develop metabolic diseases including NAFLD. IR drives hepatic de novo lipogenesis in NAFLD.61 IR is implicated both in pathogenesis of NAFLD and in disease progression from steatosis to NASH.62 Thus, it has become a consensus that modulation of IR represents a potential strategy for NAFLD treatment.3 Metformin is a clinically recognized hypoglycemic and weight-loss agent commonly used for the treatment of NAFLD (with hyperglycemia). In addition, metformin has been shown to have antioxidant and anti-inflammatory effects.63,64 In our study, NAFLD mice were accompanied by significant hyperglycemia and IR. Considering the significant role of IR in the development of NAFLD, it is important to take this factor into account when selecting therapeutic agents and aligning with the clinical medication strategy. For instance, using metformin as a reference, we can utilize the blood glucose-lowering and IR-improving abilities of PDS-C to further analyze its mechanism of action in treating NAFLD. In this study, NAFLD mice with PDS-C treatment, blood glucose was decreased to a certain extent from early stage 8 weeks, until 12 weeks, but the reduction degree of PDS-C was not as good as the positive control of metformin, suggesting that PDS-C was effective to improve IR and glucose homeostasis, although its effects and specificity were not as good as metformin. Based on the potential of PDS-C for lipid-lowering, anti-inflammatory, and antioxidant, it is expected that the combination of PDS-C and metformin will be a synergistic effect and useful to improve the efficacy of metformin and enhance the lipid-lowering, glucose-lowering, anti-inflammatory, and antioxidant effects.
The liver is the primary site for lipid synthesis and decomposition. When hepatocytes become overwhelmed with processing free fatty acids, it can lead to lipotoxicity, which involves various cytotoxic events such as oxidative stress and inflammation.65 Unsaturated OA (oleic acid) and saturated PA are the two most abundant free fatty acids in plasma. Normally, their concentrations remain within the physiological range. However, in patients with hyperlipidemia, the concentrations of PA and OA were increased in different degrees, resulting in the accumulation of a large amount of TG in hepatocytes and accelerating the progression of fatty liver disease.66In vitro, PA or OA as inducers are often used to mimic hepatocyte injury caused by excessive fatty acids. However, some studies have shown that unsaturated OA is less toxic than saturated PA, and OA can even prevent PA-induced hepatotoxicity.67,68 Evidence suggests that the excessive intake of saturated fatty acids (SFA) is more likely to cause liver steatosis, oxidative damage, and dysfunction compared to unsaturated fatty acids (USFA).69 Therefore, reducing hepatic lipotoxicity may be an effective strategy to attenuate NAFLD induced by HFD. In this study, we used saturated PA as a cellular additive to observe the protective effect of PDS-C against PA-induced hepatocyte injury in vitro.
Consistent with previous reports, we observed an increased level of MDA and decreased activity of SOD in liver tissues of NAFLD mice, while the ROS activity was also significantly enhanced in PA-HepG2 model cells, which are considered to be markers of oxidative stress.70 Oxidative stress, as the initiator of the “second strike” of NAFLD, promotes hepatic steatosis and inflammatory injury, accelerating the development of NAFLD.71 In this study, PDS-C corrected the aberrant status of MDA and SOD in the livers of NAFLD mice and reversed the excessive ROS levels in PA-HepG2 cells. More importantly, the expression of nuclear Nrf2 and HO1 proteins was significantly upregulated, which was induced by PDS-C in vivo and in vitro. As an important antioxidant transcription factor, Nrf2 plays a crucial role in the body and cells against oxidative stress and inflammation.72,73 In NAFLD mice with Nrf2 deficiency, oxidative stress and inflammatory response were more severe, and NAFLD was more likely to develop into NASH.74 On the contrary, the activation of Nrf2 can reverse insulin resistance and hepatic steatosis and alleviate NASH and liver fibrosis.75,76 The antioxidant pathway Nrf2/HO1 plays a key role in protecting the liver from oxidative damage.77 In view of the above results, PDS-C could inhibit oxidative stress by means of enhancing the antioxidant properties of Nrf2/HO1, indicating that the function of PDS-C was similar to the antioxidant.
The impact of inflammation on NAFLD is complex, and inflammation can lead to lipid deposition and insulin resistance; furthermore, inflammation was stimulated by free fatty acids to accelerate the disease process from NAFLD to NASH.78–80 NFκB is a key factor in the inflammatory response, which can mediate the expression and secretion of various inflammatory factors such as IL-6, IL-1β, and TNF-a.38 Research has demonstrated the involvement of PPARγ in the regulation of NFκB and the inflammatory response.81 Moreover, the overactivation of NLRP3 inflammasome could trigger the release of numerous pro-inflammatory proteins, thereby promoting the progression of NAFLD. However, inhibiting the nuclear translocation of NFκB can effectively suppress NLRP3 activation and alleviate the inflammatory damage associated with NAFLD.82 PDS-C treatment significantly reduced hepatic and systemic inflammatory responses by suppressing the levels of inflammatory cytokines above.83 Furthermore, PDS-C inhibited the phosphorylation status and nuclear translocation of NFκB by activating AMPK-mediated Nrf2/HO1 signaling pathway both in vitro and in vivo.
The Nrf2 transcription factor is a downstream signal of AMPK in regulating oxidative stress and inflammation in NAFLD.13,84,85 To clarify that PDS-C played its anti-oxidative and anti-inflammatory role by means of the regulated Nrf2/HO1 and AMPK pathway, PA-HepG2 cells were treated with PDS-C, followed by supplementation with the AMPK inhibitor Compound C. Interestingly, the regulatory effects of PDS-C on Nrf2/HO1 and NFκB could be partially counteracted by the inhibitor CpC, implying that the role of PDS-C participated in the regulation of AMPK and Nrf2/HO1 pathway since the NFκB pathway could be regulated by AMPK and Nrf2.20 In the Nrf2-mediated oxidative stress, PDS-C treatment showed a significantly downregulated expression of NFκB protein in the nucleus, which was consistent with increasing NFκB protein of cytoplasm. However, the Nrf2 inhibitor ML385 partially increased the level of nuclear NFκB protein; meanwhile, the content of NFκB protein in the cytoplasm was correspondingly reduced. Besides, PDS-C was useful in inhibiting the translocation of NFκB protein from the cytoplasm into the nucleus. It confirmed that PDS-C was useful in alleviating oxidative stress and inflammation via the regulating of the signal pathway of AMPK/Nrf2/NFκB.
In our study, PDS-C at a dosage of 80 mg kg−1 demonstrated significant hepatoprotective effects in NAFLD mice. These effects included the reduction of blood lipids, anti-inflammatory, and antioxidant properties as well as improvement in insulin resistance. Our previous studies also support the wide range of physiological and pharmacological effects of PDS-C (80 mg kg−1) in various animal models. For instance, in aplastic anemia model mice, PDS-C at a dosage of 80 mg kg−1 effectively stimulated the proliferation of erythroid, granulocyte, and megakaryocytic hematopoietic progenitor cells. In addition, 80 mg kg−1 PDS-C successfully regulated spleen T lymphocyte subsets, leading to a normalization of the abnormal CD4+/CD8+ cell ratio.23,24,31 Similarly, PDS-C demonstrated the ability to suppress abnormal immunity and promoted platelet production in ITP model mice, with the optimal performance observed at a dosage of 80 mg kg−1.29,86 These studies highlight the dual effects of 80 mg kg−1 PDS-C in promoting hematopoiesis and regulating immunity. Importantly, considering the dose relationship between animal models and clinical application as well as the results of Phase I clinical trials,87 the recommended safe dosage range for clinical use is 6–10 PND (or PDS-C) capsules (40 mg per capsule), equivalent to 240–400 mg day−1. In a multi-center clinical study, a dosage of 320 mg day−1 of PDS-C was used to replace half the amount of androgen in the treatment of chronic aplastic anemia.27 This treatment approach showed good efficacy, demonstrating that the dosage utilized in animal model experiments can be effectively applied in clinical treatment.
Although the current study has demonstrated the favorable hepatoprotective effects of PDS-C, there are still a few deficiencies in this study. For example, the effects of PDS-C on daily food intake and improvement obesity in mice were not observed. The changes in gut microbiota and its metabolites have a significant effect on the progression of NAFLD,88 whether PDS-C alleviates NAFLD by ameliorating intestinal barrier dysfunction is the focus of our next research. In conclusion, PDS-C exhibited a certain degree of hypoglycemic ability and anti-hepatic steatosis, ameliorating oxidative stress and relieving inflammatory damage by activating AMPK (Fig. 9). Our research illustrated that PDS-C was a potential hepatoprotective adjuvant or dietary supplement for NAFLD and provides experimental evidence for the treatment of NAFLD in the future.
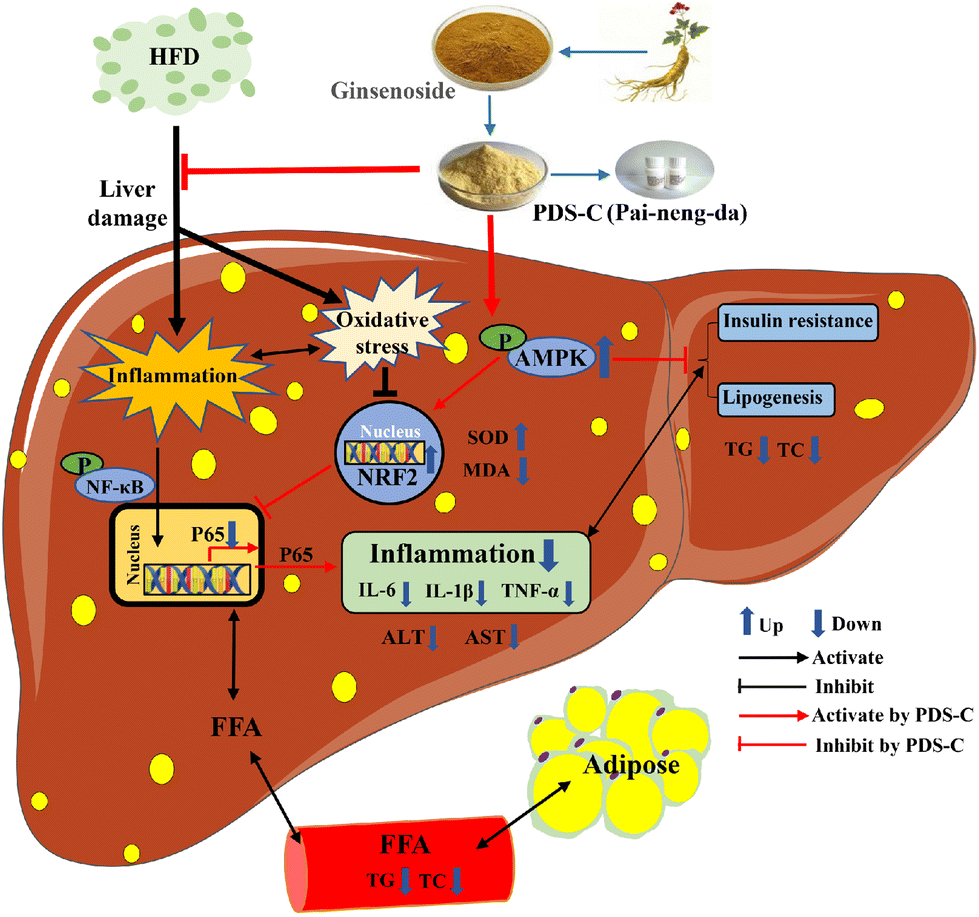 | ||
| Fig. 9 Effectiveness of PDS-C on NAFLD and its associated mechanisms. PDS-C protects against HFD-induced hepatic steatosis, improves glucose homeostasis, and suppresses oxidative stress and inflammatory responses via the AMPK signaling pathway. | ||
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| PDS-C | Panaxadiol saponin component |
| MET | Metformin |
| PA | Palmitic acid |
| PAS | Periodic acid-schiff |
| AMPK | AMP-activated protein kinase |
| ROS | Reactive oxygen species |
| SREBP-1C | Sterol regulatory element-binding protein 1C |
| FASN | Fatty acid synthase |
| ACC | Acetyl-CoA carboxylase |
| FSP 27 | Fat-specific protein of 27 |
| CPT1 | Carnitine palmitoyl transferase I |
| CPT2 | Carnitine palmitoyl transferase II |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| Nrf2 | NF-E2-related factor 2 |
| HO1 | Heme oxygenase 1 |
| NFκB | Nuclear factor kappa-B |
| IL-1β | Interleukin-1-beta |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor-α |
| ITP | Immune thrombocytopenia |
| MDA | Malonic dialdehyde |
| SOD | Superoxide dismutase |
| GTT | Glucose tolerance test |
| ITT | Insulin tolerance test |
Author contributions
Ai Mi: methodology, software, formal analysis, and writing – original draft. Qinxue Hu: validation and writing – original draft. Ying Liu and Fenglin Shen: validation and visualization. Jinjian Lan and Keren Lv: investigation. Yanna Zhao and Bolin Wang: formal analysis. Xiaoling Yu and Ruilan Gao: data curation, funding acquisition, resources and writing – review & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was funded by National Major Project for the Innovative New Drugs of the 13th Five-Year Plan (No. 2016ZX09101071), and The National Central Government Funded Local Projects – Inheritance and Innovation of Traditional Chinese Medicine (2022), State Administration of Traditional Chinese Medicine of China (No. 2A62201).References
- J. V. Lazarus, H. E. Mark, Q. M. Anstee, J. P. Arab, R. L. Batterham and L. Castera, et al., Advancing the global public health agenda for nafld: A consensus statement, Nat. Rev. Gastroenterol. Hepatol., 2022, 19(01), 60–78 CrossRef CAS PubMed.
- L. A. Ban, N. A. Shackel and S. V. McLennan, Extracellular vesicles: A new frontier in biomarker discovery for non-alcoholic fatty liver disease, Int. J. Mol. Sci., 2016, 17(3), 376 CrossRef PubMed.
- S. L. Friedman, B. A. Neuschwander-Tetri, M. Rinella and A. J. Sanyal, Mechanisms of nafld development and therapeutic strategies, Nat. Med., 2018, 24(7), 908–922 CrossRef CAS PubMed.
- C. D. Williams, J. Stengel, M. I. Asike, D. M. Torres, J. Shaw and M. Contreras, et al., Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study, Gastroenterology, 2011, 140(1), 124–131 CrossRef PubMed.
- C. P. Day and O. F. James, Steatohepatitis: A tale of two “hits”, Gastroenterology, 1998, 114(4), 842–845 CrossRef CAS PubMed.
- M. Eslam, A. J. Sanyal, J. George and International Consensus Panel, MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease, Gastroenterology, 2020, 158(7), 1999–2014 CrossRef CAS PubMed.
- R. Loomba, S. L. Friedman and G. I. Shulman, Mechanisms and disease consequences of nonalcoholic fatty liver disease, Cell, 2021, 184(10), 2537–2564 CrossRef CAS PubMed.
- E. A. Day, R. J. Ford and G. R. Steinberg, Ampk as a therapeutic target for treating metabolic diseases, Trends Endocrinol. Metab., 2017, 28(8), 545–560 CrossRef CAS PubMed.
- X. Zhou, L. He, S. Zuo, Y. Zhang, D. Wan and C. Long, et al., Serine prevented high-fat diet-induced oxidative stress by activating ampk and epigenetically modulating the expression of glutathione synthesis-related genes, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864(2), 488–498 CrossRef CAS PubMed.
- C. R. Lindholm, R. L. Ertel, J. D. Bauwens, E. G. Schmuck, J. D. Mulligan and K. W. Saupe, A high-fat diet decreases ampk activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats, J. Physiol. Biochem., 2013, 69(2), 165–175 CrossRef CAS PubMed.
- K. Zimmermann, J. Baldinger, B. Mayerhofer, A. G. Atanasov, V. M. Dirsch and E. H. Heiss, Activated AMPK boosts the Nrf2/HO-1 signaling axis–A role for the unfolded protein response, Free Radicals Biol. Med., 2015, 88(Pt B), 417–426 CrossRef CAS PubMed.
- B. E. Schaffer, R. S. Levin, N. T. Hertz, T. J. Maures, M. L. Schoof and P. E. Hollstein, et al., Identification of ampk phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction, Cell Metab., 2015, 22(5), 907–921 CrossRef CAS PubMed.
- E. Petsouki, S. N. S. Cabrera and E. H. Heiss, Ampk and nrf2: Interactive players in the same team for cellular homeostasis?, Free Radicals Biol. Med., 2022, 190, 75–93 CrossRef CAS PubMed.
- S. Kroller-Schon, T. Jansen, T. L. P. Tran, M. Kvandova, S. Kalinovic and M. Oelze, et al., Endothelial alpha1ampk modulates angiotensin ii-mediated vascular inflammation and dysfunction, Basic Res. Cardiol., 2019, 114(2), 8 CrossRef PubMed.
- T. A.-O. Jansen, M. A.-O. Kvandová, I. A.-O. Schmal, S. A.-O. Kalinovic, P. Stamm and M. A.-O. X. Kuntic, et al., Lack of endothelial α1ampk reverses the vascular protective effects of exercise by causing enos uncoupling, Antioxidants, 2021, 10(12), 1974 CrossRef CAS PubMed.
- D. Garcia, K. Hellberg, A. Chaix, M. Wallace, S. Herzig and M. G. Badur, et al., Genetic liver-specific ampk activation protects against diet-induced obesity and nafld, Cell Rep., 2019, 26(1), 192–208 CrossRef CAS PubMed.
- C. C. Jin and R. A. Flavell, Innate sensors of pathogen and stress: Linking inflammation to obesity, J. Allergy Clin. Immunol., 2013, 132(2), 287–294 CrossRef CAS PubMed.
- S. Vallabhapurapu and M. Karin, Regulation and function of NF-kappaB transcription factors in the immune system, Annu. Rev. Immunol., 2009, 27, 693–733 CrossRef CAS PubMed.
- J. Li, T. N. Sapper, E. Mah, S. Rudraiah, K. E. Schill and C. Chitchumroonchokchai, et al. , Mol. Nutr. Food Res., 2016, 60(4), 858–870 CrossRef CAS PubMed.
- A. Salminen and K. Kaarniranta, Amp-activated protein kinase (ampk) controls the aging process via an integrated signaling network, Ageing Res. Rev., 2012, 11(2), 230–241 CrossRef CAS PubMed.
- M. F. Wu, Q. H. Xi, Y. Sheng, Y. M. Wang, W. Y. Wang and C. F. Chi, et al., Antioxidant Peptides from Monkfish Swim Bladders: Ameliorating NAFLD In Vitro by Suppressing Lipid Accumulation and Oxidative Stress via Regulating AMPK/Nrf2 Pathway, Mar. Drugs, 2023, 21(6), 360 CrossRef CAS PubMed.
- Y.-M. Wang, X. Pan, Y. He, C.-F. Chi and B. Wang, Hypolipidemic activities of two pentapeptides (viapw and irwww) from miiuy croaker (miichthys miiuy) muscle on lipid accumulation in hepg2 cells through regulation of ampk pathway, Appl. Sci., 2020, 10(3), 817 CrossRef CAS.
- R. L. Gao and B. H. Chong, Research and development of the effective components of panaxdiol saponin as new Chinese patent medicine for treating hemocytopenia, Chin. J. Integr. Med., 2012, 18(12), 897–902 CrossRef PubMed.
- T. Y. Dai, J. J. Lan, R. L. Gao, Y. N. Zhao, X. L. Yu and S. X. Liang, et al., Panaxdiol saponins component promotes hematopoiesis by regulating gata transcription factors of intracellular signaling pathway in mouse bone marrow, Ann. Transl. Med., 2022, 10(2), 38 CrossRef CAS PubMed.
- G. L. Fang, R. L. Gao, X. J. Lin and J. M. Jin, Effects of ginseng panaxadiol saponin on proliferation and differentiation of human bone marrow cd34+ cells, Zhongguo Shiyan Xueyexue Zazhi, 2007, 15(4), 776–779 CAS.
- G. L. Fang, R. L. Gao, X. J. Lin and J. M. Jin, Research and development of new chinese materia medica for treatment of refractory hematopathy by establishment and application of multiple technique platforms, Chin. J. Integr. Med., 2007, 13(2), 95–97 CrossRef PubMed.
- Y. M. Kuang, Y. Zhu, R. L. Gao, J. Hu, Z. Y. Jiang and L. Huang, et al., Clinical study of pai-neng-da capsule in the treatment of chronic aplastic anemia, Chin. J. Integr. Med., 2016, 22(2), 124–129 CrossRef PubMed.
- Z. Y. Jiang, F. Q. Yu, R. L. Gao, Y. M. Kuang, Y. Zhu and Y. H. Chen, et al., Treatment of chronic aplastic anemia with chinese patent medicine pai-neng-da capsule, Chin. J. Integr. Med., 2022, 28(1), 20–27 CrossRef CAS PubMed.
- X. Lin, L. Yin, R. Gao, Q. Liu, W. Xu and X. Jiang, et al., The effects of panaxadiol saponins on megakaryocytic maturation and immune function in a mouse model of immune thrombocytopenia, Exp. Hematol., 2015, 43(5), 364–373 CrossRef CAS PubMed.
- W. B. Liu, Z. W. Tan, Y. C. Zhao, Y. N. Zhao, X. L. Yu and B. L. Wang, et al., Panaxadiol saponin ameliorates ferroptosis in iron-overload aplastic anemia mice and meg-01 cells by activating nrf2/ho-1 and pi3k/akt/mtor signaling pathway, Int. Immunopharmacol., 2023, 118, 110131 CrossRef CAS PubMed.
- Z. Y. Zheng, X. L. Yu, T. Y. Dai, L. M. Yin, Y. N. Zhao and M. Xu, et al., Panaxdiol saponins component promotes hematopoiesis and modulates t lymphocyte dysregulation in aplastic anemia model mice, Chin. J. Integr. Med., 2019, 25(12), 902–910 CrossRef CAS PubMed.
- X. Sun, Y. N. Zhao, S. Qian, R. L. Gao, L. M. Yin and L. P. Wang, et al., Ginseng-derived panaxadiol saponins promote hematopoiesis recovery in cyclophosphamide-induced myelosuppressive mice: Potential novel treatment of chemotherapy-induced cytopenias, Chin. J. Integr. Med., 2018, 24(3), 200–206 CrossRef CAS PubMed.
- J. Sun, Y. Liu, J. Yu, J. Wu, W. Gao, L. Ran and R. Jiang, et al., APS could potentially activate hepatic insulin signaling in HFD-induced IR mice, J. Mol. Endocrinol., 2019, 63(1), 77–91 CAS.
- D. E. Kleiner, M. Brunt, M. Van Natta and C Behling, et al., Design and validation of a histological scoring system for nonalcoholic fatty liver disease, Hepatology, 2005, 41(6), 1313–1321 CrossRef PubMed.
- L. Ran, X. Wang, A. Mi, Y. Liu, J. Wu and H. Wang, et al., Loss of adipose growth hormone receptor in mice enhances local fatty acid trapping and impairs brown adipose tissue thermogenesis, iScience, 2019, 16, 106–121 CrossRef CAS PubMed.
- Y. Wu, C. Liu, H. Sun, A. Vijayakumar, P. R. Giglou, R. Qiao and J. Oppenheimer, et al., Growth hormone receptor regulates β cell hyperplasia and glucose-stimulated insulin secretion in obese mice, J. Clin. Invest., 2011, 121(6), 2422–2426 CrossRef CAS PubMed.
- F. Pacifici, R. Arriga, G. P. Sorice, B. Capuani, M. G. Scioli and D. Pastore, et al., Peroxiredoxin 6, a novel player in the pathogenesis of diabetes, Diabetes, 2014, 63(10), 3210–3220 CrossRef CAS PubMed.
- M. Chen, J. Xing, D. Pan, X. Peng and P. Gao, Chinese herbal medicine mixture 919 syrup alleviates nonalcoholic fatty liver disease in rats by inhibiting the nf-kappab pathway, Biomed. Pharmacother., 2020, 128, 110286 CrossRef CAS PubMed.
- C. L. Lyons, H. M. Roche, C. L. Lyons and H. M. Roche, Nutritional modulation of AMPK-impact upon metabolic-inflammation, Int. J. Mol. Sci., 2018, 19, 3092 CrossRef PubMed.
- Z. Younossi, Q. M. Anstee, M. Marietti, T. Hardy, L. Henry and M. Eslam, et al., Global burden of nafld and nash: Trends, predictions, risk factors and prevention, Nat. Rev. Gastroenterol. Hepatol., 2018, 15(1), 11–20 CrossRef PubMed.
- R. Raeman, Inflammation: The Straw that Broke the NAFLD Liver, Cell. Mol. Gastroenterol. Hepatol., 2022, 13(4), 1273–1274 CrossRef CAS PubMed.
- E. E. Powell, V. W. Wong and M. Rinella, Non-alcoholic fatty liver disease, Lancet, 2021, 397(10290), 2212–2224 CrossRef CAS PubMed.
- Y. Wan, J. Wang, J. F. Xu, F. Tang, L. Chen and Y. Z. Tan, et al., Panax ginseng and its ginsenosides: Potential candidates for the prevention and treatment of chemotherapy-induced side effects, J. Ginseng Res., 2021, 45(6), 617–630 CrossRef PubMed.
- N. Sathishkumar, S. Sathiyamoorthy, M. Ramya, D. U. Yang, H. N. Lee and D. C. Yang, Molecular docking studies of anti-apoptotic bcl-2, bcl-xl, and mcl-1 proteins with ginsenosides from panax ginseng, J. Enzyme Inhib. Med. Chem., 2012, 27(5), 685–692 CrossRef CAS PubMed.
- L. Bai, J. Gao, F. Wei, J. Zhao, D. Wang and J. Wei, Therapeutic Potential of Ginsenosides as an Adjuvant Treatment for Diabetes, Front. Pharmacol., 2018, 9, 423 CrossRef PubMed.
- L. Shen, Y. Xiong, D. Q. Wang, P. Howles, J. E. Basford and J. Wang, et al., Ginsenoside rb1 reduces fatty liver by activating amp-activated protein kinase in obese rats, J. Lipid Res., 2013, 54(4), 1430–1438 CrossRef CAS PubMed.
- Y. Lai, Q. Tan, S. Xv, S. Huang, Y. Wang and Y. Li, et al., Ginsenoside rb1 alleviates alcohol-induced liver injury by inhibiting steatosis, oxidative stress, and inflammation, Front. Pharmacol., 2021, 12, 616409 CrossRef CAS PubMed.
- Y. Liu, N. Liu, Y. Liu, H. He, Z. Luo, W. Liu, N. Song and M. Ju, Ginsenoside Rb1 Reduces D-GalN/LPS-induced Acute Liver Injury by Regulating TLR4/NF-κB Signaling and NLRP3 Inflammasome, J. Clin. Transl. Hepatol., 2022, 10(3), 474–485 CrossRef PubMed.
- J. Wang, Y. L. Qiao, F. Li, G. Y. Li, F. Yang and G. Yang, Ginsenoside rb1 attenuates intestinal ischemia-reperfusion- induced liver injury by inhibiting nf-kappab activation, Exp. Mol. Med., 2008, 40(6), 686–698 CrossRef CAS PubMed.
- S. Lee, M. S. Lee, C. T. Kim, I. H. Kim and Y. Kim, Ginsenoside rg3 reduces lipid accumulation with amp-activated protein kinase (ampk) activation in Hepg2 cells, Int. J. Mol. Sci., 2012, 13(5), 5729–5739 CrossRef CAS PubMed.
- X. X. Liu, X. J. Mi, Z. Wang, M. Zhang, J. G. Hou and S. Jiang, et al., Ginsenoside rg3 promotes regression from hepatic fibrosis through reducing inflammation-mediated autophagy signaling pathway, Cell Death Dis., 2020, 11(6), 454 CrossRef CAS PubMed.
- Q. Huang, T. Wang, L. Yang and H. Y. Wang, Ginsenoside rb2 alleviates hepatic lipid accumulation by restoring autophagy via induction of sirt1 and activation of ampk, Int. J. Mol. Sci., 2017, 18(5), 1063 CrossRef PubMed.
- J. Zhang, X. Ma and D. Fan, Ginsenoside ck ameliorates hepatic lipid accumulation via activating the lkb1/ampk pathway in vitro and in vivo, Food Funct., 2022, 13(3), 1153–1167 RSC.
- J. Li, Q. Huang, Y. Yao, P. Ji, E. Mingyao and J. Chen, et al., Biotransformation, pharmacokinetics, and pharmacological activities of ginsenoside rd against multiple diseases, Front. Pharmacol., 2022, 13(3), 909363 CrossRef CAS PubMed.
- Y. Wang, W. Fu, Y. Xue, Z. Lu, Y. Li and P. Yu, et al., Ginsenoside rc ameliorates endothelial insulin resistance via upregulation of angiotensin-converting enzyme 2, Front. Pharmacol., 2021, 12, 620524 CrossRef CAS PubMed.
- G. R. Steinberg and D. G. Hardie, New insights into activation and function of the AMPK, Nat. Rev. Mol. Cell Biol., 2023, 24(4), 255–272 CrossRef CAS PubMed.
- H. Li, J. Sun, B. Li, A. Jiang, J. Tao and C. Ning, et al., Ampk-ppargamma-cidec axis drives the fasting-induced lipid droplet aggregation in the liver of obese mice, Front. Nutr., 2022, 9, 917801 CrossRef PubMed.
- S. Karki, Fsp27 and links to obesity and diabetes mellitus, Curr. Obes. Rep., 2019, 8(3), 255–261 CrossRef PubMed.
- J. X. Zhang, W. X. Zhang, L. Yang, W. J. Zhao, Z. J. Liu and E. R. Wang, et al., Phytochemical gallic acid alleviates nonalcoholic fatty liver disease via ampk-acc-ppara axis through dual regulation of lipid metabolism and mitochondrial function, Phytomedicine, 2023, 109, 154589 CrossRef CAS PubMed.
- Q. Yao, S. Li, X. Cheng, Y. Zou, Y. Shen and S. Zhang, Yin Zhi Huang, a traditional chinese herbal formula, ameliorates diet-induced obesity and hepatic steatosis by activating the ampk/srebp-1 and the ampk/acc/cpt1a pathways, Ann. Transl. Med., 2020, 8(5), 231 CrossRef CAS PubMed.
- R. S. Khan, F. Bril, K. Cusi and P. N. Newsome, Modulation of insulin resistance in nonalcoholic fatty liver disease, Hepatology, 2019, 70(2), 711–724 CrossRef CAS PubMed.
- G. I. Smith, M. Shankaran, M. Yoshino, G. G. Schweitzer, M. Chondronikola and J. W. Beals, et al., Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease, J. Clin. Invest., 2020, 130(3), 1453–1460 CrossRef CAS PubMed.
- M. Foretz, B. Guigas and B. Viollet, Metformin: Update on mechanisms of action and repurposing potential, Nat. Rev. Endocrinol., 2023, 19(8), 460–476 CrossRef CAS PubMed.
- Z. A. Saad, D. M. Khodeer, S. A. Zaitone, A. A. M. Ahmed and Y. M. Moustafa, Exenatide ameliorates experimental non-alcoholic fatty liver in rats via suppression of toll-like receptor 4/nfκb signaling: Comparison to metformin, Life Sci., 2020, 253, 117725 CrossRef CAS PubMed.
- P. A.-O. Rada, Á. González-Rodríguez, C. García-Monzón and Á. M. Valverde, Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver?, Cell Death Dis., 2020, 11(9), 802 CrossRef CAS PubMed.
- KS Lee, Y. Cho, H. Kim, H. Hwang, JW Cho and YH Lee, et al., Association of Metabolomic Change and Treatment Response in Patients with Non-Alcoholic Fatty Liver Disease, Biomedicines, 2022, 10(6), 1216 CrossRef CAS PubMed.
- A. Eynaudi, F. Diaz-Castro, J. C. Borquez, R. Bravo-Sagua, V. Parra and R. Troncoso, Differential Effects of Oleic and Palmitic Acids on Lipid Droplet-Mitochondria Interaction in the Hepatic Cell Line HepG2, Front. Nutr., 2021, 8, 775382 CrossRef PubMed.
- M. Ricchi, M. R. Odoardi, L. Carulli, C. Anzivino, S. Ballestri and A. Pinetti, et al., Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes, J. Gastroenterol. Hepatol., 2009, 24(5), 830–840 CrossRef CAS PubMed.
- R. C. R. Meex and E. E. Blaak, Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD, Mol. Nutr. Food Res., 2021, 65(1), e1900942 CrossRef PubMed.
- Y. Wu, F. Zhou, H. Jiang, Z. Wang, C. Hua and Y. Zhang, Chicory (cichorium intybus l.) polysaccharides attenuate high-fat diet induced non-alcoholic fatty liver disease via ampk activation, Int. J. Biol. Macromol., 2018, 118(Pt A), 886–895 CrossRef CAS PubMed.
- Y. Ma, G. Lee, S. Y. Heo and Y. S. Roh, Oxidative stress is a key modulator in the development of nonalcoholic fatty liver disease, Antioxidants, 2021, 11(1), 91 CrossRef PubMed.
- S.-L. Zheng, Y.-Z. Wang, Y.-Q. Zhao, C.-F. Chi, W.-Y. Zhu and B. Wang, High fischer ratio oligopeptides from hard-shelled mussel: Preparation and hepatoprotective effect against acetaminophen-induced liver injury in mice, Food Biosci., 2023, 53, 102638 CrossRef CAS.
- J. Kong, X. M. Hu, W. W. Cai, Y. M. Wang, C. A.-O. Chi and B. A.-O. Wang, Bioactive Peptides from Skipjack Tuna Cardiac Arterial Bulbs (II): Protective Function on UVB-Irradiated Hacat Cells through Antioxidant and Anti-Apoptotic Mechanisms, Mar. Drugs, 2023, 21(2), 105 CrossRef CAS PubMed.
- P. J. Meakin, S. Chowdhry, R. S. Sharma, F. B. Ashford, S. V. Walsh and R. J. McCrimmon, et al., Susceptibility of nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance, Mol. Cell. Biol., 2014, 34(17), 3305–3320 CrossRef PubMed.
- R. S. Sharma, D. J. Harrison, D. Kisielewski, D. M. Cassidy, A. D. McNeilly and J. R. Gallagher, et al., Experimental non-alcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of nrf2 (nf-e2 p45-related factor 2), Cell. Mol. Gastroenterol. Hepatol., 2018, 5(3), 367–398 CrossRef PubMed.
- S. Chowdhry, M. H. Nazmy, P. J. Meakin, A. T. Dinkova-Kostova, S. V. Walsh and T. Tsujita, et al., Loss of nrf2 markedly exacerbates nonalcoholic steatohepatitis, Free Radicals Biol. Med., 2010, 48(2), 357–371 CrossRef CAS PubMed.
- J. M. Zhou, Q. X. Zheng and Z. Chen, The nrf2 pathway in liver diseases, Front. Cell Dev. Biol., 2022, 10, 826204 CrossRef PubMed.
- J. Khoo, J. Hsiang, R. Taneja, N. M. Law and T. L. Ang, Comparative effects of liraglutide 3 mg vs structured lifestyle modification on body weight, liver fat and liver function in obese patients with nonalcoholic fatty liver disease: A pilot randomized trial, Diabetes Obes. Metab., 2017, 19(12), 1814–1817 CrossRef CAS PubMed.
- M. Peiseler, R. F. Schwabe, J. Hampe, P. Kubes, M. Heikenwaelder and F. Tacke, Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits, J. Hepatol., 2022, 77(4), 1136–1160 CrossRef CAS PubMed.
- A. R. Mridha, A. Wree, A. A. B. Robertson, M. M. Yeh, C. D. Johnson and D. M. Van Rooyen, et al., Nlrp3 inflammasome blockade reduces liver inflammation and fibrosis in experimental nash in mice, J. Hepatol., 2017, 66(5), 1037–1046 CrossRef CAS PubMed.
- Q. Shen, Y. E. Chen, J. X. Shi, C. Y. Pei, S. X. Chen and S. Huang, et al., Asperuloside alleviates lipid accumulation and inflammation in hfd-induced NAFLD via ampk signaling pathway and nlrp3 inflammasome, Eur. J. Pharmacol., 2023, 942, 175504 CrossRef CAS PubMed.
- H. Thomas, NAFLD: A critical role for the NLRP3 inflammasome in NASH, Nat. Rev. Gastroenterol. Hepatol., 2017, 14(4), 197 CAS.
- D. Kelly, J. I. Campbell, T. P. King, G. Grant, E. A. Jansson and A. G. Coutts, et al., Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of ppar-gamma and rela, Nat. Immunol., 2004, 5(1), 104–112 CrossRef CAS PubMed.
- C. F. Mo, L. Wang, J. Zhang, S. Numazawa, H. Tang and X. Q. Tang, et al., The crosstalk between nrf2 and ampk signal pathways is important for the anti-inflammatory effect of berberine in lps-stimulated macrophages and endotoxin-shocked mice, Antioxid. Redox Signaling, 2014, 20(4), 574–588 CrossRef CAS PubMed.
- X. Ding, T. Jian, J. Li, H. Lv, B. Tong and J. Li, et al., Chicoric acid ameliorates nonalcoholic fatty liver disease via the ampk/nrf2/nfkappab signaling pathway and restores gut microbiota in high-fat-diet-fed mice, Oxid. Med. Cell. Longevity, 2020, 2020, 9734560 Search PubMed.
- W. W. Wen, X. Sun, H. F. Zhuang, X. J. Lin, Z. Y. Zheng and R. L. Gao, et al., Effects of panaxadiol saponins component as a new Chinese patent medicine on proliferation, differentiation and corresponding gene expression profile of megakaryocytes, Chin. J. Integr. Med., 2016, 22(1), 28–35 CrossRef CAS PubMed.
- C. Zou, L. L. Xiong and M. Jiang, Phase I human tolerability study of Petundra capsules, Chin. Tradit. Pat. Med., 2015, 37(11), 2383–2386 Search PubMed.
- H. Tilg, T. E. Adolph and M. Trauner, Gut-liver axis: Pathophysiological concepts and clinical implications, Cell Metab., 2022, 34(11), 1700–1718 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work and share the first author. |
| This journal is © The Royal Society of Chemistry 2024 |