
Self-assembled porous salt crystals for solar-powered crystallization†
Jie
Yu
a,
Lenan
Zhang
b,
Jintong
Gao
a,
Wenyu
Han
a,
Ruzhu
Wang
 a and
Zhenyuan
Xu
*a
a and
Zhenyuan
Xu
*a
aEngineering Research Center of Solar Power and Refrigeration (MOE), Institute of Refrigeration and Cryogenics, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: xuzhy@sjtu.edu.cn
bSibley School of Mechanical and Aerospace Engineering, Cornell University, Ithaca, NY 14853, USA
First published on 12th November 2024
Abstract
Thermally localized solar evaporation has been recognized as an efficient and suitable pathway for desalination and brine treatment. However, the annoying salt crystallization inside the porous evaporator poses challenges in hypersaline brine evaporation. Here, we propose that the salt crystals can serve as an ideal structure for evaporation with proper manipulations. Taking advantage of the self-amplifying salt creeping and efflorescence effects, the salt crystals self-assemble to form a hierarchical porous salt evaporator (HPSE), enabling passive liquid supply and efficient evaporation. With a low-cost, solar-powered device based on HPSE, we achieve stable evaporation and crystallization when treating the hypersaline brine with 27.3 wt% salinity under one-sun illumination, resulting in a high evaporation rate of 18.78 kg m−2 h−1. This work bridges the important knowledge gap between the fundamental salt crystallization and brine treatment, paving a path toward sustainable water economy.
Broader contextThermally localized solar evaporation is a promising technology for effective brine treatment with a low carbon footprint. However, the confined salt crystallization that occurs within the evaporator, when treating hypersaline brine, strongly degrades the evaporation performance. Therefore, the conventional understandings typically believe that salt crystallization is detrimental to evaporation and aim to limit its impact, deviating from the goal of complete crystallization in brine treatment. In this work, we demonstrate that the salt crystals can serve as an ideal structure for evaporation with proper manipulations. Leveraging the self-amplifying salt creeping and efflorescence effects, the salt crystals self-assemble to form a hierarchical porous salt evaporator (HPSE), enabling passive brine transport, fast vapor escape, and efficient heat supply. With the HPSE, we achieved a high evaporation rate of 18.78 kg m−2 h−1 when treating the 27.3 wt% concentrated seawater. Additionally, the HPSE effectively treated concentrated seawater with complete crystallization and maintained stable evaporation performance during a continuous 10-day operation. To demonstrate scalability, we deployed a 50-evaporator array operating under outdoor field condition, achieving an average evaporation rate of 38.11 kg m−2 h−1. With the capability to achieve high-performance crystallization and feasibility under realistic weather conditions, the HPSE holds significant promise for brine treatment toward sustainable water economy. |
Introduction
Water scarcity is one of the most critical global challenges due to growing populations and decreasing water supplies.1,2 Desalination technologies provide a viable solution to freshwater shortage.3 However, producing every liter of freshwater through desalination creates an additional 1.5 liters of hypersaline concentrate discharge (brine) as a byproduct, leading to more than 140 million m3 of brine production per day.4 Most of this brine is directly discharged into the aquatic environment without proper treatment,5 posing a huge threat to the ecosystem health and sustainable water supply.6 Zero liquid discharge (ZLD), a strategy for the complete separation of solid solute from brine, holds significant promise to maximize freshwater recovery while mitigating the potential environmental risks of desalination.7 A core process to realize ZLD relies on a crystallization system, which takes the pre-concentrated brine as the input and reduces it to solid waste.7 In contrast to multiple techniques available for desalination and pre-concentration, crystallization is mainly driven by thermal evaporation owing to the operation of near-saturated brine, which suffers from undesirable efficiency,8 high cost,9 intensive energy consumption,3,10 and large emission.7Our goal of sustainable hypersaline brine treatment highlights the opportunity to power evaporation and crystallization processes using natural sunlight.11–13 Leveraging solar absorption, thermal insulation, and water transport, the recent advances of thermally localized solar evaporator have demonstrated a high-performance, low-cost, and low-carbon footprint solar-to-vapor conversion for several applications constrained by water-energy nexus.14–23 However, for typical thermally localized solar evaporators, dealing with the near-saturated brine can be even more challenging than conventional thermal evaporation. This is because salt crystallization and accumulation in the porous evaporator reduce solar absorption, block the water supply, and inhibit vapor transport, strongly degrading the evaporation rate.12,24–30 In addition, the crystallization of multiple salts when dealing with real seawater makes the salt clogging a key bottleneck for achieving reliable operation.12 Due to this common understanding of the detrimental role of salt, instead of promoting the crystallization, conventional design strategies of thermally localized solar evaporators aim to limit salt crystallization and avoid its impact.31–33 Current strategies for solar-powered brine evaporation include separating the solar-absorbing surface and crystallization zone to maintain high solar absorption,12,33–38 enhancing the back flow of salt ions to the bulk solution or promptly removing crystallized salt to prevent salt accumulation,39–41 constructing a confined water layer to increase the salt tolerance of the evaporator,24,42 and selectively extracting water from the brine while rejecting salt ions to mitigate salt crystallization.43,44 Although these methods exhibit superior evaporation performance, they become more challenging to sustain when treating near-saturated brine, where rapid crystallization intensifies the impact of salt. In such conditions, both the crystallization and salt collection processes are hindered by severe salt clogging and strong salt adhesion to the evaporator, limiting the previous strategies from achieving ZLD brine treatment in real-world applications.
Here, we develop a high-performance solar evaporator to enable continuous salt crystallization for hypersaline brine treatment. In contrast to the common understanding, we recognize that salt crystals can serve as a highly favorable structure for evaporation, owing to the rapid creeping, intrinsic wetting, and hierarchical porous nature. With a dendrite-structured design, we separate the solar-thermal conversion from salt crystallization, and hence enable an unconfined salt creeping and growth. We accelerate the directional salt creeping along the dendritic structure by engineering the surface wettability. Taking advantage of the self-amplifying salt creeping and efflorescence effects, a hierarchical porous salt evaporator (HPSE) that consists of two salt layers with distinct crystal sizes is self-assembled on the dendritic structure. This HPSE significantly promotes solar evaporation by enlarging the evaporation area. More importantly, the inner layer of HPSE with smaller salt crystals (tens of microns in size) creates a capillary force to drive the passive brine supply, while the outer layer of HPSE with larger salt crystals (hundreds of microns in size) ensures fast vapor transport. The HPSE design exhibits stable solar evaporation of the near-saturated brine with continuous salt crystallization. Using real concentrated seawater (27.3 wt%; from Bohai Sea, China) as an example, we demonstrate a high evaporation rate of 18.78 kg m−2 h−1 under one-sun illumination, together with a remarkable salt collection efficiency of 91.1%–99.4%. More notably, an ultrahigh average evaporation rate of 38.11 kg m−2 h−1 for a 50-evaporator array was achieved in the outdoor field test, showing favorable performance under realistic weather conditions and scalable applications. This work provides an in-depth understanding of salt crystallization, enabling a new perspective to realize high-performance, low-cost, and low-carbon footprint hypersaline brine treatment through a simple, solar-powered device.
Results and discussion
Self-assembled hierarchical porous salt crystals for solar-powered crystallization
Fig. 1a shows the schematic of a conventional thermally localized solar evaporator, which is limited by salt crystallization. In the typical design, a solar-absorbing porous capillary wick is used to supply liquid and promote evaporation. Since hypersaline brine is confined within the evaporator, heterogeneous nucleation of salt crystals is initiated inside pores (zoom-in illustration of Fig. 1a), where the fixed pore structure further restricts crystal displacement, leading to compact salt clogging. This confined salt crystallization reflects the incident sunlight, reduces the permeability of the porous structure, and induces an early dry-out of the evaporator (zoom-in illustration of Fig. 1a), resulting in a low evaporation rate when treating hypersaline brine. | ||
| Fig. 1 Self-assembled hierarchical porous salt crystals for solar-powered crystallization. (a) Confined salt crystallization in conventional thermally localized solar evaporation. The confined salt crystallization inside the porous structure reduces light absorption and permeability, resulting in undesirable evaporation performance. (b) Unconfined salt crystallization for enhanced brine evaporation. The unconfined salt crystallization is enabled by a flat surface, with solar absorption on the backside of the evaporator. (c) Salt creeping on the flat surface. The salt creeping is enhanced by increasing the hydrophilicity of the surface and aligning it along the direction of gravity, creating a uniform layer with small salt crystals. Zoom-in illustration: liquid front of creeping salts driven by capillary action. (d) Self-assembled hierarchical porous salt evaporator on the flat surface. Zoom-in illustration: hierarchical structure of salt crystals with the inner and outer layers. The inner layer, consisting of small crystals (tens of microns in size), enables a passive and rapid liquid supply. Driven by the efflorescence effect, small crystals close to the ambient air transform into large crystals (hundreds of microns in size) in the outer layer, providing access for vapor escape. (e) Dendrite-structured evaporation device for brine treatment. The device consists of a solar absorber, a solution supplier with a capillary wick, and multiple heat distributors. | ||
We introduce the concept of unconfined salt crystallization to address the above challenges, which comprises two design strategies (Fig. 1b). First, the interference of salt crystallization on solar absorption is eliminated by separating the functionalities of the solar absorber and evaporator. Using a nonpermeable solar absorber and attaching an additional evaporator from the other side, we can maintain the high solar absorption because no salt crystallization occurs on top of the solar absorber (Fig. 1b). The effectiveness of this design procedure has been demonstrated in several recent works.45–47 Second, we replace the porous evaporator by a flat surface to initiate the unconfined salt crystallization, which exhibits two characteristics, i.e., (1) salt creeping and (2) the formation of hierarchical crystals. Salt creeping describes the crystal precipitation from the meniscus of an evaporating solution film.48 By increasing the hydrophilicity of the surface and aligning it along the direction of gravity, we can accelerate the spread of the liquid film and reduce its thickness, leading to the rapid formation of a uniform crystal layer with small crystal size (tens of microns, Fig. 1c). More interestingly, the salt creeping can be a self-amplifying process because small salt crystals create a strong capillary force to promote liquid spreading along the surface. Meanwhile, the continuous evaporation of the liquid film induces more salt crystals. With the accumulation of salt crystals, small-sized crystals close to the ambient air transform into large-sized (hundreds of microns) crystals due to the efflorescence effect,49 leading to a self-assembled hierarchical porous salt layer on the flat surface (Fig. 1d). This hierarchical porous salt layer is intrinsically an ideal evaporator because (1) the self-amplifying salt creeping creates an enlarged area for evaporation, (2) the inner layer with small crystals enables a passive and rapid liquid supply, and (3) the outer layer with large crystals exhibits low vapor transport resistance. It should be noted that this hierarchical porous salt layer is enabled by allowing the unconfined salt crystals to self-assemble in open space, which is significantly different from the compact salt layer resulting from confined crystallization.
To fully exploit the potential of the unconfined salt crystallization, we integrate the hierarchical porous salt layer into a dendrite-structured design. Fig. 1e shows the schematic of the dendrite-structured device for brine treatment, which consists of a solar absorber, a solution supplier with a capillary wick, and multiple heat distributors. During the device operation, brine flows from the bottom reservoir to the back of the solar absorber, and then spreads outward to the top part of the heat distributor through the continuous solution supplier, driven by capillary action (Fig. 1e and Fig. S1, ESI†). With proper hydrophilic treatment, the brine then spreads and forms a meniscus at the top part of each heat distributor. The flow path along the solution supplier and solar absorber is covered by an evaporation preventer to avoid the early crystallization-induced clogging of liquid transport. Meanwhile, the incident sunlight is converted to heat by the solar absorber, and then transferred to heat distributors. Evaporation of the meniscus occurs, driving salt creeping and self-assembly of the HPSE on each heat distributor. The HPSE can maintain a high evaporation rate during the continuous operation, enabling the complete separation of salt from brine.
Manipulating salt creeping for optimized crystallization
Salt creeping plays a critical role in initiating the self-assembly of the HPSE. Taking advantage of the surface wettability and gravity, we can enhance the coverage, uniformity, and capillarity of salt creeping, and hence accelerate the formation of the HPSE. To justify the design proposed in Fig. 1e, we quantified the impact of surface wettability and gravity on salt creeping. To better elucidate the governing physics associated with the formation of HPSE, 25.0 wt% NaCl solution was first used in our characterizations, whereas key results were then confirmed using the concentrated seawater (with a total salt concentration of 27.3 wt%, collected from Bohai Sea, China, see Experimental section, Table S1, and Supplementary Note 1 for details, ESI†). The 25.0 wt% NaCl solution was introduced by a fabric paper and spread onto the aluminum surface (Fig. 2a and Fig. S2 for experimental setup, ESI†). Three experimental configurations with different salt creeping directions and surface wettability were considered. All experiments were performed at 35 °C and 50% relative humidity (RH) in an environmental chamber (KMF 115, BINDER) to simulate the realistic conditions during solar evaporation. | ||
| Fig. 2 Salt creeping enhancement. Photograph (a), surface morphology (b), and scanning electron microscopy image (c) of salt creeping opposite the gravity direction on a bare aluminum surface (configuration 1). Inset of (a) shows the contact angle of a 25.0 wt% NaCl solution droplet on the bare aluminum plate (105.1°). Photograph (d), surface morphology (e), and scanning electron microscopy image (f) of salt creeping along the gravity direction on a bare aluminum surface (configuration 2). A larger salt coverage was achieved with a smaller thickness and crystal size compared with configuration 1. Photograph (g), surface morphology (h), and scanning electron microscopy image (i) of salt creeping along the gravity direction on a hydrophilic aluminum surface (configuration 3) with a CA less than 5°. A much larger surface coverage of the salt crystal layer was achieved, with a much smaller thickness of ≈180 μm. (j) Schematics of the crystallization processes inside the solution meniscuses for the three configurations. The slow salt creeping in configuration 1 is attributed to the large CA-induced weak capillary force and gravity-induced decelerated liquid spread. The salt creeping of configuration 2 was accelerated by the gravity-enhanced liquid spread and crystal migration. The salt creeping of configuration 3 was further accelerated by the hydrophilic surface-induced large capillary force and better liquid spread, which induces the uniform and self-amplifying growth of small salt crystals. | ||
Fig. 2a shows the salt creeping opposite to the gravity direction (i.e., the upward direction) on a bare aluminum surface (i.e., configuration 1) at the 0th hour and 4th hour. The contact angle (CA) of a 25.0 wt% NaCl solution droplet on the bare aluminum surface was 105.1° (inset of Fig. 2a), indicating low hydrophobicity. In general, the salt creeping of configuration 1 was slow, leading to a nonuniform and thick crystal layer with large salt crystals. After a 4-hour test, the far edge of the salt crystal layer was only 2 mm above the fabric paper due to the insufficient spread of the NaCl solution (Fig. 2a). We characterized the surface morphology of the salt crystal layer using an optical microscope (VHX 5000, Keyence, Fig. 2b). A significant uplift with ≈1500 μm thickness was observed at the edge of the salt crystal layer (inset of Fig. 2b). Using a scanning electron microscope (SEM, Sirion 200, FEI), we observed large salt crystals (≈ 25 μm in size) formed in this configuration (Fig. 2c).
In configuration 2, we performed the salt creeping experiment along the gravity direction (i.e., the downward direction) on the bare aluminum surface (Fig. 2d). Compared with configuration 1, a larger salt coverage was achieved, where the far edge of the salt crystal layer extended to ≈6 mm away from the fabric paper after 4 hours (Fig. 2d). Meanwhile, the salt crystal layer also exhibited better uniformity (Fig. 2e) with a maximum thickness of ≈350 μm (inset of Fig. 2e) and smaller crystal size of ≈15 μm (Fig. 2f). In configuration 3, we coated silica nanoparticles on the aluminum surface, which became superhydrophilic with a CA of less than 5° (inset of Fig. 2g, see Experimental section for details of the sample preparation). Salt creeping occurred along the downward direction (Fig. 2g). An even more significant change of the salt crystal layer was shown in this configuration. A much larger surface coverage of the salt crystal layer was observed after 4 hours, which was more than 10 and 4 times larger than that of configurations 1 and 2, respectively. Accordingly, the salt crystal layer was more uniform as well (Fig. 2h), with a maximum thickness of ≈180 μm only (inset of Fig. 2h). The grain boundary among the salt crystals became unobservable (Fig. 2i), indicating more compact salt crystals in this configuration.
