
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh photocatalytic yield in the non-oxidative coupling of methane using a Pd–TiO2 nanomembrane gas flow-through reactor†
Victor
Longo
a,
Luana
De Pasquale
a,
Francesco
Tavella
a,
Mariam
Barawi
 b,
Miguel
Gomez-Mendoza
b,
Víctor
de la Peña O’Shea
b,
Claudio
Ampelli
a,
Siglinda
Perathoner
a,
Gabriele
Centi
a and
Chiara
Genovese
*a
b,
Miguel
Gomez-Mendoza
b,
Víctor
de la Peña O’Shea
b,
Claudio
Ampelli
a,
Siglinda
Perathoner
a,
Gabriele
Centi
a and
Chiara
Genovese
*a
aDepartment of Chemical, Biological, Pharmaceutical and Environmental Sciences and CASPE (INSTM), University of Messina, Viale F. Stagno D’Alcontres 31, 98166 Messina, Italy. E-mail: chiara.genovese@unime.it
bPhotoactivated Processes Unit, IMDEA Energy, Avda. Ramón de la Sagra, 3, Móstoles, 28935 Madrid, Spain
First published on 3rd July 2024
Abstract
The photocatalytic non-oxidative coupling of methane (NOCM) is a highly challenging and sustainable reaction to produce H2 and C2+ hydrocarbons under ambient conditions using sunlight. However, there is a lack of knowledge, particularly on how to achieve high photocatalytic yield in continuous-flow reactors. To address this, we have developed a novel flow-through photocatalytic reactor for NOCM as an alternative to the conventionally used batch reactors. Me/TiO2 photocatalysts, where Me = Au, Ag and Pd, are developed, but only those based on Pd are active. Interestingly, the preparation method significantly impacts performance, going from inactive samples (prepared by wet impregnation) to highly active samples (prepared by strong electrostatic adsorption – SEA). These photocatalysts are deposited on a nanomembrane, and the loading effect, which determines productivity, selectivity, and stability, is also analysed. Transient absorption spectroscopy (TAS) analysis reveals the involvement of holes and photoelectrons after charge separation on Pd/TiO2 (SEA) and their interaction with methane in ethane formation, reaching a production rate of about 1000 μmol g−1 h−1 and a selectivity of almost 95% after 5 hours of reaction. Stability tests involving 24 h of continuous irradiation are performed, showing changes in productivity and selectivity to ethane, ethylene and CO2. The effect of a mild oxidative treatment (80 °C) to extend the catalyst's lifetime is also reported.
Broader contextThe utilization of methane as a low-cost raw material to produce valuable chemicals, represents an appealing strategy for the transition towards a competitive climate-neutral chemical industry. However, due to the very high stability of C–H bonds, the main industrial process for methane valorisation is still strongly constrained by the energetically costly step of syngas production. Consequently, it has been nearly half a century since several strategies for methane valorisation, alternatives to the syngas route, have been investigated. An ideal and promising approach, among the different methane conversion technologies developed so far, is the non-oxidative coupling of methane (NOCM). The option of using solar energy to perform this reaction, is a recent possibility although still challenging due to (i) the difficulty in the C–H cleavage and (ii) the generally low ethane productivity that usually requires the accumulation of the products operating under batch conditions. In the current study, we reported the design of an optimized Pd-based nanomembrane photocatalyst to be used in an innovative flow-through photoreactor, to achieve high ethane yield and selectivity in light-assisted NOCM. |
Introduction
Direct catalytic valorisation of natural gas (methane) to higher hydrocarbons is still a main challenge,1 although studied for many years. Methane is a clean raw material with enormous reserves, particularly those stranded, and is available at low cost. Besides, methane hydrates will be additional important sources in the long term. Methane produced from biogas represents a further valuable opportunity. Not surprisingly, the European chemical industry indicated that natural gas is a vital energy and feedstock source for the chemical industry to transition towards a competitive, climate-neutral chemical sector by 2050.2However, the transport of methane is limited by the fact that it remains in a gaseous state even at very low temperatures. The low reactivity of the C–H bond hinders its direct use as a chemical. Today, methane valorisation (for easier storage/transport) requires passing through the economically and energetically costly step of high-temperature syngas production by steam reforming or similar technologies.3,4
It has been nearly half a century since several strategies for methane valorisation, alternatives to the syngas route, have been studied. They can be shortly described as (i) direct (low-temperature) conversion paths, such as methane to methanol conversion, characterised by extremely low productivity and (ii) direct high-temperature routes.1 The latter can be either oxidative, leading to C2+ hydrocarbons and often indicated by the acronym OCM (oxidative coupling of methane), or non-oxidative, leading mainly to aromatics but with severe problems of deactivation.5 Even after four decades of extensive studies,6–8 the OCM route is still not commercially applied due to high temperatures and the formation of carbon oxide. The non-oxidative route (methane dehydro-aromatization) has also been known for a long time. Despite the recent progress,9,10 thermodynamic limitations and fast catalyst deactivation due to coking, remain great challenges to its application.
An ideal approach to methane valorisation would be a non-oxidative route (to avoid the presence of O2 and formation of COx) operating under ambient conditions and using renewable energy. Non-thermal (catalytic) plasma conversion of methane11–13 to H2 and C2+ hydrocarbons has such characteristics, but (i) the reaction is not selective, leading to many products difficult to separate, (ii) the energy efficiency is low (a few percentages), (iii) productivity is low and (iv) scalability is difficult.14 Electrocatalytic methane conversion under mild conditions15–17 is an interesting approach, but different oxygenated products are formed, faradaic efficiency is low, and in situ H2O2 generation (or consumption of other oxidants) is required.
Photocatalytic methane conversion offers the advantage of direct use of solar energy,18–22 but it usually requires co-reactants (O2, H2O, CO2, NH3, etc.)15 leading to costly downstream operations. In addition, it exhibits poor selectivity control and low productivity. The photocatalytic non-oxidative conversion of methane (NOCM) would be preferable because (i) it does not require co-reactants, (ii) it leads to the selective formation of ethane and also H2 as valuable coproducts, and (iii) it minimises downstream operations,23–34 being therefore an area of growing research interest.
Since the pioneering work of Kato et al.,35 several photocatalysts have been developed, mainly based on semiconductors that would promote C–H cleavage and ethane desorption by creating acid/base surface pairs,27 nanoclusters or single sites in ordered porous supports.36,37 In a recent study, a high conversion rate (>330 μmol g−1 h−1) and superior coke resistance were reported by using a wurtzite GaN:ZnO solid solution.38 While alternative strategies were developed, such as chemical looping,39 the best results in terms of ethane productivity were obtained by the creation of a strong synergy between a semiconductor support and a metallic co-catalyst, either in the form of nanoparticles40 or single atoms.31
However, since ethane productivity is low, most studies on light-driven NOCM were carried out in batch reactors to accumulate the reaction product, and only a few studies have explored continuous flow conditions.27,37,41 A batch-type reaction would also affect the selectivity because it promotes secondary reactions of oxidation.19,33,42 Data obtained by using a batch photocatalytic reactor, cannot be reliably translated to flow conditions, due to the change in many aspects influencing the mechanism and not only fluid dynamics phenomena.43
In addition, appropriate design of metal nanoparticle/semiconductor systems and interfaces to suit the specific requirements of gas phase operation and the choice of photoreactor type, remain challenging issues. In fact, due to the absence of a liquid solvent/electrolyte in contact with the semiconductor interface, the charge transfer processes governing the photocatalytic reactivity upon charge separation are significantly different than those under liquid-phase conditions.44–47 This is a less explored but promising direction in the photocatalysis field.
Gas-phase photocatalysis avoids the issue of solubility of the reactant and thus increases its concentration at the photocatalyst surface.48 This aspect is crucial for the target reaction because methane is typically less soluble in most of the liquid solvents/electrolytes used in photocatalysis. In addition, a high surface concentration is important to favour C–C coupling and the formation of C2+ hydrocarbons. On the other hand, the beneficial effects of stabilisation of the charge transfer products by solvation and the role of the double layer in stabilising the charged semiconductor interface, are less effective in gas-phase photocatalysis. For example, in the photo-driven NOCM, it was observed that metallic nanoparticles might favour the surface reaction of holes with molecules49 (methane) for their oxidation (CH4 → ˙CH3 + H+).36 However, the characteristics of these metallic nanoparticles progressively changed due to the accumulation of photogenerated charges in TiO2, as well as changes in the titania surface during photoreaction. There is, thus, a need to design photocatalysts specific to the type of gas-phase photoreactor.
In this study, TiO2 (P25), one of the most studied and active photocatalytic supports, was used, facilitating the comparison with the existing literature.50,51 Au, Ag and Pd metallic nanoparticles were then deposited on this TiO2 substrate. The as-prepared photocatalysts were then loaded (in different amounts) onto a flow-through cellulosic nanomembrane to investigate the influence of the nature of the photocatalyst and its distribution on the nanomembrane.
It is worth mentioning the innovative character of this flow-through nanomembrane (FTNM) photocatalytic reactor, which is significantly different from conventional or advanced photocatalytic reactors designed so far.48,52,53 The FTNM operates (i) in the gas phase and (ii) in a flow-through mode, with the nanomembrane behaving as a confined photocatalytic reactor. This approach was already successfully adopted for CO2 photocatalytic conversion,54 with selectivity mostly towards C2+ products instead of CO or CH4 commonly formed by a conventional liquid route.
Results and discussion
Influence of the nature of metal nanoparticles
Au, Ag, and Pd-loaded TiO2 samples, were synthesised by photo-deposition to investigate the influence of the nature of catalytic sites on the photocatalytic properties. Then, they were loaded in different amounts onto the flow-through nanomembrane54 and tested in the NOCM reaction. The testing conditions of this FTNM photocatalytic reactor were ambient conditions, with a feed flow rate of 0.6 L h−1 having a composition of 50% CH4 in Ar. A Xe lamp of 300 W was used for light irradiation. A scheme of the membrane preparation and set-up for the tests, with a picture of the photo-reactor, is reported in the ESI† (Fig. S1).The crystal structures of all samples were analysed by X-ray diffraction (XRD). The crystalline structure of TiO2 P25 (a mix of rutile and anatase polymorphs (JCDPS no. 88-1175 – JCPDS no. 84-1286) has not been altered by the synthesis procedure, as evidenced in Fig. S2 (ESI†). No additional peaks of metallic gold and silver were observed, likely due to the combination of low loading (from 0.2 to 1% wt.) and high dispersion. Differently, for Pd-containing samples, the XRD reflections of metallic Pd, in particular the main reflection at 40.2°(111) (JCPDS no. 87-0638),55 can be detected, although rather weak. Instead, no indications for the presence of PdO, whose main reflection is expected at 34.2°,56 were observed for all Pd-loaded samples.
The nature of the metal forming the catalytic site is of critical importance. As evidenced in Fig. S3 (ESI†), among all samples synthesised through photo-deposition, only the Pd-loaded sample exhibits significant ethane production, which can be likely ascribed to the ability of Pd to stabilise methyl radicals. However, this would require an O*-saturated Pd surface,57,58 while we operate under non-oxidative conditions. Au and Ag-loaded samples were found to over-oxidise CH4 to CO2 under these conditions, although they may produce ethane under oxidative conditions, as evidenced by the results reported in Fig. S4 and S5 (ESI†) for the Au/TiO2 sample, attesting the significantly different mechanisms between OCM (most studied) and our NOCM conditions.19,59
Influence of the nature of Pd species
Pd-loaded samples were synthesised via three different methodologies: (i) wetness impregnation (WI) to obtain larger Pd nanoparticles, (ii) photo-deposition (Photo), which allows obtaining relatively small nanoparticles, and (iii) strong electrostatic adsorption (SEA) to obtain smaller clusters/single atoms (SA). All samples contain a low amount of Pd (0.2 wt% by atomic absorption spectroscopy, AAS) to maximise the dispersion. The different preparation methods thus enable us to obtain a variety of Pd nanoparticle characteristics, allowing to determine their role in the photo-driven NOCM by using the novel FTNM photocatalytic reactor.The dispersion of Pd was confirmed by HR-TEM and HAADF-STEM studies. Fig. 1 shows the electron microscopy images of the Pd/TiO2 (SEA) sample, the most active photocatalyst. HR-TEM (Fig. 1a) shows the d spacing of lattice fringes related to TiO2 (101, 0.350 nm). Almost no Pd can be observed in agreement with the XRD results. High-magnification high-angle annular dark-field (HAADF) STEM images reveal the presence of very small Pd particles instead (Fig. 1b). The elemental maps (Fig. 1c) suggest a homogeneous distribution of a mixture of Pd nanocluster/SA for the sample synthesised by SEA. Pd nanoclusters of 2–4 nm could be instead detected in the sample prepared by the photo-deposition method (Fig. S6, ESI†), while large particles of Pd (in the range of 5–20 nm) were present in the sample prepared by the WI method (Fig. S7, ESI†).
 | ||
| Fig. 1 (a) HR-TEM image. (b) HAADF-STEM and (c) the corresponding elemental mapping (Ti, O, and Pd) images of Pd/TiO2 (SEA). | ||
Fig. 2a shows that using large Pd nanoparticles (samples prepared by WI), almost no ethane is produced. In contrast, the Pd/TiO2 samples prepared by photo-deposition and SEA methods, show an effective capability to activate the C–H bond with high ethane productivity. The Pd/TiO2 (SEA) sample specifically exhibits an ethane yield of 3539 μmol g−1 (micromoles per gram of the catalyst) after 5 hours of irradiation.
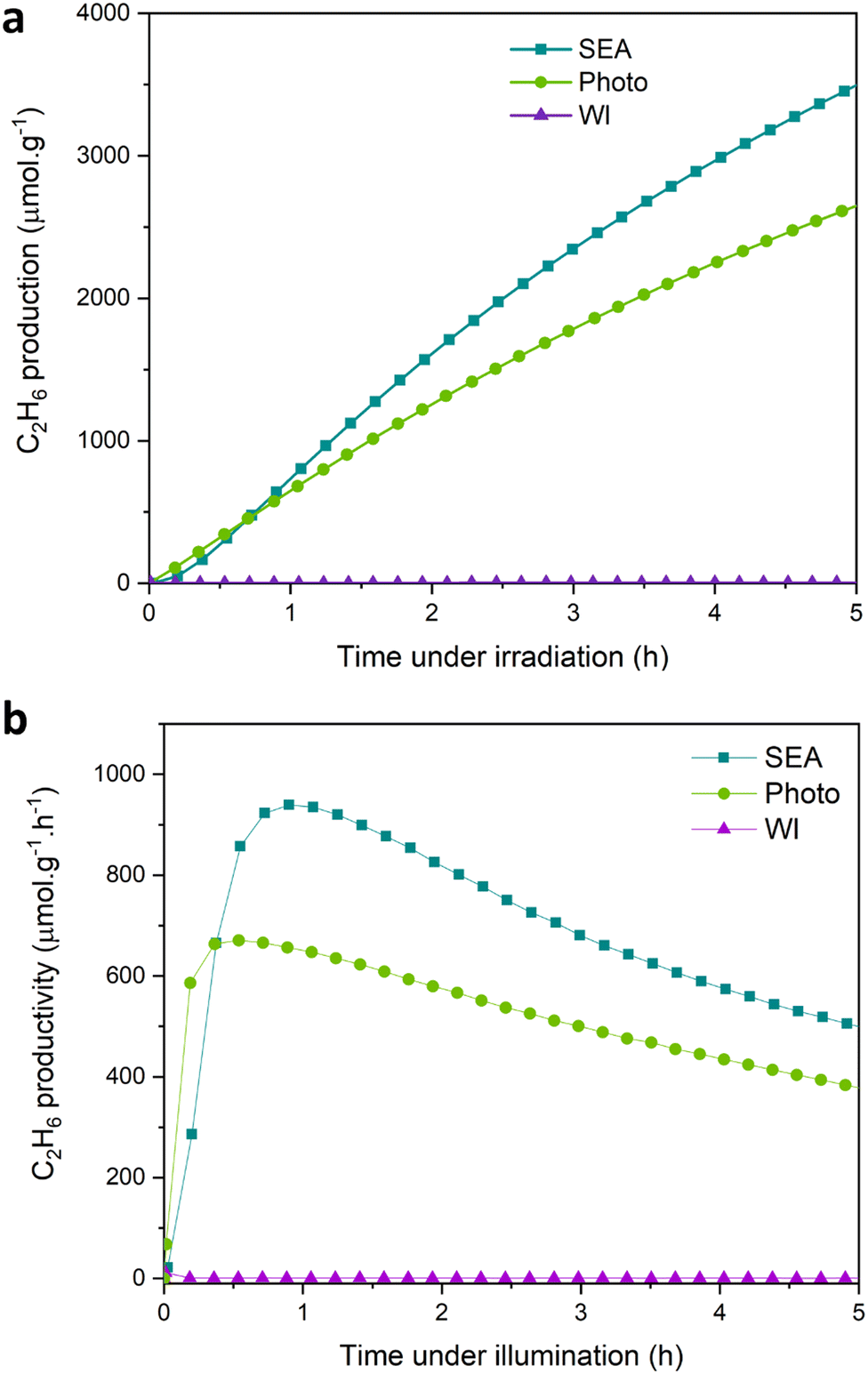 | ||
| Fig. 2 (a) Ethane yield (μmol g−1) for photocatalytic NOCM over Pd/TiO2 prepared by SEA, photo-deposition, and WI. 0.2% wt Pd loading. (b) Ethane production rate (μmol g−1 h−1) for photocatalytic NOCM over Pd/TiO2 prepared by SEA, photo-deposition and WI. Reaction conditions: flow rate 0.6 L h−1, CH4 50% in Ar, light irradiation for 5 hours at room temperature. | ||
A maximum production rate of 939 μmol h−1 g−1 is obtained after about 1 h of irradiation (Fig. 2b), which is among the highest values reported so far in the literature. In addition to ethane, a small amount of ethylene is obtained together with propane and propylene. A summary of all detected C-products for the photocatalytic NOCM test using the Pd/TiO2 (SEA) sample is provided in the ESI† (Table S1). Note that C-based products are not detected without methane or light irradiation through blank tests. All photocatalytic experiments were performed at room temperature. The temperature rise due to lamp irradiation was minimal, thus having no significant catalytic effects.
The presence of a maximum in the productivity as a function of the time of irradiation, with the exact position depending on the distribution of Pd nanoparticles (difference between Photo and SEA preparations), is a crucial aspect whose interpretation deserves additional studies. Some insights will be provided below by transient absorption spectroscopy (TAS) and further photocatalytic tests.
It can be commented that the decay after reaching the maximum productivity may suggest catalyst deactivation either by sintering or deposition of carbonaceous species during irradiation. This is likely not the correct interpretation, as the effect is related to modification of TiO2 during photoirradiation and the related influence on charge separation, diffusion to the surface and reactivity,60 as commented later. The photoexcited charges in TiO2 form localised states (polarons) stabilised by accompanying lattice distortion.61
UV-vis spectroscopy and photoelectrochemical measurements were also carried out for all samples (Au, Ag and Pd/TiO2) to correlate the catalytic activity with the optoelectrical properties.
UV-vis spectra (Fig. S8, ESI†) show two absorption peaks in the visible range for Au- and Ag-loaded samples that are indicative of surface plasmon resonance (SPR), which is particularly evident for gold. The plasmonic resonance is absent in the Pd-containing samples, evidencing that the photocatalytic performances cannot be attributed to the SPR effect.
All Pd-loaded samples present increased absorption in the visible range with respect to the blank TiO2. This is attributed to the generation of intermediate energy levels in the titania band gap, resulting from oxygen defects62 or the incorporation of Pd atoms in the lattice.63 However, by comparing these results with the photocatalytic reactivity (Fig. 2), it is evident that the absorption in the visible region does not correlate with the activity.
The photoelectrochemical properties were measured by chronoamperometry (CA) under dark and different lighting conditions and the results are reported in Fig. 3. The first observation is the very large initial photocurrent density, which decays to a nearly steady state in about one minute of photoirradiation, revealing greater light absorption but also high charge recombination. Note that this effect is rather intense in the most active photocatalyst (Pd/TiO2 SEA). At the same time, this behaviour is not observed in the Pd/TiO2 samples prepared by impregnation (WI), for which the current density is lower and increases over exposure time. There is, thus, a different transient mechanism of charge trapping and accumulation during the initial exposure to light. A similar initial overshooting in the current density, although much less intense, is present in plasmonic samples (Au and Ag/TiO2) and partially in the undoped TiO2. This overshoot in current density can be attributed to the capacitive charging of the surface state.64–66
 | ||
| Fig. 3 Chronoamperometric measurements of all synthesised samples and bare TiO2. Signals were recorded in 0.5 M Na2SO4, at 0.2 V against Ag/AgCl as the reference electrode; the light was switched ON and OFF every 50 s. | ||
The surface states on TiO2 act initially as traps for photo-holes coming from the bulk. In parallel, the photogenerated electrons flow to the counter electrode and are responsible for the current overshoot. After saturation of these hole traps, the new photogenerated holes recombine with the electrons and lead to a decrease in the photocurrent, because the reaction of the photo-holes is generally the rate-limiting step.
In translating these photocurrent results to the photo-reactivity tests, it must be considered that the photogenerated charges react with methane, generating methyl radical species by reacting with holes. The protons formed in this reaction react with the photoelectrons, generating H2 from the counter electrode.19,67 However, these charge-transfer reactions may not have the same rate and charge accumulation occurs on the metal nanoparticles or defect sites of TiO2. Thus, when using CA results to interpret the photocatalytic data, it is essential to consider these additional factors beyond the common aspects of the mechanism.
In Pd/TiO2 SEA, the intense overshooting in current density indicates that there is an efficient initial trap of photo holes, which may explain why there is an initial increase in the rate of ethane photogeneration. At the same time, to have a coupling between methyl radicals, they must accumulate on the surface in a sufficient amount to give rise to C–C bond formation. Still, there is also the need for a fast reaction of protons (generated in the reaction of methane with holes) because otherwise, electrons (accumulated on metal nanoparticles) may give rise to a fast backreaction. Thus, how trapping and surface accumulation of the photogenerated charges influence their reactivity and reaction paths, is equally relevant to discuss charge separation processes. However, the time scale in which these processes establish and reach equilibrium is likely faster than the process of change in C2 productivity (Fig. 2b). It should be a process of progressive surface modification, which may alter the physicochemical processes and, in turn, the photocatalytic behaviour. These aspects will be discussed later.
It must be noted that, although a mechanism of methane activation via surface plasmons is possible,19 the fact that plasmonic catalysts (Au and Ag/TiO2 samples) are not effective under conditions investigated in our study, suggests that this path is not relevant in our case.
For Pd-loaded samples, the significant decrease in photocurrent response under UV-B/C cut-off light irradiation, indicates that most of the generated photocurrent originates from the UV-B wavelengths. With the light energy input required to cleave the C–H bond being about 275 nm,68 the absorption at a longer wavelength (i.e., in the visible range) might be useless for the activation of the C–H bond or might yield a lower faradaic efficiency. The Pd/TiO2 (SEA) sample has shown, under the open spectrum, the greatest photocurrent response, with most of it coming from the deep-UV region, which can be associated with its higher ethane productivity. The small nanoclusters/SA obtained (as evidenced from STEM images) may allow a lower resistance to interface charge transfer,69 hence facilitating better utilisation of the photogenerated charge for C–H cleavage.
Influence of loading the photocatalyst in the nanomembrane
Confinement of the photocatalytic reaction within the nanomembrane channels, would also play a role in determining the performances. The loading of the Pd/TiO2 photocatalyst on the nanomembrane would be thus an important factor. Samples with Pd/TiO2 photocatalyst loading in the 0.5–2.0 mg cm−2 range were prepared to investigate this effect.Unexpectedly, Fig. 4a shows a strong decrease in ethane productivity upon increasing the loading of Pd/TiO2 (SEA) samples. The greatest ethane yield, after 5 hours of irradiation, is achieved with a loading of 0.5 mg cm−2 and is over two times greater than that achieved with the sample having a two times higher loading. Controlling the secondary backreactions of the generated methyl radicals appears to be the crucial aspect in determining the performance. This fact can explain why a very limited loading is necessary.
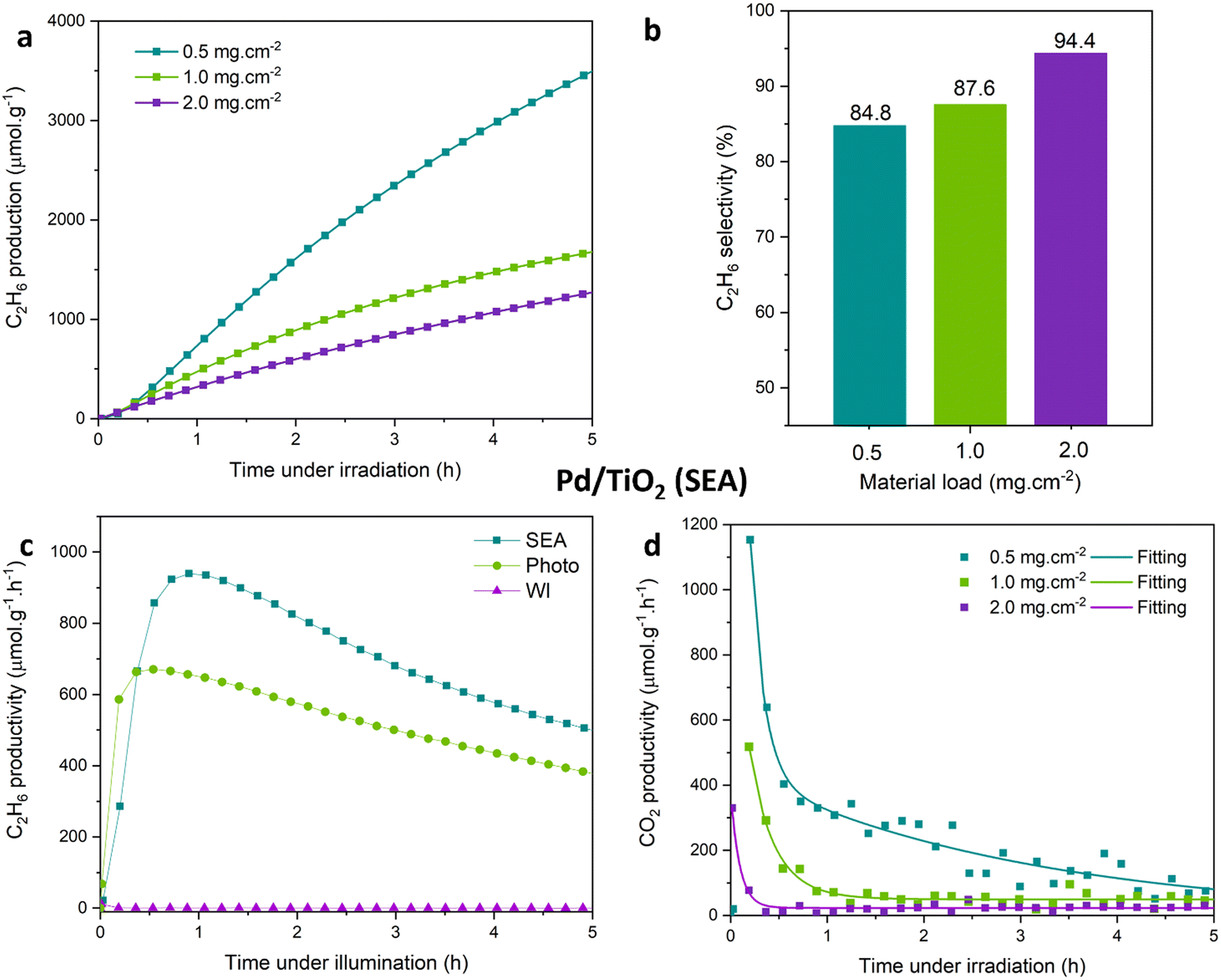 | ||
| Fig. 4 (a) Ethane yield (μmol g−1), (b) ethane selectivity (%), and (c) ethane and (d) CO2 production rates (μmol g−1 h−1) by photocatalytic NOCM over Pd/TiO2 (SEA) samples with loadings of 0.5, 1.0 and 2.0 mg cm−2. Reaction conditions as in Fig. 2. | ||
In terms of ethane selectivity, however, an opposite trend is observed (Fig. 4b). The sample Pd/TiO2 (SEA) with a loading of 2.0 mg cm−2 reaches an average ethane selectivity of almost 95% over 5 hours of reaction. Although the increase in selectivity is limited, it is not negligible.
The close to 100% selectivity is due to the formation of small amounts of CO2, related to the reaction of methyl radicals with surface hydroxyls (or lattice oxygens) of TiO2 or the nanomembrane. Increasing the loading decreases the fraction of nanomembrane “exposed” surface. The increase in selectivity could be reasonably associated with this aspect.
The ethane production rates pass through a maximum as a function of the irradiation time, as observed before. The maximum production rate nearly halves upon doubling the loading and further decreases, reaching 2.0 mg cm−2 loading (Fig. 4c). The initial increase corresponds to a strong change in the selectivity because the formation of CO2 is largely depressed (Fig. 4d). Note that these tests were performed without feeding oxygen, but there are surface hydroxyl groups (likely of the nanomembrane itself), which may lead to unselective methane conversion.70
The correspondence between the increased initial formation of CO2 and photocatalyst activity, upon both decreasing and increasing the loading from 0.5 to 2.0 mg cm−2, indicates that this is a secondary reaction of unselective conversion after methane activation, likely in the form of a methyl radical species. Still, by considering ethane and CO2 formation and accounting that two methane molecules are required to produce one ethane molecule, there is a progressive decrease in the rate of methane conversion, passing from an initial value of about 1400 μmol g−1 h−1 to around 1000 μmol g−1 h−1 after 5 h (for the most active sample with 0.5 mg cm−2 loading). The removal of surface hydroxyls generates likely surface sites that can trap photogenerated holes (generating phonon species, as commented before) and reduce their reactivity with methane to form methyl radicals.
Blank tests in Ar (Fig. S9, ESI†) evidence that irradiating the Pd/TiO2 (SEA) sample for 5 h, without feeding methane (only Ar flow), there is the formation of H2 but not of ethane. This derives from the photodissociation of strongly chemisorbed water or surface hydroxyl groups with the formation of O2. The mechanism of CO2 initial formation, is associated with the photo-splitting of chemisorbed water or reduction of surface hydroxyl groups, which generates O2 that reacts with methyl radicals, giving CO2. Likely, these species that photo dissociate, generating H2 and O2, are due to the nanomembrane itself because the formation of CO2 decreases when the photocatalyst loading increases. Note, however, that water (or hydroxyl groups) may also have a beneficial role in the stabilisation of methyl radicals.71
A positive effect of the loading is to improve the stability. In 1 h after reaching the maximum, the productivity loss decreases from 121 μmol g−1 h−1 for 0.5 mg cm−2 loading, to 41 μmol g−1 h−1 for 2.0 mg cm−2 loading (Fig. 4c). Therefore, calibrating the amount of photocatalyst in the nanomembrane is important to optimise the performance.
Transient absorption spectroscopy studies
Further insight into the interaction mechanism between methane and photocatalyst was obtained by transient absorption spectroscopy (TAS) experiments. The Pd/TiO2 (or bare TiO2) photocatalysts were supported on thin films of Fluorine Tin Oxide (FTO) covered glass substrates to perform these tests.The TAS spectrum (λexc = 355 nm, 300 mJ) of de-aerated bare TiO2 shows a main band at around 450 nm with a shoulder at 650 nm, which corresponds to the hole and electron contributions, respectively (Fig. 5a).72–75 The Pd/TiO2 samples show the same type of bands, but that at 650 nm is more intense (Fig. 5b). The intensity of this band increases from Photo to SEA samples, while it is similar to that of the bare TiO2 in the sample prepared by impregnation (WI). Note that there is good correspondence with the photocatalytic activity, e.g. with an improved e−–h+ charge separation.76 However, the asymmetric increase in the intensity of the 650 nm vs. 450 nm bands, indicates that electron trapping is especially enhanced.
 | ||
| Fig. 5 (a) Transient absorption spectra (TAS, λexc = 355 nm, 300 mJ) of bare TiO2 under Ar. (b) TAS (λexc = 355 nm, 300 mJ) of Pd/TiO2 (WI), Pd/TiO2 (Photo) and Pd/TiO2 (SEA) under Ar. (c) Normalised decay traces (λexc = 355 nm, λmon = 450 or 650 nm, 300 mJ) for bare TiO2 under Ar. (d) Normalised decay traces (λexc = 355 nm, λmon = 450 nm, 300 mJ) for Pd/TiO2 (WI), Pd/TiO2 (Photo) and Pd/TiO2 (SEA) under Ar. Bare TiO2 is included for comparison. | ||
Bare TiO2 shows a transient lifetime (τ) of 4 μs (λobs at 460 and 650 nm, Fig. 5c). Pd/TiO2 (Photo) and Pd/TiO2 (SEA) exhibit instead a long-lived lifetime in three consecutive transient contributions, with a huge increase of up to 17.5 and 26 μs, respectively, compared to bare TiO2 (Fig. 5d).77 The Pd/TiO2 (Photo) and Pd/TiO2 (SEA) samples, differently from bare TiO2 and Pd/TiO2 (WI) samples, show the presence of efficient trapping sites that slow down the electron–hole pair recombination rate. There is good correspondence between this effect and the photocatalytic activity in NOCM and CA experiments. The effect is related to the method of introduction of Pd rather than the presence of Pd itself. It is likely associated with the presence of Pd nanoclusters and/or Pd SA.
The influence of the presence of methane on these photophysical processes was also evaluated. While the transient spectra of bare TiO2 or Pd/TiO2 (WI) remain essentially unaltered in the presence of CH4, Pd/TiO2 (Photo) and Pd/TiO2 (SEA) were significantly quenched, particularly the latter (Fig. 6).
 | ||
| Fig. 6 Transient absorption spectra under a CH4 atmosphere (TAS, λexc = 355 nm, 300 mJ) for (a) TiO2, (b) Pd/TiO2 (WI), (c) Pd/TiO2 (Photo), and (d) Pd/TiO2 (SEA). | ||
More specifically, the hole contribution (400–500 nm range) was strongly decreased, indicating that photoexcited holes are scavenged by methane,22,40 in agreement with the previous comment and literature indications.24,28 In our case, moderate photoelectron contribution remained (from 550 nm). As reported, photoelectrons from the metal particles could play a role in the stabilisation of the CH3 radicals.78 Regarding the transient lifetimes, decay traces for Pd/TiO2 (Photo) and Pd/TiO2 (SEA) in the presence of methane, exhibited a very fast contribution (up to the first 500 ns), followed by a main contribution on the microsecond scale and, finally, a noticeable τ asymptotically reaching zero (Fig. 7). The τ of second contribution of Pd/TiO2 (Photo) and Pd/TiO2 (SEA) decreased up to 14 (Fig. 7a) and 10.8 μs (Fig. 7b), respectively. These strong differences could explain the different behaviours observed in the photocatalytic ethane production rate, confirming that Pd/TiO2 (SEA) exhibits an improved e−–h+charge separation, which promotes a more efficient activity and interaction with the methane substrate.79,80
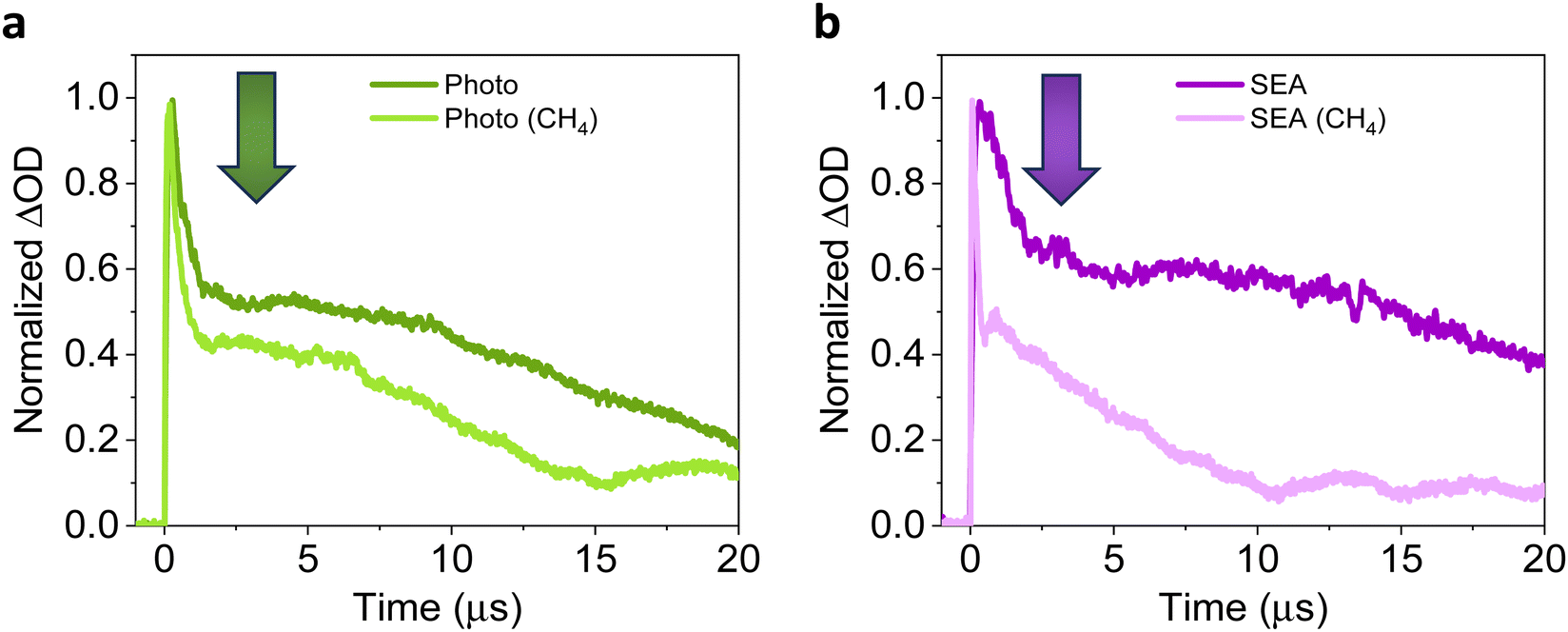 | ||
| Fig. 7 Normalised transient decay traces (λexc = 355 nm, λmon = 450 nm, 300 mJ) for (a) Pd/TiO2 (Photo) and (b) Pd/TiO2 (SEA) under a methane atmosphere. | ||
Long-term tests
The Pd/TiO2 (SEA) sample was evaluated through 24-hour continuous testing to analyse the long-term behaviour. Tests were made for loadings of 0.5 and 2.0 mg cm−2. The results of the ethane production rate are shown in Fig. S10 (ESI†). The evolution of alkane/alkene hydrocarbon selectivity is shown in Fig. S11 (ESI†). To our knowledge, this is the first time that these trends have been reported for this reaction.The alkane hydrocarbon selectivity slightly decreases with time under irradiation, although in general remaining high, except for the very initial part of the experiments due to the formation of CO2, as commented before. The sample with higher loading shows a better selectivity to ethane, but due essentially to lower formation of alkene, which instead is enhanced in the sample with a lower loading. There is, thus, a change in the relative formation of ethane vs. ethylene with the time of irradiation. Likely, this is due to a progressive decrease in the coverage of Pd nanoclusters by chemisorbed H, favouring the photo-assisted dehydrogenation of ethane. By increasing the amount of SA or low nuclearity Pd sites, the energetic stability of absorbed H in Pd decreases,81 consistently, with the increase in the ethylene formation (Fig. S11b, ESI†).
While the ethylene formation could also indicate deactivation by the formation of carbon deposits, we attribute this change in ethane productivity to the combination of surface modifications, due to the consumption of surface hydroxyl groups (or water) and/or lattice oxygen atoms and related influence on the mechanism of stabilisation and surface accumulation of charged species, as commented before.
The light irradiation in the presence of methane, creates a reductive environment that slowly reduces titania, creating oxygen vacancies, which alter the mechanism of charge trapping and availability of these photogenerated charges for reaction with methane (h+) and protons (e−), with the protons being the result of the reaction of methane with surface holes. An oxidative treatment at very mild temperature (80 °C) is proposed to remove these defects. Under these conditions, surface carbonaceous species are not removed, and the analysis of the outlet stream does not reveal the formation of COx.
Fig. 8 shows the application of this regeneration strategy to the sample Pd/TiO2 (SEA) after different times of irradiation. This procedure is effective in regenerating the photocatalyst up to about 5 h of irradiation (Fig. 8d). Still, for longer times (24 h), the treatment is essentially ineffective (Fig. 8b) or only partially effective (Fig. 8c). The same behaviour was observed with the sample Pd/TiO2 (photo) (Fig. S12, ESI†). It is worth noting that, when the Pd samples have been kept under Ar for the same time, no recovery of the original ethane productivity has been observed, proving the beneficial effect of mild reoxidation. This regeneration strategy allows the full recovery of the sample productivity only if regenerated early enough, when likely the photoinduced reduction of TiO2 does not lead to a significant surface reconstruction.
 | ||
| Fig. 8 (a) Regeneration strategy cycle, (b) regeneration cycles after 24 h of photocatalytic test on samples Pd/TiO2 (SEA) with 0.5 mg cm−2 loading and (c) Pd/TiO2 (SEA) with 2.0 mg cm−2 loading, and (d) regeneration cycles after 5 h of test on the sample Pd/TiO2 (SEA) with 2.0 mg cm−2 loading. Reaction conditions as in Fig. 2. | ||
Experimental
Materials and methods
All commercially available compounds were purchased from Sigma-Aldrich and used without further purification. Commercial Aeroxide P25 TiO2 was used as a support for all samples. Powder X-ray diffraction (XRD) patterns were recorded by using a D2 Phaser Bruker diffractometer equipped with a Ni β-filtered Cu-Kα radiation source.UV-vis data were recorded in the spectral range of 200–800 nm with a Thermo Fisher Evolution (220) spectrophotometer with an integrating sphere for solid samples.
Atomic absorption spectroscopy (AAS) was performed with a PerkinElmer Analyst 200.
Photocurrent profiles were recorded in 0.5 M Na2SO4, under a continuous flow of N2 (5 mL min−1), using a Pt wire as counter-electrode at a potential of 0.2 V against an Ag/AgCl reference electrode (Amel). The samples, acting as working electrodes, were deposited over FTO-coated glasses (active area = 2 cm2) to perform the measurements. Three different filters were applied to the lamp (AM 1.5 G, UVC blocking filter, UVB/C blocking filter) to evaluate the response in different wavelength regions.
High-resolution transmission electron microscopy (HRTEM), scanning transmission electron microscopy (STEM) and energy-dispersive X-ray (EDX) spectroscopy were carried out using a transmission electron microscope JEOL-JEM300F equipped with an ISIS 300 X-ray microanalysis system (Oxford Instruments) with a LINK “Pentafet” EDS detector.
Transient absorption spectroscopy (TAS) measurements were carried out with LP980 equipment from Edinburgh Instruments (LP980) based on an optical parametric oscillator (OPO) pumped by the third harmonic of a Nd:YAG laser (EKSPLA). The selected excitation wavelength for monitoring the signals was 355 nm with single low energy pulses of 300 μJ per pulse for ca. 5 ns duration. At the same time, a pulsed xenon flash lamp (150 W) was employed as a probe source. The probe light is dispersed through a monochromator (TMS302-A, grating 150 lines per mm) after it has passed through the sample and then reached a PMT detector (Hamamatsu Photonics) to obtain the temporal profile. The thin films of Fluorine Tin Oxide (FTO) containing the photocatalyst, were measured employing cuvettes with four clear sides and deaerated for 15 min with argon (Ar) and methane (CH4) before acquisition. All transient signals were recorded at room temperature. The transient spectra were measured after only one pulse laser, and the corresponding kinetics were measured after three pulse laser averages to avoid degradation of the sample.
Synthetic procedures
The M/TiO2 (Photo) samples were prepared by photo-deposition. Typically, a determined amount of metal precursor (HAuCl4, PdCl2, and Ag(NO3)) was dissolved in the minimum quantity of water and then added into a TiO2 P25 dispersion (5 g L−1)) with isopropanol (0.3 M). The mixture was left under agitation under nitrogen flow to remove oxygen, in the dark for 30 minutes, and then irradiated with a Xe lamp (150 mW cm−2) for 2 h. The resulting solution was filtered, washed with water 3 times, and dried in air overnight at 80 °C.The Pd/TiO2 (SEA) sample was prepared by strong electrostatic adsorption (SEA). First, 100 mg of TiO2 P25 were ultrasonically dispersed in 25 mL of dilute ammonia (pH 11.5). 100 μL of a solution of Pd acetate (20 mM) dissolved in 15 mL of dilute ammonia was then slowly introduced into the TiO2 dispersion with a syringe pump at a rate of 3 mL h−1. The solution was left under agitation for 3 hours; the powder was collected through centrifugation, washed with distilled water and finally calcined at 300 °C for 2 h.
For the synthesis of Pd/TiO2 (WI), 16.7 mg of PdCl2 were dissolved in a minimum quantity of water and added to a solution of P25 (1.0 g) dispersed in 20 mL of water. After 30 minutes of stirring, the solution was heated to 80 °C until dryness. Finally, the remaining powder was calcined at 400 °C for 2 h.
Photocatalytic tests
Catalysts were suspended in isopropanol and homogeneously deposited over a paper filter membrane by spray coating, to evaluate the photocatalytic activity in the NOCM reaction. The membrane was kept at 80 °C for 18 h before tests to remove humidity absorbed by the filter paper. Before irradiation, Ar was flown to remove air from the reactor, and then the reaction gas (50% CH4, balance Ar) was introduced into the reactor (flow rate = 0.6 L h−1, controlled by mass flow controllers). After 1 hour (to reach equilibrium conditions), the samples were irradiated with a 300 W Xe lamp (Quantum Design Europe, Darmstadt, Germany) for 5 hours. The products were quantified using a gas chromatograph (Agilent GC 8890 Ar/He carriers) equipped with 30m Rt®-Q-Bond columns and flame ionisation detectors for analysis of hydrocarbons and Poropak-Q and MS-13X columns and a thermal conductivity detector to quantify hydrogen and CO2.Conclusions
In summary, we have reported the first example of a continuous photocatalytic NOCM process in a novel type of flow-through photoreactor with very high C2 productivity. The nature of metallic electrocatalysts is essential in the selective coupling of methane to ethane, to address the high stability of the C–H bond. Pd ultrafine clusters/SA well dispersed on a TiO2 support (the sample prepared by SEA) resulted in an increased yield of C2 products (maximum production rate of 939 μmol g−1 h−1) with respect to the other Pd loaded catalysts, with a selectivity to ethane of almost 95% after 5 hours of reaction. TAS experiments revealed the involvement of the holes and photoelectrons after charge separation and their interaction with methane in the ethane production reaction. The change in ethane productivity is related to modification of the TiO2 surface during photoirradiation, due to the consumption of surface hydroxyl groups (or water) and/or lattice oxygen atoms which, by altering the physicochemical processes, may affect the photocatalytic behaviour.The material's loading is also essential to maximise the performance. The proposed regeneration strategy, based on very mild reoxidation treatment (80 °C), allows effective regeneration only when the time of exposure to irradiation is limited to a few hours. However, the results of this study allow us to obtain insight into the mechanism of the change in the photocatalytic behaviour and suggest some possible strategies, such as optimisation of the nanomembrane characteristics, doping to accelerate the reaction of protons with photogenerated electrons, and co-feeding of small amounts of water to enhance and stabilise the photocatalyst performances. Even if further work should be necessary, these results are already very promising and open new prospects in terms of novel possibilities for the photocoupling of methane.
Author contributions
VL: investigation, data curation, formal analysis, methodology, writing – original draft; LdP, FT, MG: investigation, data curation; MB: conceptualization, validation, investigation, data curation; VdlP: project administration, funding acquisition; CA: conceptualization, validation, supervision; SP: project administration, funding acquisition; GC: conceptualization, validation, writing – reviewing and editing, project administration, funding acquisition; CG: methodology, conceptualization, validation, supervision, writing – original draft, writing – reviewing and editing, project administration, funding acquisition.Data availability
Data are available upon request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Union through the ERC SCOPE project (ID 810182) and the MIUR (Italy) through the PRIN Project MAPEC (No. 2022599NR3) funded this work, and are gratefully acknowledged. MB thanks the NOVACO2 project (PID2020-118593RB-C22) and the RYC2022-038157-I grant both funded by MCIN/AEI/10.13039/501100011033.References
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- CEFIC, Natural Gas: A Vital Energy And Feedstock Source For The Chemical Industry To Transition Towards A Competitive, Climate-Neutral, 2050, https://cefic.org/media-corner/newsroom/natural-gas-a-vital-energy-and-feedstock-source-for-the-chemical-industry-to-transition-towards-a-competitive-climate-neutral-2050/, (accessed 03/05/2024) Search PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- P. Wang, R. Shi, Y. Zhao, Z. Li, J. Zhao, J. Zhao, G. I. N. Waterhouse, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304301 CrossRef CAS PubMed.
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6, 2497–2514 CAS.
- J. Deng, P. Chen, S. Xia, M. Zheng, D. Song, Y. Lin, A. Liu, X. Wang, K. Zhao and A. Zheng, Atmosphere, 2023, 14, 1538 CrossRef CAS.
- C. A. Ortiz-Bravo, C. A. Chagas and F. S. Toniolo, J. Nat. Gas Sci. Eng., 2021, 96, 104254 CrossRef CAS.
- P. Wang, X. Zhang, R. Shi, J. Zhao, Z. Yuan and T. Zhang, Energy Fuels, 2022, 36, 11627–11635 CrossRef CAS.
- Y. Liu, M. Ćoza, V. Drozhzhin, Y. van den Bosch, L. Meng, R. van de Poll, E. J. M. Hensen and N. Kosinov, ACS Catal., 2023, 13, 1–10 CrossRef CAS.
- U. Menon, M. Rahman and S. J. Khatib, Appl. Catal., A, 2020, 608, 117870 CrossRef CAS.
- S. Meng, W. Li, Z. Li and H. Song, Catal. Sci. Technol., 2023, 13, 4665–4672 RSC.
- M. Zhou, Z. Yang, J. Ren, T. Zhang, W. Xu and J. Zhang, Int. J. Hydrogen Energy, 2023, 48, 78–89 CrossRef CAS.
- P.-A. Maitre, M. S. Bieniek and P. N. Kechagiopoulos, J. Phys. Chem. C, 2022, 126, 19987–20003 CrossRef CAS.
- V. Longo, G. Centi, S. Perathoner and C. Genovese, Curr. Opin. Green Sustainable Chem., 2024, 46, 100893 CrossRef CAS.
- M. S. A. Sher Shah, C. Oh, H. Park, Y. J. Hwang, M. Ma and J. H. Park, Adv. Sci., 2020, 7, 2001946 CrossRef CAS PubMed.
- J. Jang, K. Shen and C. G. Morales-Guio, Joule, 2019, 3, 2589–2593 CrossRef.
- M. Ma, B. J. Jin, P. Li, M. S. Jung, J. I. Kim, Y. Cho, S. Kim, J. H. Moon and J. H. Park, Adv. Sci., 2017, 4, 1700379 CrossRef.
- Q. Li, Y. Ouyang, H. Li, L. Wang and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202108069 CrossRef CAS PubMed.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617–632 CrossRef CAS.
- P. Zhou, S. Tang, Z. Ye, I. A. Navid, Y. Xiao, K. Sun and Z. Mi, Chem. Sci., 2024, 15, 1505–1510 RSC.
- G. Yuniar, W. H. Saputera, D. Sasongko, R. R. Mukti, J. Rizkiana and H. Devianto, Molecules, 2022, 27, 5496 CrossRef CAS.
- P. Wang, R. Shi, J. Zhao and T. Zhang, Adv. Sci., 2024, 11, 2305471 CrossRef CAS.
- S. Wu, L. Wang and J. Zhang, J. Photochem. Photobiol., C, 2021, 46, 100400 CrossRef CAS.
- Q. Zhan, Y. Kong, X. Wang and L. Li, Chem. Commun., 2024, 60, 2732–2743 RSC.
- Y. Liu, Y. Chen, W. Jiang, T. Kong, P. H. C. Camargo, C. Gao and Y. Xiong, Research, 2022, 9831340 CAS.
- J. Wang, Y. Peng and W. Xiao, Sci. China: Chem., 2023, 66, 3252–3261 CrossRef CAS.
- Z. Chen, Y. Ye, X. Feng, Y. Wang, X. Han, Y. Zhu, S. Wu, S. Wang, W. Yang, L. Wang and J. Zhang, Nat. Commun., 2023, 14, 2000 CrossRef CAS PubMed.
- Z. Liu, B. Xu, Y. J. Jiang, Y. Zhou, X. Sun, Y. Wang and W. Zhu, ACS Environ. Au, 2023, 3, 252–276 CrossRef CAS PubMed.
- D. Hu, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2021, 286, 119913 CrossRef CAS.
- Z. Zhu, W. Guo, Y. Zhang, C. Pan, J. Xu, Y. Zhu and Y. Lou, Carbon Energy, 2021, 3, 519–540 CrossRef CAS.
- W. Zhang, C. Fu, J. Low, D. Duan, J. Ma, W. Jiang, Y. Chen, H. Liu, Z. Qi, R. Long, Y. Yao, X. Li, H. Zhang, Z. Liu, J. Yang, Z. Zou and Y. Xiong, Nat. Commun., 2022, 13, 2806 CrossRef CAS.
- J. Ma, X. Tan, Q. Zhang, Y. Wang, J. Zhang and L. Wang, ACS Catal., 2021, 11, 3352–3360 CrossRef CAS.
- S. P. Singh, A. Anzai, S. Kawaharasaki, A. Yamamoto and H. Yoshida, Catal. Today, 2021, 375, 264–272 CrossRef CAS.
- Y. Xu, E. Chen and J. Tang, Carbon Future, 2024, 1, 9200004 CrossRef.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, J. Am. Chem. Soc., 2019, 141, 6592–6600 CrossRef CAS PubMed.
- Z. Chen, S. Wu, J. Ma, S. Mine, T. Toyao, M. Matsuoka, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11901–11909 CrossRef CAS PubMed.
- G. Wang, X. Mu, J. Li, Q. Zhan, Y. Qian, X. Mu and L. Li, Angew. Chem., Int. Ed., 2021, 60, 20760–20764 CrossRef CAS.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- J. Zhang, J. Shen, D. Li, J. Long, X. Gao, W. Feng, S. Zhang, Z. Zhang, X. Wang and W. Yang, ACS Catal., 2023, 13, 2094–2105 CrossRef CAS.
- J. Lang, Y. Ma, X. Wu, Y. Jiang and Y. H. Hu, Green Chem., 2020, 22, 4669–4675 RSC.
- X. Wang, N. Luo and F. Wang, Chin. J. Chem., 2022, 40, 1492–1505 CrossRef CAS.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. - Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed.
- M. Schreck and M. Niederberger, Chem. Mater., 2019, 31, 597–618 CrossRef CAS.
- B. Hauchecorne and S. Lenaerts, J. Photochem. Photobiol., C, 2013, 14, 72–85 CrossRef CAS.
- Y. Ma, S. Wang and X. Duan, Chem. Eng. J., 2023, 455, 140654 CrossRef CAS.
- S. Ali, M. C. Flores, A. Razzaq, S. Sorcar, C. B. Hiragond, H. R. Kim, Y. H. Park, Y. Hwang, H. S. Kim, H. Kim, E. H. Gong, J. Lee, D. Kim and S.-I. In, Catalysts, 2019, 9, 727 CrossRef CAS.
- A. Visan, J. R. van Ommen, M. T. Kreutzer and R. G. H. Lammertink, Ind. Eng. Chem. Res., 2019, 58, 5349–5357 CrossRef CAS.
- Y. Sun and Z. Tang, MRS Bull., 2020, 45, 20–25 CrossRef CAS.
- T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2007, 111, 5259–5275 CrossRef CAS.
- L. De Pasquale, F. Tavella, V. Longo, M. Favaro, S. Perathoner, G. Centi, C. Ampelli and C. Genovese, Molecules, 2023, 28, 3378 CrossRef CAS PubMed.
- R. Binjhade, R. Mondal and S. Mondal, J. Environ. Chem. Eng., 2022, 10, 107746 CrossRef CAS.
- K. P. Sundar and S. Kanmani, Chem. Eng. Res. Des., 2020, 154, 135–150 CrossRef CAS.
- D. Giusi, C. Ampelli, C. Genovese, S. Perathoner and G. Centi, Chem. Eng. J., 2021, 408, 127250 CrossRef CAS.
- R. Wojcieszak, M. J. Genet, P. Eloy, P. Ruiz and E. M. Gaigneaux, J. Phys. Chem. C, 2010, 114, 16677–16684 CrossRef CAS.
- T. Kumari, R. Gopal, A. Goyal and J. Joshi, J. Inorg. Organomet. Polym. Mater., 2019, 29, 316–325 CrossRef CAS.
- Y.-H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 15425–15442 CrossRef CAS.
- Y.-H. Chin and E. Iglesia, J. Phys. Chem. C, 2011, 115, 17845–17855 CrossRef CAS.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- S. Lettieri, M. Pavone, A. Fioravanti, L. Santamaria Amato and P. Maddalena, Materials, 2021, 14, 1645 CrossRef CAS PubMed.
- S. Selçuk and A. Selloni, J. Phys. D: Appl. Phys., 2017, 50, 273002 CrossRef.
- T. Thathsara, C. J. Harrison, R. K. Hocking and M. Shafiei, Chemosensors, 2023, 11, 409 CrossRef CAS.
- S. M. Rasul, D. R. Saber and S. B. Aziz, Results Phys., 2022, 38, 105688 CrossRef.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
- J. Rongé, D. Nijs, S. Kerkhofs, K. Masschaele and J. A. Martens, Phys. Chem. Chem. Phys., 2013, 15, 9315–9325 RSC.
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CrossRef CAS.
- X.-Y. Lin, J.-Y. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Catal. Commun., 2021, 159, 106346 CrossRef CAS.
- J. Baltrusaitis, I. Jansen and J. D. Schuttlefield Christus, Catal. Sci. Technol., 2014, 4, 2397–2411 RSC.
- A. Khandelwal, D. Maarisetty and S. S. Baral, in Handbook of Smart Materials, Technologies, and Devices: Applications of Industry 4.0, ed. C. M. Hussain and P. Di Sia, Springer International Publishing, Cham, 2022, pp. 2751–2787 DOI:10.1007/978-3-030-84205-5_85.
- K. Shimura, S. Kato, T. Yoshida, H. Itoh, T. Hattori and H. Yoshida, J. Phys. Chem. C, 2010, 114, 3493–3503 CrossRef CAS.
- S. Rudić, J. M. Merritt and R. E. Miller, Phys. Chem. Chem. Phys., 2009, 11, 5345–5352 RSC.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- A. Kafizas, X. Wang, S. R. Pendlebury, P. Barnes, M. Ling, C. Sotelo-Vazquez, R. Quesada-Cabrera, C. Li, I. P. Parkin and J. R. Durrant, J. Phys. Chem. A, 2016, 120, 715–723 CrossRef CAS PubMed.
- J. Li, R. Chen, K. Wang, Y. Yang, J. Wang, W. Yang, S. Wang, G. Yang and F. Dong, Research, 2023, 6, 0055 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- J. Schneider and M. Curti, Photochem. Photobiol. Sci., 2023, 22, 195–217 CrossRef CAS.
- S. Naniwa, K. Kato, A. Yamakata, A. Yamamoto and H. Yoshida, ACS Catal., 2023, 13, 15212–15218 CrossRef CAS.
- X. Huang, D. Eggart, G. Qin, B. B. Sarma, A. Gaur, J. Yang, Y. Pan, M. Li, J. Hao, H. Yu, A. Zimina, X. Guo, J. Xiao, J. D. Grunwaldt, X. Pan and X. Bao, Nat. Commun., 2023, 14, 5716 CrossRef CAS PubMed.
- N. Feng, H. Lin, H. Song, L. Yang, D. Tang, F. Deng and J. Ye, Nat. Commun., 2021, 12, 4652 CrossRef CAS.
- Z. Gong, L. Luo, C. Wang and J. Tang, Solar RRL, 2022, 6, 2200335 CrossRef CAS.
- S. M. Kozlov, H. A. Aleksandrov and K. M. Neyman, J. Phys. Chem. C, 2015, 119, 5180–5186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00112e |
| This journal is © The Royal Society of Chemistry 2024 |