
Unveiling the mechanism of visible light-assisted peroxymonosulfate activation and carbamazepine degradation using NH2-MIL-125(Ti)@MIL-53(Fe/Co) heterojunction photocatalyst†
Debashis
Roy
,
Sayak
Saha Chowdhury
and
Sirshendu
De
 *
*
Department of Chemical Engineering, Indian Institute of Technology Kharagpur, Kharagpur – 721302, India. E-mail: sde@che.iitkgp.ac.in; Fax: 91 3222 255303; Tel: 91 3222 283926
First published on 3rd January 2024
Abstract
Herein, a facile hydrothermal synthesis method was used to create the visible light-responsive heterojunction composite NH2-MIL-125(Ti)@MIL-53(Fe/Co) (AMIL@MIL). This composite catalyst was investigated for the mineralization of carbamazepine (CBZ) in aqueous solution with a visible light-assisted activation of peroxymonosulfate (PMS). CBZ (initial concentration: 10 mg L−1) could be totally degraded by 0.05 g L−1 of composite containing 10 wt% NH2-MIL-125(Ti) (AMIL(10)@MIL) and 0.25 g L−1 of PMS within 1 h reaction under visible light. The enhanced degradation of CBZ was attributed to the efficient activation of PMS, and subsequently the generation of SO4˙−, ˙OH and O2˙− radicals, together with the remarkable light harvesting and spatial charge separation in the direct Z-scheme heterojunction configuration. The composite catalyst could effectively treat contaminated surface and groundwater samples, overcoming the minimal to moderate inhibitory effects of coexisting ions and organics, while also exhibiting good reusability and structural integrity. The adsorbed PMS molecules are activated by several surface-confined redox cycles, including Fe2+/Fe3+, Co2+/Co3+ and Ti3+/Ti4+, aiding in the production of reactive radicals in the medium and efficient charge transfer during the reaction. A thorough study on the intermediates was carried out to determine the plausible CBZ mineralization and degradation pathway in the AMIL(10)@MIL/PMS/vis system. Overall, the suggested catalytic method is applicable and feasible for the remediation of contaminated surface and groundwater when exposed to visible light radiation.
Environmental significanceSulfate radical-based AOPs (SR-AOPs) involve highly reactive SO4˙− radicals towards the degradation of different persistent organic contaminants. Herein, a visible light-responsive MOF based heterojunction photocatalyst, NH2-MIL-125@MIL-53, was developed to activate peroxymonosulfate (PMS) and degrade carbamazepine (CBZ) in aqueous medium. The composite involves redox cycles, such as Fe2+/Fe3+, Co2+/Co3+ and Ti3+/Ti4+, which are essential towards the activation of PMS and generation of various reactive radicals, i.e., SO4˙−, ˙OH, and O2˙−. A detailed intermediate analysis was conducted and the CBZ degradation pathway was proposed. The synthesized composite possessed excellent stability and reusability with low leaching of its constituent elements. Moreover, CBZ-spiked natural water was treated with catalyst/PMS and rapid CBZ degradation was observed. Overall, the synthesized composite catalyst can be suitably applied for practical water treatment. |
1. Introduction
Antibiotics, pharmaceuticals and personal care products (PPCPs) are extensively used in recent years and are potential threats to the environment when discarded in aqueous medium. Among the numerous antibiotics, carbamazepine (5H-dibenzo[b,f]azepine-5-carboxamide, CBZ) is an iminostilbene derivative with a tricyclic structure, which is frequently used as an anticonvulsant for the treatment of certain psychiatric disorders, such as epilepsy, depression and neuromorphic pain.1–4 However, due to the inability of the human body to completely metabolize CBZ, it is discharged in aqueous systems, and consequently it has been widely detected in wastewater treatment plant effluent, municipal sewage treatment system discharge water, groundwater, surface water and even drinking water. Due to its unique symmetrical aromatic heterocyclic structure, CBZ is a highly stable refractory pharmaceutical contaminant with strong persistence and bio-accumulative property. Consequently, it is extremely difficult to remove CBZ by traditional sewage treatment techniques involving different physical and biological methods, such as adsorption, activated sludge process, membrane separation, and biodegradation.5–8 Low doses of CBZ in the human body may lead to severe physical discomfort, including drowsiness and nausea. Alternatively, higher CBZ doses (>40 mg L−1) can lead to a coma, and eventually death. Therefore, the bio-toxicity of this micropollutant is a serious threat to public health and environment and considerable attention is required towards the treatment of CBZ-containing wastewater.Advanced oxidation processes (AOPs), involving hydroxyl radicals (˙OH), are frequently used for the complete mineralization and removal of toxic persistent endocrine-disrupting chemicals.9–15 However, the conventional oxidants, such as O3 and H2O2, are highly corrosive, difficult to store and handle, and generate large volumes of sludge, limiting the applicability of this method.16 Alternatively, sulfate radical (SO4˙−)-based AOPs (SR-AOPs) are beneficial and promising technologies given that SO4˙− radical (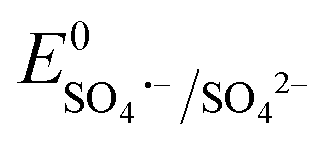 : 2.6–3.1 V vs. NHE) is much more reactive than ˙OH (
: 2.6–3.1 V vs. NHE) is much more reactive than ˙OH (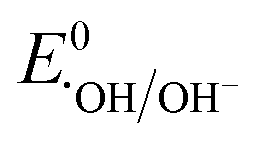 : 1.9 V vs. NHE and
: 1.9 V vs. NHE and 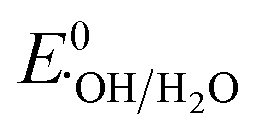 : 2.7 V vs. NHE), has a longer half-life (
: 2.7 V vs. NHE), has a longer half-life (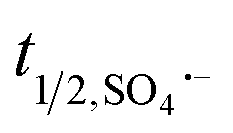 : 30–40 μS and
: 30–40 μS and 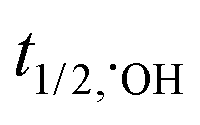 : <1.0 μS, for freely diffusible SO4˙− and ˙OH, respectively, allowing excellent mass transfer and contact between radicals and target organic contaminants in the medium, i.e., deionized water, without any external ions and organics17–20) and active over a wide pH range.21,22 Moreover, SO4˙− radicals react selectively with unsaturated organic compounds (rate constants: 106 to 109 M−1 s−1) compared to ˙OH, which is not very selective towards unsaturated organic contaminants and dissolved organic matter (DOM).23 In this case, peroxymonosulfate (PMS) and peroxydisulfate (PDS) can be effectively activated to generate SO4˙− radicals via energy and electron transfer reactions.23 Generally, the activation methods include exposure to ultraviolet (UV) light, heat, ultrasound, transition metal ions and metal oxides.24–30 The activation of PMS and PDS by UV light and ultrasound is energy intensive, whereas that by heat is only applicable for small-scale processes. In contrast, visible light-driven catalytic processes for the activation of PMS/PDS is a suitable technique for the facile generation of SO4˙− and ˙OH radicals. Between PDS and PMS, it is easier to rupture the peroxo (–O–O–) bond of the latter due to its asymmetric molecular structure. Moreover, the easy handling and non-corrosiveness of both PMS/PDS make these processes much easier to carry out.
: <1.0 μS, for freely diffusible SO4˙− and ˙OH, respectively, allowing excellent mass transfer and contact between radicals and target organic contaminants in the medium, i.e., deionized water, without any external ions and organics17–20) and active over a wide pH range.21,22 Moreover, SO4˙− radicals react selectively with unsaturated organic compounds (rate constants: 106 to 109 M−1 s−1) compared to ˙OH, which is not very selective towards unsaturated organic contaminants and dissolved organic matter (DOM).23 In this case, peroxymonosulfate (PMS) and peroxydisulfate (PDS) can be effectively activated to generate SO4˙− radicals via energy and electron transfer reactions.23 Generally, the activation methods include exposure to ultraviolet (UV) light, heat, ultrasound, transition metal ions and metal oxides.24–30 The activation of PMS and PDS by UV light and ultrasound is energy intensive, whereas that by heat is only applicable for small-scale processes. In contrast, visible light-driven catalytic processes for the activation of PMS/PDS is a suitable technique for the facile generation of SO4˙− and ˙OH radicals. Between PDS and PMS, it is easier to rupture the peroxo (–O–O–) bond of the latter due to its asymmetric molecular structure. Moreover, the easy handling and non-corrosiveness of both PMS/PDS make these processes much easier to carry out.
Metal organic frameworks (MOFs) with chromophoric groups in their organic ligands have been studied as efficient photocatalysts for various applications.31–38 However, functionalized MOFs may have limits in practical photocatalytic applications due to their weaker ability to capture visible light, rapid recombination of photoexcited e−/h+ pairs, and structural instability at extreme pH. In this case, most of these problems can be overcome through different post-synthetic modification techniques, such as tuning the structure of the chromophore, formation of heterojunction composites, and doping of metal and non-metallic elements.39–44 Among them, the formation of heterojunction composites is an effective method, where different semiconductors are introduced in the MOF structure to improve their photo-responsive properties, structural integrity and overall electronic conductivity.45,46 Also, the growth of another MOF on a base MOF crystal is a unique way to prepare heterojunction composites, which can act as efficient photocatalysts with the minimum loss of porosity and uniform distribution of active sites. Moreover, the application of bimetallic MOFs coupled with another aminated MOF is highly beneficial towards incident visible light harvesting and e− transport due to the generation of suitable surface-bound redox pairs and elongated excited states upon photonic irradiation. Among the different photo-responsive and semiconducting MOFs, MIL-53(Fe/Co) and MIL-125(Ti) are both chromophoric and photo-responsive and their aminated derivatives have high surface heterogeneities, sensitivity to visible light irradiation, abundant active sites and photo-responsive –NH2 groups on their surface.
Therefore, based on the above-mentioned considerations, herein, an MOF@MOF heterojunction photocatalyst (NH2-MIL-125@MIL-53, abbreviated as AMIL@MIL) was designed and synthesized through a facile hydrothermal technique. The produced composite was used to activate PMS under visible light, and subsequently CBZ was mineralized in synthetic and contaminated surface water samples. Furthermore, based on the intermediate study, the thorough pathway for the degradation of CBZ was suggested. The main objectives of this study are as follows: (i) investigate the effects of different operational parameters on the catalytic activities of the AMIL@MIL/PMS combination; (ii) study the mechanism of radical generation, heterojunction formation and light harvesting, leading to superior CBZ degradation; (iii) identify the different radical species and generated intermediates in the medium and propose the detailed CBZ degradation pathway; and (iv) examine the structural stability and reusability of the AMIL@MIL composite.
2. Synthesis of materials
The detailed methods for the synthesis of MIL-53(Fe/Co), MIL-125(Ti) and their aminated counterparts, i.e., NH2-MIL-53(Fe/Co) and NH2-MIL-125(Ti), are described in Section S1.2 of the ESI.†The hydrothermal method was employed to prepare the binary composites. For example, in the case of the AMIL@MIL composite, a specific weight of MIL-53 was uniformly dispersed in a mixture of 72 mL methanol and 8 mL DMF. Then, a predetermined amount of NH2-bdc and Ti-isopropoxide were added to the dispersion. The resultant mixture was stirred for 1 h, and then the autoclave reactor assembly containing the Teflon cup with the reaction mixture was hydrothermally treated at 150 °C for 24 h. The resultant light brownish AMIL@MIL composite was thoroughly washed using DI water and ethanol and dried to obtain the material. The predetermined amount of the precursors of NH2-MIL-125 were suitably varied to obtain the binary composite with different wt% of NH2-MIL-125 on MIL-53. The resultant composites were designated as AMIL(x)@MIL, where x = 5, 10, 15 and 20, based on the wt% of NH2-MIL-125 on MIL-53. For the other composites, the base MOF powder was initially dispersed in a specific solvent, and then the required precursors of the other MOF were added to the mixture. The resultant mixture was treated at a high temperature for the required duration in a sealed autoclave reactor assembly to generate the MOF@MOF heterojunction composites. The final composite was washed thoroughly, and then dried before collection.
The various materials required for the synthesis, the characterization methods, and the experimental and analytical procedures are summarized in Sections S1.1–S1.5 of the ESI.†
3. Results and discussion
3.1. Details of the materials
The pristine MIL-53(Fe/Co) crystals possessed either a small rhombohedral or elongated hexagonal prismatic shape (Fig. 1(a and b) and S1(a and b)†), with surface heterogeneities, which is probably due to the in situ-formed metallic oxides, i.e., Fe2O3 and Co3O4. In contrast, the pristine MIL-125(Ti) crystals possessed a pebble-like shape (Fig. 1c and S1c†). Upon amination, NH2-MIL-53(Fe/Co) retained its hexagonal prismatic shape (Fig. 1d and S1d†), with NH2-MIL-125(Ti) generating a mixture of pebble- and clinopinacoid-shaped particles (Fig. 1e and S1e†). The binary AMIL(10)@MIL crystals were irregularly shaped (Fig. 1f), with the growth of the shell MOF layer from the tip to center (Fig. S1(f–n)†) and in situ-grown TiO2 particles on its surface, which is also evident from the HRTEM analysis. Specifically, lattice fringes having d-spacings of 0.19 nm and 0.20 nm, were identified, corresponding to the (200) and (020) planes of anatase TiO2, respectively, together with a smooth heterojunction interface between both components (Fig. 1(g–i)). Both planes were also evident in the SAED spectrum (Fig. 1j). | ||
| Fig. 1 FESEM images of (a and b) pristine MIL-53(Fe/Co), (c) MIL-125(Ti), (d) NH2-MIL-53(Fe/Co), (e) NH2-MIL-125(Ti) and (f) AMIL(10)@MIL. (g) STEM image, (h and i) HRTEM images and (j) SAED pattern of AMIL(10)@MIL. | ||
The results of the XRD analysis of all the materials are shown in Fig. 2a. In the case of pure MIL-125(Ti) with an orthorhombic crystal structure, the characteristic peaks were observed at the 2θ values of 5.87°, 9.34°, 12.67°, 17.58° and 25.4°, which are ascribed to the (101), (200), (211), (222) and (422) planes, respectively.47–49 Additionally, the peak at the 2θ value of 35.4° is ascribed to the (004) plane of anatase TiO2, implying its presence on the MOF structure.50 Another strong peak was generated at the 2θ value of 44.74°, which was also present in the other binary composites containing MIL-125(Ti). In contrast, for NH2-MIL-125, several peaks were identified at the 2θ values of 5.63°, 10.2°, 15.48°, 18.36°, 21.46°, 27.47° and 44.98°, corresponding to the (101), (200), (211), (310), (213), (204) and (111) planes.47,51 Using the Scherrer equation, the crystallite size was calculated for the peak at the 2θ of 27.47° to be 0.339 nm. In the case of pure MIL-53, prominent peaks were generated at the 2θ values of 9.36°, 12.67°, 15.88°, 17.64°, 25.25°, 28.1°, 29.92° and 39.77°, with three weak peaks at 2θ values of 31.9°, 32.7° and 41.25°.33,34 The size of the crystallites was determined to be 0.319 nm based on the peak at the 2θ of 28.2° employing the Scherrer equation. Specifically, the peaks at the 2θ values of 25.25°, 32.7° and 39.77° were assigned to the (012), (104) and (113) planes of α-Fe2O3 (JCPDS card no: 33-0664),52–54 together with the peaks at the 2θ values of 28.1° and 31.9°, corresponding to the (022) and (113) planes of Co3O4, respectively (JCPDS card no: 96-900-5889).55,56 The XRD spectrum of AMIL(10)@MIL showed distinct peaks for both major components, showing an increase in intensity with an increase in the content of AMIL. In the case of AMIL(10)@MIL, the peaks were identified at the 2θ values of 9.09°, 16.26°, 18.04°, 23.8°, 25.4°, 28.9°, 32.11°, 34.03° and 46.72°, corresponding to both primary components, i.e., NH2-MIL-125(Ti) and MIL-53(Fe/Co). Therefore, pure-phase MOFs were successfully synthesized. Moreover, the weak peaks at the 2θ values of 25.4° and 32.11° are ascribed to the (012) and (113) planes of Fe2O3 and Co3O4, respectively, implying the minor presence of these compounds in AMIL(10)@MIL.
 | ||
| Fig. 2 (a) XRD spectra and (b) UV-vis DRS of all the composites. (c) PL analysis of the original components and binary composites. (d) Nyquist plots of the original components and AMIL(10)@MIL. (e) Photocurrent analysis of the different primary components and binary composites. (f) Schematic illustration of the charge transfer pathway of type-II and direct Z-scheme heterojunctions. | ||
The FTIR spectra of the pristine MOFs (i.e., MIL-53, MIL-125, NH2-MIL-53 and NH2-MIL-125) and the relevant composites (Fig. S1i†) exhibit the presence of the characteristic peaks of the primary MOFs in the corresponding binary composites, implying successful formation of the heterojunction materials and retention of their surface functional groups. A detailed discussion is provided in Section S1.6.1 of the ESI.†
The photo-responsive properties of the different primary components (pristine MIL-53 and NH2-MIL-125(Ti)), and AMIL(10)@MIL were studied using UV-vis DRS (Fig. 3a). The spin-permitted d–d transition [6A1g → 4A1g + 4Eg(G)] of the octahedrally coordinated Fe3+ moieties of the pristine MIL-53(Fe/Co) generated an absorption edge at ∼490 nm due to the ligand to metal charge transfer (LMCT) from O(II) to Fe(III) and absorption-induced n–π and π–π shifting. In the case of the NH2-MIL-53(Fe/Co) MOF, the introduction of chromophoric –NH2 groups extended its absorption edge to 625 nm. The higher visible light responsiveness is due to the enhanced charge transfer from the –COO linkers and redox pairs (i.e., Co2+/Co3+|surf. and Fe2+/Fe3+|surf.) to the –NH2-containing aromatic framework. MIL-125(Ti) showed a sharp absorption edge at 450 nm and the introduction of –NH2 groups resulted in a red-shift towards the visible region, with an extended absorption edge at around 500 nm. The AMIL(10)@MIL composite showed the highest UV-vis light-harvesting capability due to the presence of –NH2 groups on its outer surface and tuning of the bandgap structure of the shell MOF. Specifically, the absorption spectrum of AMIL(10)@MIL included a sharp peak with an extended adsorption edge at 530 nm. The introduction of –NH2 groups in both primary components and the binary composite enhanced the overall sensitivity to UV-vis irradiation and corresponding excitation of charge carriers. Moreover, the presence of –NH2 groups lowered the bandgap energy of the aminated MOF due to the increase in the upper energy level of the valence band (HOMO, composed of C, O and N2p orbitals), which was localized on the organic motifs, while maintaining the lower energy level of conduction band (LUMO, composed of Ti3d and O2p orbitals).47 According to the Tauc plot analysis,33,34,57 the bandgap energy (Eg) of NH2-MIL-53 and MIL-53 was calculated to be 2.5 eV and 2.65 eV (Fig. S2a and c†), respectively. Similarly, the Eg of MIL-125 was modified from 3.23 eV to 2.45 eV for NH2-MIL-125(Ti) (Fig. S2b and S2d†), respectively. The edge potential (EVB) of the valence bands (HOMO) of the developing components was calculated using the valence-band (VB) XPS analysis. The computed EVB for MIL-53 and NH2-MIL-125 was 2.34 and 1.86 eV (Fig. S2(e and f)†), respectively. Further, using the relation: ECB = Eg − EVB, the corresponding CB (LUMO) edge potentials (ECBs) were determined to be −0.31 eV and −0.59 eV, for MIL-53 and NH2-MIL-125, respectively.
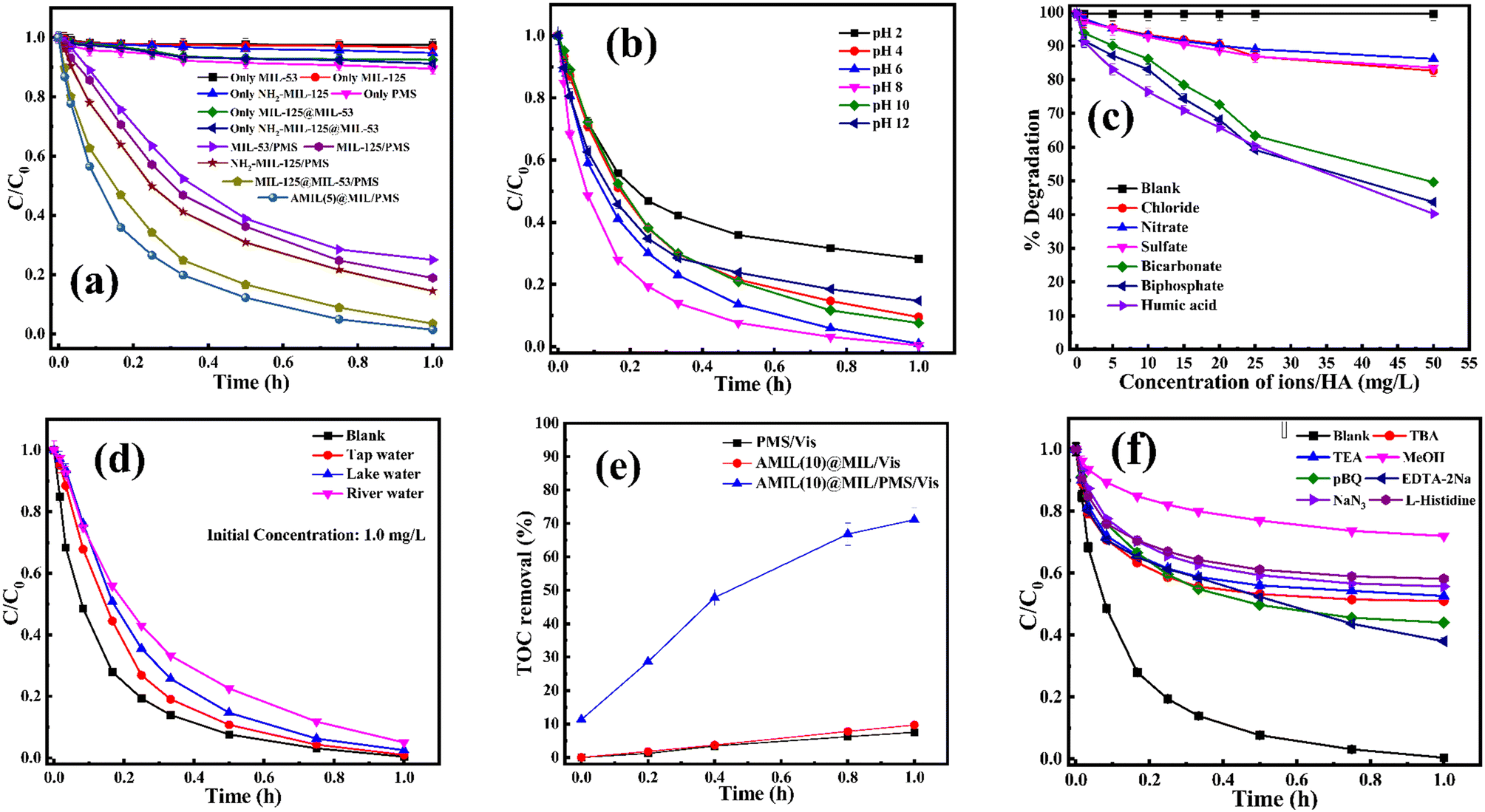 | ||
| Fig. 3 Variation in residual CBZ concentration for (a) different catalyst/PMS combinations, (b) different initial solution pH, in presence of (c) different coexisting anions and organics and (d) various contaminated surface water sources, (e) variation in residual TOC in different catalytic systems, and (f) different radical scavengers (experimental conditions: catalyst dose: 0.05 g L−1 (except for a, catalyst dose: 0.01 g L−1), PMS dose: 0.25 g L−1, operating temperature: 30 °C, solution initial pH: 6.5, initial CBZ concentration: 10 mg L−1, and dose of each scavenger: 5 mM). | ||
Finally, to assess the effectiveness of charge carrier separation in the materials, photoluminescence (PL) analysis was performed (Fig. 2c). Among the materials including the primary MOFs, AMIL(10)@MIL generated a PL spectrum with the weakest peak intensity, indicating the most efficient e−/h+ separation ability and charge carrier migration to the MOF surface and active sites. The HOMO of both MIL-125(Ti) and NH2-MIL-125(Ti) resides on the organic linker (i.e., terephthalate or amino-terephthalate, respectively), whereas the lowest unoccupied crystal orbital (LUCO) is localized largely on the Ti-oxo clusters. Therefore, upon light absorption, a long-lived excited charge separation state occurs by transferring an e− from the organic linker to Ti4+, which is reduced to Ti3+. Moreover, the –NH2 groups can act as h+ stabilizers and prolong the excited states of NH2-MIL-125 compared to its non-aminated counterpart (MIL-125).58,59 According to the EIS analysis (Fig. 2d), the smallest Nyquist radius of AMIL(10)@MIL implies the maximum resistance towards e−/h+ recombination and excellent spatial separation due to the formation of a heterojunction compared to both primary components. Moreover, transient photocurrent response analysis was carried out for MIL-53(Fe/Co), NH2-MIL-125(Ti), and AMIL(10)@MIL. Compared to each of the primary component, AMIL(10)@MIL generated a stronger photocurrent (Fig. 2e), which is more direct evidence for its efficient charge separation and longer lifetime of excited e−/h+ pairs.
