
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA minireview on electrochemical CO2 conversion based on carbonate/bicarbonate media
Tiehuai
Li
a and
Minhua
Shao
 *abc
*abc
aDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: kemshao@ust.hk
bEnergy Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
cChinese National Engineering Research Center for Control & Treatment of Heavy Metal Pollution, The Hong Kong University of Science and Technology, Kowloon, Hong Kong, China
First published on 18th January 2024
Abstract
Direct electrochemical CO2 conversion in carbonate/bicarbonate based CO2 capture media has emerged as a promising technology for integrating carbon capture and CO2 electroreduction processes in recent years, garnering significant attention from researchers owing to its high energy efficiency and carbon efficiency. For a holistic understanding of the development status of this field, this minireview summarizes a series of studies on the mechanism of carbonate/bicarbonate electrolyzers. Detailed mechanisms of the electrochemical conversion of carbonate/bicarbonate, the evolution of electrolyzers, and factors influencing the performance of electrolyzers are introduced. A summary of carbonate/bicarbonate electrolyzers' performance is also provided. Representative systems and materials for regulating the selectivity towards various products (e.g., CO, formate, methane, ethylene, and ethanol) and the cell voltage are highlighted. Furthermore, the challenges and future opportunities in this research area are also discussed.
 Minhua Shao | Minhua Shao earned his BS (1999) and MS (2002) degrees in Chemistry from Xiamen University, and a PhD degree in Materials Science and Engineering from the State University of New York at Stony Brook (2006). He is now the head and chair professor of the Department of Chemical and Biological Engineering and the director of the Energy Institute of the Hong Kong University of Science and Technology. He is a Cheong Ying Chan Professor of Energy Engineering and Environment. His research mainly focuses on fuel cells, advanced batteries, electrocatalysis, advanced materials, and modeling. |
Broader contextThe CO2 electrochemical reduction (CO2RR) is a promising technology for controlling the atmospheric CO2 concentration and producing renewable fuels. In recent years, significant progress has been made in this field with impressive achievements in product selectivity and current density. However, high energy consumption and low CO2 utilization remain major challenges that hinder the industrial application of this technology. The traditional CO2RR technology typically uses high-pressure and high-purity CO2 gas, which undoubtedly increases energy consumption during the recovery and utilization of waste CO2. Additionally, gas-phase products often contain a large amount of unreacted CO2, which increases energy consumption during product purification and reduces the overall carbon efficiency. Carbonate/bicarbonate electrochemical reduction technologies, which integrate the processes of carbon capture and electrochemical utilization, avoid the aforementioned issues by directly using carbon capture media as the carbon source. This provides a feasible solution for the industrial application of electrochemical utilization of CO2. In this minireview, we summarized recent advances in carbonate/bicarbonate electrochemical reduction, including studies of reaction mechanisms and the latest developments in catalysts and electrode configuration design. Scale-up of such technologies will drive significant growth in commercial electrochemical utilization of CO2. |
1. Introduction
Extensive use of fossil fuels has caused excessive emissions of carbon dioxide (CO2), and with the increase in atmospheric CO2 concentration comes changes in the global ecological environment, which in turn have negative impacts on human livelihoods and production.1,2 Compared with other CO2 conversion and utilization technologies, electrochemical utilization technology of CO2 has attracted much attention because of its mild reaction conditions and the high added value of the products.3 In recent years, significant progress has been achieved in the study of CO2 electroreduction under alkaline conditions. Notably, as one of the significant products yielded from the reduction of CO2, ethylene demonstrates a remarkable faradaic efficiency (FE) exceeding 80% in alkaline environments.4,5 Additionally, the partial current densities can exceed 1 A cm−2.6,7 Although the selectivity and yield are capable of satisfying the standards for industrial applications, the energy efficiency and carbon efficiency have become obstacles to the industrial application. In alkaline CO2 electrolyzers, CO2 will not only be reduced by the catalysts, but also form carbonate (CO32−) under strong alkaline conditions. The formation of CO32− results in a higher full-cell voltage, and the CO32− crossover through an anion exchange membrane (AEM) is the culprit of carbon loss.8 Moreover, Alerte et al.9 also pointed out that the removal and recovery of CO2 represents the most energy-intensive step in the downstream product separation process in AEM-based CO2 electroreduction systems. Thus, avoiding the losses caused by the formation of CO32− and further promoting the energy efficiency and carbon efficiency of CO2 reduction systems are crucial for the industrial application of CO2 electroreduction.Equipping an alkaline membrane CO2 electrolysis cell with a CO2 recovery component, or using acidic catholyte, can effectively alleviate the problems of CO32− formation.10–12 However, these technologies only focus on the process after the CO2 enters the electrolyzer. In order to obtain high-purity compressed CO2, CO2 in the atmosphere has to undergo a series of processes such as capture, precipitation, calcination, and compression, which significantly increases the cost of CO2 conversion.13 Thus, a new route integrating traditional CO2 capture and CO2 electroreduction technologies was proposed. As shown in Fig. 1, the device can directly convert the CO2 in CO2 capture media into products, which provides an encouraging strategy to promote the energy efficiency and carbon efficiency of the CO2 utilization process.
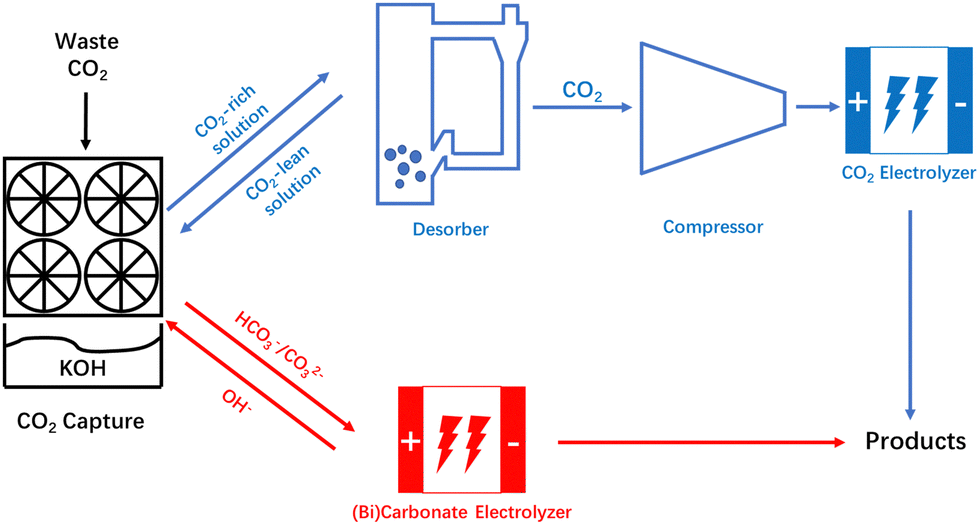 | ||
| Fig. 1 Schematic representation of the entire CO2 valorization process of CO2 electrolyzer and CO32−/HCO3− electrolyzer. | ||
Sullivan et al.14 summarized three different levels of combining CO2 capture and CO2 electroreduction, including independent, subsequent, and fully coupled CO2 conversion and capture processes. The fully coupled mode, which directly utilizes the CO2 capture media that absorbs CO2 as the carbon source of the electroreduction device, was described in detail. Four different media, including amines, carbonates/bicarbonates (CO32−/HCO3−), ionic liquids, and covalent organic frameworks (COFs), were introduced and compared with those in the literature. Zhang et al.15 summarized and evaluated amines and CO32−/HCO3− systems as media for the fully coupled mode mentioned above. They indicated that although both amines and CO32−/HCO3− suffer from low CO2 concentration at the electrode/electrolyte interface, using the amine media it is more difficult to achieve high performance due to the C–N bond cleavage challenge. Moreover, the amine medium also has to face the problem of metal corrosion. Welch et al.16 compared the CO32−/HCO3− direct conversion process with commercial syngas synthesis processes such as coal pyrolysis and steam methane reform in terms of total energy requirements and environmental sustainability. The conclusion demonstrated that the CO32−/HCO3− systems not only have the potential to achieve lower energy consumption, but also generate less pollution during the production process. In this review, we will focus on the CO32−/HCO3− systems as media for coupling CO2 capture and electrochemical conversion in terms of mechanism studies, catalyst selection, and product distributions.
2. Mechanism studies
As previously mentioned, using CO2 capture solvents directly as a feedstock for CO2 electrochemical utilization is an effective method for improving energy efficiency and carbon efficiency. One of the commonly employed CO2 capture techniques is atmospheric pressure CO2 capture using inorganic alkaline solutions. When using KOH as the capture medium, the main product obtained is K2CO3.13 On the other hand, when saturated solutions of K2CO317 or Na2CO318 are chosen as capture media, the predominant products obtained are KHCO3 and NaHCO3, respectively. It is notable that whether in carbonate or bicarbonate solutions, the dominant reactant for electroreduction is molecular CO2 from the CO2-bicarbonate equilibrium. In typical carbonate and bicarbonate electrolyzers, the in situ generated CO2 at the membrane surface from the acid/alkaline equilibriums with protons transported by the membrane viaeqn (1) and (2), respectively.19,20 It is worth noting that for electrolyzers equipped with a CEM (cation exchange membrane), protons are supplied by the anodic reaction. In contrast, for electrolyzers based on a BPM (bipolar membrane), protons come from the water dissociation within the interlayer of the membrane. Then the in situ generated CO2 is converted to common CO2 electroreduction products such as CO, formate, methane, ethylene, etc. at the surface of catalysts.19–22| CO32− + 2H+ ⇌ CO2 + H2O | (1) |
| HCO3− + H+ ⇌ CO2 + H2O | (2) |
In 2019, the membrane electrode assembly (MEA) system was first introduced into bicarbonate electrolyzer by Li et al.19 A typical bicarbonate electrolyzer consists of a catholyte chamber, an anolyte chamber, and a membrane (CEM or BPM). Subsequent research on carbonate/bicarbonate electrolyzers has generally adopted similar cell configurations. The bicarbonate electrolyzer was equipped with a BPM and used 3 M KHCO3 as the catholyte. In situ generated CO2 bubbles were observed near the membrane, which strongly supported the reaction pathway where the HCO3− was first dissociated to produce CO2. Due to the flow mode of the electrolyte and the MEA system, the cell achieved a CO faradaic efficiency of 81% at 25 mA cm−2 and 37% at 100 mA cm−2 with a total cell voltage of 3.4 V. The high CO selectivity, low cell voltage, and relatively high current density opened up a new perspective for researchers, who could now see the potential for industrial applications of HCO3− solution electrolysis. On the basis of the flow HCO3− electrolyzer, a series of studies on the mechanism about electrochemical conversion of CO32−/HCO3− solutions were conducted. Zhang et al.27 made use of operando Raman spectroscopy to monitor the surface pH of the catalyst in a flow HCO3− electrolyzer. The results demonstrate that as the current increases, the pH on the catalyst surface also rises. Furthermore, increasing the temperature not only accelerates the decomposition of HCO3−, but also increases the local pH of the catalyst surface, which has a significant inhibitory effect on the competing hydrogen evolution reaction (HER). Since acidic conditions are favorable for the in situ generation of CO2, whereas CO2 electrochemical reduction tends to prefer alkaline conditions, the pH issue plays an important role in carbonate/bicarbonate electrolyzers. Consequently, the control of pH distribution between the cathode and membrane, along with research on the underlying mechanisms, holds paramount importance in enhancing the performance of carbonate/bicarbonate electrolyzers. As the formation and transportation of CO2 in the HCO3− electrolyzer exhibit notable differences from conventional CO2-fed electrolyzer, Kas et al.28 developed a novel mass transport model to quantify the local concentration of species during HCO3− conversion process under the conditions of 3 M KHCO3 and porous electrode. The model indicates that the actual concentration of CO2 present in the catalyst layer is quite low, leading to a concentration overpotential. The concentration overpotential, the reduced use of protons for generating CO2, and the existence of intermediate H2CO3 may be responsible for the decline in product selectivity at high current density. The authors further suggest that a promising approach to enhancing the performance of the HCO3− electrolyzer is to improve the transport and decomposition conditions of HCO3− by modifying the structure of the electrolyzer. Lees et al.29 also built up an experimentally validated model for the HCO3− electrolyzer which focuses on the mass transfer processes and reaction chemicals in the catalyst layer and cation exchange layer. The result mutually confirmed by experiment and model demonstrates that the thickness of the catalyst layer and cation exchange layer affect the selectivity of the CO product and current density by influencing the concentration of CO32−, HCO3−, and CO2.
Besides the structure of the cell, the composition of the electrolyte also plays an essential role in the performance of a bicarbonate electrolyzer. The effect of cations on bicarbonate electrolysis was investigated by Fink et al.30 By altering the alkali metal ion species while maintaining a fixed HCO3− concentration, it was found that the selectivity for the CO product increases in correspondence with an increasing atomic number of the alkali metal element. In-depth research revealed that the type of anion did not affect the in situ generation of CO significantly. Instead, it primarily influenced the conversion of CO2 to CO. Pimlott et al.31 took into account the scenario of CO2 capture from flue gas and examined the effects of nitrogen oxides (NOx) and sulfur oxides (SOx) existing in the flue gas on the HCO3− electrolyzers. The results indicated that SOx did not have a significant impact on the generation of CO. However, for NOx impurities, the selectivity for CO was found to be significantly reduced as a result of the preferential reduction of nitrite (NO2−) and nitrate (NO3−) over CO2. These findings demonstrate the importance of considering the potential presence of impurities in the abandoned CO2 source when designing processes for CO2 capture and conversion.
3. Performance of bicarbonate electrolyzers
3.1 Selective production of CO
Lees et al.32 systematically investigated the effect of gas diffusion electrode (GDE) preparation methods on the performance of a silver composite electrode in producing CO in a 3 M KHCO3 electrolyte. The test results indicate that both hydrophobic treatment (using polytetrafluoroethylene (PTFE)) and the addition of a microporous layer (MPL) have a negative impact on the performance of CO generation. This is due to hindrance in the permeation of the electrolyte, which leads to a decrease in the amount of in situ generated CO2. Compared to the traditional spray-coating method, the physical vapor deposition (PVD) increased the silver surface coverage from 72% to 92%. Furthermore, when both methods were applied (hybrid), the silver surface coverage was able to achieve 95% (Fig. 2a). The results of this study demonstrate that a higher silver surface coverage results in improved CO selectivity and CO2 utilization. On the hybrid GDE with a high Ag coverage, the maximum FE of CO reached 82% at 100 mA cm−2, and a full cell voltage of 3.4 V. To address the issues of low stability and poor electrolyte permeability caused by the traditional carbon-based GDE, Zhang et al.33 fabricated a porous silver electrode by etching a silver foam in a dilute nitric acid solution. The FE of CO at a current density of 100 mA cm−2 was approximately 60%. This number could be improved to 95% at the same current density when the pressure was increased to 4 atmospheres. In terms of stability, the silver foam electrode was able to run stably with around 80% CO selectivity at a current density of 65 mA cm−2 for 80 hours, with timely replacements of the electrolyte. In contrast, the performance of the carbon-based GDE began to decline after 5 hours of operation under the same conditions. Larrea et al.34 discussed the effects of different ion exchange membranes on the performance of a bicarbonate electrolyzer and the selectivity towards CO with a CEM, AEM, and BPM were compared. The conclusion is that using a BPM can achieve the best CO selectivity. However, due to the large overpotential required for water splitting inside the membrane, the full cell voltage of this type of electrolyzer is significantly larger than those of the AEM and CEM equipped ones. To improve the CO selectivity and stability of HCO3− electrolyzers, the same group introduced a surfactant dodecyltrimethylammonium bromide (DTAB) into 2 M KHCO3 to prepare the catholyte.35 With the help of DTAB, the FE of CO increased from 65% to 85% at a current density of 50 mA cm−2. Further research suggests that DTAB can reduce the surface tension of the solution, making it easier for HCO3− to be transferred to the cathode. The majority of bicarbonate electrolyzers mentioned above have utilized BPM, which suffers from the large overpotential required for water splitting inside the BPM, severely diminishing overall energy efficiency. To enhance the energy efficiency of bicarbonate electrolyzers, Zhang et al.36 employed a CEM as a substitute for the BPM and utilized the hydrogen oxidation reaction (HOR) instead of the conventional oxygen evolution reaction (OER) at the anode (Fig. 2b). Compared to the traditional BPM-OER system, the advantage of the novel CEM-HOR system is that at the same high CO partial current density, the cell voltage of CEM-HOR is significantly lower, resulting in a higher energy efficiency. Taking the achievement of 80 mA cm−2 CO partial current density as an example, the cell voltage of the CEM-HOR system is about 2.2 V, and the overpotential is nearly 1.2 V lower than that of the BPM-OER system. Furthermore, increasing pressure has been proven to be an effective method of improving performance. When the pressure is increased from 1 atm to 3.5 atm, the partial current density of CO can increase to 220 mA cm−2 at a cell voltage of 2.2 V (Fig. 2c).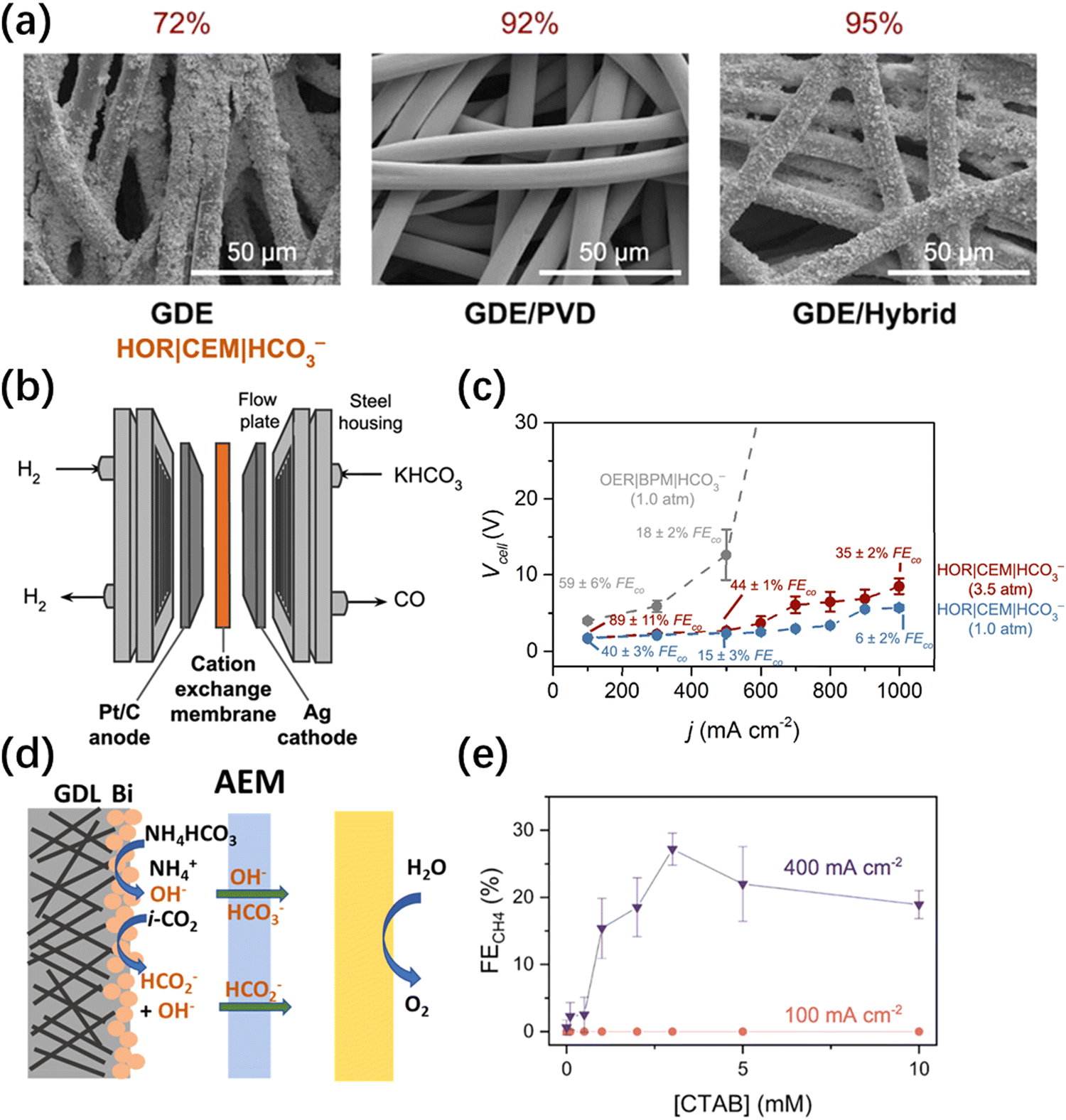 | ||
| Fig. 2 (a) SEM images of the silver coated GDEs fabricated using spray-coating (GDE), PVD (GDE/PVD), or both techniques (GDE/hybrid).32 Copyright © 2020, American Chemical Society. (b) Schematics of the electrolyzer using a reactive carbon solution feedstock, a CEM, and hydrogen oxidation at the anode.36 (c) Voltage and current characteristics of an electrolyzer that couples bicarbonate conversion with hydrogen oxidation under 1.0 and 3.5 atm of pressure.36 (d) Schematic illustration of the local environment at the cathode for the electrolyzers with a NH4HCO3 feed when using an AEM.38 Copyright © 2022, American Chemical Society. (e) FE for CH4 measured during electrolysis for bicarbonate solutions doped with CTAB at current densities ranging from 100 to 400 mA cm−2.22 Copyright © 2022, American Chemical Society. | ||
3.2 Other products
Compared to CO, higher-value products such as formate, as well as advanced products such as methane and ethylene, are more attractive to researchers. Similar to the development of traditional CO2 electroreduction, after achieving a certain level of CO selectivity and current density, some researchers have turned their attention to the production of other products from HCO3− electrolyzers. Li et al.21 fabricated the electrodeposited Bi electrode by immersing carbon paper in an aqueous Bi3+ solution and the electrodeposited was performed at a constant current of 72 mA for 5 minutes using a two-electrode setup. In 3 M KHCO3 electrolyte, the FE of formate achieved 64% at a current density of 100 mA cm−2 and a full cell voltage of −4 V. Gutiérrez-Sánchez et al.37 systematically investigated the performance of commercial Sn and SnO2 nanoparticles in generating formate in a bicarbonate electrolyzer. They loaded the nanoparticles onto porous carbon paper to fabricate working electrodes using the spray-coating method. The results demonstrated that SnO2 outperformed Sn nanoparticles in producing formate. The study also examined the effects of electrolyte flow rate, concentration of KHCO3, use of binders, and reaction temperature on formate selectivity. It was found that a high electrolyte flow rate and high KHCO3 concentration were favorable for formate generation, while the application of acidic or basic binders had a negative impact on catalyst performance. Interestingly, unlike the CO production in HCO3− electrolyzers, increasing the reaction temperature did not necessarily improve formate selectivity and could even have a negative effect. Under optimized conditions, the FE of formate achieved 58% at a current density of 100 mA cm−2 under ambient conditions. In order to address the issue of the difficulty in situ CO2 release from KHCO3, Liu et al.38 replaced it with NH4HCO3. The experiment showed that at 40 °C, the in situ decomposition of 2.5 M NH4HCO3 produced about 3.4 times more CO2 than KHCO3. The formate generation performance was first evaluated by electrodeposited Bi electrode and BPM. The results demonstrated that at a current density of 100 mA cm−2, regardless of the temperature, the selectivity of formate in NH4HCO3 was about 20% higher than that in KHCO3 at the same concentration. It should be noted that the decomposition reaction of NH4HCO3 involves the generation of hydroxide ions (OH−). Thus, removing OH− is beneficial for the in situ generation of CO2. An AEM, which can transfer OH− to the anolyte, can replace the BPM (Fig. 2d). After using AEM, the selectivity of formate reached 82% at a current density of −100 mA cm−2, and the cell voltage was merely 2.4 V. Analysis of the anolyte indicated that the crossover of HCO3− was only 4.7%. Lees et al.22 fabricated a copper foam electrode using acid etching and applied it in an experiment of 3 M KHCO3 electrochemical conversion. The predominant reduction product of HCO3− on the copper foam surface was CH4, with a FE of 27% at a current density of 400 mA cm−2. In this work, introducing CTAB into the catholyte was found to be an essential condition for CH4 generation via electroreduction. Almost no CH4 was detected in the products when CTAB was absent in the electrolyte, and the optimal concentration of the CTAB additive was determined to be 3 mM (Fig. 2e). Further investigations revealed that the hydrophobic alkyl group of CTAB increased the local CO2 concentration near the catalyst surface, thereby promoting CH4 production. Lee et al.39 first reported a case for direct electrochemical conversion of HCO3− solution into C2+ products by a Cu/Ag bilayer electrode. The C2+ products mainly include C2H4, acetate, ethanol, and 1-propanol and the maximum FE of C2+ reached 41.6% at a current density of 100 mA cm−2 and a cell voltage of 3.9 V.In summary, the bicarbonate electrolyzer has achieved high selectivities for primary products such as CO and formate. The reported maximum FE for CO and formate reached 95% and 82%, respectively. To improve the selectivity of the product, researchers employed a series of strategies, including heating, pressurization, improved electrode design, and the utilization of electrolyte additives such as CTAB and DTAB. Heating and pressurization can significantly improve product selectivity but require additional energy input. Additives are suitable for scenarios where the target product is in the gas phase. Improving electrode design is a versatile method that does not necessitate extra energy consumption. Therefore, we believe that the improvement of electrodes (including catalyst selection and electrode structure design) is an important direction for future research on improving the performance of bicarbonate electrolyzers. It is worth noting that carbonate/bicarbonate electrolyzers offer a distinct advantage over CO2 electrolyzers in terms of significantly lower CO2 content in the product. This not only enhances carbon efficiency but also reduces the energy required for downstream CO2 separation. However, it is important to highlight that this advantage primarily applies to gaseous products, whereas for liquid-phase products such as formate, separation processes like distillation are still necessary. Moreover, the description regarding product separation applies equally to carbonate electrolyzers. Thus, CO production from bicarbonate electrolyzers has more promising prospects. As for other advanced products such as methane and ethylene, looking for suitable catalysts to enhance product selectivity is currently an important task.
4. Performance of carbonate electrolyzers
Compared with HCO3− electrolyzers, CO32− electrolyzers suffer more from low product selectivity due to the significantly greater difficulty in converting CO32− to CO2 compared to HCO3−. Therefore, the carbonate electrolyzers are primarily focused on producing syngas at the current stage. Li et al.20 evaluated the electroreduction performance of Ag and Cu nanoparticles in a carbonate electrolyzer equipped with a BPM. The experimental results demonstrated that when Ag nanoparticles were used as the catalyst, the dominant product was CO, and the selectivity of CO decreased from 28% to 14% as the current density increased from 100 to 300 mA cm−2. Upon using a Cu film with loaded Cu nanoparticles as the working electrode, the primary products generated from carbonate reduction were ethylene and ethanol. The peak total selectivity for C2+ products reached 14.4%, occurring at a current density of 250 mA cm−2. A significant drawback of BPM is that, at high current densities, it necessitates a considerable potential, resulting in a lower energy efficiency for the systems.40 Thus, Xiao et al.41 replaced the BPM with CEM to decrease the overpotential of the membrane. However, the use of CEM resulted in an acidic reaction environment so that it did not improve CO selectivity. To provide an alkaline reaction environment, the researchers added a CO2 diffusion layer (CDL) by spray-coating TiO2 nanoparticles between the catalyst layer and the membrane. The results demonstrated that the CDL was most effective when its thickness was 25 μm, with the highest selectivity for CO reaching 46.4%. Lee et al.42 reported a novel interposer and a Cu/CoPc-CNTs electrocatalyst joint design for the carbonate electrolyzer. 47% selectivity of the C2+ product (the ethylene selectivity is 34%) was achieved at a current density of 300 mA cm−2 and a cell voltage of 4.1 V.Compared to bicarbonate electrolyzers, one major advantage of using carbonates as the feedstock is that carbonate solutions can be obtained from direct air capture, while bicarbonate solutions are mainly obtained from flue gas capture. As a result, carbonate electrolyzers have advantages in terms of application areas and feedstock costs. However, a significant challenge for carbonate electrolyzers is the low selectivity of their reduction products. As mentioned above, the highest CO and ethylene selectivities are around 46% and 34%, respectively. Therefore, we believe that developing suitable catalysts, improving the structure of the electrolyzers, and thereby increasing the faradaic efficiency of the product are the main research directions for carbonate electrolyzers in the future.
5. Conclusion and outlook
Compared to traditional alkaline CO2 electroreduction systems, the integration of carbon capture and electroreduction steps is a major advantage of CO32−/HCO3− electrolyzers, which simplifies the system. By directly employing carbon capture media as the carbon source in the electroreduction step, the demand for a series of steps involving the recovery and compression of CO2 from the capture solution is eliminated, leading to a significant improvement in overall energy efficiency. Furthermore, as the system does not need CO2 flow for reaction, it largely avoids the losses caused by excessive CO2 and saves the energy required to separate CO2 from the product gas. From the perspective of both energy and carbon efficiency, CO32−/HCO3− electrolyzers have great potential. As exhibited in Table 1, HCO3− electrolyzers for CO and formate production can achieve high selectivity and current density. However, studies on the generation of high-value-added products (e.g., C2H4) and the stability of the system are still limited.| Cathode | Catholyte | Membrane | Full cell voltage (V) | Current density (mA cm−2) | Main products | FE (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Tested at 40 °C. b Prepared by purging CO2 at 80 sccm into 85 mL of 2 M KOH for 40 minutes. | |||||||
| Ag composite electrode | 3 M KHCO3 | BPM | −3.4 | 100 | CO | 82 | 32 |
| Porous Ag electrode | 3 M KHCO3 | BPM | −3.7 | 100 | CO | 60 | 33 |
| Ag nanoparticles | 2 M KHCO3 | BPM | −3.8 | 200 | CO | 46 | 34 |
| Electrodeposited Ag electrode | 2 M KHCO3 with 0.02 M DTAB | BPM | ∼−4 | 100 | CO | 70 | 35 |
| Ag foam electrode | 3 M KHCO3 | CEM | −2.2 | 500 | CO | 15 | 36 |
| Electrodeposited Bi electrode | 3 M KHCO3 | BPM | −4 | 100 | HCOO− | 64 | 21 |
| SnO2 nanoparticles | 3 M KHCO3 | BPM | −4.1 | 100 | HCOO− | 58 | 37 |
| Electrodeposited Bi electrode | 2.5 M NH4HCO3 | AEM | −2.4 | 100 | HCOO− | 82a | 38 |
| Cu foam electrode | 3 M KHCO3 with 3 mM CTAB | BPM | −7.2 | 400 | CH4 | 27 | 22 |
| Cu/Ag bilayer electrode | 3 M KHCO3 | BPM | −3.9 | 100 | C2+ | 41.6 | 39 |
| Ag nanoparticles on Ag films | 1 M K2CO3 | BPM | −3 | 100 | CO | 28 | 20 |
| Cu nanoparticles on Cu films | 1 M K2CO3 | BPM | −4 | 250 | C2+ | 14.4 | 20 |
| Ag nanoparticles | CO2-capture solutionb | CEM | −3.8 | 200 | CO | 46.4 | 41 |
| Cu/CoPc-CNTs | 1.5 M K2CO3 | CEM | −4.1 | 300 | C2+ | 47 | 42 |
To push CO32−/HCO3− electrolyzers towards industrial applications, more research efforts should still be contributed. High-performance catalysts and electrodes need to be designed aiming at CO32−/HCO3− electrolyzers. At the current stage, the electrocatalysts for the reaction are still based on commercial nanoparticles or porous metal electrodes. A promising direction is to develop electrocatalysts with more affinity towards CO32−/HCO3−. For instance, theoretical research indicated that for Fe-Porphyrin, CO2 reduction is hindered kinetically, while the reduction of H2CO3 and HCO3− is both kinetically and thermodynamically favorable.44 Thus, Fe-Porphyrin is a promising electrocatalyst for CO32−/HCO3− electrolyzers. Moreover, the theoretical research on Fe-Porphyrin also indicated that the direct electrochemical conversion of CO32−/HCO3− is feasible. Actually, there have been reports indicating that both carbonate45 and bicarbonate46 could be directly electrochemically reduced into formate. Compared to CO32−/HCO3− electrolyzers that rely on the in situ generation of CO2, direct CO32−/HCO3− electrolysis technology makes it easier to achieve full utilization of carbon. While the selectivity of direct reduction is still relatively low at present, this technology exhibits significant potential. The design of working electrodes is also an important direction. Traditional carbon-based GDL suffers from poor durability in CO32−/HCO3− electrolyzers, while when using more stable porous metal electrodes it is difficult to ensure product selectivity. We believe that the carbon-based single-atom catalyst electrodes can be a feasible solution. As shown in Fig. 3a, this Ni single-atom/porous carbon fiber membrane (NiSA/PCFM) catalyst uses a single piece of carbon-based single-atom material directly as the working electrode.43 The high FE of CO can be ensured by the Ni single-atoms, and the Ni single atoms distributed throughout the entire carbon fiber membrane effectively prevent the traditional carbon-based GDL from experiencing the side reaction of the HER caused by immersion (Fig. 3b). Meanwhile, for the industrial application of CO32−/HCO3− electrolyzers, the choice of electrolyte for electroreduction cannot be ignored. Although high selectivity and current density have been achieved for CO and formate generation in HCO3− electrolyzers, the reactions have to be carried out at high HCO3− concentrations.32,38 In order to enhance the practical applicability of HCO3− electrolyzers, research on low concentration HCO3− solution electrochemical conversion needs to be conducted. On the other hand, introducing additives such as organic amines, carbonic anhydrase, or frothers into the capture media to overcome the poor CO2 capture kinetics of bicarbonate solutions is also a feasible solution.18,47,48 However, the presence of additives can sometimes impede the electroreduction of CO2.49,50 For instance, Fink et al.49 found that the carbonic anhydrase would decrease the rate of CO formation of the electrolyzer if the enzyme is not filtered out upstream. Further research revealed that the decrease in CO selectivity was attributed to the deactivation of the catalyst surface caused by carbonic anhydrase and a carbon microporous layer was found to effectively suppress this deactivation. Improving the efficiency of CO2 capture in carbonate capture solutions is of significant importance for systems utilizing bicarbonate solutions as the reaction medium. Therefore, exploring the performance of additives and their impact on the bicarbonate electrolysis process is a worthwhile research direction. In carbon capture systems that utilize bicarbonate as the product, the primary source of feedstock is CO2-rich flue gas. In contrast, carbonate solutions can be obtained through direct air capture, which not only diversifies the feedstock sources but also has significant importance in reducing atmospheric CO2 concentrations and mitigating the greenhouse effect.13,51 Thus, directly applying CO32− solution as the electrolyte is clearly more promising.16 Therefore, developing a CO32− electrolyzer with high product selectivity is also a meaningful direction. As previously mentioned, the in situ generation of CO2 heavily relies on the high H+ flux transported by the membrane (BPM or CEM). Hence, research on ion exchange membranes is also conducive to promoting the development of carbonate/bicarbonate electrolyzers. In the case of BPM, a significant challenge to address is its large membrane overpotential. Researchers found that adding a water dissociation catalyst into the BPM can effectively reduce the membrane overpotential.52 Introducing this design into BPM-based carbonate/bicarbonate electrolyzers to reduce cell voltage is a potential research direction. As for CEM-based carbonate/bicarbonate electrolyzers, improving the product selectivity is the top priority. Recently, it has been reported that introducing nitrogen sources into CO2 electroreduction can expand the product species, and the possible products include urea, acetamides, and amines.53 Considering that captured gases, such as flue gas, contain a certain amount of NOx. Using suitable catalysts to co-reduce CO32−/HCO3− and NO3− in the capture solution to produce nitrogen-containing products is also a promising research field.
 | ||
| Fig. 3 (a) Digital images of the NiSA/PCFM membrane. (b) Schematics of the NiSA/PCFM membrane directly used as the GDE.43 | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Research Grants Council (16310419 and 16308420), Innovation and Technology Commission via the Chinese National Engineering Research Center for Control & Treatment of Heavy Metal Pollution, and Hong Kong Branch of the National Precious Metals Material Engineering Research Center of the Hong Kong Special Administrative Region.References
- S. S. Myers, A. Zanobetti, I. Kloog, P. Huybers, A. D. Leakey, A. J. Bloom, E. Carlisle, L. H. Dietterich, G. Fitzgerald and T. Hasegawa, Nature, 2014, 510, 139–142 CrossRef CAS PubMed.
- P. N. Pearson and M. R. Palmer, Nature, 2000, 406, 695–699 CrossRef CAS PubMed.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen and D. Tan, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat Catal., 2020, 3, 478–487 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, Science, 2020, 367, 661–666 CrossRef PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O’Brien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer and H. L. MacLean, ACS Energy Lett., 2021, 6, 4405–4412 Search PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen, C.-T. Dinh, J. E. Huang, Q. Xu, C. M. Gabardo and G. Lee, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- S. Zhang, C. Chen, K. Li, H. Yu and F. Li, J. Mater. Chem. A, 2021, 9, 18785–18792 RSC.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- K. A. Mumford, K. H. Smith, C. J. Anderson, S. Shen, W. Tao, Y. A. Suryaputradinata, A. Qader, B. Hooper, R. A. Innocenzi and S. E. Kentish, Energy Fuels, 2012, 26, 138–146 CrossRef CAS.
- S. Valluri and S. K. Kawatra, Fuel Process. Technol., 2021, 212, 106620 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh and O. Voznyy, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- E. W. Lees, A. Liu, J. C. Bui, S. Ren, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 1712–1718 CrossRef CAS.
- Y. Hori and S. Suzuki, J. Electrochem. Soc., 1983, 130, 2387 CrossRef CAS.
- N. Osetrova, V. Bagotzky, S. Guizhevsky and Y. M. Serov, J. Electroanal. Chem., 1998, 453, 239–241 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- R. Kas, K. Yang, G. P. Yewale, A. Crow, T. Burdyny and W. A. Smith, Ind. Eng. Chem. Res., 2022, 61, 10461–10473 CrossRef CAS.
- E. W. Lees, J. C. Bui, D. Song, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 834–842 CrossRef CAS.
- A. G. Fink, E. W. Lees, Z. Zhang, S. Ren, R. S. Delima and C. P. Berlinguette, ChemElectroChem, 2021, 8, 2094–2100 CrossRef CAS.
- D. J. Pimlott, A. Jewlal, B. A. Mowbray and C. P. Berlinguette, ACS Energy Lett., 2023, 8, 1779–1784 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- C. Larrea, D. Torres, J. R. Avilés-Moreno and P. Ocón, J. CO2 Utilization, 2022, 57, 101878 CrossRef CAS.
- C. Larrea, J. R. Avilés-Moreno and P. Ocón, Molecules, 2023, 28, 1951 CrossRef CAS PubMed.
- Z. Zhang, E. W. Lees, S. Ren, B. A. Mowbray, A. Huang and C. P. Berlinguette, ACS Cent. Sci., 2022, 8, 749–755 CrossRef CAS PubMed.
- O. Gutiérrez-Sánchez, B. De Mot, M. Bulut, D. Pant and T. Breugelmans, ACS Appl. Mater. Interfaces, 2022, 14, 30760–30771 CrossRef PubMed.
- H. Liu, Y. Chen, J. Lee, S. Gu and W. Li, ACS Energy Lett., 2022, 7, 4483–4489 CrossRef CAS.
- J. Lee, H. Liu and W. Li, ChemSusChem, 2022, e202201329 CrossRef CAS PubMed.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- Y. C. Xiao, C. M. Gabardo, S. Liu, G. Lee, Y. Zhao, C. P. O’Brien, R. K. Miao, Y. Xu, J. P. Edwards and M. Fan, EES Catal., 2023, 1, 54–61 RSC.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi and H. Liu, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang and J. Liu, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- R. Khakpour, D. Lindberg, K. Laasonen and M. Busch, ChemCatChem, 2023, 15(6), e202201671 CrossRef CAS.
- H. Ma, E. Ibáñez-Alé, R. Ganganahalli, J. Pérez-Ramírez, N. López and B. S. Yeo, J. Am. Chem. Soc., 2023, 145(45), 24707–24716 CAS.
- N. Sreekanth and K. L. Phani, Chem. Commun., 2014, 50, 11143–11146 RSC.
- A. Y. Shekh, K. Krishnamurthi, S. N. Mudliar, R. R. Yadav, A. B. Fulke, S. S. Devi and T. Chakrabarti, Crit. Rev. Environ. Sci. Technol., 2012, 42, 1419–1440 CrossRef CAS.
- J. T. Cullinane and G. T. Rochelle, Chem. Eng. Sci., 2004, 59, 3619–3630 CrossRef CAS.
- A. G. Fink, E. W. Lees, J. Gingras, E. Madore, S. Fradette, S. A. Jaffer, M. Goldman, D. J. Dvorak and C. P. Berlinguette, J. Inorg. Biochem., 2022, 231, 111782 CrossRef CAS PubMed.
- J. H. Kim, H. Jang, G. Bak, W. Choi, H. Yun, E. Lee, D. Kim, J. Kim, S. Y. Lee and Y. J. Hwang, Energy Environ. Sci., 2022, 15, 4301–4312 RSC.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- L. Chen, Q. Xu, S. Z. Oener, K. Fabrizio and S. W. Boettcher, Nat. Commun., 2022, 13, 3846 CrossRef CAS PubMed.
- Z. Tao, C. L. Rooney, Y. Liang and H. Wang, J. Am. Chem. Soc., 2021, 143, 19630–19642 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |