
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the mechanistic complexity of the oxygen evolution reaction and Ir dissolution in highly dimensional amorphous hydrous iridium oxides†
Marianne
van der Merwe
 *a,
Yonghyuk
Lee
*b,
Romualdus Enggar
Wibowo
a,
Tathiana
Kokumai
a,
Anna
Efimenko
ac,
Mauricio D.
Arce
ad,
Catalina E.
Jimenez
a,
Benjamin
Howchen
a,
Rosario
Suarez Anzorena
ae,
Ilaria
Lucentini
f,
Carlos
Escudero
f,
Götz
Schuck
g,
Zdravko
Kochovski
h,
Marco
Favaro
i,
David E.
Starr
i,
Karsten
Reuter
b,
Christoph
Scheurer
bj,
Marcus
Bär
ackl and
Raul
Garcia-Diez
*a
*a,
Yonghyuk
Lee
*b,
Romualdus Enggar
Wibowo
a,
Tathiana
Kokumai
a,
Anna
Efimenko
ac,
Mauricio D.
Arce
ad,
Catalina E.
Jimenez
a,
Benjamin
Howchen
a,
Rosario
Suarez Anzorena
ae,
Ilaria
Lucentini
f,
Carlos
Escudero
f,
Götz
Schuck
g,
Zdravko
Kochovski
h,
Marco
Favaro
i,
David E.
Starr
i,
Karsten
Reuter
b,
Christoph
Scheurer
bj,
Marcus
Bär
ackl and
Raul
Garcia-Diez
*a
aInterface Design, Helmholtz Zentrum Berlin für Materialien und Energie GmbH (HZB), Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: marianne.vdm@helmholtz-berlin.de; raul.garcia_diez@helmholtz-berlin.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, 14195 Berlin, Germany. E-mail: ylee@fhi-berlin.mpg.de
cEnergy Materials In situ Laboratory Berlin (EMIL), HZB, Albert-Einstein-Str. 15, 12489 Berlin, Germany
dDepartamento Caracterización de Materiales, INN-CNEA-CONICET, Centro Atómico Bariloche, Av. Bustillo 9500, S. C. de Bariloche, Rio Negro, 8400, Argentina
eUNIDEF, CONICET, MINDEF, Departamento de Investigaciones en Sólidos, CITEDEF, J.B. de La Salle 4397, B1603ALO Villa Martelli, Pcia. de Buenos Aires, Argentina
fALBA Synchrotron Light Source, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Barcelona, Spain
gDepartment Structure and Dynamics of Energy Materials, HZB, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
hInstitute of Electrochemical Energy Storage, HZB, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
iInstitute for Solar Fuels, HZB, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
jIET-1 Fundamental Electrochemistry, FZJ, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
kDepartment of Chemistry and Pharmacy, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Egerlandstr. 3, 91058 Erlangen, Germany
lDepartment of X-ray Spectroscopy at Interfaces of Thin Films, Helmholtz Institute Erlangen-Nürnberg for Renewable Energy (HI ERN), Albert-Einstein-Str. 15, 12489 Berlin, Germany
First published on 29th November 2024
Abstract
Understanding the oxygen evolution reaction (OER) and Ir dissolution mechanisms in amorphous, hydrous iridium oxides (am-hydr-IrOx) is hindered by the reliance on crystalline iridium oxide theoretical models to interpret its behaviour. This study presents a comprehensive investigation of hydrous iridium oxide thin films (HIROFs) as a model for am-hydr-IrOx to elucidate electronic and structural transformations under OER conditions of proton exchange membrane water electrolyzers (PEM-WE). Employing in situ and operando Ir L3-edge X-ray absorption spectroscopy supported by density functional theory calculations, we introduce a novel surface H-terminated nanosheet model that better characterizes the short-range structure of am-hydr-IrOx compared to previous crystalline models, which exhibits elongated Ir–O bond lengths compared to rutile-IrO2. This atomic model unveils the electronic and structural transformations of am-hydr-IrOx, progressing from H-terminated nanosheets to structures with multiple Ir vacancies and shorter bond-lengths at OER potentials. Notably, Ir dissolution emerges as a spontaneous, thermodynamically driven process, initiated at potentials lower than OER activation, which requires a parallel mechanistic framework describing Ir dissolution by Ir defect formation. Moreover, our results provide mechanistic insights into the activity-stability relationship of am-hydr-IrOx by systematically screening the DFT-calculated OER activity of diverse Ir and O chemical environments. This work challenges conventional perceptions of iridium dissolution and OER mechanisms in am-hydr-IrOx, providing an alternative perspective within a dual-mechanistic framework.
Broader contextWater electrolysis is crucial for effective harnessing energy coupling with intermittent renewable energy sources like wind and solar energy, being iridium the state-of-the-art anode catalyst for the sluggish oxygen evolution reaction (OER). However, its scarcity drives efforts towards low loading designs with high catalyst activities and low Ir dissolution rates, where the catalyst-specific activity-stability trade-off is particularly relevant. A preeminent candidate, amorphous, hydrous iridium oxide (am-hydr-IrOx), exhibits higher activity but poorer stability compared to benchmark anhydrous rutile-IrO2, urging for the understanding of the intricate relationship between these processes and for the atomistic description of the underlying (electro)chemical mechanisms. While mechanistic insights based on rutile-IrO2 models have been proposed, these explanatory frameworks derived from highly ordered materials are insufficient for describing the amorphous and hydroxylated suboxide nature of the highly active am-hydr-IrOx. In this study, we combine results derived from in situ and operando characterization techniques (e.g., synchrotron-based X-ray spectroscopies) with density functional theory calculations to derive a comprehensive model that better describes the highly dimensional structure of am-hydr-IrOx. The proposed theoretical approach is capable of disentangling the OER-limiting steps from the precious metal dissolution process and arises as an efficient model to better describe highly active, amorphous catalysts for water splitting. |
Introduction
Water electrolysis (WE) is vital for global sustainable and renewable energy systems.1 Efficient and stable anode materials for the oxygen evolution reaction (OER) are essential for the advancement of proton exchange membrane (PEM) WE technology, with iridium-based materials being state-of-the-art.2,3 A preeminent OER candidate material is highly active amorphous, hydrous iridium oxide (am-hydr-IrOx). However, its technological application is hindered by high iridium dissolution rates.4,5 Thus, the realization of stable am-hydr-IrOx based catalysts relies crucially on understanding the mechanisms underlying OER and Ir dissolution. Numerous studies have been devoted to deciphering activity-stability relationships in iridium oxides,6–9 where the crystal structure was reported to strongly influence its properties, with am-hydr-IrOx exhibiting superior OER activity but inferior stability compared to its crystalline-IrO2 counterparts.10–12 Complementary mechanisms have been proposed to explain the different OER performance of these two material classes,13–17 namely: the adsorbate evolution mechanism (AEM)18–20 and the lattice oxygen evolution mechanism (LOER).21–23 These classical mechanisms describe transitions in the oxidation state (coordination) of the active Ir species during the 4-electron transfer process, with AEM prevailing in crystalline oxides and LOER in amorphous (hydrous) iridium oxides. For the latter, the participation of lattice oxygen in the OER mechanism is suggested to be directly related to the higher dissolution of Ir atoms, supposedly via unstable valent states,24,25 which paved the way to the widely accepted entangled mechanism explaining the strong activity-instability correlation in these Ir oxides. Yet the use of crystal-based theoretical model interpretations26–30 limits the ability to comprehensively describe iridium oxide variants without any long-range order, like am-hydr-IrOx. The high structural complexity of these amorphous materials, linked to the presence of bulk defects, constitutes an insurmountable translational gap for these crystalline models, which we aim to address in this study.Consequently, we investigated hydrous iridium oxide thin films (HIROFs) as a model system for am-hydr-IrOx exhibiting a singular iridium suboxide species typically related to a high OER activity.31–33 The complex structure of this hydroxylated porous electrochemically grown oxide requires appropriate theoretical models to interpret its OER activity and stability4,33 beyond conventional crystalline-IrO2 approaches. Furthermore, lack of long-range order necessitates the use of experimental techniques capable of probing the local electronic and geometric structural configurations of the material under relevant operational conditions. In situ X-ray absorption spectroscopy in combination with density functional theory (DFT) emerges as a suitable tool to unveil the complementary mechanisms governing the OER activity of am-hydr-IrOx and its low stability caused by high Ir dissolution. Using an overarching dual-mechanistic theoretical framework, unique insights into Ir dissolution occurring already below the onset of OER, and the predicted high activity of electrophilic O-species26,28–30,34,35 and metal Ir-sites are revealed and discussed in this study. The study's integrated approach offers a valuable methodology for understanding and optimizing OER and Ir dissolution mechanisms, thereby advancing the development of efficient and stable am-hydr-IrOx-based anode materials.