We explained the distinct salt creeping behaviors in the above three configurations. In general, gravity can affect both the liquid spread and salt crystal migration. Capillarity not only drives the liquid spread, but also dictates the liquid film thickness and salt crystal size. The slow salt creeping in configuration 1 primarily originated from the weak capillary force due to the large CA on the bare aluminum surfaces (Fig. 2j). Since gravity acted along the opposite direction, it further decelerated the liquid spread. During salt creeping, crystallization was first initiated at the liquid-air interface (Fig. S3, ESI†). Salt crystals were then driven away from the meniscus by gravity and accumulated on the edge of the porous media (Fig. 2j), where they continued to grow, resulting in the large crystals and thick crystal layer observed in Fig. 2a–c. In configuration 2, gravity was along the direction of liquid spread, leading to the slightly larger salt coverage compared with configuration 1 (Fig. 2d). Meanwhile, salt crystals migrated toward the meniscus because of gravity (Fig. 2j). Since the liquid film was relatively thin at the edge of the meniscus, it confined the growth of salt crystals and caused the more uniform salt crystal layer and smaller crystal size, as shown in Fig. 2d–f. In contrast to configurations 1 and 2, a much larger capillary driving force and stronger confinement effect were created by the superhydrophilic surface in configuration 3, which induced the uniform growth of small salt crystals (Fig. 2j). Moreover, configuration 3 enabled the self-amplifying salt creeping (see Fig. S4, ESI†) because small salt crystals can generate a large capillary force to further extend the liquid film.50,51 With the detailed characterizations of salt creeping, we confirmed that the heat distributors with the downward orientation and superhydrophilic treatment are highly desirable for the self-amplifying salt creeping, enabling the rapid formation of a high-quality salt crystal layer for solar evaporation.
HPSE on a dendrite-structured device
The insights gained from the salt creeping guided our design of a proof-of-concept setup. Fig. 3a shows the prototype of the dendrite-structured device for solar-powered brine treatment. We chose the dendritic structure to enhance mass transfer during evaporation. A 31.0 × 31.0 mm2 aluminum plate with a carbon black (CB) coating was used as the solar absorber52 (see Fig. S5 and Experimental section for details of the fabrication process, ESI†). The CB coating was based on a solvent evaporation process, which created nanocavities (≈100 nm–1 μm diameter) to enhance the light trapping (Fig. S6, ESI†). Owing to both intrinsically high absorption of CB and nanocavity-induced light trapping, the solar absorber exhibited a high solar absorptance of 97% (Fig. S7, ESI†). On each side of the solar absorber, there are five aluminum heat distributors (34.5 mm × 2.0 mm × 0.8 mm, Fig. 3a and Fig. S5, ESI†). To enable the self-amplifying salt creeping, all of the heat distributors were vertically aligned and treated with the hydrophilic coating. Brine can spread along the heat distributor and then evaporate, creating the HPSE (Fig. 3b). Using numerical simulations, we confirmed that the design with five heat distributors exhibited a higher evaporation rate than a plate with the same projected area (i.e., 0.8 × 31.0 mm2) due to the fin effect19 (see Supplementary Note 2 for details of the numerical simulations, ESI†). The solar absorber was supported by a ≈45 mm tall solution supplier, which contained a fabric paper as the capillary wick. To avoid salt crystallization on the fabric paper or beneath the solar absorber, polystyrene foam was used as an evaporation preventer to cover the flow path (Fig. 3a). The entire dendrite-structured device was placed on a reservoir containing brine. The reservoir was covered by aluminum foil to avoid solar heating. | ||
| Fig. 3 HPSE on a dendrite-structured device. (a) The prototype of the dendrite-structured evaporation device for solar-powered brine treatment. The evaporation device consists of a solar absorber with five branches on each side and a solution supplier. (b) Salt crystal layer used for XRM characterization. The salt crystals were peeled entirely off the heat distributor. (c) Section view XRM image of the salt crystal layer crossing its thickness direction. The salt crystals showed a hierarchical structure, with the inner layer consisting of compact small salt crystals, and the outer layer comprising loose large salt crystals. (d) Formation of the HPSE. A thin layer of small salt crystals first covered the entire heat distributor (stage 1, salt creeping), and then the multilayer structure of small salt crystals was created (stage 2, transition). The salt crystals on top of the multilayer structure merged into large salt crystals due to the efflorescence effect (stage 3, efflorescence), and finally formed the HPSE. The evaporation interface was eventually pinned between the inner-layer and outer-layer salt crystals. The thickness of the inner layer remained steady, while the outer layer increased with time. | ||
We performed solar evaporation tests of both near-saturated NaCl solution (25.0 wt%) and real concentrated seawater (27.3 wt%) to understand the formation of HPSE on the dendrite-structured heat distributors. The experimental setup was placed under a solar simulator (CEL-S500R3, CEAuLight), which provided one-sun uniform illumination (1000 W m−2) on the solar absorber. After a 6-hour continuous test, we peeled the entire salt crystal layer off the heat distributor (Fig. 3b) and characterized its microstructure using an X-ray microscope (XRM, Xradia 520 Versa, ZEISS). Fig. 3c shows the section view XRM image of the salt crystal layer crossing its thickness direction by evaporating the 25.0 wt% NaCl solution. A hierarchical structure with two distinct crystal layers was confirmed, where the inner layer close to the heat distributor consists of compact small salt crystals (≈ 10–50 μm in size, Fig. 3c) and the outer layer near the ambient air comprises loose large salt crystals (≈ 100–400 μm in size, Fig. 3c). We estimated the porosities of the inner (≈ 0.52) and outer (≈ 0.35) layers by imaging the salt crystal distribution with XRM (Fig. S8, ESI†). A similar hierarchical structure of salt crystals was also observed during the evaporation of real seawater on the hydrophilic heat distributors (Fig. S9, ESI†).
Formation of the HPSE can be attributed to the combined effects of salt creeping and efflorescence, which lead to three distinct stages of salt crystallization (Fig. 3d), i.e., salt creeping (stage 1), transition (stage 2), and efflorescence (stage 3). Due to the evaporation of brine, a thin layer of small salt crystals (tens of microns in size) first covers the heat distributors (stage 1). Meanwhile, the strong capillarity created by the small salt crystals further drives the propagation of the liquid front along the heat distributors, resulting in the self-amplifying salt creeping. With continuous crystallization, the multilayer structure of small salt crystals was created (stage 2). However, the multilayer structure cannot keep increasing because of the efflorescence effect, where multiple small salt crystals on the top of the multilayer structure merged into a large salt crystal to lower the surface energy (stage 3). With the interplay between the salt creeping and efflorescence, the multilayer structure finally became the inner layer of the HPSE with a steady thickness ≈500 μm, while the large crystals comprising the outer layer of the HPSE increased with time. The evaporation interface was eventually pinned at the interface between the inner and outer layers (stage 3), as the smaller pores of the inner layer can induce stronger capillarity. These structural characteristics are also favorable for heat and mass transfer, as liquid confined within the inner layer of the HPSE has a steady thickness. As a result, the thermal resistance of the liquid layer does not increase with the growth of the HPSE, which is essential to maintain a steady evaporation rate during a long-time operation. Meanwhile, vapor diffusion occurs on the outer layer of the HPSE, which has low mass transport resistance because of the high permeability.
Enhanced transport via the HPSE
In contrast to the conventional understanding that salt crystallization is detrimental, we provided theoretical and experimental evidence for the enhanced liquid transport mediated by the HPSE. Salt crystals are desirable for liquid transport due to the intrinsically wetting nature. Fig. 4a shows that the CAs of water and saturated NaCl solution on the bulk salt crystal were only ≈11°, indicating a highly hydrophilic wetting state. As a result, the micropores in the HPSE can create a large capillary pressure to enable the passive liquid supply. Considering the hierarchical structure of the salt crystal layer, liquid could flow through both the gap between the heat distributor and inner layer, as well as the inner layer (Fig. 3d), leading to two potential flow paths (Fig. 4b). We estimated the water transport properties of each flow path using Darcy's law (see Supplementary Note 3 for detailed analysis, ESI†). The water transport velocity inside the inner layer (![[l with combining dot above]](https://www.rsc.org/images/entities/i_char_006c_0307.gif) 1) is calculated as eqn (1), whereas the highest transport velocity inside the gap (2) can be estimated as eqn (2) by assuming an ideal capillary tube with a diameter equal to the gap width (Supplementary Note 3, ESI†).
