 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
To form or not to form a reaction complex: exploring ion–molecule reactions between C3H4 isomers and Xe+ and O2+
C.
Zagorec-Marks
 *ab,
G. S.
Kocheril
ab,
O. A.
Krohn
ab,
T.
Kieft
ab,
A.
Karpinska
abc,
T. P.
Softley
d and
H. J.
Lewandowski
ab
*ab,
G. S.
Kocheril
ab,
O. A.
Krohn
ab,
T.
Kieft
ab,
A.
Karpinska
abc,
T. P.
Softley
d and
H. J.
Lewandowski
ab
aDepartment of Physics, University of Colorado, Boulder, CO 80309, USA. E-mail: chase.zagorec-marks@colorado.edu
bJILA, National Institute of Standards and Technology and the University of Colorado, Boulder, CO 80309, USA
cDepartment of Chemistry and Applied Life Sciences, ETH Zürich, 8093 Zürich, Switzerland
dSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK
First published on 23rd January 2024
Abstract
Ion–molecule reactions are an essential contributor to the chemistry of a diverse range of environments. While a great deal of work has been done to understand the fundamental mechanisms driving these reactions, there is still much more to discover. Here, we expand upon prior studies on ion–molecule reactions involving two isomers of C3H4, allene (H2C3H2) and propyne (H3C3H). Specifically, we probe the previously observed isomeric dependent reactivity of these molecules by reacting them with two ions with nearly identical ionization potentials, Xe+ and O2+. Our goal is to determine if the isomer-dependent reaction mechanisms previously observed are universal for C3H4 or if they depend on the ion character as well. Through the combination of experimental measurements and theoretical calculations, we found that both isomeric structure and identity of the ion contribute to the propensity of a reaction complex forming or for only long-range charge transfer to occur.
1 Introduction
Cold ion–molecule reactions provide a unique view of the interactions found in many diverse chemical environments, such as the Earth’s upper atmosphere and interstellar gas clouds. The long-range interaction between the charged ion and the neutral molecule results in large scattering cross-sections for inelastic or reactive processes.1 This makes ion–molecule reactions relevant to a variety of environments, creating significant interest in predicting their fundamental behavior.2 At the single molecule level, the outcomes or products of these ion–molecule reactions are thought to be determined by two primary mechanisms, direct charge transfer and complex formation.3 As the name suggests, direct charge transfer largely results in a simple transfer of an electron between an ion and molecule, while complex formation results in the reactants forming an intermediate that has a sufficiently long lifetime for equilibration of vibrational energy in the complex, often resulting in the formation of new chemical bonds.3 While complex formation is often attributed to the observation of distinct product distributions,4–7 it is still unclear whether the formation of a complex occurs consistently for reactions of the same neutral with different ions or if the identity of the ion also plays a role. Cold ion techniques, used in conjunction with reactant-ion purification and product time-of-flight analysis, have enabled significant progress towards addressing these questions for reactions with an increasing degree of complexity.Previously, our lab studied the relatively complex reaction between acetylene ions, C2H2+, and two structural isomers of C3H4, allene (H2C3H2) and propyne (H3C3H), and found that product formation is determined by the isomeric structure of the neutral.8,9 Specifically, c-C3H3+ is the sole observed product in the reaction with allene, whereas in the reaction with propyne, c-C3H3+, C3H4+, and C5H5+ are all observed in appreciable amounts originating from complex formation/dissociation. For the allene reaction with C2H2+, it was found that, following charge exchange, the C2H2 was inhibited from forming a reaction complex with the allene cation, likely due to energy localization on the cation that originated from a Jahn–Teller distortion of the allene upon ionization. In contrast, propyne had no structural changes upon ionization, thereby allowing for C2H2 to approach and form a complex. This was an interesting finding as it showed that the isomeric structure was in fact the dominant influence on the formation of the reaction complex for a set of reactions on the same global potential energy surface.
Herein, we report an investigation where we extend these studies on reactions with C3H4 isomers to simpler non-hydrocarbon ions as the reaction partners to see if the same isomer dependent product distributions are observed for both neutral isomers. O2+ and Xe+ were chosen due to their similar ionization potentials, 12.0697 and 12.12987 eV respectively, and are expected to behave similarly in terms of energetics.10,11 However, they are expected to follow different reaction mechanisms. While it has been suggested that Xe+ can perform charge transfer resulting from complex formation,12,13 it has been shown that Xe+ rarely forms covalently bound reaction complexes,14–16 so products of Xe+ reactions typically arise from direct charge transfer alone.17–21 Reactions with O2+, on the other hand, have been shown in Selected Ion Flow Tube (SIFT) studies to both react via complex formation and via direct charge transfer.22,23 Using these two ions in tandem allows us to further explore the mechanisms of reactivity between the C3H4 isomers. Surprisingly, we find here that the ion is now the dominant influence on the reaction mechanisms, and no isomer dependent behavior is observed for either system, which is in direct contrast to the previous reactions involving C2H2+. All Xe+ reactions with C3H4 isomers undergo exclusively direct charge transfer reactions, whereas O2+ reactions show clear complex formation that leads to different product distributions from those observed in the Xe+ reactions.
2 Methods
The experimental setup has been previously described elsewhere, and only a brief description will be given here for clarity.24 Ca+ ions formed via non-resonant ionization of an effusive Ca beam using the third harmonic of a Nd:YAG laser were loaded into a linear Paul trap. The Ca+ ions were then laser cooled to secular temperatures of <1 K using the frequency doubled and fundamental outputs of two Ti-Sapphire lasers (∼4 mW at 397 nm and ∼20 mW at 866 nm). The resultant Coulomb crystal (comprised typically of ∼1000 ions) was used for sympathetically cooling co-trapped reactant ions. Fluorescence emitted by the Ca+ Coulomb crystal was imaged by an electron multiplying charge-coupled device (EMCCD) camera to qualitatively observe reactions. Reactions were quantitatively monitored via time-of-flight mass spectrometry by observing a reduction in reactant ion peaks and correlated growth of product ion peaks. Neutral products were not detected.2.1 Reactant ion loading
After the Ca+ Coulomb crystal had formed, Xe was introduced into the chamber via a pulsed, skimmed supersonic expansion of Xe (2%) in a buffer gas of ∼1030 Torr He. Xe+ ions were formed in the center of the trap via a 2+1 resonance enhanced multi-photon ionization (REMPI) scheme using a focused beam from a pulsed dye laser at 249.6 nm (2 mJ per pulse) overlapped spatio-temporally with the Xe atomic beam.25 It was expected that approximately 22% of the Xe+ ions were formed in the excited 2P1/2 state using this scheme. However, this excited state has a lifetime of ∼49 ms, and the Xe+ ions were held in the trap for 60 seconds prior to introduction of neutrals to ensure that all the Xe+ ions were maximally cooled before reacting; therefore, none of the reacting Xe+ ions were in the excited state.26O2 was also introduced into the chamber via a pulsed, skimmed supersonic expansion of O2 (12%) in a buffer gas of ∼1030 Torr He prior to the formation of the Ca+ Coulomb crystal. O2+ ions were formed in the center of the trap via a 2+1 REMPI scheme using a focused 287.5 nm beam overlapped spatio-temporally with the molecular beam.27 All O2+ ions were believed to have been formed in the ground electronic state, X2Πg, and are efficiently cooled by Ca+, thereby allowing for the reaction to commence once the Coulomb crystal had fully formed.
2.2 Reacting with neutrals
Reactions were initiated by admitting either neutral allene (H2C3H2) or propyne (H3C3H) (14–16% in N2) into the chamber via a pulsed-leak valve to a fixed chamber pressure (with partial pressures at 300 K of 0.7–2.5 × 10−10 Torr and 4 × 10−10 Torr for O2+ and Xe+ reactions, respectively) for fixed periods of time.28,29 Pressures were measured using a Bayard–Alpert hot cathode ion gauge in the same chamber as the ion trap. Typical pressures in the chamber prior to admittance of gas were 1.5 × 10−9 Torr. Because Bayard–Alpert hot cathode ion gauges have reduced accuracy below 10−8 Torr, absolute reaction rates have considerable uncertainty.30 As such, we report only products and branching ratios for the reactions measured here.To determine the branching ratios for a reaction, we measured a reaction curve by varying the length of time the neutral gas was admitted into the chamber. The opening of the pulsed leak valve corresponded to the zero-time point. At each reaction time point, the contents of the trap were ejected into a time-of-flight mass spectrometer (TOF-MS), and a new Coulomb crystal was formed for the next measurement. The contents of the trap at each time point were measured at least 10 times, and the mean and standard error of these measurements were then plotted as a function of time to produce a reaction curve. The reaction curve was fit according to a reaction model representative of the reaction being studied. These reaction models assume that ion numbers within the trap are conserved throughout the course of a reaction.
To verify that no ions were lost from the trap during the reaction, the total ion number was determined for each time step. For the O2+ reactions, the total ion number remained constant during a reaction. However, the case was more complicated for Xe+. We found that not all of the trapped Xe+ ions were detected on our micro-channel plate (MCP) detector. (When analyzing the Xe+ data, all isotopes of Xe+ were summed together and reported as just the total number of Xe+ ions.) This underestimate of the number of Xe+ ions is because they have a much larger mass than Ca+, which results in inefficient sympathetic cooling and a weaker trapping potential. These two factors combine to decrease the efficiency of detecting Xe+ by our TOF-MS, as compared to all other ions present in these reactions. Further, simulations of the Xe+ ions’ extraction trajectories suggested that a fair number of these ions collided with the trap rods or grounded plate at the entrance of the TOF-MS flight tube and were thus not detected. Heavy ions have previously been observed to be more difficult to detect.31 While we estimate the detection efficiency of Xe+ ions to be 50% less than lighter mass ions, none of the reaction analysis conducted relied on the measured Xe+ numbers and was determined solely on product ion numbers, which, being more similar in mass to Ca+, are cooled more efficiently and detected with the same efficiency.
