
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing the LAGP/Li interface and in situ visualizing the interfacial structure evolution for high-performance solid-state lithium metal batteries†
Jin
Li
a,
Junjie
Chen
a,
Xiaosa
Xu
a,
Jing
Sun
*a,
Baoling
Huang
*a and
Tianshou
Zhao
 *ab
*ab
aDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: jsunav@connect.ust.hk; mebhuang@ust.hk; zhaots@sustech.edu.cn
bDepartment of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen, 518055, China
First published on 28th June 2024
Abstract
Direct tracking of the structure and composition evolution at the solid-state electrolyte/electrode interface and properly addressing the interfacial issues are crucial for the performance improvement of solid-state lithium metal batteries (SSLMBs). In this study, we investigate the structure evolution of the interface between Li1.5Al0.5Ge1.5(PO4)3 (LAGP) and the lithium anode using in situ transmission electron microscopy (TEM). It is found that the reaction between lithium and pristine LAGP results in a continuous volume expansion and contact loss, even without applying voltage. To stabilize the interface, we construct a multi-layer solid electrolyte where the LAGP is coated with the polymer electrolyte (P-DOL), enabling the interface layer to maintain its pristine morphology throughout the lithiation process. In addition, P-DOL promotes the formation of rich LiF at the interface, inhibiting the electron transport and volume expansion of LAGP, as further confirmed by the cryo-TEM and simulation analysis. The effectiveness and cyclability of the unique multi-layer electrolyte are demonstrated in various cells, even under harsh testing conditions, such as a high rate (10 C), a high active material loading (11.7 mg cm−2), a wide voltage range (2.8–4.45 V), and temperatures ranged from −20 to 50 °C. By applying the same interfacial modification method, LLZTO-based (Li6.4La3Zr1.4Ta0.6O12) electrolytes with both high ionic conductivity and interfacial stability are also prepared. This work provides valuable guidance for investigations of contact reactions and failure mechanisms at solid–solid interfaces, ultimately facilitating the design of high-performance SSLMBs.
Broader contextCurrently, solid state lithium metal batteries are being researched intensively on account of their merits of superior energy density and safety. Oxide types solid electrolytes such as LLZTO (Li6.4La3Zr1.4Ta0.6O12) and LAGP (Li1.5Al0.5Ge1.5(PO4)3) have garnered significant attention from researchers due to their high ionic conductivity, excellent mechanical properties, and ease of synthesis. Among them, LLZTO is easily degraded by exposure to air, forming lithium-phobic Li2CO3 which leads to increased interfacial impedance. In comparison, LAGP exhibits good air stability, eliminating the need for stringent synthesis and storage requirements, and is a promising solid electrolyte candidate. However, it is crucial to acknowledge the pivotal role in determining the performance and stability of lithium metal batteries, especially to understand the mechanisms of interfacial reaction. In our study, we utilize real-time TEM imaging techniques, combined with cryo-TEM and TOF-SIMS analysis to look into the structure evolution and composition change of both original and stabilized LAGP/Li interface. The findings presented in this study contribute to the development of more efficient and durable lithium metal batteries, as well as interfacial modification for various alkali metal batteries. |
1. Introduction
Rechargeable lithium metal batteries (LMBs), with their high theoretical capacity and energy density, have garnered significant research attention for meeting portable electronic devices and electric vehicles demands.1–3 However, the practical application of LMBs is hindered by the growth of lithium dendrites, which can lead to short circuits and rapid capacity degradation.4,5 To address these challenges, the use of inorganic solid electrolytes (ISEs) to form solid-state electrolytes has emerged as an effective approach due to their high shear modulus, which physically inhibits lithium dendrite growth.6–9 Among ISEs, the oxide type (such as LAGP and LLZTO) has attracted considerable interest due to its favorable properties, including high lithium conductivity (10−3–10−4 S cm−1 at 25 °C), wide electrochemical window (up to 6 V), and straightforward synthesis.10 However, LLZTO tends to degrade upon exposure to air, forming lithium-phobic Li2CO3, which increases interfacial impedance.11–13 In comparison, LAGP exhibits good air stability, eliminating the need for stringent synthesis and storage requirements.14,15 Leveraging these advantages, LAGP has emerged as a promising candidate for large-scale applications of high-energy-density solid-state lithium-metal batteries (SSLMBs).Although LAGP exhibits a wide electrochemical window that can be paired with various high-voltage cathodes, it is prone to unstable interfaces when coupled with lithium metal.16–18 At the Li/LAGP interface, the side reaction between Li and LAGP leads to the formation of a porous solid electrolyte interphase (SEI) layer and volume expansion (Fig. 1a), which hampers Li+ transportation and fails to block the electron transfer.19 The continuous growth of the SEI layer leads to uneven Li+/e− flux, resulting in increased interfacial resistance and local current density (hotpots).20 The presence of hotspots and volume changes further exacerbates the interfacial chemistry, promoting the growth of lithium dendrites and ultimately causing cell short-circuiting.21,22 To address these challenges, researchers have reported strategies for regulating interfacial chemistry, such as introducing a lithophilic and flexible polymer interlayer to enhance wettability and reduce interfacial resistance.23–27 However, most polymer electrolytes exhibit insufficient room-temperature ionic conductivity, and many lithophilic interlayers (i.e., Ge and ZnO) require preparation at high temperatures (>200 °C), incurring additional costs.28,29 Jiang et al. constructed LiF-rich intermediate layers which is in situ self-generated through electrochemical processes in lean liquid electrolyte to alleviate LATP/Li interface incompatibility.10 Also, Xu et al. indicated that 1,3 dioxolane is conducive to LiPF6 decomposition to LiF, achieving excellent interface compatibility and lithium deposition/stripping.30 Moreover, the understanding of the evolution process of the Li/LAGP interface structure and its impact on interfacial properties remains unclear. Conventional ex situ experimental methods, such as XPS and SEM, only provide information of the initial and final interface states. Therefore, real-time tracking of interfacial electrochemical processes using in situ electron microscopy characterization is crucial for gaining knowledge about interface evolution and understanding performance degradation mechanisms.
 | ||
| Fig. 1 Schematic illustrations of the working principle of the SSLMBs with (a) pristine LAGP and (b) LAGP@P-DOL. (c) and (d) Schematic diagram of the in situ TEM nano cell. Time-lapse in situ TEM images during lithiation and delithiation process with (e) pristine LAGP and (f) LAGP@P-DOL. The scale in the picture is 500 nm. | ||
Herein, we investigated the structure and composition change at the anode/electrolyte interfaces of SSLMBs with Li/LAGP and Li/LAGP@P-DOL using in situ TEM. The main cause of instability at the LAGP/Li interface is determined to be the increased interfacial contact loss resulting from the expansion of LAGP volume during lithiation and delithiation processes. To address this issue, we used a polymer electrolyte (referred to as P-DOL) to stabilize the Li/LAGP interface in SSLMBs due to its high ionic conductivity, ease of preparation, and good wettability. In situ observations reveal that P-DOL played a crucial role as a protector of the LAGP interfacial layer (Fig. 1b). The presence of P-DOL suppresses interfacial side reactions and promotes the formation of a LiF-rich interface, significantly improving the stability of interface between the electrolyte and lithium anode and extending the operating life of the SSLMBs. Electrochemical results demonstrate that lithium symmetric batteries using P-DOL modified LAGP electrolyte can withstand high critical current densities (CCD) up to 2.4 mA cm−2 and provide an ultra-long cycle life over 2100 hours at 0.2 mA cm−2. Additionally, the assembled LiFePO4 (LFP) solid-state batteries exhibit stable operation within the temperature range from −20 to 50 °C, and even demonstrates 1450 cycles at a hash current of 10.0 C at 30 °C. These findings highlight the effectiveness of P-DOL in suppressing side reactions at the Li/LAGP interface, regulating electron transfer and ion migration at the interface, and contributing to uniform lithium deposition, thus, significantly enhancing the performance of SSLMBs based on LAGP.
2. Results and discussion
In situ TEM techniques offer valuable visual insights into the structural evolution and phase transitions of materials during cycling.31,32 In this study, nanoscale battery configurations are designed for in situ TEM visualization where a LAGP@P-DOL or LAGP sphere was attached to a gold wire as the working electrode and a tungsten wire with a piece of Li/Li2O served as the counter electrode (Fig. 1c and d). By applying a negative bias to the tungsten wire, LAGP or LAGP@P-DOL underwent lithiation, followed by delithiation when the bias was reversed to a positive value. Time-lapse TEM images captured the transient morphological changes of the LAGP sample during the cycling process (Fig. 1e and Video S1, ESI†). The results reveal that chemical reactions occur between LAGP and lithium, even without the application of voltage. Notably, LAGP experiences significant volume expansion when a negative bias is applied, nearly doubling its volume compared to the unbiased state. Even after the bias is switched to a positive value, the volume expansion of LAGP continues, indicating a poor interface compatibility between LAGP and lithium metal. Due to the influence of steric hindrance, the morphology of LAGP in the in direct contact with Li/Li2O shows unobvious change in the longitudinal direction but expands laterally, thereby affecting the volume of surrounding LAGP particles. In contrast, LAGP@P-DOL preserves morphology throughout the entire lithiation/delithiation process, demonstrating that P-DOL layer can buffer the volume expansion of the LAGP@P-DOL/Li heterogeneous structure (Fig. 1f and Video S2, ESI†). The results of Electrochemical impedance spectroscopy (EIS) tests in lithium symmetrical battery with different resting times show that the interface impedance gradually increases for the cell with LAGP directly contacting lithium. In comparison, when adopting LAGP@P-DOL with lithium, interface impedance of the cell almost does not change over the same period (Fig. S1a and b, ESI†). The structural stability of LAGP@P-DOL was further confirmed through repeated lithiation/delithiation cycles under a constant bias (−5/+5 V). The results highlight the excellent structural and mechanical stability of LAGP@P-DOL, enabling remarkable electrochemical reversibility in SSLMBs.After determining the protective functions of P-DOL, we constructed a multi-layer quasi composite solid-state electrolytes (referred to as QCPE@P-DOL) using an in situ ring-opening polymerization method, as shown in Fig. 2a. The relative concentrations of DOL monomer, (1,3-dioxolane) and LAGP powder, solvent diethyl carbonate (DEC) and fluoroethylene carbonate (FEC), LiTFSI (lithium bis(triffuoromethanesulfonyl)imide), EMImNTF2 (1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide) and initiator LiPF6 (lithium hexafluorophosphate) were systematically adjusted to achieve the highest ionic conductivity. The FEC was added in the solvent to induce the formation of a fluorine-rich interphase, thus enhancing interfacial stability. The QCPE@P-DOL was designed to achieve stable electrolyte/electrode interfaces and enable high-energy-density SSLMBs with excellent electrochemical performance over a wide temperature range. In contrast, electrolytes without P-DOL nanolayers, referred to as QCPE, were prepared using the same materials but with different addition sequences of the ingredients (see the details in ESI† and schematic setup of the cells with different CPEs as exhibited in Fig. S2, ESI†). X-ray computed tomography characterization (Fig. 2b) demonstrates the successful construction of the multilayer structure QCPE@P-DOL through selective adsorption on the GF separator, resulting in the formation of LAGP@P-DOL particles on the anode side to improve the electrolyte-lithium anode interface. On the cathode side, the in situ formed polymer layer (P-DOL) efficiently wets the cathode and reduces interfacial impedance.33,34 The asymmetric structure of QCPE@P-DOL film was characterized in Fig. 2c and d from disassembled cells. After the in situ polymerization the gaps between GF fibers are filled with P-DOL electrolyte while the side with content of LAGP@P-DOL particles is installed facing the lithium anode due to the high mechanical strength. Moreover, the coated P-DOL effectively prevents direct contact between LAGP and the lithium metal anode, reducing side reactions at the interface. TEM investigation was conducted to assess the microstructure of LAGP@P-DOL. As depicted in Fig. 2e, LAGP particles are surrounded by non-uniform P-DOL nanolayers which formed by in situ polymerization. The highly homogeneous distribution of different elements, as shown in EDS mapping (Fig. 2f and Fig. S3, ESI†), indicates that P-DOL, containing C, N, S, and F elements, is coated onto LAGP. The in situ polymerization of DOL was characterized by Fourier-transform infrared (FTIR) spectroscopy (Fig. 2g). The FTIR spectrum of pure DOL exhibit characteristic peaks corresponding to C–H out-of-plane bending vibration (915 cm−1) and C–O–C vibration (1029 cm−1).35 In contrast, QCPE and QCPE@P-DOL show the characteristic peak of poly-DOL, with a significant intensity reduction in the C–H out-of-plane bending vibration, a shift in the C–O–C vibration, and the appearance of vibrations from long-chain species (840 cm−1).36
 | ||
| Fig. 2 (a) Schematic diagram of the synthetic route. (b) X-ray computed tomography of QCPE@P-DOL. SEM and inset digital images of QCPE@P-DOL membranes from the disassembled batteries with different sides (c) near cathode. (d) near anode. (e) TEM images (f) EDS mapping of LAGP@P-DOL particles. (g) FTIR spectra of DOL solvent, QCPE, and QCPE@P-DOL. (h) The ionic conductivity heat diagram of the developed electrolytes at 25 °C with varied DOL and LAGP. (i) Arrhenius plots for the calculation of the activation energy related to the mobility of Li+. (j) EIS of symmetrical lithium metal batteries with QCPE and QCPE@P-DOL. (k) DFT calculations of the interfacial formation energies of the Li/P-DOL, LAGP/Li, and LAGP/P-DOL interfaces. Color code: Li (green), C (brown), O (red), H (white), P (light purple), Ge (purple), Al (blue). (l) Comparisons of σLi+ and tLi+ between QCPE and QCPE@P-DOL. | ||
To improve the ionic conductivity of the electrolyte, the ratios of DOL, LAGP, and solvent were optimized. EIS was employed to measure the ionic conductivity of electrolytes at room temperature (Fig. 2h), and the electrolyte compositions were provided in Table S1 (ESI†). Visual observation of the precursor solution of CPEs after polymerization was shown in Fig. S4 (ESI†), confirming their liquid-like state (when DOL content <50 wt%). The conductivity exhibits a decreasing trend with the increased DOL content. When the DOL content was 60 wt%, the electrolyte is quasi-solid state and its ionic conductivity decrease further as DOL content increased to 70 wt%, likely due to increased solidification and reduced localization length of poly-DOL.34 The ionic conductivity initially increases and then decreases with the addition of ceramic oxide (LAGP). At high concentrations (>5 wt%), LAGP particles will form agglomeration that reduces interface wettability, resulting in decreased conductivity.37 In addition, the activation energy for the QCPE and QCPE@P-DOL system is 0.16 and 0.14 eV, respectively (Fig. 2i), indicating that QCPE@P-DOL can facilitate lithium-ion transport. The Li-ion transference number (tLi+), which plays a vital role in the ionic conductivity of CPEs, is increased from 0.19 to 0.56 at room temperature with P-DOL coating (Fig. S5a and b, ESI†). Therefore, it can be concluded that the LAGP@P-DOL provides high-speed transmission channels and promotes lithium-ion transport. The electrochemical stability of SSEs at room temperature was studied by linear sweep voltammetry (LSV) (Fig. S6, ESI†). QCPE@P-DOL maintains anodic stability at threshold voltages up to 4.7 V (vs. Li+/Li), meeting the requirements of most high-voltage cathode materials.
The interfacial resistance was further determined through EIS test as shown in the Nyquist plots of symmetric lithium cells with different electrolytes (Fig. 2j). It is revealed that the impedance of the QCPE@P-DOL cell is significantly reduced from 1200 Ω of the QCPE cell to 450 Ω, indicating the coating of P-DOL layers facilitates interfacial ion migration. The interfacial formation energies of the Li/P-DOL, LAGP/Li, and LAGP/P-DOL interfaces are calculated to be −0.16, −1.14, and −0.3 J m−2, respectively (Fig. 2k and Table S2, the molecular models were in Fig. S7, ESI†). The interfacial formation energies of the Li/P-DOL and LAGP/P-DOL interfaces are more positive than those of LAGP/Li, indicating that the introduction of P-DOL coating layers can effectively suppress the interfacial reaction between the lithium anode and LAGP. Overall, as summarized in Fig. 2l, the aforementioned analyses demonstrate that the designed QCPE@P-DOL exhibits higher lithium-ion conductivity and transference number compared to QCPE in SSLMBs.
The electrochemical performance of QCPE@P-DOL is investigated in full LMBs with LFP based cathode. The EIS of the full cell was conducted to characterize the Li+ and electron transfer resistance (Fig. 3a).38,39 The cell with QCPE@P-DOL exhibit much lower charge transfer resistance compared to those with QCPE, primarily due to the improved conductivity of the electrolyte and enhanced wettability of the anode interface. To further investigate ion transport in the batteries, the Li+ diffusion coefficient of QCPE@P-DOL is calculated to be 1.11 × 10−14 cm2 s−1, which is more than quadruple that of QCPE (2.33 × 10−15 cm2 s−1), as shown in Fig. 3b. The higher Li+ diffusion coefficient of QCPE@P-DOL promotes the transport of Li+ in the cells, resulting in improved discharge capacity in SSLMBs. Rate performance tests ranging from 0.2 to 5.0 C were conducted (Fig. S8a–c, ESI†), and the LMBs with QCPE@P-DOL exhibits higher discharge capacity at each rate compared to those with QCPE. Specifically, LMBs with QCPE@P-DOL achieves a discharge capacity of 113.3 mA h g−1 at 5.0 C, while the capacity of the LMBs with QCPE drops to nearly 10 mA h g−1. In addition, Fig. 3c and d show that the cell with QCPE@P-DOL (LFP|QCPE@P-DOL|Li) exhibits a stable charge/discharge profile and lower polarization during battery operation. On the other hand, the cell with QCPE (LFP|QCPE|Li) displays extremely unstable coulombic efficiency (CE) during cycling due to poor contact and continuous side reactions at the LAGP/Li interface, which is consistent with the interfacial evolution observed using in situ TEM in Fig. 1. Meanwhile, EIS measurements after different cycle numbers further confirmed that QCPE@P-DOL effectively improved the electrode interface stability compared with QCPE (Fig. S9, ESI†). The LFP|QCPE@P-DOL|Li battery delivers a discharge capacity of 105.1 mA h g−1 with a capacity retention of 81.7% after 1300 cycles with cutoff voltages being 2.5 and 3.8 V at 2.0 C and 30 °C (Fig. 3e). Even at higher current rates of 5.0 C and 10.0 C, the battery still exhibits high initial capacity and stable reversible capacity (113.1 mA h g−1 at 5.0 C and 106.4 mA h g−1 at 10.0 C), as shown in Fig. 3f and Fig. S10 (ESI†).
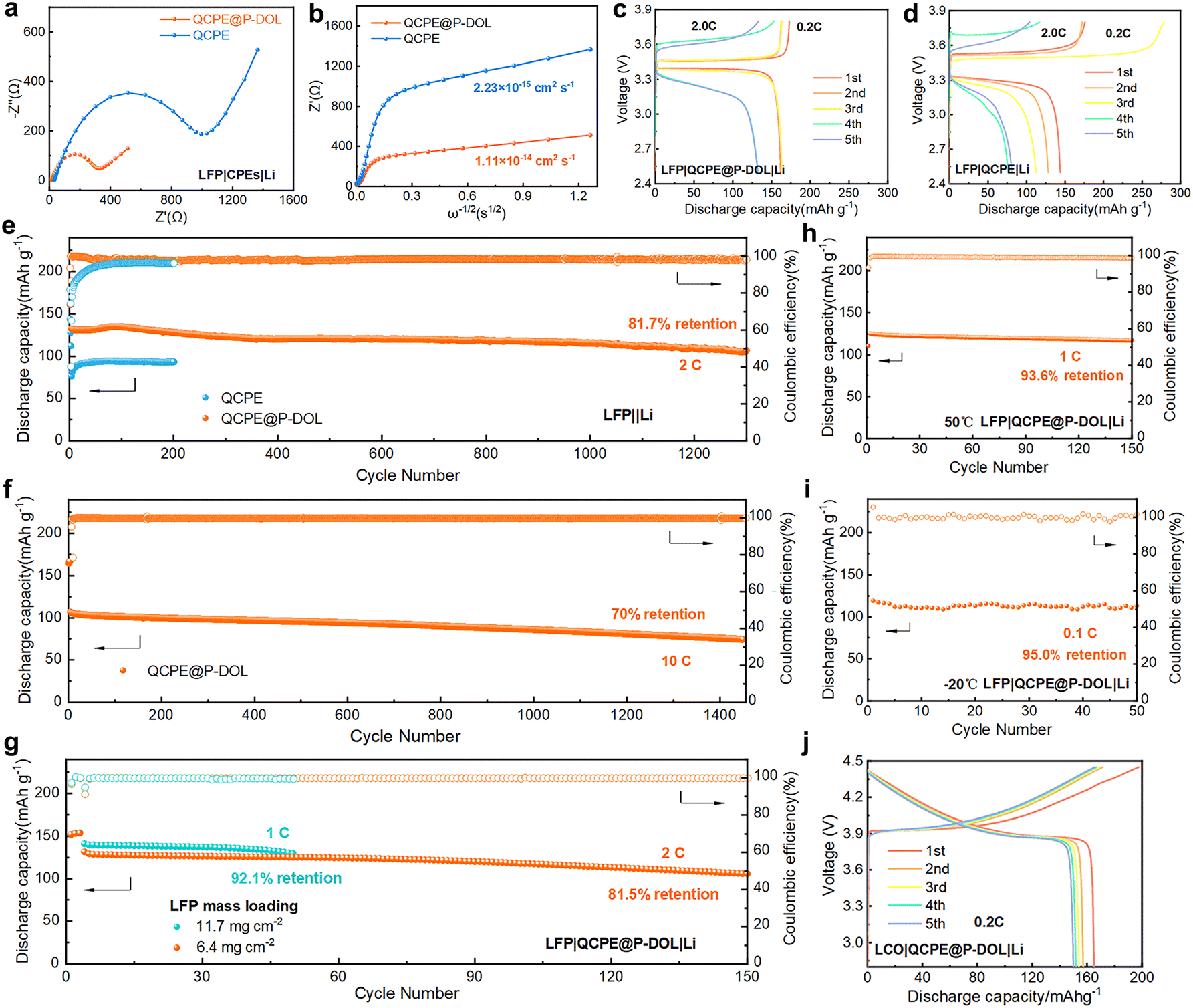 | ||
| Fig. 3 (a) EIS of LFP-based LMBs with QCPE and QCPE@P-DOL. (b) The calculation of Li+ diffusion coefficient with different CPEs. The corresponding charge and discharge voltage profiles of cells with (c) QCPE and (d) QCPE@P-DOL under different cycles. (e)–(g) Long cycling performances of LFP batteries with different CPEs under different current rates at 30 °C. (h) and (i) Cycling performance of the LFP|QCPE@P-DOL|Li cell at 50 °C and −20 °C. (j) Charge–discharge curves of the first five cycles for LCO|QCPE@P-DOL|Li batteries. | ||
Furthermore, it is found that the LFP|QCPE@P-DOL|Li batteries with high loading of LFP (Fig. 3g) can also demonstrate good reversible capacity and stable cycling performance. With an active material loading of 6.4 mg cm−2, an initial discharge capacity of 131.5 mA h g−1 is obtained with capacity decay of 0.123% per cycle at 2.0 C. Even with a loading as high as 11.7 mg cm−2, an initial discharge capacity of 141.2 mA h g−1 is achieved with capacity decay of 0.158% per cycle at 1.0 C. Similarly, since the applicable temperature range is a crucial factor for the next-generation energy storage devices, the LFP|QCPE@P-DOL|Li battery is tested at both a high temperatures of 50 °C and low temperatures of −20 °C. A discharge capacity of 129.8 mA h g−1 at 50 °C and stable cycling over 150 cycles at 1.0 C is successfully demonstrated in Fig. 3h. Moreover, the battery is capable of being operated at low temperatures as low as −20 °C while maintaining high initial discharge capacity and cycle stability. For example, the battery exhibits a discharge capacity of 119.1 mA h g−1 for 50 cycles at 0.1 C (Fig. 3i). The decrease in performance at low temperatures is attributed to a reduction in ionic conductivity. Overall, the assembled LFP|QCPE@P-DOL|Li battery exhibits excellent cycling performance and electrochemical stability even at high loadings and across a wide temperature range (−20 °C to 50 °C). To demonstrate the compatibility of QCPE@P-DOL with high-voltage cathodes, a LiCoO2 (LCO) cathode was chosen. Cycling performance tests were conducted at 0.2 C and 30 °C in the voltage range of 2.8–4.45 V. The results show that the characteristic plateau of the LCO cathode could be observed in the initial five cycles, exhibiting a high specific capacity of 165.1 mA h g−1 obtained in the first cycle (Fig. 3j and Fig. S11, ESI†). Further investigation of the composition evolution at the Li/QCPE@P-DOL interface is discussed in the following sections.
To gain a depth understanding of the structure evolution at the Li/QCPE@P-DOL interface during cycling, cryo-TEM was used to observe the morphology and composition of the interface at the atomic level.40,41 A schematic diagram of the sample preparation process for cryo-TEM was shown in Fig. 4a. A copper (Cu) grid was placed between the QCPE@P-DOL and the lithium foil, and the copper grid was collected after 20 cycles for cryo-TEM analysis. Fig. 4b illustrates the morphology of the QCPE@P-DOL electrolyte after lithium deposition. The interface layer appears smooth, indicating that the P-DOL layer hinders the continuous reaction between lithium and LAGP. STEM maps in Fig. 4c and Fig. S12 (ESI†) depict the distribution of elements such as C, F, N, O, and S in the interface layer, and the TEM image reveals the amorphous phases and embedded nanocrystals of Li, Li2O, Li2CO3, and LiF in mosaic structure of the interfaces shown in Fig. 4d. Selective area electron diffraction (SAED) in Fig. 4e further confirms the presence of these nanocrystals. The crystal structure of the main component in the interface layer was characterized with high-resolution TEM (HRTEM) images. The calibrated plane spacing of 0.23 nm correspond well to the (111) plane of LiF and electron energy-loss spectroscopy (EELS) elemental mapping further confirms the product of LiF (Fig. 4f and Fig. S13, ESI†).42 Therefore, as a major component of the SEI, LiF acts as an excellent electronic insulator, facilitating uniform Li+ transport and protecting the lithium anode during cycling, which contributes to high reversibility, high-rate capability, and long runtime in SSLMBs. Overall, the cryo-TEM analysis provides valuable insights into the morphology and composition of the Li/QCPE@P-DOL interface, highlighting the presence of various nanocrystals and the beneficial role of LiF in enhancing battery performance. In parallel with the experimental observations, COMSOL simulations were conducted to gain further insights into the Li+ flux at the anode/CPEs interface.43,44Fig. 4g and Fig. S14a, b (ESI†) present the distribution of electric field, current density, and surface concentration at different time intervals. Initially, during the Li+ plating stage, the tip protrusion of both QCPE and QCPE@P-DOL displays the highest current density, indicating these protrusions are the most active reaction region. However, in the final stage, cells using QCPE@P-DOL demonstrates uniform Li+ flux, effectively suppressing high reactivity at the protrusions. On the contrary, cells using QCPE exhibits uneven Li+ transport and high current density due to significant ion concentration polarization and uneven Li+ flux. Based on the cryo-TEM and simulation, a hypothesis can be put forward to explain the inhomogeneous Li+ transport in cells using QCPE. It is suggested that the small amount of LiF in the SEI layer of QCPE-based cells lead to increased lithium nucleation density and facilitate the growth of small lithium nuclei. However, QCPE@P-DOL based cells with rich LiF interface exhibits high surface conduction electrodynamics and lithium-ion affinity, which achieves homogenous current density distribution and uniform dendrite-free lithium deposition. To validate this hypothesis and identify the SEI composition, ex situ XPS and TOF-SIMS analyses were performed in the subsequent section.
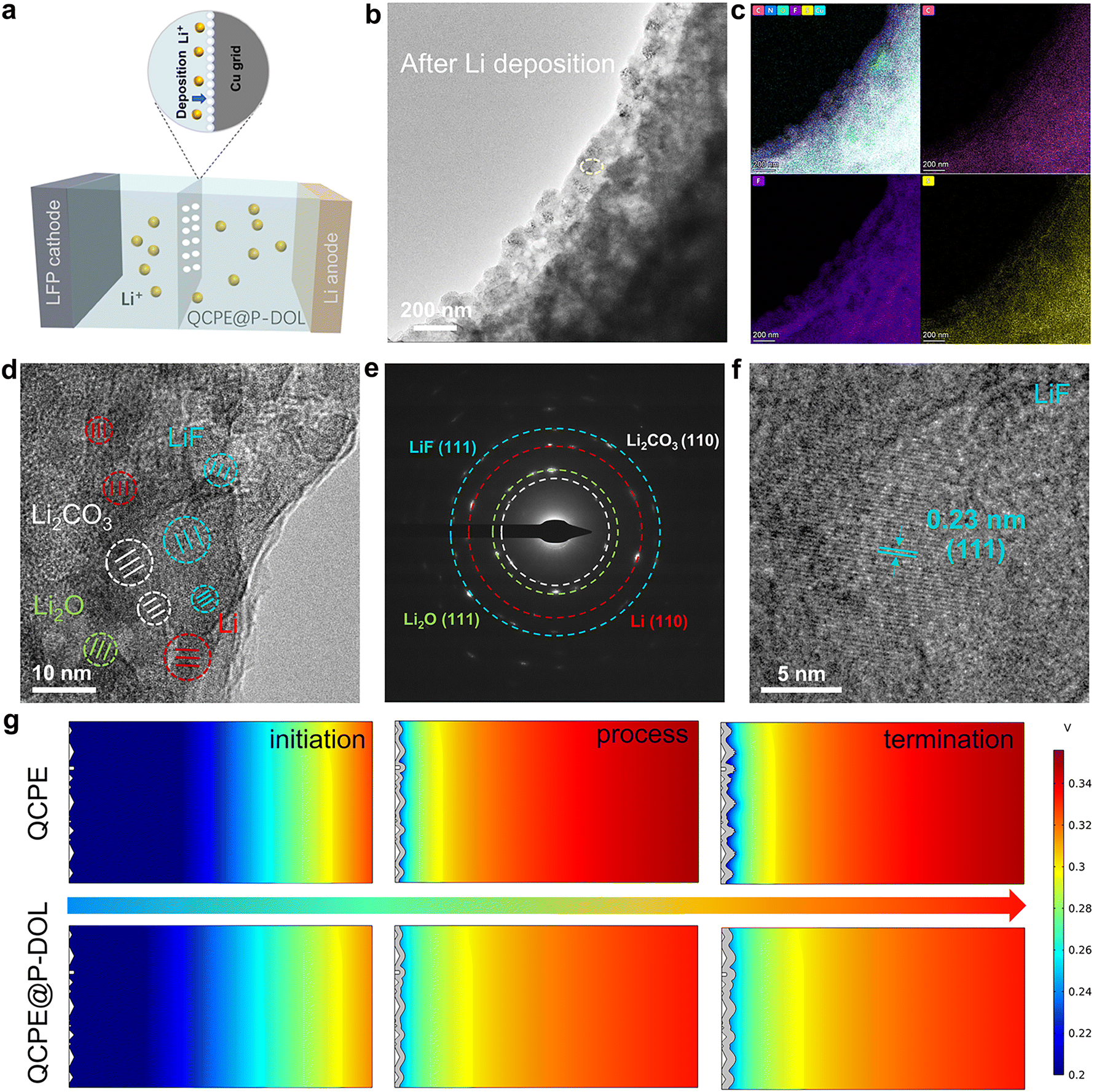 | ||
| Fig. 4 Cryo-TEM characterization of morphology and solid-stated electrolyte interphase investigation of plated Li metal in QCPE electrolyte. (a) Schematic diagram of the sample preparation process for cryo-TEM observation. (b) TEM images illustrating the morphology of LFP|QCPE@P-DOL|Li cell after Li deposition. (c) The corresponding distributions of C, F, and S elements. (d) HRTEM image of the interface where the typical interfacial mosaic morphology and crystalline regions are shown. (e) SAED images. (f) HRTEM images of LiF showing long-range ordering lattice. (g) Electric field simulation distributions at the time of QCPE and QCPE@P-DOL on Li anode. | ||
To assess the interfacial stability and the ability to inhibit the growth of lithium dendrites in the electrolyte film, symmetric lithium batteries were assembled and studied for their lithium plating/stripping behavior at 30 °C. Tafel profiles (Fig. 5a) were tested to calculate the exchange current density of symmetric cells with QCPE and QCPE@P-DOL which are 0.133 and 0.171 mA cm−2, respectively, indicating that QCPE@P-DOL effectively reduces the interfacial charge transfer barrier between the electrolytes and the lithium metal anode. Moreover, rate performance for symmetric cells with current density ranging from 0.1 to 1.0 mA cm−2 (Fig. S15, ESI†) demonstrates that the cell with Li|QCPE@P-DOL|Li exhibits lower polarization voltages compared to that with Li|QCPE|Li. The critical current densities (CCD) test with each cycle lasting for one hour was further carried out in the symmetric cell, showing that the cell with QCPE@P-DOL exhibited a high CCD of up to 2.4 mA cm−2 (Fig. 5b). To further investigate the anode stability, long-term cycling tests were performed on lithium symmetric cells. As depicted in Fig. 5c–e, Li|QCPE@P-DOL|Li batteries exhibited a relatively small overpotential of 250 mV at 0.5 mA cm−2 and could operate for over 1100 hours, whereas Li|QCPE|Li cells only lasted for 60 hours. The short cycle life of Li|QCPE|Li indicates that the continuous and vigorous side reaction between LAGP and Li gradually accumulates, leading to the formation of dead lithium and subsequent short-circuiting of SSLMBs. Additionally, the overpotential of Li|QCPE@P-DOL|Li batteries remain stable for over 2100 hours at 0.2 mA cm−2 with an overpotential of 80 mV and for 300 hours at 1.0 mA cm−2 with an overpotential of 370 mV. Overall, these findings demonstrate that QCPE@P-DOL exhibits enhanced interfacial stability with lithium metal, inhibits lithium dendrite growth, thus enabling long-term cycling with improved performance.
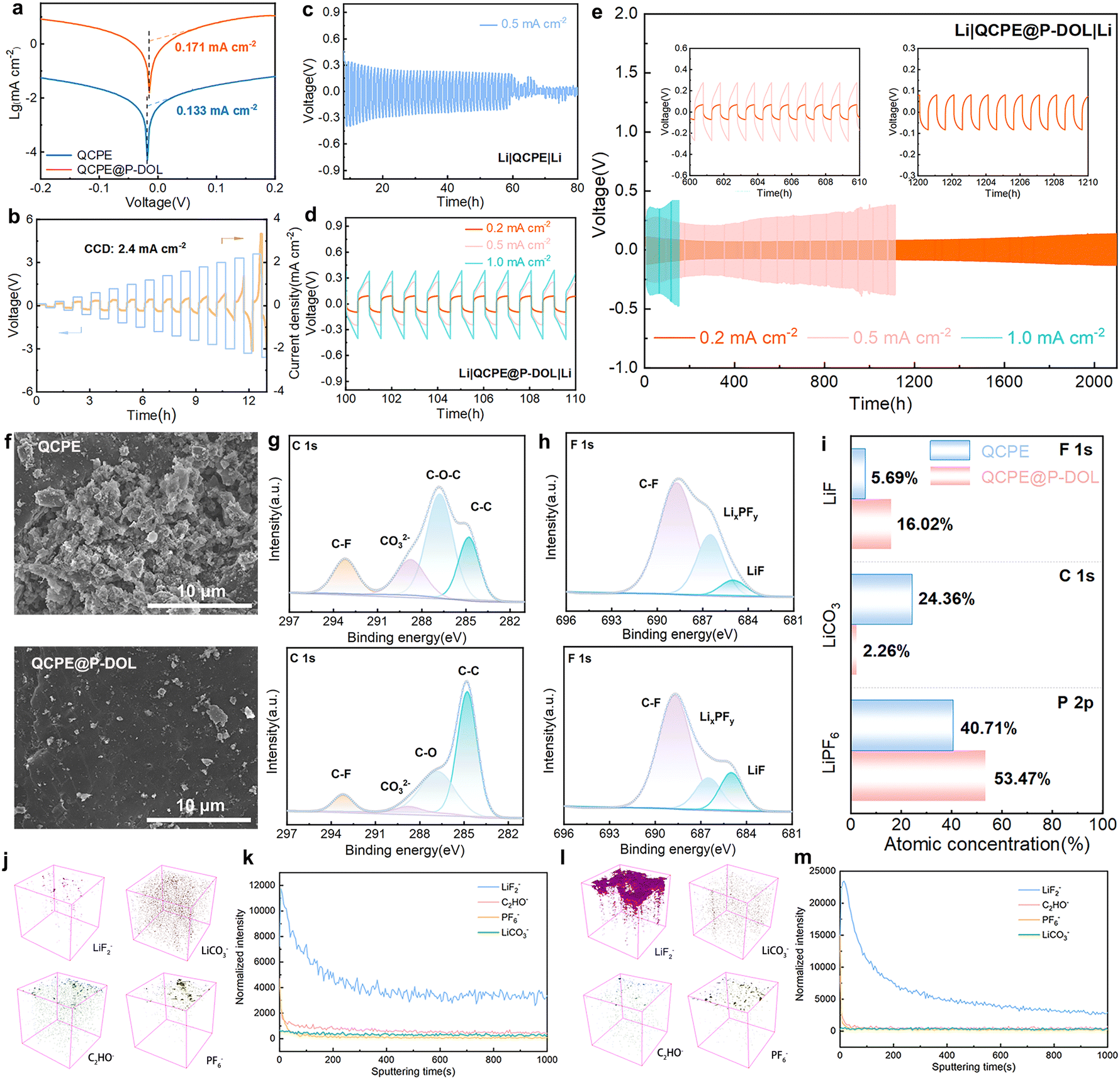 | ||
| Fig. 5 (a) Tafel plots of lithium symmetric cells with QCPE and QCPE@P-DOL. (b) Rate performance tests were performed on Li|QCPE@P-DOL|Li cells to determine their critical current densities. (c) Galvanostatic cycling curves at 0.5 mA cm−2 of Li|QCPE|Li. (d) and (e) Long-term galvanostatic cycling profile of the Li|QCPE@P-DOL|Li cell measured at 0.2, 0.5, and 1.0 mA cm−2. (f) SEM images of lithium metal surface with QCPE and QCPE@P-DOL after 50 h. XPS spectra of cycled Li discs with different CPEs (g) C 1 s, (h) F 1 s (upper is QCPE, bottom is QCPE@P-DOL). (i) The quantified atomic ratios of LiF, Li2CO3, and LiPF6 in F 1s, C 1s and P 2p spectra from CPEs. Corresponding 3D reconstruction (LiF2−, LiCO3−, C2HO− and PF6−) of Li anode with (j) QCPE and (l) QCPE@P-DOL. TOF-SIMS negative ion depth profiles on the cycled Li electrode disassembled from (k) Li|QCPE|Li and (m) Li|QCPE@P-DOL|Li cells. | ||
The morphology characterization of the lithium metal after cycling is carried out using ex situ SEM, as shown in Fig. 5f. It is revealed that the cycled lithium anode in Li|QCPE@P-DOL|Li cells exhibit a uniform and dendrite-free surface. In contrast, cells using QCPE show random and rough lithium dendrite growth on the lithium metal surface. The results further confirm that QCPE@P-DOL leads to improved stability of the anode interface and effective inhibition of lithium dendrite growth. To further investigate the chemical composition of the interfacial layer on the cycled lithium anode, XPS and TOF-SIMS analysis were performed to compare the composition of the SEI in the lithium symmetric cells after 20 cycles at 0.5 mA cm−2. The C 1s XPS spectrum (Fig. 5g) indicates that the surface of SEI layer is composed of a combination of organic and inorganic compounds and the proportion of Li2CO3 is much lower on the lithium metal with QCPE@P-DOL compared to that with QCPE. Typically, Li2CO3 has low Li+ conductivity, leading to the formation of an unstable SEI layer.45 The F 1s XPS spectrum (Fig. 5h) shows peaks corresponding to C–F, LiPxFy, and LiF, and the SEI formed with QCPE@P-DOL exhibits a higher ratio of LiF, which is beneficial for inhibiting lithium dendrite growth in Li|QCPE@P-DOL|Li batteries. The P 2p XPS spectrum (Fig. 5i and Fig. S16, ESI†) reveals a smaller number of products such as LixPOy and LiPFyOz on the lithium metal with QCPE@P-DOL, further confirming the positive role of QCPE@P-DOL in inhibiting the decomposition of LiPF6. The chemical composition of anode interface was further analyzed using the TOF-SIMS. The distribution of LiF2−, C2HO−, LiCO3−, PO2−, PO3−, PO2F2− and PF6− in a 3D view (Fig. 6j, l, and Fig. S17a, b, ESI†) show that a significant amount of LiF on the cycled lithium anode surface of Li|QCPE@P-DOL|Li batteries compared to that with QCPE. The depth profile of chemical component (Fig. 6k, m, and Fig. S18a, b, ESI†) indicated that the surface of the cycled lithium anode with QCPE@P-DOL exhibited a high content of PF6−, while other elements and groups (PO2−, PO3−, PO2F2−) have relatively low content. These results suggest that QCPE@P-DOL has a favorable inhibitory effect on LiPF6 decomposition, consistent with the XPS results.46 Furthermore, ex situ XPS and TOF-SIMS analysis verified that the surface of the interface is rich in LiF and scarce in Li2CO3, consistent with the cryo-TEM observations. Notably, the QCPE modified by P-DOL exhibits a LiF-enriched interface (Fig. S19, ESI†), which helps prevent side reactions between Li and LAGP while promoting ion diffusion in the interface layer. In summary, the presence of a LiF-rich interfacial layer contributes to the formation of a dense and stable electrolyte/Li anode interface, ensuring the long-term stability of SSLMBs.
 | ||
| Fig. 6 (a) and (b) Schematic illustration of Li+ transport and LLZTO/Li interfacial evolution in SSLMBs with QCPE1 and QCPE1@P-DOL. (c) C-rate performances at various rates from 0.2 to 10.0 C at 30 °C. (d)–(f) Long cycling performances of LiFePO4 batteries with different CPEs under different current rates. (g) Charge–discharge curves of the first five cycles for solid-state LiCoO2 batteries with QCPE1@P-DOL. | ||
The concept of constructing in situ P-DOL protective layers can be applied to other inorganic oxides, such as LLZTO, using a similar preparation process. The resulting integrated electrolytes were named QCPE1@P-DOL and QCPE1, respectively. Although no side reactions were observed between LLZTO and Li, the poor physical contact at the solid–solid interface led to high interfacial impedance and limited ionic diffusion. Therefore, modifying the LLZTO interface is crucial to enhance interfacial compatibility and ionic conductivity in solid-state batteries (Fig. 6a and b). LFP|QCPE1@P-DOL|Li batteries exhibit high discharge capacities of approximately 166.0, 158.5, 149.4, 137.9, 116.9, and 101.2 mA h g−1 at rates of 0.2, 0.5, 1.0, 2.0, 5.0, and 10.0 C, respectively (Fig. 6c). In comparison, LFP|QCPE1|Li cells show specific capacities of approximately 152.1, 145.5, 131.4, 124.9, 104.9, and 51.6 mA h g−1 at the same rates. Stable charge/discharge profiles and low internal polarization were observed in cells with QCPE1@P-DOL for over 1200 cycles at 5 C and 30 °C (Fig. 6d). However, the LFP|QCPE1|Li cells can only operate normally for 300 cycles due to dendrite formation on the lithium anode that leads to soft short circuit as seen from the fluctuation in coulombic efficiency (CE). Therefore, it is concluded that QCPE1@P-DOL electrolyte with a P-DOL nanolayer enables higher discharge capacity and improves operating life. The SSLMBs utilizing QCPE1@P-DOL electrolyte achieve a high reversible capacity with a capacity decay of only 0.021% per cycle under 10.0 C after 1400 cycles, as shown in Fig. 6f. Furthermore, an LFP|QCPE1@P-DOL|Li cell with a high LFP loading (5.2 mg cm−2) demonstrate a good initial discharge capacity of 133.7 mA h g−1 at 2.0 C and stable cycling performance, with a capacity retention of 81.6% after 250 cycles (Fig. 6e). To evaluate the compatibility with high-voltage cathodes, batteries with a LCO cathode operating from 2.8 to 4.45 V were assembled. The LCO|QCPE1@P-DOL|Li cell exhibits a high initial discharge capacity of 168.5 mA h g−1 at 0.2 C and 30 °C (Fig. 6g). These results demonstrate that the strategy of designing a unique multilayer electrolyte to construct a P-DOL protective layer can be widely applied to interface engineering in various types of inorganic oxides.
3. Conclusion
In summary, the interface reactions and structural changes of electrolytes/lithium in SSLMBs at the nanoscale is visualized with in situ TEM, revealing minimal morphological alterations of LAGP@P-DOL particles during charge and discharge processes. Cryo-TEM and TOF-SIMS confirm the formation of a LiF-rich layer at the LAGP@P-DOL/Li interface, promoting uniform distribution of Li+ ions and facilitating dense and uniform lithium deposition. QCPE@P-DOL-based lithium metal batteries exhibit exceptional cycling stability in a temperature range from −20 to 50 °C, which demonstrates an impressive cycle life of over 1450 cycles at 30 °C with a discharge rate of 10.0 C. This highlights the effectiveness of the in situ polymerization-derived P-DOL buffer layer in mitigating chemical and physical degradation at the LAGP/Li interface. These findings offer a theoretical basis for understanding the evolution of interface structure and components between lithium and inorganic oxides in SSLMBs, providing valuable insights for designing interface layers for improving the performance of SSLMB systems and various alkali metal batteries.Author contributions
T. S. Zhao supervised this work. J. Li and J. Sun conducted the concept design. J. Li provided material characterizations, conducted the experiments and simulations. J. Li, J. Chen, B. Huang and X. Xu did the data analysis. J. Li wrote the paper. T. S. Zhao, J. Chen, B. Huang, X. Xu and J. Sun revised the manuscript. All authors commented on the final manuscript.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was fully supported by grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. R6005-20) and Guangdong Major Project of Basic and Applied Basic Research(2023B0303000002). We would like to acknowledge Prof. Khalil Amine for providing valuable suggestions and advice on the characterization of the solid–solid interface.References
- Y. J. Liu, X. Y. Tao, Y. Wang, C. Jiang, C. Ma, O. W. Sheng, G. X. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- Y. P. Guo, H. Q. Li and T. Y. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- C. C. Fang, J. X. Li, M. H. Zhang, Y. H. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Y. C. Yang, B. Y. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. F. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- C. B. Jin, T. F. Liu, O. W. Sheng, M. Li, T. C. Liu, Y. F. Yuan, J. W. Nai, Z. J. Ju, W. K. Zhang, Y. J. Liu, Y. Wang, Z. Lin, J. Lu and X. Y. Tao, Nat. Energy, 2021, 6, 378–387 CrossRef CAS.
- A. Manthiram, X. W. Yu and S. F. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- D. H. S. Tan, Y. T. Chen, H. D. Yang, W. Bao, B. Sreenarayanan, J. M. Doux, W. K. Li, B. Y. Lu, S. Y. Ham, B. Sayahpour, J. Scharf, E. A. Wu, G. Deysher, H. E. Han, H. J. Hah, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Science, 2021, 373, 1494–1499 CrossRef CAS PubMed.
- J. H. Wu, S. F. Liu, F. D. Han, X. Y. Yao and C. S. Wang, Adv. Mater., 2021, 33, 2000751 Search PubMed.
- D. W. Zeng, J. M. Yao, L. Zhang, R. N. Xu, S. J. Wang, X. L. Yan, C. Yu and L. Wang, Nat. Commun., 2022, 13, 1909 Search PubMed.
- P. F. Jiang, J. Q. Cao, B. Wei, G. Y. Qian, S. F. Wang, Y. S. Shi, G. Y. Du, X. Y. Lu, C. Y. Ouyang, F. H. Cao and X. Lu, Energy Storage Mater., 2022, 48, 145–154 CrossRef.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. P. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- J. Li, H. T. Zhang, Y. Y. Cui, H. R. Da, Y. J. Cai and S. J. Zhang, Chem. Eng. J., 2022, 450, 138457 CrossRef CAS.
- J. W. Meng, Y. Zhang, X. J. Zhou, M. Lei and C. L. Li, Nat. Commun., 2020, 11, 3716 Search PubMed.
- Q. Ye, H. Y. Liang, S. H. Wang, C. Cui, C. Zeng, T. Y. Zhai and H. Q. Li, J. Energy Chem., 2022, 70, 356–362 Search PubMed.
- R. Zhao, L. Gao, M. R. Song, Y. Ye, Z. B. Liang, J. C. Bian, J. L. Zhu, S. Li, R. Q. Zou and Y. S. Zhao, ACS Energy Lett., 2021, 6, 3141–3150 Search PubMed.
- H. Chung and B. Kang, Chem. Mater., 2017, 29, 8611–8619 CrossRef CAS.
- J. A. Lewis, F. J. Q. Cortes, M. G. Boebinger, J. Tippens, T. S. Marchese, N. Kondekar, X. M. Liu, M. F. Chi and M. T. McDowell, ACS Energy Lett., 2019, 4, 591–599 CrossRef CAS.
- Q. Liu, Q. P. Yu, S. Li, S. W. Wang, L. H. Zhang, B. Y. Cai, D. Zhou and B. H. Li, Energy Storage Mater., 2020, 25, 613–620 CrossRef.
- S. Y. Sun, J. A. Wang, S. R. Zong, Q. Y. Ma, H. X. Li, X. Chen, X. M. Cui, K. Yang, Q. Cai, Y. L. Zhao and W. Yan, Adv. Funct. Mater., 2023, 33, 2304929 CrossRef CAS.
- X. L. Fan, X. Ji, F. D. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. J. Jiang and C. S. Wang, Sci. Adv., 2018, 4, eaau9245 Search PubMed.
- S. Lee, K. S. Lee, S. Kim, K. Yoon, S. Han, M. H. Lee, Y. Ko, J. H. Noh, W. Kim and K. Kang, Sci. Adv., 2022, 8, eabq0153 Search PubMed.
- Q. Liu, J. H. Yu, W. Q. Guo, Y. F. Pan, C. P. Han, H. B. Liu and B. H. Li, Ecomat, 2023, 5, e12322 CrossRef CAS.
- J. Y. Liang, X. X. Zeng, X. D. Zhang, T. T. Zuo, M. Yan, Y. X. Yin, J. L. Shi, X. W. Wu, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 9165–9169 Search PubMed.
- Q. Liu, D. Zhou, D. Shanmukaraj, P. Li, F. Y. Kang, B. H. Li, M. Armand and G. X. Wang, ACS Energy Lett., 2020, 5, 1456–1464 CrossRef CAS.
- T. R. Wang, J. Duan, B. Zhang, W. Luo, X. Ji, H. H. Xu, Y. Huang, L. Q. Huang, Z. Y. Song, J. Y. Wen, C. S. Wang, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2022, 15, 1325–1333 RSC.
- W. Y. Wu, Z. Y. Song, Y. M. Dai, X. Y. Zheng, G. Y. Chai, J. B. Yang and W. Luo, Adv. Energy Mater., 2022, 12, 2200999 Search PubMed.
- J. H. Yu, Q. Liu, X. Hu, S. W. Wang, J. R. Wu, B. Liang, C. P. Han, F. Y. Kang and B. H. Li, Energy Storage Mater., 2022, 46, 68–75 CrossRef.
- X. G. Hao, Q. Zhao, S. M. Su, S. Q. Zhang, J. B. Ma, L. Shen, Q. P. Yu, L. Zhao, Y. Liu, F. Y. Kang and Y. B. He, Adv. Energy Mater., 2019, 9, 1901604 Search PubMed.
- Y. J. Liu, C. Li, B. J. Li, H. C. Song, Z. Cheng, M. R. Chen, P. He and H. S. Zhou, Adv. Energy Mater., 2018, 8, 1702374 Search PubMed.
- K. X. Mu, D. Wang, W. L. Dong, Q. Liu, Z. N. Song, W. J. Xu, P. P. Yao, Y. A. Chen, B. Yang, C. H. Li, L. Tian, C. Z. Zhu and J. Xu, Adv. Mater., 2023, 35, 2304686 CrossRef CAS PubMed.
- J. Li, Y. J. Cai, Y. Y. Cui, H. Wu, H. R. Da, Y. J. Yang, H. T. Zhang and S. J. Zhang, Nano Energy, 2022, 95, 107027 Search PubMed.
- J. Q. Li, Y. Y. Yi, X. T. Zuo, B. B. Hu, Z. H. Xiao, R. Q. Lian, Y. Kong, L. M. Tong, R. W. Shao, J. Y. Sun and J. Zhang, ACS Nano, 2022, 16, 3163–3172 Search PubMed.
- Y. Chen, F. Huo, S. M. Chen, W. B. Cai and S. J. Zhang, Adv. Funct. Mater., 2021, 31, 2102347 Search PubMed.
- Q. Zhao, X. T. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 Search PubMed.
- J. Yu, X. D. Lin, J. P. Liu, J. T. T. Yu, M. J. Robson, G. D. Zhou, H. M. Law, H. R. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2022, 12, 2102932 Search PubMed.
- J. W. Xiang, Y. Zhang, B. Zhang, L. X. Yuan, X. T. Liu, Z. X. Cheng, Y. Yang, X. X. Zhang, Z. Li, Y. Shen, J. J. Jiang and Y. H. Huang, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- N. Y. Zhang, G. X. Wang, M. Feng and L. Z. Fan, J. Mater. Chem. A, 2021, 9, 4018–4025 Search PubMed.
- Z. D. Hao, C. J. Wang, Y. Wu, Q. Q. Zhang, H. Xu, Y. H. Jin, J. B. Liu, H. Wang and X. M. He, Adv. Energy Mater., 2023, 13, 2204007 Search PubMed.
- J. Y. Shih, G. Y. Lin, Y. J. J. Li, T. F. Hung, R. Jose, C. Karuppiah and C. C. Yang, Electrochim. Acta, 2022, 419, 140356 Search PubMed.
- J. Li, Y. J. Cai, F. J. Zhang, Y. Y. Cui, W. H. Fang, H. Da, H. T. Zhang and S. J. Zhang, Nano Energy, 2023, 118, 108985 Search PubMed.
- Y. Z. Li, Y. B. Li, A. L. Pei, K. Yan, Y. M. Sun, C. L. Wu, L. M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 CrossRef CAS PubMed.
- O. W. Sheng, J. H. Zheng, Z. J. Ju, C. B. Jin, Y. Wang, M. Chen, J. W. Nai, T. F. Liu, W. K. Zhang, Y. J. Liu and X. Y. Tao, Adv. Mater., 2020, 32, 2000223 CrossRef CAS PubMed.
- J. J. Li, H. M. Hu, W. H. Fang, J. W. Ding, D. Yuan, S. J. Luo, H. T. Zhang and X. Y. Ji, Adv. Funct. Mater., 2023, 33, 2303718 Search PubMed.
- J. P. Liu, B. T. Yuan, N. A. D. He, L. W. Dong, D. J. Chen, S. J. Zhong, Y. P. Ji, J. C. Han, C. H. Yang, Y. P. Liu and W. D. He, Energy Environ. Sci., 2023, 16, 1024–1034 Search PubMed.
- F. Li, J. He, J. Liu, M. Wu, Y. Hou, H. Wang, S. Qi, Q. Liu, J. Hu and J. Ma, Angew. Chem., Int. Ed., 2021, 60, 6600–6608 Search PubMed.
- Q. Ma, J. P. Yue, M. Fan, S. J. Tan, J. Zhang, W. P. Wang, Y. Liu, Y. F. Tian, Q. Xu, Y. X. Yin, Y. You, A. Luo, S. Xin, X. W. Wu and Y. G. Guo, Angew. Chem., Int. Ed., 2021, 60, 16554–16560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02075h |
| This journal is © The Royal Society of Chemistry 2024 |