1) is calculated as eqn (1), whereas the highest transport velocity inside the gap (2) can be estimated as eqn (2) by assuming an ideal capillary tube with a diameter equal to the gap width (Supplementary Note 3, ESI†). | (1) |
 | (2) |
 | ||
| Fig. 4 Enhanced transport via the HPSE. (a) The CAs of water and saturated NaCl solution on the bulk salt crystal. The CAs were only ≈11°, indicating a highly hydrophilic wetting state. (b) Two potential flow paths inside the HPSE. The blue and red arrows indicate the flows through the gap between the heat distributor and inner layer, as well as the inner layer, respectively. (c) Theoretically calculated liquid transport velocity as a function of the transport distance through different flow paths. The liquid transport velocity through the inner layer is only less than one order of magnitude lower than the most ideal condition with a perfect capillary tube, indicating a reasonably high liquid transport performance of the HPSE. (d) Time-lapse images of the liquid transport in fabric papers (top row) and HPSE (bottom row). Both fabric papers and HPSE exhibited fast liquid transport. (e) Rapid liquid transport confined within the inner layer of the HPSE. The entire inner layer of the HPSE on the middle branch became black-colored at 18 min, while the majority of the outer layer still remained uncolored. (f) Liquid transport velocities from the propagation of the colored fronts and theoretical calculation. The transport in the inner layer of the HPSE was comparable with that in the fabric paper, and showed a reasonable agreement with the theoretical estimation. | ||
We confirmed the high liquid transport performance with experimental characterizations. To better quantify the performance of HPSE, we used the black ink transport in fabric paper as a reference, which has been recognized as a high-performance wicking material and widely applied to solar evaporation.45,47 By dripping black ink from the top of the fabric paper and HPSE, we visualized the liquid transport by tracking the evolution of the colored front. Fig. 4d shows the time-lapse images of the liquid transport in fabric paper (the top row) and HPSE (the bottom row), where the same dendrite-structured devices were used. In general, both the fabric paper and HPSE exhibited fast liquid transport. It took approximately 3.5–5.7 min and 6.0 min for the fabric paper and HPSE to transport the black ink from the top to the bottom, respectively. More interestingly, we directly observed that the rapid liquid transport was confined within the inner layer of the HPSE, validating our former analysis (Fig. 3d). As shown in Fig. 4e, at 18.0 min, the entire inner layer of HPSE on the middle branch became black-colored, while the majority of the outer layer still remained uncolored. The outer layer ultimately became black as well at 30.0 min (Fig. 4d). This is mainly because the black-colored small crystals within the inner layer finally transformed into the large crystals due to the efflorescence effect. We measured the liquid transport velocities from the propagation of the colored fronts, and summarized them in Fig. 4f. The transport velocity in the inner layer of the HPSE (≈35 cm h−1) was comparable with that in the fabric paper (≈48 cm h−1), indicating a high liquid transport performance of the HPSE. The experimental characterization (≈35 cm h−1) also showed a reasonable agreement with our theoretical estimation (≈45 cm h−1).
High-performance solar evaporation of hypersaline brine
Taking advantage of the favorable liquid, vapor, and heat transport within the HPSE, we demonstrated the efficient and stable solar evaporation of near-saturated brine (25.0 wt% NaCl solution and 27.3 wt% concentrated seawater) in a laboratory condition. Fig. 5a shows the time-lapse images of continuous salt crystallization from 25.0 wt% NaCl solution during a 6-hour test under one-sun illumination. The evolution of salt crystallization was consistent with the three stages depicted in Fig. 3d. Specifically, stage 1, i.e., the salt creeping stage, occurred in the first hour with a thin layer of small salt crystals covering heat distributors (Fig. 3d). Stage 2, i.e., the transition stage, was seen from the ≈1st hour to the 3rd hour, associated with a thicker layer due to the accumulation of salt crystals (Fig. 3d). After the third hour, stage 3, i.e., the efflorescence stage, started, as featured by the highly rough morphology owing to the formation of large crystals (Fig. 3d). Fig. 5b shows the corresponding temperature profiles at the 1st hour and 6th hour captured by an infrared (IR) camera (T630sc, FLIR). The solar absorber exhibited the highest temperature of ≈33 °C, whereas the temperature of the heat distributors was only ≈5 °C higher than the ambient temperature (≈25 °C), indicating low heat loss to the ambient air and effective solar energy utilization.12,38,42,53,54 It should be noted that the amount of water in the HPSE can affect the accuracy of the temperature measurement using an IR camera. Since water has a larger IR emissivity (≈ 0.95) than aluminum and salt crystals, HPSE with lower water content can exhibit weaker IR emission, which may result in an inaccurate “lower temperature” region in the IR image. Therefore, the slightly lower temperature of a few heat distributors shown in the IR images can be attributed to the lower amount of water.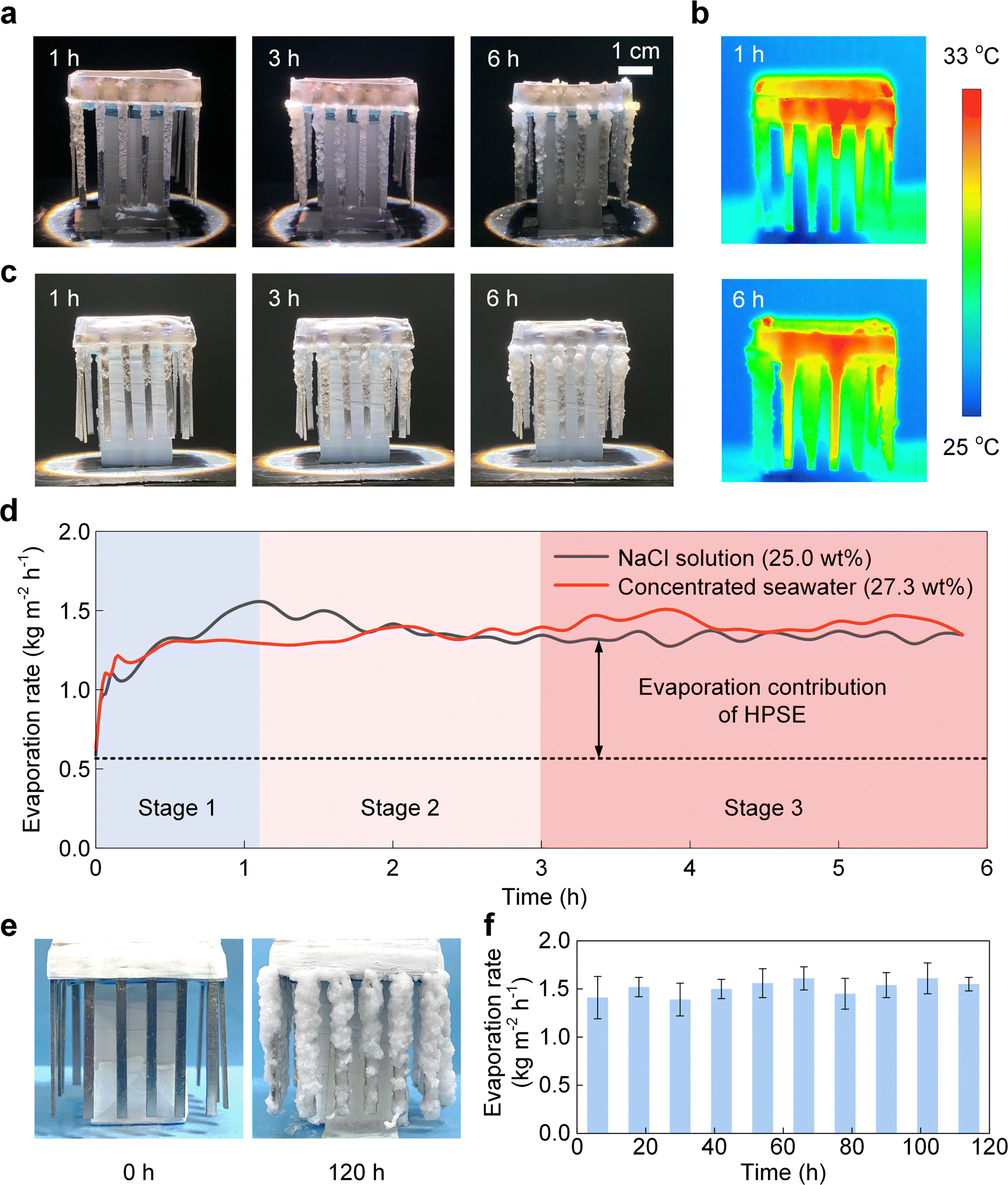 | ||
| Fig. 5 Solar evaporation performance of the near-saturated NaCl solution (25.0 wt%) and concentrated seawater (27.3 wt%). (a) Time-lapse images of continuous salt crystallization (25.0 wt% NaCl solution) during a 6-hour test under one-sun illumination. The evolution of salt crystallization could be divided into three stages. Stage 1 occurred in the first hour, stage 2 occurred from ≈1st hour to the 3rd hour, and stage 3 occurred after the third hour, which were consistent with the salt creeping stage, transition stage, and efflorescence stage in Fig. 3d, respectively. (b) Temperature profiles for evaporation of the NaCl solution at the 1st hour and 6th hour captured by an infrared camera. The temperature of the heat distributors was ≈5 °C higher than the ambient temperature and close to that of the solar absorber, indicating an effective heat transfer. (c) Time-lapse images of continuous salt crystallization (27.3 wt% concentrated seawater) during a 6-hour test under one-sun illumination. (d) Solar evaporation rates of 25.0 wt% NaCl solution (black curve) and 27.3 wt% concentrated seawater (red curve) as functions of time. The device finally reached steady-state evaporation rates of 1.34 kg m−2 h−1 and 1.42 kg m−2 h−1 for 25.0 wt% NaCl solution (black curve) and 27.3 wt% concentrated seawater (red curve) evaporation, respectively. The rapid increase (stage 1), slight drop (stage 2), and steady state (stage 3) of the evaporation rate for 25.0 wt% NaCl solution correspond to the salt creeping stage, transition stage, and efflorescence stage, respectively. (e) Images of salt crystallization during the 120-hour continuous evaporation test with 27.3 wt% concentrated seawater. (f) Evaporation rate of the 27.3 wt% concentrated seawater during the 120-hour continuous evaporation test. There was no degradation in the evaporation performance throughout the test, and an average evaporation rate of 1.39–1.61 kg m−2 h−1 was achieved. The evaporation rates are presented as the mean value ± standard deviation in every 12-hour duration. | ||
To further demonstrate the feasibility of our design for real brine treatment, we performed solar-powered evaporation experiments using concentrated seawater (27.3 wt%). Fig. 5c shows the time-lapse images of continuous salt crystallization during a 6-hour test. Both the salt creeping (0th hour–3rd hour) and HPSE formation (3rd hour–6th hour) were observed, similar to the test with 25.0 wt% NaCl solution (Fig. 5a). Fig. 5d shows the solar evaporation rates of 25.0 wt% NaCl solution and 27.3 wt% concentrated seawater as functions of time. We measured the evaporation rates from the mass changes of the device using a digital balance (ME503TE, Mettler Toledo). In general, for the evaporation rate of NaCl solution (black curve in Fig. 5d), it first increased from 0.59 kg m−2 h−1 to more than 1.50 kg m−2 h−1 within the first hour, followed by a slight decrease of ≈0.10 kg m−2 h−1 from the 1st hour to the 3rd hour (Fig. 5d). The device finally reached a steady-state evaporation rate of 1.34 kg m−2 h−1 with continuous salt crystallization after the third hour, which was 127% higher than the initial evaporation rate (0.59 kg m−2 h−1). This significant increment originated from the enhanced evaporation due to the HPSE. Specifically, the change of the evaporation rate with time can be well explained by the liquid transport mechanisms in the three stages of salt crystallization (Fig. 3d). The rapid increase of the evaporation rate in the first hour operation was attributed to the enlarging evaporation area induced by the self-amplifying salt creeping (stage 1 of Fig. 3d and 5a, d). The slight drop of the evaporation rate after the 1st hour indicates the formation of the multilayer structure in the transition stage (stage 2 of Fig. 3d and 5a, d), which created slightly higher thermal resistance because of the thicker liquid film. After the 3-hour operation, the evaporation continued at a steady rate despite the continuous increase of salt crystals (stage 3 of Fig. 3d and 5a, d). This is because in stage 3, the liquid film was confined to the inner layer of the HPSE with a steady thickness, providing a constant thermal resistance for solar evaporation. On the other hand, evaporation of the concentrated seawater (27.3 wt%) exhibited similar performance (see Supplementary Note 4 for details, ESI†), with an average steady-state evaporation rate of 1.42 kg m−2 h−1 from the 3rd hour to the 6th hour (red curve in Fig. 5d). The evaporation rate was further increased to 4.85 kg m−2 h−1 in an outdoor field test (see Supplementary Note 5 for details, ESI†), which originated from the mass transfer enhancement under the natural wind. We demonstrated a complete separation of all solid salts and liquid water with concentrated seawater through a 36-hour continuous test (see Supplementary Note 6 for details, ESI†). More notably, we further performed a 120-hour continuous test using concentrated seawater (Fig. 5e and Supplementary Note 7, ESI†). Throughout the entire test, there was no degradation in the evaporation performance and an average evaporation rate of 1.39–1.61 kg m−2 h−1 was achieved (Fig. 5f), indicating a highly reliable performance of HPSE when treating the real concentrated seawater containing multiple species of salts. This remarkable durability can be attributed to the unique hierarchical structure of the HPSE, which ensures a uniform distribution of Mg and Ca-related salts on the NaCl crystals, and thereby prevents the clogging at the evaporation interface (see detailed discussion in Supplementary Note 8, ESI†). In contrast, the conventional capillary wick evaporators frequently suffer from severe Mg and Ca-related salt clogging between the NaCl crystals at the evaporation interface.12 In addition, the HPSE is capable of treating the brine containing a high concentration of minor species after undergoing the well-established pre-softening process55 (see details in Supplementary Note 8, ESI†). This superior anti-clogging property of the HPSE makes it well-suited for practical applications involving real brine treatment.
Performance enhancement with HPSE
The favorable liquid and vapor transport capability of the HPSE allows for the optimization of the evaporation device to achieve a higher evaporation rate. We further fabricated a device with a high evaporation area index (EAI, ratio of the total area available for evaporation relative to the projected ground area),53 and carried out laboratory tests under one-sun illumination using 27.3 wt% concentrated seawater. Although a higher EAI could be obtained by simply reducing the solar absorption area, an EAI value of 70 (original EAI = 4), which is not higher than previous studies, was adopted for fair comparison of the evaporation rate (Fig. S10, ESI†). As shown in Fig. 6a, HPSE was successfully self-assembled, while the whole process was slower compared with that in Fig. 5 due to there being less energy input from sunlight. At the 1st hour, high temperature on the bottom of the heat distributor was observed in the temperature profile (Fig. 6a), as the salt creeping did not reach the bottom of the heat distributor. At the 8th hour, the high-temperature region vanished. This is due to the coverage of salt crystals on the whole heat distributor. As shown in Fig. 6b, c, the evaporation rates of our device were 18.78 kg m−2 h−1 under one-sun illumination and 13.75 kg m−2 h−1 under dark evaporation. These evaporation rates are significantly higher than those observed in evaporators with low EAI design, which is due to the additional environmental energy extraction enabled by the sub-ambient evaporation temperature under both solar and dark conditions in our design (Fig. 6a). It should be noted that the evaporation rate difference between the solar and dark evaporation here exceeds the theoretical evaporation rate limit under one-sun illumination, which is due to the dynamic growth of the salt evaporator. Experimentally, the solar and dark evaporation rates can only be measured consecutively, during which the salt evaporator itself undergoes a continuous self-assembly process (see Supplementary Note 9 for detailed analysis, ESI†) and leads to different evaporator configurations. On the other hand, the ambient condition also kept changing, which affected the evaporation performance. This made it difficult to directly compare the solar evaporation rate and dark evaporation rate. In this case, we performed an energy balance analysis instead, showing a solar energy utilization efficiency of 97% (Supplementary Note 9, ESI†). | ||
| Fig. 6 Performance enhancement with HPSE. (a) The photographs and corresponding IR images of HPSE (EAI = 70) during a 12-hour evaporation test under one-sun illumination with 27.3 wt% concentrated seawater. A HPSE temperature lower than the ambient air was observed during the whole test, showing effective environmental energy utilization. (b) Mass change curve of the 12-hour solar evaporation and subsequent 1-hour dark evaporation. (c) Solar evaporation rate, dark evaporation rate, and their difference at the end stage, which are 18.78 kg m−2 h−1, 13.75 kg m−2 h−1, and 5.03 kg m−2 h−1, respectively. (d) Image of the 50-evaporator array after 6-hour outdoor field evaporation test. (e) Evaporation rate of the 50-evaporator array (left axis) and the real-time variation of solar irradiance (right axis). A peak evaporation rate of 79.95 kg m−2 h−1 and average evaporation rate of 38.11 kg m−2 h−1 were achieved with an average solar irradiance intensity of 606.81 W m−2. | ||
The demonstrated evaporation rates and operating salinities of our HPSE represent high values compared with the recent representative works,12,35–38,41,42,53,56,57 with no higher EAI value than previous studies and a moderate environmental condition (Fig. S11 and Table S2, ESI†). By achieving high values in these two important aspects at the same time, our device achieved an ultra-high theoretical salt production rate of 7.05 kg m−2 h−1. While the salt production rate here is a value calculated based on the evaporation rate and brine salinity, we have performed additional salt collection experiments to validate these calculations. Remarkably, the mass of salt collected in these experiments shows a good agreement with the calculation, demonstrating a high salt collection efficiency of 91.1%–99.4% (see Supplementary Note 10 for details, ESI†) and a minimum salt collection loss enabled by HPSE. This high salt collection efficiency also facilitates the regeneration process of the HPSE by providing a fresh surface for restarted salt creeping process (see Supplementary Note 10 for details, ESI†). Both the high-performance evaporation and regeneration capabilities of the HPSE were then validated via a 10-day (240-hour) continuous evaporation test with salt collection, which demonstrated a stable evaporation rate without degradation and rapid recovery after salt collection (Supplementary Note 11, ESI†).
Moreover, to demonstrate the feasibility in a realistic weather condition and the scalability of our strategy, we further carried out an outdoor field test with a group of 50 evaporators (Fig. 6d and Supplementary Note 12, ESI†). In general, the HPSE was formed on every evaporator, resulting in a peak evaporation rate of 79.95 kg m−2 h−1 and average evaporation rate of 38.11 kg m−2 h−1, with an average solar irradiance intensity of 606.81 W m−2 (Fig. 6e). These significantly increased evaporation rates further validate the sufficient liquid supply capability of the HPSE, and they are expected to improve further by extending the length of the salt evaporator, with the maximum transport length being 10 times higher than that used in our experiment (Supplementary Note 3, ESI†). The high evaporation rate, salt production rate, operating salinity, and feasibility under realistic weather conditions highlight the significant role of the HPSE in enhancing solar evaporation of hypersaline brine with complete crystallization.
Conclusion
We have demonstrated the capability of a high-performance solar-powered hypersaline brine treatment approach that is fundamentally grounded with an in-depth understanding of salt crystallization. We found that the commonly believed adverse impacts of salt crystallization, such as the clogging of the evaporator, can be eliminated with proper manipulations. This is because we identified that salt crystals can form an ideal structure for high-performance evaporation, due to the intrinsic highly wetting nature. Therefore, passive liquid transport during solar evaporation does not necessarily rely on an additional capillary wick, such as fabric papers or membranes. Instead, a porous structured salt crystal layer can replace the conventional solar evaporator. The removal of capillary wicks for evaporation creates a significant engineering space to utilize the salt crystallization. We introduced the concept of unconfined salt crystallization on a flat surface, where we took advantage of two basic phenomena associated with crystallization, i.e., salt creeping and efflorescence. Leveraging gravity and hydrophilic treatment, we triggered a rapid self-amplifying salt creeping, enabling the full surface coverage of small salt crystals (tens of microns in size). Salt creeping benefits the enhanced evaporation in two aspects: (1) ensuring the largest evaporating area and (2) creating a large capillary pressure for the passive liquid supply. Efflorescence, featured by the transformation of multiple small crystals into a large crystal (hundreds of microns in size), occurred after salt creeping. The interplay of salt creeping and efflorescence led to the self-assembly of the hierarchical porous salt crystals for enhanced solar evaporation and crystallization. The inner layer of the HPSE consisted of small salt crystals, enabling a rapid liquid transport because of the large capillary pressure and high porosity. Liquid film was confined within the inner layer due to the receding and pinning of the meniscus. Since the thickness of the inner layer (≈ 500 μm) did not change with continuous crystallization, a steady evaporation rate was achieved. The outer layer of the HPSE was featured by loosely packed large salt crystals with high permeability, which is highly favorable for vapor transport. With the above self-assembled hierarchical porous salt layer, we have shown that the liquid transport performance in the HPSE was comparable with that of the conventional highly wicking porous materials, such as the fabric paper. The enhanced liquid, heat, and mass transport of HPSE was further confirmed using the real seawater sample. Moreover, the HPSE exhibited superior anti-clogging property compared to conventional capillary wick evaporators when dealing with concentrated seawater containing multiple ions (Supplementary Notes 8 and 13, ESI†).We designed a dendrite-structured device to exploit the full potential of the HPSE for brine treatment. With insights into the salt crystallization process, we designed vertically aligned heat distributors with hydrophilic treatment to accelerate the formation of HPSE. Meanwhile, at the device level, the dendritic structure was carefully optimized to take advantage of the fin effect for mass transfer enhancement. With this simple, solar-powered device, we demonstrated high-performance solar evaporation of concentrated seawater (27.3 wt% salinity), where the evaporation rate and theoretical salt production rate were 18.78 kg m−2 h−1 and 7.05 kg m−2 h−1, respectively. In addition, our design allows the utilization of an anti-corrosion alloy as a heat distributor, which not only showed comparable evaporation and crystallization performance, but also ensured even longer evaporator reliability when dealing with hypersaline brine (see Supplementary Note 14 for details, ESI†). Taking advantage of the HPSE, for the first time, we demonstrate the complete solar-powered salt-water separation of real seawater without any additives, further showing the significant potential of our approach for sustainable brine treatment.
Moreover, the utilization of salt crystals as evaporator further simplifies the device design. Using commercially available materials, the water treatment cost of our prototype (functioning as crystallizer in the ZLD system) is ≈$ 1.49 m−3, which is less than half cost of the evaporation pond in conventional ZLD technologies,5 as the brine treatment capacity is significantly increased with a higher evaporation rate enabled by our design (see Supplementary Note 15 for details, ESI†). This cost value is even lower than that of a commercial crystallizer,12 further showcasing the potential toward sustainable water economy (see Supplementary Note 15 for details, ESI†).9,58 In addition, owing to the simple structure, our design can be easily scaled up by placing multiple dendrite-structured devices into an array and enlarging the area of each individual device (Supplementary Note 12, ESI†). For example, considering a large-scale plant (≈ 100 m–1 km) with moderate desalination capacity of 5000 m3 day−1, which represents the average capacity of a desalination plant,59 an approximately 100 × 100 m2 area covered by our devices as crystallizer will enable the ZLD treatment of the produced brine from this desalination plant. This smaller footprint, compared to the desalination plant itself, indicates that the solar-powered crystallization device could be installed on the rooftop of the factory, eliminating the need for additional land space. In summary, this work provides a new perspective on the role of salt crystallization in solar evaporation, and directs a pathway toward the complete separation of salt and water by utilizing salt crystallization. Our device developed in this work enables a high-performance and low-cost solar evaporation and crystallization of brine, which shows meaningful impacts on various water treatment and production technologies.
Experimental section
Chemicals
The aluminum plate and admiralty brass were purchased from Alibaba. NaCl and ethanol were purchased from SINOPHARM. The hydrophilic paint (silica nanoparticles, NC-3082) was purchased from Changzhou Nanocoatings Co., Ltd. The CB nanoparticles were purchased from TIMICAL (particle size of 45 nm). n-Hexane and 1H,1H,2H,2H-perfluorooctyl-trichlorosilane were purchased from Macklin. The ethylcellulose and terpilenol were purchased from MERYER. The black ink was purchased from Hero-ink.Salt creeping experiments
A salt creeping setup was built to explore the enhanced salt creeping process on aluminum surfaces. Aluminum plates with dimensions of 30.0 mm × 30.0 mm × 0.5 mm were prepared as the substrates. The porous fabric papers (OUCM cotton tissue) were used for the liquid supply.Fabrication of the hydrophilic aluminum plate
The hydrophilic aluminum plate with a dimension of 30.0 mm × 30.0 mm × 0.5 mm was prepared. The aluminum plate was firstly cleaned in an ultrasonic bath with ethanol for 10 minutes and then rinsed with deionized water. Then, the aluminum plate was dried in a convection oven under 80 °C for 30 minutes. The hydrophilic coating was formed by swabbing the commercial hydrophilic paint onto the aluminum plate. The coated aluminum plate was finally dried in the convection oven under 80 °C for 1 hour.Fabrication of the heat distributor
An aluminum plate with dimension of 100.0 mm × 100.0 mm × 0.8 mm was cut by laser beam (YC-MPLC6045, Yunco Precision) into a shape, as shown in Fig. S5 and S10 (ESI†). The obtained structure was then built into the shape shown in Fig. 3a to form the heat distributor. The vertically aligned dendritic heat distributors were then coated with the hydrophilic nanocoating to form the hydrophilic heat distributors.Fabrication of the solar absorber
The top surface of the as-fabricated heat distributor was coated with CB paint and hydrophobic coating to form the solar absorber. The CB nanoparticles were prepared in a solution composed of CB, ethylcellulose, terpilenol, and ethanol (CB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylcellulose:terpilenol:ethanol = 1:1.5:3:60 wt%). The mixed solution was stirred in the water bath with a temperature of 60 °C for 1 hour, and then ultrasonically dispersed for 20 minutes. The CB paint was swabbed onto the top surface of the heat distributor. Before drying, the CB coating was further modified with 0.5 mM n-hexane solution of 1H,1H,2H,2H-perfluorooctyl-trichlorosilane to form a hydrophobic coating. The coating was smoothed by a plate. Finally, the coated heat distributor was dried in the convection oven under 120 °C for 1 hour to form the solar absorber on top.
:ethylcellulose:terpilenol:ethanol = 1:1.5:3:60 wt%). The mixed solution was stirred in the water bath with a temperature of 60 °C for 1 hour, and then ultrasonically dispersed for 20 minutes. The CB paint was swabbed onto the top surface of the heat distributor. Before drying, the CB coating was further modified with 0.5 mM n-hexane solution of 1H,1H,2H,2H-perfluorooctyl-trichlorosilane to form a hydrophobic coating. The coating was smoothed by a plate. Finally, the coated heat distributor was dried in the convection oven under 120 °C for 1 hour to form the solar absorber on top.
Characterizations
The optical reflectance (R) of the prepared solar absorber was measured in the spectral range of 0.3–2.5 μm using a spectrophotometer (Lamda 950, PerkinElmer). As the solar absorber is opaque, the absorptance (A) is obtained by A = 1 – R. The contact angles of the samples were measured using a droplet shape analyzer (DSA 100, Kruss).Preparation of concentrated seawater
The concentrated seawater was prepared by adding sea salts collected from Bohai Sea (Weifang, Shandong, China) to the deionized water. To create a concentrated solution, the amount of sea salts exceeded the maximum solubility of water so that the sea salts were not fully dissolved. Then, the solution was mixed for at least 3 hours and finally filtrated to remove the excessive solid salts. The total salt concentration of the concentrated seawater was 27.3 wt% (see Supplementary Note 1 for details of concentration measurement, ESI†).Solar evaporation experiments
A proof-of-concept device was built to test the evaporation performance of the HPSE. Laboratory solar evaporation experiments with 25.0 wt% NaCl solution and evaporator EAI = 4 were conducted with a controlled ambient temperature of 24–25 °C and RH of 55%–60% for 6 hours. The laboratory solar evaporation experiments with 27.3 wt% concentrated seawater and evaporator EAI = 4 were conducted with an ambient temperature of 22–24 °C and RH of 40%–45% for 6 hours. The laboratory evaporation experiments with 27.3 wt% concentrated seawater and evaporator EAI = 70 were conducted for 13 hours, with an ambient temperature of 23–26 °C and RH of 40%–55% for the aluminum-based evaporator, as well as ambient temperature of 23–27 °C and RH of 34%–56% for the admiralty brass-based evaporator. The 36-hour continuous evaporation test with concentrated seawater was performed with an ambient temperature of 22–24 °C and RH of 40%–55%. The 120-hour continuous evaporation test with concentrated seawater was performed with an ambient temperature of 25 °C and RH of 50%. The 10-day (240-hour) continuous evaporation test with concentrated seawater was performed with salt collection after the first 5-day operation (ambient condition of 26 °C, 42% RH before salt collection, and 24 °C, 44% RH after salt collection). The outdoor field tests were conducted in the Minhang campus of Shanghai Jiao Tong University on May 30, 2021 (evaporator EAI = 4) and October 16, 2023 (evaporator EAI = 70, 50-evaporator array).Solution transport experiments
The solution transport performances were tested for both HPSE and fabric paper. The HPSE was obtained after 6-hour solar driving evaporation and crystallization of 25.0 wt% NaCl solution. The fabric paper was attached to the heat distributor surface. For the solution transport test, the porous media were first dried under natural condition, and then 2 mL of black ink was dripped to the top of the dendritic structures. The solution transport experiments were conducted with an ambient temperature of 22–24 °C and RH of 40%–50%. The durations of the solution transport from top to bottom of the dendritic structure with a distance of 34.5 mm were recorded, and the solution transport velocities were obtained by calculating the average velocities of such solution transport processes.Data availability
The data that support the findings of this study are available in the ESI.† Additional data are available from the corresponding author on request.Author contributions
Z. X. conceived the initial idea. J. Y. and Z. X. developed the experimental device. J. Y., J. G., and W. H. conducted the experiments. J. Y. developed the models. Z. X., J. Y., and L. Z. analyzed the results. All authors prepared the manuscript. Z. X., R. W., and L. Z. supervised the research.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We gratefully acknowledge the funding support from the National Natural Science Foundation of China (Grant No. 52376200, 51976123).References
- M. M. Mekonnen and A. Y. Hoekstra, Sci. Adv., 2016, 2, e1500323 CrossRef PubMed.
- P. Poredoš and R. Wang, Science, 2023, 380, 458–459 CrossRef.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS.
- E. Jones, M. Qadir, M. T. H. van Vliet, V. Smakhtin and S. Mu Kang, Sci. Total Environ., 2019, 657, 1343–1356 CrossRef CAS PubMed.
- A. Panagopoulos, K. J. Haralambous and M. Loizidou, Sci. Total Environ., 2019, 693, 133545 CrossRef CAS PubMed.
- K. Elsaid, M. Kamil, E. T. Sayed, M. A. Abdelkareem, T. Wilberforce and A. Olabi, Sci. Total Environ., 2020, 748, 141528 CrossRef CAS.
- T. Tong and M. Elimelech, Environ. Sci. Technol., 2016, 50, 6846–6855 CrossRef CAS PubMed.
- M. Ahmed, W. H. Shayya, D. Hoey, A. Mahendran, R. Morris and J. Al-Handaly, Desalination, 2000, 130, 155–168 CrossRef CAS.
- A. K. Menon, I. Haechler, S. Kaur, S. Lubner and R. S. Prasher, Nat. Sustainability, 2020, 3, 144–151 CrossRef.
- M. Yaqub and W. Lee, Sci. Total Environ., 2019, 681, 551–563 CrossRef PubMed.
- C. Chen, Y. Kuang and L. Hu, Joule, 2019, 3, 683–718 CrossRef CAS.
- C. Zhang, Y. Shi, L. Shi, H. Li, R. Li, S. Hong, S. Zhuo, T. Zhang and P. Wang, Nat. Commun., 2021, 12, 998 CrossRef CAS.
- N. Xu, J. Li, C. Finnerty, Y. Song, L. Zhou, B. Zhu, P. Wang, B. Mi and J. Zhu, Nat. Water, 2023, 1, 494–501 CrossRef.
- P. Tao, G. Ni, C. Song, W. Shang, J. Wu, J. Zhu, G. Chen and T. Deng, Nat. Energy, 2018, 3, 1031–1041 CrossRef.
- Z. Wang, Y. Liu, P. Tao, Q. Shen, N. Yi, F. Zhang, Q. Liu, C. Song, D. Zhang, W. Shang and T. Deng, Small, 2014, 10, 3234–3239 CrossRef CAS.
- H. Ghasemi, G. Ni, A. M. Marconnet, J. Loomis, S. Yerci, N. Miljkovic and G. Chen, Nat. Commun., 2014, 5, 4449 CrossRef CAS PubMed.
- L. Zhou, Y. Tan, J. Wang, W. Xu, Y. Yuan, W. Cai, S. Zhu and J. Zhu, Nat. Photonics, 2016, 10, 393–398 CrossRef CAS.
- F. Zhao, X. Zhou, Y. Shi, X. Qian, M. Alexander, X. Zhao, S. Mendez, R. Yang, L. Qu and G. Yu, Nat. Nanotechnol., 2018, 13, 489–495 CrossRef CAS.
- X. Li, J. Li, J. Lu, N. Xu, C. Chen, X. Min, B. Zhu, H. Li, L. Zhou, S. Zhu, T. Zhang and J. Zhu, Joule, 2018, 2, 1331–1338 CrossRef CAS.
- Y. Shi, R. Li, Y. Jin, S. Zhuo, L. Shi, J. Chang, S. Hong, K. C. Ng and P. Wang, Joule, 2018, 2, 1171–1186 CrossRef CAS.
- H. Shan, Z. Ye, J. Yu, R. Wang and Z. Xu, Device, 2023, 1, 100065 CrossRef.
- L. Yang, T. Sun, J. Tang, Y. Shao, N. Li, A. Shen, J. Chen, Y. Zhang, H. Liu and G. Xue, Nano Energy, 2021, 87, 106163 CrossRef CAS.
- P. Poredoš, J. Gao, H. Shan, J. Yu, Z. Shao, Z. Xu and R. Wang, Nat. Commun., 2024, 15, 7890 CrossRef.
- N. Xu, J. Li, Y. Wang, C. Fang, X. Li, Y. Wang, L. Zhou, B. Zhu, Z. Wu, S. Zhu and J. Zhu, Sci. Adv., 2019, 5, eaaw7013 CrossRef CAS.
- G. Ni, S. H. Zandavi, S. M. Javid, S. V. Boriskina, T. A. Cooper and G. Chen, Energy Environ. Sci., 2018, 11, 1510–1519 RSC.
- L. Zhang, X. Li, Y. Zhong, A. Leroy, Z. Xu, L. Zhao and E. N. Wang, Nat. Commun., 2022, 13, 849 CrossRef CAS.
- Y. Zhang, H. Zhang, T. Xiong, H. Qu, J. J. Koh, D. K. Nandakumar, J. Wang and S. C. Tan, Energy Environ. Sci., 2020, 13, 4891–4902 RSC.
- Z. Xu, J. Yu, H. Shan, J. Wang, J. Gao, Z. Ye and R. Wang, Energy Environ. Sci., 2023, 16, 5325–5338 RSC.
- J. Gao, L. Zhang, J. You, Z. Ye, Y. Zhong, R. Wang, E. N. Wang and Z. Xu, Joule, 2023, 7, 2274–2290 CrossRef.
- Y. Song, S. Fang, N. Xu, M. Wang, S. Chen, J. Chen, B. Mi and J. Zhu, Science, 2024, 385, 1444–1449 CrossRef CAS PubMed.
- L. Zhang, Z. Xu, L. Zhao, B. Bhatia, Y. Zhong, S. Gong and E. N. Wang, Energy Environ. Sci., 2021, 14, 1771–1793 RSC.
- W. Xu, X. Hu, S. Zhuang, Y. Wang, X. Li, L. Zhou, S. Zhu and J. Zhu, Adv. Energy Mater., 2018, 8, 1702884 CrossRef.
- Y. Xia, Q. Hou, H. Jubaer, Y. Li, Y. Kang, S. Yuan, H. Liu, M. W. Woo, L. Zhang, L. Gao, H. Wang and X. Zhang, Energy Environ. Sci., 2019, 12, 1840–1847 RSC.
- M. A. Abdelsalam, M. Sajjad, A. Raza, F. AlMarzooqi and T. J. Zhang, Nat. Commun., 2024, 15, 874 CrossRef CAS.
- H. Li, W. Zhang, X. Liao and L. Xu, Sci. Adv., 2024, 10, eado1019 CrossRef PubMed.
- Y. Wang, X. Wu, P. Wu, H. Yu, J. Zhao, X. Yang, Q. Li, Z. Zhang, D. Zhang, G. Owens and H. Xu, J. Mater. Chem. A, 2022, 10, 14470–14478 RSC.
- S. Sun, C. Shi, Y. Kuang, M. Li, S. Li, H. Chan, S. Zhang, G. Chen, A. Nilghaz, R. Cao and J. Tian, Water Res., 2022, 226, 119279 CrossRef CAS PubMed.
- Y. Shi, C. Zhang, R. Li, S. Zhuo, Y. Jin, L. Shi, S. Hong, J. Chang, C. Ong and P. Wang, Environ. Sci. Technol., 2018, 52, 11822–11830 CrossRef CAS.
- Y. Kuang, C. Chen, S. He, E. M. Hitz, Y. Wang, W. Gan, R. Mi and L. Hu, Adv. Mater., 2019, 31, 1–8 CrossRef.
- Y. Wang, W. Zhao, Y. Lee, Y. Li, Z. Wang and K. C. Tam, Nat. Commun., 2024, 15, 6157 CrossRef CAS PubMed.
- N. Xu, H. Zhang, Z. Lin, J. Li, G. Liu, X. Li, W. Zhao, X. Min, P. Yao, L. Zhou, Y. Song, B. Zhu, S. Zhu and J. Zhu, Nat. Sci. Rev., 2021, 8, nwab065 CrossRef.
- L. Wu, Z. Dong, Z. Cai, T. Ganapathy, N. X. Fang, C. Li, C. Yu, Y. Zhang and Y. Song, Nat. Commun., 2020, 11, 521 CrossRef CAS.
- W. Zhao, H. Gong, Y. Song, B. Li, N. Xu, X. Min, G. Liu, B. Zhu, L. Zhou, X. X. Zhang and J. Zhu, Adv. Funct. Mater., 2021, 31, 2100025 CrossRef CAS.
- X. Hao, H. Yao, P. Zhang, Q. Liao, K. Zhu, J. Chang, H. Cheng, J. Yuan and L. Qu, Nat. Water, 2023, 1, 982–991 CrossRef.
- F. Wang, N. Xu, W. Zhao, L. Zhou, P. Zhu, X. Wang, B. Zhu and J. Zhu, Joule, 2021, 5, 1602–1612 CrossRef CAS.
- W. Wang, S. Aleid, Y. Shi, C. Zhang, R. Li, M. Wu, S. Zhuo and P. Wang, Joule, 2021, 5, 1873–1887 CrossRef.
- Z. Xu, L. Zhang, L. Zhao, B. Li, B. Bhatia, C. Wang, K. L. Wilke, Y. Song, O. Labban, J. H. Lienhard, R. Wang and E. N. Wang, Energy Environ. Sci., 2020, 13, 830–839 RSC.
- M. J. Qazi, H. Salim, C. A. W. Doorman, E. Jambon-Puillet and N. Shahidzadeh, Sci. Adv., 2019, 5, eaax1853 CrossRef CAS PubMed.
- S. Veran-Tissoires and M. Prat, J. Fluid Mech., 2014, 749, 701–749 CrossRef CAS.
- E. W. Washburn, Phys. Rev., 1921, 17, 273–283 CrossRef.
- J. Bear and Y. Bachmat, Introduction to modeling of transport phenomena in porous media, 1990.
- J. S. Jiang, R. H. Guo, Y. S. Chiu and C. C. Hua, Soft Matter, 2018, 14, 9786–9797 RSC.
- C. T. K. Finnerty, A. K. Menon, K. M. Conway, D. Lee, M. Nelson, J. J. Urban, D. Sedlak and B. Mi, Environ. Sci. Technol., 2021, 55, 15435–15445 CrossRef CAS PubMed.
- K. Yang, T. Pan, S. Dang, Q. Gan and Y. Han, Nat. Commun., 2022, 13, 6653 CrossRef CAS.
- Y. Shao, J. Tang, N. Li, T. Sun, L. Yang, D. Chen, H. Zhi, D. Wang, H. Liu and G. Xue, EcoMat, 2020, 2, e12018 CrossRef.
- C. Finnerty, L. Zhang, D. L. Sedlak, K. L. Nelson and B. Mi, Environ. Sci. Technol., 2017, 51, 11701–11709 CrossRef CAS PubMed.
- X. Chen, M. Yang, S. Zheng, F. Temprano-Coleto, Q. Dong, G. Cheng, N. Yao, H. A. Stone, L. Hu and Z. J. Ren, Nat. Water, 2023, 1, 808–817 CrossRef.
- Q. Chen, M. Burhan, M. W. Shahzad, D. Ybyraiymkul, F. H. Akhtar, Y. Li and K. C. Ng, Desalination, 2021, 502, 114928 CrossRef CAS.
- J. Eke, A. Yusuf, A. Giwa and A. Sodiq, Desalination, 2020, 495, 114633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04741a |
| This journal is © The Royal Society of Chemistry 2025 |