2.3 Theoretical
Possible global potential energy surfaces for Xe+ and O2+ reactions were determined via quantum chemical calculations performed using Gaussian 16 at the unrestricted ωb97xd/aug-cc-pvtz level of theory.32 Each surface was computed starting with the reactants at infinite separation, with stationary points along the surface identified until the system reached the final observed product. Scans over bond lengths, angles, and dihedrals were used for identifying minima and potential transition states. These potential maxima were explored using optimization and frequency calculations. Transition states were identified by the presence of a vibrational mode with an imaginary frequency and through use of intrinsic reaction coordinate calculations. An effective core potential (ECP) was included for calculations directly involving Xe and Xe+ to account for relativistic effects.33 Only barrierless pathways were considered because of the energetic constraints imposed by the low temperatures of the reactions.3 Results
3.1 Xe+ with C3H4 isomers
A typical TOF-MS trace for the Xe+ reactions is shown in Fig. 1. Reaction progress was monitored by the growth of the 39 m/z signal (c-C3H3+) and decay of the various Xe+ isotope signals, 128–136 m/z. As seen in the inset of the figure, the mass resolution of the experimental setup was sufficient to resolve the seven most abundant isotopes of Xe. Secondary product growth was observed by the delayed growth of peaks at 77 and 79 m/z (C6H5+ and C6H7+). The signals for these secondary products are not shown in the figure for clarity. The peak observed at 57 m/z corresponded to CaOH+, which was formed by a side reaction of Ca+ and trace H2O left in the vacuum chamber. This reaction was accounted for by adding the number of ions observed in the 57 m/z channel to the 40 m/z (Ca+, C3H4+, and C2O+) channel during the analysis.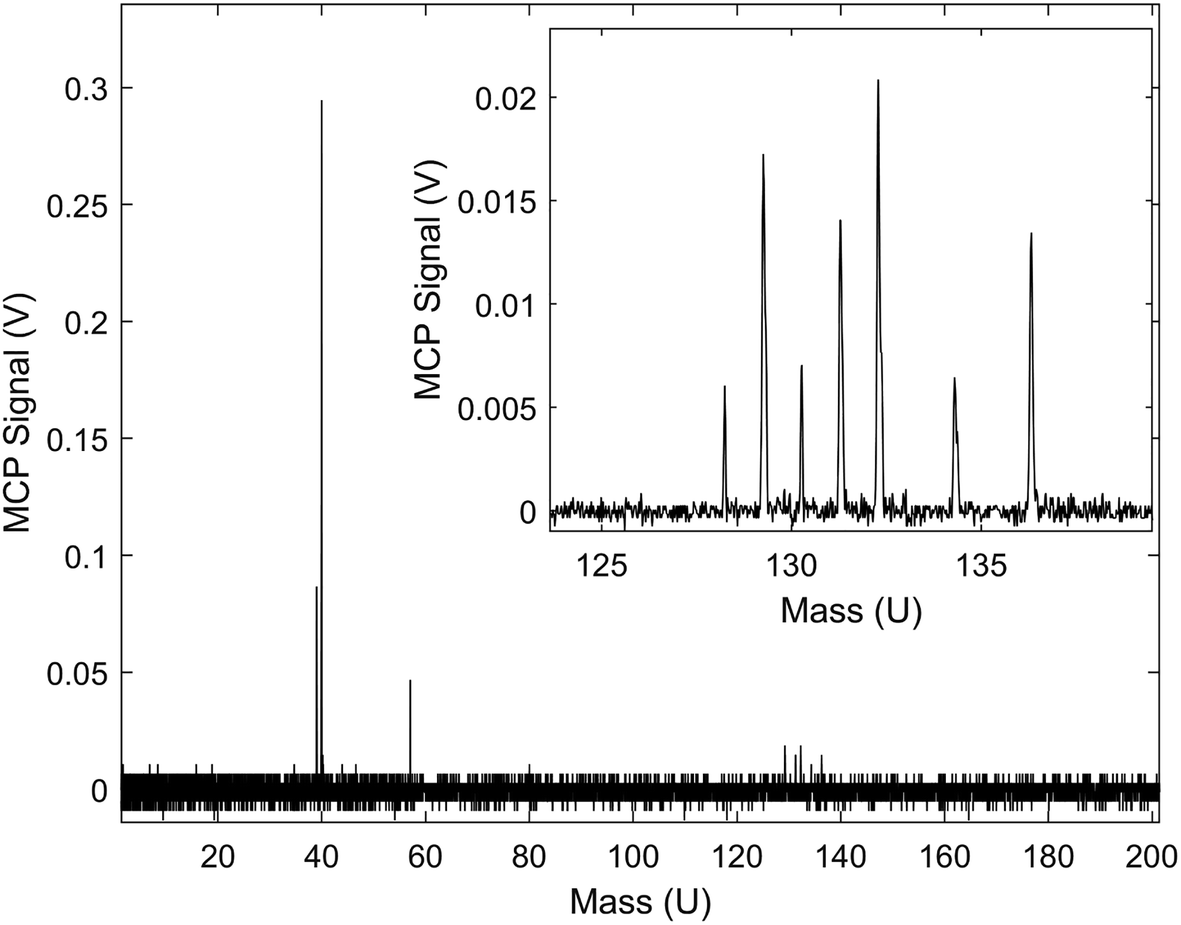 | ||
| Fig. 1 Typical TOF-MS trace observed from the reaction of Xe+ with C3H4. Primary plot displays the trace’s full signal intensity. Peaks observed in the primary plot, from left to right, correspond to 39 m/z (c-C3H3+, product), 40 m/z (Ca+), 57 m/z (CaOH+), 128–136 m/z (Xe+). Inset displays the Xe+ signals when viewed with greater voltage resolution. | ||
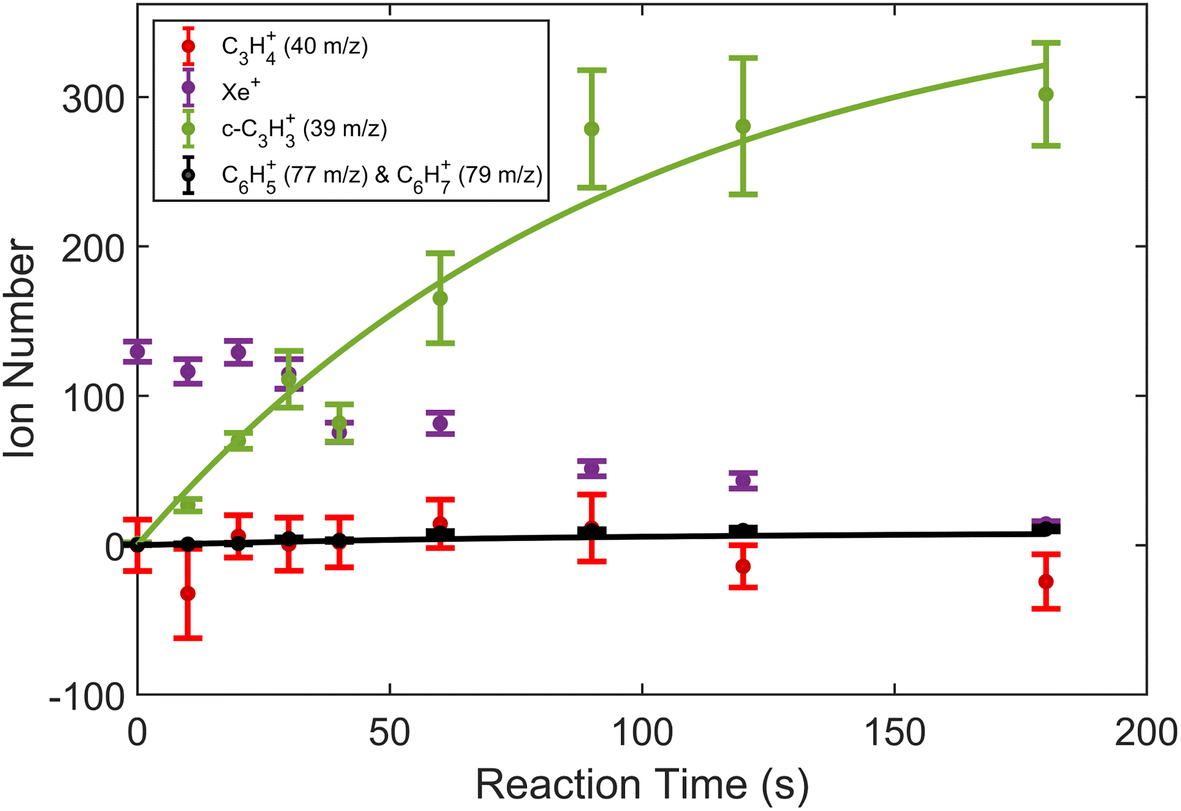 | ||
| Fig. 2 Product growth in reaction of Xe+ with allene (H2C3H2) as a function of time. Data points correspond to mean and standard error of 11 measurements. Time was measured from the opening of the pulsed leak valve and subsequent inlet of allene into the chamber. Ion counts corresponding to 40 m/z were adjusted by subtracting the initial number of Ca+ ions for clarity. Note that the parasitic reaction of Ca+ + H2O → CaOH+ + OH was accounted for by adding ions observed in the 57 m/z channel back to the 40 m/z channel. Additionally, lines plotted correspond to fits produced from eqn (1) and (2). | ||