Results
In situ synthesis of a highly porous am-hydr-IrOx
This work studies the structure of am-hydr-IrOx and its OER and Ir dissolution mechanisms by investigating hydrous iridium oxide thin film (HIROF) as a model system. HIROF is produced by the controlled electrochemical oxidation of a metallic Ir (Ir0) substrate by continuous cycling between reductive and oxidative potentials, resulting in the monolayer-by-monolayer growth of am-hydr-IrOx31 (Fig. 1(a)). The fast oxidation kinetics coupled with slower reduction kinetics36 during the rapid cycling results in the formation of highly porous, hydrated am-hydr-IrOx with large number of bulk defects.33,37 This ‘3-dimensional’ electrocatalyst features a high density of catalytically active sites allowing for near-complete participation of all Ir atoms in redox reactions,4,33 as indicated by the strong redox peaks in the pseudo-capacitive region of the cyclic voltammograms (CVs), increasing with the number of growth cycles (Fig. 1(b)). Interestingly, the double-layer region and hydrogen underpotential deposition (HUPD) regions remain mostly unchanged during the growth, proportional only to the constantly accessible surface of the underlying metallic Ir substrate.11,31,38 The am-hydr-IrOx layer acts as a switchable conductor, becoming active for anodic reactions (such as redox and OER) and inactive for cathodic reactions (like HUPD), which are only characterized by the underlying metallic Ir substrate (see Fig. S1 in Supplementary Note 1, ESI†). The poor electronic transport of highly hydrated iridium oxides at potentials below 0.6 VRHE is likely hindering the contribution of the evolving surface at this lower potential window. Cryogenic Transmission Electron Microscopy (cryo-TEM) ought to show the highly disordered nature of am-hydr-IrOx measurements in its native acidic aqueous environment (Fig. 1(c)), responsible for its amorphous porous structure and low density32 (∼1–2 g cm−3 compared to ρrutile-IrO2 = 11.6 g cm−3, refer to Supplementary Note 2 for calculation details, ESI†). More details on the electrochemical behaviour of am-hydr-IrOx as well as the comparison with metallic Ir and rutile-IrO2 can be found in Supplementary Note 3 (ESI†) and extensively elsewhere.4,11,13,33 | ||
| Fig. 1 Electronic synthesis and characterization of am-hydr-IrOx. (a) Schematic of the electrochemical oxidation of Ir0 into HIROFn, where n is the number of growth cycles. (b) Cyclic voltammograms (CVs) acquired at intervals of 100 cycles at 50 mV s−1 during HIROF growth to 1000 cycles (HIROF1000), exhibiting distinct redox processes. (c) Cross-section scanning electron microscopy (SEM) image of HIROF grown to 600 cycles from a 100 nm metallic Ir substrate. (d) Cryogenic transmission electron microscopy (cryo-TEM) image of am-hydr-IrOx. (e) Ir 4f spectra of am-hydr-IrOx obtained by Al Kα ultra-high vacuum X-ray photoelectron spectroscopy (UHV XPS, light blue) and in situ “dip-and-pull” near-ambient pressure hard X-ray photoelectron spectroscopy (NAP-HAXPES, dark blue) at 12 mbar using 3 keV, see Supplementary Notes 5 and 6 (ESI†), respectively, for further details. Spectra were normalized to the Ir 4f7/2 peak maximum. UHV XPS Ir 4f spectra of metallic Ir (Ir0, grey), Ir 4f7/2 at 60.8 eV binding energy, and rutile-IrO2 reference (Ir4+, black), Ir 4f7/2 at 61.5 eV, is shown for comparison. The dotted vertical lines mark the maximum intensity of the 4f7/2 peaks. (f) In situ Ir L3-edge X-ray absorption near edge structure (XANES) recorded at intervals during HIROF growth to 1000 cycles. Spectra of Ir0 (grey) and rutile-IrO2 (black) are shown as references for the white-line (WL) positions of Ir0 and Ir4+. (g) In situ k2-weighted Fourier transforms (FTs) of the Ir L3-edge Extended X-ray Absorption Fine Structure (EXAFS) recorded in intervals during the HIROF growth to 1000 cycles. Ir0 (grey) and IrO2 (black) references are shown for comparison, with vertical lines marking the ‘Ir–O’ and ‘Ir0–Ir0’ first coordination shell scattering peaks. FT analysis highlights the unique local geometric structure of am-hydr-IrOx (Ir–O scattering peak at longer bond lengths compared to rutile-IrO2). (h) (i) In situ recorded Ir L3-edge WL position, (ii) mole fraction determined from EXAFS fittings, and (iii) am-hydr-IrOx film thickness determined by CV charge integration as a function of growth cycles. Horizontal dashed lines in (i) indicate the Ir0 and rutile-IrO2 reference WL positions. The solid grey line in (ii) indicates the fitted second-order polynomial trend line. Plotted in black squares in (iii) are the am-hydr-IrOx film thicknesses determined from cross-section SEM images, for HIROF grown to 600 and 800 cycles. | ||
The chemical nature of am-hydr-IrOx was probed under hydrated conditions by “dip-and-pull” technique39–41in situ near ambient pressure hard X-ray photoelectron spectroscopy (NAP-HAXPES), which exhibited a single electronic state with a Ir 4f binding energy (BE) position higher than anhydrous rutile-IrO2 and similar to that predicted for the trivalent Ir-defect site in rutile-IrO226 (Fig. 1(d), and complementary O 1s spectra in Fig. S8 in Supplementary Note 4, ESI†). This observation aligns with previous studies on highly hydroxylated Ir(III)OOH nanosheets42 and other am-IrOx.29,43 Under ultra-high vacuum (UHV) conditions, am-hydr-IrOx exhibits a broader Ir 4f peak shape (light blue spectra in Fig. 1(d)) explained by a higher contribution of Ir4+ in the analytical fit and lattice-O in the O 1s spectrum (Fig. S11 in Supplementary Note 5, ESI†), associated with its (oxy)hydroxide nature (refer to Supplementary Notes 4 and 5 for more details of the analytical fits, ESI†). Presumably, UHV conditions cause dehydration44,45 resulting in mixed iridium states (Fig. S12 in Supplementary Note 5, ESI†), whereas hydrated conditions preserve am-hydr-IrOx's hydroxylated form in a single electronic state. This hydroxylated iridium suboxide species in am-hydr-IrOx is of prime interest, as it has often been associated with sites with high intrinsic OER activity.9,26 These catalytic sites involve coordinatively unsaturated electrophilic O-species situated at Ir defect sites.26,29,30
The electronic and local geometric structure of am-hydr-IrOx was further investigated under hydrated conditions by means of bulk-sensitive in situ Ir L3-edge X-ray absorption near-edge structure (XANES) (Fig. 1(e)) and extended X-ray absorption fine structure (EXAFS) measurements (Fig. 1(f)) during the growth of HIROF to 1000 cycles. The evolving XANES and EXAFS spectra confirm near-complete conversion of Ir0 into am-hydr-IrOx. Am-hydr-IrOx exhibits an Ir L3-edge white-line (WL) position at lower excitation energies than rutile-IrO2 (Fig. 1(e) and (g)-(i)), confirming its unique suboxide state, consistent with our in situ NAP-HAXPES observations. Complementarily, Fourier transform (FT) analyses of the k2-weighted Ir L3-edge EXAFS spectra (Fig. S14, ESI†) reveal the unique geometrical structure of am-hydr-IrOx (Fig. 1(f)). The evolving FT spectra (green to blue in Fig. 1(f)) reveal the conversion of the metallic Ir substrate (scattering peak at ∼2.26 Å attributed to the first-shell Ir0–Ir0 in the metallic Ir reference) to am-hydr-IrOx, with a characteristic Ir–O scattering peak at ∼1.68 Å (blue spectra in Fig. 1(f)). This indicates that am-hydr-IrOx has a longer Ir–O bond length compared to crystal rutile-IrO2, which has an Ir–O scattering peak at ∼1.61 Å. This demonstrates the distinctly different structure of am-hydr-IrOx in comparison to rutile-IrO2, which we consider as important as the Ir–O bond length is proposed to be a fundamental factor influencing OER activity.46,47
The characteristic, dominating spectral fingerprint of am-hydr-IrOx after 1000 cycles of HIROF growth confirms its successful preparation with a thickness sufficient to dominate the Ir L3-edge XAS spectroscopic signal. Quantitative analysis of the first-shell Ir0–Ir0 and Ir–O scattering paths in the EXAFS spectra (see Supplementary Note 6 for the fits of the EXAFS data, ESI†) was used to determine the molar fractions of the Ir0 and am-hydr-IrOx in the total generated HIROF sample after 1000 cycles. These analyses reveal an ∼80% molar contribution of am-hydr-IrOx in HIROF after 1000 cycles (Fig. 1(g)-(ii)), corresponding to a thickness of ∼300 nanometres (Fig. 1(g)-(iii)), as calculated from charge integration of the main redox couple (refer to Supplementary Note 1 for CV integration details, ESI†) and cross-validated by cross-section SEM images. CVs reveal negligible differences in the characteristic features after ∼800 cycles (Fig. 1(b)), pointing towards increased mass transport limitations within the film.33 Continued growth beyond 1000 cycles is undesirable as this would lead to eventual am-hydr-IrOx layer detachment due to mechanical instability.48
Our results collectively highlight the necessity for a revised structural model tailored to am-hydr-IrOx, considering its unique electronic and structural properties (singular, hydroxylated Ir suboxide species, ‘3-dimensional’ nature, high active site density, and longer Ir–O bond length) challenging the use of conventional rutile-IrO2 based theoretical interpretations. Utilization of highly periodic structures would be unsuitable for modelling materials like am-hydr-IrOx which exhibit only short-range order, and also possess some amount of defects. This also introduces the likelihood of a diverse array of distinct possible active sites associated with different chemical environments. Therefore, novel structures, more accurately representing the physicochemical properties of am-hydr-IrOx, which dictate its OER behaviour, are required.
Revealing the structural configuration of am-hydr-IrOx by theory-guided evaluation of in situ Ir L3-edge XAS data
A comprehensive screening of structures that could model am-hydr-IrOx was performed by comparing the experimental in situ Ir L3-edge EXAFS data of HIROF1000 with various atomistic models. These models range from more periodic rutile-types to flexible structures, with well-defined edges like stacked-sheets or nanosheets, potentially forming water containing “hydrogel” networks, similar to previous work on Ru oxides.49 Given the highly porous nature, large surface area, and the monolayer-by-monolayer growth31 of am-hydr-IrOx, we expect highly flexible arrangements allowing for water intercalation to best describe am-hydr-IrOx. For this reason, sheet-like structures have been proposed to complement the crystal-like periodic models. Providing enough lateral periodicity to mirror the short-range order observed experimentally, finite sheet structures offer a variety of local motifs at the edges that can mimic the defective environments and coordinatively unsaturated O-edge sites found in bulk vacant sites associated with high activity.9,29 With sufficient size to bridge the mesoscale of the studied material with the computational limitations of current quantum chemistry, these nanosheets exhibit high mobility of charge carriers, allowing for good electronic and ionic transport in the material, as expected from a highly active OER electrocatalyst as am-hydr-IrOx. For all the proposed models, various surface terminations (e.g., *H, *O, *OH, *OOH, and their numerous combinations) and different degrees of surface coverages (Θi) were considered. All the structures have been pre-selected by ab initio thermodynamics (i.e., most stable structures from a large structural manifold with a formal theoretical basis for their existence).Individual fits of the experimental EXAFS data were performed using every atomic model, constraining their interatomic distances and coordination numbers obtained after structural relaxation (see Supplementary Note 7 (ESI†) for details on the EXAFS fittings and on the figure-of-merit parameter – 1/r-factor – used to quantify the EXAFS goodness-of-fit). This systematic approach, focusing on EXAFS fitting of the short-range order, served as an efficient filter to select the most probable structure families and surface terminations to describe am-hydr-IrOx. Fig. 2(a) shows the EXAFS goodness-of-fit of the different structure-termination-permutations considered, which describes their ability to mimic the elongated Ir–O bond of am-hydr-IrOx.
 | ||
| Fig. 2 Electronic and local geometric structure of am-hydr-IrOx. (a) EXAFS goodness of fit values (1/r-factor from the EXAFS fit results) for a library of atomic models differentiated by atomic model families and surface termination types. Atomic model families include rutile-IrO2, stacked sheets, and nanosheet structures. Surface termination types were divided into H-terminated, bare, and oxo, peroxo, superoxo groups. The radii of the circles correspond to goodness of EXAFS fit values. Overall, nanosheets describe the experimental data the best. See Supplementary Note 11 (ESI†) for more details of EXAFS fittings. (b) Ball and stick perspective and top model views of the nanosheet with a 0.50 monolayer H-coverage (ΘH = 0.50 ML). The threefold-coordinated O3f and sixfold-coordinated Ir6f surface species constituting the nanosheet surface are highlighted. At the nanosheet edges, the coordinatively unsaturated (cus) Ircus, onefold-coordinated terminal Ocus, and twofold-coordinated bridging (br) Obr species are highlighted. (c) EXAFS goodness of fit values (1/r-factor from the EXAFS fit results) as a function of H-coverage (ΘH) for EXAFS fits using nanosheet structures with varying H-coverages. (d) in situ k2-weighted FT spectra of HIROF1000 with EXAFS fits performed using an H-terminated (ΘH = 0.50 ML) nanosheet structure (in blue) compared to employing the conventional bulk rutile (tetragonal) IrO2 structure (in red) for the am-hydr-IrOx component (molar fraction = 80%). Elemental fcc (face-centered cubic) Ir was used for the metallic Ir substrate component (molar fraction = 20%). | ||
Nanosheet structures exhibited the highest goodness-of-fit (largest circle in Fig. 2(a)), and thus is the best atomistic model to describe the structural properties of am-hydr-IrOx, along with stacked-sheet structures. These results suggest a structured network composed of pores and channels, where single nanosheets likely describe the walls, and stacked sheets are indicative of the joints. This amorphous architecture likely facilitates efficient water transport between interconnected nanostructures, mirroring the highly porous structure of am-hydr-IrOx. Nanosheets consist of hydrogen atoms adsorbed preferentially atop the threefold-coordinated O atoms (O3f), which are bonded to sixfold-coordinated Ir atoms (Ir6f) (Fig. 2(b)). At the nanosheet edges, coordinatively unsaturated Ir sites (Ircus) are present with O existing as both onefold-coordinated terminal (Ocus) species and as twofold-coordinated bridging O-atoms (Obr). These edge sites can be used as a proxy to simulate bulk defective environments likely found in am-hydr-IrOx, which are also analogous to electrophilic μ1-O and μ2-O species in the Ir-defect site of the rutile-IrO2 structural model as discussed elsewhere.9,29,30,35 The optimal H-coverage (ΘH) was determined to be 0.50 monolayer (ML) (Fig. 2(c) and (d)). Structural models with higher ΘH values were thermodynamically unfavourable, e.g., undergoing Ir–O bond breaking during the structural relaxation; therefore, these models were not considered in the analysis of the experimental data. In fact, the low current densities measured at the double-layer region of the CV (Fig. 1(b)) likely originate from the low electronic conductivity typically associated with highly hydrated structures, as the nanosheet found to best fit our data. The use of the nanosheet atomistic model with ΘH = 0.50 ML in the EXAFS fitting (Fig. 2(d)) illustrates its improved description of the electronic and structural properties of am-hydr-IrOx in comparison with conventionally adopted rutile-IrO2. This supports experimental observations by in situ NAP-HAPXES and in situ Ir L3-edge XANES and EXAFS measurements (Fig. 1), highlighting the distinctly different chemical environment in am-hydr-IrOx (e.g., hydroxylated Ir suboxide and longer Ir–O bond, 2.04 Å) in comparison to rutile-IrO2 (e.g., anhydrous Ir4+ and shorter Ir–O bond, 1.98 Å). Our results bridge a critical gap in literature by unveiling the (until now) undefined structure of highly hydroxylated iridium suboxide materials, better describing the higher surface area and flexible morphology of am-hydr-IrOx.
This advances our ability to investigate and understand the electronic and structural changes of am-hydr-IrOx under potential application, providing a coherent theoretical–experimental framework to interpret the intricate mechanisms that define its OER electrocatalytic behaviour (i.e., activity and stability). The unique structure of the nanosheet suggests the presence of various potential active sites (Ir6f, O3f and the edge-related Ircus, Ocus and Obr sites mimicking bulk defects) and thus hinting at the co-existence of multiple OER reaction pathways in am-hydr-IrOx, beyond the commonly assumed mechanisms based on rutile-IrO2 surfaces.9,13,22,26,34,35 The following section aims to interpret the potential-induced transformations of am-hydr-IrOx using this newly introduced H-terminated nanosheet model.
Electronic and structural transformations of am-hydr-IrOx under potential application
Electronic and structural changes of am-hydr-IrOx under potential application were investigated by operando Ir L3-edge XANES and EXAFS, along with Fixed Energy X-ray Absorption Voltammetry (FEXRAV),50 with three distinct regimes: “pre-redox”, “redox” and “OER” (Fig. 3). | ||
| Fig. 3 Electronic and local geometric structural transformation of am-hydr-IrOx under potential application. (a) Operando Ir L3-edge white-line (WL) position as a function of applied potential. Horizontal dashed grey lines indicate the Ir0 and rutile-IrO2 reference WL positions. Fixed energy X-ray voltammetry (FEXRAV) measurement at the WL position of Ir4+ (11219.5 eV) is shown in orange in (a) and in (b) its corresponding CVs acquired at 2 mV s−1 is shown in blue. The FEXRAV 1st derivative is shown in orange in (b). (c) First coordination shell Ir–O distance of am-hydr-IrOx as a function of applied potential, obtained by operando Ir L3-edge EXAFS fits using the H-terminated nanosheet (ΘH = 0.50 ML) structure. The horizontal dashed grey line indicates the first coordination shell Ir–O distance of the rutile-IrO2 reference. | ||
At potentials (<∼0.60 VRHE), characterized by near-featureless CV response indicating minimal charge transfer processes, am-hydr-IrOx exhibits subtle changes in its electronic and structural configuration, evidenced by a slight WL shift (Fig. 3(a)) and minor changes in its Ir–O bond length (Fig. 3(c)).
A more significant WL shift occurs between ∼0.60 VRHE and ∼1.23 VRHE (Fig. 3(a)). This potential region is typically attributed to a Ir3+ ↔ Ir4+ redox couple with a maximum around 0.97 VRHE and a smaller pre-peak at 0.70 VRHE with various attributions51,52 (Fig. 3(b)). The potentiodynamic FEXRAV measurement (orange curve in Fig. 3(a)), which monitors the spectral intensity change at a fixed excitation energy of 11219.5 eV (WL of rutile-IrO2) during cyclic voltammetry,50 provides insights into the reversibility of the main redox couple, associated with the first major electronic transformation of am-hydr-IrOx. The FEXRAV 1st derivative signal (orange line in Fig. 3(b)) closely aligns with the oxidation/reduction profiles of the main redox peak (blue line in Fig. 3(b)), highlighting the reversibility of the oxidation/reduction process. Furthermore, the Ir–O bond length also starts to decrease within this regime (Fig. 3(c)).
A third distinctive region is observed under OER relevant potentials (>1.23 VRHE), where the WL energy position does not significantly change but a stronger contraction of the Ir–O bond length is evident (Fig. 3(c)).
The observed trends in the WL position shifts and Ir–O bond length contraction of am-hydr-IrOx agree with the trends found in other studies on amorphous iridium oxides,53–59 however, limited interpretations of their electronic behaviour are provided, and mainly based on rutile-IrO2 models. Our alternative approach, using the highly dimensional, amorphous and flexible H-terminated nanosheet model, establishes a dual-mechanistic theoretical framework based on DFT-predicted structural transformations of the nanosheet (Fig. 4). The proposed model supports the experimental WL position and Ir–O bond length data trends of am-hydr-IrOx and attempts to unify in a unique explanatory framework the dissolution of Ir with the oxidation behaviour extensively observed in the redox and OER regions, thereby providing deeper insights into the electronic and structural transformation processes occurring across the three potential regions and the correlations between both mechanisms.
 | ||
| Fig. 4 DFT supported analysis of the in situ Ir L3-edge XAS of am-hydr-IrOx at applied potentials. (a) and (b) Phase diagrams showing the stability of the different terminations of the iridium nanosheet surface as a function of applied potential, following the dual-mechanism framework of (a) the chemical behaviour (i.e., deprotonation and oxidation) and (b) Ir defect formation. The vertical dotted lines indicate the OER thermodynamic equilibrium potential of U = 1.23 V. At the top of each of the plots are marked the potential regions associated with the pre-redox, redox and OER regimes. Perspective atomistic views of the deprotonation of the nanosheet (ΘH = 0.50–0 ML) and spontaneous Ir dissolution (1VIr, 2VIr, to 3VIr) in the nanosheet are shown under (a) and (b), respectively, highlighting the fully coordinated Ir6f and O3f species, and dangling O2f and O1f species. (c) Ir 5d derived partial density of states (pDOS) of the most stable structures predicted by the phase diagrams in (a) and (b). For comparison, the pDOS of rutile-IrO2 is also shown. Vertical dashed lines give the energy position of the weighted centroid of the unoccupied states. (d) Comparison of the experimental and theoretical Ir–O interatomic distances. The operando experimental Ir–O bond lengths are reproduced from Fig. 3(c) and are shown as green triangles with error bars. DFT obtained distributions of Ir–O bond distances from the most stable structures predicted by the phase diagrams are shown by the coloured violin plots. The respective mean values are shown as circles. Black bars delimit the potentials related to the most relevant chemico-structural changes of am-hydr-IrOx. | ||
Dual-mechanistic framework: chemical transformations and spontaneous Ir dissolution
Ab initio thermodynamic analysis was employed to reveal the structural progression of the nanosheet with changing applied potentials. DFT-calculated phase diagrams (Fig. 4(a) and (b)) illustrate the nanosheet structures’ Gibbs free energy of formation (ΔG) versus electrode potential, with lower ΔG indicating more thermodynamically favourable structures. Within the proposed dual-mechanistic framework, our analysis juxtaposes Fig. 4(a) the chemical transformations of the nanosheet in the pre-redox and redox potential regions, and Fig. 4(b) the spontaneous loss of iridium atoms from am-hydr-IrOx during OER and at lower applied potentials. For this, Fig. 4(a) focuses on the chemical behaviour (deprotonation and oxidation), crucial for understanding the structural changes under redox potentials and insights into the distinct OER activity of the different sites co-existing in am-hydr-IrOx. Complementary, Fig. 4(b) presents structures related to Ir defect site formation within the pristine nanosheet. Combined, Fig. 4 encapsulates the dual-mechanistic framework providing theoretical interpretations of the activity-stability trade-off in am-hydr-IrOx.4,11,33 Inherent bulk defects in am-hydr-IrOx are modelled by the finite nanosheet edges. Moreover, a small quantity of Ir defects is expected during HIROF growth, given notable Ir dissolution quantities within this potential region (>1 VRHE).4,11,33 Their contribution to deprotonation behaviour is modelled using a nanosheet with a single Ir vacant site, i.e., 1VIr. The vertical lines at U = 1.23 VRHE mark the thermodynamic OER onset potential.
Fig. 4(a) illustrates the DFT-predicted progressive deprotonation of distinct O-sites in the nanosheet, reaching complete deprotonation at high redox-relevant potentials. According to the reaction:
| *O–H → *O + H+ + e− |
Considering the second aspect of the dual-mechanistic framework, i.e., the structural transformations of the nanosheet, our DFT-guided study suggests that the formation of Ir defect sites:
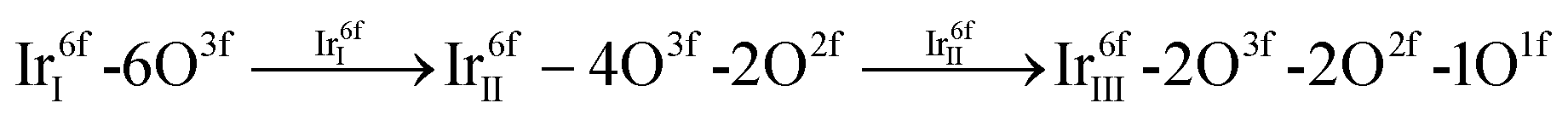
Assuming that the nanosheet atomistic model best describes the electronic and structural properties of am-hydr-IrOx, our DFT-predicted dual-mechanistic framework provides explanatory theories for both chemical transformations (deprotonation and OER intermediate formation) and Ir dissolution processes, and is to the best of our knowledge, the first atomistic model able to describe the process of pronounced Ir dissolution at OER potentials. Based on these premises, this framework predicts several mechanisms (e.g., deprotonation, Ir dissolution and oxidation) occurring with the three potential regions (pre-redox, redox and OER), which align well with the potential-dependent electronic and structural transformations of am-hydr-IrOx observed experimentally by operando Ir L3-edge XANES and EXAFS (Fig. 3).
Beyond using the simplified average Ir oxidation state formalism often adopted in several studies,8,65,66 interpretations of the experimental WL shifts (e.g., electronic transformations of am-hydr-IrOx) presented in this work are based on calculations of the Ir 5d partial density of states (pDOS) (Fig. 4(c)), which correlate with energy reconfigurations of the unoccupied states as probed by the XAS process. This approach avoids assumptions of one-to-one linearity between Ir L3-edge WL position and formal Ir 5d occupancy describing Ir oxidation states, but rather interprets WL position shifts relative to modifications of the Ir 5d energy configurations with respect to the whole electronic structure. Further details on the correlation between the formal oxidation state of the active sites derived from simple electron-counting and the calculated electronic structure (based on the Bader charge analysis) are presented in Supplementary Note 8 (ESI†). O 2p pDOS are shown in Fig. S18 (ESI†) for completeness, although strong Ir–O hybridization in the nanosheets suggests similar trends to those observed in Ir 5d pDOS.
Interestingly, the expected low electronic conductivity of highly hydrated oxides could explain the insulating properties of am-hydr-IrOx at low applied potentials e.g., why at cathodic potentials the electrochemical signal is dominated by the underlying metallic Ir substrate (Fig. 1(b)). In the nanosheet structures with high H-coverages (e.g., ΘH = 0.50 ML in Fig. 4(c)), there appears a large gap in the Ir 5d pDOS, indicating insulating behaviour. As H-coverage decreases with increasing applied potential, the gap closes and Ir 5d states extend across the Fermi level, giving transition to a more conductive state.
The experimentally determined average Ir–O bond lengths obtained by EXAFS fittings correlate well with the trend of the different Ir–O bond lengths of the DFT-predicted nanosheet structures over the three potential regions (bond length distributions presented as violin plots in Fig. 4(d)).
In the pre-redox region below ∼0.60 VRHE, characterized by minimal charge transfer processes, the experimental average Ir–O bond length (Fig. 3(c)) closely aligns with the DFT-predicted mean Ir–O bond lengths of the H-terminated nanosheet (Fig. 4(d)), longer than those of rutile-IrO2. Likewise, the difference in the WL position of am-hydr-IrOx and rutile-IrO2 (Fig. 3(a)) agrees well with the anticipated difference in their Ir 5d electronic configurations. Analysis of the Ir 5d pDOS reveals a large difference in the energy positions of unoccupied states between the H-terminated nanosheet and rutile-IrO2 (Fig. 4(c)), thus validating their large WL position difference.
The strong WL position shift over the redox regime (Fig. 3(a)) can be explained by the dramatic change of the electronic configuration as the H-terminated nanosheet becomes fully deprotonated. The trend in the large shift of the centroids of the Ir 5d unoccupied states towards higher energies following the deprotonation process (vertical lines in Fig. 4(d)) correlates well with the observed Ir L3-edge WL position shift over ∼0.80 to 1.60 VRHE. The centroids of the Ir 5d unoccupied states of the bare nanosheet and rutile-IrO2 appear similar, confirming their expected similar Ir L3-edge WL positions. Both experimentally observed WL position shift and initial Ir–O bond contraction (Fig. 3(a)) agree with the nanosheet structural transformation during deprotonation, culminating at the end of the redox potential region (Fig. 4(d)). The effect of a single Ir vacancy formation on the expected WL position is of roughly the same order of magnitude as arising from deprotonation (Fig. 4(c)). This further highlights the difficulty to deconvolute co-existing mechanisms in the redox regime and supports the need for a dual-mechanism to comprehensively explain the am-hydr-IrOx behaviour.
Moreover, the rapid decrease in the experimentally observed Ir–O bond length within the redox and OER potential regimes aligns well with the DFT-predicted potential range for multiple Ir vacancy site formation. The 1VIr, 2VIr, and 3VIr nanosheet structures exhibit wide distributions of Ir–O bond lengths (Fig. 4(d)), with the shorter bonds emerging from edge termination effects and dangling bonds at the Ir defect sites (refer to Supplementary Note 9 (ESI†) for the different contributions of Ir–O bond distributions). Consequently, the observed reduction in Ir–O bond length can be related to (increasing) concentration of dangling bonds and edge terminations resulting from Ir vacancy site formation. Contraction of the Ir–O bond length increases with increasing OER potentials. In this potential regime, Ir defect site formation and formation of oxidized OER intermediates both influence Ir–O bond lengths.
Our dual-mechanistic DFT-guided framework (Fig. 4) reliably predicts the observed electronic and structural transformations of am-hydr-IrOx across the pre-redox, redox, and OER potential regions, convincingly explaining trends in the shifts in the Ir L3-edge WL position shifts and EXAFS determined Ir–O bond length contraction (Fig. 3). Thus, our study provides, for the first time, a comprehensive framework that can simultaneously explain all aspects of the activity-stability trade-off in am-hydr-IrOx.
In the following section, we delve into the exploration of the different chemical environments that could participate in the OER to gain more insights into the underlying mechanisms in am-hydr-IrOx.
DFT-guided understanding of the OER mechanism of am-hydr-IrOx
The previous section outlined the DFT-predicted dual-mechanistic structural progression, suggesting that under OER potentials, finite nanosheets with highly accessible surface Ir6f and O3f sites, and coordinatively unsaturated Ocus and Obr edge sites (mimicking bulk defects) undergo Ir dissolution resulting in the formation of multiple Ir defect sites with additional coordinatively unsaturated O2f and O1f sites. These unsaturated O-sites have already been proposed to be responsible for the high activity of amorphous, hydrous iridium oxides.8,29,30,35Supported by ab initio calculations and employing the widely adopted OER pathway,67 our investigation primarily seeks to explore the underlying chemistry of each of these sites in the nanosheet to unveil the nature of the OER mechanism(s) in am-hydr-IrOx.
According to the classic DFT peroxide pathway proposed by Rossmeisl et al.,67 the reactions can be described as follows for the O (e.g., O3f/2f/1f) and Ir (e.g., Ir6f) active sites:

where the charge carrier (H+/e−) balance is implicit and the catalyst resting state is underlined, constituting the starting reaction coordinate. The functional group (i.e., *O or *Ir) is separated from the site by a “-_”, and in the case of the bare site (e.g., “*”) the underlying site is denoted between “[]” (e.g., [Ircus]*). The sites explored in this work include the readily accessible surface Ir6f sites, O1f sites at Ir defect sites (modelled by 2VIr and 3VIr) likely arising from Ir dissolution, and Ocus sites at nanosheet edges absorbed to Ircus sites, which mimic bulk defects. For the various plausible active sites in the nanosheet, only the edge site has a different resting state and its corresponding reaction is:


Fig. 5 illustrates the DFT-predicted thermodynamic stabilities of these OER intermediates (Fig. 5(a)) and their subsequent ΔG barriers for O2 evolution alongside the marked potential determining step (PDS) (Fig. 5(b)) for the various potential active sites (Ir6f, O3f, O2f, O1f and Ocus). Oxygen vacancies ([Ircus]*) have not been included since their DFT-predicted ΔG at relevant potentials are much higher than the considered motifs without O-defects (Fig. S25 in Supplementary Note 10, ESI†). The most stable structure (i.e., catalyst resting state) among reaction intermediates at each of the sites at 1.23 VRHE (lowest ΔG) constitutes the starting reaction coordinate (Fig. 5(a)). The PDS describes the largest ΔG change among all four proton-coupled electron transfer (PCET) steps, which is the minimum required for OER to become exergonic (Fig. 5(b)). The prospective likelihood of the chemical species to act as active sites in am-hydr-IrOx is evaluated based on their thermodynamic favourability and energy barriers of O2 generation pathways using the nanosheet atomistic model.
 | ||
| Fig. 5 DFT peroxide OER mechanism at multiple active sites within the nanosheet atomistic model. The top panels show the different active sites for which the energy requirements of the peroxide pathway were calculated: the O3f and Ir6f sites at the nanosheet surface, O2f sites at the 1VIr defect sites, O1f sites at 2VIr defect sites, and OcusH sites at the nanosheet edges. (a) Phase diagram showing the stability of different terminations of the nanosheet surface as a function of applied potential. The bare nanosheet with O3f and Ir6f sites is shown by the solid horizontal black line in the phase diagrams. Intermediate OER species (*O, *OH, *OOH, *—) at the coordinatively saturated Ir6f sites are shown in blue, those at the coordinatively unsaturated O2f site in the 2VIr atomistic model are shown in orange, and those at the Ocus edge site are shown in purple. The coordinatively saturated O3f site with an *OOH (*O3fOH) termination is shown by the black dashed line. The thermodynamic stability of the 3VIr structure is shown in yellow. The vertical dashed line indicates the OER thermodynamic equilibrium onset potential U = 1.23 V, and the potentials regions associated with the pre-redox, redox and OER regimes are marked at the top of the plot. (b) The reaction coordinate energy diagram for each active site is shown at U = 1.23 V, along with the reaction steps below. The potential determining step (PDS) indicates the highest free energy change and is indicated by the thicker lines. The thermodynamic overpotential (η) for each pathway is annotated in the energy diagram. (c) Comparison of the experimental Ir–O bond lengths derived from EXAFS fittings and the theoretical Ir–O interatomic distances of the OER intermediate species for the edge site (OcusH). Horizontal lines indicate the experimental Ir–O bond lengths within the OER region (1.25–1.6 VRHE). The resting state (most thermodynamically stable) is shown by the star and the PDS is indicated by the square. | ||
Based on this theoretical framework, at 1.23 VRHE, coordinatively unsaturated O-species (O1f at Ir defect sites and Ocus at edge sites) are the most thermodynamically favourable sites for water oxidation (purple and orange lines in Fig. 5(a)). Surprisingly, atop site of fully coordinated Ir6f is more thermodynamically favourable for the stabilization of higher oxidized OER intermediates (blue lines in Fig. 5(a)) than higher coordinated O-species (i.e., O3f and O2f) showing higher ΔG values at 1.23 VRHE. Fully coordinated O3f atoms arise as the least reactive site, even compared to Obr or Ocus sites in rutile-IrO2 surfaces.60,61 Likely, the rigid, upstanding bond character of *O3f–OH and their even distribution hinders the additional stabilization effects through hydrogen bond networks (large ΔG difference between *O3f–OH and bare nanosheet lines in Fig. 5(a)).
Consequently, O3f and O2f sites exhibit the highest energy barriers for OER, with the first reaction steps of nucleophilic H2O attack having the highest free energy changes of η = 1.45 VRHE and η = 1.54 VRHE, respectively (Fig. 5(b)). At the cost of irreversible Ir loss leading to the formation of multiple adjacent Ir vacant sites, highly uncoordinated dangling O1f bonds at Ircus defect sites are one of the most energetically favourable OER sites (e.g., orange 2VIr lines in Fig. 5(a)), with η = 0.81 VRHE (Fig. 5(b)). Simulating finite structures like the studied nanosheets in this work offers a unique advantage in modelling the edge sites’ contributions to OER (i.e., mimicking bulk defects). The mixture of *Ocus–H/*Ocus edge sites (Fig. S30 in Supplementary Note 10, ESI†) exhibited the lowest energy barrier for OER (η = 0.52 VRHE, Fig. 5(b)) of all the investigated O-sites, consistent with findings on RuOx nanosheets.49
While untangling contributions of parallel mechanisms to describe the experimentally measured average Ir–O bond length under OER potentials is challenging (given predicting kinetic effects with DFT is error-prone), comparing the theoretical and experimental Ir–O bond lengths already provides some insights into the potential dominating active sites and/or OER intermediates which best fit the experimental data. For instance, *O–H (*O1f/cus–H) and *O–OH (*O1f/cus–OH) species at the O1f/cus sites best agree with the experimentally observed Ir–O bond lengths at OER potentials. These two species represent the thermodynamic resting state (*O–H) and potential determining step (*O–OH), which according to the reaction scheme, would be present with highest concentrations given their lower energy barriers compared to the other reaction steps. These theoretical Ir–O bond lengths correlate well with the average Ir–O bond length observed experimentally (Fig. 5(c)), and further support the validity of our theoretical framework.
Our DFT calculations suggest OER-active sites in am-hydr-IrOx extend beyond only the participation of O-sites – a perspective that has not previously been considered or integrated into current mechanistic schematics of OER.13,35,68 Metal Ir6f surface sites also offer a highly feasible O2 formation pathway with η = 0.71 VRHE (Fig. 5(b)). Activation of these sites occurs at much higher OER potentials, implying Ir participation in OER likely occurs at higher potentials (hinted by the additional small shift of the WL position >1.55 VRHE), complementing current theories of a different OER and Ir dissolution mechanisms at higher potentials.64,69
While oxidation of edge sites is less thermodynamically favourable than generating Ir defect sites in the nanosheet (higher ΔG of purple edge versus orange defect lines at 1.23 VRHE in Fig. 5(a)), edges and bulk defects are inherent in am-hydr-IrOx due to the nature of HIROF preparation. Therefore, alongside highly active Ir defect site, O1f bonds and surface Ir6f-sites in the nanosheet, the higher activity of Ocus edge sides must be considered when describing am-hydr-IrOx activity.
Our DFT calculations, which rely on the nanosheet atomistic model capturing the electronic and structural characteristics of am-hydr-IrOx, propose new insights into the prospective active and inactive sites in am-hydr-IrOx, revealing the likely species responsible for the higher activity of am-hydr-IrOx compared to rutile-IrO2 (Fig. S4, ESI†). Furthermore, our results substantiate the importance of modelling finite structures, morphologically analogous to the investigated am-hydr-IrOx electrocatalyst and more closely mimicking real-world materials.
Discussion
Understanding the mechanisms underlying Ir dissolution and OER activity of an amorphous, highly dimensional iridium oxide catalyst, such as am-hydr-IrOx, demands novel, applicable atomistic models beyond crystalline IrO2 structures, capturing its high active surface area, unique hydroxylated Ir suboxide state, and multiple active sites. Our proposed nanosheet-based model aims to clarify factors limiting am-hydr-IrOx performance, particularly its intrinsic trade-off between low stability against Ir dissolution and high OER activity. DFT-guided interpretations of experimental Ir L3-edge XANES and EXAFS data, supported by the new nanosheet model, predicted the mechanisms behind the electronic and structural transformations of am-hydr-IrOx across three potential regions (pre-redox, redox, and OER) with distinct thermodynamically driven transformations. Our dual-mechanistic nanosheet model offers a more comprehensive view, integrating chemical transformations (e.g., deprotonation and OER intermediate formation) with spontaneous Ir dissolution both prior to and under OER-relevant potentials, proposing parallel, complementary mechanisms operating in the redox and OER regions. This advances our understanding of the complex behaviour of am-hydr-IrOx under OER conditions.The nanosheet model's structural progression provides insights into am-hydr-IrOx's comparatively inferior stability against Ir dissolution versus rutile-IrO2.4,11,33 Previous ICP-MS experiments on am-hydr-IrOx reveal Ir dissolution beginning in the redox region, below 1.23 VRHE, and peaking at ∼1.40 VRHE.4,11 In contrast to mechanistic explanations of kinetic-driven Ir dissolution via unstable solvated Ir3+ OER intermediate,64 our study proposes a purely thermodynamically driven process of Ir loss, independent of OER mechanisms. Our theoretical model predicts Ir dissolution both below OER potentials in the redox region and the higher dissolution rates observed under OER potentials. Notably, the DFT-predicted potential (∼1.35 VRHE) for rapid formation of multiple adjacent Ir vacant sites (i.e., a drastic loss of iridium atoms from the nanosheet atomistic model) aligns closely with the experimental maximum rate of Ir dissolution at ∼1.40 VRHE.4,11
On the other hand, a few ordered layers of hydrous iridium oxides have shown higher stability against Ir dissolution,42,70 likely related to their extended long-range ordering. Enhanced structural order would likely limit Ir defect site formation, via stabilizing surface terminations, however, further investigations of periodic systems using the presented theoretical model are required. Our atomistic model approach was also able to complement previous predictions about the thermodynamic instability of metal oxide couples under OER, which assumed the presence of OER pathways in the theoretical framework.68,71
The creation of new sites during the thermodynamically driven Ir defect formation process under OER potentials can prospectively trigger the participation of these new sites, beyond the inherent highly active edge sites. In fact, O-dangling bonds formed at these Ir defect sites are among the most active sites according to our calculations presented in Fig. 5. Since the O-dangling bonds, along with highly active edge sites, possess the shortest Ir–O bond lengths in the atomistic model, we propose that the degree of Ir–O bond contraction under OER potentials could be a good descriptor of the contribution of these highly active O-species, as captured by Fig. 4(d).
Though limited research has addressed the energetic behaviour and contribution to the OER activity of the Ir6f metal sites, the nanosheet atomistic model presented in this study proposes their role to be significant in am-hydr-IrOx and comparable to the active O-sites studied. In fact, nucleophilic H2O attack is the bottleneck for the Ir6f sites as well as for the defect-related O1f sites and *Ocus–H edge sites in the nanosheet, consistent with previous studies on the rate-limiting nature of the H2O attack on the electrophilic OI species8,29,72 (i.e., edges and O1f sites).
Generally, our results highlight the critical role of defect sites in the electrocatalytic activity of am-hydr-IrOx. Thus, we suggest that hydrous iridium oxide-based electrocatalysts synthesized with high concentrations of pre-existing defect sites would yield a dense array of catalytically active sites, like O dangling bonds or edges. Recent literature reports promising approaches to tailor such materials, achieving a successful trade-off between OER activity and stability, such as core–shell IrOx@IrNiOx, where Ni leaching enhances defect density65 or amorphous iridium (oxy) hydroxides where Li facilitates long-range order breaking.73,74 Another strategy for long-term stabilization could involve beneficial interaction with the catalyst's support, e.g., thin iridium oxide films on TiO2 with core–shell morphology.61
Conclusions
This study introduces new key insights into the structural dynamics and electrocatalytic behaviour of am-hydr-IrOx materials, alongside proposing a novel Ir dissolution mechanism. By examining a highly active and porous catalyst with a singular hydroxylated Ir suboxide species, comparing experimental in situ and operando Ir L3-edge data with theory-predicted structures and surface terminations, we have developed a nanosheet atomistic model more representative of the physiochemical properties of am-hydr-IrOx, surpassing conventional crystal-based models. DFT-supported analyses of experimentally observed electronic and structural changes reveal under redox potentials (∼0.60–1.23 VRHE) parallel deprotonation and spontaneous Ir loss via vacancy formation mechanisms, and under OER potentials (>1.23 VRHE) rapid generation of multiple Ir defect sites, crucial for creating additional active sites despite the high cost of Ir loss, delving into previously unaddressed iridium dissolution mechanisms. Notably, the most active sites cluster around Ir defect sites and at edges, pointing to the importance of bulk defects in dictating electrocatalytic behaviour of am-hydr-IrOx. Our results and approach challenge the traditional understanding of OER mechanisms in am-hydr-IrOx and offer an alternative atomistic perspective, shedding light on the delicate relationship between OER activity and durability. In addition, we expect our findings to be broadly applicable to interpretations of the behaviour of other amorphous, hydrous iridium oxides, potentially guiding the development of more efficient and stable anode materials.Experimental methods
Preparation of HIROF and electrochemical measurements
Electrochemical measurements were done in 0.5 M H2SO4, using a 1 × 1 cm Pt mesh (52 mesh woven, 0.1 mm diameter wire, 99.9% Alfa Aesar) as the counter electrode and a Mini HydroFlex reversible hydrogen electrode (Gaskatel) as the reference electrode (all potentials reported are relative to RHE) with a Biologic SP-300 potentiostat. HIROF synthesis involved continuous potential cycling of the Ir0 substrate prepared by sputter deposition (see Supplementary Note 11 for preparation details, ESI†) between 0.05–1.5 VRHE at 500 mV s−1 for a specified number of complete cycles (referred to as “HIROFn” where ‘n’ indicates the number of cycles of growth), leading to the formation of highly porous am-hydr-IrOx layer on top of the Ir0 substrate. Cyclic voltammograms (CVs) from 0.05–1.5 VRHE at 50 mV s−1 were conducted to assess the electrochemical behaviour of am-hydr-IrOx in HIROF after different growth cycles (see Supplementary Note 1, ESI†). The OER activity of am-hydr-IrOx in HIROF, was compared to the Ir0 and rutile-IrO2 references (see Supplementary Note 11 for preparation details, ESI†) using linear sweep voltammetry from 1.2 VRHE to 1.6 VRHE at a scan rate of 10 mV s−1 (see Supplementary Note 3, ESI†). Am-hydr-IrOx possesses a higher activity (i.e., higher current density at 1.6 VRHE) than its anhydrous rutile-IrO2 counterpart and the Ir0 substrate from which it is grown.Scanning electron microscopy (SEM)
Cross-section SEM images were obtained using a Hitachi S-1400 microscope operating at 5 kV. Thicknesses were measured using imageJ,75 and the density estimation calculations are shown in Supplementary Note 2 (ESI†).Cryogenic transmission electron microscopy (cryo-TEM)
Cryo-TEM imaging was performed using a JEOL JEM-2100 electron microscope (JEOL GmbH, Eching, Germany) operated at 200 keV. Deposition of am-hydr-IrOx on TEM grids was performed by growing HIROF to 2000 cycles in 0.5 M H2SO4 and by ultra-sonication for 40 min, and a subsequent delamination of the am-hydr-IrOx layer; a 4 μL drop was taken from the aqueous solution and drop-casted onto a lacey carbon-coated copper TEM grid (200 mesh, Electron Microscopy Sciences, Hatfield, PA). The TEM grid was then plunge-frozen in liquid ethane using a vitrobot (FEI Mark IV) set at 4 °C and 95% humidity. The TEM grid was transferred to the cryo-transfer holder of the microscope (Gatan 914, Gatan, Munich, Germany) and imaging was performed using a bottom-mounted 4*4k CMOS camera (TemCam-F416, TVIPS, Gauting, Germany). Imaging was carried out at temperatures around 93 K. For further information refer to Supplementary Note 12 (ESI†).X-ray photoelectron spectroscopy (XPS)
In situ near-ambient pressure hard X-ray photoelectron spectroscopy (NAP-HAXPES) at 3 keV and 12 mbar was carried out at the SpAnTeX end-station40 at the KMC-1 beamline76 of BESSY II using a PHOIBOS 150 HV NAP electron analyser from SPECS. The electron analyser and the potentiostat (Biologic SP-300) shared a common grounding. Using the ‘dip-and-pull’ method with a 3-electrode configuration,39–41 a thin film of electrolyte on the surface of the electrode was formed for the in situ growth of am-hydr-IrOx, as shown in Fig. S6 in Supplementary Note 4 (ESI†). Thus, NAP-HAXPES measurements provided information of the hydrated condition of am-hydr-IrOx.The electronic structure of am-hydr-IrOx in HIROF was measured by XPS conducted in ultra-high vacuum (UHV) with an Al Kα excitation X-ray source (1486.58 eV, PREVAC RS40B1) and a ScientaOmicron Argus CU analyzer in the energy materials in situ laboratory Berlin (EMIL).77 The HIROF electrodes were rinsed with deionized water and dried in the Ar-filled glovebox antechamber (10−3 mbar) for ∼2 hours prior to measurements (see Supplementary Note 5 for additional details, ESI†).
The in situ NAP-HAXPES and ex situ UHV Al Kα XPS Ir 4f spectra were fitted using the LMFIT python package78 using an asymmetric finite Lorentzian79 lineshapes for metallic Ir (Ir0), rutile-IrO2 (Ir4+), and am-hydr-IrOx, and a Shirley background. Further details on the fitting procedures and the complementary O 1s spectra can be found in the Supplementary Notes 4 and 5 (ESI†), respectively.
Ir L3-edge X-ray absorption spectroscopy
The growth of HIROF to 1000 cycles and its response upon applied potentials (0.3–1.6 VRHE) was studied by Ir L3-edge X-ray absorption spectroscopy (XAS). Measurements were carried out at two different beamlines: KMC-3 at BESSY II, Germany80,81 (Supplementary Note 13, ESI†) and NOTOS at ALBA, Spain (Supplementary Note 14, ESI†). Ir L3-edge X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were collected to obtain simultaneous electronic and local geometric information. The in situ Ir L3-edge XANES and EXAFS collected during HIROF growth presented in Fig. 1 were obtained at the KMC-3 beamline (raw XAS data and in situ electrochemical growth is shown in Supplementary Note 15 (ESI†), along with discussions of the correlation between WL position and WL intensity). The in situ Ir L3-edge XANES and EXAFS of am-hydr-IrOx under potential applications presented in Fig. 3 were collected at the NOTOS beamline (raw XAS data shown in Supplementary Note 16, ESI†). The experimental data collected from both beamlines exhibit strong correlations (Fig. S41 and S42 in Supplementary Note 17, ESI†), indicating a high level of agreement between the results obtained from different experimental setups. In addition, Fixed Energy X-ray Absorption Voltammetry (FEXRAV)50 measurements at the KMC-3 beamline were also performed at the Ir4+ white-line energy with CVs recorded at 2 mV s−1 between 0.05 and 1.5 VRHE.For each XANES and EXAFS measurement, three spectra were collected and averaged. Some minor deglitching of small intensity spikes due to small bubbles and/or vibrations was removed using the Athena software,82,83 a program in the IFEFFIT package. The signal-to-noise ratio of the oscillations was determined to be sufficient until k = 11 Å−1, therefore EXAFS fittings were performed on k2-weighted EXAFS spectra over a k-range of k = 3–11 Å−1 (Fig. S38 in Supplementary Note 15, ESI†). Reasonable quantitative parameters could therefore be determined until R = ∼4 Å for the first coordination shells of the Ir0 and am-hydr-IrOx components, which is sufficient for this work as it focusses on the short-range investigation of the local geometric structure of am-hydr-IrOx.
The Ir L3-edge XANES and EXAFS data were averaged and normalized using the Athena software.82,83 For handling of the EXAFS data, the Larch software package84 was used. The ab initio atomic scattering paths were constructed using the FEFF 8.0 code85 within the Larch package.84
EXAFS fit analysis of the Ir0 and rutile-IrO2 references were used to determine the passive electron reduction factors (S02) of the metallic and oxide iridium components used in the EXAFS fits of the in situ Ir L3-edge EXAFS spectra of the HIROF system (Supplementary Note 18, ESI†). Fittings of the in situ Ir L3-edge EXAFS collected during HIROF growth to 1000 cycles were performed using a two-component structural model consisting of fcc-elemental Ir to model the Ir0 substrate in HIROF, along with rutile-IrO2 as an initial approximation for the am-hydr-IrOx in HIROF (Supplementary Note 6, ESI†). A linear combination of the first coordination scattering paths was employed to determine the relative weight of the Ir0 scattering path, which can be approximated to the nominal concentration of the Ir0 within the sample environment. The EXAFS spectra were fitted over R = 1–3.5 Å using the first coordination shell scattering paths, with relaxed energy shift (ΔE0) and Debye–Waller (σ) parameters, and constrained interatomic distances (R) and coordination numbers (CNs). The EXAFS fits are reported in Fig. S14 (ESI†) along with the fitting parameters in Table S3 in Supplementary Note 6 (ESI†). These EXAFS fittings were later repeated using the H-terminated nanosheet and stacked nanosheet structures (best fit structures), yielding the same mole fraction trend but with better r-factor fit values (Fig. S15 in Supplementary Note 6, ESI†).
Fittings of the in situ Ir L3-edge EXAFS HIROF1000 spectra were performed to obtain the atomic model that best describes am-hydr-IrOx. Larch's scripting capabilities84 enabled the efficient analysis of all structural options maintaining consistent input parameters, as opposed to the approach of using Artemis82,83 which requires manual structure importing and fitting. This expedited the analysis and enabled tailored workflows to efficiently explore all structural options in the library provided by DFT. In these fittings, the parameters related to the Ir0 substrate were fixed (i.e., R, CN, and the mole fraction, 0.2 ± 0.1). All the oxygen scattering paths in the first coordination shell(s) of the library structures were included within the fit, and with their respective CN and R fixed, while only the ΔE0 and σ parameters were allowed to vary within reason during the fitting process. Exclusion of structural options was made based on poor EXAFS fit parameters such as large r-factor values, large ΔE0 shifts and negative σ values. See Supplementary Note 7 (ESI†) for further details.
Fittings of the in situ Ir L3-edge EXAFS data of am-hydr-IrOx after 1000 cycles of HIROF growth under potential application (Supplementary Note 16, ESI†) were focused on monitoring changes in the first coordination shell Ir–O bond length. A single component model, H-terminated nanosheet (ΘH = 0.50 ML), was used to fit the region R = 1–2.1 Å, where the CN and R were also allowed to vary.
Density functional theory
All DFT calculations are performed using the projector augmented wave (PAW) method86 as implemented in the VASP code.87,88 The 5d and 6s states of Ir, and the 2s and 2p states of O are explicitly considered as the valence states. Electronic exchange and correlation (xc) are treated at the level of the semi-local generalized gradient approximation (GGA) functional due to Perdew–Burke–Ernzerhof (PBE)89 and including the van der Waals (vdW) correction method of Tkatchenko and Scheffler (TS).90 In addition, to estimate the contribution of an aqueous environment to the structures, energetics, and electronic structures of the iridium oxides, we apply an implicit solvation model in our surface calculations with a relative permittivity (εb) value of 80 to mimic the immediate water environment (Supplementary Note 20, ESI†). This is achieved via the VASPsol91 subroutine, which is augmented to the VASP code. The kinetic energy cut-off of the planewave basis set is set to 700 eV for all calculations. The Brillouin zone integrations are carried out with reciprocal distances of 0.2/2π Å−1, producing (12 × 12 × 8) k-grids with 131 irreducible k-points for bulk-layered IrO2. Optimized lattice parameters for bulk-layered IrO2 are determined by iteratively minimizing lattice stress and internal atomic displacements until the residual external pressure falls below 0.5 kbar. All 2D single-sheet structures are fully relaxed in the lateral directions with constraining the perpendicular lattice vector.Supercell setup
All 2D nanosheet calculations are performed using periodic boundary condition supercell geometries, with a minimum vacuum separation of 15 Å. Symmetric layer models are employed within the sub-space of the supercell, including 16 IrO2 units. In order to obtain the most stable hydrogen adsorption configurations within this supercell setup, we calculate all symmetry inequivalent configurations for each hydrogen coverage without an implicit solvation model and select the most stable configurations for further calculations. Here, we note that ΔG can differ by up to 0.5 eV f.u.−1 depending on configurations within the same hydrogen coverage (see Fig. S46, ESI†). Vacancies and surface reaction intermediates are calculated under assumption of OER operando conditions, all hydrogens at sheet surface are deprotonated consequently. We extend to explore this direction by considering edge sites. Similar to the procedure for the nanosheet surface, we initially identify the most stable edge termination under applied onset potentials. To achieve this, we employ a periodic supercell in which the edges of repeating ribbons representing the iridate nanosheets are separated by a 15 Å spacing. Each ribbon is 21 atomic rows wide to decouple its two edges, and a doubled periodicity along the sheet edge allows us to assess the stability of OER reaction intermediates at 0%, 50%, and 100% coverages. For comparison, we calculate rhombus-like (2·1) supercells of rutile (110) surfaces with various terminations. Periodic slabs are employed at the thickness of seven IrO2 trilayers with a minimum vacuum separation of 20 Å.61
supercell, including 16 IrO2 units. In order to obtain the most stable hydrogen adsorption configurations within this supercell setup, we calculate all symmetry inequivalent configurations for each hydrogen coverage without an implicit solvation model and select the most stable configurations for further calculations. Here, we note that ΔG can differ by up to 0.5 eV f.u.−1 depending on configurations within the same hydrogen coverage (see Fig. S46, ESI†). Vacancies and surface reaction intermediates are calculated under assumption of OER operando conditions, all hydrogens at sheet surface are deprotonated consequently. We extend to explore this direction by considering edge sites. Similar to the procedure for the nanosheet surface, we initially identify the most stable edge termination under applied onset potentials. To achieve this, we employ a periodic supercell in which the edges of repeating ribbons representing the iridate nanosheets are separated by a 15 Å spacing. Each ribbon is 21 atomic rows wide to decouple its two edges, and a doubled periodicity along the sheet edge allows us to assess the stability of OER reaction intermediates at 0%, 50%, and 100% coverages. For comparison, we calculate rhombus-like (2·1) supercells of rutile (110) surfaces with various terminations. Periodic slabs are employed at the thickness of seven IrO2 trilayers with a minimum vacuum separation of 20 Å.61
Ab initio thermodynamics
To determine the relative stabilities of possible nanosheet terminations, we employed the ab initio thermodynamics approach.92,93 This yields the free energy change ΔG in comparison to the reference state of bulk layered IrO2 as: | (1) |
 , μH2O,
, μH2O,  and μe− represent the Gibbs free energies of the nanosheet system and bulk layered IrO2, as well as the chemical potentials of water, protons, and electrons, respectively. ni (i = Ir, O, and H) is the number of each species in the periodic supercell used to describe the nanosheet system. To circumvent the computational description of
and μe− represent the Gibbs free energies of the nanosheet system and bulk layered IrO2, as well as the chemical potentials of water, protons, and electrons, respectively. ni (i = Ir, O, and H) is the number of each species in the periodic supercell used to describe the nanosheet system. To circumvent the computational description of  , we adopted the computational hydrogen electrode (CHE) approach by Nørskov et al.94 The CHE leverages the equivalence of
, we adopted the computational hydrogen electrode (CHE) approach by Nørskov et al.94 The CHE leverages the equivalence of  to the chemical potential of hydrogen gas (μH2) under standard conditions and the applied external potential U in relation to the reduced hydrogen electrode (RHE) at standard conditions under acidic environment.
to the chemical potential of hydrogen gas (μH2) under standard conditions and the applied external potential U in relation to the reduced hydrogen electrode (RHE) at standard conditions under acidic environment.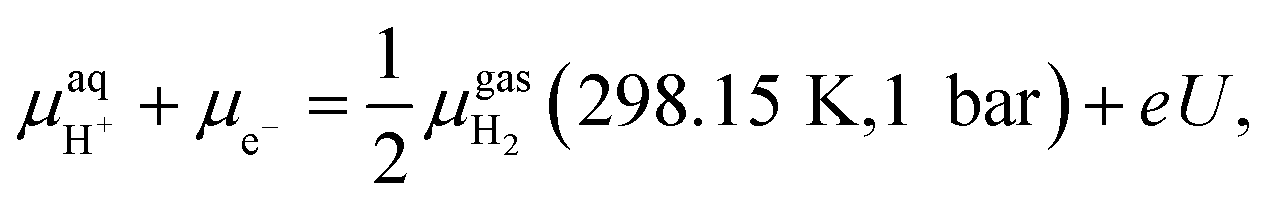 | (2) |
As a result, we reformulated eqn (1) as,
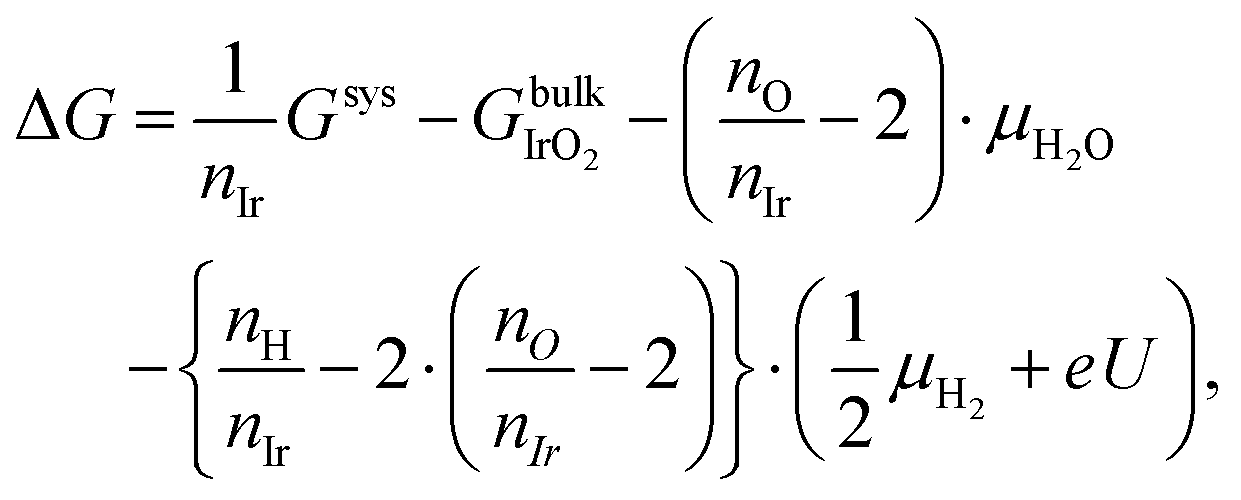 | (3) |
 using zero-point energy (ZPE)-corrected total energies.95 ZPE and entropic contributions to the chemical potentials of molecules (μH2O and μH2) were obtained from experimental reference tables.96–98 Through the relationship between relative stabilities ΔG and applied potential U, relevant terminations at a certain U value can be determined in a phase diagram. The absolute value of the thermodynamic potentials derived from the CHE approach (e.g., overpotential) should not be compared directly to experimentally measured values.
using zero-point energy (ZPE)-corrected total energies.95 ZPE and entropic contributions to the chemical potentials of molecules (μH2O and μH2) were obtained from experimental reference tables.96–98 Through the relationship between relative stabilities ΔG and applied potential U, relevant terminations at a certain U value can be determined in a phase diagram. The absolute value of the thermodynamic potentials derived from the CHE approach (e.g., overpotential) should not be compared directly to experimentally measured values.
Author contributions
Marianne van der Merwe: conceptualization, methodology, investigation, data curation, formal analysis, visualization, writing – original draft. Yonghyuk Lee: methodology, investigation, data curation, formal analysis, visualization, writing – original draft. R. Enggar Wibowo: investigation, data curation, writing – review & editing. Tathiana Kokumai: investigation, data curation, formal analysis, writing – review & editing. Anna Efimenko: investigation, data curation, writing – review & editing. Mauricio Arce: investigation, data curation, writing – review & editing. Catalina E. Jimenez: investigation, data curation, writing – review & editing. Benjamin Howchen: investigation, data curation, writing – review & editing. Rosario Suarez Anzorena: investigation, data curation, writing – review & editing. Ilaria Lucentini: investigation, data curation, writing – review & editing. Carlos Escudero: investigation, data curation, writing – review & editing. Götz Schuck: investigation, data curation, writing – review & editing. Zdravko Kochovski: investigation, data curation, writing – review & editing. Marco Favaro: investigation, data curation, writing – review & editing. David E. Starr: investigation, data curation, writing – review & editing. Karsten Reuter: funding acquisition; resources, writing – review & editing. Christoph Scheurer: methodology, investigation, data curation, formal analysis, visualization, writing – original draft, resources, supervision. Marcus Bär: funding acquisition; resources, supervision, writing – review & editing. Raul Garcia-Diez: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the German Federal Ministry of Education and Research (BMBF) in the framework of the Kopernikus P2X project (ID: 03SFK2X0). Raul Garcia-Diez and Benjamin Howchen acknowledge support from BMBF in the framework of the project CatLab, Germany (03EW0015A/B). Marianne van der Merwe acknowledges the support from the Graduate School Materials for Solar Energy Conversion (MatSEC), as part of Dahlem Research School. The authors are grateful to the KMC-3 beamline at BESSY II (proposal: 221-10927), and the NOTOS beamline at ALBA (proposal: 2022097090) for providing access to beamtime for in situ Ir L3-edge XAS measurements. The SpAnTeX end-station at the BESSY II synchrotron facility was financed by the Helmholtz Association through the Helmholtz Energy Materials Foundry (HEMF, GZ 714-48172-21/1). The authors thank Pip C. J. Clark and Michael J. Sear for their experimental assistance during the in situ NAP-HAXPES measurements at the SpAnTeX endstation, and Michael Haumann and Paul Beyer from FU Berlin for their assistance during the in situ Ir L3-edge XAS measurements at KMC-3 beamline. The authors thank Alexander Steigert from the Institute of Nanospectroscopy, HZB for magnetron sputtering deposition of Ir0 and IrO2 thin films, and Carola Klimm from the Department Solution-Processing of Hybrid Materials and Devices, HZB for the SEM measurements. The Energy Materials In Situ Laboratory Berlin (EMIL) is acknowledged for providing the infrastructure for magnetron sputter thin film deposition, lab-source X-ray analytic measurements, and the use of the Chemistry Lab for electrochemical measurements. Open Access funding provided by the Max Planck Society.References
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef.
- M. Möckl, M. F. Ernst, M. Kornherr, F. Allebrod, M. Bernt, J. Byrknes, C. Eickes, C. Gebauer, A. Moskovtseva and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 064505 CrossRef.
- M. Bernt, C. Schramm, J. Schröter, C. Gebauer, J. Byrknes, C. Eickes and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 084513 CrossRef CAS.
- O. Kasian, S. Geiger, T. Li, J. P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J. J. Mayrhofer, Energy Environ. Sci., 2019, 12, 3548–3555 RSC.
- M. Fathi Tovini, A. M. Damjanovic, H. A. El-Sayed, J. Speder, C. Eickes, J.-P. Suchsland, A. Ghielmi and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 064521 CrossRef.
- C. Massué, V. Pfeifer, M. van Gastel, J. Noack, G. Algara-Siller, S. Cap and R. Schlögl, ChemSusChem, 2017, 10, 4786–4798 CrossRef.
- V. A. Saveleva, L. Wang, D. Teschner, T. Jones, A. S. Gago, K. A. Friedrich, S. Zafeiratos, R. Schlögl and E. R. Savinova, J. Phys. Chem. Lett., 2018, 9, 3154–3160 CrossRef CAS PubMed.
- J. J. Velasco-Vélez, E. A. Carbonio, C.-H. Chuang, C.-J. Hsu, J.-F. Lee, R. Arrigo, M. Hävecker, R. Wang, M. Plodinec, F. R. Wang, A. Centeno, A. Zurutuza, L. J. Falling, R. V. Mom, S. Hofmann, R. Schlögl, A. Knop-Gericke and T. E. Jones, J. Am. Chem. Soc., 2021, 143, 12524–12534 CrossRef.
- L. J. Frevel, R. Mom, J. J. Velasco-Vélez, M. Plodinec, A. Knop-Gericke, R. Schlögl and T. E. Jones, J. Phys. Chem. C, 2019, 123, 9146–9152 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, J. Electroanal. Chem., 2016, 773, 69–78 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- S. Cherevko, T. Reier, A. R. Zeradjanin, Z. Pawolek, P. Strasser and K. J. J. Mayrhofer, Electrochem. Commun., 2014, 48, 81–85 CrossRef CAS.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- C. Liang, R. Rao, K. L. Svane, J. H. L. Hadden, B. Moss, S. Scott, M. Sachs, J. Murawski, A. M. Frandsen, D. J. Riley, M. P. Ryan, J. Rossmeisl, J. Durrant and I. E. L. Stephens, Nat. Catal., 2024, 7, 763–775 CrossRef CAS.
- S. B. Scott, J. E. Sørensen, R. R. Rao, C. Moon, J. Kibsgaard, Y. Shao-Horn and I. Chorkendorff, Energy Environ. Sci., 2022, 15, 1988–2001 RSC.
- J. W. Zhao, K. Yue, H. Zhang, S. Y. Wei, J. Zhu, D. Wang, J. Chen, V. Y. Fominski and G. R. Li, Nat. Commun., 2024, 14, 2928 CrossRef.
- M. Retuerto, L. Pascual, J. Torrero, M. A. Salam, Á. Tolosana-Moranchel, D. Gianolio, P. Ferrer, P. Kayser, V. Wilke, S. Stiber, V. Celorrio, M. Mokthar, D. G. Sanchez, A. S. Gago, K. A. Friedrich, M. A. Peña, J. A. Alonso and S. Rojas, Nat. Commun., 2022, 13, 7935 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal.: Sci. Technol., 2014, 4, 3800–3821 RSC.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- A. Grimaud, A. Demortiere, M. Saubanere, W. Dachraoui, M. Duchamp, M. L. Doublet and J. M. Tarascon, Nat. Energy, 2017, 2, 16189 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Velasco Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7, 6791–6795 RSC.
- O. Kasian, J. P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS.
- K. Schweinar, B. Gault, I. Mouton and O. Kasian, J. Phys. Chem. Lett., 2020, 11, 5008–5014 CrossRef CAS PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- E. Willinger, C. Massué, R. Schlögl and M. G. Willinger, J. Am. Chem. Soc., 2017, 139, 12093–12101 CrossRef CAS.
- J. J. Velasco-Velez, L. J. Falling, D. Bernsmeier, M. J. Sear, P. C. J. Clark, T. S. Chan, E. Stotz, M. Hävecker, R. Kraehnert, A. Knop-Gericke, C. H. Chuang, D. E. Starr, M. Favaro and R. V. Mom, J. Phys. D: Appl. Phys., 2021, 54, 124003 CrossRef CAS.
- R. V. Mom, L. J. Falling, O. Kasian, G. Algara-Siller, D. Teschner, R. H. Crabtree, A. Knop-Gericke, K. J. J. Mayrhofer, J. J. Velasco-Vélez and T. E. Jones, ACS Catal., 2022, 12, 5174–5184 CrossRef CAS.
- J. J. Velasco Vélez, D. Bernsmeier, R. V. Mom, P. Zeller, Y. Shao-Horn, B. Roldan Cuenya, A. Knop-Gericke, R. Schlögl and T. E. Jones, Adv. Energy Mater., 2024, 1–17 Search PubMed.
- P. G. Pickup and V. I. Birss, J. Electroanal. Chem., 1987, 220, 83–100 CrossRef CAS.
- L. D. Burke and E. J. O’Sullivan, J. Electroanal. Chem., 1981, 117, 155–160 CrossRef CAS.
- S. Geiger, O. Kasian, B. R. Shrestha, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2016, 163, F3132–F3138 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- J. J. Velasco-Vélez, T. E. Jones, V. Streibel, M. Hävecker, C. H. Chuang, L. Frevel, M. Plodinec, A. Centeno, A. Zurutuza, R. Wang, R. Arrigo, R. Mom, S. Hofmann, R. Schlögl and A. Knop-Gericke, Surf. Sci., 2019, 681, 1–8 CrossRef.
- L. D. Burke, N. S. Naser and B. M. Ahern, J. Solid State Electrochem., 2007, 11, 655–666 CrossRef CAS.
- L. Ouattara, S. Fierro, O. Frey, M. Koudelka and C. Comninellis, J. Appl. Electrochem., 2009, 39, 1361–1367 CrossRef CAS.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem. Interfacial Electrochem., 1978, 86, 89–104 CrossRef CAS.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 1–12 Search PubMed.
- M. Favaro, P. C. J. Clark, M. J. Sear, M. Johansson, S. Maehl, R. van de Krol and D. E. Starr, Surf. Sci., 2021, 713, 121903 CrossRef CAS.
- R. E. Wibowo, R. Garcia-Diez, T. Bystron, M. Prokop, M. van der Merwe, M. D. Arce, C. E. Jiménez, T.-E. Hsieh, J. Frisch, A. Steigert, M. Favaro, D. E. Starr, R. G. Wilks, K. Bouzek and M. Bär, ACS Appl. Mater. Interfaces, 2023, 15, 51989–51999 CrossRef CAS PubMed.
- D. Weber, L. M. Schoop, D. Wurmbrand, S. Laha, F. Podjaski, V. Duppel, K. Müller, U. Starke and B. V. Lotsch, J. Mater. Chem. A, 2018, 6, 21558–21566 RSC.
- L. J. Frevel, R. Mom, J.-J. Velasco-Vélez, M. Plodinec, A. Knop-Gericke, R. Schlögl and T. E. Jones, J. Phys. Chem. C, 2019, 123, 9146–9152 CrossRef CAS.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem. Interfacial Electrochem., 1978, 86, 89–104 CrossRef CAS.
- R. Kötz, J. Electrochem. Soc., 1984, 131, 72 CrossRef.
- A. Zagalskaya and V. Alexandrov, ACS Catal., 2020, 10, 3650–3657 CrossRef CAS.
- A. Zagalskaya, I. Evazzade and V. Alexandrov, ACS Energy Lett., 2021, 6, 1124–1133 CrossRef CAS.
- B. E. Conway and J. Mozota, Electrochim. Acta, 1983, 28, 9–16 CrossRef CAS.
- S. Laha, Y. Lee, F. Podjaski, D. Weber, V. Duppel, L. M. Schoop, F. Pielnhofer, C. Scheurer, K. Müller, U. Starke, K. Reuter and B. V. Lotsch, Adv. Energy Mater., 2019, 9, 1803795 CrossRef.
- A. Minguzzi, O. Lugaresi, C. Locatelli, S. Rondinini, F. D’Acapito, E. Achilli and P. Ghigna, Anal. Chem., 2013, 85, 7009–7013 CrossRef CAS PubMed.
- V. I. Birss, C. Bock and H. Elzanowska, Can. J. Chem., 1997, 75, 1687–1693 CrossRef CAS.
- I. T. E. Fonseca, M. I. Lopes and M. T. C. Portela, J. Electroanal. Chem., 1996, 415, 89–96 CrossRef.
- M. Hüppauff, J. Electrochem. Soc., 1993, 140, 598 CrossRef.
- Y. Mo, I. C. Stefan, W. Bin Cai, J. Dong, P. Carey and D. A. Scherson, J. Phys. Chem. B, 2002, 106, 3681–3686 CrossRef CAS.
- A. R. Hillman, M. A. Skopek and S. J. Gurman, Phys. Chem. Chem. Phys., 2011, 13, 5252–5263 RSC.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Chem. Sci., 2014, 5, 3591–3597 RSC.
- A. Minguzzi, C. Locatelli, O. Lugaresi, E. Achilli, G. Cappelletti, M. Scavini, M. Coduri, P. Masala, B. Sacchi, A. Vertova, P. Ghigna and S. Rondinini, ACS Catal., 2015, 5, 5104–5115 CrossRef CAS.
- D. F. Abbott, D. Lebedev, K. Waltar, M. Povia, M. Nachtegaal, E. Fabbri, C. Copéret and T. J. Schmidt, Chem. Mater., 2016, 28, 6591–6604 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- D. Opalka, C. Scheurer and K. Reuter, ACS Catal., 2019, 9, 4944–4950 CrossRef CAS.
- Y. Lee, C. Scheurer and K. Reuter, ChemSusChem, 2022, 15, 1–10 Search PubMed.
- L. D. Burke and D. P. Whelan, J. Electroanal. Chem., 1981, 124, 333–337 CrossRef CAS.
- L. D. Burke and D. P. Whelan, J. Electroanal. Chem., 1984, 162, 121–141 CrossRef CAS.
- O. Kasian, J. P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS PubMed.
- H. N. Nong, T. Reier, H. S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- A. H. Reksten, A. E. Russell, P. W. Richardson, S. J. Thompson, K. Mathisen, F. Seland and S. Sunde, Phys. Chem. Chem. Phys., 2020, 22, 18868–18881 RSC.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kötz and T. J. Schmidt, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- T. Naito, T. Shinagawa, T. Nishimoto and K. Takanabe, Inorg. Chem. Front., 2021, 8, 2900–2917 RSC.
- Z. Liu, J. Qi, H. Zeng, Y. Zeng, J. Wang, L. Gu, E. Hong, M. Yang, Q. Fu, J. Chen and C. Yang, ACS Appl. Energy Mater., 2022, 5, 6869–6877 CrossRef CAS.
- T. Binninger, G. C. Moss, Z. S. H. S. Rajan, R. Mohamed and M. H. Eikerling, ChemCatChem, 2024, 16(17), 202400567 CrossRef.
- L. J. Falling, W. Jang, S. Laha, T. Götsch, M. W. Terban, S. Bette, R. Mom, J.-J. Velasco Vélez, F. Girgsdies, D. Teschner, A. Tarasov, C.-H. Chuang, T. Lunkenbein, A. Knop-Gericke, D. Weber, R. Dinnebier, B. V. Lotsch, R. Schlögl and T. E. Jones, J. Am. Chem. Soc., 2024, 146, 27886–27902 CrossRef CAS PubMed.
- J. Gao, C. Q. Xu, S. F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. M. Chen, H. Xiao, J. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS.
- J. Ruiz Esquius, D. J. Morgan, G. Algara Siller, D. Gianolio, M. Aramini, L. Lahn, O. Kasian, S. A. Kondrat, R. Schlögl, G. J. Hutchings, R. Arrigo and S. J. Freakley, J. Am. Chem. Soc., 2023, 145, 6398–6409 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- F. Schaefers, M. Mertin and M. Gorgoi, Rev. Sci. Instrum., 2007, 78, 123102 CrossRef CAS PubMed.
- S. Hendel, F. Schäfers, M. Hävecker, G. Reichardt, M. Scheer, J. Bahrdt and K. Lips, AIP Conf. Proc., 2016, 1741, 030038 CrossRef.
- M. Newville, T. Stensitzki, D. B. Allen, M. Rawlik, A. Ingargiola and A. Nelson, Astrophysics Source Code Library, 2016 DOI:10.5281/zenodo.11813.
- G. H. Major, D. Shah, V. Fernandez, N. Fairley and M. R. Linford, Vac. Tech. Coat., 2020, 35–40 Search PubMed.
- G. Schuck and I. Zisak, J. Large-Scale Res. Facilities, 2020, 6, 139 CrossRef.
- I. Zizak and P. Gaal, J. Large-Scale Res. Facilities, 2017, 3, A123 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 1995, 12, 537–541 CrossRef PubMed.
- B. Ravel and M. Newville, Phys. Scr., 2005, 1007–1010 CrossRef CAS.
- M. Newville, J. Phys.: Conf. Ser., 2013, 430, 012007 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140(8) DOI:10.1063/1.4865107.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Q. Sun, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205424 CrossRef.
- W. S. Benedict, N. Gailar and E. K. Plyler, J. Chem. Phys., 1956, 24, 1139–1165 CrossRef CAS.
- K. K. Irikura, J. Phys. Chem. Ref. Data, 2007, 36, 389–397 CrossRef CAS.
- M. W. Chase, J. L. Curnutt, J. R. Downey, R. A. McDonald, A. N. Syverud and E. A. Valenzuela, J. Phys. Chem. Ref. Data, 1974, 11, 311–480 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02839b |
| This journal is © The Royal Society of Chemistry 2025 |