Therefore, based on the Eg and corresponding EVB/ECB values, the band structure of AMIL@MIL was designed, as shown in Fig. 2f. A direct Z-scheme configuration is formed and e− from the CB (HOMO) of MIL-53 and h+ from the VB (LUMO) of NH2-MIL-125 combine. This results in abundant e− in the VB (LUMO) of NH2-MIL-125 and h+ in the CB (HOMO) of MIL-53. A space-charge zone is produced at the interface upon attaining Fermi level equilibrium. Band-bending in the appropriate directions (upwards for NH2-MIL-125(Ti) due to the formation of a charge depletion region near the interface and downwards for MIL-53(Fe/Co) due to the generation of s charge-accumulation region at the interface) limits the possibility of the recombination of transferred charge carriers. By adopting this structure, the e− in the CB of NH2-MIL-125 can achieve the maximum reduction capacity (ECB: −0.59 eV vs. NHE) and convert the adsorbed O2 molecules into O2˙− radicals (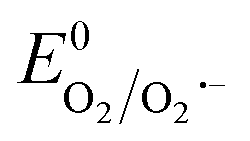 = −0.33 eV vs. NHE). Alternatively, h+ in the VB (HOMO) of MIL-53(Fe/Co) is highly oxidative (EVB: 2.34 eV) and can easily oxidize OH− and H2O into ˙OH radicals (
= −0.33 eV vs. NHE). Alternatively, h+ in the VB (HOMO) of MIL-53(Fe/Co) is highly oxidative (EVB: 2.34 eV) and can easily oxidize OH− and H2O into ˙OH radicals (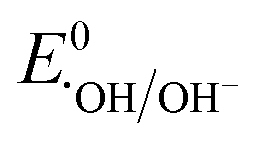 : 1.9 V vs. NHE and
: 1.9 V vs. NHE and 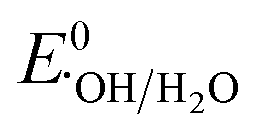 : 2.7 V vs. NHE).60 Through traditional radical scavenging tests and EPR analysis, these radicals were extensively detected in the reaction medium. Therefore, a direct Z-scheme heterojunction is likely to develop instead of a type-II one, where the accumulated e− (h+) in the CB (VB) of MIL-53 (NH2-MIL-125) is not reductive (oxidative) enough to generate O2˙− (˙OH) radicals from the adsorbed O2 molecules (OH− ions and H2O molecules).
: 2.7 V vs. NHE).60 Through traditional radical scavenging tests and EPR analysis, these radicals were extensively detected in the reaction medium. Therefore, a direct Z-scheme heterojunction is likely to develop instead of a type-II one, where the accumulated e− (h+) in the CB (VB) of MIL-53 (NH2-MIL-125) is not reductive (oxidative) enough to generate O2˙− (˙OH) radicals from the adsorbed O2 molecules (OH− ions and H2O molecules).
3.2. Photocatalytic degradation of CBZ
CBZ degradation experiments were carried out with various catalyst/oxidant combinations (Fig. 3a). Initially, adsorption on the catalyst and PMS self-oxidation were investigated. The removal of CBZ due to adsorption on all the catalysts was negligible (1% to 1.5%). Similarly, in the presence of visible light, PMS alone could only remove only 10.5% CBZ (kapp: 2.34 × 10−3 min−1). After adsorption, the catalyst/visible light combination could degrade a minor amount of CBZ, i.e., 5.2%, 2.6% and 8.8% for pristine NH2-MIL-125, MIL-53(Fe/Co) and AMIL(5)@MIL, with kapp: 1.07 × 10−3 min−1, 0.6 × 10−3 min−1 and 0.026 min−1, respectively. Upon the addition of PMS, the catalytic activities of all the materials, i.e., primary and binary composites, improved. The CBZ removal efficiencies were enhanced up to 98.6% for AMIL(5)@MIL/PMS (kapp: 0.0715 min−1) compared to 75% and 85.5% degradation for MIL-53(Fe/Co)/PMS and NH2-MIL-125/PMS, with kapp: 0.03 min−1 and 0.035 min−1, respectively. Among the binary composite combinations, AMIL(5)@MIL/PMS/vis showed the highest activity (98.6% CBZ removal, with kapp: 0.072 min−1), followed by MIL-125@MIL-53(Fe/Co)/PMS (96.5% CBZ removal, with kapp: 0.058 min−1). The highest reactivity of AMIL(5)@MIL was due to its most efficient light harvesting by its surface-anchored NH2 groups and in situ-generated TiO2 particles, which imparted antenna effects to transmit the photonic irradiation energy in the lattice structure, resulting in superior charge carrier excitation, separation and transport across the lattice compared to the other composites with no –NH2 groups. Next, the optimum composition of the AMIL@MIL composite was determined. An increase in the NH2-MIL-125 content in AMIL@MIL to 10 wt% boosted the CBZ degradation to 99% (kapp: 0.084 min−1) and the further loading of NH2-MIL-125 up to 15 wt% slightly suppressed the catalytic activity (98.3% CBZ removal with kapp, 0.068 min−1) (Fig. S3a†). These results are due to the agglomeration of the layered structure, which decreased the availability of accessible active sites and charge transfer routes and introduced more charge-recombination centers, diminishing the light-harvesting and activation of the PMS species in the medium. Hence, AMIL(10)@MIL was chosen as the best catalyst for the following studies.The effects of various AMIL(10)@MIL and PMS doses on the degradation of CBZ are provided in Fig. S3(b and c†). Only 10.5% CBZ was degraded by PMS alone in the absence of a catalyst (Fig. 3a). The CBZ removal gradually increased to 99.1% up to a catalyst dose of 0.05 g L−1 (Fig. S3b†). Subsequently, the extent of degradation remained constant (∼99%) up to the catalyst dose of 0.075 g L−1, and a further increase in the AMIL(10)@MIL dose to 0.1 g L−1 slightly decreased the CBZ removal to 97.6%. These results can be explained based on the availability of more active sites and the ability to generate a greater number of free reactive oxidative species (ROS) (˙OH and SO4˙−) in the medium at the optimal catalyst dose, which resulted in superior CBZ removal. Alternatively, an excessive catalyst dose may result in the agglomeration of particles, which decreases the number of accessible active sites and increases the turbidity of the medium, thus reducing the catalytic activity. Hence, 0.05 g L−1 AMIL(10)@MIL was selected as the optimum catalyst dose at which a steady increase in PMS dose up to 0.25 g L−1 resulted in nearly total CBZ removal (99.6%) (Fig. S3c†). The optimal PMS dose was assessed to be 0.25 g L−1 given that an increase in the dose to 0.5 g L−1 slightly decreased the CBZ removal extent (98.5%). At this PMS dose, adequate ˙OH and SO4˙− radicals were generated for the complete degradation of the CBZ molecules. Excessive PMS can adversely affect the catalytic process by consuming generated SO4˙− and ˙OH to produce less reactive SO5˙− radicals (eqn (S1)–(S4)†).61–63 Next, the variation in initial CBZ concentration (C0) from 1.0 mg L−1 to 25 mg L−1 resulted in almost complete CBZ removal in the presence of the optimal catalyst and PMS dose (Fig. S3d†). In the case of lower concentrations (up to 10 mg L−1), the kinetics of degradation was very fast and it gradually slowed down as the CBZ concentration increased, i.e., kapp: 0.147 min−1 and 0.089 min−1 for C0: 1.0 mg L−1 and 10 mg L−1, respectively. Thus, the optimum dose of catalyst/PMS could remove CBZ almost completely up to 10 mg L−1 initial concentration of CBZ, with substantial mineralization (∼71% removal of total organic carbon).
To study the applicability of the AMIL(10)@MIL catalyst at different solution pH, its stability was determined through crystallographic analysis. The catalyst, treated at different pH, in the presence of PMS (0.5 g L−1) and CBZ (10 mg L−1), was analyzed using XRD and the comparative results are provided in Fig. S3f.† No significant variation in peak positions was observed, although a slight variation in the intensity of the peaks was evident, which is due to the leaching of the constituent atoms at different pH. Overall, the AMIL(10)@MIL catalyst was stable at different solution pH and is applicable in the study involving effect of pH on CBZ degradation. The effect of a variation in initial pH (pHi) from 2 to 12 on CBZ degradation is shown in Fig. 3b. The self-hydrolysis of PMS caused pHi to decrease to the acidic range and neutral to slightly alkaline final pH (pHf) was obtained only for highly alkaline pHi (10–12). The highest CBZ degradation was obtained at near neutral pHi (99.1% and 99.6% removal for pHi: 6 and 8, with kapp: 0.073 min−1 and 0.089 min−1, respectively) and the CBZ degradation was moderately affected at highly acidic and alkaline pHi (71.8% and 85.3% CBZ removal at pHi: 2 and 12, with kapp: 0.027 min−1 and 0.04 min−1, respectively). Based on the protonation–deprotonation of the –NH2 groups (eqn (1)), the CBZ molecule has two pKa values of pKa1: 2.3 and pKa2: 13.9. Therefore, in the pHf range of 2.5 to 9.5, CBZ molecules exist in zwitterionic and neutral forms. Also, the catalyst has pHPZC of 5.3 and was positively (negatively) charged below (above) pHf of 5.3. Moreover, the PMS molecules exist as HSO5− within 2.5 < pHf < 9.5 and beyond pHf > 9.5, HSO5− ions are converted into less reactive SO52−. An increase in pHf to 8.5–9.0 can lead to a transition from SO4˙− to ˙OH as the primary ROS in the medium, through the one e− oxidation of OH− by SO4˙− (k = 6.5 × 107 M−1 s−1), which is kinetically favored over the reverse reaction, i.e., ˙OH + HSO4− → SO4˙− + H2O, (k = 6.9 × 105 M−1 s−1).23 In the highly acidic region, excessive H+ ions form H-bonding and diminish the dissociation of PMS, together with strong charge repulsion between the catalyst and CBZ molecules. In addition, Co, Fe, and Ti hydroxide complexes are produced on the catalyst surface in highly alkaline environments, which decrease the catalytic activity and oxidation potentials, also affecting the charge transfer processes.64,65 Moreover, at higher pHf, SO4˙− is transformed into ˙OH, together with unfavorable electrostatic repulsion between the catalyst and PMS species, which lowers the overall reactivity and corresponding CBZ degradation (eqn (S5)–(S15)†). Therefore, the optimum pHi for PMS activation and CBZ degradation by AMIL(10)@MIL is in the range of 6–8.
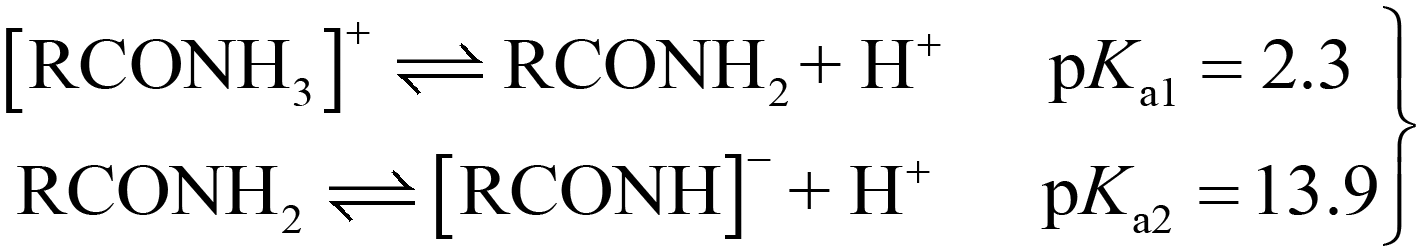 | (1) |
The effect of operating temperature on the degradation of CBZ is shown in Fig. S3e.† An increase in temperature from 293 K to 313 K accelerated the rate of CBZ degradation (kapp,293: 0.078 min−1 and kapp,313: 0.095 min−1, respectively). The faster CBZ removal at higher temperature is attributed to the thermal activation of the PMS molecules and corresponding generation of ROS in the medium,22,25 together with the enhanced randomness of the reactive species. Further, according to the activation energy (Ea) of the process of 13.3 kJ mol−1, surface chemical reactions were established as the rate-determining step compared to the convective transport of the reactants to the catalyst surface and their subsequent diffusion in the catalyst pores.
The effects of common inorganic anions (HPO42−, HCO3−, NO3−, SO42−, Cl−) and dissolved organic matter (DOM, humic acid, and HA) on the CBZ degradation are shown in Fig. 3c. NO3− and SO42− weakly affected the CBZ removal and 86.2% and 83.6% CBZ was degraded for 50 mg L−1 of NO3− and SO42−, respectively. NO3− could scavenge a small amount of SO4˙− (k = 2.1 × 106 M−1 s−1) and ˙OH (k = 1.8 × 106 M−1 s−1), and the overall activity was marginally affected due to the generation of slightly less reactive ˙NO3 (E0 = 2.3 V vs. NHE) and ˙NO2 (E0 = 1.03 V vs. NHE), which are also able to remove CBZ molecules.66,67 The weak ROS scavenging tendency and slight dimerization of SO4˙− radicals may lead to minor inhibition (eqn (S16)–(S22)†).34,68 Although SO42− ions cannot react with SO4˙− radicals, they can react with ˙OH to generate SO4˙− radicals. Alternatively, the minor inhibition towards CBZ degradation was also due to the decrease in the reduction potential of the SO4˙− radicals in the presence of SO42− ions.23,67 However, 90.5% and 82.7% CBZ degradation were obtained for 20 mg L−1 and 50 mg L−1 Cl−, respectively. Cl− generates Cl˙ and Cl2˙− with lower redox potentials 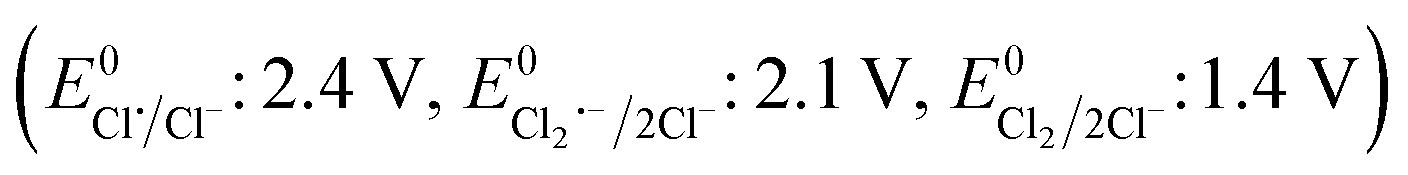 than SO4˙−
than SO4˙−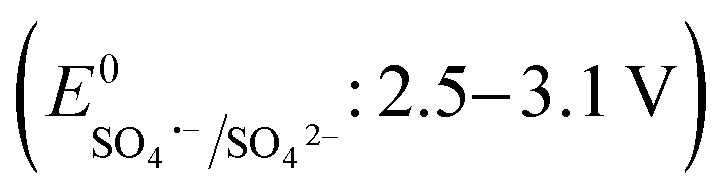 . The generated Cl˙ and Cl2˙− can react with organic contaminant molecules via single e− transfer (SET), H-abstraction and addition pathways.23 In the case of CBZ, Cl˙ reacts through the SET mechanism, as inferred from the higher rate constant (kCBZ/Cl˙ = (3.3–5.6) × 1010 M−1 s−1).23 Alternatively, Cl2˙− reacts through either the addition or H-abstraction pathways,23 with a slower rate constant of
. The generated Cl˙ and Cl2˙− can react with organic contaminant molecules via single e− transfer (SET), H-abstraction and addition pathways.23 In the case of CBZ, Cl˙ reacts through the SET mechanism, as inferred from the higher rate constant (kCBZ/Cl˙ = (3.3–5.6) × 1010 M−1 s−1).23 Alternatively, Cl2˙− reacts through either the addition or H-abstraction pathways,23 with a slower rate constant of 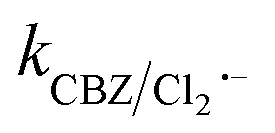 = 4.3 × 107 M−1 s−1. Therefore, both Cl˙ and Cl2˙− can degrade CBZ at comparable rates as that of ˙OH (kCBZ/˙OH = (3.07–8.8) × 109 M−1 s−1) and SO4˙− (
= 4.3 × 107 M−1 s−1. Therefore, both Cl˙ and Cl2˙− can degrade CBZ at comparable rates as that of ˙OH (kCBZ/˙OH = (3.07–8.8) × 109 M−1 s−1) and SO4˙− (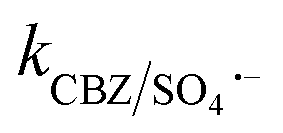 = (0.68–1.92) × 109 M−1 s−1).23,69 Alternatively, at a higher Cl− concentration, the two e− oxidation of Cl− ions by PMS (kCl−/PMS = 2.1 × 10−3 M−1 s−1) leads to the direct formation of HOCl, which has a much longer lifetime.69 HOCl can oxidize selected e−-rich organics such as CBZ and the overall oxidizing capacity of the reaction medium remains almost unaffected. In the case of Cl−, the different reactions involved are summarized as eqn (S23)–(S33).†61,62 HCO3− and HPO42− adversely affected the CBZ degradation and 49.6% and 43.7% CBZ were removed for 50 mg L−1 of HCO3− and HPO42−, respectively. Oxyanions such as phosphate (HPO42−/H2PO4−) and bicarbonate/carbonate (HCO3−/CO32−) exist as ion-pairs and can cause a shift in solution pH towards the alkaline range23 due to the strong buffering effects of H2CO3/HCO3− (pKa: 6.4) and H2PO4−/HPO42− (pKa: 7.2) equilibrium. Specifically, after the addition of HCO3− and HPO42− in the solution, the final pH was measured as 8.7 ± 0.5 and 8.3 ± 0.3, respectively. In the case of phosphate, beyond pH > pKa: 7.2, HPO42− can scavenge SO4˙− more readily than H2PO4− (ref. 23) (
= (0.68–1.92) × 109 M−1 s−1).23,69 Alternatively, at a higher Cl− concentration, the two e− oxidation of Cl− ions by PMS (kCl−/PMS = 2.1 × 10−3 M−1 s−1) leads to the direct formation of HOCl, which has a much longer lifetime.69 HOCl can oxidize selected e−-rich organics such as CBZ and the overall oxidizing capacity of the reaction medium remains almost unaffected. In the case of Cl−, the different reactions involved are summarized as eqn (S23)–(S33).†61,62 HCO3− and HPO42− adversely affected the CBZ degradation and 49.6% and 43.7% CBZ were removed for 50 mg L−1 of HCO3− and HPO42−, respectively. Oxyanions such as phosphate (HPO42−/H2PO4−) and bicarbonate/carbonate (HCO3−/CO32−) exist as ion-pairs and can cause a shift in solution pH towards the alkaline range23 due to the strong buffering effects of H2CO3/HCO3− (pKa: 6.4) and H2PO4−/HPO42− (pKa: 7.2) equilibrium. Specifically, after the addition of HCO3− and HPO42− in the solution, the final pH was measured as 8.7 ± 0.5 and 8.3 ± 0.3, respectively. In the case of phosphate, beyond pH > pKa: 7.2, HPO42− can scavenge SO4˙− more readily than H2PO4− (ref. 23) (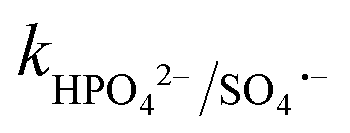 = 1.2 × 106 M−1 s−1,
= 1.2 × 106 M−1 s−1, 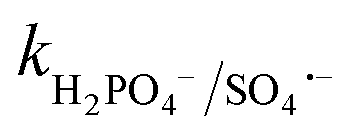 ≤ 7 × 104 M−1 s−1), forming both HPO4˙− and H2PO4˙ radicals. In the case of bicarbonate, HCO3− and CO32− can scavenge SO4˙− almost at the same rates to form HCO3˙ and CO3˙− radicals23 (
≤ 7 × 104 M−1 s−1), forming both HPO4˙− and H2PO4˙ radicals. In the case of bicarbonate, HCO3− and CO32− can scavenge SO4˙− almost at the same rates to form HCO3˙ and CO3˙− radicals23 (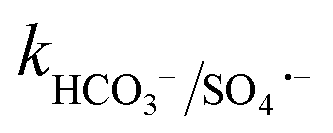 = 1.6 × 106 M−1 s−1 and
= 1.6 × 106 M−1 s−1 and 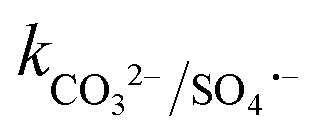 = 6.1 × 106 M−1 s−1), respectively. Moreover, beyond pH > 10.3, the primary reactive species changes to ˙OH from SO4˙− and HCO3−/CO32− can readily quench ˙OH to form HCO3˙ and CO3˙− radicals23 (
= 6.1 × 106 M−1 s−1), respectively. Moreover, beyond pH > 10.3, the primary reactive species changes to ˙OH from SO4˙− and HCO3−/CO32− can readily quench ˙OH to form HCO3˙ and CO3˙− radicals23 (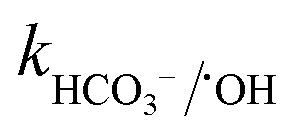 = 8.5 × 106 M−1 s−1 and
= 8.5 × 106 M−1 s−1 and 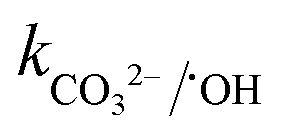 = 3.9 × 108 M−1 s−1), respectively. In addition, it has been reported that Fe2+/Fe3+|surf. and Co2+/Co3+|surf. can form a complex with (HPO42−/H2PO4−) and (HCO3−/CO32−), and the overall catalytic activity is adversely affected due to the interruption in charge transfer during the reaction.23 Overall, the presence of (HCO3−/CO32−) and H2PO4−/HPO42− can lower the catalytic activity and extent of CBZ degradation. Lastly, HA was used as a representative DOM, which showed a very strong inhibitory effects towards the removal of CBZ, i.e., 40.2% degradation in the presence of 50 mg L−1 of HA. The strong inhibition by DOM is a combined effect of various factors. DOMs are large supramolecular assemblies held together by van der Waals interaction, which can interact with unsaturated organic compounds via electronic interaction, H-bonding and hydrophobic interactions. DOM molecules have both polar and non-polar domains, and consequently, based on the solution pH and ionic strength, the three-dimensional structure and charge-density of DOMs vary. Therefore, H-bonding can occur among its deprotonated carboxylic and phenolic moieties and unsaturated organic contaminants. Hence, DOMs can affect the ionic CBZ molecules in the reaction medium. Alternatively, DOMs contain phenolic, quinone, amine and ketonic groups, which are redox active and can form complexes with metals, inducing and mediating e− transfer, and affecting the overall charge transfer pathways. Moreover, DOM molecules can be readily adsorbed on the catalytic active sites and block the pores, reducing the available intraparticle surface area of the catalyst. Lastly, DOM molecules are quite efficient in scavenging the reactive species,70 including ˙OH, SO4˙−, O2˙− and O21 with k˙OH/DOM = 106–109 M−1 s−1,
= 3.9 × 108 M−1 s−1), respectively. In addition, it has been reported that Fe2+/Fe3+|surf. and Co2+/Co3+|surf. can form a complex with (HPO42−/H2PO4−) and (HCO3−/CO32−), and the overall catalytic activity is adversely affected due to the interruption in charge transfer during the reaction.23 Overall, the presence of (HCO3−/CO32−) and H2PO4−/HPO42− can lower the catalytic activity and extent of CBZ degradation. Lastly, HA was used as a representative DOM, which showed a very strong inhibitory effects towards the removal of CBZ, i.e., 40.2% degradation in the presence of 50 mg L−1 of HA. The strong inhibition by DOM is a combined effect of various factors. DOMs are large supramolecular assemblies held together by van der Waals interaction, which can interact with unsaturated organic compounds via electronic interaction, H-bonding and hydrophobic interactions. DOM molecules have both polar and non-polar domains, and consequently, based on the solution pH and ionic strength, the three-dimensional structure and charge-density of DOMs vary. Therefore, H-bonding can occur among its deprotonated carboxylic and phenolic moieties and unsaturated organic contaminants. Hence, DOMs can affect the ionic CBZ molecules in the reaction medium. Alternatively, DOMs contain phenolic, quinone, amine and ketonic groups, which are redox active and can form complexes with metals, inducing and mediating e− transfer, and affecting the overall charge transfer pathways. Moreover, DOM molecules can be readily adsorbed on the catalytic active sites and block the pores, reducing the available intraparticle surface area of the catalyst. Lastly, DOM molecules are quite efficient in scavenging the reactive species,70 including ˙OH, SO4˙−, O2˙− and O21 with k˙OH/DOM = 106–109 M−1 s−1, 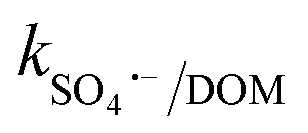 = 107–108 M−1 s−1,
= 107–108 M−1 s−1,  = (1.8–2.2) × 103 M−1 s−1 and
= (1.8–2.2) × 103 M−1 s−1 and 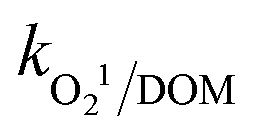 = (0.41–1.6) × 106 M−1 s−1, respectively, together with increasing the solution turbidity. This adversely affects the penetration of photoirradiation in the medium through the inner filter effect and generates other byproducts and intermediates. Overall, DOM showed the maximum inhibitory effect towards CBZ degradation followed by HPO42− and HCO3−.
= (0.41–1.6) × 106 M−1 s−1, respectively, together with increasing the solution turbidity. This adversely affects the penetration of photoirradiation in the medium through the inner filter effect and generates other byproducts and intermediates. Overall, DOM showed the maximum inhibitory effect towards CBZ degradation followed by HPO42− and HCO3−.
The findings of the study on the application of the catalyst/PMS combination towards the removal of CBZ in polluted real-water matrixes are displayed in Fig. 3d. CBZ was added to maintain a concentration of 1.0 mg L−1 in water samples taken from various locations, including municipal taps, lakes, and rivers. Before the experiment, each of the water samples was filtered through a filter paper (pore size: 0.42 μm) to remove the suspended and dispersed particles. The overall degradation performance was compared with that in DI water. Specifically, for tap water, river water and lake water, 99%, 95% and 97.5% CBZ degradation was obtained (with kapp: 0.075 min−1, 0.05 min−1 and 0.063 min−1, respectively) compared to 99.8% CBZ removal (kapp: 0.089 min−1) for DI water. For all cases, the final residual concentration of CBZ was lower than the permissible limit of CBZ in treated effluent water, i.e., 0.04–0.1 mg L−1.71,72 The slight to moderate inhibition in real-life water matrixes directly demonstrates the scavenging effect of various anions and organics in water (Table S1† showing the detailed physiochemical properties of the water samples). Thus, the above-mentioned study proved that CBZ-contaminated real-life surface water and groundwater can be suitably treated with the residual CBZ concentration within the permissible limit. Therefore, with the appropriate doses of catalyst and PMS, the AMIL(10)@MIL/PMS/vis combination is effective for CBZ removal in real-water matrixes.
The AMIL(10)@MIL catalyst achieved quite higher TOC removal (∼71%) compared to the other combinations, i.e., 7.5% for PMS/vis and 9.7% for AMIL(10)@MIL/vis (Fig. 3e). However, its degree of mineralization was lower than the corresponding degradation (>99%), indicating the production of numerous smaller intermediates, which contribute to the total TOC content.
4. Mechanism of PMS activation and removal of CBZ in the AMIL(10)@MIL/PMS system
4.1. Identification of reactive species
Traditional radical scavenging studies were conducted to ascertain the relative relevance of different reactive radicals in the medium,73 followed by EPR analysis. The results are shown in Fig. 3f and S4(a–d),† respectively. Among the different scavengers, each at a concentration of 5 mM, methanol showed the maximum inhibition, with 28% CBZ degradation (kapp: 7.0 × 10−3 min−1), implying the strong contribution of the ˙OH and SO4˙− radicals (due to the presence of three α-H atoms, methanol can effectively scavenge ˙OH and SO4˙− radicals with k˙OH/MeOH = 1 × 109 M−1 s−1 and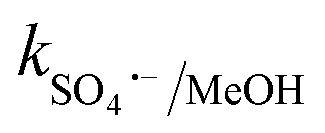 = 1.1 × 107 M−1 s−1), respectively.70 After methanol, L-histidine (His) showed high inhibition, with 41.8% CBZ removal (kapp: 0.013 min−1). L-Histidine is a strong scavenger of ˙OH, SO4˙− and O21 with k˙OH/His = 4.8 × 109 M−1 s−1,
= 1.1 × 107 M−1 s−1), respectively.70 After methanol, L-histidine (His) showed high inhibition, with 41.8% CBZ removal (kapp: 0.013 min−1). L-Histidine is a strong scavenger of ˙OH, SO4˙− and O21 with k˙OH/His = 4.8 × 109 M−1 s−1, 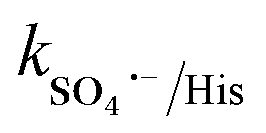 = 2.5 × 109 M−1 s−1 and
= 2.5 × 109 M−1 s−1 and 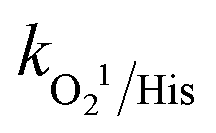 = 1.5 × 107 M−1 s−1, implying the strong contributions of ˙OH and SO4˙− radicals towards CBZ degradation, respectively.23 Also, the selective ˙OH removal by tert-butyl alcohol (TBA) and tri-ethanol amine (TEA) (with no α-H atoms in each case) and the corresponding CBZ removal (49% and 47.4% with kapp: 0.016 min−1 and 0.015 min−1) imply the dominance of SO4˙− compared to ˙OH in the medium, given that both TBA and TEA can preferentially scavenge ˙OH (k˙OH/TBA = 5.9 ± 0.5 × 108 M−1 s−1, k˙OH/TEA = 7.2 × 108 M−1 s−1) over SO4˙− (
= 1.5 × 107 M−1 s−1, implying the strong contributions of ˙OH and SO4˙− radicals towards CBZ degradation, respectively.23 Also, the selective ˙OH removal by tert-butyl alcohol (TBA) and tri-ethanol amine (TEA) (with no α-H atoms in each case) and the corresponding CBZ removal (49% and 47.4% with kapp: 0.016 min−1 and 0.015 min−1) imply the dominance of SO4˙− compared to ˙OH in the medium, given that both TBA and TEA can preferentially scavenge ˙OH (k˙OH/TBA = 5.9 ± 0.5 × 108 M−1 s−1, k˙OH/TEA = 7.2 × 108 M−1 s−1) over SO4˙− (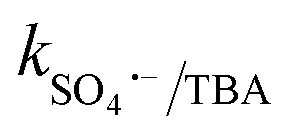 = 8.0 × 105 M−1 s−1,
= 8.0 × 105 M−1 s−1, 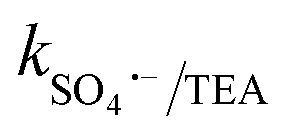 = 4.5 × 106 M−1 s−1), respectively.23 Additionally, p-benzoquinone (pBQ) and EDTA-2Na exhibited potent inhibitory actions (56% and 62% CBZ removal with kapp: 0.018 min−1 and 0.019 min−1, respectively). This finding implies that O2˙− and h+ are also highly active in CBZ degradation, together with the dominance of ˙OH and SO4˙− radicals. Lastly, NaN3 was used to determine the contribution of O21, together with ˙OH and SO4˙− radicals, and the strong inhibition (44.3% CBZ removal with kapp: 0.0138 min−1) indicates the presence of excess ˙OH and SO4˙−, together with O21 given that NaN3 can scavenge ˙OH, SO4˙− and O21 with
= 4.5 × 106 M−1 s−1), respectively.23 Additionally, p-benzoquinone (pBQ) and EDTA-2Na exhibited potent inhibitory actions (56% and 62% CBZ removal with kapp: 0.018 min−1 and 0.019 min−1, respectively). This finding implies that O2˙− and h+ are also highly active in CBZ degradation, together with the dominance of ˙OH and SO4˙− radicals. Lastly, NaN3 was used to determine the contribution of O21, together with ˙OH and SO4˙− radicals, and the strong inhibition (44.3% CBZ removal with kapp: 0.0138 min−1) indicates the presence of excess ˙OH and SO4˙−, together with O21 given that NaN3 can scavenge ˙OH, SO4˙− and O21 with 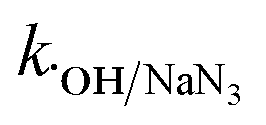 = 1.2 × 1010 M−1 s−1,
= 1.2 × 1010 M−1 s−1, 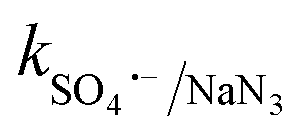 = 2.4 × 109 M−1 s−1 and
= 2.4 × 109 M−1 s−1 and 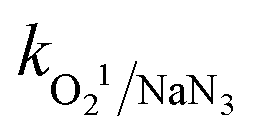 = 1.8 × 108 M−1 s−1, respectively.23 A similar conclusion was obtained from the corresponding EPR analysis, which implies that O2˙−, SO4˙− and ˙OH predominantly contribute towards the degradation of CBZ, with moderate contribution from h+. A detailed discussion is provided in Section S2 of the ESI.†
= 1.8 × 108 M−1 s−1, respectively.23 A similar conclusion was obtained from the corresponding EPR analysis, which implies that O2˙−, SO4˙− and ˙OH predominantly contribute towards the degradation of CBZ, with moderate contribution from h+. A detailed discussion is provided in Section S2 of the ESI.†
4.2. Activation mechanism
XPS analysis was performed to study the oxidation states and surface-functionalities of the initial and reacted AMIL(10)@MIL composites, as well as the associated charge transfer during the reaction (Fig. 4(a–g)). The peaks of the different constituent elements, namely, Fe, Co, Ti, C, N, and O, were evident in the survey spectra of both the initial and reacted AMIL(10)@MIL (Fig. 4a). The core-level peaks in the Co2p, Fe2p and Ti2p spectra of the initial AMIL(10)@MIL shifted slightly to higher binding energies compared to the reacted AMIL(10)@MIL (Fig. 4(b–d)). These results imply the slight interconversion of Co2+|surf. (Fe2+|surf.) into Co3+|surf.(Fe3+|surf.) and involvement of Co2+/Co3+|surf. and Fe2+/Fe3+|surf. in the charge transfer process during the reaction, together with the generation of the Ti3+/Ti4+|surf. redox pair. Therefore, Co2+/Co3+|surf., Fe2+/Fe3+|surf. and Ti3+/Ti4+|surf. are involved in the e−-transfer process from the metal nodes to the adsorbed HSO5− ions, generating SO4˙− and ˙OH radicals. In addition, the comparison of the deconvoluted C1s and N1s spectra of the initial and reacted AMIL(10)@MIL implies that charge transfer occurs during the reaction (Fig. 4(e and f)). Finally, three peaks were fitted in the O1s spectrum of the initial (reacted) composite, corresponding to lattice oxygen (OL, M–O, M: Fe, Co and Ti), framework oxygen (OF, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and –COO) and adsorbed oxygen (Oa, from adsorbed H2O and generated hydroxyl intermediates), respectively (Fig. 4g). The relatively higher amount of surface-bound hydroxyl moieties in the reacted composite implies the generation of transient, high-spin, metal-hydroxyl intermediates during the reaction.
O, and –COO) and adsorbed oxygen (Oa, from adsorbed H2O and generated hydroxyl intermediates), respectively (Fig. 4g). The relatively higher amount of surface-bound hydroxyl moieties in the reacted composite implies the generation of transient, high-spin, metal-hydroxyl intermediates during the reaction.
 | ||
| Fig. 4 (a) Survey spectra and XPS core-level spectra of (b) Co2p, (c) Fe2p, (d) Ti2p, (e) C1s, (f) N1s, and (g) O1s of the initial and reacted AMIL(10)@MIL composites. | ||
Based on the results of the XPS analysis and radical identification experiments, the detailed e− transport and PMS activation mechanism is proposed. It is widely considered that the formation of transient metal hydroxyl (M–OH) complexes, such as Co–OH, Fe–OH, and Ti–OH, is the key step in the activation of PMS and corresponding generation of ROS in the medium.74 In the case of the heterogeneous activation of PMS, the surface-bound metal ions (initial AMIL(10)@MIL, together with a small amount of Fe2O3 and Co3O4 generated on the surface) capture H2O molecules to form [Co(II/III)–(OH)]+/2+, [Fe(II/III)–(OH)]+/2+ and [Ti(III/IV)–(OH)]2+/3+ complexes, which react with the adsorbed HSO5− ions to form [Co(III)–(OH)(OSO3−)]+, [Fe(III)–(OH)(OSO3−)]+ and [Ti(IV)–(OH)(OSO3−)]2+. Subsequently, these high-spin intermediates generate SO4˙− and ˙OH through e− transfer from the metal nodes to the adsorbed PMS moieties. Meanwhile, Co(III), Fe(III) and Ti(IV) are reduced to Co(II), Fe(II) and Ti(III), and the regeneration steps of Fe(II) and Ti(III) involve the Co(III)/Co(II) redox pair and O2˙− radicals in the medium (eqn (S46) and (S47) and (S68) and (S69)†), respectively. The lower standard oxidation potentials of Ti(IV)/Ti(III) (E0: 0.95 V vs. NHE) and Fe(III)/Fe(II) (E0: 0.71 V vs. NHE) do not allow the regeneration step to occur in the presence of only HSO5− ions (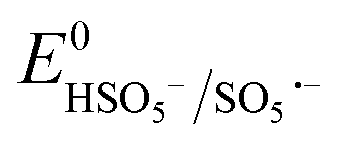 = 1.1 V,
= 1.1 V, 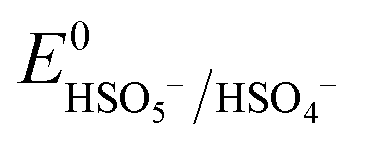 = 1.78 V and
= 1.78 V and 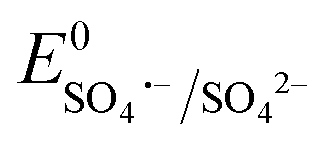 = 2.5–3.1 V vs. NHE), respectively. Therefore, Co(III)/Co(II) (E0: 1.91 V vs. NHE) and O2˙− (E0: −0.33 V vs. NHE) are gradually consumed during the reaction. The regenerated Co(II), Fe(II) and Ti(III) again become involved in the e− transfer from HSO5− to generate SO4˙− radicals, which subsequently generate ˙OH in the medium. Also, the adsorbed CBZ molecules can donate e− to the metal centers via hydroxyl complex synthesis (e− bridge production), leaving them progressively e−-deficient and vulnerable to radical (O2˙−, SO4˙− and ˙OH) electrophilic attack.
= 2.5–3.1 V vs. NHE), respectively. Therefore, Co(III)/Co(II) (E0: 1.91 V vs. NHE) and O2˙− (E0: −0.33 V vs. NHE) are gradually consumed during the reaction. The regenerated Co(II), Fe(II) and Ti(III) again become involved in the e− transfer from HSO5− to generate SO4˙− radicals, which subsequently generate ˙OH in the medium. Also, the adsorbed CBZ molecules can donate e− to the metal centers via hydroxyl complex synthesis (e− bridge production), leaving them progressively e−-deficient and vulnerable to radical (O2˙−, SO4˙− and ˙OH) electrophilic attack.
Additionally, the e−-mediated mechanism can also be active during lattice-bridged e− transfer from the adsorbed CBZ molecules to the surface metal ions. Thus, in this case, an e− bridge configuration is formed between the adsorbed CBZ (e− donor) and catalyst (e− acceptor). However, the contribution due to this non-radical-based pathway is very weak, as can be explained from the strong effects of the different ions in the medium. Therefore, the radical-based inner-sphere mechanism is the primary contributor to the transfer of e− from the transient, high-spin intermediates to the adsorbed HSO5− ions. Hence, for each metal redox involved, the adsorbed HSO5− ions acquire two e−, i.e., one from each of the Fe2+/Fe3+|surf., Co2+/Co3+|surf. and Ti3+/Ti4+|surf., followed by another e− from the adsorbed CBZ molecules to generate SO4˙− in the medium. Consequently, a large amount of ROS is generated, which can further attack the e−-deficient CBZ molecules on the catalyst surface to degrade them into smaller intermediates. The relevant equations for PMS activation and ROS generation are presented as eqn (S42)–(S73) in the ESI.†
5. Degradation pathway of CBZ
Based on the LCMS analysis of different intermittently collected CBZ samples during the degradation process, a total of 31 intermediates was identified. The chemical formulas, molecular structures and m/z ratios are presented in Table S2,† together with the LCMS results (Fig. S5(a–t)†). Fig. 5 depicts the predicted CBZ degradation route. The CBZ molecule has an uneven electronic distribution and based on the Fukui electrophilic (f−) and radical attack (f0) indices,74,75 the most susceptible/reactive sites of the CBZ molecule were identified as the ‘N’ site and the C10–C11 carbon atoms of the seven-membered heterocyclic ring with an olefinic double bond. Therefore, the O2˙−, SO4˙− and ˙OH radicals in the medium can attack the CBZ molecule through three pathways, namely, deamidation, hydroxylation and e− transfer. Firstly, in pathway-1, direct attack of the ROS on the N atom can remove the –CONH2 group and form intermediate P(1,1) (m/z: 193.05), which is further attacked at its C10–C11 double bond to form unstable epoxide intermediate P(1,2) (m/z: 209.1).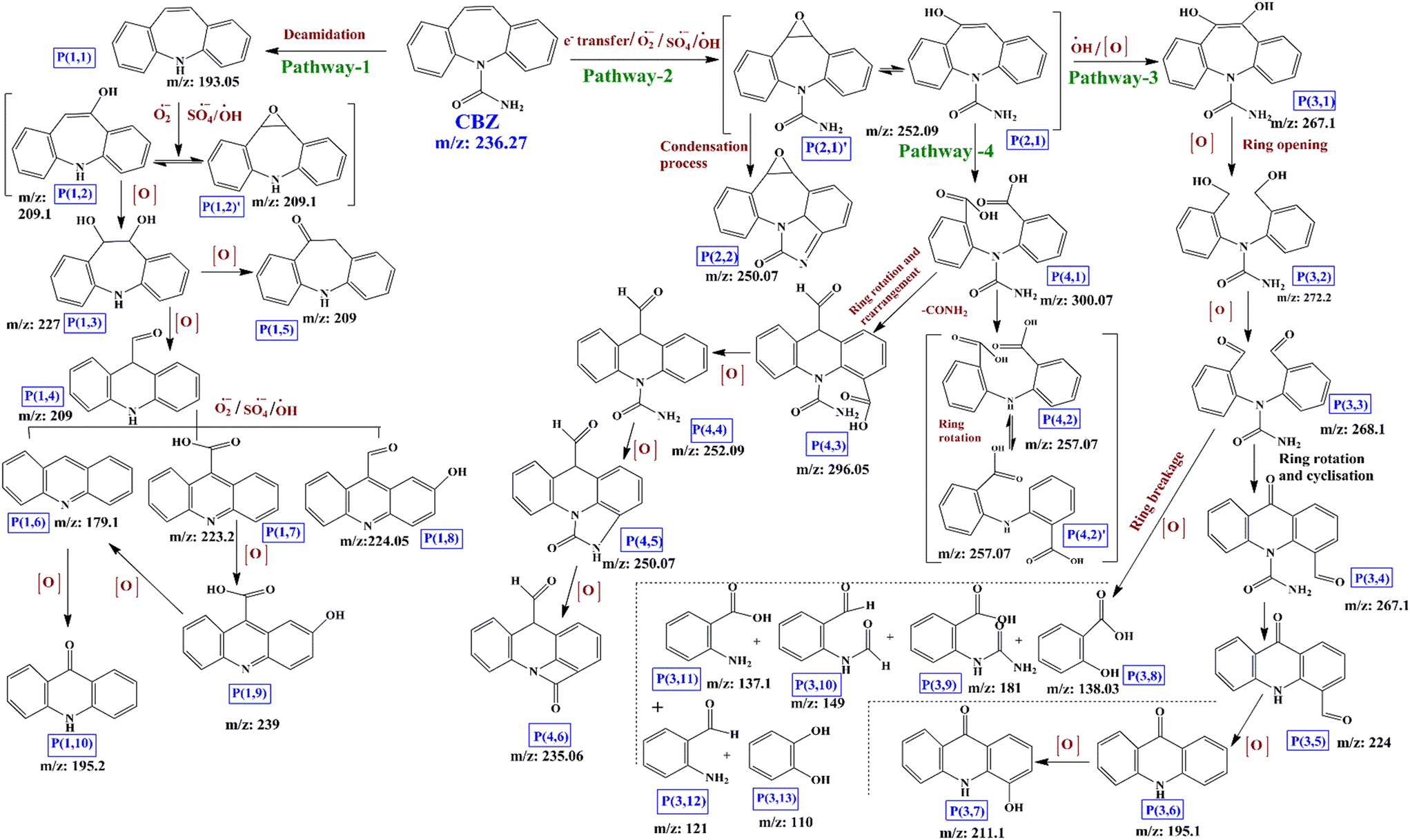 | ||
| Fig. 5 Degradation pathway of CBZ in AMIL(10)@MIL/PMS/vis system. | ||
This intermediate exists in dynamic equilibrium with the corresponding hydroxide P′(1,2) (m/z: 209.1). Further hydroxylation of P(1,2)/P′(1,2) leads to dihydroxyl intermediate P(1,3) (m/z: 227), which can undergo heterocyclic ring opening and further rearrangement to form intermediate P(1,4) (m/z: 209.0). In parallel, P(1,3) can undergo dehydroxylation and ketonation to form P(1,5) (m/z: 209.0). Intermediate P(1,4) can be further oxidized and hydroxylated to generate P(1,6) (m/z: 179.1), P(1,7) (m/z: 223.2) and P(1,8) (m/z: 224.05). Next, hydroxyl addition and further rearrangement of P(1,6) generate P(1,10) (m/z: 195.2). Further, P(1,7) can be hydroxylated to form intermediate P(1,9) (m/z: 239.0), which again undergoes decarboxylation to form P(1,6) (m/z: 179.1). Alternatively, SO4˙− radicals can react with organic molecules through a single e− transfer process. Therefore, in pathway-2, the attack of the CBZ molecule by SO4˙− at its C10–C11 sites will initially generate a transient carbon–carbon free radical cation, which can be subsequently attacked by the O2˙− radicals to form epoxide intermediates P′(2,1) and P(2,1) (m/z: 252.09), which remain in dynamic equilibrium. Ring closure of intermediate P′(2,1) through the –CONH2 group on the aromatic ring will generate intermediate P(2,2) (m/z: 250.07). Next, hydroxide intermediate P(2,1) can be further hydroxylated through pathway-3 to generate P(3,1) (m/z: 267.1). Further, oxidation leads to breakage of the C10–C11 bond to generate P(3,2) (m/z: 272.2), and the corresponding aldehyde P(3,3) (m/z: 268.1). Now, the second highest reactivity of the ortho C-atom of the N-atom leads to N–C bond rotation and further cyclization generates intermediate P(3,4) (m/z: 267.1), which can be deamidated into P(3,5) (m/z: 224). Further oxidation of P(3,5) leads to the formation of P(3,6) (m/z: 195.1) through the loss of an aldehyde group. P(3,6) can produce P(3,7) (m/z: 211.1) through hydroxylation. Simultaneously, P(2,1) can undergo ring breakage to generate dicarboxylic acid intermediate P(4,1) (m/z: 300.07), which loses the –CONH2 group to generate isomeric intermediates P(4,2) and P′(4,2) (m/z: 257.07). Alternatively, C–N bond rotation and further ring closure, deamidation and N-ring cyclization lead to intermediates P(4,3) (m/z: 296.05), P(4,4) (m/z: 252.09), P(4,5) (m/z: 250.07) and P(4,6) (m/z: 235.06). Intermediates P(1,6), P(1,10) and P(3,3) are further degraded into smaller molecules, including P(3,8) (m/z: 138.03), P(3,9) (m/z: 181.0), P(3,10) (m/z: 149.0), P(3,11) (m/z: 137.1), P(3,12) (m/z: 121.0) and P(3,13) (m/z: 110.0). These are intermediates are further mineralized into CO2, H2O, NH4+ and other lower molecular weight compounds, which are less toxic than the CBZ molecule.
6. Comparative study
A detailed comparative study was undertaken to evaluate the usefulness of the developed catalytic system with respect to other recently developed materials. Table S3 in the ESI† provides a summary of the results. In all cases, the required catalyst dose was higher than that needed for AMIL(10)@MIL (i.e., 0.05 g L−1) to remove >99.9% CBZ in water, with a rate constant (kapp) of 0.089 min−1.76–81 The kinetics of CBZ degradation is comparable with the reported catalytic processes. Moreover, all the other systems employ either conventional catalysts (i.e., spinel metal oxides and metal chalcogenides) or biomaterial-derived catalysts, which require high temperature annealing for their synthesis. Moreover, MOF@MOF-based composite photocatalysts have rarely been employed towards catalytic water decontamination. Therefore, the present study demonstrates several merits compared to reported catalytic systems towards the mineralization of CBZ in aqueous medium.The efficiency of the AMIL(10)@MIL/PMS/vis combination towards the degradation of other emerging contaminants was also investigated using tetracycline (TC) and rhodamine-B (RhB) as model pollutants. The optimized dose of AMIL(10)@MIL (0.05 g L−1) and PMS (0.25 g L−1) was employed towards the removal of TC (C0: 25 mg L−1) and RhB (C0: 100 mg L−1), under visible light irradiation and the variation in the corresponding residual concentration is shown in Fig. S3g.† The kinetics of RhB degradation was faster (kapp: 0.114 s−1) than that of TC (kapp: 0.078 s−1) and CBZ (kapp: 0.089 s−1). Compared to 99.3% and 99.8% degradation of TC and CBZ, under 1 h of visible light irradiation, complete (100%) RhB removal was obtained, respectively. Therefore, the AMIL(10)@MIL/PMS/vis system was remarkably effective towards excellent removal of different emerging contaminants from aqueous medium.
7. Regeneration and stability of the catalyst
The reacted AMIL(10)@MIL catalyst particles were collected from the reaction medium and further analyzed using FESEM and XRD. In parallel, the collected particles from each cycle were subjected to the regeneration process involving multiple washings using DI water and ethanol, followed by drying, and further employed in the next set of degradation cycles. The removal of CBZ in each cycle is shown in Fig. 6a. Up to the 5th degradation cycle, 83.4% CBZ removal was obtained. This gradual loss in catalytic activity can be attributed to the sequential deposition of external moieties on the catalyst surface, blocking its active sites and pore structure. Moreover, with multiple reuses, the constituent metal ions were slightly leached from the catalyst structure. As a combined effect, the overall charge transfer and redox processes were affected during the degradation reaction, together with the adsorption and diffusive transport of the reactants inside the catalyst. In addition, the morphological and crystallographic analysis of the reacted catalyst exhibited no distinct variations, indicating the structural integrity of the material during the reaction. In the XRD spectra, the slight variation and lowering of the peak intensities imply the gradual adsorption of organic molecules on the catalyst surface and subsequent loss of surface-bound metal atoms due to leaching (Fig. 6(b–d)). Moreover, the concentration of leached Co and Fe, after each catalytic cycle, was measured using AAS and the profiles are shown in Fig. 6e. In both cases, the leached ion concentration gradually decreased with cycles. Nonetheless, the initial and final leached concentration of Co (1st: 0.048 mg L−1 and 5th: 0.01 mg L−1) and Fe (1st: 0.27 mg L−1 and 5th: 0.1 mg L−1) were lower than their corresponding permissible limits, i.e., 0.05 mg L−1 for Co and 0.3 mg L−1 for Fe, respectively.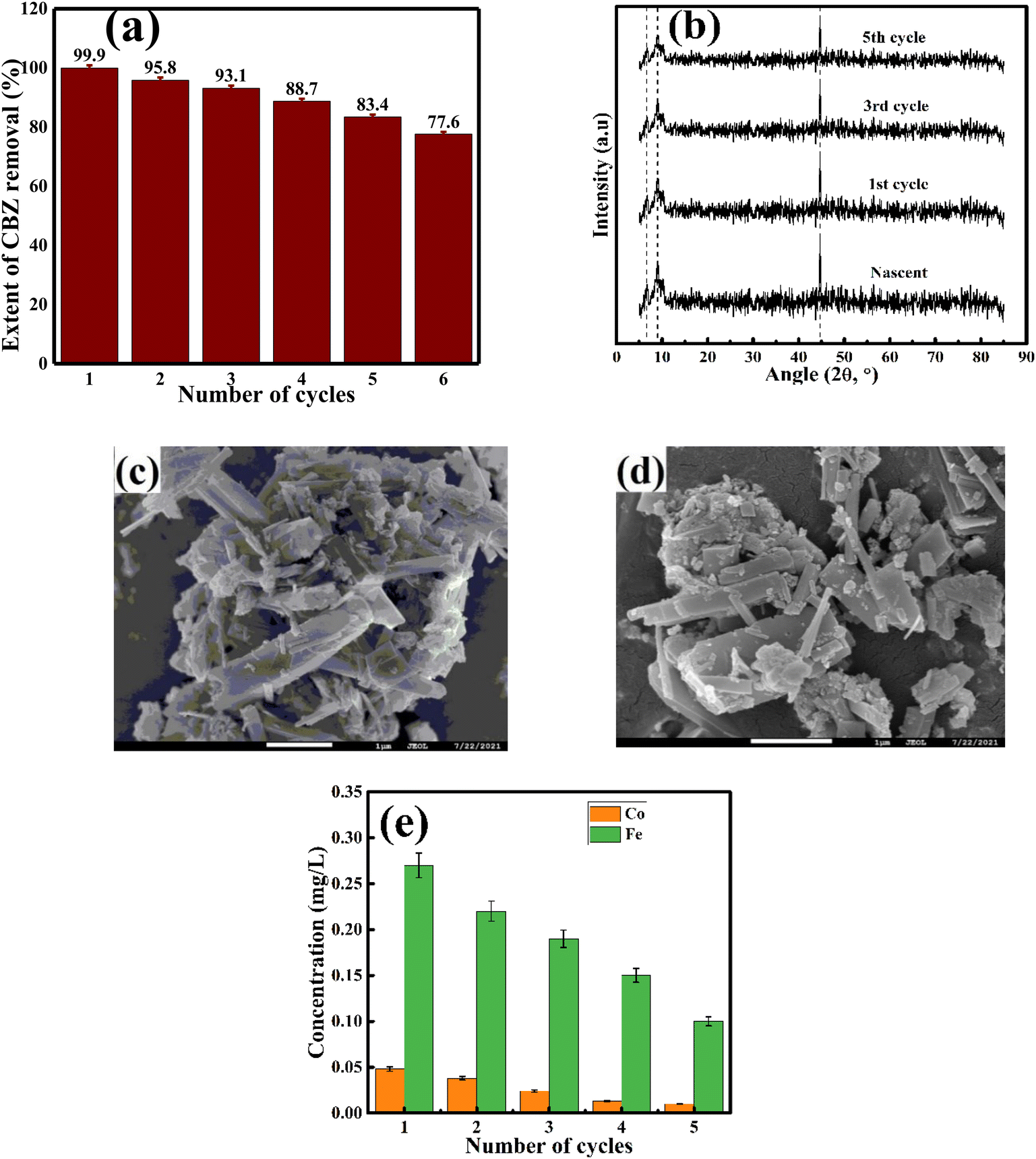 | ||
| Fig. 6 (a) Regeneration cycle analysis. (b) XRD spectra of reused composites. FESEM analysis of (c) fresh and (d) reused AMIL(10)@MIL composite. (e) Variation in the concentration of leached metals (Fe, Co) with regeneration cycles. | ||
8. Conclusion
NH2-MIL125(Ti) was hydrothermally grown on MIL-53(Fe/Co) to generate the AMIL@MIL composite for the activation of PMS and degradation of CBZ under visible light. The efficient separation of photoexcited e−/h+ pairs in the direct Z-scheme configuration of AMIL(10)@MIL and generation of SO4˙−, ˙OH and O2˙− resulted in the excellent degradation of CBZ. Specifically, 99.9% CBZ degradation (together with ∼71% mineralization) was obtained in presence of 0.05 g L−1 AMIL(10)@MIL and 0.025 g L−1 PMS within 1 h visible light exposure at neutral pH (6–8). HA, HPO42− and HCO3− induced moderate inhibition due to the strong radical quenching, pH buffering and inner filter effects. A large number of intermediates was detected in the LCMS analysis, and the corresponding probable degradation pathway of CBZ proposed. Three mechanisms, i.e., deamidation, hydroxylation and e− transfer, were involved in the mineralization of the CBZ molecule. Also, 83.4% CBZ was removed after 5 consecutive degradation cycles, with marginal morphological and crystallographic variation in the composite. The AMIL(10)@MIL/PMS/vis combination together with negligible leaching of the constituent metals, coupled with excellent CBZ removal potential can be recommended as a viable option for the removal of emerging micropollutants from the contaminated surface water.Nomenclature
Alphabetic symbols
| A | Band tailing parameter |
| h | Planck constant |
| E g | Bandgap energy (eV) |
| E CB | Conduction band potential (eV) |
| E VB | Valence band potential (eV) |
| T | Absolute temperature (K) |
| t | Time (h) |
Abbreviation
| AMIL | Aminated MIL-125(Ti) |
| AOP | Advanced oxidation processes |
| CB | Conduction band |
| CBZ | Carbamazepine |
| DMF | Dimethyl formamide |
| EDC | Endocrine disrupting chemicals |
| HA | Humic acid |
| His | L-Histidine |
| MOF | Metal organic framework |
| NHE | Normal hydrogen electrode |
| NOM | Natural organic matter |
| pBQ | para-Benzoquinone |
| PMS | Peroxymonosulfate |
| PPCP | Pharmaceuticals and personal care products |
| ROS | Reactive oxygen species |
| TBA | tert-Butyl alcohol |
| TOC | Total organic carbon |
| VB | Valence band |
Greek symbols
| α | Absorption coefficient |
| ν | Irradiation frequency |
Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
This work is supported by a grant from the Department of Science and Technology, Government of India, under the scheme no. DST/TM/WTI/WIC/2K17/84(G), Dt. 15-01-2019. Any opinions, findings and conclusions expressed in this paper are those of the authors. The authors would also like to acknowledge Dr. Poulomi Sarkar for helping with the LCMS/MS analysis and insightful discussion.References
- S. Fan, J. Shang, S. Kulan, X. He, X. Wang, B. Nasen, J. Nie, D. Feng and X. Cheng, Enhanced degradation of carbamazepine in water over SC-modified NiFe2S4 nanocomposites by peroxymonosulfate activation, Chem. Eng. J., 2022, 450, 138190 CrossRef CAS.
- Z. Cheng, L. Ling, J. Fang and C. Shang, Visible light-driven g-C3N4 peroxymonosulfate activation process for carbamazepine degradation: Activation mechanism and matrix effects, Chemosphere, 2022, 286, 131906 CrossRef CAS PubMed.
- Y. Hong, H. Zhou, Z. Xiong, Y. Liu, G. Yao and B. Lai, Heterogeneous activation of peroxymonosulfate by CoMgFe-LDO for degradation of carbamazepine: Efficiency, mechanism and degradation pathways, Chem. Eng. J., 2020, 391, 123604 CrossRef CAS.
- J. Wang, L. Chu, L. Wojnárovits and E. Takács, Occurrence and fate of antibiotics, antibiotic resistant genes (ARGs) and antibiotic resistant bacteria (ARB) in municipal wastewater treatment plant: An overview, Sci. Total Environ., 2020, 744, 140997 CrossRef CAS.
- J. Cui, Y. Pei, J. Hou, Y. Zheng, Y. Zhang, K. Wei, X. He and B. Xi, Construction of a carbon dots-based Z-scheme photocatalytic electrode with enhanced visible-light-driven activity for Cr(VI) reduction and carbamazepine degradation in different reaction systems, Chem. Eng. J., 2021, 420, 127571 CrossRef CAS.
- J. Cui, Y. Zhang, Y. Zheng, Y. Pei, X. He and B. Xi, Catalytic degradation of carbamazepine by a novel granular visible-light-driven photocatalyst activating the peroxydisulfate system, Chem. Eng. J., 2021, 421, 127867 CrossRef CAS.
- Q.-T. Sun, B.-D. Xu, J. Yang, T.-T. Qian and H. Jiang, Layered oxides supported Co-Fe bimetal catalyst for carbamazepine degradation via the catalytic activation of peroxymonosulfate, Chem. Eng. J., 2020, 400, 125899 CrossRef CAS.
- J. Wang and S. Wang, Removal of pharmaceuticals and personal care products (PPCPs) from wastewater: A review, J. Environ. Manage., 2016, 182, 620–640 CrossRef CAS PubMed.
- O. Primo, M. J. Rivero and I. Ortiz, Photo-Fenton process as an efficient alternative to the treatment of landfill leachates, J. Hazard. Mater., 2008, 153, 834–842 CrossRef CAS.
- J. Wang and R. Zhuan, Degradation of antibiotics by advanced oxidation processes: An overview, Sci. Total Environ., 2020, 701, 135023 CrossRef CAS.
- J. L. Wang and L. J. Xu, Advanced Oxidation Processes for Wastewater Treatment: Formation of Hydroxyl Radical and Application, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251–325 CrossRef CAS.
- J. Tang and J. Wang, Metal Organic Framework with Coordinatively Unsaturated Sites as Efficient Fenton-like Catalyst for Enhanced Degradation of Sulfamethazine, Environ. Sci. Technol., 2018, 52, 5367–5377 CrossRef CAS.
- J. Wang and J. Tang, Fe-based Fenton-like catalysts for water treatment: Preparation, characterization and modification, Chemosphere, 2021, 276, 130177 CrossRef CAS PubMed.
- J. Tang and J. Wang, Iron-copper bimetallic metal-organic frameworks for efficient Fenton-like degradation of sulfamethoxazole under mild conditions, Chemosphere, 2020, 241, 125002 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao and J. Wang, Fenton/Fenton-like processes with in situ production of hydrogen peroxide/hydroxyl radical for degradation of emerging contaminants: Advances and prospects, J. Hazard. Mater., 2021, 404, 124191 CrossRef CAS PubMed.
- J. Wang and H. Chen, Catalytic ozonation for water and wastewater treatment: Recent advances and perspective, Sci. Total Environ., 2020, 704, 135249 CrossRef CAS PubMed.
- T. Olmez-Hanci and I. Arslan-Alaton, Comparison of sulfate and hydroxyl radical based advanced oxidation of phenol, Chem. Eng. J., 2013, 224, 10–16 CrossRef CAS.
- P. Hu and M. Long, Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- W. Da Oh, Z. Dong and T.-T. Lim, Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects, Appl. Catal., B, 2016, 194, 169–201 CrossRef.
- C. Liu, L. Chen, D. Ding and T. Cai, Sulfate radical induced catalytic degradation of metolachlor: Efficiency and mechanism, Chem. Eng. J., 2019, 368, 606–617 CrossRef CAS.
- J. Wang and S. Wang, Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- Y. Peng, H. Tang, B. Yao, X. Gao, X. Yang and Y. Zhou, Activation of peroxymonosulfate (PMS) by spinel ferrite and their composites in degradation of organic pollutants: A Review, Chem. Eng. J., 2021, 414, 128800 CrossRef CAS.
- J. Lee, U. von Gunten and J.-H. Kim, Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS.
- C. Qi, G. Yu, J. Huang, B. Wang, Y. Wang and S. Deng, Activation of persulfate by modified drinking water treatment residuals for sulfamethoxazole degradation, Chem. Eng. J., 2018, 353, 490–498 CrossRef CAS.
- J. Wang and S. Wang, Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- G. Wang, D. Huang, M. Cheng, S. Chen, G. Zhang, L. Du, L. Lei, Y. Chen, R. Li and Y. Liu, An insight into the bridging role of Co3O4 in MOF-derived binary metal oxide modified sheet-like g-C3N4 for photo-assisted peroxymonosulfate activation, Environ. Sci.: Nano, 2022, 9, 4393–4410 RSC.
- J. Li, J. Wei, M. Xu, G. Pan, Y. Zhang, L. Xing, Y. Li, J. Li and Z. Jiang, A porous graphitic biochar wrapped Co9S8 core–shell composite enables pH-universal activation of peroxymonosulfate for highly efficient and rapid antibiotics degradation, Environ. Sci.: Nano, 2022, 9, 3629–3645 RSC.
- M. Li, Z. He, H. Zhong, L. Hu and W. Sun, Oxygen vacancy enhances the catalytic activity of trimetallic oxide catalysts for efficient peroxymonosulfate activation, Environ. Sci.: Nano, 2022, 9, 1037–1051 RSC.
- X. Jiang, W. Yan, Z. Xiong and L. Zhao, A highly dispersed magnetic polymetallic catalyst to activate peroxymonosulfate for the degradation of organic pollutants in wastewater, Environ. Sci.: Nano, 2022, 9, 2939–2953 RSC.
- S. Liu, B. Jing, C. Nie, Z. Ao, X. Duan, B. Lai, Y. Shao, S. Wang and T. An, Piezoelectric activation of peroxymonosulfate by MoS2 nanoflowers for the enhanced degradation of aqueous organic pollutants, Environ. Sci.: Nano, 2021, 8, 784–794 RSC.
- Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, From molecular metal complex to metal-organic framework: The CO2 reduction photocatalysts with clear and tunable structure, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS.
- D. Roy, S. Neogi and S. De, Mechanistic investigation of photocatalytic degradation of Bisphenol-A using MIL-88A(Fe)/MoS2 Z-scheme heterojunction composite assisted peroxymonosulfate activation, Chem. Eng. J., 2022, 428, 131028 CrossRef CAS.
- D. Roy, S. Neogi and S. De, Degradative removal of Sulfamethoxazole through visible light driven peroxymonosulfate activation by direct Z-scheme MIL-53(Co/Fe)/MoS2 heterojunction composite: Role of dual redox mechanism and efficient charge separation, Process Saf. Environ. Prot., 2022, 161, 723–738 CrossRef CAS.
- D. Roy, S. Neogi and S. De, Visible light assisted activation of peroxymonosulfate by bimetallic MOF based heterojunction MIL-53(Fe/Co)/CeO2 for atrazine degradation: Pivotal roles of dual redox cycle for reactive species generation, Chem. Eng. J., 2022, 430, 133069 CrossRef CAS.
- T. Zhang and W. Lin, Metal–organic frameworks for artificial photosynthesis and photocatalysis, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- H. Luo, Z. Zeng, G. Zeng, C. Zhang, R. Xiao, D. Huang, C. Lai, M. Cheng, W. Wang, W. Xiong, Y. Yang, L. Qin, C. Zhou, H. Wang, Y. Zhou and S. Tian, Recent progress on metal-organic frameworks based- and derived-photocatalysts for water splitting, Chem. Eng. J., 2020, 383, 123196 CrossRef CAS.
- D. A. Reddy, Y. Kim, M. Gopannagari, D. P. Kumar and T. K. Kim, Recent advances in metal–organic framework-based photocatalysts for hydrogen production, Sustainable Energy Fuels, 2021, 5, 1597–1618 RSC.
- J. Du, F. Li and L. Sun, Metal–organic frameworks and their derivatives as electrocatalysts for the oxygen evolution reaction, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, Graphitic Carbon Nitride: Synthesis, Properties, and Applications in Catalysis, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Graphitic Carbon Nitride (g-C 3 N 4 )-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability?, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- T. Li, P. Cui, X. Wang, C. Liu, Y. Zeng, G. Fang, Y. Zhao, J. Gao, Y. Wang and D. Zhou, Efficient activation of peroxymonosulfate by C3N5 doped with cobalt for organic contaminant degradation, Environ. Sci.: Nano, 2022, 9, 2534–2547 RSC.
- X. Xie, R. Xie, Z. Suo, H. Huang, M. Xing and D. Lei, A highly dispersed Co–Fe bimetallic catalyst to activate peroxymonosulfate for VOC degradation in a wet scrubber, Environ. Sci.: Nano, 2021, 8, 2976–2987 RSC.
- C. Gu, Y. Zhang, P. He, J. Zhu and M. Gan, Insights into biochar supported atomically dispersed cobalt as an efficient peroxymonosulfate activator for sulfamethoxazole degradation: robust performance, ROS and surface electron-transfer pathways, Environ. Sci.: Nano, 2022, 9, 3551–3561 RSC.
- C. Guo, M. Cheng, G. Zhang, W. Xiong, C. Zhou, B. Song, L. Du, L. Li, C. Tang, G. Wang and H. Liu, Degradation of organic contaminants by peroxymonosulfate activated with Zeolitic imidazolate frameworks-based catalysts: performances, mechanisms and stability, Environ. Sci.: Nano, 2023, 10, 1528–1552 RSC.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, Graphitic carbon nitride materials: variation of structure and morphology and their use as metal-free catalysts, J. Mater. Chem., 2008, 18, 4893 RSC.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Graphitic carbon nitride materials: controllable synthesis and applications in fuel cells and photocatalysis, Energy Environ. Sci., 2012, 5, 6717 RSC.
- S. Zhang, M. Du, J. Kuang, Z. Xing, Z. Li, K. Pan, Q. Zhu and W. Zhou, Surface-defect-rich mesoporous NH2-MIL-125 (Ti)@Bi2MoO6 core-shell heterojunction with improved charge separation and enhanced visible-light-driven photocatalytic performance, J. Colloid Interface Sci., 2019, 554, 324–334 CrossRef CAS PubMed.
- M. Govarthanan, R. Mythili, W. Kim, S. Alfarraj and S. A. Alharbi, Facile fabrication of (2D/2D) MoS2@MIL-88(Fe) interface-driven catalyst for efficient degradation of organic pollutants under visible light irradiation, J. Hazard. Mater., 2021, 414, 125522 CrossRef CAS PubMed.
- A. M. Omer, E. M. Abd El-Monaem, G. M. El-Subruiti, M. M. Abd El-Latif and A. S. Eltaweil, Fabrication of easy separable and reusable MIL-125(Ti)/MIL-53(Fe) binary MOF/CNT/Alginate composite microbeads for tetracycline removal from water bodies, Sci. Rep., 2021, 11, 23818 CrossRef CAS.
- A. Sharma, P. Negi, R. J. Konwar, H. Kumar, Y. Verma, Shailja, P. C. Sati, B. Rajyaguru, H. Dadhich, N. A. Shah and P. S. Solanki, Tailoring of structural, optical and electrical properties of anatase TiO2 via doping of cobalt and nitrogen ions, J. Mater. Sci. Technol., 2022, 111, 287–297 CrossRef CAS.
- M. Wang, L. Yang, J. Yuan, L. He, Y. Song, H. Zhang, Z. Zhang and S. Fang, Heterostructured Bi 2 S 3 @NH 2 -MIL-125(Ti) nanocomposite as a bifunctional photocatalyst for Cr(VI) reduction and rhodamine B degradation under visible light, RSC Adv., 2018, 8, 12459–12470 RSC.
- S. Xu, A. H. Habib, S. H. Gee, Y. K. Hong and M. E. McHenry, Spin orientation, structure, morphology, and magnetic properties of hematite nanoparticles, J. Appl. Phys., 2015, 117, 17A315 CrossRef.
- M. Qayoom, K. A. Shah, A. H. Pandit, A. Firdous and G. N. Dar, Dielectric and electrical studies on iron oxide (α-Fe2O3) nanoparticles synthesized by modified solution combustion reaction for microwave applications, J. Electroceram., 2020, 45, 7–14 CrossRef CAS.
- M. Hjiri, N. Zahmouli, K. Khouzami, L. El Mir, M. S. Aida, K. Moulaee, O. M. Lemine, S. G. Leonardi and G. Neri, A comparison of NO2 sensing characteristics of α- and γ-iron oxide-based solid-state gas sensors, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 788 CrossRef CAS.
- J. Chang, J. Sun, C. Xu, H. Xu and L. Gao, Template-free approach to synthesize hierarchical porous nickel cobalt oxides for supercapacitors, Nanoscale, 2012, 4, 6786 RSC.
- A. L. Bhatti, U. Aftab, A. Tahira, M. I. Abro, M. Kashif Samoon, M. H. Aghem, M. A. Bhatti and Z. HussainIbupoto, Facile doping of nickel into Co 3 O 4 nanostructures to make them efficient for catalyzing the oxygen evolution reaction, RSC Adv., 2020, 10, 12962–12969 RSC.
- G. E. M. Schukraft, B. Moss, A. G. Kafizas and C. Petit, Effect of Band Bending in Photoactive MOF-Based Heterojunctions, ACS Appl. Mater. Interfaces, 2022, 14, 19342–19352 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, An Amine-Functionalized Titanium Metal-Organic Framework Photocatalyst with Visible-Light-Induced Activity for CO2 Reduction, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. M. Spoor, K. Rock, L. D. A. Siebbeles, F. Kapteijn, F. Grozema, A. Houtepen, J. Gascon, J. Hunger and M. A. van der Veen, Organic Linker Defines the Excited-State Decay of Photocatalytic MIL-125(Ti)-Type Materials, ChemSusChem, 2016, 9, 388–395 CrossRef CAS.
- P. Sarkar, S. Neogi and S. De, Activation of peroxymonosulfate by S-scheme Bi2S3/doped gCN heterostructure photocatalyst for highly efficient visible light driven tetracycline degradation: Insights into reaction mechanisms, Sep. Purif. Technol., 2023, 308, 122908 CrossRef CAS.
- P. Sarkar, D. Roy, B. Bera, S. De and S. Neogi, Efficient photocatalytic degradation of ciprofloxacin using novel dual Z-scheme gCN/CuFe2O4/MoS2 mediated peroxymonosulphate activation, Chem. Eng. J., 2022, 430, 132834 CrossRef CAS.
- P. Sarkar, S. De and S. Neogi, Microwave assisted facile fabrication of dual Z-scheme g-C3N4/ZnFe2O4/Bi2S3 photocatalyst for peroxymonosulphate mediated degradation of 2,4,6-Trichlorophenol: The mechanistic insights, Appl. Catal., B, 2022, 307, 121165 CrossRef CAS.
- P. Sarkar, D. Roy, B. Bera, S. De and S. Neogi, Enhanced photodegradation of reactive dyes in textile effluent with CoFe2O4/g-CN heterostructure–mediated peroxymonosulphate activation, Environ. Sci. Pollut. Res., 2022, 29, 50566–50583 CrossRef CAS.
- P. Sarkar, S. Neogi and S. De, Accelerated radical generation from visible light driven peroxymonosulfate activation by Bi2MoO6/doped gCN S-scheme heterojunction towards Amoxicillin mineralization: Elucidation of the degradation mechanism, J. Hazard. Mater., 2023, 451, 131102 CrossRef CAS.
- P. Sarkar, S. Neogi and S. De, Activation of peroxymonosulfate by S-scheme Bi2S3/doped gCN heterostructure photocatalyst for highly efficient visible light driven tetracycline degradation: Insights into reaction mechanisms, Sep. Purif. Technol., 2023, 308, 122908 CrossRef CAS.
- J. Wang and S. Wang, Effect of inorganic anions on the performance of advanced oxidation processes for degradation of organic contaminants, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- J. Wang and S. Wang, Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism, Chem. Eng. J., 2020, 401, 126158 CrossRef CAS.
- D. Roy, N. Poddar, M. Singh, S. Neogi and S. De, Photocatalytic degradation of Rhodamine-B by visible light assisted peroxymonosulfate activation using Z-scheme MIL-100(Fe)/Bi2S3 composite: a combined experimental and theoretical approach, New J. Chem., 2022, 46, 10728–10745 RSC.
- Y. Lei, S. Cheng, N. Luo, X. Yang and T. An, Rate Constants and Mechanisms of the Reactions of Cl • and Cl 2 •– with Trace Organic Contaminants, Environ. Sci. Technol., 2019, 53, 11170–11182 CrossRef CAS PubMed.
- X. Yang, F. L. Rosario-Ortiz, Y. Lei, Y. Pan, X. Lei and P. Westerhoff, Multiple Roles of Dissolved Organic Matter in Advanced Oxidation Processes, Environ. Sci. Technol., 2022, 56, 11111–11131 CrossRef CAS PubMed.
- S. Bhattacharya, P. Banerjee, P. Das, A. Bhowal, S. K. Majumder and P. Ghosh, Removal of aqueous carbamazepine using graphene oxide nanoplatelets: process modelling and optimization, Sustainable Environ. Res., 2020, 30, 17 CrossRef CAS.
- V. L. Cunningham, C. Perino, V. J. D'Aco, A. Hartmann and R. Bechter, Human health risk assessment of carbamazepine in surface waters of North America and Europe, Regul. Toxicol. Pharmacol., 2010, 56, 343–351 CrossRef CAS.
- Y. Lei, Y. Yu, X. Lei, X. Liang, S. Cheng, G. Ouyang and X. Yang, Assessing the Use of Probes and Quenchers for Understanding the Reactive Species in Advanced Oxidation Processes, Environ. Sci. Technol., 2023, 57, 5433–5444 CrossRef CAS PubMed.
- Q. Ma, Y. Zhang, X. Zhu and B. Chen, Hollow multi-shelled Co3O4 as nanoreactors to activate peroxymonosulfate for highly effective degradation of Carbamazepine: A novel strategy to reduce nano-catalyst agglomeration, J. Hazard. Mater., 2022, 427, 127890 CrossRef CAS.
- F. Pan, H. Ji, P. Du, T. Huang, C. Wang and W. Liu, Insights into catalytic activation of peroxymonosulfate for carbamazepine degradation by MnO2 nanoparticles in situ anchored titanate nanotubes: Mechanism, ecotoxicity and DFT study, J. Hazard. Mater., 2021, 402, 123779 CrossRef CAS PubMed.
- H. Zhou, L. Lai, Y. Wan, Y. He, G. Yao and B. Lai, Molybdenum disulfide (MoS2): A versatile activator of both peroxymonosulfate and persulfate for the degradation of carbamazepine, Chem. Eng. J., 2020, 384, 123264 CrossRef CAS.
- S. Liu, C. Zhao, Z. Wang, H. Ding, H. Deng, G. Yang, J. Li and H. Zheng, Urea-assisted one-step fabrication of a novel nitrogen-doped carbon fiber aerogel from cotton as metal-free catalyst in peroxymonosulfate activation for efficient degradation of carbamazepine, Chem. Eng. J., 2020, 386, 124015 CrossRef CAS.
- Y. Li, S. Zhang, Y. Qin, C. Yao, Q. An, Z. Xiao and S. Zhai, Preparation of cobalt/hydrochar using the intrinsic features of rice hulls for dynamic carbamazepine degradation via efficient PMS activation, J. Environ. Chem. Eng., 2022, 10, 108659 CrossRef CAS.
- S.-F. Jiang, L. Wang, W.-F. Hu, K. Tian and H. Jiang, Preparation of Flower-like CuFe 2 O 4 by a Self-Templating Method for High-Efficient Activation of Peroxymonosulfate To Degrade Carbamazepine, Ind. Eng. Chem. Res., 2021, 60, 11045–11055 CrossRef CAS.
- G. Gou, S. Kang, H. Zhao, C. Liu, N. Li, B. Lai and J. Li, Efficient peroxymonosulfate activation through a simple physical mixture of FeS2 and WS2 for carbamazepine degradation, Sep. Purif. Technol., 2022, 290, 120828 CrossRef CAS.
- Y. Zhang, Y. Cheng and H. Qi, Synergistic degradation of organic pollutants on CoFe2O4/rGO nanocomposites by peroxymonosulfate activation under LED irradiation, Appl. Surf. Sci., 2022, 579, 152151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3en00741c |
| This journal is © The Royal Society of Chemistry 2024 |