To extract branching ratios from these data, they were fit assuming pseudo-first order kinetics with allene in excess. Because the detection efficiency of Xe+ was lower than for the other ions in the trap, the growth of products were fit independently of the Xe+ data using the following reaction model:
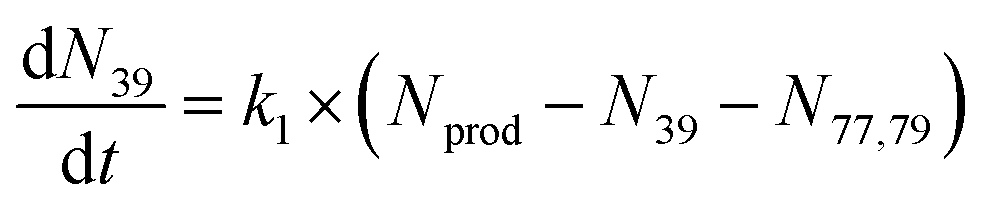 | (1) |
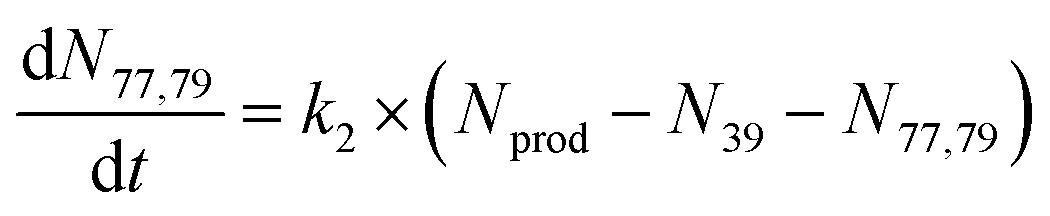 | (2) |
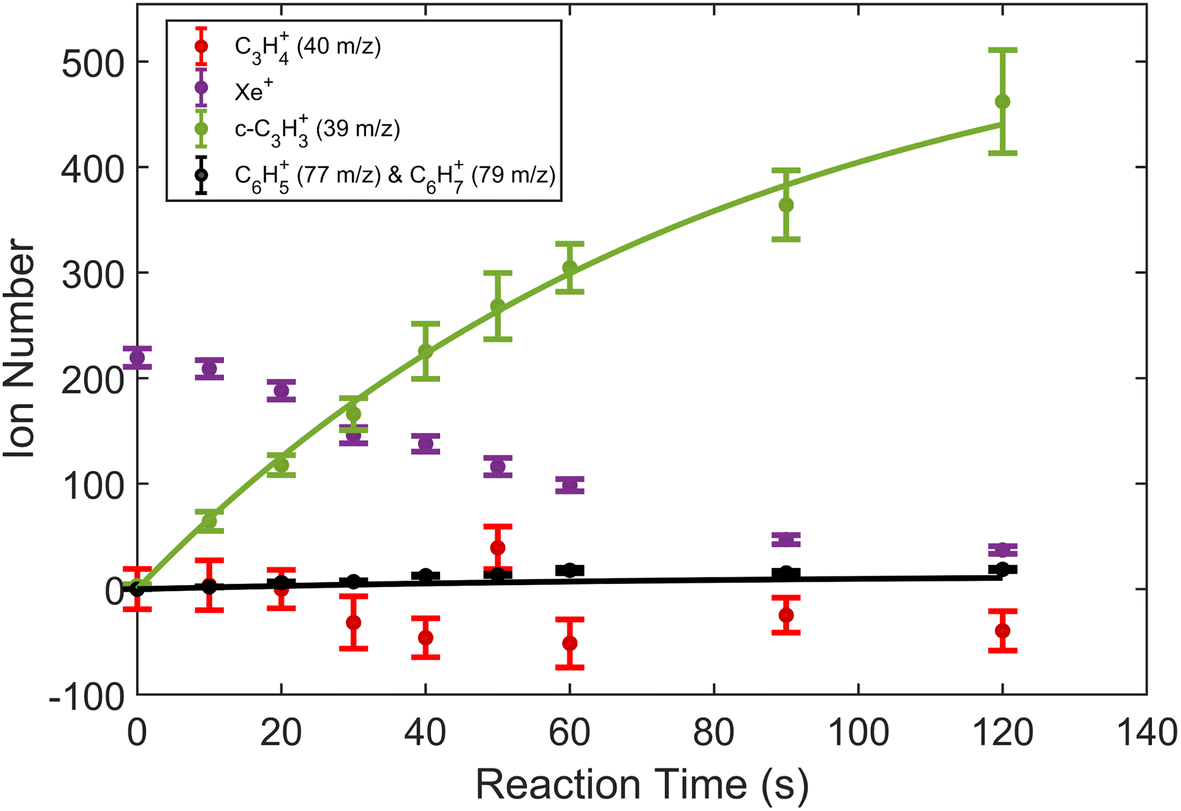 | ||
| Fig. 3 Product growth in reaction of Xe+ with propyne (H3C3H) as a function of time. Data points correspond to mean and standard error of 12 measurements. Time was measured from the opening of the pulsed leak valve and subsequent inlet of propyne into the chamber. Ion counts corresponding to 40 m/z were adjusted by subtracting the initial number of Ca+ ions for clarity. Note that the parasitic reaction of Ca+ + H2O → CaOH+ + OH was accounted for by adding ions observed in the 57 m/z channel back to the 40 m/z channel. Additionally, lines plotted correspond to fits produced from eqn (1) and (2). | ||
3.2 O2+ with C3H4 isomers
In addition to reactions of Xe+ with two isomers of C3H4, we also studied reactions of these two with O2+ ions. In these reactions, we measured both the growth of the 39 m/z signal and decay of the O2+ signal at 32 m/z. In addition to these channels, secondary product growth was observed by the delayed growth of peaks at 77 and 79 m/z. In contrast to the Xe+ reactions, all reactant and product ions were detected with the same efficiency. This detection efficiency was determined by comparing the total number of ions present in the trap across all time points of the reaction and confirming that the number of charges was conserved. With the knowledge that no ions were lost over the course of the reaction, it was possible to determine the number of 40 m/z product ions obfuscated by the large Ca+ signal. Specifically, the number of 40 m/z product ions can be calculated by:| N40,prod(t) = O2+(t = 0) − (O2+(t) + N39(t) + N77,79(t)) | (3) |
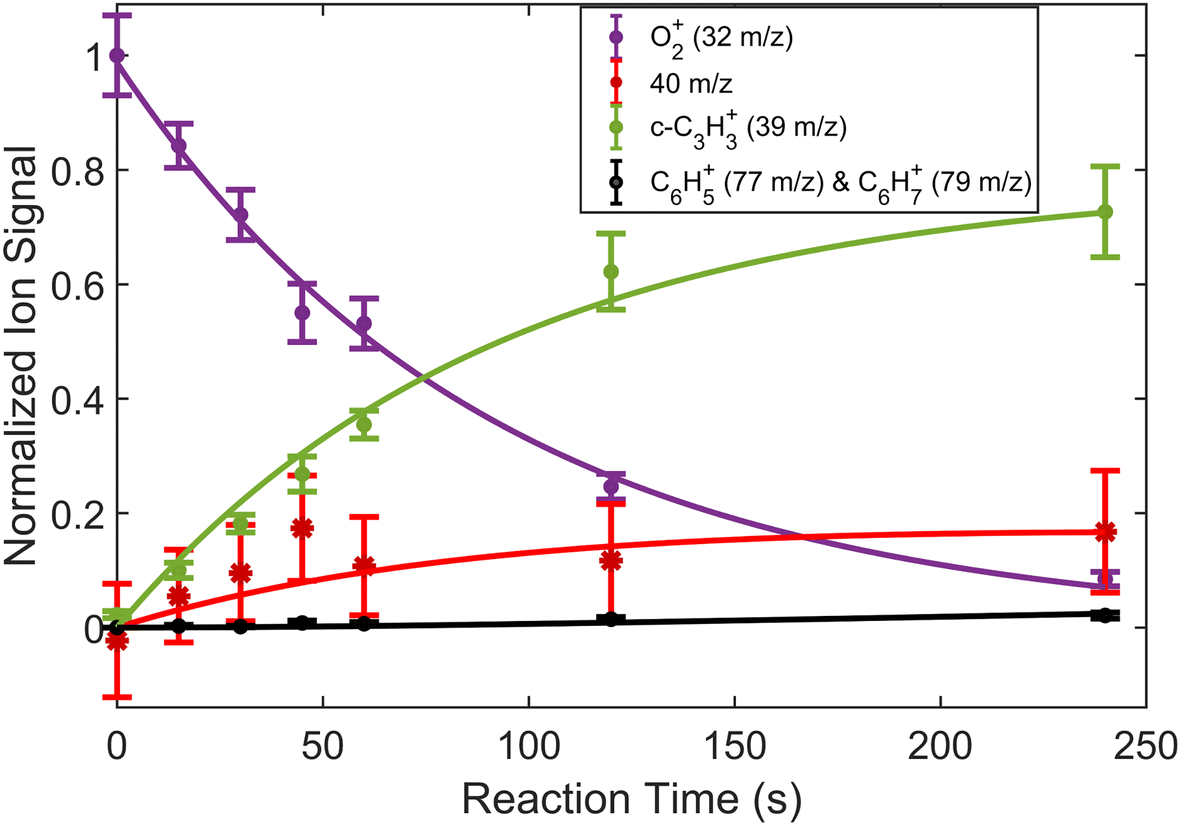 | ||
| Fig. 4 Fraction of reactant and product ions in the reaction of O2+ with H2C3H2 as a function of time. Circular data points correspond to mean and standard error of 14 measurements. Asterisk data points correspond to calculated mean and standard error of 40 m/z products using eqn (3). All data points have been normalized to the initial number of O2+ ions (∼180 ions). Time was measured from the opening of the pulsed leak valve and subsequent inlet of allene into the chamber. Plotted lines correspond to calculated fits for the products and reactants using eqn (4)–(6). | ||
The reaction data were fit assuming pseudo-first order kinetics with allene in excess and the following reaction model:
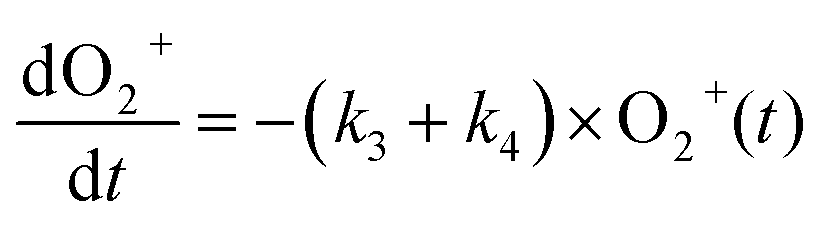 | (4) |
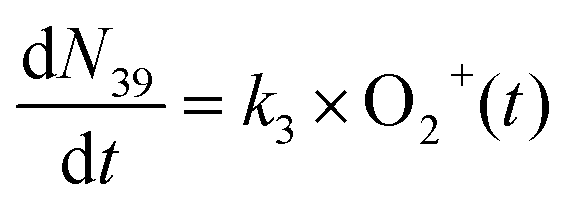 | (5) |
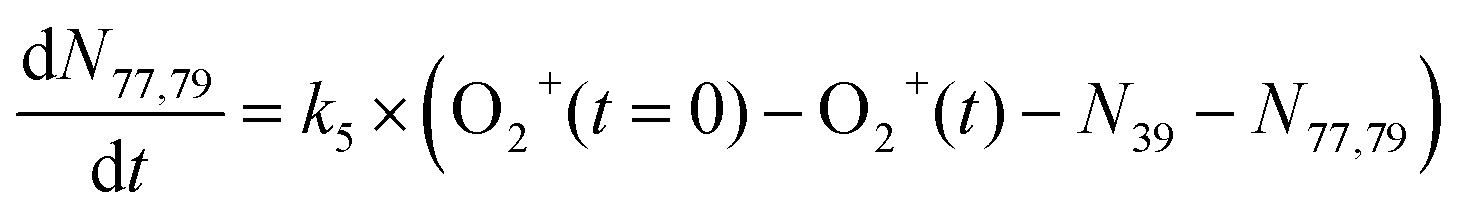 | (6) |
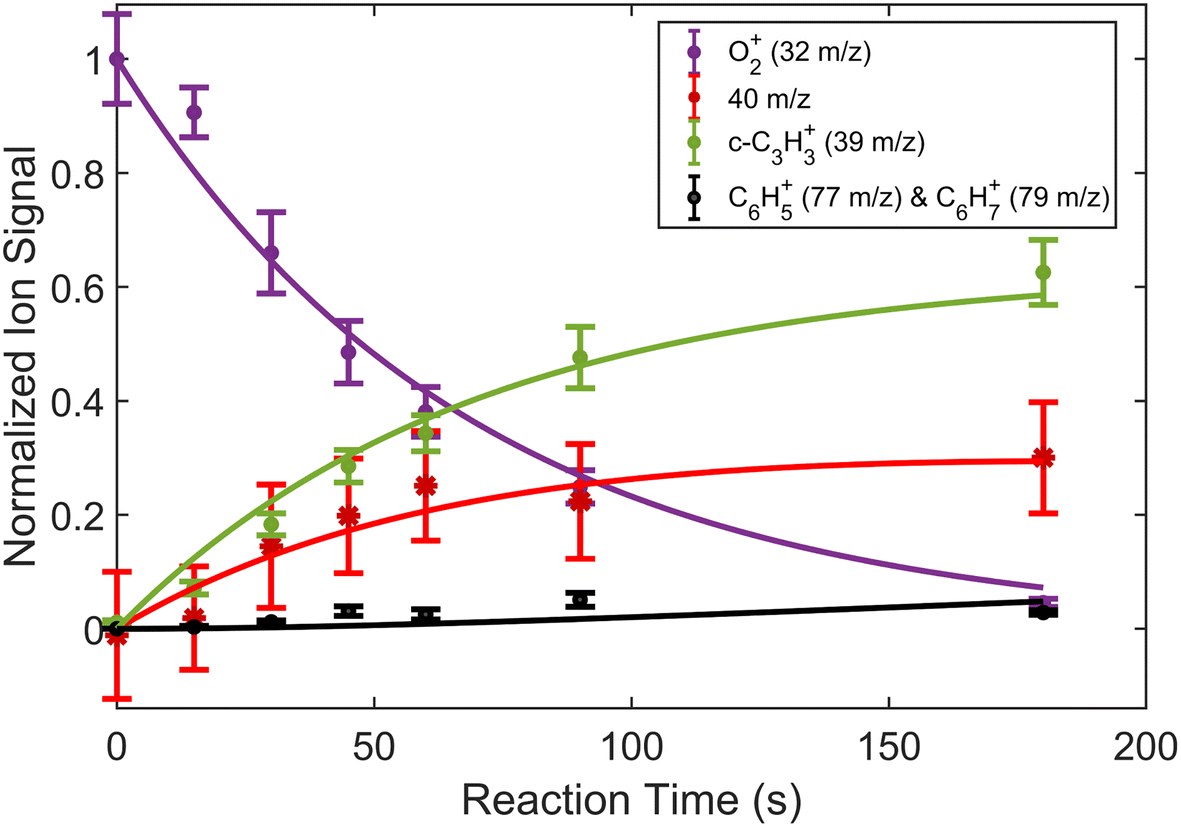 | ||
| Fig. 5 Fraction of reactant and product ions in the reaction of O2+ with H3C3H as a function of time. Circular data points correspond to mean and standard error of 13 measurements. Asterisk data points correspond to calculated mean and standard error of 40 m/z products using eqn (3). All data points have been normalized to the initial number of O2+ ions (∼190 ions). Time was measured from the opening of the pulsed leak valve and subsequent inlet of propyne into the chamber. Plotted lines correspond to calculated fits for the reactants and products using eqn (4)–(6). | ||
4 Discussion
Our overall goal for this study was to determine if isomer-specific reactivity previously observed with two isomers of C3H4, allene (H2C3H2) and propyne (H3C3H), was a universal trait for ion–molecule reactions involving these molecules, or if the ion character plays an important role for complex formation as well. To explore this question, we studied reactions of allene and propyne with two ions, Xe+ and O2+, chosen because the energy available to the reaction is nearly identical in the two cases due to the very similar ionization potentials of Xe and O2.The results of the reactions with Xe+ and O2+ clearly show distinct product distributions, demonstrating that the ionic reactant can be at least partially responsible for differences in branching ratios. In the following, we investigate how these distinct product distributions provide evidence for these reactions progressing via direct charge transfer or via complex formation.
4.1 Charge transfer driven reactions with Xe+
For the Xe+ reactions with both C3H4 isomers studied here, c-C3H3+ is the prevailing product. This result is similar to the case of acetylene (C2H2+) with allene (H2C3H2), which was shown to progress via direct charge transfer. The dominance of the c-C3H3+ product for both Xe+ reactions strongly suggests that these reactions proceed along the same reaction pathway regardless of isomer and theoretical calculations validated this conclusion.In the Xe+ reactions, there are two possibilities for how c-C3H3+ could be formed as a product, hydride abstraction by Xe+ to form XeH or C–H bond dissociation of C3H4+. The former relies on the formation of a complex between the Xe+ and C3H4, but the latter does not as C3H4+ can be formed via direct charge transfer. While Xe+ has been shown to occasionally, but rarely, form reaction complexes, the lack of any XeH+ ions detected in our experiments suggests that complex formation is very unlikely.14–16 Further, it was determined via quantum chemical calculations that the formation of XeH and C3H3+ is highly endothermic (>2 eV), making these products impossible to form in the current experimental system. Calculations were also conducted to determine if complex formation is energetically accessible, but all attempts led to endothermic pathways inaccessible for this experiment. Therefore, we find neither experimental nor theoretical support for complex formation in the reaction of Xe+ + C3H4.
