
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput design of complex oxides as isothermal, redox-activated CO2 sorbents for green hydrogen generation†
Runxia
Cai‡
a,
Kunran
Yang‡
 a,
Xijun
Wang‡
ab,
Mahe
Rukh
a,
Azin Saberi
Bosari
a,
Eric
Giavedoni
a,
Alexandra
Pierce
a,
Leo
Brody
a,
Wentao
Tang
a,
Phillip R.
Westmoreland
a and
Fanxing
Li
*a
a,
Xijun
Wang‡
ab,
Mahe
Rukh
a,
Azin Saberi
Bosari
a,
Eric
Giavedoni
a,
Alexandra
Pierce
a,
Leo
Brody
a,
Wentao
Tang
a,
Phillip R.
Westmoreland
a and
Fanxing
Li
*a
aDepartment of Chemical and Biomolecular Engineering, North Carolina State University, 911 Partners Way, Raleigh, North Carolina 27695-7905, USA. E-mail: fli5@ncsu.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois 60208, USA
First published on 25th July 2024
Abstract
Sorption-enhanced reforming and gasification (SERG) offers a promising approach to intensify hydrogen production from carbonaceous feedstocks. However, conventional sorbents require substantial temperature increases for the endothermic CO2 release step and are prone to deactivation. This study introduces a new class of redox-activated sorbents capable of stable isothermal operation and tunable heats of reactions, thereby facilitating an efficient reactive separation scheme. Using plane-wave density functional theory (DFT) calculations of structures and free energies, we screened 1225 perovskite-structured sorbent candidates, followed with extensive experimental validation. An effective descriptor, (ΔGabs + ΔGreg), was identified to expedite sorbent optimization. The advanced sorbents showed reversible, isothermal carbonation of up to 78% of the A-site cation, permitting isothermal SERG or “iSERG”. Their versatility was demonstrated in a fluidized bed for woody biomass gasification and a packed bed for biogas conversion, yielding hydrogen-enriched (73 vol%) syngas from biomass and 95+% pure H2 from biogas. Our results also support integrated CO2 capture to produce carbon-negative hydrogen products.
Broader contextHydrogen is a clean energy carrier with significant potential to advance global efforts towards carbon neutrality. Among the various production methods, green hydrogen generated from sorption-enhanced reforming and gasification (SERG) of biogenic feedstocks stands out as a promising approach. However, conventional SERG technologies face critical challenges, including sorbent deactivation, substantial temperature fluctuations, and the requirement for large energy inputs to facilitate CO2 release. This study rationally designs and validates a unique family of redox-activated sorbents that are capable for isothermal CO2 capture and release with excellent efficiency and stability for woody biomass and biogas conversion. Furthermore, they facilitate in situ CO2 capture, leading to efficient hydrogen production with negative life cycle carbon emissions. Through integration with conventional Fischer–Tropsch and methanol synthesis technologies, the isothermal SERG technology can be extended to produce carbon negative methanol and liquid fuel. Our results represent a significant advancement in sustainable hydrogen production, paving the way for broader adoption and impactful carbon reduction. |
1. Introduction
Hydrogen is widely recognized as a clean-energy vector that could contribute substantially to the global effort to achieve carbon neutrality.1–3 However, less than 1% of the 94 million tons of hydrogen produced in 2021 was derived from non-fossil fuel sources.4,5 Given the pressing need to address global climate change and the expected growth in hydrogen demand, it is highly desirable to develop new technologies that can produce carbon-free, cost-competitive hydrogen from renewable sources.6,7 Biogenic feedstocks, such as biomass and biogas, represent abundant and sustainable alternatives to coal and natural gas for hydrogen production.8–13 Nevertheless, conventional gasification and reforming processes are energy-intensive, and the distributed nature of biomass and biogas sources significantly hinders the ability to leverage economies of scale. Additionally, the compositional complexity of biogenic feedstocks further inflates system complexity and costs.14–16 Consequently, the development of innovative technologies that markedly intensify the hydrogen production process is essential for achieving cost-competitive renewable hydrogen generation.Sorption-enhanced reforming and gasification (SERG) represents a promising approach to simplify hydrogen production from biogenic resources. This objective is achieved by integrating gasification/reforming, the water–gas-shift reaction, and CO2 capture into a single cyclic operation. This two-step operation comprises a reforming/gasification step (with in situ CO2 capture) for H2-rich syngas production, followed by a decarbonation step for sorbent regeneration.17–20
Practical implementation of SERG, however, is challenged by substantial energy consumption for the decarbonation step and lack of sorbent stability over repeated cycles. Notably, all high-temperature CO2 sorbents reported in the literature to date require a large temperature swing (≥100 °C) to drive the highly endothermic CO2 release reaction (ΔH = 167.6 kJ mol−1 for CaCO3 at 800 °C), a crucial condition for effective sorbent regeneration for practical applications.21,22 However, such temperature swing operations incur considerable exergy loss and thermal stress on the particles and reactors. Moreover, the prevailing CaO-based sorbents typically lose 50–80% of their initial CO2 sorption capacity after just a few dozen cycles.23–26 While steam re-activation at a lower temperature proves effective in regenerating the CaO sorbents, it introduces additional process complexity and cost.27
Advanced sorbents with engineered structures have also shown promise by exhibiting significantly lower deactivation rates.28–35 Nonetheless, the high cost of manufacturing these sorbents hinders their practical application, and the loss of activity, albeit slow, is generally irreversible without an additional reactivation step. Mixed oxide sorbents such as Ca3Co4O9 and Sr1−xCaxFe1−yNiyO3−δ have been explored in the context of sorption-enhanced steam reforming of glycerol. While these sorbents exhibit excellent cyclic stability through reversible phase transitions,36–39 large temperature swings are still required for the decarbonization step. Our recent study indicated that perovskite sorbents such as Sr0.25Ba0.75Fe0.375Co0.625O3−δ has the potential to operate isothermally for toluene reforming in the presence of a Ni-based reforming catalyst.39 Nevertheless, ∼100 °C temperature increase is still necessary for complete sorbent regeneration.
To address these challenges, this study proposes and systematically validates new families of redox-activated mixed-oxide sorbents capable of isothermal SERG (iSERG) operation with tunable heat of reaction and excellent cyclic stability. As depicted in Fig. 1, a redox-active, perovskite-structured (ABO3−δ) sorbent plays a central role in this two-step process. During the reforming or gasification step, the strongly reducing environment would trigger the reduction of the B-site metal cation (e.g., Fe3/4+), which induces phase segregation. As a result, the highly basic A-site cation (e.g., Sr2+) would bind with CO2 to form carbonates. In the subsequent regeneration step, the carbonated and reduced sorbent is put in contact with air or O2 to trigger the oxidation of the B-site metal, leading to spontaneous release of CO2 and reformation of the ABO3−δ structure. The variation in external oxygen chemical potentials would provide significantly higher thermodynamic driving force for carbonation and decarbonation reactions, when compared to temperature swings in conventional processes. Moreover, the heats for the reduction and oxidation reactions would counterbalance those of the exothermic carbonation and endothermic decarbonation reactions. Consequently, the considerable structural and compositional flexibility of perovskite oxides, as illustrated in this study, also opens a unique avenue for tuning the thermal energy distribution between the reforming/gasification and regeneration/decarbonation steps, thereby minimizing the exergy loss for hydrogen generation.
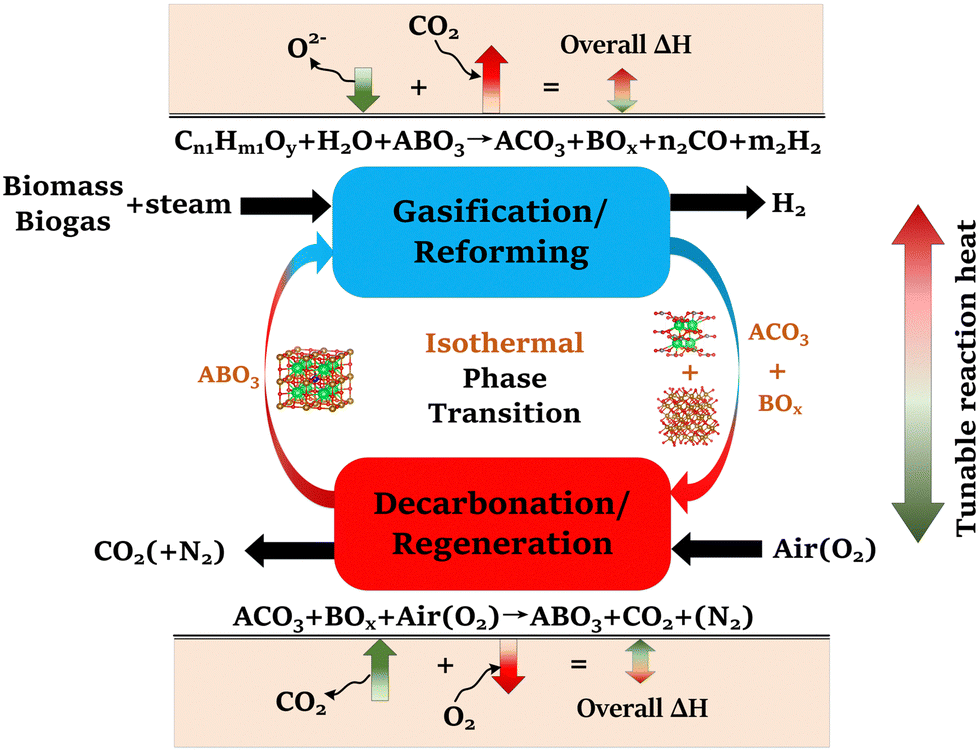 | ||
| Fig. 1 Schematic of the isothermal sorption-enhanced reforming and gasification (iSERG) concept utilizing perovskite-structured isothermal sorbents. | ||
Taking advantage of the inherent flexibility of perovskite structures, we conducted a comprehensive screening of 1225 A/B-site doped perovskite oxides (SrxA1−xFeyB1−yO3−δ) using plane wave density functional theory (DFT) calculations of structures and free energies. The effectiveness of our redox-activated sorbent concept and the proposed computational framework was validated experimentally. The advanced sorbents developed in this research achieved high capacity and versatility for biomass gasification, biogas, and methane reforming. The use of concentrated O2 in the regeneration step can facilitate integrated CO2 capture, resulting in a life-cycle CO2 emission as low as −2.18 kg CO2-eq per kg of H2.
2. Results and discussion
2.1. High throughput computational screening
In all, 1225 sorbent compositions, with a general formula of SrxA1−xFeyB1−yO3−δ (A = Ca, K, or Ba and B = Mn, Ti, Co, Cu, Ni, or Mg), were considered in this study. The workflow for the computational screening is summarized in Fig. 2a. Initial steps in the workflow focus on ensuring the pre-requisites for the sorbent compositions, namely (i) charge neutrality and (ii) structural stability.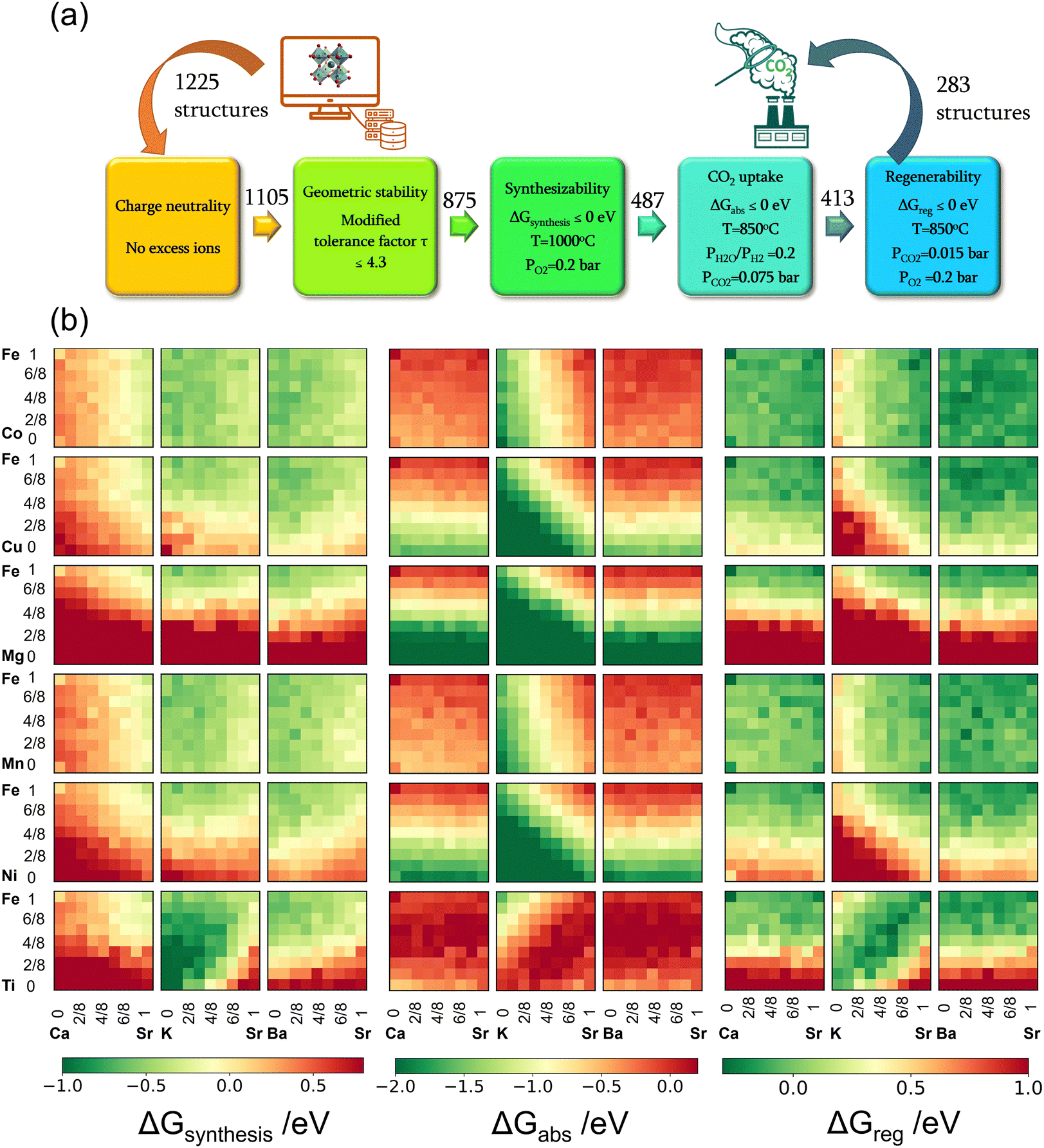 | ||
| Fig. 2 Computational screening approach and results. (a) Screening workflow for iSERG sorbents. The values above each arrow represent the count of perovskite structures that successfully passed the preceding screening step. (b) Heatmaps of the thermodynamic descriptors of the 1225 candidates. The values of the x and y axes denote the concentrations of Sr and Fe in SrxA1−xFeyB1−yO3−δ, respectively. | ||
Firstly, the charge neutrality of various A and B-site cation combinations in the SrxA1−xFeyB1−yO3−δ structures were assessed. The total positive charge of all cations must be able to balance the total negative charge of all anions (i.e., O2−).40 Out of the initial set, 120 compositions failing to satisfy the charge neutrality criterion were excluded from further consideration, with most of these containing a large fraction of Mg (≥50%). Subsequently, the structural stability of the remaining perovskite compositions was investigated using the modified tolerance factor (τ) proposed by Bartel et al.41 Derived from extensive data sets, this descriptor has demonstrated superior predictive capability for perovskite stability compared to the geometric-based Goldschmidt tolerance factor. A relaxed criterion of τ < 4.3, as reported in our recent work,40 was used to screen out 230 compositions that are likely to be unstable in a perovskite structure. Many of them contained a large fraction of K on the A-site and were shown to be unstable based on our previous synthesis experience. These screening steps narrowed down the sorbent candidates to 875 compositions, summarized in Fig. S1(a) (see ESI†).
Beyond structural stability, a suitable perovskite sorbent for isothermal SERG must satisfy the following criteria:
(i) Synthesizability: the mixed oxide should be more stable than a simple mixture of its monometallic oxide precursors in an oxidizing environment (e.g., air);
(ii) Sorption capability: the sorbent should undergo spontaneous reduction and carbonation under a reducing environment and in the presence of CO2;
(iii) Regenerability: the sorbent should spontaneously release CO2 and reform the ABO3−δ structure under an oxidizing environment.
The reactions corresponding to these criteria are listed below. Given the reducing atmosphere in the gasifier/reformer, a Brownmillerite phase with a general formula of ABO2.5 was considered in these reactions. Operational parameters compatible with actual operating conditions were used as the input parameters for the analysis. For instance, the synthesis condition was set at 1000 °C and PO2 = 0.2 bar. The sorption step condition was 850 °C with PH2O/PH2 = 0.2 and PCO2 = 0.075 bar. The regeneration step condition was PO2 = 0.2 bar and PCO2 = 0.015 bar. We also note that these sorbent particles absorb CO2via a carbonation reaction. Therefore, the sorbents are chemically specific for CO2 absorption and does not adsorb or absorb H2, N2, or CH4.
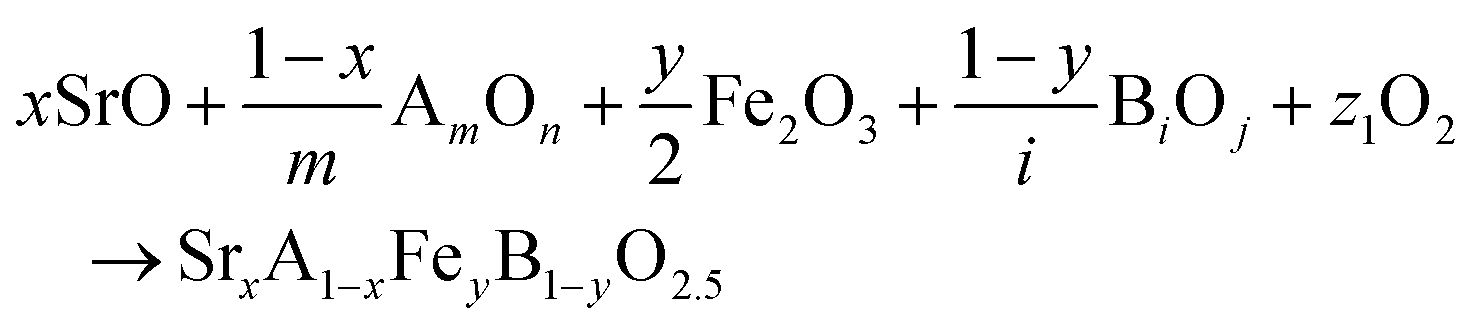 | (1) |
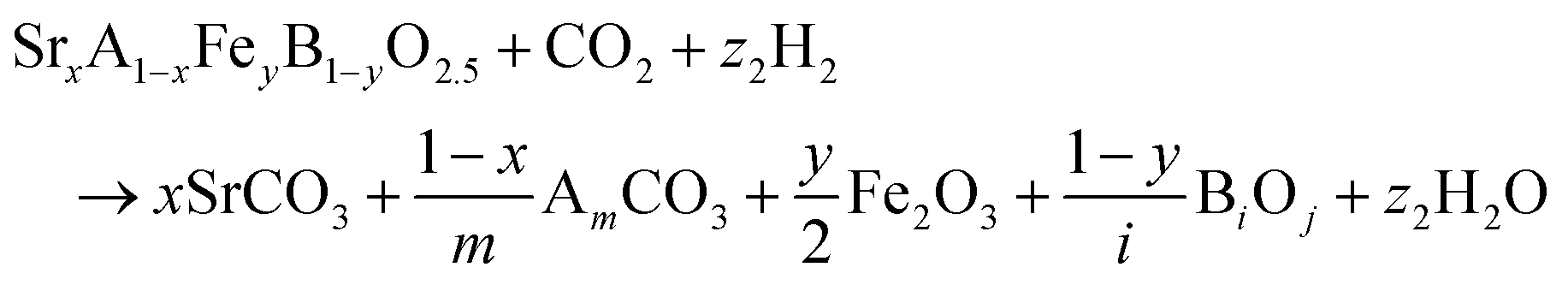 | (2) |
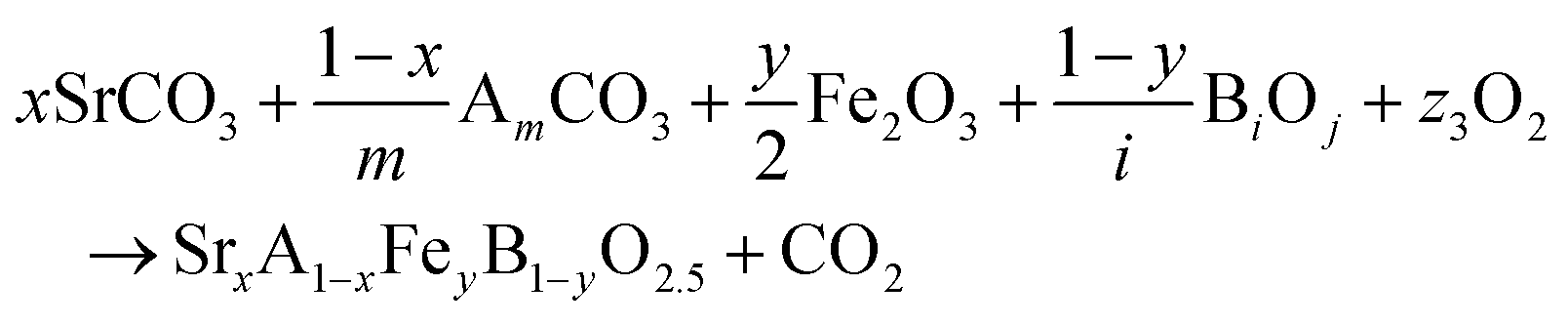 | (3) |
The Gibbs free energies of reactions (1)–(3), denoted as ΔGsyn(thesis), ΔGabs(orption), and ΔGreg(eneration), respectively, are delineated in Fig. 2b for the 875 compositions under examination. To meet the screening criteria, these ΔGs should be ≤0 eV to ensure adequate thermodynamic driving forces. Applying this criterion, 283 compositions were identified as potentially suitable candidates for isothermal SERG operations. Computational details, based on a combination of DFT calculations of the perovskite oxides and the bulk thermodynamic properties of both the monometallic oxides and carbonates, are provided in the methodology section. Table 1 highlights several promising sorbent composition families, whereas Fig. S1e (see ESI†) summarizes all the suitable candidates predicted by the simulation. It is noted that over 90% of the SrBaFeCo and SrBaFeMn compositions were predicted to be favorable for iSERG. Therefore, our experimental investigation prioritized the SrBaFeCo and SrBaFeMn families of sorbents.
| Compositions | General dopant concentrations at A-site and B-site |
|---|---|
| SrBaFeMn | Ba: 0–1; Mn: 0–1 |
| SrBaFeCo | Ba: 0–1; Co: 0–1 |
| SrBaFeCu | Ba: 0–1; Cu: 0–0.5 |
| SrCaFeCo | Ca: 0–0.375; Co: 0–1 |
| SrBaFeNi | Ba: 0–1; Ni: 0–0.375 |
2.2. Experimental evaluation of sorbent isothermal performance
To assess the computational screening results, 100 sorbent compositions were prepared and characterized. Among these, 80 were deemed thermodynamically suitable for iSERG applications based on the computation results. As depicted in Fig. 3a and detailed in Table S1 and Fig. S2 (see ESI†), X-ray diffraction (XRD) analyses revealed that 72 of the samples contained a single perovskite phase, while an additional 13 displayed minimal phase impurities. The remaining 15 samples presented at least one prominent secondary phase, although perovskite phase(s) were observed in all the samples. This confirms the effectiveness of computational screening in predicting the stability and synthesizability of the perovskite sorbents. Notably, most cobalt-containing materials, i.e., SrxA1−xFeyCo1−yO3−δ, were phase-pure. This may have resulted from the flexible valence states of Co and Fe as well as the similarities in ionic radii. On the other hand, Mn-doped materials tended to segregate into two distinct perovskite phases. Ni, Mg, and Cu-containing samples also tend to phase segregate when the doping level surpassed 12.5% (1/8) on the B-site, due to their instability at a valence state above 2+. It is noted that our model estimates phase stability and synthesizability based on the relative stability of perovskite and respective monometallic oxides (reaction (1)), it does not preclude the formation of alternative, more stable mixed oxide phases.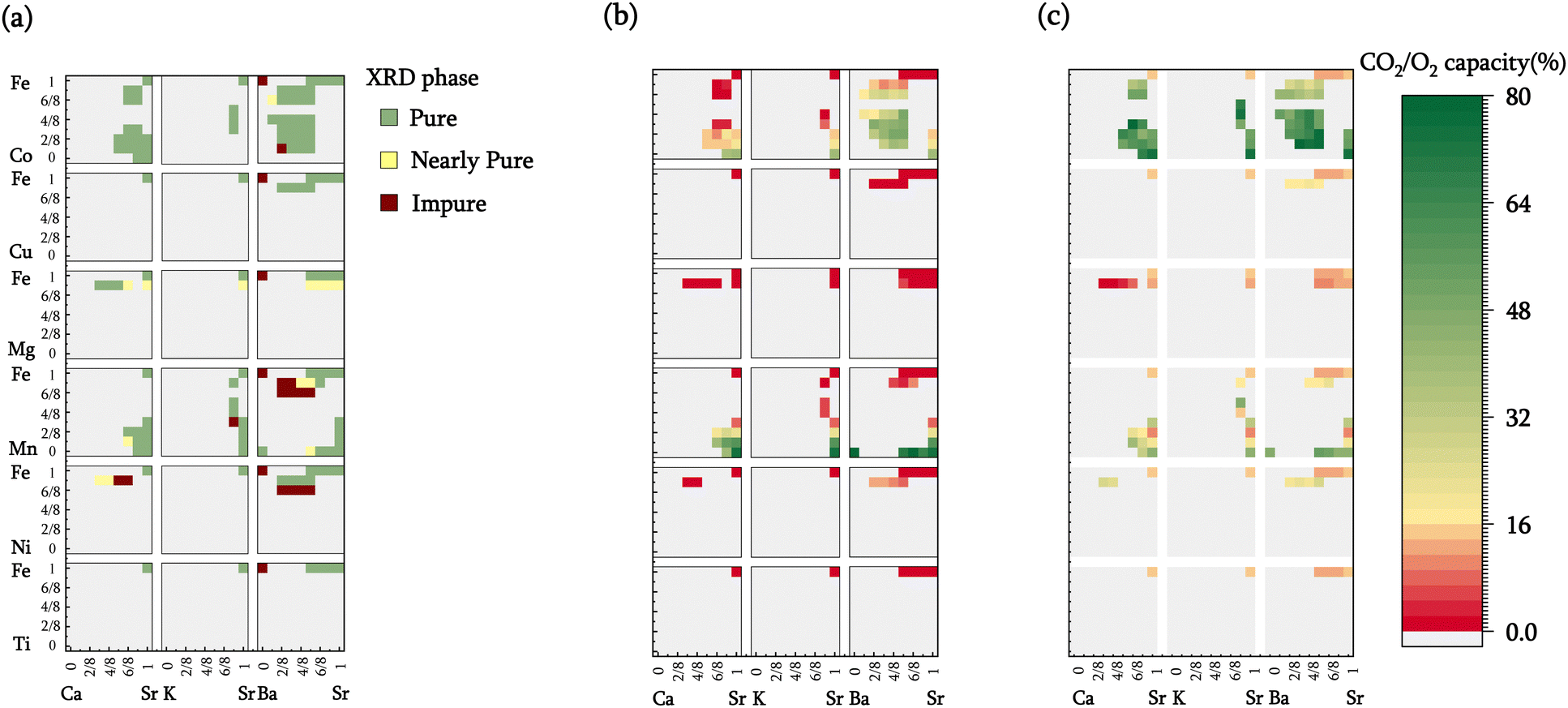 | ||
| Fig. 3 Experimental evaluation of the sorbents. (a) Phase purity of the synthesized perovskites: green-, yellow- and burgundy-colored cells represent pure, nearly pure, and impure samples, respectively. (b) CO2 sorption capacities and (c) oxygen capacities of pure and nearly pure materials measured in the TGA. The rightmost color bar represents the CO2 capacity for (b) and the O2 capacity for (c). | ||
Fig. 2b displays the isothermal sorption capacities of the screened materials, defined as the percentage of A-site cations being reversibly carbonated, determined by thermogravimetric analysis (TGA). A relatively high operating temperature of 850 °C is chosen since it is commensurate with typical methane reforming and biomass gasification temperatures. This choice of operating temperature underscores an additional distinguishing feature of the redox-activated iSERG sorbents: whereas conventional sorbents are ineffective for CO2 sorption at >700 °C due to thermodynamic constraints, the iSERG sorbents do not face this limitation. This thermodynamic flexibility allows them to maintain effectiveness in the conversion of biomass and biogas. In terms of sorbent performance, it was observed that calcium doping in the A-site reduced the CO2 capacity, while barium doping resulted in an increase. This trend can be explained by the instability of CaCO3 at 850 °C, whereas BaCO3 exhibits better stability than SrCO3. Additionally, all samples with <25% B-site doping exhibited low sorption capacity, indicating that the B-site cation composition also plays a critical role in sorbent performance. This observation lends further credence to the proposed iSERG concept (Fig. 1), emphasizing the link between the redox behavior of the B-site cations and the carbonation and decarbonation reactions. For instance, SrFeO3−δ showed a negligible (∼0.13%) CO2 sorption capacity, but upon doping the B-site with 75% Mn, the CO2 capacity increased to 21%. Further increasing the Mn fraction to 87.5% boosted the CO2 capacity to 59%. This finding aligns with DFT simulations, which revealed a decrease in ΔGabs of 0.3 eV as Mn doping increased from 12.5% to 87.5%. More detailed fitting results (see ESI:† Fig. S3(a–c)) unveiled robust correlations (R2 between 0.7–0.9) between the dopant fractions of A-site and B-site elements and the CO2 capacity. Additionally, doping the B-site with Ni, Mg, and Cu in amounts greater than 12.5% leads to phase segregation and are thus not suitable for iSERG.
Of all the samples tested, 21 perovskite oxides exhibited excellent sorption capacities (>35%). These high-performing sorbents primarily belong to three families: SrxBa1−xFeyCo1−yO3−δ, SrxCa1−xFeyMn1−yO3−δ, and SrxBa1−xFeyMn1−yO3−δ. Among these, Sr0.75Ba0.25MnO3−δ registered the highest CO2 capacity of 78%, closely followed by BaMnO3−δ and SrMnO3−δ with capacities of 77% and 75%, respectively. Within the tested SrxBa1−xFeyCo1−yO3−δ samples, Sr0.625Ba0.375Fe0.5Co0.5O3−δ displayed the highest CO2 capacity of ∼49%. Additionally, SrFeO3−δ samples with elevated levels of cobalt doping also exhibited significant sorption capacities, such as SrCoO3−δ, which had a CO2 capacity of 40%. However, the cost of cobalt may limit their economic attractiveness.
In addition to the sorption capacity, the redox-based oxygen capacity, defined as the percentage of reducible lattice oxygen removed during the gasification/reforming step, is an important parameter for redox-activated sorbents. The oxygen capacity influences (i) the overall heat of reaction for the two-step process, as the redox reaction facilitates in situ combustion of a fraction of the feedstock to counterbalance the endothermic gasification or reforming processes; and (ii) the distribution of reaction heat between the two steps. Specifically, the exothermic oxidation reaction would partially offset the heat required for carbonate decomposition, while the endothermic reduction reaction mitigates heat release during the carbonation reaction. Oxygen capacities for the screened materials are depicted in Fig. 3c.
Generally, within a specific sorbent family (e.g., SrxBa1−xFeyCo1−yO3−δ), compositions exhibiting higher sorption capacities also tend to have higher oxygen capacities. This relationship suggests a positive correlation between the carbonation and reduction reactions, aligning with the proposed iSERG concept. Oxygen capacities are also greatly influenced by both the type and levels of dopants at the B site. For instance, materials doped with cobalt generally exhibit greater oxygen capacity than other dopants. Increasing the B-site dopant level also significantly enhanced the oxygen capacities and reducibility of the sorbents. These distinctive characteristics provide us a broad design space for choosing redox-activated CO2 sorbents, allowing for the design of materials that optimally balance CO2 sorption and redox properties, thereby improving iSERG performance.
2.3. Correlating computational results and experimental performance
Although our screening method primarily focused on evaluating the thermodynamic feasibility and driving forces associated with sorption–desorption reactions, it is nevertheless instructive to explore the correlations between experimentally measured sorption capacities and the reaction free energies predicted by DFT. Fig. 4a depicts the relationships between CO2 sorption capacity and various ΔGs for the 85 samples that are either pure or nearly phase-pure. Meanwhile, Fig. 4b presents a confusion matrix and highlights the key parameters of the matrix to underscore the model's accuracy. Considering the potential experimental errors that can arise during CO2 capacity measurement, we adopted 2% sorbent capacity as a threshold for determining thermodynamic feasibility for iSERG applications. As can be seen, the screening model achieved a precision rate of 80% and an F1 score of 0.82, confirming the effectiveness of our model and the screening criteria.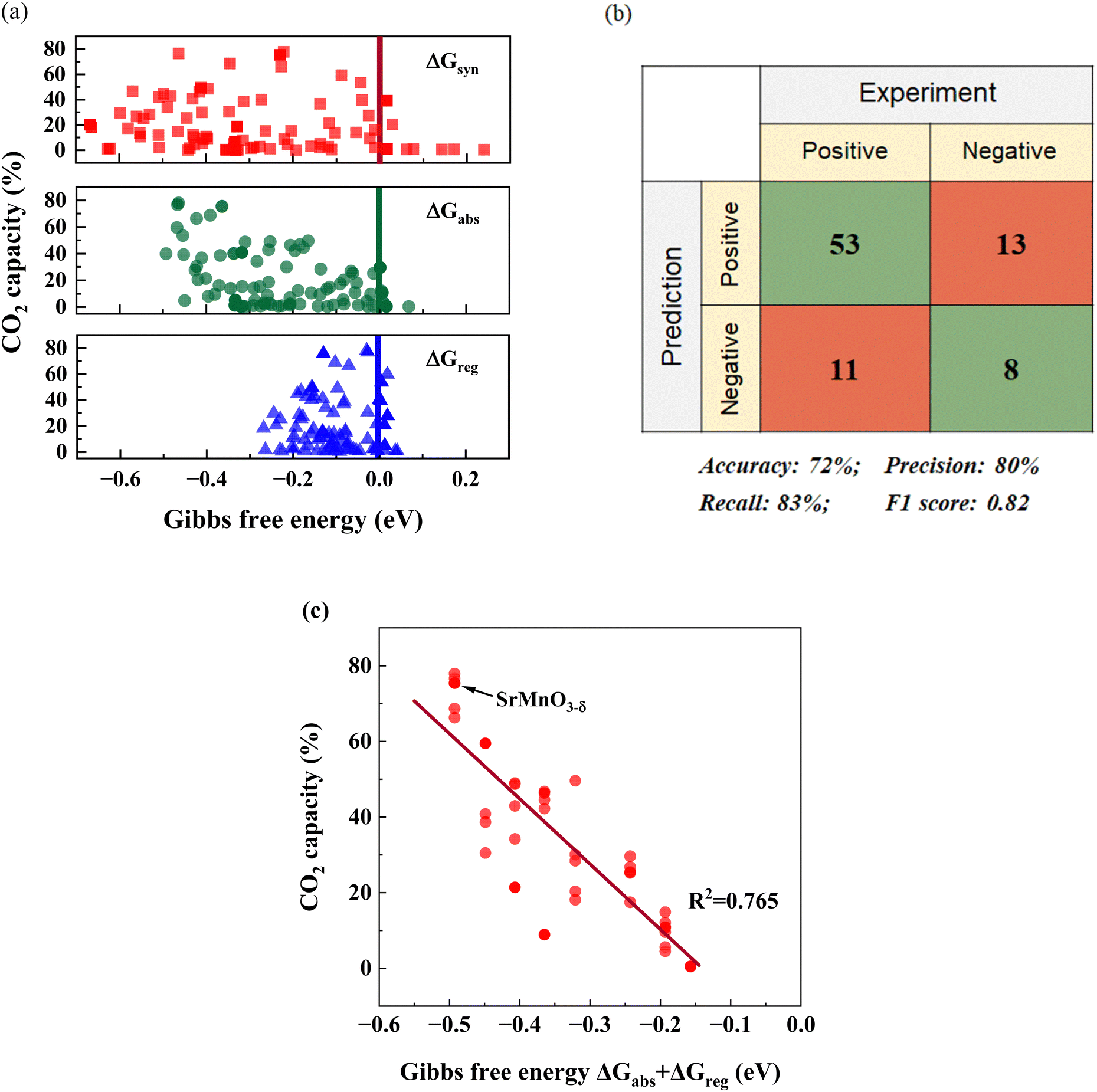 | ||
| Fig. 4 Correlation between computational and experimental results. (a) Relationships between the CO2 capacity and the different computed ΔGs. (b) Confusion matrix and the performance of the screening model. A sample is classified as “Positive” if it exhibits a CO2 capacity greater than 2%, since this confirms thermodynamic feasibility. Conversely, it is labeled as “Negative” if the capacity is less than 2%. For the prediction aspect, a “Positive” classification indicates the candidate meets all the criteria in Fig. 2a and otherwise it is classified as “Negative”. (c) Correlation between the CO2 capacity and ΔGabs + ΔGreg for SrxBa1−xFeyB1−yO3−δ (B = Co/Mn) perovskites (40 materials total). | ||
While predicting a sorbent's thermodynamic feasibility is crucial, identifying an effective descriptor for CO2 capacity holds greater practical value. It is therefore informative to investigate the correlation between the computationally derived thermodynamic parameters and the experimentally derived sorbent capacity. We note that ΔGsyn, ΔGabs, or ΔGreg, when used as a single fitting parameter, exhibited a weak correlation to CO2 capacity. Given the tendency for CaCO3 to decompose at the temperature of interest (850 °C) and the uncertainty related to Ca-containing solid solutions, the following fitting focused on the SrxBa1−xFeyB1−yO3−δ (B = Co/Mn) based materials (40 in total). Among all the fitting correlations, which are detailed in Table S2 (ESI†), the one involving (ΔGabs + ΔGreg) displayed a relatively strong relationship with CO2 capacity, exhibiting a statistical R2 value of 0.765. We also note that multiple regressions provided similar correlations (Table S2, ESI†). As a descriptor, (ΔGabs + ΔGreg) also holds physical significance: materials that exhibit low (ΔGabs + ΔGreg), which correspond to higher overall thermodynamic favorability for the carbonation and decarbonation steps tend to exhibit higher sorption capacities. This concise descriptor can notably improve the effectiveness for sorbent optimization, since it strikes a good balance among efficiency, accuracy, and practicality. Although the current model does not consider kinetic parameters, the sorption and regeneration rates are both sufficient for efficient reforming/gasification and fast regeneration, as demonstrated in the next section.
2.4. Demonstration of isothermal hydrogen generation
Considering its simplicity, lower raw material cost, and relative ease of scalable synthesis, we selected SrMnO3−δ for comprehensive performance evaluations. As shown in Fig. 5a, SrMnO3 is highly effective for iSERG of various carbonaceous feedstocks, producing ∼95% pure H2 from both biogas and methane, and yielding hydrogen enriched syngas with 73 vol% H2 from biomass under its more selective regime. Fig. 5b shows the sorbent stability under repeated iSERG cycles using simulated biogas (CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 = 2:1). The XRD of reduced and regenerated samples also affirmed the reversible phase transitions (Fig. S4, ESI†). Detailed data for the SrMnO3−δ sorbent with respect to the typical product gas profiles and cyclic stability with methane, biogas, and biomass are summarized in Fig. S5 (see ESI†). We note that during the initial stage of the reaction, the SrMnO3−δ sorbent tends to be less selective. This is likely related to the less selective oxygen species associated with the high initial oxidation state of Mn in SrMnO3−δ. In fact, a pre-reduction of the sorbent can substantially improve selectivity (Fig. S5(h), ESI†). Following a transition to SrMnO2.5, concentrated hydrogen with low CO2 concentration was produced. As depicted in Fig. S5(a/b) (see ESI†), even with a small sorbent bed containing 1.5 g of sorbent particles, the hydrogen production stage from methane was longer than 10 minutes, with ∼95% H2 purity before the CO2 breakthrough. The presence of CO2 in biogas accelerates the saturation of the sorbent, leading to a shorter yet still significant pre-breakthrough period (>5 min) for hydrogen production with ∼95% purity (Fig. S5(c/d), ESI†). The sorbent also exhibited promising performance for biomass gasification in a fluidized bed reactor (Fig. S5(e–g), ESI†). Again, the initial stage for SrMnO3 reduction was accompanied by CO2 generation. This is followed by high selectivity for hydrogen generation with effective in situ CO2 removal.
:CO2 = 2:1). The XRD of reduced and regenerated samples also affirmed the reversible phase transitions (Fig. S4, ESI†). Detailed data for the SrMnO3−δ sorbent with respect to the typical product gas profiles and cyclic stability with methane, biogas, and biomass are summarized in Fig. S5 (see ESI†). We note that during the initial stage of the reaction, the SrMnO3−δ sorbent tends to be less selective. This is likely related to the less selective oxygen species associated with the high initial oxidation state of Mn in SrMnO3−δ. In fact, a pre-reduction of the sorbent can substantially improve selectivity (Fig. S5(h), ESI†). Following a transition to SrMnO2.5, concentrated hydrogen with low CO2 concentration was produced. As depicted in Fig. S5(a/b) (see ESI†), even with a small sorbent bed containing 1.5 g of sorbent particles, the hydrogen production stage from methane was longer than 10 minutes, with ∼95% H2 purity before the CO2 breakthrough. The presence of CO2 in biogas accelerates the saturation of the sorbent, leading to a shorter yet still significant pre-breakthrough period (>5 min) for hydrogen production with ∼95% purity (Fig. S5(c/d), ESI†). The sorbent also exhibited promising performance for biomass gasification in a fluidized bed reactor (Fig. S5(e–g), ESI†). Again, the initial stage for SrMnO3 reduction was accompanied by CO2 generation. This is followed by high selectivity for hydrogen generation with effective in situ CO2 removal.
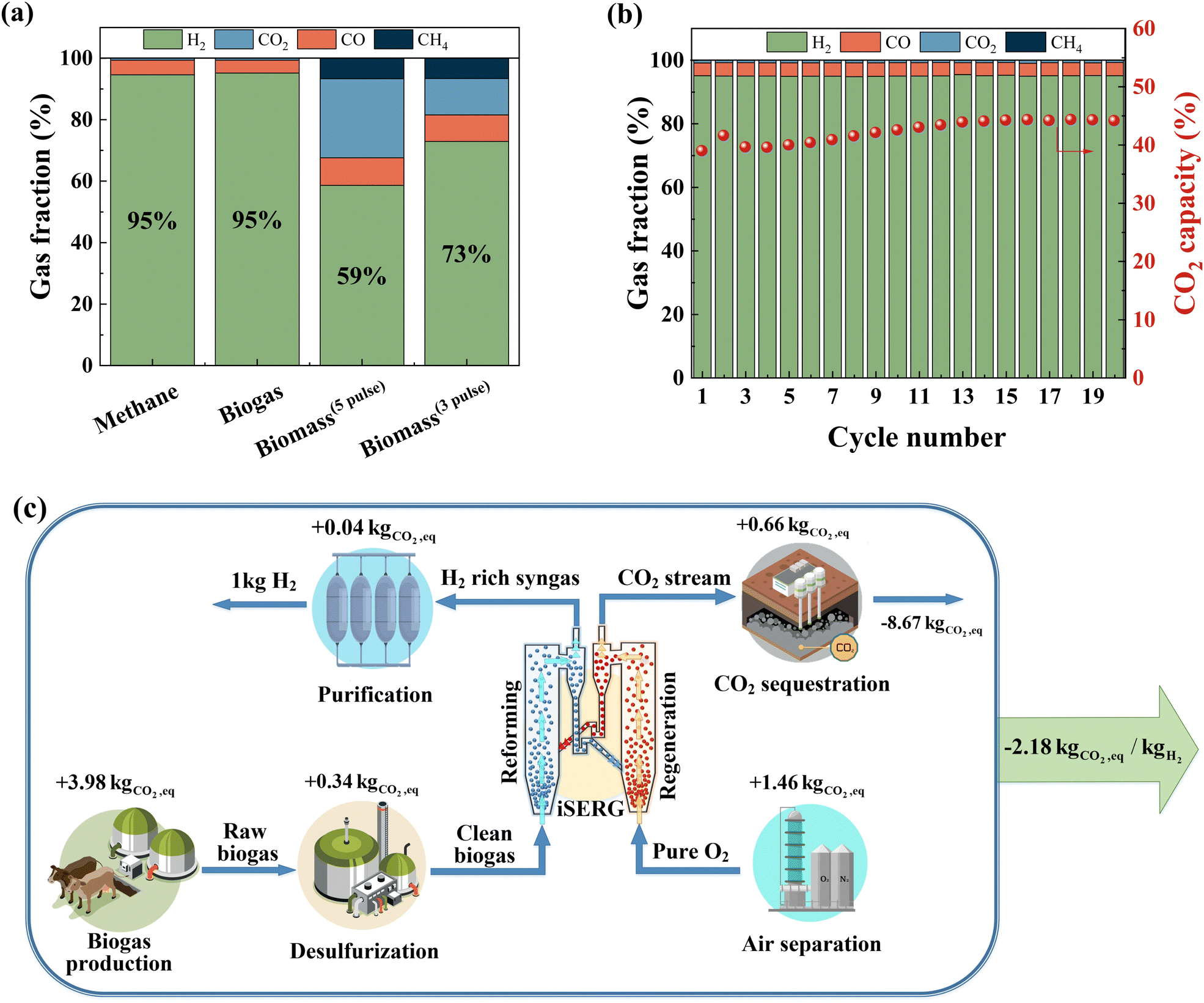 | ||
| Fig. 5 iSERG with a SrMnO3 sorbent. (a) Product gas compositions from iSERG using methane, biogas (CH4:CO2 = 2:1), and biomass feedstocks. Biomass(5pulse) represents the average gas composition of five consecutive injections of biomass samples into the fluidized bed whereas Biomass(3pulse) represents the average gas composition from the last three injections, i.e., the more selective regime. (b) Cyclic stability of the sorbent over 20 cycles using simulated biogas (CH4:CO2 = 2:1). Gas fraction is shown in a dry and Ar-free basis during the low-CO2 period. (c) Schematic of the life-cycle analysis (LCA) for hydrogen production via the iSERG process. The global warming potential (GWP) was calculated based on a per kg H2 basis. Recycle of the tail gas from the hydrogen purification step is not shown in this simplified schematic. | ||
An additional attractive feature of the iSERG is its capability for integrated CO2 capture. By modulating the duration of the regeneration step with the SrMnO3 sorbent, pure CO2 can be produced without a breakthrough of O2 (Fig. S5(i/j), ESI†). For practical implementation in a circulating fluidized bed based iSERG gasifier, we anticipate a better control over the degree of regeneration of the sorbent particles to further decrease CO2 selectivity in the gasification step while producing concentrated CO2 in the regeneration step. The latter is accomplished using concentrated oxygen from an air separation unit as the oxidant. This will eliminate the need for the energy-intensive CO2 separation step. Sequestering the captured carbon yields a carbon-negative hydrogen product with a life-cycle global warming potential (GWP) of −2.18 kg CO2eq per kg H2 (Fig. 5c and Table S3, ESI†).
3. Conclusions
This study reports a new approach that integrates both simulation and experimental methods to engineer a unique class of perovskite-structured, redox-activated CO2 sorbents. These sorbents are characterized by their stable isothermal operation and adjustable heats of reactions, enabling an efficient isothermal sorption-enhanced reforming and gasification (iSERG) process for green hydrogen production. Specifically, a high-throughput computational screening of 1225 A/B-site doped SrFeO3−δ was conducted using first-principles calculations to assess their structural stability and thermodynamic favorability for iSERG. Complementary to the simulation efforts, extensive experiments were carried out to validate the effectiveness of the computation-guided sorbent design framework. Out of the 100 sorbent candidates synthesized, 85 displayed predominantly pure perovskite phases. Moreover, 80% perovskite phases predicted to be active exhibited at least 2% CO2 sorption capacities. Among these, Sr0.75Ba0.25MnO3−δ demonstrated the highest CO2 capacity of 78%, followed by BaMnO3−δ of 77% and SrMnO3−δ of 75%. An effective thermodynamic descriptor, ΔGabs + ΔGreg, was also identified, displaying strong correlation with the CO2 capacity of sorbents. Finally, the SrMnO3−δ sorbent was extensively demonstrated for hydrogen generation through sorption-enhanced steam reforming of methane and biogas, as well as sorption-enhanced gasification of biomass. Biomass gasification in a lab-scale fluidized bed yielded nearly 73% pure H2, while both methane and biogas produced approximately 95% pure H2. This reinforces the versatility and effectiveness of the iSERG sorbents developed in this study. Our findings also support the feasibility of integrating CO2 capture in iSERG to yield carbon-negative hydrogen products from biogas with a life-cycle global warming potential as low as −2.18 kg CO2eq per kg H2.4. Experimental section
4.1. DFT calculations
Density functional theory (DFT) calculations were conducted using VASP software.42–45 The projector-augmented wave (PAW) method was used to calculate the interaction between electrons and atomic cores.46,47 The generalized gradient approximation (GGA) approach48 combined with the PBE exchange–correlation functional was applied to calculate the electronic structure and corresponding energies. The cutoff energy was 450 eV. The energy threshold was set to 10−5 eV and the force threshold to 0.01 eV Å−1. Here spin polarization effect was considered with the initial spin moments of Co, Fe, Mn and Ni to be 5, 4, 4 and 5, respectively. In addition, and the DFT+U method was utilized to give accurate description of the d electrons for Co, Cu, Fe, Mn, Ni and Ti with the effective Hubbard U values of 3.4, 4, 4, 3.9, 6 and 3 (unit in eV).49,50All the 2 × 2 × 1 supercell structures were built based on the brownmillerite SrFeO2.5 structure. We used the Monte Carlo quasi-random structures (MCSQS) algorithm implemented in the Alloy Theoretic Automated Toolkit (ATAT) code to generate the structures under different doping conditions.51 We consider the A-site dopant elements: Ba, Ca, and K, and the B-site dopant elements: Co, Cu, Fe, Mn, Ni, and Ti. It is noted that our previous work considered other A-site metals such as La, Sm, and Y. However, their carbonates are unstable at the reaction temperature of interest. These A-site elements are probably unsuitable for CO2 capture under high-temperature conditions, and they are not considered in this study. For A-site and B-site doping, the doping concentrations vary from 0 to 1 with an interval of 0.125, i.e., SrxA1−xFeyB1−yO2.5 with x and y to be 0, 0.125, …, 0.875, and 1. By following the SQS structure generation algorithm,51 1225 SrxA1−xFeyB1−yO2.5 structures were obtained. In addition, we further conducted vibrational calculations for each structure and analyzed the results by using Phonopy, which could be further used to obtain free energies.52,53 Similarly, calculations were performed on the metal oxides by the same procedures. Due to the complex spin state of O2, its free energies were obtained by using the CBS-QB3 method implemented in Gaussian16,54 and the energies were reported before.40 The other gas phase species, such as CO2, and H2O, were calculated in a cubic box with a length of 10 Å.
4.2. Thermodynamic calculations
A reaction generator was developed to balance the reaction stoichiometry for the eqn (1)–(3). The Gibbs free energies of the perovskite structures and other relevant species are obtained using the methods in Section 4.1. The transition between the perovskite phase and the brownmillerite phase is spontaneous under the reduction and oxidation conditions. Therefore, the brownmillerite structure was chosen as the initial state of eqn (2) and the end state of eqn (3) in thermodynamic analysis. This calculation approach is expected to provide more accurate results compared to using perovskites. To minimize the error arising from the inaccurate description of the free energies of species such as carbonates, experimental Gibbs free energies from HSC Chemistry software were utilized for solid reactions. These reactions include carbonation reactions (e.g., SrO + CO2 → SrCO3) for eqn (2), decarbonation reactions for eqn (3), and hydrogen reduction to water for eqn (2). The reaction free energy ΔG ≤ 0 eV was used as the initial criteria for eqn (1)–(3). The energies of oxides, oxygen, etc. are from DFT VASP calculations.The first thermodynamic screening step was aligned with typical perovskite synthesis conditions (1000 °C and PO2 = 0.2 bar). Fig. 2b presents the heatmap displaying the ΔGsyn of all the structures. Notably, the stability of perovskites decreases with the increase in the doping concentrations of Cu, Mg, Ni, and Ti. However, this trend is not observed in samples doped with Co and Mn, making them more suitable for the SERG process due to their stability. A typical SERG temperature of 850 °C was selected as the reaction temperature of eqn (2) and (3). During the reduction/carbonation step, it is assumed that the system reaches equilibrium, with the partial pressure ratio PH2O/PH2 set at 0.2 and PCO2 at 0.075 bar. During the regeneration step, the obtained mixed metal oxides and carbonates are expected to regenerate under oxidation conditions with PO2 = 0.2 bar and PCO2 = 0.015 bar.
4.3. Sample synthesis and characterization
The perovskite materials for both the screening and reactor experiments were prepared using a solid-state reaction method. For a typical synthesis of SrxCa1−xFeyCo1−yO3−δ for the screening experiment in the thermogravimetric analyzer (TGA), stoichiometric amounts of SrCO3, CaCO3, Fe2O3, and Co3O4 were added in a 5 mL Teflon vial and then mixed with 2 mm yttrium-stabilized ZrO2 balls with a mass ratio of 1:5. 2–3 mL ethanol (Fisher Scientific, CDA 19 (Histological)) was further added to the mixture to promote the mixing performance and reduce the particle size. A stainless-steel sample jar holding four vials with four different materials was balled milled at 1200 rpm for 3 h or 24 h using a high-energy ball mill (Vivtek Instrument: VBM-V80). The resulting wet slurry was dried in an oven at 95 °C for 30 min and 130 °C for another 30 min to remove ethanol. The dried powders were separated from ZrO2 beads and then calcined at 1000 °C in a muffle furnace for 10 h to form the perovskite structure. The ramping rates for both heating and cooling steps were all set to 3 °C min−1. Several Mn-doped materials were subjected to calcination at 1200 °C in a tube furnace to improve the phase purity. The fresh perovskite sorbents were sieved to two desired particle size ranges, i.e., 180–250 μm for TGA experiments and 0–180 μm for XRD. A total of 100 different materials were synthesized for the screening experiments in the TGA. A large batch of SrMnO3 was prepared for the reactor experiments. The precursors were mixed in two 150 mL and two 100 mL Teflon jars and ball-milled with 3 mm, 6 mm, and 10 mm ZrO balls and then milled at 250 rpm at a 45° angle for 12 h in a planetary ball mill (Columbia International, CIT-XBM4X-2.0L). The mixture of precursor powders after the drying step was first pelletized at 20 tons before calcined at 1100 °C in the muffle furnace. Afterwards, the resulting sorbent particles were sieved into a size of 180–425 μm. The rest of the synthesis procedures were the same as the screening study.
The precursors used in this study were SrCO3 (Sigma Aldrich, >99.9%), CaCO3 (Sigma Aldrich, >99%), BaO (Thermo Scientific, >99.5%), Co3O4 (Aldrich, >99%), Fe2O3 (Noah Chemicals, >99.9%), MnO2 (Materion, >99.9%), CuO (Noah Chemicals, >99%), KNO3 (Noah Chemicals, >99%), NiO (Noah Chemicals, >99%), and MgO (Materion, >99.5%). The phase structures of the prepared fresh sorbents were characterized on a PANalytical X'Pert PRO X-ray diffraction (XRD) operating at 45 kV and 40 mA. The sample was scanned from 2θ of 15° to 80° with a step size of 0.0262° and a hold time of 0.2 s for each step. The analyses and identifications of the XRD phases of all the samples were accomplished using Highscore Plus software.
4.4. Experimental reactivity evaluation
To mitigate the less selective regime, another experiment was implemented with the incorporation of a pre-reduction step. The sample was pre-reduced with a gas mixture of 6 sccm H2 and 30 sccm Ar for 10 min, followed by an Ar purge prior to the normal reaction cycle. Additionally, another experiment aimed at producing pure CO2 without O2 breakthrough for sequential carbon capture involved a shortened regeneration time. To control the reaction environment, 1.75 g sorbents, 4 sccm of CH4, 2.3 sccm of CO2, 5.88 μL min−1 of deionized water, and an 8 min regeneration time were utilized. The methane-to-carbon dioxide ratio was adjusted to match a typical biogas composition in the literature.55 Throughout these experiments, steam was condensed and separated downstream, and the remaining gaseous products were analyzed using an MKS Cirrus II quadrupole mass spectrometer (QMS). The reduced and regenerated samples from the biogas experiments were collected and tested using XRD to validate the reversibility for phase transition. Due to mass transfer limitations in the pack bed, the regenerated samples taken from the reactors were treated in TGA at 850 °C under a continuous flow of 40 sccm O2 and 160 sccm Ar prior to XRD characterization.
Author contributions
Conceptualization, F. L.; methodology, R. C., K. Y., X. W., F. L.; investigation, R. C., K. Y., X. W., M. R., A.-S. B., E. G., A. P., L. B., W. T.; writing – original draft, R. C., K. Y.; writing – review & editing, R. C., K. Y., X. W., L. B., W. T., P. W., F. L.; funding acquisition, F. L.; resources, F. L.; supervision, F. L.Data availability
The data supporting the findings are available within the main text and ESI.† More detailed data can be made available upon request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the U.S. Department of Energy Office of Energy Efficiency & Renewable Energy (no. EE0008809) and the US National Science Foundation (CBET-1923468). We acknowledge the use of the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation. We also acknowledge the computing resources provided by North Carolina State University High Performance Computing Services Core Facility (RRID:SCR_022168). We also thank Dr Fan Yang and Dr Yuan Yao (Yale University) for their suggestions with respect to the life-cycle analysis.References
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2020, 45(5), 3396–3406 CrossRef CAS.
- T. da Silva Veras, T. S. Mozer, D. da Costa Rubim Messeder dos Santos and A. da Silva César, Int. J. Hydrogen Energy, 2017, 42(4), 2018–2033 CrossRef CAS.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- International Energy Agency, Global hydrogen review 2022, Paris, https://www.iea.org/reports/global-hydrogen-review-2022, (Access. May. 6th, 2024), 2022.
- A. Zapantis, Circular carbon economy series: blue hydrogen., 2021, https://www.globalccsinstitute.com/wp-content/uploads/2021/04/Circular-Carbon-Economy-series-Blue-Hydrogen.pdf, (Access. May 6th, 2024) Search PubMed.
- N. Sazali, Int. J. Hydrogen Energy, 2020, 45(38), 18753–18771 CrossRef CAS.
- F. Dawood, M. Anda and G. M. Shafiullah, Int. J. Hydrogen Energy, 2020, 45(7), 3847–3869 CrossRef CAS.
- L. Cao, I. K. M. Yu, X. Xiong, D. C. W. Tsang, S. Zhang, J. H. Clark, C. Hu, Y. H. Ng, J. Shang and Y. S. Ok, Environ. Res., 2020, 186, 109547 CrossRef CAS PubMed.
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9(10), 2939–2977 RSC.
- U.S. Department of Energy. DOE national clean hydrogen strategy and roadmap, Jun. 2023, https://www.hydrogen.energy.gov/library/roadmaps-vision/clean-hydrogen-strategy-roadmap (Access May 6th, 2024).
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10(9), 1885–1910 RSC.
- Y. Gao, J. Jiang, Y. Meng, F. Yan and A. Aihemaiti, Energy Convers. Manage., 2018, 171, 133–155 CrossRef CAS.
- T. Mendiara, F. García-Labiano, A. Abad, P. Gayán, L. F. de Diego, M. T. Izquierdo and J. Adánez, Appl. Energy, 2018, 232, 657–684 CrossRef CAS.
- J. Andersson and J. Lundgren, Appl. Energy, 2014, 130, 484–490 CrossRef CAS.
- Y. Richardson, J. Blin and A. Julbe, Prog. Energy Combust. Sci., 2012, 38(6), 765–781 CrossRef CAS.
- M. Puig-Gamero, J. Argudo-Santamaria, J. L. Valverde, P. Sánchez and L. Sanchez-Silva, Energy Convers. Manage., 2018, 177, 416–427 CrossRef CAS.
- A. M. Parvez, S. Hafner, M. Hornberger, M. Schmid and G. Scheffknecht, Renewable Sustainable Energy Rev., 2021, 141, 110756 CrossRef CAS.
- D. Chen and L. He, ChemCatChem, 2011, 3(3), 490–511 CrossRef CAS.
- M. Broda, A. M. Kierzkowska, D. Baudouin, Q. Imtiaz, C. Copéret and C. R. Müller, ACS Catal., 2012, 2(8), 1635–1646 CrossRef CAS.
- C. Dang, L. Liu, G. Yang, W. Cai, J. Long and H. Yu, Chem. Eng. J., 2020, 383, 123204 CrossRef CAS.
- J. Udomsirichakorn, P. Basu, P. Abdul Salam and B. Acharya, Fuel Process. Technol., 2014, 127, 7–12 CrossRef CAS.
- H. An, T. Song, L. Shen, C. Qin, J. Yin and B. Feng, Int. J. Hydrogen Energy, 2012, 37(19), 14195–14204 CrossRef CAS.
- B. Jiang, B. Dou, K. Wang, C. Zhang, M. Li, H. Chen and Y. Xu, Chem. Eng. J., 2017, 313, 207–216 CrossRef CAS.
- J. C. Abanades, Chem. Eng. J., 2002, 90(3), 303–306 CrossRef CAS.
- F. D. M. Daud, K. Vignesh, S. Sreekantan and A. R. Mohamed, New J. Chem., 2016, 40(1), 231–237 RSC.
- G. S. Grasa and J. C. Abanades, Ind. Eng. Chem. Res., 2006, 45(26), 8846–8851 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2007, 41(4), 1420–1425 CrossRef CAS PubMed.
- H. K. Rusten, E. Ochoa-Fernández, H. Lindborg, D. Chen and H. A. Jakobsen, Ind. Eng. Chem. Res., 2007, 46(25), 8729–8737 CrossRef CAS.
- H. R. Radfarnia and M. C. Iliuta, Chem. Eng. Process., 2014, 86, 96–103 CrossRef CAS.
- G. Wu, C. Zhang, S. Li, Z. Huang, S. Yan, S. Wang, X. Ma and J. Gong, Energy Environ. Sci., 2012, 5(10), 8942 RSC.
- A. Armutlulu, M. A. Naeem, H. J. Liu, S. M. Kim, A. Kierzkowska, A. Fedorov and C. R. Muller, Adv. Mater., 2017, 29(41), 1702896 CrossRef PubMed.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Muller, Chem. Rev., 2021, 121(20), 12681–12745 CrossRef CAS PubMed.
- V. Manovic and E. J. Anthony, Fuel, 2011, 90(1), 233–239 CrossRef CAS.
- F.-C. Yu, N. Phalak, Z. Sun and L.-S. Fan, Ind. Eng. Chem. Res., 2011, 51(4), 2133–2142 CrossRef.
- H. Sun, J. Wang, J. Zhao, B. Shen, J. Shi, J. Huang and C. Wu, Appl. Catal., B, 2019, 244, 63–75 CrossRef CAS.
- L. Brody, R. Cai, A. Thornton, J. Liu, H. Yu and F. Li, ACS Sustainable Chem. Eng., 2022, 10(19), 6434–6445 CrossRef CAS.
- C. Dang, Y. Li, S. M. Yusuf, Y. Cao, H. Wang, H. Yu, F. Peng and F. Li, Energy Environ. Sci., 2018, 11(3), 660–668 RSC.
- C. Dang, H. Yu, H. Wang, F. Peng and Y. Yang, Chem. Eng. J., 2016, 286, 329–338 CrossRef CAS.
- L. Brody, M. Rukh, R. Cai, A. Saberi Bosari, R. Schomäcker and F. Li, J. Phys. Energy, 2023, 5(3), 035004 CrossRef.
- X. Wang, Y. Gao, E. Krzystowczyk, S. Iftikhar, J. Dou, R. Cai, H. Wang, C. Ruan, S. Ye and F. Li, Energy Environ. Sci., 2022, 15(4), 1512–1528 RSC.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5(2), eaav0693 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- A. Jain, G. Hautier, C. J. Moore, S. Ping Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2011, 50(8), 2295–2310 CrossRef CAS.
- G. Hautier, S. P. Ong, A. Jain, C. J. Moore and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(15), 155208 CrossRef.
- A. van de Walle, P. Tiwary, M. de Jong, D. L. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L. Q. Chen and Z. K. Liu, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2013, 42, 13–18 CrossRef CAS.
- A. Togo, J. Phys. Soc. Jpn., 2023, 92(1), 012001 CrossRef.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- N. Hajjaji, S. Martinez, E. Trably, J.-P. Steyer and A. Helias, Int. J. Hydrogen Energy, 2016, 41(14), 6064–6075 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02119c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |