
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Epitaxial heterointerfacial electron bridge synchronizes oxygen evolution activity and stability on a layered double hydroxide surface†
Jia
Wang
ab,
Zelin
Zhao
b,
Min
Guo
c,
Liang
Xiao
 b,
Haolin
Tang
de,
Jiantao
Li
*f,
Zongkui
Kou
*d and
Junsheng
Li
*be
b,
Haolin
Tang
de,
Jiantao
Li
*f,
Zongkui
Kou
*d and
Junsheng
Li
*be
aSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
bSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: li_j@whut.edu.cn
cSchool of Mechanical and Electronic Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
dState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan 430070, P. R. China. E-mail: zongkuikou@whut.edu.cn
eFoshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory, Foshan 528200, P. R. China
fChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA. E-mail: jiantao.li@anl.gov
First published on 2nd April 2024
Abstract
Scalable green hydrogen production via electrocatalytic water splitting is largely restricted by the insufficient activity and stability of oxygen evolution reaction (OER) catalysts at the anode. As a class of the most active OER catalysts in alkaline electrolyzers, the application of layered double hydroxides (LDHs) remains a main challenge owing to the unstable lattice oxygen dissolution due to the dominant lattice oxygen-involving OER mechanism during long-term operation. Herein, we found that using an epitaxial hetero-interfacing nickel hydroxide (namely Ni(OH)2) as an electron bridge between an active FeCo LDH and Ni foam support to form an LDH*/NFO catalyst, the electronic storage capacity around the Fermi level (−0.5 to +0.5 eV, e-DFE) sharply increases from 0.93 per cell to 1.51 per cell. Subsequently, we demonstrate that this high e-DFE enables ceaseless and fast power injection into the kinetic process of intermediate species conversion and inhibits lattice oxygen dissolution in the active FeCo LDH. Consequently, it demonstrated a low OER overpotential of 246 mV at a current density of 100 mA cm−2 and ultrahigh stability for up to 3500 hours with an ultraslow overpotential increase rate of 9.4 × 10−3 mV h−1. Therefore, we developed an epitaxial hetero-interfacial electron bridging strategy to synchronize the activity and stability of available catalysts for scalable green hydrogen production via electrocatalytic water splitting.
Broader contextThe green production of hydrogen via water electrolysis is a promising solution to sustain a zero-carbon emission society. However, owing to the high activation barrier (requiring a large overpotential) and harsh corrosive environment of the anodic oxygen evolution reaction (OER), the cost-effectiveness of electrocatalytic water splitting is considerably hampered by the insufficient activity and stability of OER catalysts. In this work, highly active and durable LDH-based catalysts (LDH*/NFO) were designed for the anodic OER through the introduction of hetero-interfacing Ni(OH)2, acting as an electron bridge between LDHs and the Ni support. This innovative strategy successfully enhanced the durability and activity of the LDH*/NFO catalyst during the OER process, particularly at increasing current densities, resulting in more cost-effective hydrogen production. This study serves as the first-ever demonstration of an electron bridge strategy, showcasing its effectiveness in synchronizing the activity and stability of available catalysts for scalable green hydrogen production. |
1. Introduction
Global carbon emissions, which have triggered serious climate change and environmental issues, are largely associated with the combustion of carbon-based fossil fuels.1–3 In this case, hydrogen is widely considered a perfect alternative energy carrier due to its high energy density (33.6 kW h kg−1) and environmental friendliness.4–7 Alkaline water electrolysis is the most widely employed technique for the production of high-purity hydrogen nowadays. However, alkaline water electrolysis faces the limitation of sluggish kinetics in the anodized oxygen evolution reaction (OER).7–10 Theoretically, the OER exhibits a high activation barrier due to the limitation of the “scaling relation”.8,11,12 In addition, a catalyst undergoes dynamic structural changes, which may lead to its rapid deterioration, especially in the case of the lattice oxygen mechanism (LOM)-dominated OER process.13,14 Practically, water splitting on an industrial scale should be conducted at a large current density (i.e. >500 mA cm−2) to make hydrogen production cost-effective.15 Thus far, most of the investigations on OER catalysts have focused on the exploration, fabrication and understanding the mechanism of novel catalysts under laboratory conditions (a low current density of 10–50 mA cm−2 over a short period), indicating a large gap in industrial application.16 Consequently, bridging the gap between laboratory investigations and industrial application is critical for the future development of OER catalysts.17–19Layered double hydroxides (LDHs) are considered promising catalysts for the practical alkaline OER owing to their unique electronic states in the 3d-band of metal sites and 2D micro-structure, which lead to moderate adsorption and desorption energy towards the OER intermediates and improved mass transfer of reactants and products, respectively.20–23 However, due to the coordinatively saturated nature of most metal sites in LDHs, they cannot directly participate in the multi-step adsorption and desorption of oxygen intermediates.24–26 Consequently, the electro-transfer process during an LDH-catalyzed OER primarily follows the oxygen redox chemistry referring to the LOM, rather than metal redox chemistry, which is known as the adsorbate evolution mechanism (AEM).27 However, LOM-based electrocatalysts will undergo surface reconstruction, given that the cations are prone to leaching to achieve charge equilibrium due to the slower speed of refilling of oxygen vacancies compared to their formation.27,28 The LOM-introduced surface reconstruction will lead to an unstable structure and a sharp performance decay, especially at a large current density and during long-term operation.29 In practical alkaline water electrolysis, the anode should function properly at a large current density for thousands of hours at a high potential.20,30,31 A pressing issue in the development of LDH electrocatalysts is improving their long-term stability at large current densities because typical LDH-based catalysts suffer from rapid performance decay guided by LOM.9,32,33 Thus far, only few studies focused on enhancing the long-term durability of LDH electrodes. Moreover, most of the reported LDH catalysts exhibit a reasonable OER performance at a current density lower than 100 mA cm−2. Currently, the apparent OER activity of LDH is typically regulated through cation doping, anion intercalation, defect engineering, etc. However, these strategies usually lead to adverse crystal destruction and lattice distortion, which sacrifice the long-term stability of LDHs under strong oxidative OER conditions.
Given that the surface of the catalyst experiences possible compositional and morphological changes during OER, immobilization of the catalyst on the electrode is critical to ensure the robustness of the anode.34–36 In the case of the commonly used integrated electrodes with the metal substrate and catalyst in situ grown on the substrate, fine control of the heteroepitaxy growth process of catalytic species on the substrate is of vital importance for the performance of the electrode.37 On the one hand, constructing a conformal interface not only helps to generate a strong linkage between the catalyst and substrate but may also improve the crystallinity of the catalyst.38,39 On the other hand, the “electron bridge” (electron density around the Fermi level [−0.5 eV, 0.5 eV], e-DEF) of the catalyst can be also modulated by the underlying substrate to optimize the adsorption/desorption of the OER intermediates on the catalysts.40–44 Currently, active LDH layers are introduced directly on the surface of nickel foam.45–47 It has been noted that a high in-plane lattice mismatch (12.8%) exists between the cubic Ni(001) and typical facets of hexagonal/rhombic LDH. This lattice mismatch may limit both the stability and activity of the electrode.48–51 From this perspective, optimization of the interface between LDH and Ni foam is expected to improve the performance of the electrode. Herein, an in situ-grown intermediate Ni(OH)2 layer as an electron bridge was rationally designed and introduced between Ni foam and Fe/Co LDH. The ultra-low in-plane lattice mismatch (0.04%) between Ni(OH)2 and FeCo-LDH reduced the strain from the in-plane lattice mismatch and suppressed the lattice distortion. Specifically, the capacity of the electron pocket increased from 0.63 per cell for LDH and 0.93 per cell for LDH/NF to 1.51 per cell of LDH*/NFO, indicating more electrons being enriched at the active site, show then inject more power into the kinetic progression and restrain the leaching of the lattice oxygen (Fig. S1, ESI†). The above-mentioned benefits altogether resulted in the electrocatalyst exhibiting outstanding OER activity (η100 = 246 mV) and ultra-high stability (stable for at least 3500 h at 100 mA cm−2).
2. Results and discussion
2.1 The role of Ni(OH)2 electron bridge on LDH*/NFO identified by DFT calculations
The degree of lattice matching between the catalyst and the support is an important factor that governs both the activity and stability of the catalyst.41,43 Thus, to demonstrate the favorable regulation of the electron bridge Ni(OH)2 on the heterojunction structure and the electron structure, density functional theory (DFT) calculations were conducted. The formation energy of FeCo-LDH on NFO and NF substrates was analyzed using the optimal configuration of LDH*/NFO and LDH/NF, respectively (Fig. 1a). The result revealed a much lower formation energy of FeCo-LDH on NFO than that on NF (−27.10 eV vs. −9.06 eV), suggesting that LDH*/NFO is more thermodynamically stable. In addition, the optimized molecular configuration demonstrated high interfacial structural ordering for LDH*/NFO, expressing a high lattice matching between LDH* and NFO, which will restrain the metal cation leaching caused by the LOM-introduced surface reconstruction.34,39 Furthermore, the formation energy of Ni/Ni(OH)2 was calculated to be lower, i.e. −28.45 eV, indicating the excellent thermodynamical stability of the Ni/Ni(OH)2 interface (NFO). Considering the effect of the electronic structure on the intrinsic activity of catalysts, the total density of states (DOS) was compared. As shown in Fig. 1b–d, the electron density around the Fermi level (−0.5 eV, 0.5 eV) of LDH*/NFO (1.51 per cell) is higher than that in LDH (0.63 per cell) and LDH/NF (0.93 per cell), indicating a large electron capacity in LDH*/NFO. The large electron capacity around the Fermi level (Ef) is beneficial for electron enrichment and electron storage at the active sites, which will inject more power into the kinetic progression. To gain a comprehensive understanding of the effect of the electron bridge on the interaction between the reaction intermediates and the metal sites, we computed the position of the d-band center (εd) of the LDH*/NFO. The εd value of the Fe sites (Fig. S2a–c, ESI†) was calculated to be −0.25, −2.55 and −1.74 eV for LDH, LDH/NF and LDH*/NFO, respectively. According to the d-band center theory, when the εd shifts to approach the Ef, the electrons in the antibonding orbitals transfer to the bonding orbital, resulting in the increased adsorption of intermediates on the metal sites.5,6,50 Therefore, the intermediate εd value for LDH*/NFO (1.74 eV) will contribute to regulating the adsorption and desorption capacities of the reaction intermediates on the Fe sites. In the case of the Co sites (Fig. S2d and e, ESI†), LDH*/NFO exhibited the lowest εd value (−2.02 eV) compared to LDH (−1.78 eV) and LDH/NF (−1.64 eV). This discrepancy will lead to an increase in filled electrons in the antibonding orbital, consequently facilitating O2 desorption. Thus, according to the DFT calculations, the Ni(OH)2 electron bridge demonstrates feasibility to enhance the OER performance via the regulation of its electron structure.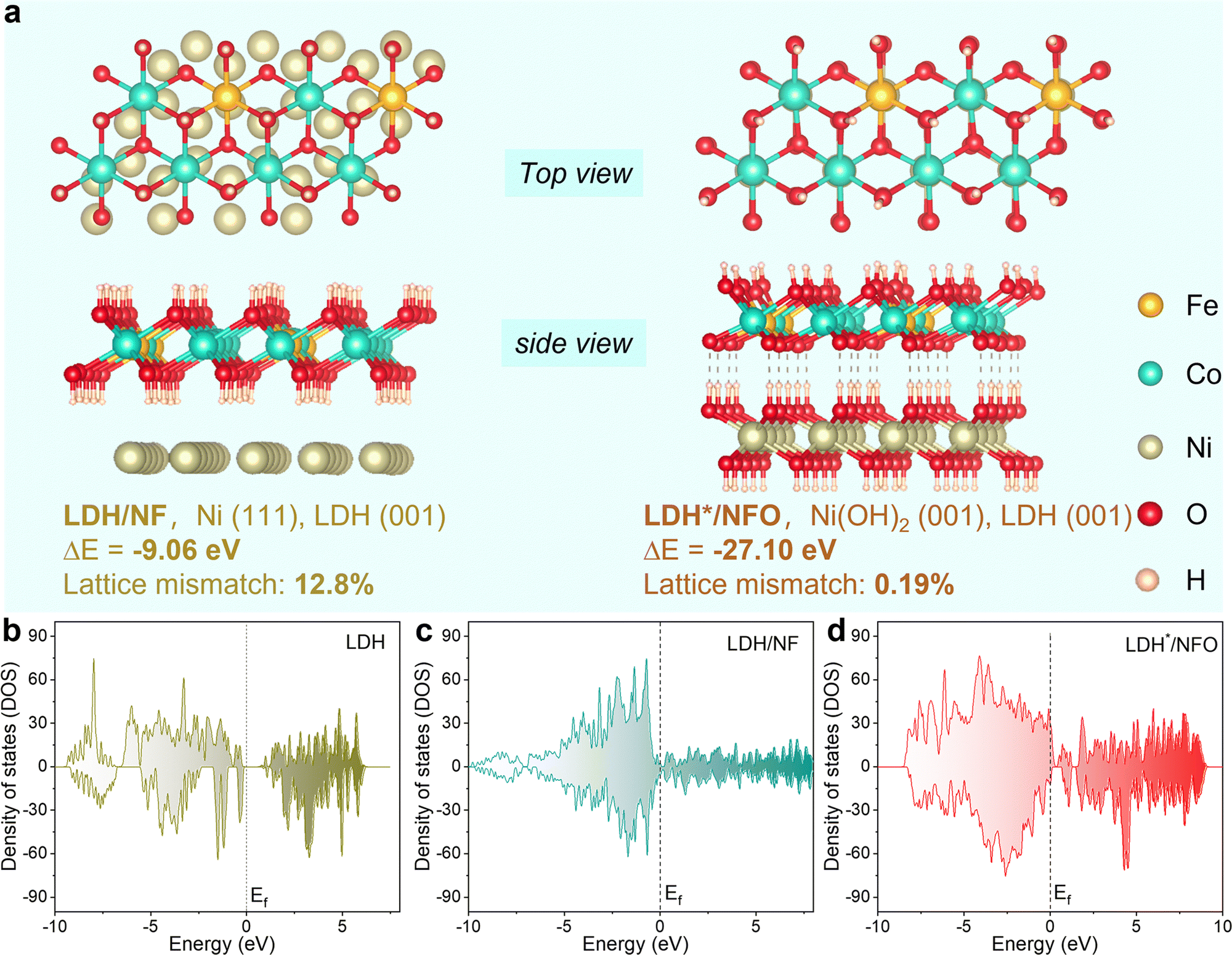 | ||
| Fig. 1 Thermodynamic analysis of the catalyst. (a) Geometric structure of LDH/NF and LDH*/NFO. DOS curves of (b) LDH, (c) LDH/NF and (d) LDH*/NFO. | ||
2.2 Material synthesis and characterization
The two-step fabrication process of LDH*/NFO is exhibited in Fig. 2a, where the in situ oxidization growth of Ni(OH)2 is the key to constructing the electron bridge between LDH and NF. For comparison, FeCo-LDH (LDH) was also prepared directly on NF. The optical photos showed a salient color evolution in NF after electrochemical oxidation and a distinct color difference between LDH*/NFO and LDH/NF (Fig. S3, ESI†). This color difference suggests that the epitaxial growth behavior of FeCo-LDH may be influenced by the underlying support. Powder X-ray diffraction (PXRD) measurements were performed to analyze the lattice matching of LDH* and LDH with their underlying substrates. In the case of LDH/NF (Fig. 2b and Fig. S4a, ESI†), its characteristic diffraction peaks can be indexed to standard cubic Ni phase (PDF#04-0850) with the lattice constant of 3.52 Å and hexagonal FeCo-LDH (PDF#50-0235) with the in-plane lattice constant of 3.120 Å, revealing a large in-plane lattice mismatch exceeding 12.8% between NF and LDH. According to the epitaxial growth theory, the epilayer quality and the stability of the heterointerface will deteriorate when the lattice mismatch exceeds 10% because of the presence of strain and the formation of a vulnerable semi-collegial structure between the epilayers and substrates.49,51,52 In the PXRD spectrum of LDH*/NFO (Fig. 2b), the diffraction peaks centered at 19.2°, 33.0° and 38.5° can be indexed to the (001), (100) and (101) facets of hexagonal Ni(OH)2 (PDF#14-0117). The lattice constant of Ni(OH)2 is 3.126 Å, which yields significantly lower lattice mismatch (∼0.19%) between FeCo-LDH and the underlying substrate for LDH*/NFO. The higher intensity of the diffraction peaks in the PXRD spectrum of LDH*/NFO compared to that of LDH/NF indicates that the introduction of the electron bridge contributed to reducing the lattice mismatch. This observation is consistent with the DFT results. The poor crystallinity of LDH on LDH/NF is due to the interfacial stress induced by the prominent lattice mismatch between LDH and NF, disrupting the formation of an in-plane periodic arrangement of LDH.53–55 The (003) diffraction peak of LDH* shifted toward a lower diffraction angle compared to that of pure FeCo-LDH synthesized without NF support (Fig. 2c), indicating a larger interlayer space, which is beneficial for mass transfer in the electrocatalytic process. LDH/NF displayed a broad diffraction peak at 11.75° (Fig. 2c), indicating its lower crystallinity. Additionally, this diffraction peak (2θ = 11.75°) for LDH/NF shifted positively compared to that for pure FeCo-LDH, suggesting the smaller interlayer space of LDH/NF. The positive shift in peak position may be attributed to the compressive strain induced by the NF substrate. The DFT analysis revealed this lattice distortion of FeCo-LDH in LDH/NF, with the in-plane lattice constant changing from 3.12 Å to 3.04 Å (Table S1, ESI†), thereby altering the interlayer space.56,57 These findings demonstrate the advantageous role of the Ni(OH)2 electron bridge in regulating the microstructure of epitaxial-grown FeCo-LDH toward a more efficient catalytic process.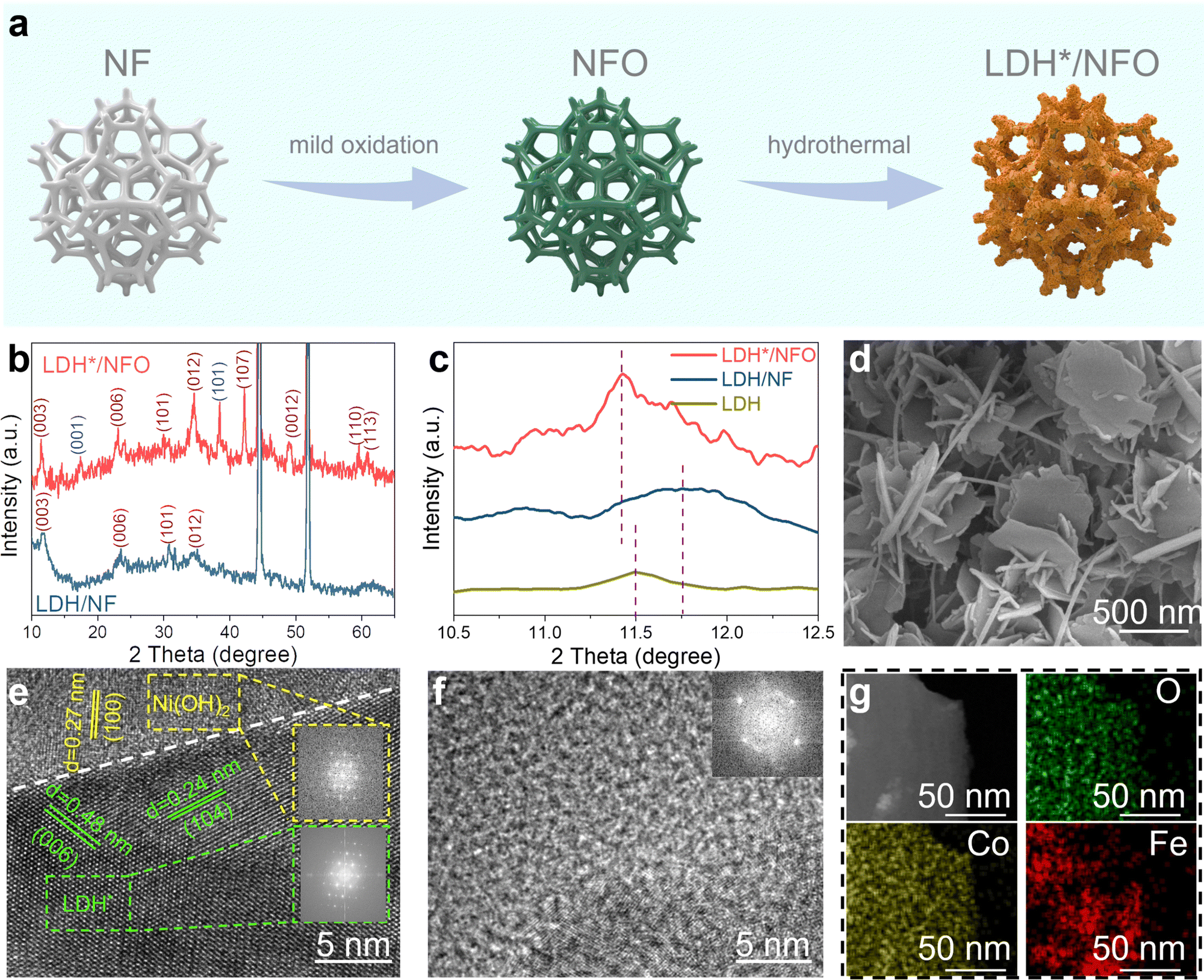 | ||
| Fig. 2 Structural characterization of LDH*/NFO. (a) Schematic illustration of the fabrication process of LDH*/NFO. (b) and (c) XRD patterns of LDH, LDH/NF and LDH*/NFO. (d) SEM images of LDH*/NFO. HRTEM images (e) LDH*/NFO and (f) LDH/NF. (g) EDX-mapping images of LDH*/NFO. | ||
The characteristic 3D porous configuration of NFO and the evenly cross-distributed ultrathin nanosheet of LDH* on the framework of NFO were observed in the SEM images (Fig. 2d and Fig. S5a, ESI†). A single LDH* nanosheet was gently isolated from the substrate by ultrasound sonication and further characterized. The ultrathin nanosheet of LDH* possessed a size of about 360–380 nm, as revealed in the TEM images (Fig. S5b–d, ESI†). Moreover, a distinct hetero-interface between Ni(OH)2 and LDH* could be observed (Fig. 2e). The crystal fringes exhibit interplanar spacings of 0.48 nm and 0.24 nm, corresponding to the (006) and (104) facets of FeCo-LDH, respectively. The fringe with the spacing of 0.27 nm can be attributed to the (100) facets of Ni(OH)2, confirming the successful synthesis of the Ni(OH)2 and FeCo-LDH heterostructure. The TEM images of LDH/NF (Fig. 2f and Fig. S6, ESI†) show a blurry lattice fringe, indicating the poor crystallinity of LDH/NF, in agreement with the broadened diffraction peaks in its XRD spectra. The interfacial stress introduced by lattice mismatch could be responsible for the atomic arrangement and low crystallinity of LDH/NF.58,59 This difference in crystallinity proved that the introduction of Ni(OH)2 as an electron bridge regulated the ordered epitaxial growth of FeCo-LDH. Besides, EDS-mapping analysis showed the uniformly distributed Fe, Co and O both on the surface of LDH* (Fig. 2g) and LDH (Fig. S7c–f, ESI†) on NFO and NF, respectively.
XPS was performed to elucidate the regulation of the Ni(OH)2 electron bridge on the electron structure. All the binding energies in the XPS spectra were calibrated using the peak of the C–C component at 284.8 eV. As depicted in Fig. S8a and b (ESI†), Fe, Co, Ni and O can be observed in the XPS survey spectra of LDH*/NFO and LDH/NF. In the case of LDH/NF, the Fe 2p spectra could be deconvoluted into two sets of spin–orbit doublets of 2p3/2 (711.6 eV) and 2p1/2 (724.1 eV) and two satellite peaks (716.7 eV and 734.8 eV) (Fig. 3a), which are assigned to Fe3+ in FeCo-LDH.32,60 The peaks for Co species related to Co2+ could be also fitted with binding energies of 780.5 eV (2p3/2) and 796.4 eV (2p1/2) in the Co 2p spectra (Fig. 3b).61–63 The peaks for Fe 2p and Co 2p shifted to a higher binding energy in the XPS spectra of LDH*/NFO compared to LDH/NF, providing the successful introduction of the Ni(OH)2 electron bridge and its role in facilitating efficient electron transfer.16 To analyze the origin of the change in the oxidation states of Fe and Co in LDH*/NFO, the XPS spectra of NF, NFO and NF after hydrothermal treatment (without the growth of FeCo-LDH, denoted as NFh) were also collected and compared. As shown in Fig. S8d (ESI†), the signal of Ni0 is detected in the Ni 2p fine spectra of the pure NF, which was not observed in the spectrum NFO. The disappearance of Ni0 peaks suggests the formation of a compact Ni(OH)2 covering layer on the surface of NF. The position of the Ni 2p1/2 and Ni 2p3/2 peaks for LDH*/NFO shifted negatively compared to that of NFO, NFh or LDH/NF (Fig. S8e and f, ESI†).34 Furthermore, the valence band spectra (VBS) of LDH*/NFO were measured by XPS. The d-band center of VBS was calculated to be 5.6838 eV and 5.8044 eV for LDH and LDH*/NFO, respectively (Fig. S9, ESI†). Careful inspection of the VBS revealed a clear increase in the density of states near the Fermi level of LDH/NFO, corresponding with the proven DFT result of the higher electron capacity around e-DFE. According to these results, it can be concluded that a strong electronic interaction exists between the Ni(OH)2 electron bridge and the epitaxial layer of LDH*. DFT analyses were conducted to understand the electronic properties of the electron bridge. The charge density differential diagram showed the electron transfer from the FeCo-LDH layer to the electron bridge (Fig. S10, ESI†). Also, Bader charge analysis was performed to gain deep insight into the charge transfer introduced by the electron bridge (Table S2, ESI†). In the case of the Fe sites, the average number of transferred electrons to neighboring O is 1.38, 1.59 and 1.70 for LDH, LDH/NF and LDH*/NF, while 1.31, 1.27 and 1.44 electrons are subtracted from Co for LDH, LDH/NF and LDH*/NF, respectively. LDH*/NF shows the highest electron deficiency for both Fe and Co. Due to the high intrinsic OER activity of the high-oxidation-state metal site, LDH* with electron-deficient Fe and Co sites is expected to show a promising OER performance.64–66 This result further confirms that the Ni(OH)2 electron bridge contributes to the superior OER performance by modulating the electron structure of FeCo-LDH. The O 1s spectra shown in Fig. S6c (ESI†) could be deconvoluted into three peaks at 530.5 eV, 531.1 eV and 531.8 eV, which are ascribed to the oxygen species in the interlayer anion CO32−, M–O and –OH, respectively.67,68 Compared to LDH/NF, the CO32− and M–O contents in LDH*/NFO are higher than that in LDH/NF, indicating an increase in the interlayer anion concentration. The enlarged interlayer spacing, suggested by the PXRD results, is responsible for the increase in CO32− contents. As shown in Fig. 3c, Raman peaks centered at 449.2 cm−1 and 529.1 cm−1, which are the characteristic peaks for the stretching vibration of M–O (M denotes Fe and Co, respectively), were observed for LDH*/NFO.16,64,69 Meanwhile, these characteristic peaks of M–O were found at higher wavenumbers for LDH/NF (456.7 cm−1 and 531.6 cm−1) and unsupported LDH (458.3 cm−1 and 530.6 cm−1), respectively. The Raman shift for the M–O bands toward a lower wavenumber reflects a longer M–O bond,64,65 which agrees with the DFT calculations (the average length of Fe–O bond for LDH*/NFO, LDH/NF and FeCo-LDH is 2.18, 2.11 and 2.08 Å, respectively). The results confirm the influence of the electron bridge on the LDH*/NF lattice structure, which may contribute to optimizing the intermediate conversion process.
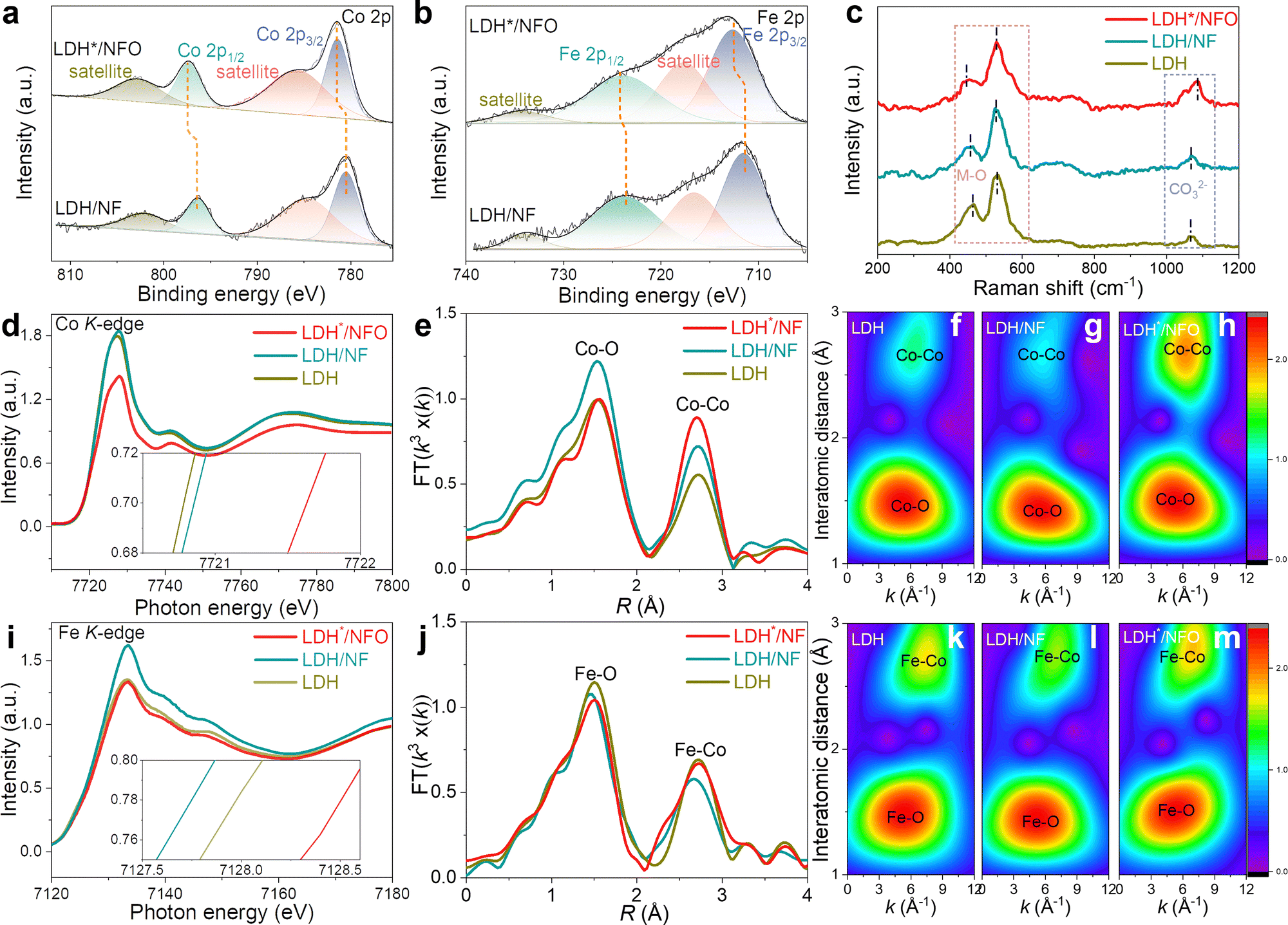 | ||
| Fig. 3 XPS and XAS characterizations of LDH*/NFO. High-resolution XPS spectra of (a) Fe 2p and (b) Co 2p of LDH/NF and LDH*/NFO. (c) Raman spectra of LDH, LDH/NF and LDH*/NFO. (d) Co K-edge XANES curves, (e) FT-EXAFS and (f)–(h) Wavelet transform EXAFS (WT-EXAFS) of LDH, LDH/NF and LDH*/NFO, respectively. (i) Fe K-edge XANES, (j) FT-EXAFS and (k)–(m) WT-EXAFS of LDH, LDH/NF and LDH*/NFO, respectively. | ||
Subsequently, we conducted a further investigation into the local coordination environment and electronic structure of the LDH*/NFO heterointerfaces using X-ray absorption spectroscopy (XAS). Fig. 3d displays the Co K-edge XANES of LDH*/NFO in comparison with LDH and LDH/NF. An energy shift was observed in the edge absorption energy of the XANES spectrum. Further amplification (the inset of Fig. 3d) revealed that the K-edge of LDH*/NFO shifted to a higher energy than that of LDH and LDH/NF, implying the high oxidation state of Co in LDH*/NFO.70 A similar result was observed in Fe K-edge XANES (Fig. 3i), corresponding with the XPS results. These results further verify the significant modulation of the Ni(OH)2 electron bridge on the electron structure LDH*/NFO, leading to the formation of an electron-deficient metal site. The unique electronic structure strengthens the covalency of Co–O and Fe–O, which is beneficial for suppressing the dissolution of Co and Fe during the OER process and enhancing the durability of the catalyst.71 The local coordination of Co and Fe was further characterized by the corresponding Fourier transformations of EXAFS (FT-EXAFS). As shown in Fig. 3e, the peaks at 1.56 Å and 2.69 Å correspond to the Co–O bond and the Co–Co distances between two octahedral units, respectively. Similarly, the peaks at 1.50 Å and 2.71 Å in Fig. 3j correspond to the Fe–O bond and Fe–Co distances, respectively. Notably, the signals of the Co–O and Fe–O peaks in LDH*/NFO exhibit a weak positive shift compared to that in LDH/NFO, which is consistent with the valence change in XANES,72,73 providing additional confirmation of the regulatory effect of the interaction between the Ni(OH)2 electron bridge and FeCo-LDH epitaxial layer on the electronic structure. The corresponding wavelet transform EXAFS (WT-EXAFS) spectra of Co (Fig. 3f–h) and Fe (Fig. 3k–m) were also calculated. A positive shift in the peaks corresponding to Co–O and Fe–O was observed in WT-EXAFS as well. More importantly, the peak intensity of the Co–Co bond and Fe–Co in LDH*/NFO was significantly higher than that of LDH/NF, demonstrating the ordered structure of LDH*/NFO.73 This finding is consistent with the XRD result, further confirming the lattice matching for high-quality epitaxial growth. Thus, the characterization results suggest that LDH*/NFO guided by DFT calculations can be synthesized. The impact of the Ni(OH)2 electron bridge on the OER performance, through the regulation of the large electron capacity around e-DFE and ordered lattice structure, was further confirmed by the electrochemical results.
2.3 Electrocatalytic performance of LDH*/NFO in OER
The OER performance of the catalysts was evaluated in 1 M KOH electrolyte using the standard three-electrode system. The polarization curves were recorded in a steady state at a low scan rate of 1 mV s−1 without iR compensation (Fig. 4a). Compared to NF, NFO showed a reduction in overpotential, indicating that the introduction of Ni(OH)2 optimized the OER performance of the carrier itself. In addition, NFO exhibited a slightly higher ECSA than that of NF, further confirming the formation of Ni(OH)2. However, it should be noted that the OER activity was significantly lower than that for LDH*/NFO, demonstrating the neglectable contribution of NFO in the catalytic process. Noteworthy, LDH*/NFO exhibited the optimal OER activity with an overpotential (η) of 201 mV and 246 mV to achieve a current density of 10 mA cm−2 and 100 mA cm−2, respectively. This performance of LDH*/NFO is superior to that of LDH/NF (256 mV and 346 mV) and RuO2 (301 mV and 448 mV). To confirm that the recorded current was generated from oxygen evolution entirely, LSV curves were also recorded from a higher potential to lower potential at the same scan rate to avoid the effect of metal oxidation (Fig. 4b). The minor difference between the LSV curves (a potential difference of 1.42 mV@100 mA cm−2) suggests negligible side reactions of LDH*/NFO during the OER process. The OER Tafel slopes were derived from the LSV curves. The Tafel slope of LDH*/NFO is 38.24 mV dec−1 (Fig. 4c), which is much lower than that of LDH/NF (60.26 mV dec−1), RuO2 (86.65 mV dec−1) and NF (142.13 mV dec−1), suggesting a lower energy barrier for the OER rate-limiting step on LDH*/NFO. The results indicate that the introduction of the Ni(OH)2 electron bridge, with an enlarged electron capacity around e-DFE, injects more power into the kinetic progression, resulting in excellent OER activity. The comparison of the OER performance in terms of overpotential and Tafel slope between LDH*/NFO and representative LDH catalysts showed that the LDH*/NFO outperformed most of its counterparts (Fig. 4d and Table S3, ESI†). The electrochemical impedance spectroscopy (EIS) curves of the catalysts were measured at 1.53 V (vs. RHE) in the range of 100 kHz to 0.01 Hz to achieve a mechanistic understanding of the OER kinetics. The Nyquist plots are shown in Fig. 4e. The Nyquist plots of all the samples demonstrate a typical semicircle structure, indicating that the limitation for the OER kinetics is the electron transfer rather than mass transfer in the self-supported electrode. Compared with the charge transfer resistance (Rct) of LDH/NF (0.50 Ω) and RuO2 (0.67 Ω), a small Rct of 0.21 Ω was identified for LDH*/NFO, verifying the fast electron transfer in LDH*/NFO. The smaller Rct is probably caused by several reasons, as follows: (1) the higher lattice matching between FeCo-LDH and Ni(OH)2 reduces the interface resistance, facilitating the electron transfer at the interface. (2) The introduction of Ni(OH)2 as an electron bridge modulates the electron structure, creating a larger electron capacity around e-DFE and suitable position of the d-band center. This modulation optimizes the adsorption and desorption capacities of the reaction intermediates on the metal sites, resulting in fast electron transfer on the metal sites.8,74 The electrochemical active surface areas (ECSA) of the electrodes were calculated based on the double-layer capacitance (Cdl) measured in the non-faradaic region (1.0–1.1 V vs. RHE). As shown in Fig. S12 (ESI†), LDH*/NFO showed a larger Cdl than that in LDH/NF, which could be a result of its enlarged interlayer space. The intrinsic activity of the electrodes, analyzed by normalizing the LSV curves to the corresponding ECSA value, was compared (Fig. S13a, ESI†), and the results also proved the excellent OER activity of LDH*/NFO. The turnover frequency (TOF) was calculated based on the LSV curves after ECSA-normalization to gain a quantitative view of the intrinsic activity. As shown in Fig. S13b (ESI†), LDH*/NFO demonstrated a higher TOF value of 0.5 s−1 at an overpotential of 270 mV and 2.65 s−1 at 300 mV, which is about 2.6-times and 2.2-times higher than that for LDH/NF (0.195 s−1 and 1.19 s−1) under identical conditions, respectively. Moreover, LDH* and LDH were separated from LDH*/NFO and LDH/NF, respectively, by ultrasonication and characterized to gain further insight into their intrinsic activity. The corresponding LSV curves and Nyquist plots of the samples with a loading of 0.2 mg cm−2 (Fig. S14 and S15, ESI†) showed that LDH* still exhibited the highest OER performance. In addition, the faradaic efficiency (FE) was tested by collecting the gas volume at the anode. The results (Fig. S16, ESI†) showed that the actual value of O2 volume agrees with the theoretical value and a half of the H2 volume, indicating the nearly 100% FE of LDH*/NFO for OER. Therefore, we concluded that the excellent intrinsic OER activity of LDH*/NFO originated from the unique lattice structure induced by the intermediate Ni(OH)2 layer.43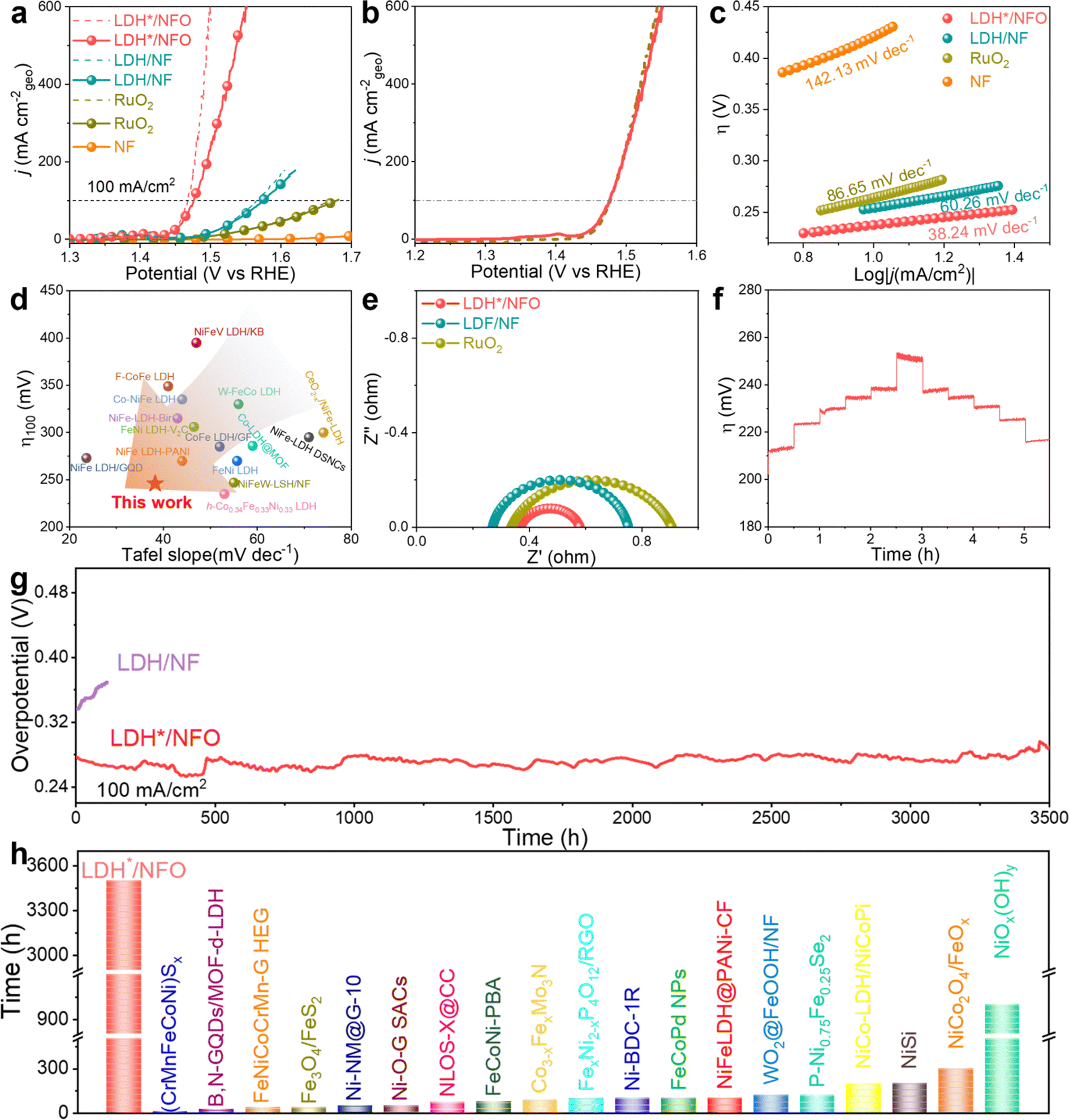 | ||
| Fig. 4 Electrochemical evaluation of LDH*/NF towards the OER. (a) and (b) OER LSV curves (dash lines represent uncompensated and solid lines represent 85% iR-corrected) and (c) corresponding Tafel plots of NF, LDH, LDH/NF, LDH*/NF and RuO2 in 1 M KOH electrolyte. (d) Comparison of overpotential at 100 mA cm−2 (η100) and Tafel slope of LDH*/NFO with other representative LDH-based catalysts. (e) Nyquist plots at 1.53 V (vs. the RHE). (f) Step stability from 10 mA cm−2 to 100 mA cm−2 of LDH*/NFO. (g) Chronopotentiometric curve of LDH*/NFO at a high current density of 100 mA cm−2. (h) Comparison of the stability of LDH*/NFO with other representative OER catalysts at 100 mA cm−2. | ||
The stability of the electrodes was first examined via multi-step chronopotentiometric measurements from 10 to 100 mA cm−2. As shown in Fig. 4f, the potential of LDH*/NFO responded quickly as the current density changed and no obvious decay was observed. To evaluate the potential of LDH/NF for practical application, its durability was examined at a high current density of 100 mA cm−2. As shown in Fig. 4g, LDH*/NFO displayed outstanding stability with an ultraslow overpotential increase rate of 9.4 × 10−3 mV h−1 during 3500 h OER tests, proving its outstanding stability under conditions relevant to practical application. Meanwhile, there was no evident degradation in the LSV curves of LDH*/NFO after the stability test (Fig. S10c, ESI†). The ultraslow overpotential increase for LDH*/NFO may originate from the higher lattice matching between Ni(OH)2 and FeCo-LDH, which contributes to enhancing the thermal stability and restraining the metal cation leaching during the OER process. The Nyquist plot of LDH*/NFO after the stability test (A-LDH*/NFO) showed that the Rct of the electrode remained almost unchanged, further confirming the advantageous stability of LDH*/NFO (Fig. S13d, ESI†). In the case of LDH/NF, a drastic decay in potential was observed after the 110 h test at 100 mA cm−2 (Fig. 4g). The ICP results exhibited a lower metal leaching rate (7.03 × 10−4 μg h−1, 3.70 × 10−4 μg h−1 and 1.92 × 10−4 μg h−1 of Fe, Co and Ni) for LDH*/NFO than that in LDH/NF (2.01 × 10−2 μg h−1, 1.34 × 10−2 μg h−1 and 4.55 × 10−4 μg h−1 of Fe, Co and Ni) after the stability test, respectively (Fig. S17, ESI†). The in situ Raman spectra (Fig. S18, ESI†) showed that with the application of voltage, neither the peak position or relative peak intensity had a dramatic change, indicating the excellent stability of LDH*/NFO. This indicates the larger electron pocket for electron storage and electron enrichment, which retarded the LOM-induced cation leaching for charge equilibrium. The excellent stability is a combined result of the excellent lattice matching of LDH*/NFO and larger electron density around the Fermi level caused by the introduction of Ni(OH)2 as an electron bridge. The comparison of the stability at 100 mA cm−2 between LDH*/NFO and representative catalysts exhibited the excellent durability of LDH*/NFO (Fig. 4h and Table S4, ESI†).
2.4 Insights into the OER mechanism
SEM, XRD, XPS and Raman characterizations were conducted with the electrodes after the stability test to gain further insights into the structure stability of the samples. The SEM results (Fig. S19, ESI†) revealed that LDH*/NFO retained its nanosheet morphology after the stability test, and the corresponding EDS mapping also proved the uniform distribution of Fe, Co, Ni and O. The XRD pattern (Fig. S20, ESI†) of LDH*/NFO remained almost unchanged, proving the retention of the LDH structure. Besides, the XPS survey spectra for LDH*/NFO after the stability measurement (Fig. S21, ESI†) were similar to that of the fresh electrode, except for the presence of characteristic peaks of K species, which can be attributed to electrolyte adsorption during the OER process. Besides, no significant differences were observed between the Raman spectra of LDH*/NF and A-LDH*/NFO (Fig. S22, ESI†), indicating the excellent stability of LDH*/NFO.To understand the origin of the enhanced OER performance on LDH*/NFO and the effect of the electron bridge on the OER activity, DFT calculations were performed to confirm the OER mechanism. Two different OER mechanisms, namely the adsorbate evolution mechanism (AEM) and LOM, were considered. The free energy barrier of AEM is 0.69 eV at 1.23 V for LDH*/NFO (Fig. S23, ESI†), which is significantly higher than that of LOM (0.65 eV), demonstrating that LDH*/NFO follows the LOM mechanism. The four-electron associative process of LOM over LDH, LDH/NF and LDH*/NFO is presented in Fig. 5a–c, respectively. As depicted in the free energy landscape (Fig. 5d), the free energy barrier of LDH*/NFO (0.65 eV) is much lower than that of LDH (1.9 eV) and LDH/NF (1.12 eV), demonstrating the higher activity of LDH*/NFO. Besides, different rate-determining steps (RDS) were found between LDH*/NFO, LDH/NF and LDH. Briefly, the RDS is the O2 desorption step for pristine LDH and LDH/NF, coinciding with previous reports that O2 desorption is the RDS for LDH.75 The RDS of LDH*/NFO transformed into the formation of an oxygen–oxygen bond. According to the d-band center, the change in RDS may be caused by the strong interaction between Ni(OH)2 and FeCo-LDH, leading to a lower energy level of εd, and consequently reducing the adsorption energy of the intermediates on the metal sites. The results indicate that the introduction of Ni(OH)2 as an electron bridge can modulate the reaction mechanism by tailoring the electron structure. Subsequently, we collected information at the atomic and electronic levels to further understand the structure of LDH*/NFO. The average bond length of Fe–O in the step of O2 desorption was calculated first to determine the reason for the change in the limiting step. Evidently, LDH*/NFO exhibits the largest average bond length of 2.15 Å (Fig. S24, ESI†), which is 2.09 Å and 2.04 Å for LDH/NF and LDH, respectively. Generally, an increase in M–O bond length suggests a weakened M–O bond, and thus facilitates O2 desorption.65
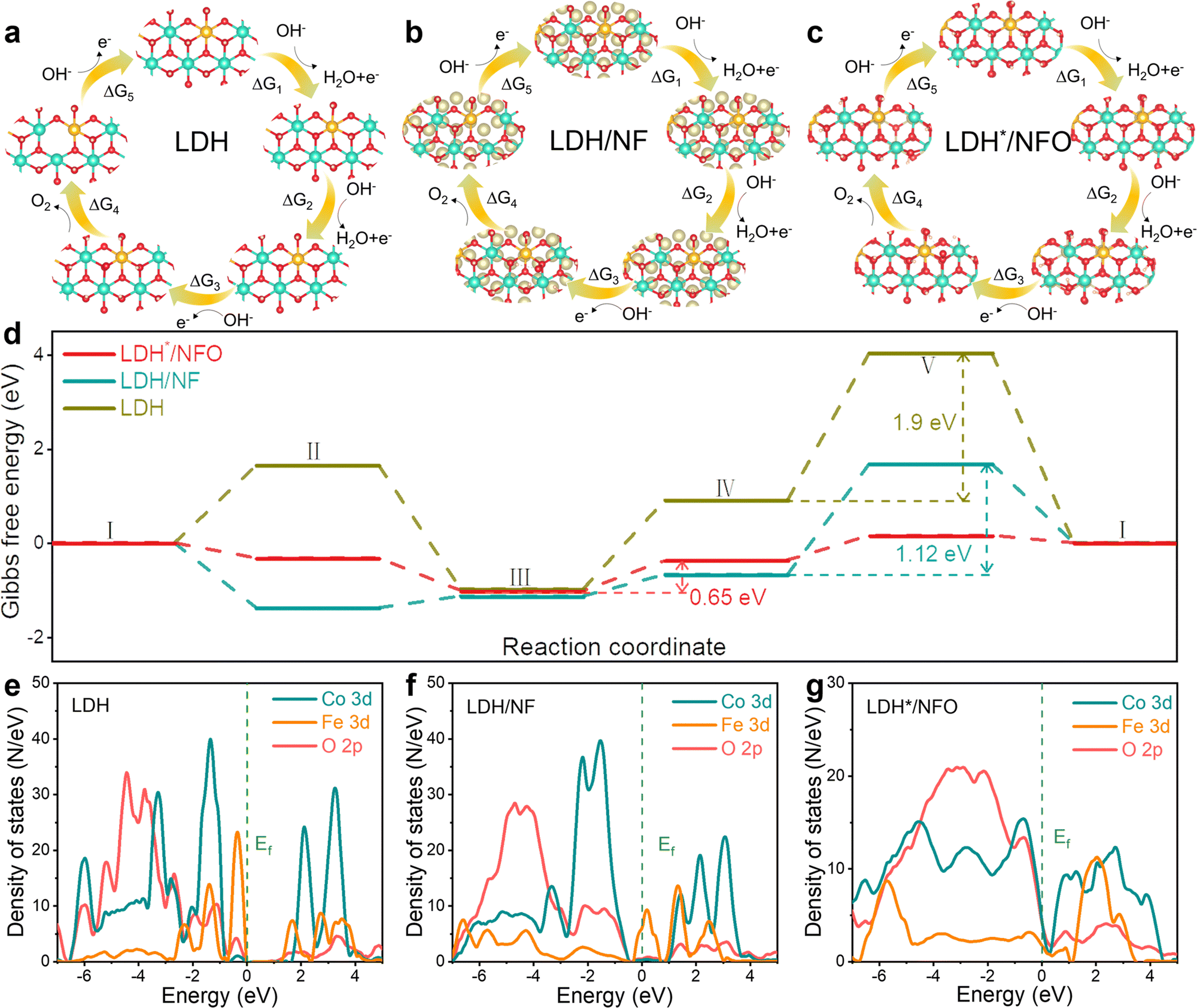 | ||
| Fig. 5 Identification of the catalytic mechanism using DFT calculations. (a)–(c) Four-electron associative process of LOM and (d) Gibbs free energy diagrams of LDH, LDH/NF and LDH*/NFO. (e)–(g) PDOS of the d orbital of Fe and Co and p orbital of O for LDH, LDH/NF and LDH*/NFO. | ||
According to the d-band center theory, the d-band center of the catalyst determines its intrinsic activity by modulating the adsorption and desorption behavior of the intermediates.76 Therefore, we estimated the hybridization between the O 2p orbital and Fe(Co) d orbital by the density of states (DOS), which was calculated to elucidate the underlying reason for the improvement in activity. The corresponding projected density of state (PDOS) of LDH, LDH/NF and LDH*/NFO is shown in Fig. 5e–g, respectively. The 3d states of the Fe and Co sites in LDH*/NFO show the strongest hybridization with the 2p states of O, which leads to enhanced OER activity.77 The Fe(Co)–O d–p hybridization for LDH/NF is slightly weaker than that in LDH, which can be caused by the electrostatic interaction of Ni. Meanwhile, the antibonding orbitals at high energy formed from the splitting of the Fe and Co d-band for LDH and LDH/NF shifted away from Ef compared with that in LDH*/NFO. Specifically, the electrons in the antibonding orbitals tend to transfer to the bonding orbitals, which leads to an enhancement in the adsorption of the OER intermediates on the surface of LDH and LDH/NF, resulting in prohibited O2 desorption.78 Hence, the strongest hybridization between the O 2p states and Fe(Co) 3d states and optimized adsorption/desorption caused by the electronic state modulation between the Ni(OH)2 electron bridge and LDH* contribute to improving the OER activity.
2.5 Application for hydrogen production from water electrolysis
Overall water electrolysis using the LDH*/NFO electrode was also demonstrated. Phosphorus-doping was conducted to endow LDH*/NFO with good HER activity (Fig. S25a–c, ESI†). Given its excellent OER performance and HER activity, a two-electrode system employing LDH*/NFO and P-doped LDH*/NFO was constructed. The LSV curves (Fig. S25d, ESI†) show that a low voltage of 1.49 V is required for the LDH*/NFO-based electrolyzer to afford a current density of 10 mA cm−2, which is 20 mV lower than that of RuO2//PtC (1.51 V). Furthermore, the current density of the LDH*/NFO-based electrolyzer showed a slight attenuation in current density, which decayed to 9.1 mA cm−2 after the 140 h chronoamperometry test at 1.49 V (Fig. S25e, ESI†), while the RuO2//PtC electrolyzer exhibited a prominent current decay from 10 mA cm−2 to 5.8 mA cm−2 after 140 h test at 1.51 V.3. Conclusions
In summary, the LDH*/NFO catalyst, with an Ni(OH)2 layer as an electron bridge between the FeCo LDH and Ni support, was delicately designed. The resulting LDH*/NFO catalyst exhibited a higher electron capacity around e-DFE (increased from 0.93 per cell of LDH/NF to 1.51 per cell of LDH*/NFO) after the introduction of the electron bridge. According to the DFT calculations, the free energy barriers decreased from 1.12 eV for LDH/NF to 0.65 eV for LDH*/NFO, demonstrating that the electron bridge accelerates the intermediate conversion kinetic process. With the help of meticulous XAS and ICP characterization, we found that the electron bridge strengthened the metal–oxygen covalency and inhibited the cation leaching. Benefiting from this synergistic effect, LDH*/NFO exhibited remarkable OER activity and exceptional durability, sustaining 3500 h at a high current density of 100 mA cm−2. Moreover, the assembled electrolyzer, featuring LDH*/NFO as the anode and PLDH*/NFO as the cathode, demonstrated superior water electrolysis capabilities, operating a low voltage of 1.49 V at 10 mA cm−2. This marks the inaugural demonstration of synergistically modulating OER activity and stability through the electron bridging strategy. This work holds significant promise for the development of efficient electrocatalysts, not only for water electrolysis but also for various other energy storage and conversion devices.Author contributions
J. W., Z. K., and J. L. conceived and directed the project. J. W. carried out the syntheses and characterizations. J. W. assisted in drawing schematic diagram. J. L. helped perform the XANES tests. Z. Z. helped perform the DFT. H. T., Z. K., and J. L. helped analyse the data. J. W., Z. Z., M. G, L. X., H. T., J. L., Z. K. and J. L. revised the manuscript and suggested for details in tests. All authors participated in the discussion of the results, commented on the implications, and fully approved the content of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 52271226, 51972254), National Key R&D Program of China (2022YFB4003500), Guangdong Basic and Applied Basic Research Foundation (2020B1515120042), and Fundamental Research Funds for the Central Universities (2022IVA197, 2022IV002h, 2021III029GX) for financial support. Prof. Z. Kou acknowledges the financial support of the Fundamental Research Funds for the Central Universities (Grant No. 2023IVA011) and National Natural Science Foundation of China (Grant No. 52202291) for the support. This work gratefully acknowledges support from the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy, Vehicle Technologies Office. This research used resources of the National Synchrotron Light Source II (beamline 7-BM), a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- G. Cai, M. Ding, Q. Wu and H. L. Jiang, Natl. Sci. Rev., 2020, 7, 37–45 CrossRef CAS PubMed.
- S. Wan, J. Xu, S. Cao and J. Yu, Interdisciplinary Mater., 2022, 1, 294–308 CrossRef CAS.
- B. Jiang, J. Zhu, Z. Xia, J. Lyu, X. Li, L. Zheng, C. Chen, S. Chaemchuen, T. Bu, F. Verpoort, S. Mu, J. Wu, J. Wang and Z. Kou, Adv. Mater., 2024, 36, 2310699 CrossRef CAS PubMed.
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583–3602 RSC.
- B. Singh, A. Singh, A. Yadav and A. Indra, Coord. Chem. Rev., 2021, 447, 214144 CrossRef CAS.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- Z. Shi, Y. Wang, J. Li, X. Wang, Y. Wang, Y. Li, W. Xu, Z. Jiang, C. Liu, W. Xing and J. Ge, Joule, 2021, 5, 2164–2176 CrossRef CAS.
- Y. Hao, Y. Li, J. Wu, L. Meng, J. Wang, C. Jia, T. Liu, X. Yang, Z. P. Liu and M. Gong, J. Am. Chem. Soc., 2021, 143, 1493–1502 CrossRef CAS PubMed.
- R. Paul, Q. Zhai, A.-K. Roy and L.-M. Dai, Interdisciplinary Mater., 2022, 1, 28–50 CrossRef CAS.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W.-F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- B. Wei, Z. Fu, D. Legut, T. C. Germann, S. Du, H. Zhang, J. S. Francisco and R. Zhang, Adv. Mater., 2021, 33, e2102595 CrossRef PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J.-M.-V. Nsanzimana, C. Wang, Z.-J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- J. Gao, H. Tao and B. Liu, Adv. Mater., 2021, 33, e2003786 CrossRef PubMed.
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2022, 34, e2108133 CrossRef PubMed.
- H. Sun, L. Chen, Y. Lian, W. Yang, L. Lin, Y. Chen, J. Xu, D. Wang, X. Yang, M.-H. Rummerli, J. Guo, J. Zhong, Z. Deng, Y. Jiao, Y. Peng and S.-Z. Qiao, Adv. Mater., 2020, 32, e2006784 CrossRef PubMed.
- L. Chong, G.-P. Gao, J.-G. Wen, H.-X. Li, H.-P. Xu, Z. Green, J.-D. Sugar, A.-J. Kropf, W.-Q. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- Y. Zhao, X. Zhang, X. Jia, G. I. N. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L. Wu, C. Tung, D. Hare and T. Zhang, Adv. Energy Mater., 2018, 8, 1703585 CrossRef.
- X. Jia, X. Zhang, J. Zhao, Y. Zhao, Y. Zhao, G. I. N. Waterhouse, R. Shi, L. Wu, C. Tung and T. Zhang, J. Energy Chem., 2019, 34, 57–63 CrossRef.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. Sun-Waterhouse, L. Shang, T. Zhang and G.-I.-N. Waterhouse, Angew. Chem., Int. Ed., 2021, 60, 24612–24619 CrossRef CAS PubMed.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- D.-P. Sahoo, K.-K. Das, S. Mansingh, S. Sultana and K. Parida, Coord. Chem. Rev., 2022, 469, 214666 CrossRef CAS.
- X. Bai, Z. Duan, B. Nan, L. Wang, T. Tang and J. Guan, Chin. J. Catal., 2022, 43, 2240–2248 CrossRef CAS.
- L. Zhang, J. Liang, L. Yue, K. Dong, J. Li, D. Zhao, Z. Li, S. Sun, Y. Luo, Q. Liu, G. Cui, A. Alshehri, X. Guo and X. Sun, Nano Res. Energy, 2022, 1, e9120028 CrossRef.
- S. A. Patil, A. C. Khot, V. D. Chavan, I. Rabani, D. Kim, J. Jung, H. Im and N. K. Shrestha, Chem. Eng. J., 2024, 480, 146545 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, J. Han, S. Cho, A. I. Inamdar, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 8989 RSC.
- X. Wang, H. Zhong, S. Xi, W. S. V. Lee and J. Xue, Adv. Mater., 2022, 34, e2107956 CrossRef PubMed.
- A. I. Inamdar, H. S. Chavan, J. H. Seok, C. H. Lee, G. Shin, S. Park, S. Yeon, S. Cho, Y. Park, N. K. Shrestha, S. U. Lee, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 20497 RSC.
- Q. Ma and S. Mu, Interdisciplinary Mater., 2023, 2, 53–90 CrossRef.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C.-J. Sun, L. Li, D. Sokaras, C. Dong, S.-Z. Qiao, X.-W. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- R. Chen, S.-F. Hung, D. Zhou, J. Gao, C. Yang, H. Tao, H.-B. Yang, L. Zhang, L. Zhang, Q. Xiong, H.-M. Chen and B. Liu, Adv. Mater., 2019, 31, e1903909 CrossRef PubMed.
- L. Zhou, C. Zhang, Y. Zhang, Z. Li and M. Shao, Adv. Funct. Mater., 2021, 31, 2009743 CrossRef CAS.
- W. Huang, J. Li, X. Liao, R. Lu, C. Ling, X. Liu, J. Meng, L. Qu, M. Lin, X. Hong, X. Zhou, S. Liu, Y. Zhao, L. Zhou and L. Mai, Adv. Mater., 2022, 34, e2200270 CrossRef PubMed.
- Y. Lin, H. Wang, C.-K. Peng, L. Bu, C.-L. Chiang, K. Tian, Y. Zhao, J. Zhao, Y. G. Lin, J.-M. Lee and L. Gao, Small, 2020, 16, e2002426 CrossRef PubMed.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, J. H. Seok, A. S. Salunke, S. Cho, A. I. Inamdar, Y. Park, S. U. Lee, H. Kim and H. Im, Mater. Today Phys., 2023, 38, 101252 CrossRef CAS.
- L. Zhang, J. Zhu, X. Li, S. Mu, F. Verpoort, J. Xue, Z. Kou and J. Wang, Interdisciplinary Mater., 2022, 1, 51–87 CrossRef.
- J. Wei, J. Wang, W. Guo, H.-L. Tang and J.-S. Li, Chem. Eng. J., 2023, 460, 141783 CrossRef CAS.
- Y. Li, Y. Zhang, K. Qian and W. Huang, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- L. Zhang, F. Yao, J. Meng, W. Zhang, H. Wang, X. Liu, J. Meng and H. Zhang, J. Mater. Chem. A, 2019, 7, 18558–18567 RSC.
- Y. Cheng, L. Zhang, S. Wang, M. Wang, C. Deng, Y. Sun, C. Yan and T. Qian, ACS Nano, 2023, 17, 15504–15515 CrossRef CAS PubMed.
- Z. Luo, J. Wang, W. Zhou and J. Li, Molecules, 2023, 28, 2262 CrossRef CAS PubMed.
- J. Wang, J. Wei, C. An, H. Tang, Q. Deng and J. Li, Chem. Commun., 2022, 58, 10907–10924 RSC.
- J. Zhang, X. Fu, F. Xia, W. Zhang, D. Ma, Y. Zhou, H. Peng, J. Wu, X. Gong, D. Wang and Q. Yue, Small, 2022, 18, e2108031 CrossRef PubMed.
- T. Wang, H. Wu, C. Feng, L. Zhang and J. Zhang, J. Mater. Chem. A, 2020, 8, 18106–18116 RSC.
- Z. Chen, M. Ju, M. Sun, L. Jin, R. Cai, Z. Wang, L. Dong, L. Peng, X. Long, B. Huang and S. Yang, Angew. Chem., Int. Ed., 2021, 60, 9699–9705 CrossRef CAS PubMed.
- R. Rajendiran, N. Muthuchamy, K.-H. Park, O.-L. Li, H.-J. Kim and K. Prabakar, J. Colloid Interface Sci., 2020, 566, 224–233 CrossRef CAS PubMed.
- S. Mondal, S. Dutta, S. Mal, S. K. Pati and S. Bhattacharyya, Angew. Chem., Int. Ed., 2023, 62, e202301269 CrossRef CAS PubMed.
- X. Zhou, Y. Liang, H. Fu, R. Zhu, J. Wang, X. Cong, C. Tan, C. Zhang, Y. Zhang, Y. Wang, Q. Xu, P. Gao and H. Peng, Adv. Mater., 2022, 34, e2202754 CrossRef PubMed.
- L. Y. Zhang, T. Zeng, L. Zheng, Y. Wang, W. Yuan, M. Niu, C. X. Guo, D. Cao and C. M. Li, Adv. Powder Mater., 2023, 2, 100131 CrossRef.
- S. Li, H. Liang, C. Li and Y. Liu, J. Mater. Sci. Technol., 2022, 106, 19–27 CrossRef CAS.
- X. Wu, Y. Shao, H. Liu, Z. Feng, Y.-L. Wang, J.-T. Sun, C. Liu, J.-O. Wang, Z.-L. Liu, S.-Y. Zhu, Y.-Q. Wang, S.-X. Du, Y.-G. Shi, K. Ibrahim and H.-J. Gao, Adv. Mater., 2017, 29, e1605407 CrossRef PubMed.
- J. Li, K.-J. Yu, X.-D. Zhang, Y.-K. Li, L. Qiao, X.-L. Peng, X. Dong, Z.-W. Wang, J. Ma, W.-D. Xiao and Y.-G. Yao, J. Phys. Chem. C, 2022, 126, 5022–5027 CrossRef CAS.
- J.-A. Vargas, V. Petkov, E.-A. Nouh, R.-K. Ramamoorthy, L.-M. Lacroix, R. Poteau, G. Viau, P. Lecante and R. Arenal, ACS Nano, 2018, 12, 9521–9531 CrossRef CAS PubMed.
- L.-Z. Bu, N. Zhang, S.-J. Guo, X. Zhang, J. Li, J.-L. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X.-Q. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- F. Ishizaka, Y. Hiraya, K. Tomioka, J. Motohisa and T. Fukui, Nano Lett., 2017, 17, 1350–1355 CrossRef CAS PubMed.
- L. Cheng, K. Meng, Z. Qiao, Y. Zhai, R. Yu, L. Pan, B. Chen, M. Xiao and G. Chen, Adv. Mater., 2022, 34, 2106380 CrossRef CAS PubMed.
- W. Li, S. Zhang, W. Zheng, J. Ma, L. Li, Y. Zheng, D. Sun, Z. Wen, Z. Liu, Y. Wang, G. Zhang and G. Cui, Adv. Funct. Mater., 2023, 33, 2300791 CrossRef CAS.
- J. Chen, G. Qian, H. Zhang, S. Feng, Y. Mo, L. Luo and S. Yin, Adv. Funct. Mater., 2022, 32, 2107597 CrossRef CAS.
- H. Zhang, M. Zhao, H. Liu, S. Shi, Z. Wang, B. Zhang, L. Song, J. Shang, Y. Yang, C. Ma, L. Zheng, Y. Han and W. Huang, Nano Lett., 2021, 21, 2255–2264 CrossRef CAS PubMed.
- R. Li, B. Hu, T. Yu, H. Chen, Y. Wang and S. Song, Adv. Sci., 2020, 7, 1902830 CrossRef CAS PubMed.
- D. He, X. Song, W. Li, C. Tang, J. Liu, Z. Ke, C. Jiang and X. Xiao, Angew. Chem., Int. Ed., 2020, 59, 6929–6935 CrossRef CAS PubMed.
- Z. Sun, A. Curto, J. Rodriguez-Fernandez, Z. Wang, A. Parikh, J. Fester, M. Dong, A. Vojvodic and J. V. Lauritsen, ACS Nano, 2021, 15, 18226–18236 CrossRef CAS PubMed.
- J.-D. Michael, E.-L. Demeter, S.-M. Illes, Q. Fan, J.-R. Boes and J.-R. Kitchin, J. Phys. Chem. C, 2015, 119, 11475–11481 CrossRef CAS.
- A.-C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M.-T.-M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- C.-F. Li, J. W. Zhao, L. J. Xie, J. Q. Wu, Q. Ren, Y. Wang and G.-R. Li, Angew. Chem., Int. Ed., 2021, 60, 18129–18137 CrossRef CAS PubMed.
- J. He, X. Zhou, P. Xu and J. Sun, Nano Energy, 2021, 80, 105540 CrossRef CAS.
- C. Wu, H. Li, Z. Xia, X. Zhang, R. Deng, S. Wang and G. Sun, ACS Catal., 2020, 10, 11127–11135 CrossRef CAS.
- D. Zhong, T. Li, D. Wang, L. Li, J. Wang, G. Hao, G. Liu, Q. Zhao and J. Li, Nano Res., 2021, 15, 162–169 CrossRef.
- K. Lee, J. Shim, H.-Y. Jang, H.-S. Lee, H. Shin, B.-H. Lee, M.-S. Bootharaju, K.-S. Lee, J. Lee, S. Lee, Y.-H. Lee, C. W. Lee, Y. Jung, G. Deng, S. Yoo, S. Back, Y.-E. Sung and T. Hyeon, Chem, 2023, 9, 1–13 Search PubMed.
- T. Wang, Z. Li, H. Jang, M. G. Kim, Q. Qin and X. Liu, ACS Sustainable Chem. Eng., 2023, 11, 5155–5163 CrossRef CAS.
- Y.-S. Park, J. Yang, J. Lee, M.-J. Jang, J. Jeong, W.-S. Choi, Y. Kim, Y. Yin, M.-H. Seo, Z. Chen and S.-M. Choi, Appl. Catal., B, 2020, 278, 119276 CrossRef CAS.
- Z. Wu, Y. Wang, D. Liu, B. Zhou, P. Yang, R. Liu, W. Xiao, T. Ma, J. Wang and L. Wang, Adv. Funct. Mater., 2023, 33, 2307010 CrossRef CAS.
- J. Wang, Q. Qin, Z. Wang, X. Zhao, Y. Chen, L. Hou, S. Liu and X. Liu, Acta Phys. Chim. Sin., 2023, 0, 2304044 Search PubMed.
- L. Hu, R. Xiao, X. Wang, X. Wang, C. Wang, J. Wen, W. Gu and C. Zhu, Appl. Catal., B, 2021, 298, 120599 CrossRef CAS.
- J.-K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef CAS PubMed.
- L. Zhang, W. Cai, N. Bao and H. Yang, Adv. Mater., 2022, 34, e2110511 CrossRef PubMed.
- T. Jiang, L. Yu, Z. Zhao, W. Wu, Z. Wang and N. Cheng, Chem. Eng. J., 2022, 433, 133525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00037d |
| This journal is © The Royal Society of Chemistry 2024 |