In contrast, direct charge transfer and resultant dissociation of a hydrogen from C3H4+ is well supported. This can be seen in the potential energy surfaces (PESs) for the reaction of Xe+ with each C3H4 isomer shown in Fig. 6. These surfaces begin with a highly exothermic charge transfer, with exothermicities of 2.35 eV and 2.96 eV for propyne (H3C3H) and allene (H2C3H2) respectively. Because of the ultrahigh vacuum environment of the system, this energy is not quenched by collisions with other ions or neutral molecules. Instead, the ion retains a significant portion of this energy in its rovibrational modes, and the ion rapidly progresses along the PES. In both reactions, the C3H4+ can inter-convert between the allene cation (identified as INT2 in Fig. 6), propyne cation, and INT4 until transitioning to TS4, where the molecule dissociates yielding a hydrogen atom and ultimately goes on to form c-C3H3+. These PESs demonstrate a barrierless pathway that leads to the production of c-C3H3+ without the formation of a complex between the Xe+ and C3H4 isomers. Further, these PESs are nearly identical to that for the reaction of acetylene (C2H2+) with allene, which was determined to progress solely through direct charge transfer.8,9 Therefore, this reveals that in the Xe+ reactions direct charge transfer can ultimately lead to formation of c-C3H3+ regardless of isomer.
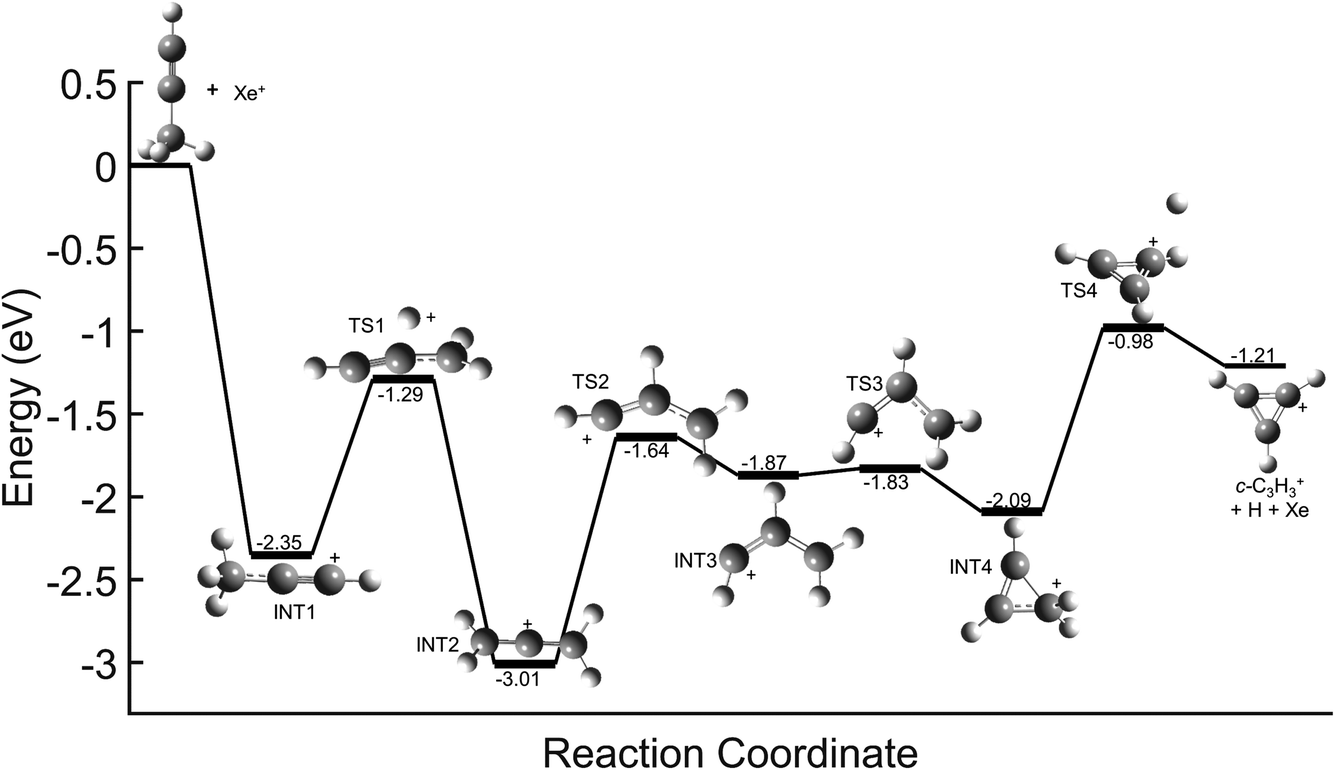 | ||
| Fig. 6 Global potential energy surface of the reaction between Xe+ and propyne (H3C3H) calculated at the ωb97x/aug-cc-pvtz level. All energies are shown relative to reactant energy at infinite separation, with INT1 corresponding to the first stationary point following charge transfer. Note that the allene (H2C3H2) cation is identified as INT2, and the PES for allene with Xe+ is identical to that shown here with energies being shifted to higher energies by 5 meV. | ||
4.2 Evidence of complex formation with O2+
Comparisons between the products in the O2+ and Xe+ reactions reveal that complex formation plays a role in the reaction mechanism for O2+. For example, in the reactions of O2+ with both C3H4 isomers studied here, c-C3H3+ is produced to a lesser extent than in the Xe+ reactions (∼60–75% of total products for O2+, versus >95% for Xe+). Further, in contrast to the Xe+ reactions, there is also noticeable growth in the 40 m/z channel for both O2+ reactions. Specifically, in the O2+ reactions, 40 m/z products are present in branching ratios of ∼17% (allene) and ∼30% (propyne) compared to the branching ratios observed in the Xe+ reactions of <5%. This discrepancy between the O2+ and Xe+ reactions suggests that direct charge transfer is not the sole mechanism present in the O2+ reactions. Additionally, the presence of multiple products is similar to the reaction of C2H2+ with propyne (H3C3H), which progressed via complex formation. Therefore, these results suggest that O2+ forms a complex with each C3H4 isomer, and this complex formation aids in the production of the 40 m/z products, C3H4+ and C2O+.The conclusion that both isomers of C3H4 form a complex with O2+ is strengthened through calculation of PESs. Plausible PESs by which complex formation can lead to formation of C3H4+ and c-C3H3+ in the reaction with propyne (H3C3H) are shown separately in Fig. 7 and 8 for clarity. The propyne with O2+ surfaces for C3H4+ and c-C3H3+ production both begin with barrierless, exothermic (∼1.32 eV) complex formation, and follow similar trajectories for the attachment of both oxygen atoms onto the complex. The two surfaces ultimately separate at INT4 at which point the complex can lead to production of C3H4+ or c-C3H3+. In the production of C3H4+, the majority of intermediate states are relatively similar in energy allowing for rapid inter-conversion and sampling of states. Further, C3H4+ can be produced in two separate isomers depending on when and how the O2 detaches from the complex. Specifically, C3H4+ can be produced either in the H2C3H2+ isomer if the O2 detaches at TS6, or it can be produced in the H3C3H+ isomer if the complex regresses at TS1 and detaches O2. In contrast to this, the surface for production of c-C3H3+ has a relatively high-energy transition state (TS5, 0.87 eV exothermic compared to reactants) necessary for transferring the hydrogen to the attached O2. c-C3H3+ production is finalized upon dissociation of HO2 from the complex. Surfaces for allene (H2C3H2) were calculated, but were not included here because of their similarity with the surfaces for propyne. The surfaces provided demonstrate a barrierless pathway for the production of both c-C3H3+ and C3H4+ given complex formation.
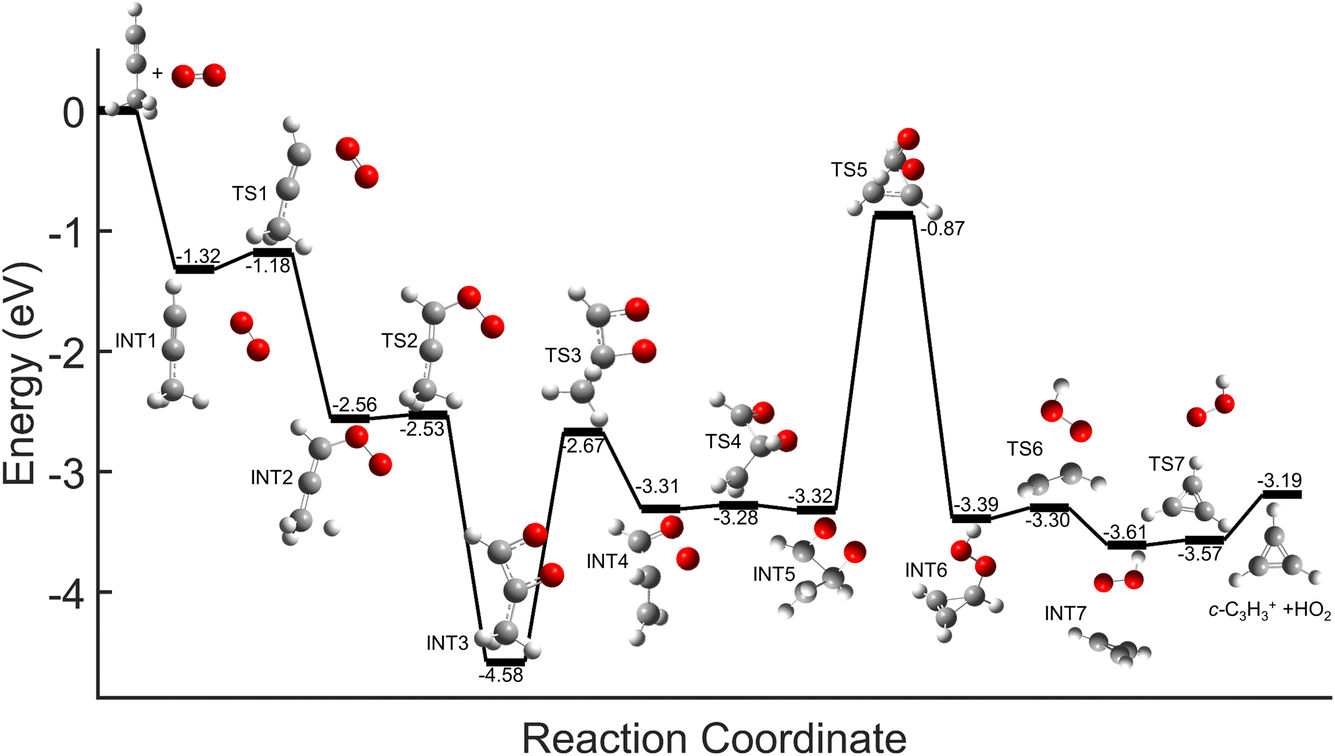 | ||
| Fig. 7 Global potential energy surface of complex formation leading to c-C3H3+ production for O2+ with propyne (H3C3H) calculated at the ωb97x/aug-cc-pvtz level. All energies are displayed relative to reactant energy at infinite separation. Note that states INT1–INT4 are identical to those shown in Fig. 8. These surfaces have been separated for clarity. | ||
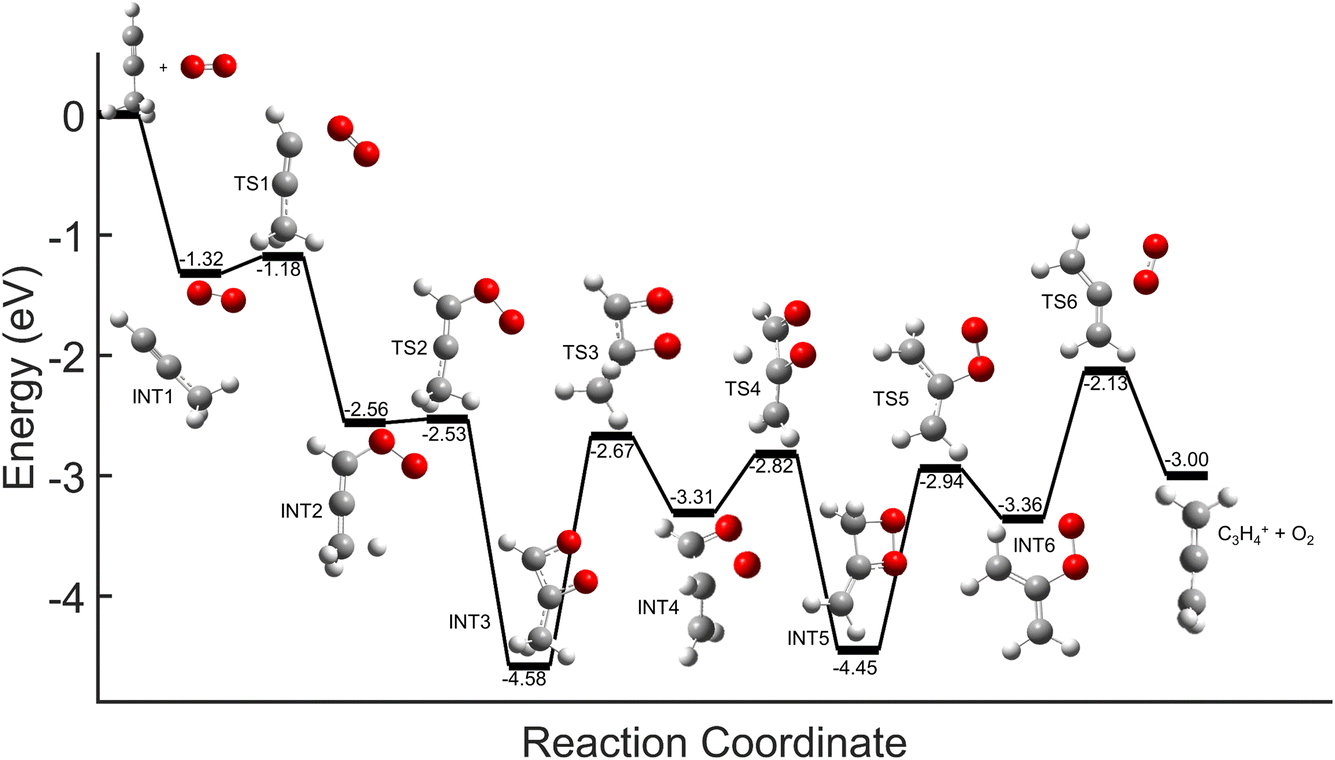 | ||
| Fig. 8 Global potential energy surface of complex formation leading to C3H4+ production for O2+ with propyne (H3C3H) calculated at the ωb97x/aug-cc-pvtz level. All energies are displayed relative to reactant energy. Note that states INT1–INT4 are identical to those shown in Fig. 7. These surfaces have been separated for clarity. | ||
However, not all of the 40 m/z products observed in the O2+ reactions can be accounted for by C3H4+. This is because it is known that C3H4+ reacts with C3H4 to form larger hydrocarbons, and many of the 40 m/z products observed here appear to be stable. While other molecules exist with a charge-to-mass ratio of 40 m/z, C2O+ is one of the most likely products. For example, C2O+ has a simple combination of atoms involved in the reaction that seem plausible given complex formation. Further, calculations find that production of C2O+ and its co-product, CH3OH, is exothermic (∼2.9 eV). Finding a plausible PES for production of C2O+ is beyond the scope of our current work. Verifying the presence of C2O+via isotopic substitution will be the subject of a future study.
5 Conclusions
The reactions of Xe+ and O2+ with two isomers of C3H4 demonstrate that the identity of the ion, not just the charge-transfer energetics or the isomer structure, contributes to the reaction pathway. Xe+ reactions proceed solely via direct charge transfer reactions, principally forming c-C3H3+, which is unsurprising. However, O2+ forms complexes with both isomers resulting in a more diverse product distribution, including c-C3H3+, C3H4+, and possibly C2O+. Combining these results with our previous work on reactions between acetylene ions (C2H2+) and C3H4 isomers, we find that Xe+ and O2+ behave as the two extremes cases where the mechanism is driven by the ion identity, whereas the acetylene cation behaves in a manner whereby the neutral isomer determines the mechanism. While Xe+ solely performs direct charge transfer and O2+ undergoes complex formation with both C3H4 isomers, acetylene performs direct charge transfer with one isomer, allene, and complex formation with the other, propyne. The ability to demonstrate a reaction mechanism that is dependent on the ion identity, independent of the ionization potential of the ion, can be uniquely demonstrated in this pair of reactions. These findings suggest that in the case of the two isomers of C3H4 studied here, either the identity of the ion, or the structure of the neutral can contribute to the propensity of a reaction complex forming or direct charge transfer occurring. Further studies will be required to better understand how each component independently influences this propensity and their relative importance in doing so.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (PHY-1734006, CHE-1900294) and the Air Force Office of Scientific Research (FA9550-16-1-0117). TPS is grateful to the Leverhulme Trust and JILA Visiting Fellow program for supporting his contributions to this work.Notes and references
- J. Meyer and R. Wester, Annu. Rev. Phys. Chem., 2017, 68, 333–353 CrossRef CAS PubMed.
- B. H. Mahan, Acc. Chem. Res., 1968, 1, 217–224 CrossRef CAS.
- I. Prigogine and S. A. Rice, Advances in chemical physics, John Wiley & Sons, 2009, vol. 250 Search PubMed.
- J. A. Luine and G. H. Dunn, Astrophys. J., 1985, 299, L67–L70 CrossRef CAS.
- H. Guo, Int. Rev. Phys. Chem., 2012, 31, 1–68 Search PubMed.
- J. Troe, J. Chem. Soc., Faraday Trans., 1994, 90, 2303–2317 RSC.
- J. Troe, Chem. Rev., 2003, 103, 4565–4576 CrossRef CAS PubMed.
- J. Greenberg, P. Schmid, J. H. Thorpe, T. L. Nguyen, K. J. Catani, O. A. Krohn, M. I. Miller, J. F. Stanton and H. J. Lewandowski, J. Chem. Phys., 2021, 154, 124310 CrossRef CAS PubMed.
- P. C. Schmid, J. Greenberg, T. L. Nguyen, J. H. Thorpe, K. J. Catani, O. A. Krohn, M. I. Miller, J. F. Stanton and H. J. Lewandowski, Phys. Chem. Chem. Phys., 2020, 22, 20303–20310 RSC.
- R. G. Tonkyn, J. W. Winniczek and M. G. White, Chem. Phys. Lett., 1989, 164, 137–142 CrossRef CAS.
- D. R. Lide, Ionization potentials of atoms and atomic ions, in Handbook of Chemistry and Physics, Taylor & Francis, 1992 Search PubMed.
- L. S. Petralia, A. Tsikritea, J. Loreau, T. P. Softley and B. R. Heazlewood, Nat. Commun., 2020, 11, 3235–3246 CrossRef PubMed.
- A. Tsikritea, K. Park, P. Bertier, J. Loreau, T. P. Softley and B. R. Heazlewood, Chem. Sci., 2021, 12, 10005–10013 RSC.
- P. Armentrout, D. Berman and J. Beauchamp, Chem. Phys. Lett., 1978, 53, 255–259 CrossRef CAS.
- A. B. Raksit and D. K. Bohme, Can. J. Chem., 1984, 62, 2123–2126 CrossRef CAS.
- P. Španěl, M. Tichy and D. Smith, Int. J. Mass Spectrom. Ion Processes, 1993, 129, 155–162 CrossRef.
- K. Giles, N. G. Adams and D. Smith, J. Phys. B: At., Mol. Opt. Phys., 1989, 22, 873–883 CrossRef CAS.
- N. G. Adams, D. Smith and E. Alge, J. Phys. B: At. Mol. Phys., 1980, 13, 3235–3246 CrossRef CAS.
- M. Chau and M. T. Bowers, Chem. Phys. Lett., 1976, 44, 490–494 CrossRef CAS.
- R. Derai, G. Mauclaire and R. Marx, Chem. Phys. Lett., 1982, 86, 275–280 CrossRef CAS.
- S. A. Ard, A. A. Viggiano, B. C. Sweeny, B. Long and N. S. Shuman, Nat. Commun., 2022, 13, 3310 CrossRef PubMed.
- V. G. Anicich, An index of the literature for bimolecular gas phase cation-molecule reaction kinetics, JPL-Publ-03-19, JPL, 2003 Search PubMed.
- P. F. Wilson, C. G. Freeman and M. J. McEwan, Int. J. Mass Spectrom., 2003, 229, 143–149 CrossRef CAS.
- P. C. Schmid, J. Greenberg, M. I. Miller, K. Loeffler and H. J. Lewandowski, Rev. Sci. Instrum., 2017, 88, 123107 CrossRef CAS PubMed.
- S. J. Bajic, R. N. Compton, X. Tang and P. Lambropoulos, Phys. Rev. A, 1991, 44, 2102–2112 CrossRef CAS PubMed.
- S. Jullien, J. Lemaire, S. Fenistein, M. Heninger, G. Mauclaire and R. Marx, Chem. Phys. Lett., 1993, 212, 340–346 CrossRef CAS.
- Y. Wu, Z. Zhang and S. F. Adams, Chem. Phys. Lett., 2011, 513, 191–194 CrossRef CAS.
- P. C. Schmid, J. Greenberg, M. I. Miller, T. L. Nguyen, J. F. Stanton and H. J. Lewandowski, Mol. Phys., 2019, 117, 3036–3042 CrossRef CAS.
- C. Q. Jiao, D. R. Ranatunga, W. E. Vaughn and B. S. Freiser, J. Am. Soc. Mass Spectrom., 1996, 7, 118–122 CrossRef CAS PubMed.
- K. Jousten, CAS-CERN Accelerator School: Vacuum in Accelerators, 2007, 65–86 Search PubMed.
- J. A. Diprose, V. Richardson, P. Regan, A. Roberts, S. Burdin, A. Tsikritea, K. Mavrokoridis and B. R. Heazlewood, Spatial and Temporal Detection of Ions Ejected from Coulomb Crystals, private communication Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- V. G. Anicich, G. A. Blake, J. K. Kim, M. J. McEwan and W. T. Huntress Jr, J. Phys. Chem., 1984, 88, 4608–4617 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |