
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the high-temperature degradation mechanism of solid oxide electrolysis cells through direct imaging of nanoscale interfacial phenomena†
Haneul
Choi
ab,
Jisu
Shin
a,
Changho
Yeon
 cd,
Sun-Young
Park
e,
Shin-Tae
Bae
f,
Ji Wan
Kim
f,
Jong-Ho
Lee
ag,
Jin-Woo
Park
b,
Chan-Woo
Lee
c,
Kyung Joong
Yoon
*a and
Hye Jung
Chang
*ag
cd,
Sun-Young
Park
e,
Shin-Tae
Bae
f,
Ji Wan
Kim
f,
Jong-Ho
Lee
ag,
Jin-Woo
Park
b,
Chan-Woo
Lee
c,
Kyung Joong
Yoon
*a and
Hye Jung
Chang
*ag
aCenter for Energy Materials Research, Korea Institute of Science and Technology, Seoul, 02792, Republic of Korea. E-mail: kjyoon@kist.re.kr; almacore@kist.re.kr
bDepartment of Materials Science and Engineering, Yonsei University, Seoul, 03722, Republic of Korea
cEnergy AI & Computational Science Laboratory, Korea Institute of Energy Research, Daejeon 34129, Republic of Korea
dDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea
eTechnological Convergence Center, Korea Institute of Science and Technology, Seoul, Republic of Korea
fGreen Energy Materials Research Team, Research & Development Division, Hyundai Motor Company, Uiwang-si, 16082, Republic of Korea
gDivision of Nano Convergence, KIST School, University of Science and Technology, Seoul 02792, Republic of Korea
First published on 22nd May 2024
Abstract
Solid oxide electrolysis cell (SOEC) technology potentially offers the most efficient means of clean H2 production. Currently, the most critical issue is the delamination of the air electrode, but its fundamental cause has long been elusive. Using cutting-edge transmission electron microscopy techniques and density functional theory calculations, we reveal nanometer-scale interfacial degradation phenomena occurring in the early stages, clarifying the entire process of delamination and the origin thereof. During SOEC operation, oxygen ions accumulate at specific locations where they cannot be released as a gas. The annihilation of oxygen vacancies modifies the unit cell structure, causing anisotropic lattice strain; further injection of excess oxygen ions creates dislocations and segmented subgrains. Subsequently, these ions initiate the formation of nanopores, which eventually develop into cracks and delaminate the electrode. These previously undiscovered structural alterations contradict the long-held but unsubstantiated notion of gas pressure build-up, providing novel guidance for future development.
Broader contextAlthough carbon-free hydrogen production by water electrolysis represents an ideal approach to achieve net-zero emissions, the associated cost is substantially higher than that of conventional fossil fuel-based processes. Among various electrolyzer types, the solid oxide electrolysis cell (SOEC), which operates at high temperatures typically in excess of 700 °C, offers the strongest potential for cost reduction owing to its unrivaled efficiency. Currently, the major challenge hindering its commercialization is rapid performance degradation, particularly owing to the delamination of the air electrode. Although significant research efforts have been devoted to understanding the fundamental cause of this degradation, they have been unsuccessful owing to the difficulty of characterizing nano-scale interfacial phenomena. In this study, we adopted novel analytical techniques including precession electron diffraction in transmission electron microscopy to elucidate the origin and evolution of interfacial degradation. Specifically, we visualized the entire delamination process involving the local accumulation of oxygen ions, change in anisotropic lattice strain, generation of dislocation and subgrain boundaries, and formation of aligned nanopores. The resulting comprehensive understanding of SOEC degradation phenomena establishes a basis for rational development strategies, and the associated detailed knowledge of atomic/nanoscale interfacial phenomena represents a valuable asset for the entire energy community. |
Introduction
Hydrogen is expected to play a critical role in decarbonizing the global energy system and realizing net-zero greenhouse gas emissions. At present, hydrogen is mostly produced by fossil-fuel-based processes, which are economical but impose heavy environmental burdens. Water electrolysis powered by renewable energy sources represents an ideal alternative for producing clean hydrogen, but its practical use has been hindered by high costs. Among various electrolysis technologies, solid oxide electrolysis cells (SOECs), which operate at high temperatures, typically over 700 °C, offer the highest energy conversion efficiency and have exhibited strong potential to compete economically with conventional fossil-fuel-based technologies.1–3 Particularly, the electrical energy demand for electrolysis, which accounts for the largest share of the overall hydrogen production cost, decreases with increasing temperature. Overpotentials are also reduced at high temperatures owing to fast reaction and transport kinetics, which enables hydrogen production with minimal energy consumption. Owing to such unique advantages associated with high operating temperature, producing hydrogen using an SOEC system is predicted to cost as little as $1–2 per kg H2,4 which would achieve cost parity with natural gas reforming.5–7 Furthermore, SOECs can efficiently electrolyze both H2O and CO2, enabling the production of various fuels and chemicals besides hydrogen. Consequently, SOECs have emerged as the most promising solution for clean fuel and chemical production and have recently experienced a dramatic expansion in R&D activities.However, SOECs have not been extensively deployed or commercialized thus far, primarily owing to degradation issues. Various high-temperature degradation phenomena have been reported in cells and stacks, including the Cr poisoning of air electrode,8 the chemical reaction between air electrode and electrolyte,9–11 the Ni depletion of fuel electrode,12 and the oxide scale growth of metallic interconnect.13 Among them, the delamination of the air electrode is considered the most fatal as it can cause the entire SOEC system to catastrophically fail.1,14–16 The delamination of air electrode was first reported in 1990's,17 and since then, significant research has attempted to resolve this issue. Nevertheless, the mechanisms underlying delamination have not been clarified to date.18
During SOEC operation, oxygen ions (O2−) migrate from the fuel electrode to the air electrode across the electrolyte and are released as oxygen gas (O2). Because the most intuitive feature of the SOEC air electrode is the evolution of O2 gas, many previous studies have explained that delamination could be caused by the build-up of gas pressure when the gas transport through the porous electrode is not sufficiently facile.16,19,20 The presence of an interfacial insulating phase, which is formed by undesired chemical reaction between air electrode and electrolyte, was also suggested as the main cause of delamination.21 An externally applied electric field that causes cation migration from air electrode to electrolyte was reported as another possible cause of interfacial degradation.22 Interfacial adhesion can also be weakened by the disintegrate of air electrode materials into nanoparticles at the interfacial region.23 Most of these mechanisms, however, were hypothesized without solid evidence because of the limitations of conventional analytical techniques. Consequently, valid solutions have not been established, and delamination remains a critical obstacle to the commercialization of SOEC technology.
Herein, we report previously undiscovered physical and chemical changes at the electrode–electrolyte interface at the beginning of delamination. Cutting-edge transmission electron microscopy (TEM) techniques provide details on the crystal structure, chemical composition, defect structure, and strain distribution in the interfacial region, and these experimental observations are theoretically interpreted by density functional theory (DFT) calculations. The resulting comprehensive understanding will inform future research on optimizing the interfacial properties.
Methods
To simulate electrode–electrolyte interfacial degradation in a well-controlled manner, we performed model studies using symmetric cells, which can be tested in a single chamber without requiring complicated sealing, as schematically illustrated in Fig. S1(a) in the ESI.† The symmetric cell is composed of identical air electrodes deposited on both sides of the electrolyte substrate; we can induce fuel cell and electrolysis reactions on each electrode by applying an electric current in a certain direction. Originally, SOECs were derived from solid oxide fuel cells (SOFCs), which convert the chemical energy of H2 into electric power. Notably, SOEC and SOFC reactions are reverse in direction. When an electric current is applied to the symmetric cell, one electrode is cathodically polarized, leading to the reduction of oxygen (eqn (1)) and incorporation of O2− into the electrolyte; this reaction corresponds to fuel cell mode operation.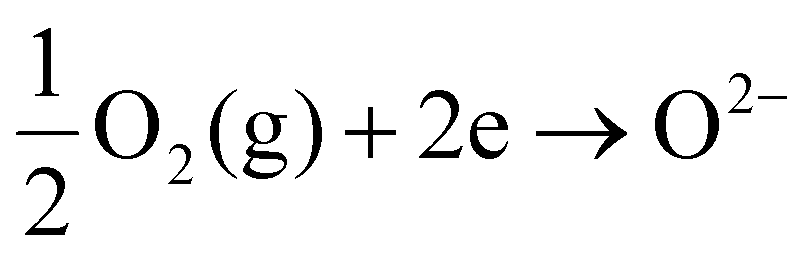 | (1) |
On the opposite side, the air electrode is anodically polarized and electrons are released from the O2−, causing O2 to evolve (eqn (2)); this reaction represents electrolysis mode operation.
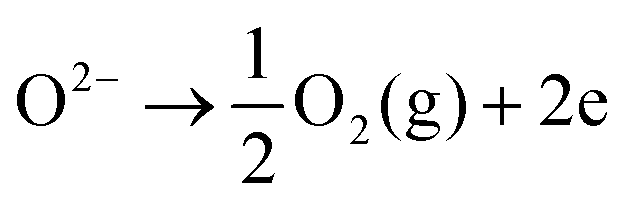 | (2) |
We focused on the physical and chemical changes occurring at the electrolysis side of symmetric cells. As an air electrode for our model study, we selected lanthanum strontium manganite (LSM; (La0.8Sr0.2)0.95MnO3), which is a standard material with well-known properties. The porous electrode was uniformly deposited on a dense substrate, and its thickness and porosity were estimated to be ∼12 μm and ∼30%, respectively (Fig. S1(b) in the ESI†).
Cell fabrication
To fabricate symmetric cells, yttria-stabilized zirconia (YSZ) discs were prepared by die pressing 8 mol% YSZ powder (TZ-8Y, Tosoh Corp.) and sintering at 1500 °C for 5 h. The diameter and thickness of the discs were ∼8 and ∼3 mm, respectively. Next, LSM ink was prepared by mixing commercial LSM powder ((La0.8Sr0.2)0.95MnO3, Fuelcellmaterials) with solvent, binder, and dispersant in the desired ratio. The LSM air electrodes were screen-printed on both sides of the YSZ disc and sintered at 1150 °C for 3 h.Electrochemical testing
The symmetric cells were electrochemically tested in an alumina tube reactor using a Solartron 1260/1287 potentiostat and frequency response analyzer. The two LSM electrodes served as working and counter electrodes, and two pieces of Pt mesh were used as current collectors. The assembly was equipped with a reference electrode in a ring-type geometry by wrapping the Pt wire around the outer rim of the electrolyte disc midway between the working and counter electrodes. Dry air was supplied, and a constant current was applied between the working and counter electrodes at 700 °C. The voltage responses of the working and counter electrodes were individually monitored using the reference electrode. Impedance spectra were collected before and after constant current operation with an AC perturbation of 20 mV in the 0.1–105 Hz frequency range.Structural and chemical analysis
The microstructure was analyzed using scanning electron microscopy (SEM; Regulus 8230, Hitachi and Inspect F50, ThermoFisher). The chemical compositions were quantitatively characterized using an electron probe micro-analyzer (EPMA; JXA-8500F, JEOL) under an electron beam at an accelerating voltage of 20 kV and a current of 20 nA. To minimize the electron interaction volume during EPMA analysis, we prepared a specimen with a thickness of ∼10 μm using a focused ion beam (FIB; Ethos NX5000, Hitachi) (Fig. S2(a) in the ESI†). High-resolution analysis was performed using a TEM (Talos F200X, ThermoFisher) equipped with an extreme brightness field emission gun and energy-dispersive X-ray spectrometer (EDS; Super-X, Bruker). The maps of crystal orientation and strain were obtained using the TEM precession electron diffraction (PED) method. Both scanning and precession were enabled through a DigiSTAR system (NanoMEGAS) installed in a TEM (Tecnai F20 G2, ThermoFisher) operated at 200 keV, and data were acquired and processed using the ASTAR and Topspin software. The precession angle of the electron beam was 1.4°, and the scanning step size was set to 5 nm with 10 precessions per frame. The diffraction pattern at each step was recorded by an external high-speed camera and indexed automatically to the calculated diffraction pattern for a cubic YSZ structure of CaF2 type, with space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (a = 5.142 Å).24,25 PED is a specialized technique for collecting electron diffraction patterns in a TEM by rotating a tilted incident electron beam around the microscope's central axis. As it has minimal dynamic scattering, PED is widely used to determine crystal structures26 by precisely mapping grain orientation and phase as well as measuring strain.27–29 The analysis processes are briefly illustrated in Fig. S3 in the ESI.† A thin lamella for cross-sectional TEM analysis was prepared using the same FIB equipped with an Ar ion gun for fine milling at the final step. Electron diffraction patterns were simulated using the free, open-source, multipurpose crystallographic software ReciPro30 and obtained using kinematic scattering approximation with the specimen thickness set to 50 nm.
m (a = 5.142 Å).24,25 PED is a specialized technique for collecting electron diffraction patterns in a TEM by rotating a tilted incident electron beam around the microscope's central axis. As it has minimal dynamic scattering, PED is widely used to determine crystal structures26 by precisely mapping grain orientation and phase as well as measuring strain.27–29 The analysis processes are briefly illustrated in Fig. S3 in the ESI.† A thin lamella for cross-sectional TEM analysis was prepared using the same FIB equipped with an Ar ion gun for fine milling at the final step. Electron diffraction patterns were simulated using the free, open-source, multipurpose crystallographic software ReciPro30 and obtained using kinematic scattering approximation with the specimen thickness set to 50 nm.
Computational details
The DFT calculations were performed using the Vienna Ab initio Simulation Package31,32 within the projector augmented wave scheme considering the spin polarization. The valence configurations of each atom were treated as 4s24p64d3 (Y), 4s24p64d4 (Zr), and 2s22p4 (O). The Perdew–Burke–Ernzerhof generalized gradient approximation33 was used for the exchange–correlation functionals. The plane-wave cutoff energy was set to 550 eV, and the Γ-only k-point mesh34 of the Brillouin zone was applied for all bulk YSZ models (Y-doped 32ZrO2). The adopted convergence criteria of the electronic and ionic energies were 10−6 eV and 0.01 eV Å−1, respectively. The stoichiometric YSZ model was constructed according to the following rules:35–38 (i) the Y–O–Y pair is stably located in the YSZ lattice, (ii) the Y atom is located in the next-nearest–neighbor sites to the oxygen vacancy (VO), and (iii) VO and interstitial oxygen are uniformly distributed in the supercell to maximize the distance between VO. In addition, non-stoichiometric models were constructed by adding oxygen atoms to the lattice oxygen sites and interstitial sites in the stoichiometric YSZ model. To elucidate the trends in the lattice strain according to the oxygen composition, three YSZ models—Y4Zr28O62, Y4Zr28O64, and Y4Zr28O80—were respectively selected to implement the following cases: 8 mol% YSZ, YSZ with no VO, and YSZ with excess oxygen, even at interstitial sites. The lattice distances and unit cell volumes were extracted from X-ray diffraction (XRD) simulations (see Supplementary Notes 1 and 2 in the ESI† for more details).Results and discussion
Electrochemical degradation upon SOEC operation
For the electrochemical tests, we applied a constant electric current of 1.5 A cm−2 at 700 °C and measured the cell voltage over time (Fig. 1(a)). The voltage gradually increased for the first ∼100 h and then irregularly surged in a stepwise manner as operation continued. We stopped passing current before complete failure at ∼220 h. The impedance of the cell was measured before and after testing (Fig. 1(b)). In the Nyquist plot of impedance spectra, the high-frequency intercept represents the Ohmic resistance, and the width of the arc corresponds to the electrode polarization resistance. After operation, the Ohmic and polarization resistances increased from 21.8 to 32.9 Ohm cm2 and from 2.1 to 10.9 Ohm cm2, respectively. These simultaneous increases indicate that both the current passage and electrode reaction were significantly interrupted during SOEC operation. Fig. 1(c) shows a cross-sectional secondary electron (SE) image of the air electrode recorded after testing using SEM. Although the electrochemical performance substantially degraded, no noticeable changes appeared in the electrode at this scale. Through TEM analysis (Fig. 1(d)–(f)), however, we detected small-scale local changes occurring in the initial stages of delamination. In the high-angle annular dark field–scanning transmission electron microscopy (HAADF–STEM) image (Fig. 1(d)), the porous LSM air electrode is shown deposited on the dense YSZ electrolyte, and the LSM particle size is estimated to be 0.5–1 μm. The elemental map (Fig. S4 in the ESI†) recorded using an EDS confirms that the constitutional elements of LSM and YSZ were homogeneously distributed without significant chemical interactions or interdiffusion at the electrode–electrolyte interface. Although no macroscale delamination appears in Fig. 1(d), the higher magnification images (Fig. 1(e) and (f)) reveal nanometer-scale defects at two different locations. In Fig. 1(e), multiple nanopores with sizes of tens of nanometers are closely aligned along the two-phase boundary between LSM and YSZ (blue square, Fig. 1(d)). In Fig. 1(f), similar nanopores are observed along the grain boundaries within the YSZ electrolyte near the interface (red square, Fig. 1(d)), implying that the aligned nanopores at the boundary between grains I and II formed the trans-granular microcracks in grain III. Cracks have been commonly observed at these two locations in delaminated cells,16,19–21,23,39–43 but their fundamental cause has not been understood. The predominant hypothesis thus far has been gas pressure build-up resulting from O2 generation, which is the most intuitive explanation considering SOEC operating principles, but such pressure-driven fracture would occur instantaneously. In contrast, our TEM analysis in Fig. 1(d) and (f) indicates that delamination is a gradual process associated with the formation of multiple nanopores.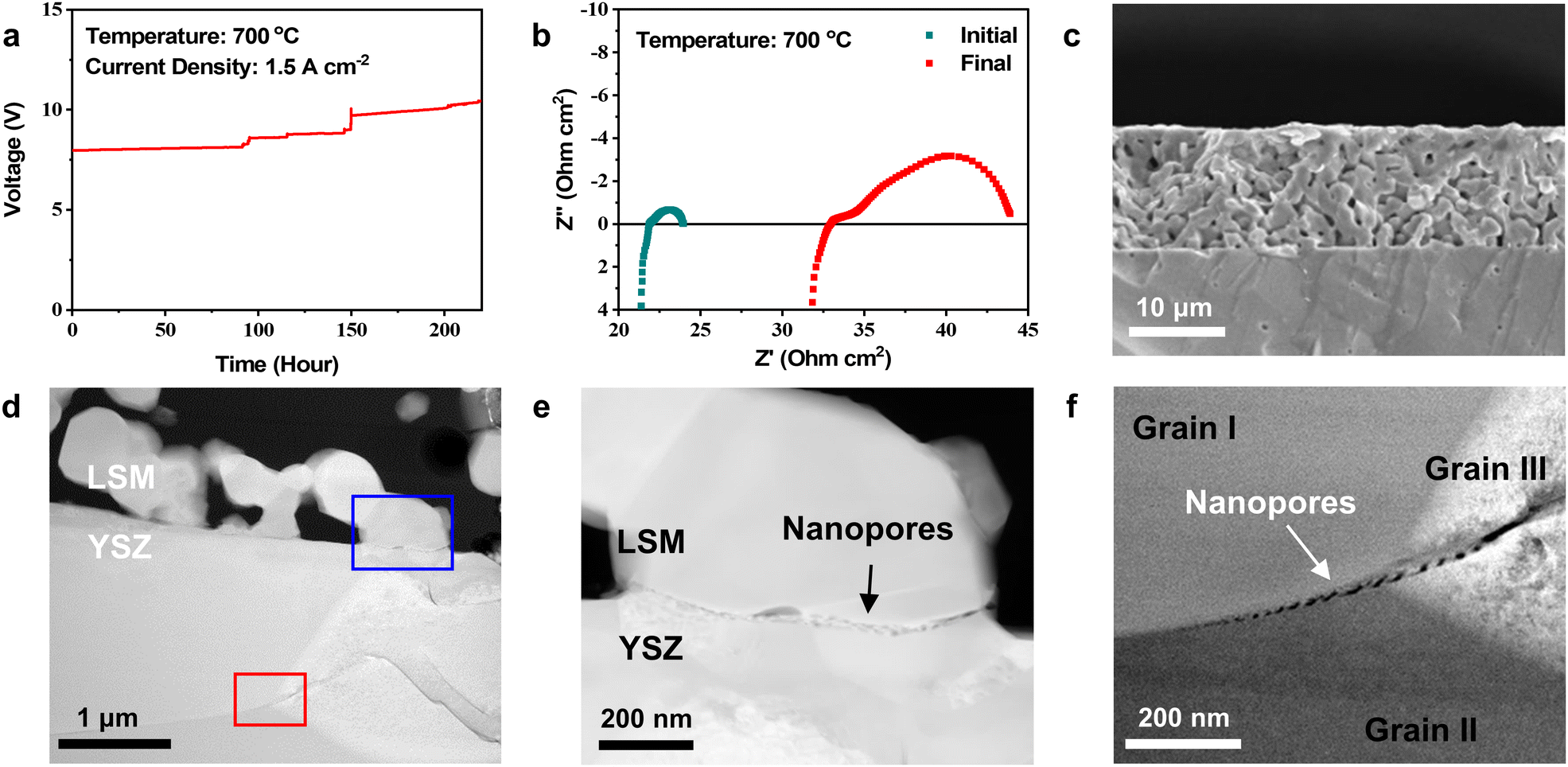 | ||
| Fig. 1 Electrochemical tests of symmetric cells. (a) Cell voltage as a function of time measured at a constant current density of 1.5 A cm−2 at 700 °C. (b) Nyquist plot of the symmetric cell impedance spectra measured before and after long-term testing. (c) SEM image of the air electrode after the SOEC test. (d) HAADF-STEM image of the interfacial region. (e) and (f) Magnified HAAFD-STEM images showing nanopores at the (e) LSM–YSZ interface (blue square, Fig. 1(d)) and (f) grain boundary within the YSZ electrolyte (red square, Fig. 1(d)). | ||
Crystal structure changes caused by the annihilation of oxygen vacancies
To understand the fundamental cause of nanopore formation, we first examined the local elemental compositions in the YSZ electrolyte near the electrode interface through a point-by-point analysis using an EPMA. In the SE image of the analyzed region (Fig. 2(a)), the linear composition profiles of Y, Zr, and O were obtained along the dots within ∼10 μm of the interface. To ensure the reliability of our data, we performed line scan along five distinct lines, as shown in Fig. S2 in the ESI,† The experimentally quantified compositions and corresponding chemical formulas are summarized in Table 1. The composition in the bulk was estimated to be Y0.165Zr0.835O1.968, which is close to the theoretical composition of 8 mol% YSZ (Y0.13Zr0.88O1.94), verifying the quantitative reliability of the EPMA. On the one hand, in the concentration line profile, the oxygen content was constant in the bulk area but abruptly increased within ∼3 μm of the interface, as clearly shown in the concentration ratio plot as well. On the other hand, the cation ratio (Y/(Y + Zr)) did not significantly change over the measured depth. These results suggest that O2− can accumulate in the interfacial region when the influx of O2− is faster than O2 evolution. The composition at the interface was measured to be Y0.157Zr0.842O2.592, indicating that the oxygen content exceeded that of stoichiometric ZrO2.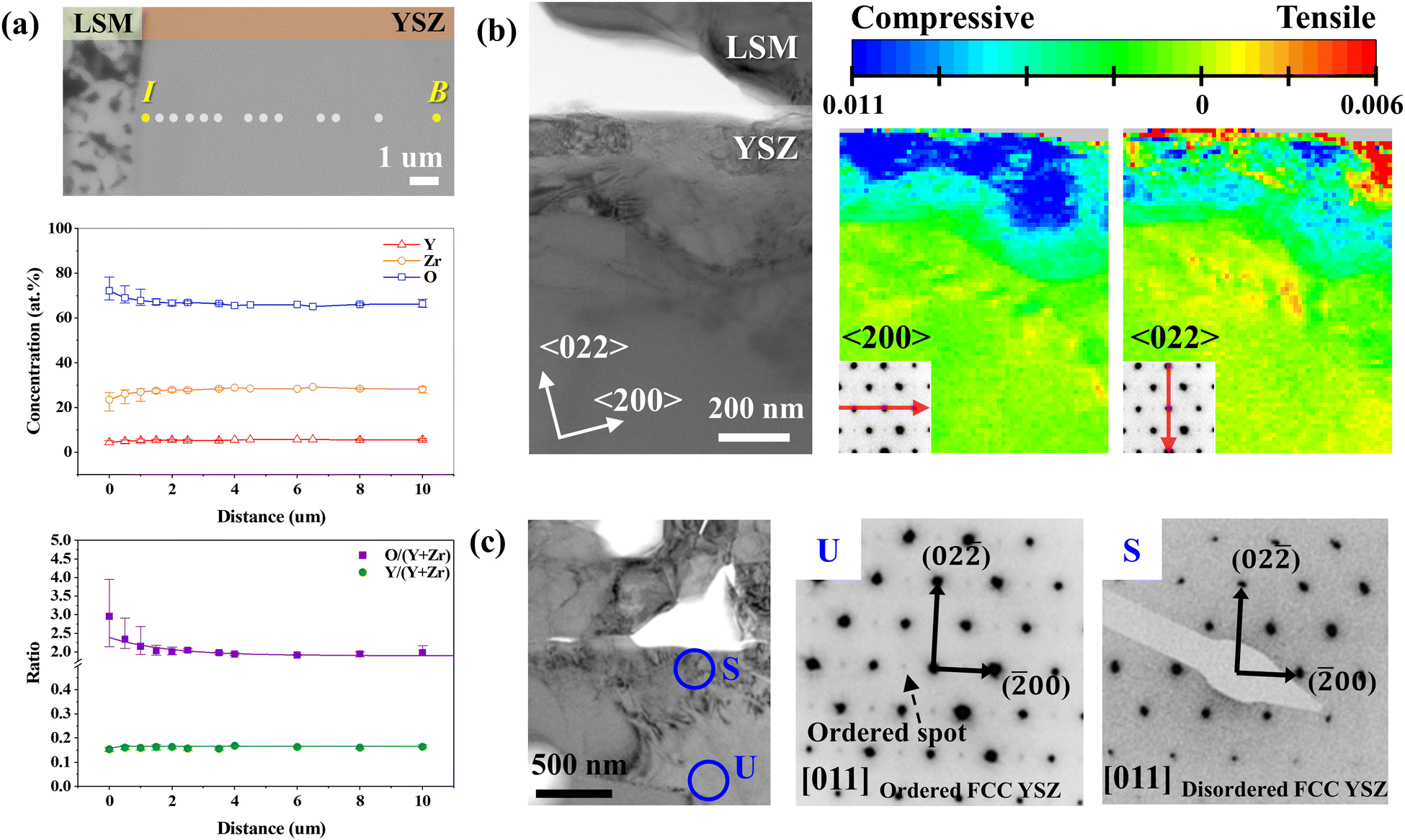 | ||
| Fig. 2 Compositional and structural changes in YSZ at the electrode–electrolyte interface. (a) SE image of YSZ electrolyte at the interfacial region showing the EPMA analysis points from bulk region “B” to interface “I.” The profiles beneath present the concentration ratios obtained by EPMA and the ratios of oxygen or yttrium relative to the sum of cations. (b) BF TEM image of the interface and corresponding strain maps obtained along the horizontal (〈200〉) and vertical (〈022〉) directions. (c) BF TEM image used for selected area diffraction analysis and SADPs obtained from the unstrained bulk region marked “U” and the highly strained interface region marked “S”. | ||
| Theoretical | Experimental | |||
|---|---|---|---|---|
| 8 mol% YSZ (0.08(Y2O3) 0.92(ZrO2)) | Bulk (B) | Near the interface (I) | ||
| Formula | Y0.148Zr0.852O1.926 | Y0.165Zr0.835O1.968 | Y0.157Zr0.842O2.592 | |
| Composition (at%) | Y | 5.06 | 5.55 | 4.39 |
| Zr | 29.12 | 28.16 | 23.45 | |
| O | 65.82 | 66.28 | 72.16 | |
We confirmed the increase in oxygen content and consistent cation concentration ratio in a narrower interfacial region of the ∼3.5 μm range using STEM–EDS with a higher spatial resolution (Fig. S5 in the ESI†). The accumulation of oxygen ions caused by electrolysis operation was further confirmed by EPMA analysis of the un-operated cell in Fig. S6 in the ESI.† Specifically, the oxygen content does not vary within the electrolyte for the un-operated cell, indicating that oxygen accumulates during the electrolysis operation. In addition, the cathodically polarized electrode, where oxygen reduction reaction occurred, was free of defects (Fig. S7 in the ESI†), confirming that the interfacial defects were caused by the anodic polarization in electrolysis reaction. Then, we performed a TEM–PED analysis to investigate the effect of the increased oxygen content on the crystal structure of YSZ after electrolysis operation. The bright-field (BF) TEM image (Fig. 2(b)) shows the strain distribution map obtained using the PED method. The TEM sample was tilted to align the (011) plane of the YSZ grain perpendicular to the electron beam direction, and the strain distribution was mapped along the 〈200〉 and 〈022〉 directions. The inset diffraction patterns at the bottom left of the strain maps show the strain directions. The unit cell information taken from the bulk region was used as a reference to calculate the strain, and the strain map was constructed by comparing individual strained diffraction patterns at each pixel with the reference. Near the interface, a large compressive (blue) strain of 1.1% was measured along the 〈200〉 direction, while strain was negligible along the 〈022〉 direction, indicating anisotropic lattice contraction. Such anisotropic lattice strain is related to changes in the crystal structure, as evidenced by the selected area diffraction patterns (SADPs; Fig. 2(c)). The SADPs of the [011] axis obtained from the intrinsic crystal structure of 8 mol% YSZ (Fig. S8 in the ESI†) and unstrained region in the bulk area (marked “U” in Fig. 2(c)) exhibit lattice ordering on the (100) and (011) planes. Specifically, the little extra spots are observed between the transmitted beam and the main diffraction spots in “U”, and their regular, repeating arrangement indicates ordered crystal structure.44 In contrast, these ordered spots disappear in the highly strained region near the interface (marked “S” in Fig. 2(c)), manifesting the change of crystal structure. Considering that positions U and S are in a single grain, it is clear that within an extremely narrow range near the interface, the strain caused by excess oxygen is correlated with the order–disorder transition of the YSZ crystal structure. In addition, the ordered structure was consistently observed in the interfacial region for the pristine cell (Fig. S8 in the ESI†), confirming that SOEC operation causes the order–disorder transition.
Theoretical interpretation of crystal structure changes by DFT
We performed DFT calculations to elucidate the intrinsic relationship between the excess O2− and crystal structure changes. First, an atomic model of YSZ was developed based on the TEM analysis of the crystal structure. In general, 8 mol% YSZ is known to have a fluorite structure with a space group of Fmm,24,25,45 and the order–disorder transition of YSZ observed in Fig. 2(c) can be attributed to a structural transition from pyrochlore to fluorite.46–50 The primary difference between these two crystal structures is the arrangement of VO, which displaces cations. However, the SADP obtained from the undeformed YSZ (Fig. 2(c)) did not match with either the fluorite or pyrochlore structure.51 Thus, we developed a structure for Y4Zr28O62, which we expanded to a 2 × 2 × 2 supercell structure (Fig. 3(a)), to simulate the composition closest to 8 mol% YSZ, which was theoretically expected and also experimentally confirmed (Table 1). The “Computational details” section in the Methods describes the rules employed for the atomic positions in the DFT calculations. The Y–O–Y pairs in the cubic cell have 〈111〉 ordering and were placed as far apart from each other as possible under periodic boundary conditions. For charge neutrality, a single VO was intrinsically applied per two yttrium atoms in stoichiometric YSZ; the VO were placed at the second-nearest–neighbor sites rather than the first-nearest sites. In the YSZ models, the oxygen composition was varied from 1.938 (stoichiometric) to 2.0 (non-stoichiometric) (Fig. 3(b)). The lowest value was set at 1.938 based on the EPMA composition analysis results (Table 1), whereas the highest value was limited to 2.0 because the DFT calculations found the oxygen interstitial in the YSZ lattice to be unfeasible, as explained in detail in Fig. S9 and Supplementary Notes 1 and 2 in the ESI.† The low and high values represent pristine 8 mol% YSZ and YSZ with fully occupied VO, respectively. The simulated electron diffraction patterns of the atomic models shown (Fig. 3(c)) matched well with the experimental results (Fig. 2(c)), validating that our atomic models successfully represented the experimentally identified YSZ structures in both the bulk and interfacial regions.
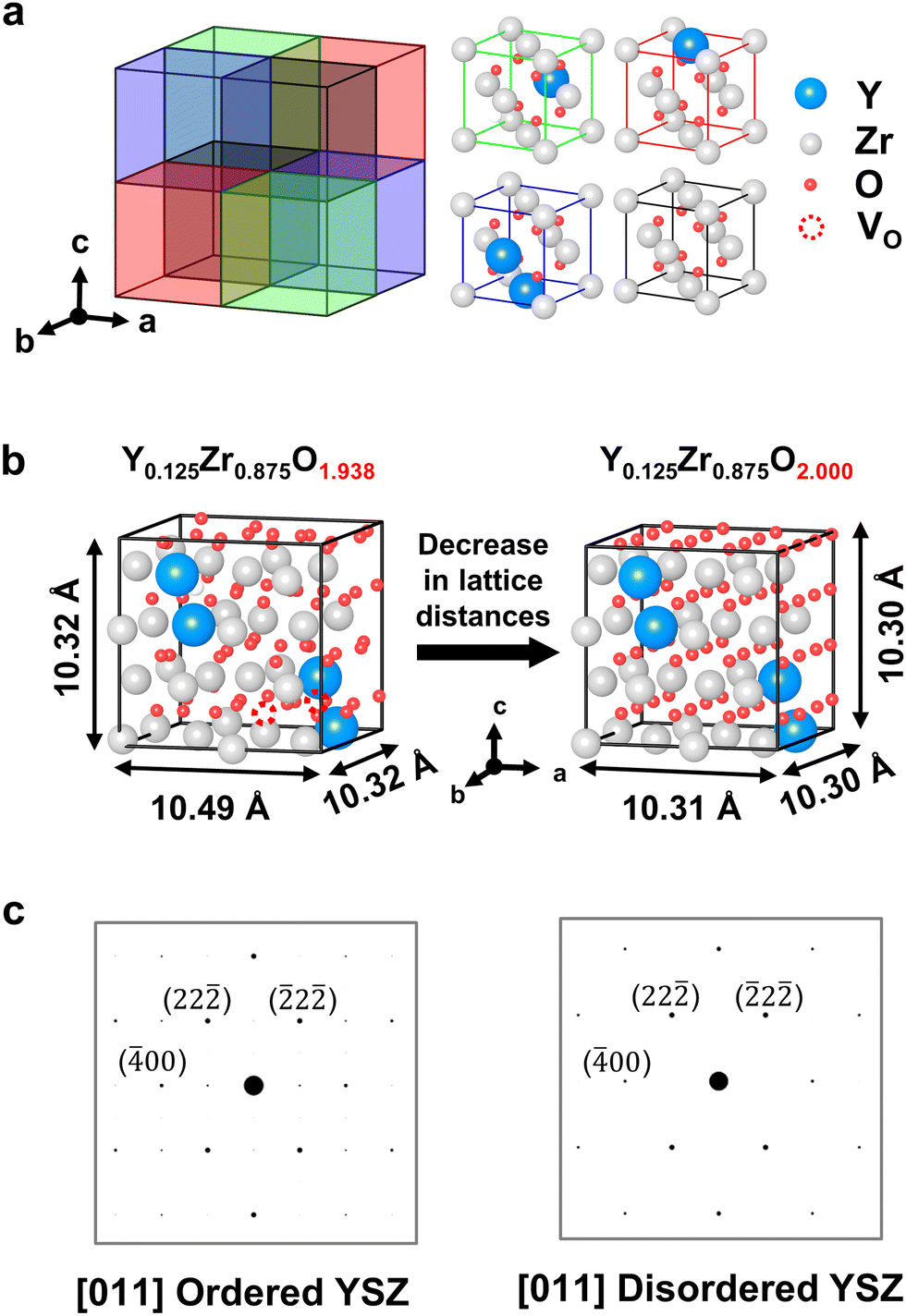 | ||
| Fig. 3 DFT calculations based on the simulated YSZ model. (a) Sublattice components of the supercell model of simulated YSZ unit cells. (b) Simulated YSZ models and unit-cell parameters along 〈100〉 calculated by DFT: (left) stoichiometric 8 mol% YSZ and (right) YSZ with completely filled oxygen vacancies. (c) Simulated electron diffraction patterns of YSZ models. | ||
The lattice distances and unit cell volumes, extracted from the XRD simulations, are summarized in Fig. S9(b) in the ESI.† The calculation results indicate that the occupation of VO in the YSZ lattice decreases the unit cell distance in a certain direction (Fig. 3(b)). Specifically, lattice strain in the [100] direction is dominant at −0.017, whereas it is negligible in the other two directions at only −0.002, which is consistent with the anisotropic lattice deformation discovered by the PED analysis (Fig. 2(b)). A lattice strain as much as 0.017 above the yield stress of YSZ52,53 can readily cause plastic deformation, as evidenced by dislocations and discussed in the following section.
Defect formation caused by the injection of excess oxygen ions
According to the EPMA analysis (Fig. 2(a)), the oxygen content of YSZ at the electrode–electrolyte interface far exceeded that of stoichiometric ZrO2. Energetically, however, interstitial oxygen is unlikely to form in the YSZ lattice (Fig. S9 and Supplementary Notes 1 and 2 in the ESI†), suggesting that further structural changes are needed to accommodate excess O2− at defects. Indeed, the BF TEM image in Fig. 4(a) shows that dislocations formed in the strained interfacial area. Clear images of dislocations were acquired by tilting slightly away from the zone axis, corresponding to a diffraction condition with maximum contrast. These dislocations indicate that local plastic deformation occurred because of the lattice strain induced by excess O2− at the interface. Thus, the dislocation distribution profile directly suggests where the O2− predominantly accumulated. In Fig. 4(a), dislocations are densely populated mostly at the YSZ free surface and YSZ–LSM adjoining boundaries in YSZ side, forming into semicircular region, whereas dislocations are nearly absent at the triple-phase boundary (TPB) points. During SOEC operation, O2− migrate toward the air electrode over the entire electrolyte area and are released in the form of O2 at the TPBs, where O2−, electrons, and gas can simultaneously contact each other (Fig. 4(b)). If the O2− influx is faster than the O2 evolution, O2− selectively accumulate in regions where oxygen cannot be released, thereby creating bottlenecks near the TPBs. At the LSM–YSZ boundaries, oxygen ions accumulate at the YSZ side because LSM cannot accommodate the large amount of extra oxygen ions into the lattice. Specifically, LSM is a predominantly electronic conductor with negligible oxygen ion conductivity, and exhibits the minimal variation of oxygen stoichiometry in a wide pO2 range.54,55 Therefore, during the SOEC operation, the large amount of oxygen ions migrates towards LSM air electrode, and the fraction of oxygen ions incorporated into LSM lattice is considered to be insignificant. Instead, the large portion of oxygen ions accumulate within YSZ electrolyte at the interface, causing the alteration of crystal structure and the creation of defects. Because the incorporation of excess oxygen ions in the bulk lattice of cubic YSZ is limited,56,57 the influx of the large amount of oxygen ions would form dislocations and grain boundaries that can act as the sink or source of charged point defects.58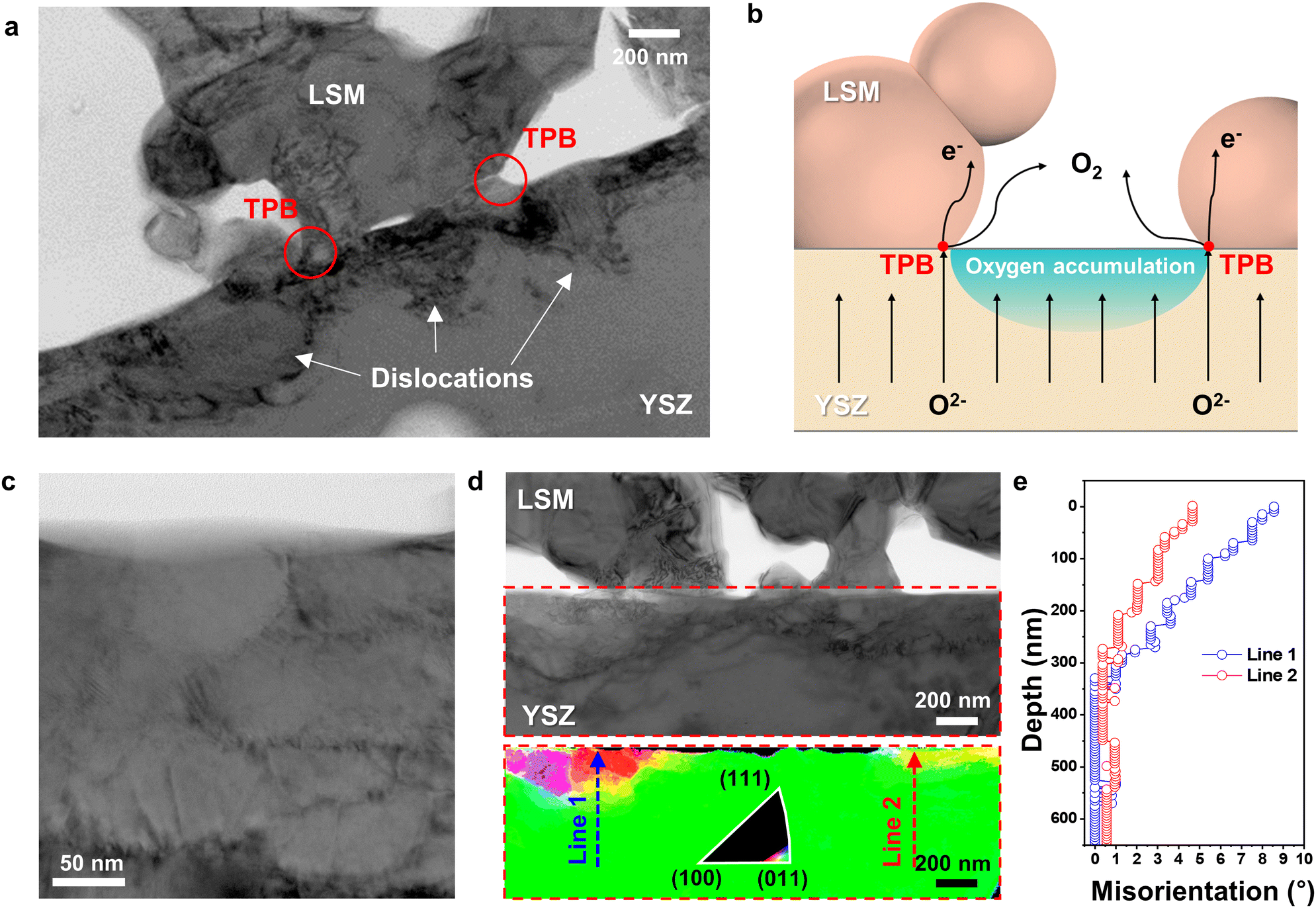 | ||
| Fig. 4 Interfacial defects formed by the injection of excess oxygen ions. (a) BF TEM image showing the dislocation profile in the interfacial region. (b) Schematic illustration of the air electrode reaction and oxygen accumulation at the electrode–electrolyte interface. (c) BF TEM image of fine-grained polycrystalline structure. (d) PED crystal orientation from the red dashed box in the BF TEM image above; the color triangle (inset) presents the crystal orientation, and the (011) plane of the YSZ grain is aligned normal to the electron beam direction. (e) Line profiles of misorientation angles from the bulk toward the interface along lines 1 and 2 in (d). | ||
The semicircular dislocation distribution in Fig. 4(a) reveals interfacial regions that are especially vulnerable to delamination. In addition, dislocations in the electrolyte may be a major source of performance degradation because positively charged dislocations could trap negatively charged O2− and impede their transport.59–61
Another distinct feature caused by the injection of O2− is that the YSZ grains segment into nanometer-scale subgrains. In the BF TEM image in Fig. 4(c), a fine-grained polycrystalline structure is observed near the interface. The original grain size of the YSZ electrolyte was estimated to be ∼8 μm, but it collapsed into small, misoriented subgrains with a size of ∼100 nm. To spatially visualize the degree of polygonization, we constructed a grain orientation map using PED (Fig. 4(d)), the process for which is shown in Fig. S3(b) in the ESI.† The YSZ grain was tilted to align the (011) plane normal to the precessed electron beam. In the orientation map, the color varies only in the interfacial region, indicating changes in the crystalline orientation. The grain refinement mostly appears in the highly strained regions, which coincides with the dislocation profile in Fig. 4(a). Fig. 4(e) shows the misorientation angles, which were quantitatively measured along lines 1 and 2 in Fig. 4(d). The total misorientation angles crossing a few grains from the bulk region to the interface were only 8.5° and 4° at most. These small misorientation angles and indistinct boundaries suggest that subgrain boundaries were formed through dislocation build-up and re-alignment, i.e., polygonization62–64 (see additional TEM results in Fig. S10 in the ESI†). Such polygonization in YSZ has been also been observed to be caused by irradiation damage.65,66 To the best of our knowledge, however, we are the first to report the formation and rearrangement of the subgrain structure in the YSZ electrolyte during SOEC operation, which is attributed to oxygen-accumulation-induced strain.
Mechanism of interfacial degradation during SOEC operation
The microstructural changes at the electrode–electrolyte interface in the early stages of delamination unveil critical information for understanding not only the origin but the entire process of delamination. Fig. 5 illustrates the sequence of degradation steps at the interface. During SOEC operation, O2− accumulate at specific locations where O2 cannot evolve when the influx of O2− is faster than the O2− consumption at the TPBs. This accumulation causes various alterations in the physical and chemical properties that degrade electrochemical performance, even before complete delamination. The annihilation of oxygen vacancies leads to an order–disorder transition in the YSZ unit cell structure, which results in anisotropic lattice strain. As compressive strain at the interface decreases the interfacial ionic conduction,35,67–70 more O2− are necessarily injected, thereby generating interfacial defects, such as dislocations and segmented subgrains, to accommodate the excess O2−. Based on the SADP analysis in Fig. S11 and Supplementary Note 3 in the ESI,† the change of the lattice parameter of LSM is negligible after SOEC operation, indicating that the majority of oxygen ions accumulate within YSZ electrolyte rather than being incorporated in LSM lattice. Dislocations and subgrain boundaries with higher concentrations of O2− serve as initiation sites for further nanopore formation, which interferes with O2− transport and the electrode reaction. Eventually, these pores connect, leading to cracks and delamination. The evolution of multiple nanopores into cracks is further supported by the TEM analysis of the cell operated at higher current density. When the cell was operated at a higher current density of 3.6 A cm−2, the voltage rapidly increased within 25 min (Fig. S12 in the ESI†), and TEM analysis in Fig. S13 in the ESI† revealed the lateral cracks and delamination resulting from the connection of multiple nanopores. Our experimental and theoretical investigations thus clarify the fundamental cause of two major types of cracks—one at the electrode–electrolyte interface and the other at the grain boundaries within the electrolyte—which have been frequently observed but not clearly explained. In particular, the interfacial accumulation of O2− was identified as the fundamental cause of delamination; thus, the balance between the influx of O2− and the evolution of O2 is the key to preventing interfacial degradation.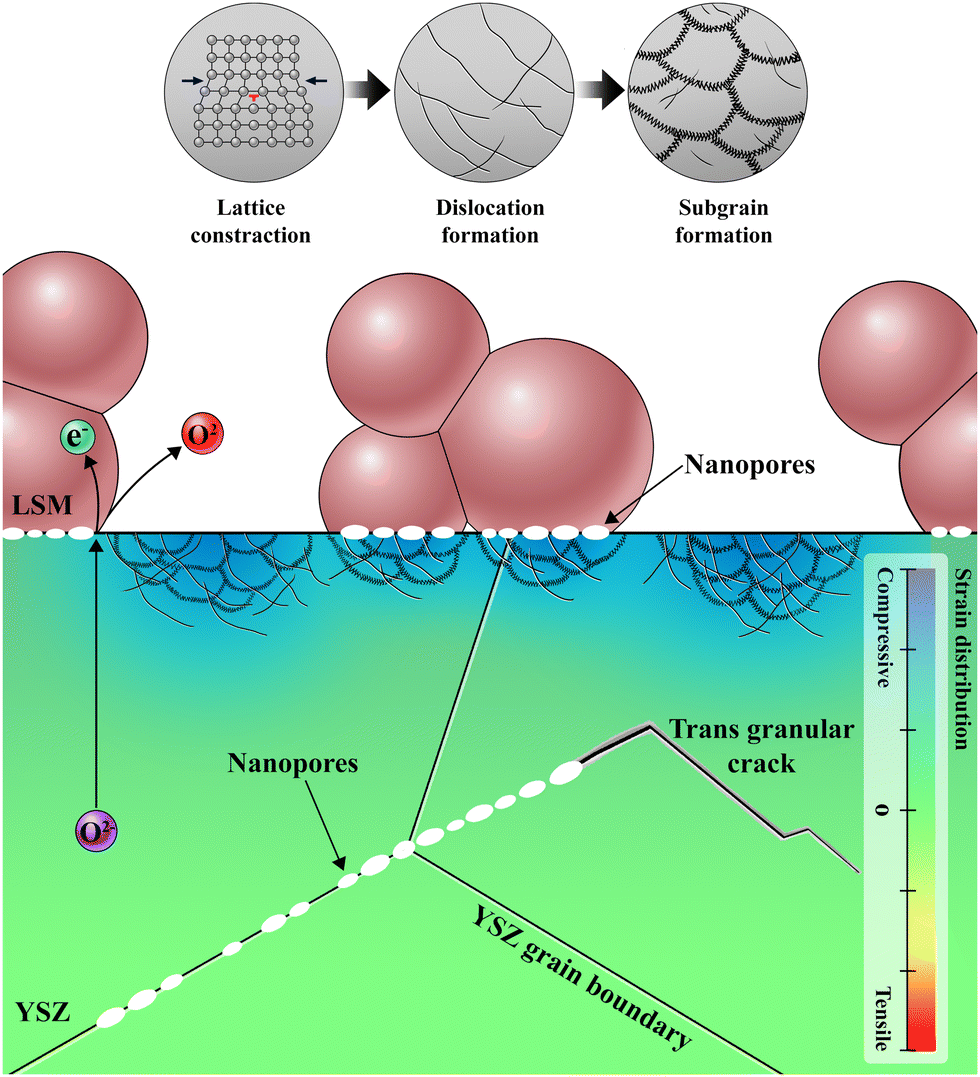 | ||
| Fig. 5 Schematic illustration of degradation process at the electrode–electrolyte interface; formation of nanopores and local lattice strain inducing dislocation and subgrain defects. | ||
Our findings provide novel perspectives on the delamination phenomena. In previous studies, degradation was frequently attributed to the gas pressure build-up resulting from the low releasing rate of oxygen gas.16,17,19,20 Such hypothesis was made simply based on the fact that oxygen gas is generated at the air electrode, and no solid evidence has been provided yet. In this research, we obtained the direct evidences of oxygen accumulation, crystal structure change, dislocation formation and grain segmentation at the interface that eventually causes crack formation. The presence of an interfacial insulating phase, La2Zr2O7, which is formed by the chemical reaction between LSM and YSZ, was also suggested as the main cause of delamination.21 The degree of La2Zr2O7 formation depends on processing conditions, and it was absent in our cells, as seen in TEM-EDS analysis in Fig. S4 in the ESI.† Therefore, the formation of insulating phase is not a direct cause of delamination, although its presence can accelerate delamination by impeding oxygen transport. The disintegration of LSM into nanoparticles at the interface was also suggested as delamination mechanism.23 We observed the formation of nanopores along the interface between LSM and YSZ in Fig. 1(e), and a further progress of nanopore formation would lead to the complete delamination with LSM nanoparticles remaining on the surface. Therefore, the disintegration of LSM observed in the previous studies is considered as one of the results of defect generation reported in this paper rather than the cause of delamination. These findings suggest that the key to resolve the delamination is to prevent the accumulation of oxygen ions at the interface. One of the potential solutions could be to use electrode materials that can accommodate larger amount of oxygen ions. To validate this hypothesis, we replaced LSM with the Ruddlesden–Popper material, La2NiO4+δ, which has a high capacity for interstitial oxygen ions. We constructed full cells using a Ni–YSZ fuel electrode, YSZ electrolyte, and either LSM or La2NiO4+δ as the air electrode. We used the processing techniques reported in our previous work for the fabrication of the cell with La2NiO4+δ as the air electrode.71 As illustrated in Fig. S14 (ESI†), the cell with the LSM air electrode failed in 40 h at a current density of 0.5 A cm−2, whereas the cell with the La2NiO4+δ air electrode operated stably up to 1.1 A cm−2 with the cell voltage up to 1.6 V. This finding serves as preliminary experimental evidence supporting our hypothesis, and we will report more systematic experiments and detailed analyses on cells with various air electrodes, assessing different oxygen accommodation capacities, in our upcoming paper.
Currently, mixed ionic- and electronic-conducting (MIEC) electrode materials, such as (La,Sr)(Co,Fe)O3−δ (LSCF), are widely used. While structural changes predominantly occur in the YSZ electrolyte at the interface in this study owing to limited oxygen ionic conductivity in LSM, for MIECs, a portion of the incoming oxygen ions will enter the air electrode, resulting in less accumulation within the electrolyte. However, these oxygen ions in the MIEC electrode will alter oxygen stoichiometry and induce chemical stress, potentially leading to delamination, as reported in previous studies.72,73 Enhancing the oxygen evolution reaction was found to be effective in mitigating this delamination.74 The physical and chemical state of the electrode–electrolyte interface is more complex for MIEC electrodes compared to LSM electrodes, primarily due to the involvement of more elements and increased interdiffusion. This work lays the groundwork for a clearer understanding of the delamination phenomena in state-of-the-art MIEC electrodes.
Conclusions
The mechanism underlying air electrode delamination has long been questioned, and the various hypotheses that have been proposed to explain it thus far have lacked solid evidence. In this study, we clarified various physical and chemical alterations at the electrode–electrolyte interface using cutting-edge electron microscopy techniques and identified vulnerable locations by directly visualizing the crystal structures and defects. This comprehensive understanding will provide rational guidelines for future research. Although more O2− tended to accumulate with increasing operating current density, high-current operation is desirable from an economic perspective. Therefore, further research is required to prevent oxygen accumulation at these two susceptible spots, the electrolyte surface, and the electrolyte–electrode interface when operating at a high current. The knowledge obtained in this research could be exploited to understand delamination mechanisms of various state-of-the-art electrode materials with more complicated interfaces, which would establish rational strategies for reforming the interfacial properties and resolving this critical issue in SOEC development. Our findings can be further supported by in situ/operando TEM analysis that visualizes the real-time progress of delamination. However, in situ/operando analysis of SOEC delamination phenomena is extremely challenging because at least three stimuli (electric potential, reactive gases and high temperature) should be simultaneously applied on the complex multi-layered samples. We will report in situ/operando TEM results and further clarify the origin and sequence of delamination in our following paper.Author contributions
H. J. C. and K. J. Y. conceived the idea and co-supervised this project. J. S., S. Y. P. and K. J. Y. fabricated and evaluated cells. H. C. and H. J. C. conducted microstructure analysis. C. Y. and C. W. L. performed the DFT calculations. S. B., J. W. K., J. L. and J. W. P. interpreted the analytical results. H. C., H. J. C., K. J. Y., C. Y., and C. W. L. wrote the paper. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Ministry of Trade, Industry, and Energy (MOTIE) of Korea [no. P0022331] supervised by the Korea Institute for Advancement of Technology (KIAT) and the institutional research program [2E33263] of the Korea Institute of Science and Technology (KIST). This work was also partially supported by Hyundai Motors and National Research Council of Science and Technology [No. CAP22072-000]. The authors would like to thank Lauren Plavisch of Editage (https://www.editage.co.kr) for English language editing.References
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- M. Ni, M. K. Leung and D. Y. Leung, Int. J. Hydrogen Energy, 2008, 33, 2337–2354 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- D. Ferrero, M. Gamba, A. Lanzini and M. Santarelli, Energy Procedia, 2016, 101, 50–57 CrossRef CAS.
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583–3602 RSC.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- B. Parkinson, P. Balcombe, J. Speirs, A. Hawkes and K. Hellgardt, Energy Environ. Sci., 2019, 12, 19–40 RSC.
- A. Nechache and S. Hody, Renewable Sustainable Energy Rev., 2021, 149, 111322 CrossRef CAS.
- S. P. Simner, J. F. Bonnett, N. L. Canfield, K. D. Meinhardt, J. P. Shelton, V. L. Sprenkle and J. W. Stevenson, J. Power Sources, 2003, 113, 1–10 CrossRef CAS.
- W.-H. Kim, H.-S. Song, J. Moon and H.-W. Lee, Solid State Ionics, 2006, 177, 3211–3216 CrossRef CAS.
- Y. Lim, J. Park, H. Lee, M. Ku and Y.-B. Kim, Nano Energy, 2021, 90, 106524 CrossRef CAS.
- A. Hauch, K. Brodersen, M. Chen and M. B. Mogensen, Solid State Ionics, 2016, 293, 27–36 CrossRef CAS.
- A. Gil, J. Wyrwa and T. Brylewski, Oxid. Met., 2016, 85, 151–169 CrossRef CAS.
- K. Chen, J. Electrochem. Soc., 2016, 163, F3070 CrossRef CAS.
- S. Y. Gómez and D. Hotza, Renewable Sustainable Energy Rev., 2016, 61, 155–174 CrossRef.
- R. Knibbe, M. L. Traulsen, A. Hauch, S. D. Ebbesen and M. Mogensen, J. Electrochem. Soc., 2010, 157, B1209 CrossRef CAS.
- A. Momma, T. Kato, Y. Kaga and S. Nagata, J. Ceram. Soc. Jpn., 1997, 105, 369–373 CrossRef CAS.
- M. S. Khan, X. Xu, R. Knibbe and Z. Zhu, Renewable Sustainable Energy Rev., 2021, 143, 110918 CrossRef CAS.
- J. R. Mawdsley, J. D. Carter, A. J. Kropf, B. Yildiz and V. A. Maroni, Int. J. Hydrogen Energy, 2009, 34, 4198–4207 CrossRef CAS.
- M. Laguna-Bercero, R. Campana, A. Larrea, J. Kilner and V. Orera, J. Power Sources, 2011, 196, 8942–8947 CrossRef CAS.
- M. Keane, M. K. Mahapatra, A. Verma and P. Singh, Int. J. Hydrogen Energy, 2012, 37, 16776–16785 CrossRef CAS.
- J. Kim, H.-I. Ji, H. P. Dasari, D. Shin, H. Song, J.-H. Lee, B.-K. Kim, H.-J. Je, H.-W. Lee and K. J. Yoon, Int. J. Hydrogen Energy, 2013, 38, 1225–1235 CrossRef CAS.
- K. Chen, Int. J. Hydrogen Energy, 2011, 36, 10541–10549 CrossRef CAS.
- H. Scott, J. Mater. Sci., 1975, 10, 1527–1535 CrossRef CAS.
- D. K. Smith and W. Newkirk, Acta Crystallogr., 1965, 18, 983–991 CrossRef CAS.
- A. Avilov, K. Kuligin, S. Nicolopoulos, M. Nickolskiy, K. Boulahya, J. Portillo, G. Lepeshov, B. Sobolev, J. Collette and N. Martin, Ultramicroscopy, 2007, 107, 431–444 CrossRef CAS PubMed.
- M. J. Hÿtch and A. M. Minor, MRS Bull., 2014, 39, 138–146 CrossRef.
- D. L. Foley, A. C. Leff, A. C. Lang and M. L. Taheri, Acta Mater., 2020, 185, 279–286 CrossRef CAS.
- A. Bashir, R. W. Millar, K. Gallacher, D. J. Paul, A. D. Darbal, R. Stroud, A. Ballabio, J. Frigerio, G. Isella and I. MacLaren, J. Appl. Phys., 2019, 126, 235701 CrossRef.
- Y. Seto and M. Ohtsuka, J. Appl. Crystallogr., 2022, 55, 397–410 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- A. Kushima and B. Yildiz, J. Mater. Chem., 2010, 20, 4809–4819 RSC.
- S.-H. Guan, K.-X. Zhang, C. Shang and Z.-P. Liu, J. Chem. Phys., 2020, 152, 094703 CrossRef CAS PubMed.
- X. Xia, R. Oldman and R. Catlow, Chem. Mater., 2009, 21, 3576–3585 CrossRef CAS.
- S.-H. Guan, C. Shang and Z.-P. Liu, J. Phys. Chem. C, 2020, 124, 15085–15093 CrossRef CAS.
- K. Chen and N. Ai, Int. J. Hydrogen Energy, 2012, 37, 10517–10525 CrossRef CAS.
- W.-Z. Li, L. Kovarik, D. Mei, J. Liu, Y. Wang and C. H. F. Peden, Nat. Commun., 2013, 4, 2481 CrossRef PubMed.
- M. S. Khan, X. Xu, J. Zhao, R. Knibbe and Z. Zhu, J. Power Sources, 2017, 359, 104–110 CrossRef CAS.
- X. I. Pereira-Hernández, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. M. Hensen, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef PubMed.
- M. S. Khan, X. Xu, R. Knibbe, A. U. Rehman, Z. Li, A. J. Yago, H. Wang and Z. Zhu, Energy Technol., 2020, 8, 2000241 CrossRef CAS.
- P. E. Champness, Electron diffraction in the transmission electron microscope, Garland Science, 2020 Search PubMed.
- N. Ishizawa, Y. Matsushima, M. Hayashi and M. Ueki, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 726–735 CrossRef PubMed.
- T. Yamashita, K.-i Kuramoto, M. Nakada, S. Yamazaki, T. Sato and T. Matsui, J. Nucl. Sci. Technol., 2002, 39, 585–591 CrossRef.
- J. W. Fergus, Metall. Mater. Trans. E, 2014, 1, 118–131 CAS.
- S. V. Ushakov, A. Navrotsky, R. J. Weber and J. C. Neuefeind, J. Am. Ceram. Soc., 2015, 98, 3381–3388 CrossRef CAS.
- A. Dworkin and M. A. Bredig, J. Phys. Chem., 1968, 72, 1277–1281 CrossRef CAS.
- C. Catlow and W. Hayes, J. Phys. C-Solid State Phys., 1982, 15, L9 CrossRef CAS.
- T. Götsch, W. Wallisch, M. Stöger-Pollach, B. Klötzer and S. Penner, AIP Adv., 2016, 6, 025119 CrossRef.
- A. Tikhonovsky, M. Bartsch and U. Messerschmidt, Phys. Status Solidi A, 2004, 201, 26–45 CrossRef CAS.
- K. Otsuka, A. Kuwabara, A. Nakamura, T. Yamamoto, K. Matsunaga and Y. Ikuhara, Appl. Phys. Lett., 2003, 82, 877–879 CrossRef CAS.
- J. Mizusaki, N. Mori, H. Takai, Y. Yonemura, H. Minamiue, H. Tagawa, M. Dokiya, H. Inaba, K. Naraya, T. Sasamoto and T. Hashimoto, Solid State Ionics, 2000, 129, 163–177 CrossRef CAS.
- J. Mizusaki, H. Tagawa, K. Naraya and T. Sasamoto, Solid State Ionics, 1991, 49, 111–118 CrossRef CAS.
- S. Middleburgh, G. Lumpkin and R. Grimes, Solid State Ionics, 2013, 253, 119–122 CrossRef CAS.
- M. J. Rushton, I. Ipatova, L. J. Evitts, W. E. Lee and S. C. Middleburgh, RSC Adv., 2019, 9, 16320–16327 RSC.
- K. Kliewer and J. Koehler, Phys. Rev., 1965, 140, A1226 CrossRef.
- M. D. Armstrong, K.-W. Lan, Y. Guo and N. H. Perry, ACS Nano, 2021, 15, 9211–9221 CrossRef CAS PubMed.
- K. K. Adepalli, J. Yang, J. Maier, H. L. Tuller and B. Yildiz, Adv. Funct. Mater., 2017, 27, 1700243 CrossRef.
- H. Schraknepper, T. E. Weirich and R. A. De Souza, Phys. Chem. Chem. Phys., 2018, 20, 15455–15463 RSC.
- J. Kim, J. W. Lim, J. K. Kim, D. H. Kim, E. S. Park and H. J. Chang, Scr. Mater., 2021, 190, 158–162 CrossRef CAS.
- Y. Miao, T. Yao, J. Lian, S. Zhu, S. Bhattacharya, A. Oaks, A. M. Yacout and K. Mo, Scr. Mater., 2018, 155, 169–174 CrossRef CAS.
- W. F. Hosford, Physical metallurgy, CRC Press, 2010 Search PubMed.
- S. Moll, L. Thomé, L. Vincent, F. Garrido, G. Sattonnay, T. Thomé, J. Jagielski and J. Costantini, J. Appl. Phys., 2009, 105, 023512 CrossRef.
- J.-M. Costantini, F. Beuneu and W. J. Weber, J. Nucl. Mater., 2013, 440, 508–514 CrossRef CAS.
- N. Schichtel, C. Korte, D. Hesse, N. Zakharov, B. Butz, D. Gerthsen and J. Janek, Phys. Chem. Chem. Phys., 2010, 12, 14596–14608 RSC.
- M. Kilo, C. Argirusis, G. Borchardt and R. A. Jackson, Phys. Chem. Chem. Phys., 2003, 5, 2219–2224 RSC.
- C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Phys. Chem. Chem. Phys., 2008, 10, 4623–4635 RSC.
- C. Korte, N. Schichtel, D. Hesse and J. Janek, Monatsh. Chem., 2009, 140, 1069–1080 CrossRef CAS PubMed.
- J. Shin, S. Yang, H.-I. Ji, S. Park, H. Kim, J.-W. Son, J.-H. Lee, B.-K. Kim, J. Hong and K. J. Yoon, J. Alloys Compd., 2021, 868, 159092 CrossRef CAS.
- Y. Wen and K. Huang, J. Electrochem. Soc., 2024, 171, 034510 CrossRef.
- K. Cook, J. Wrubel, Z. Ma, K. Huang and X. Jin, J. Electrochem. Soc., 2021, 168, 114510 CrossRef CAS.
- P. Jayapragasam, Y. Wen, K. Cook, J. A. Wrubel, Z. Ma, K. Huang and X. Jin, J. Electrochem. Soc., 2023, 170, 054509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures depicting analysis procedures and results, as well as notes detailing density functional theory calculations. See DOI: https://doi.org/10.1039/d4ee00896k |
| This journal is © The Royal Society of Chemistry 2024 |