
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitigating cathodic dissolution through interfacial water masking to enhance the longevity of aqueous zinc–ion batteries†
Wei
Zhong‡
abc,
Zeyu
Shen‡
ab,
Jiale
Mao
ab,
Shichao
Zhang
a,
Hao
Cheng
 *abc,
Yoonseob
Kim
de and
Yingying
Lu
*abc
*abc,
Yoonseob
Kim
de and
Yingying
Lu
*abc
aState Key Laboratory of Chemical Engineering, Institute of Pharmaceutical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: Bob_hao@zju.edu.cn; yingyinglu@zju.edu.cn
bZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou 311215, China
cInstitute of Wenzhou, Zhejiang University, Wenzhou 325006, China
dDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Hong Kong SAR, China
eEnergy Institute, The Hong Kong University of Science and Technology, Hong Kong SAR, China
First published on 6th February 2024
Abstract
Aqueous zinc-ion batteries (AZIBs) hold vast potential for large-scale energy storage applications due to their intrinsic safety features. Vanadium-based cathodes, known for their high specific capacity, face dissolution issues primarily due to water activity at the cathode–electrolyte interface. Herein, we introduce an innovative strategy that mitigates cathode dissolution through the use of an interfacial water-masking agent (IWMA). Taking isosorbide dimethyl ether (IDE) as a subject, it preferentially adsorbs onto the cathode interface, effectively displacing surface-active water. Simultaneously, its strong hydrogen bonding with water reduces the number of active bound water molecules in the solvation structure of Zn2+. These merits substantially strengthen the structural stability of the cathode, thereby improving both its reversible capacity and cycle stability. Impressively, this IWMA avoids involvement in the solvation structure and effectively reduces the desolvation energy of hydrated Zn2+, resulting in excellent rate performance for V2O5·nH2O and Zn0.25V2O5·nH2O cathodes, with remarkable longevity exceeding 2500 and 4000 cycles at a rate of 5 A g−1, respectively. This groundbreaking achievement marks a substantial stride in surmounting a formidable challenge in the realm of AZIBs.
Broader contextLarge-scale energy storage applications necessitate enhanced safety measures for batteries. Aqueous zinc-ion batteries (AZIBs) hold promise for scale-up applications due to their high theoretical capacity, low redox potential, safety, and environmental advantages. However, the unstable cathode–electrolyte interface often gives rise to challenges, such as dissolution, structural collapse and formation of by-products. These issues are typically interactive and dynamic in nature. Therefore, a comprehensive and continuous strategy is imperative to fortify interface stability, moving beyond localized and temporary alleviation. In this study, an interfacial water masking strategy is proposed to comprehensively enhance the stability of the cathodes, thereby achieving AZIBs with excellent rate performance. The pioneering findings of this research are anticipated to offer valuable insights into addressing cathode–electrolyte interface challenges beyond the scope of Zn. |
Introduction
Aqueous zinc-ion batteries (AZIBs) hold immense promise for revolutionizing large-scale energy storage. Their appeal is underpinned by substantial theoretical capacity (820 mA h g−1 or 5855 mA h cm−3), low redox potential (–0.762 V vs. standard hydrogen electrode, SHE), inherent safety and environmental advantages.1–3 Among the plethora of cathode materials, vanadium (V)-based compounds shine brightly due to their multi-electron redox reactions, multidimensional ionic diffusion pathways, and impressive specific capacity (>300 mA h g−1), coupled with excellent rate performance and extended cycle life.4,5 Unfortunately, these cathodes grapple with a significant challenge: pronounced dissolution in aqueous solutions, which is prone to lead to capacity decline.6,7 This issue is highly related to the desolvation process of the hydrated [Zn(H2O)x]2+ ion at the cathode–electrolyte interface,8–10 where a significant amount of active bound water is released. Different from the free water in the solvent, bound water often exhibits higher polarity and reactivity.11 During the charge/discharge process, these active water molecules continuously intercalate/extract at the cathode surface, attacking the lattice of V oxides and causing severe dissolution of V (V2O5 + 3H2O → 2VO2(OH)2− + 2H+).12–14 Therefore, measures are needed to shield the interface from water intrusion, i.e., interfacial water masking to stabilize the V-based cathode.Numerous researchers have been exploring the strategy of interface water masking agents (IWMAs) from the perspective of cathodes and electrolytes. Creating an artificial solid electrolyte interface (e.g., poly(3,4-ethylenedioxythiophene),15 HfO2,16 ZnO,17 polyaniline,18 Ti3C2Tx MXene19) on the cathode surface has emerged as an effective protection approach. By forming a surface passivation layer as a barrier between the cathode and electrolyte, this strategy mitigates the entry of water molecules into the electrode material, effectively inhibiting undesirable interfacial reactions and preventing the dissolution of active chemicals in the electrolyte. However, this method often involves intricate synthesis steps such as atomic layer deposition, limiting its broad applicability. Electrolyte engineering, by contrast, has garnered favor among researchers due to its scalability for integration into current battery production technologies. In recent years, the advent of highly concentrated electrolytes has been pivotal in electrolyte innovation. Various highly concentrated electrolyte formulations (e.g., 15 M NaClO4 + 1 M zinc trifluoromethanesulfonate (Zn(OTf)2),20 1 M Zn(OTf)2 + 20 M lithium bis(trifluoromethanesulfonyl)imide,21 0.5 M Zn(ClO4)2 + 18 M NaClO4,22 and 22 M tetraethylammonium triflate + 9 M sodium trifluoromethane sulfonate8) have effectively curbed dissolution by suppressing Zn2+–H2O interaction. Nevertheless, the challenges of elevated costs, increased viscosity, reduced ionic conductivity, and diminished energy density have hampered their widespread adoption. Some researchers have introduced organic solvents with strong solvating abilities, such as N-methyl-2-pyrrolidone (NMP),23 ethylene glycol (EG),24 diethyl carbonate (DEC),25 trimethyl phosphate (TMP),26 and N,N-dimethylacetamide (DMA),27 into aqueous electrolytes as IWMAs. By forming hydrogen bonds, these solvents capture free water molecules and modify the solvation structure of Zn2+. The reduction in the number of highly reactive water molecules attacking the vanadium oxide lattice at the interface effectively hinders the cathodic dissolution of V-based materials. Regrettably, these organic molecules also insert into the solvation structure of Zn2+, inevitably enlarging the size of solvated ion complexes, which leads to a slowdown in ion transport kinetics, an increase in interfacial desolvation energy barrier, and an increase in charge transfer resistance, thereby affecting the rate performance of the battery.
In order to safeguard cathodes against the dissolved effects of interfacially active water and to improve the battery's rate performance, innovative IWMAs should meet the following fundamental criteria: (1) they should possess the capability to physically shield interfacial water molecules on the cathode surface through specific adsorption; (2) they should be capable of forming hydrogen bonds to restrict water activity in the bulk solution; (3) they should not participate in the solvation structure of Zn2+, thus reducing desolvation energy.
Isosorbide dimethyl ether (lDE) is considered an ideal candidate for an IWMA due to its electron-rich groups, multiple hydrogen bond acceptors, substantial spatial resistance, and symmetrical charge distribution (Fig. S1 and S2, ESI†). In this study, we introduce IDE into electrolytes to reshape the electrolyte structure and electrode interface chemistry, curtailing the water activity. Both experimental and theoretical evidence unequivocally demonstrate that IDE exhibits distinct preferential specific adsorption at the cathode–electrolyte interface, effectively isolating water molecules and reducing the water density. In addition, IDE establishes robust hydrogen bonds with water, thereby disrupting the initial hydrogen bonding network among water molecules and suppressing free water activity (Fig. 1a and b). Moreover, the introduction of IDE decreases the water content of Zn2+ solvation sheath, reducing the release of interfacial bound water while decreasing the desolvation energy. Therefore, in the IDE-modified electrolytes, Zn‖V2O5·nH2O (VOH) batteries exhibit superior electrochemical performance, retaining 92.7% of reversible capacity after 2500 cycles with a maximum specific capacity of 190.3 mA h g−1, at a current density of 5 A g−1. Zn‖Zn0.25V2O5·nH2O (ZVO) batteries also demonstrate a commendable maximum specific capacity of 266.6 mA h g−1 and retain 79.4% of reversible capacity after 4000 cycles. This pioneering IWMA strategy offers a versatile approach to address cathode–electrolyte interface challenges, ushering in an era of enduring AZIBs.
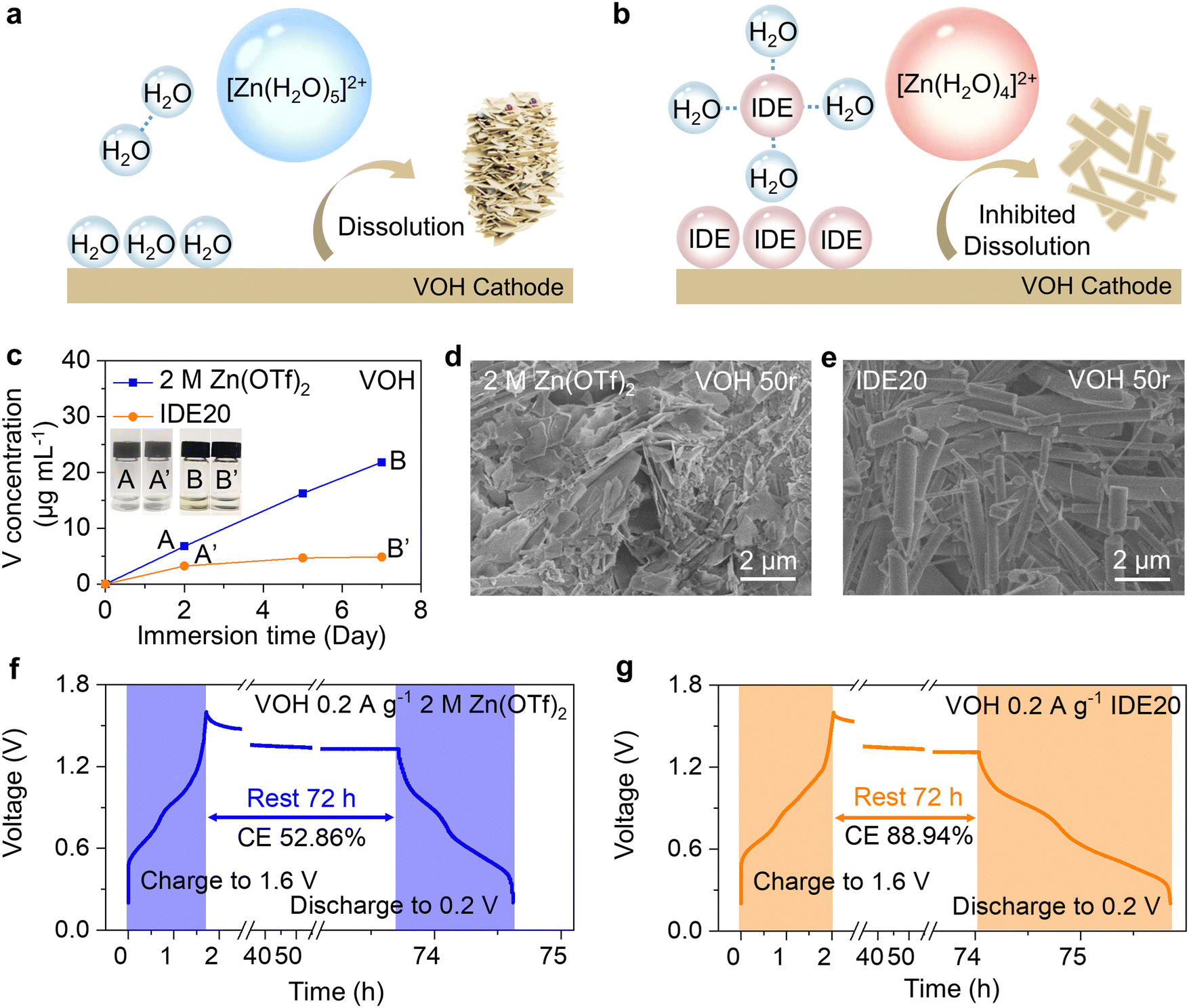 | ||
| Fig. 1 Dissolution behavior of VOH in aqueous solution. (a) and (b) Schematic illustration of IWMA strategy. (c) ICP-OES results of electrolytes with VOH cathodes immersing at various time. SEM images of VOH cathodes using (d) 2 M Zn(OTf)2 and (e) IDE20 electrolytes after 50 cycles. Self-discharge analysis using (f) 2 M Zn(OTf)2 and (g) IDE20 electrolytes. | ||
Results and discussion
To visually track V dissolution in the aqueous electrolyte, we conducted an immersion experiment. V species in solution typically exhibit a yellow coloration.28 As depicted in photograph B of Fig. 1c, the 2 M Zn(OTf)2 electrolyte manifests a noticeable yellow tint after just 7 days of contact with the VOH cathode. This visually confirms the dissolution of V species. To precisely quantify the concentration of V in the electrolyte, we performed inductively coupled plasma-optical emission spectrometer (ICP-OES) tests. The persistence of the dissolution behavior of VOH in the 2 M Zn(OTf)2 electrolyte is underscored by the nearly linear increase in V concentration with extended immersion time. A similar trend was also observed with ZVO (Fig. S3, ESI†).Scanning electron microscopy (SEM) and energy dispersive spectrometer (EDS) were used to examine the surface morphology and elemental distribution of the VOH cathode after 50 cycles (Fig. 1d and Fig. S4, ESI†). They reveal a transformation: the initially elongated cathode materials have given way to numerous thick platelets. The surface of the ZVO cathode material has also begun to exhibit roughening (Fig. S6, ESI†). Evaluating the extent of VOH dissolution in the 2 M Zn(OTf)2 aqueous electrolyte, we employed an open-circuit voltage (OCV) rest test. The battery was fully charged and allowed to remain open circuit for 72 hours before measuring the recoverable capacity. The decline in discharge capacity can be attributed primarily to structural deformation/collapse due to V dissolution, in addition to ion intercalation and deintercalation during OCV. At higher current densities, capacity loss results from structural deformation/collapse occurring rapidly during each cycle of continuous galvanostatic charge–discharge.29 Consequently, assessing the capacity loss stemming from V dissolution via the capacity retention ratio is straightforward when experiments are conducted at a low current density of 0.2 A g−1. As depicted in Fig. 1f and Fig. S7a (ESI†), following OCV-3 days of testing in the 2 M Zn(OTf)2 aqueous electrolyte, the Coulombic efficiency (CE) of batteries equipped with the VOH cathode was only 52.86%, while that of batteries with the ZVO cathode stood at a mere 65.25%. This underscores the pronounced severity of V-based cathode dissolution in aqueous electrolytes.
To mitigate cathode dissolution, reduce capacity loss, and enhance cathode stability, we introduced an IWMA, IDE, into the electrolyte. The orange line in Fig. 1c and Fig. S3 (ESI†) demonstrates that the quantity of dissolved V in the electrolyte decreased with IDE compared to the control group. This provides clear evidence that IDE contributes to inhibiting V-based cathode dissolution. After 50 cycles, the cathode crystal structure remained fine and rod-like (Fig. 1e and Fig. S5, S6, ESI†), indicating that the modified electrolyte has a positive impact on cathode stability. Following OCV-3 days, the CEs of both cathode systems increased substantially to 88.94% (VOH) and 90.25% (ZVO), signifying the significant inhibitory effect of IDE on cathodic dissolution (Fig. 1g and Fig. S7b, ESI†).
In order to observe the dissolution phenomenon on the cathode, high resolution transmission electron microscope (HRTEM) and EDS were implemented to characterize the cathode surface. From the HRTEM and fast Fourier transform (FFT) insets in Fig. 2a and b, it can be inferred that the chosen lattice spacing is 0.215 nm, corresponding to the lattice plane (400) of ZnV2O6. The elevated Zn content and decreased V/O ratio highlight the limited reversibility of the VOH cathode within the 2 M Zn(OTf)2 electrolyte, as well as the dissolution of V from the cathode during cycling. The disappearance of water pillars in the structure also promotes the collapse of the cathode structure. Further EDS line scan revealed an augmentation in O content as the V content decreased at the edge (Fig. 2c), whereas this phenomenon was not observed in the VOH original powder (Fig. S8, ESI†). The EDS mapping results in Fig. S9 and S10 (ESI†) can also observe the enrichment of edge O before modification. The lattice spacing of selected areas I and II in Fig. 2d are both 0.347 nm, which corresponds to the lattice plane (003) of Zn0.36V2O5·nH2O (Fig. 2e).30 Less Zn content indicates higher reversibility of the IDE-modified cathode. The line scan in Fig. 2f shows that no V dissolution occurs at the edge, and Fig. S11 (ESI†) shows that there is no enrichment of edge O. These demonstrate the exceptional stability of the cathode when cycled in IDE-modified electrolytes.
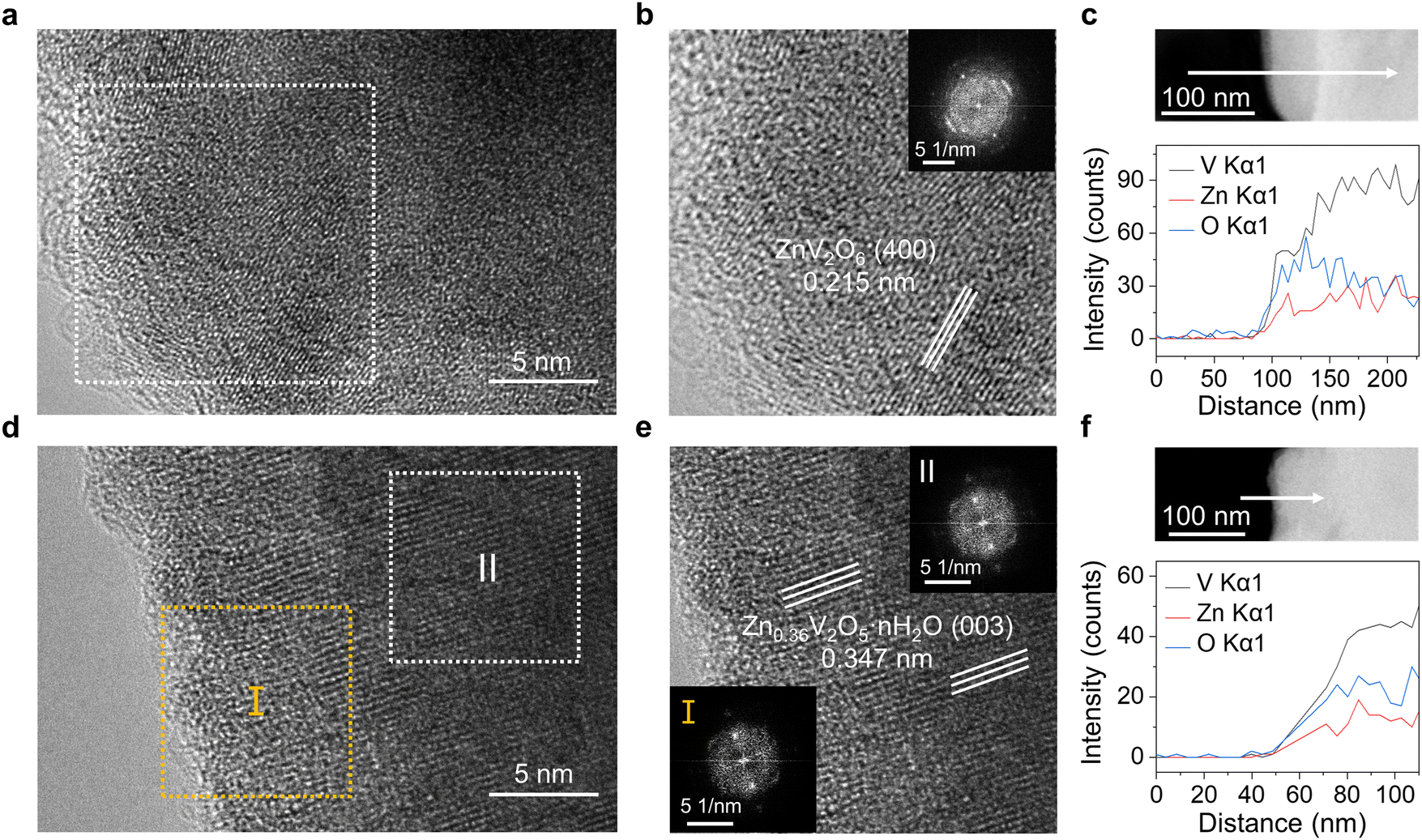 | ||
| Fig. 2 Dissolution behavior of VOH cathode surfaces. The HRTEM images of VOH cathodes near the surface after cycling with (a) 2 M Zn(OTf)2 and (d) IDE20 for 20 cycles at 5 A g−1. (b) and (e) Magnified images of (a) and (d). Insets are the FFT patterns from the selected HRTEM regions. (c) and (f) EDS line scanning of corresponding HRTEM images of VOH cathodes. | ||
To gain practical insights into the solvation structure of Zn2+ and the hydrogen bonding network within the electrolyte, we conducted Raman characterization. In the Raman spectra, which encompass the stretching vibration of O–H in the range of 3000–3800 cm−1, three characteristic peaks can be distinguished, representing strong, mediate, and weak hydrogen bonding, respectively.23,31Fig. 3a and b and Fig. S12 (ESI†) clearly demonstrate an augmentation in the proportion of robust hydrogen bonds, accompanied by a simultaneous reduction in the proportion of medium and weak hydrogen bonds with the introduction of IDE. This suggests a strong interaction between IDE and H2O, leading to the formation of robust intermolecular hydrogen bonds. Consequently, this interaction disrupts the pre-existing hydrogen bonds among water molecules, resulting in a decrease in the presence of free water molecules. The hydrogen bonding network was then validated by nuclear magnetic resonance (NMR) spectroscopy. As shown in Fig. 3c, the 1H peak from H2O shifts with increasing IDE addition, suggesting a significant deshielding effect due to the disruption of the hydrogen bonding network in the water. This disruption results in a decrease in the associated 1H electron density, indicating that IDE additives stabilize water clusters through stronger hydrogen bonding interactions and reduce the content of free water in bulk electrolytes. The 1H resonance of the IDE in Fig. 3d shows a decrease in electron density and an increase in chemical shift on the methyl hydrogen due to the formation of hydrogen bonds between the IDE and H2O.
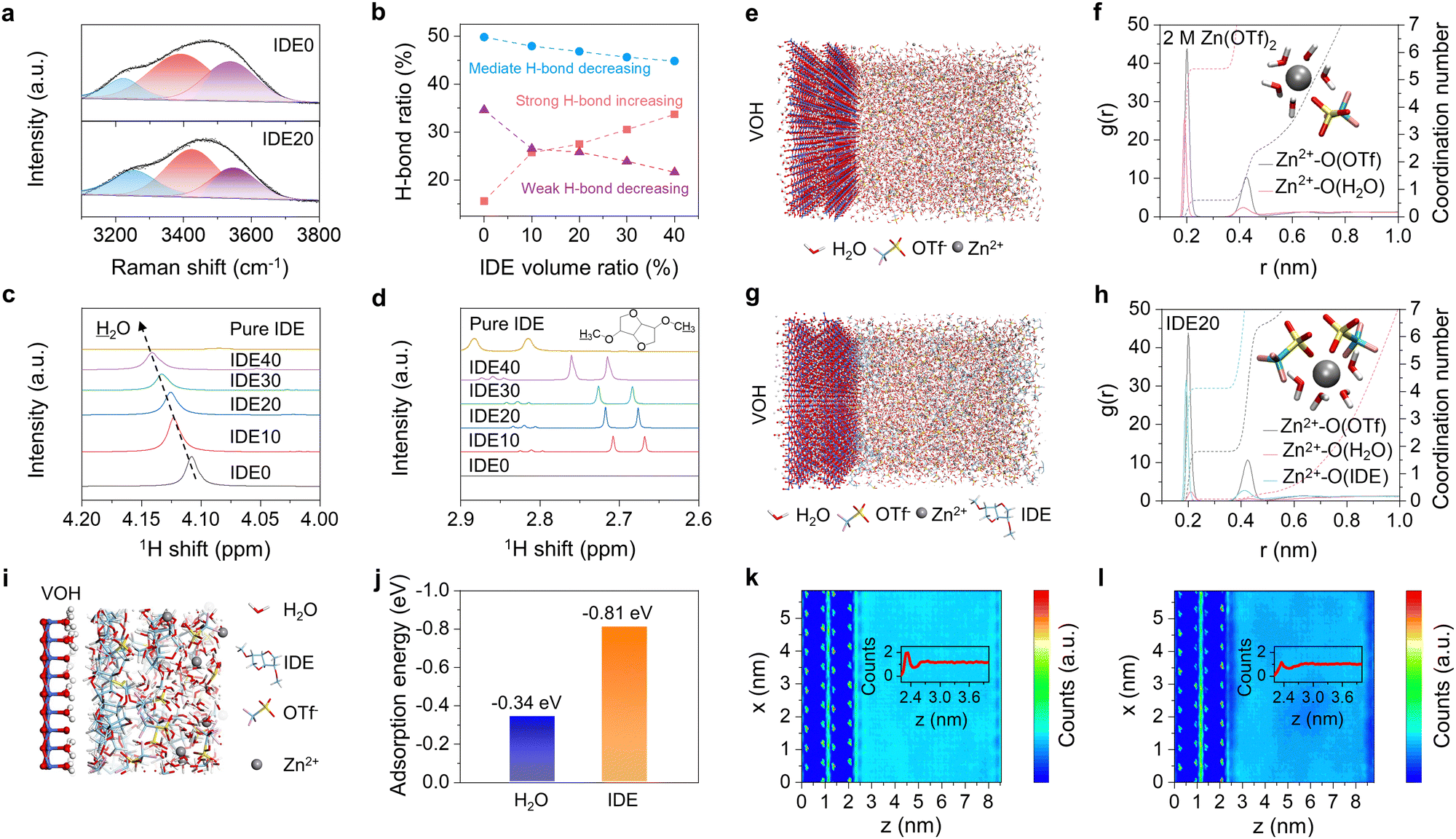 | ||
| Fig. 3 Theoretical and experimental characterizations for the impact of IDE additives on the electrolyte structure and VOH interface. (a) Fitted Raman spectra of the electrolytes in the region of O–H stretching. (b) The proportions of weak, mediate and strong H-bond in the electrolytes. (c) and (d) 1H NMR spectra of the electrolytes. The MD simulation snapshot of (e) 2 M Zn(OTf)2 and (g) IDE20 electrolytes. RDFs and corresponding coordination numbers of Zn–O bond in (f) 2 M Zn(OTf)2 and (h) IDE20 electrolytes. (i) Partial enlarged view of interface MD. (j) Adsorption energy of H2O and IDE on VOH cathode. Cross-section of the water density distribution from the surface of VOH cathode to bulk electrolytes in (k) 2 M Zn(OTf)2 and (l) IDE20 electrolytes, respectively. The inset shows the distribution of water along the z-axis at x = 3 nm. | ||
To delve deeper into the solvation structure, molecular dynamics (MD) simulations of the VOH cathode–electrolyte interface were conducted. Fig. 3e and g illustrate the geometry in MD simulations. The solvation structure is further explored using the associated radial distribution function (RDF) and coordination number (CN) distribution function. In the blank electrolyte, a prominent Zn–O(H2O) signal at approximately 0.19 nm suggests that H2O is structurally oriented around the Zn2+ in the solvation structure. The blank electrolyte features coordination of five H2O molecules and one OTf− anion with one Zn2+ (Fig. 3f). In contrast, Zn2+ solvation shell of the IDE20 electrolyte is mostly occupied by four H2O molecules, confirming that the primary ionic solvation cluster is [Zn(H2O)4]2+ (Fig. 3h). One coordinated H2O molecule is replaced by OTf−, resulting in a reduction in the amount of bound water in the solvation structure. The absence of IDE in the solvation structure of Zn2+ in the IDE20 electrolyte suggests that the IDE molecule has not entered the solvation shell. An enlarged interfacial MD diagram in Fig. 3i clearly illustrates the aggregation of IDE molecules on the cathode surface. Density functional theory (DFT) calculations provide atomic-scale insights into this interaction. The predicted adsorption energies of H2O and IDE at the VOH cathode interface are −0.34 eV and −0.81 eV, respectively, suggesting that IDE adsorbs more readily on the VOH cathode contact surface (Fig. 3j and Fig. S13, ESI†). Differential charge density diagram indicates that the electronegative O atoms in the IDE structure are more readily adsorbed on the cathode (Fig. S14, ESI†). Cross-section plots of the water density distribution from the surface of VOH cathode to bulk electrolytes show that the addition of IDE leads to the discontinuation of water at the interface (Fig. 3k, l and Fig. S15, ESI†). The two-dimensional (2D) water and IDE density plots at 0 V in Fig. S16 and S17 (ESI†) indicate that the H2O density on the VOH cathode surface decreases while original surface water sites are occupied by IDE.
Based on the aforementioned research, we can conclude that IDE preferentially adsorbs at the cathode interface, displacing the original hydrogen bonding network of water in the bulk electrolyte. IDE forms strong hydrogen bonds with water, altering the solvation structure of Zn2+, and contributing to a reduction in water activity.
To gain a deeper understanding of how the IDE inhibits the dissolution of V-based cathodes, we calculated the desolvation energy of Zn2+ on the surface of the VOH cathode using electrochemical impedance spectroscopy (EIS) at different temperatures in symmetric VOH‖VOH cells with two sets of electrolytes. After 5 cycles at 5 A g−1, cathodes from both sets of cells were disassembled, cleaned, and assembled into VOH‖VOH symmetric cells with either 2 M Zn(OTf)2 or IDE20 electrolytes. The Nyquist plots of these cells at different temperatures (Fig. 4a and b) reveal semicircles corresponding to interface resistance (Rint) and charge transfer resistance (Rct).32 These resistances were used to calculate the desolvation activation energy (Ea) using the Arrhenius equation (eqn (1)):
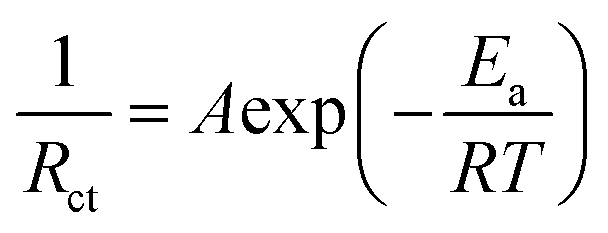 | (1) |
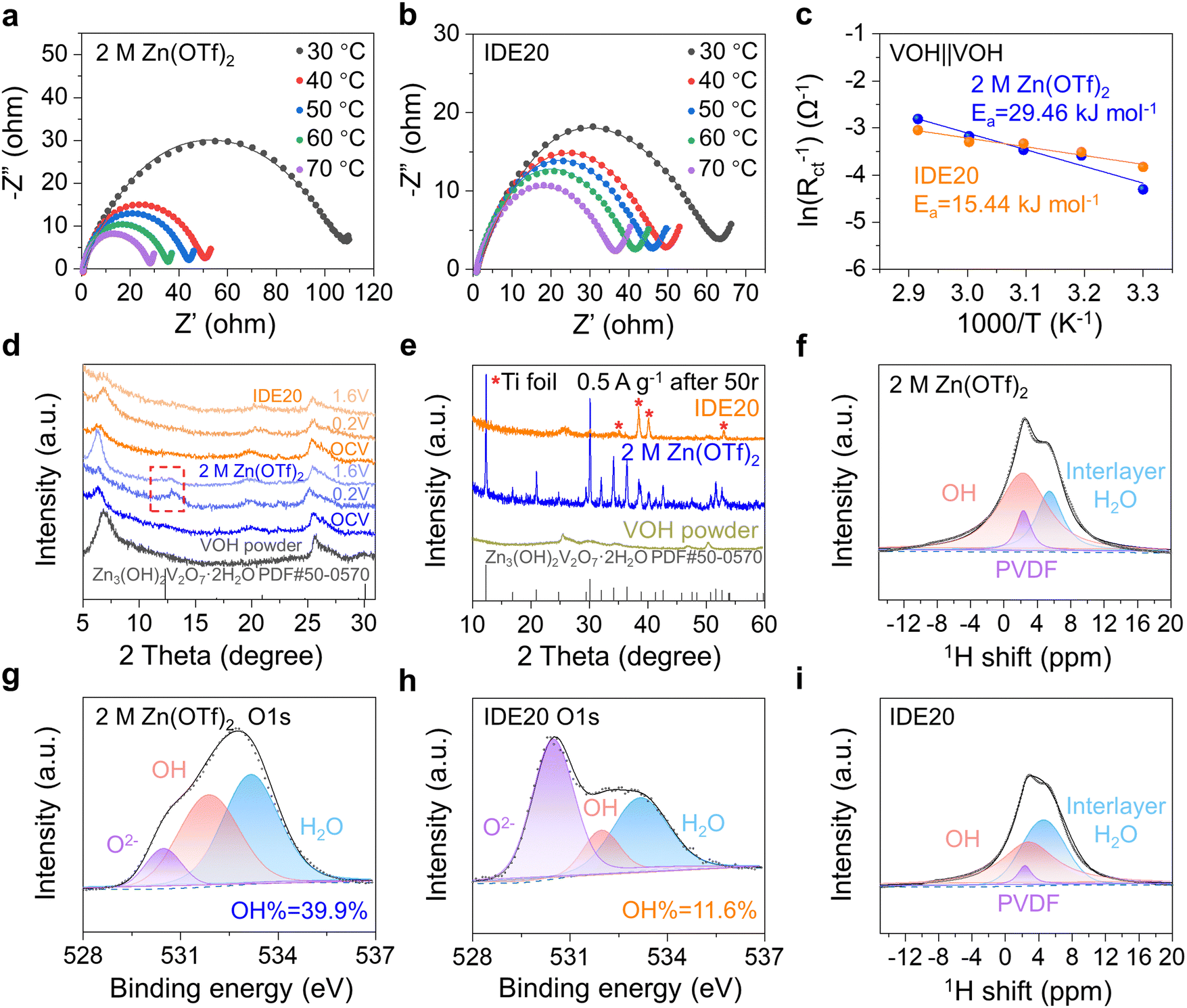 | ||
| Fig. 4 Characterization of VOH cathode interface. (a) and (b) EIS of the symmetric batteries using the cycled VOH cathodes in different electrolytes at different temperatures and (c) the desolvation activation energy that was calculated according to the Arrhenius equation. (d) Ex situ XRD patterns of the VOH cathodes in 2 M Zn(OTf)2 and IDE20 electrolytes upon the 1st discharge–charge process at selected states at 0.5 A g−1. (e) XRD patterns of VOH cathodes cycled under 2 M Zn(OTf)2 and IDE20 electrolytes after 50 cycles. 1H NMR spectra of the VOH cathodes after cycling in (f) 2 M Zn(OTf)2 and (i) IDE20 electrolytes. XPS fitted curves of the O 1s element on the VOH cathodes after cycling in (g) 2 M Zn(OTf)2 and (h) IDE20 electrolytes. | ||
X-ray diffraction (XRD) analysis was employed to investigate the impact of the two electrolytes on the cathode during charge and discharge. The results (Fig. 4d) show a clear peak increase at 12.3° in the XRD pattern, attributed to the by-product Zn3(OH)2V2O7·2H2O (PDF#50-0570) in the 2 M Zn(OTf)2 electrolyte.35 In the presence of IDE20, this peak disappears, indicating no accumulation of by-products. This is in accordance with the surface morphology of the electrodes depicted in Fig. 1d and e. Similarly, the XRD patterns of the VOH cathode in 2 M Zn(OTf)2 electrolyte reveal the presence of by-products after 50 cycles (Fig. 4e) as well as after OCV-3 days (Fig. S18, ESI†). This suggests that upon dissolution, VOH forms soluble VO2(OH)2−, which subsequently reacts with Zn2+ to produce by-products (eqn (2)).12 The addition of IDE effectively prevents the formation of these by-products.
| 2VO2(OH)2− + 3Zn2+ + 3H2O → Zn3(OH)2V2O7·2H2O + 4H+ | (2) |
1H-NMR (Fig. 4f and i) and X-ray photoelectron spectroscopy (XPS) (Fig. 4g, h and Fig. S19, ESI†) were used to analyze the cathode interface composition. The 1H-NMR spectrum revealed three components: PVDF, OH, and interlayer H2O.36 The proportion of OH peaks on the surface of the VOH cathode decreases when IDE20 is used, indicating a reduction in by-products (Fig. 4f and i). The O 1s XPS curve shows three oxygen-containing elements: O2−, OH, and H2O. In the 2 M Zn(OTf)2 electrolyte, OH% is 39.9%, while in IDE20, it is 11.6% (Fig. 4g and h). The amount of OH is proportional to the amount of by-products. Therefore, the inclusion of IDE appears to minimize the accumulation of by-products on the cathode surface. These characterizations confirm the presence of by-products on the cathode surface in the blank electrolyte and underscore the effectiveness of IDE in limiting cathode dissolution.
The IDE additives also contribute to improving the stability of Zn anodes. The researchers evaluated the cycling stability and CE of Zn‖Cu asymmetric cells using 2 M Zn(OTf)2 and IDE20 electrolytes. In a battery using the pristine Zn(OTf)2 electrolyte, it exhibited a cycle life of only 38 cycles and an average CE of 97.0% at 4 mA cm−2 and 1 mA h cm−2. However, when IDE20 was used, the battery showed remarkable improvement with a cycle life of about 2600 cycles and an average CE of 99.4%, indicating a 68-fold increase in cycle life (Fig. 5a). Zn‖Cu asymmetric cells using IDE20 electrolytes still exhibited a life of about 350 cycles at 4 mA cm−2 and 4 mA h cm−2 (Fig. S20, ESI†). We also examined the stability of galvanizing/dezincification using a Zn‖Zn symmetric cell. In the case of the symmetric cell with 2 M Zn(OTf)2 electrolyte, it failed at 17 hours, with the polarization voltage suddenly fluctuating at a current density of 4 mA cm−2 and a capacity of 1 mA h cm−2. However, when IDE was added, the symmetric cell can be cycled for 1400 hours, approximately 82 times longer than the one with the 2 M Zn(OTf)2 electrolyte (Fig. 5b and Fig. S21, ESI†).37,38 The cycling stability of Zn was further assessed under rigorous conditions (20 mA cm−2, 20 mA h cm−2). The IDE-modified symmetric cell exhibited a consistent voltage profile over 130 hours, whereas the 2 M Zn(OTf)2 electrolyte struggled to cycle under these demanding conditions (Fig. S22, ESI†).
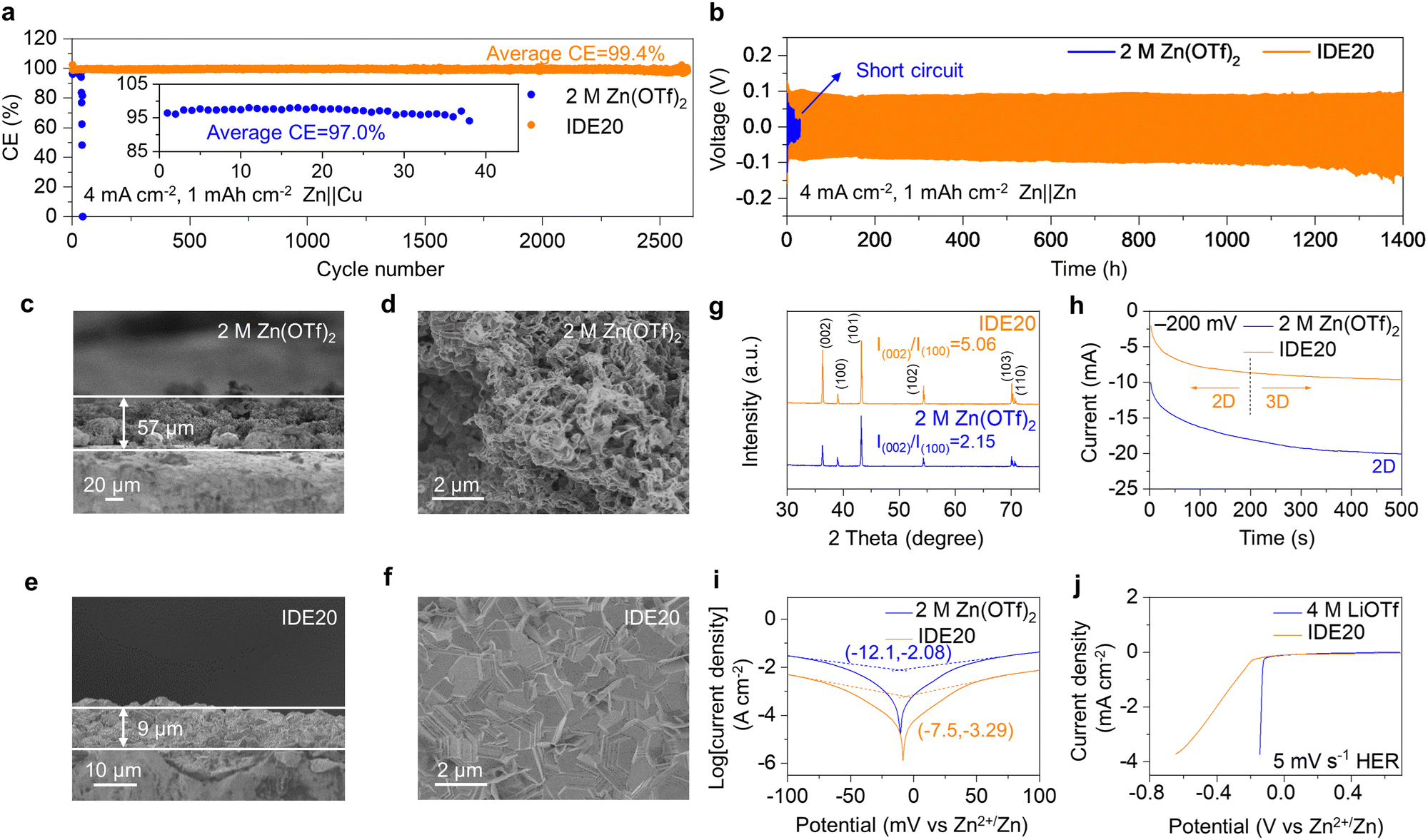 | ||
| Fig. 5 Effect of electrolyte on Zn deposition behavior and HER. (a) CE measurements of asymmetric Zn‖Cu cells with different electrolytes at 4 mA cm−2. The inset is an enlarged view of the CE of Zn‖Cu asymmetric cell using 2 M Zn(OTf)2 electrolyte. (b) Galvanostatic cycling stability of symmetric Zn‖Zn cells under 4 mA cm−2. Cross-section and surface SEM images of the deposited Zn using (c) and (d) 2 M Zn(OTf)2 and (e) and (f) IDE20 electrolytes, respectively. (g) XRD patterns of the deposited Zn electrodes on Zn substrates with an areal capacity of 4 mA h cm−2 at a current density of 4 mA cm−2. (h) Chronoamperometric curves of Zn electrodeposition in different electrolytes under −200 mV. (i) Tafel plots of Zn electrodes tested in electrolytes. (j) LSV characterization in the electrolytes. | ||
To understand the deposition behavior of Zn, SEM was used to capture cross-sectional images of 4 mA h cm−2 deposited on Zn and surface morphology of 1 mA h cm−2 deposited on Zn. The 2 M Zn(OTf)2 electrolyte led to loosely deposited Zn with dendrite formation (Fig. 5c and d). In contrast, Zn deposition in the IDE20 electrolyte was dense and uniform, with the deposited 4 mA h cm−2 Zn layer being only 9 μm thick, close to the theoretical thickness (Fig. 5e and f). The XRD results indicated a higher intensity ratio of Zn(002) to Zn(100) in the Zn deposits of the IDE20 system compared to the IDE-free electrolyte (5.06 vs. 2.15). This indicates that the Zn2+ solvation structure created by IDE favors the desired (002) crystallite orientation for Zn electrodeposition, resulting in dendrite-free behavior (Fig. 5g). Nucleation overpotential tests (Fig. S23, ESI†) demonstrated that the nucleation overpotential increased with increasing IDE concentration, suggesting that a stronger driving force was required for uniform Zn deposition, consistent with SEM results (Fig. S24, ESI†). The calculated adsorption energies of H2O and IDE at the Zn anode interface are −0.24 eV and −1.05 eV, respectively, indicating a higher affinity for IDE adsorption on the Zn anode (Fig. S25 and 26, ESI†). The differential charge density diagram reveals that the electronegative O atoms in the IDE structure exhibit a greater propensity for adsorption on the Zn anode (Fig. S27, ESI†). This interface-specific adsorption is conducive to promoting the electrocrystallization of the Zn (002) surface by regulating ion diffusion and deposition rate.2,39 Chronoamperometry (CA) tests at a constant voltage of −200 mV showed that in the presence of IDE20, Zn exhibited a consistent 3D diffusion process, maintaining a smooth surface morphology during the plating process, whereas in the 2 M Zn(OTf)2 electrolyte, Zn displayed a typical 2D diffusion mode with dendrite growth (Fig. 5h). Contact angle measurements revealed that the addition of IDE improved the electrolyte's hydrophilicity, aiding in the smooth plating behavior by facilitating fast and uniform electrolyte wetting on the Zn surface, improving the homogeneity of the Zn2+ flux (Fig. S28, ESI†). This advantageous dispersion of interfacial ionic fluxes fosters lateral growth and uniform deposition.40,41
Tafel curves were employed to investigate the effect of IDE on Zn corrosion (Fig. 5i). The presence of IDE reduced the corrosion potential and corrosion current compared to the 2 M Zn(OTf)2 electrolyte, indicating that IDE inhibited hydrogen corrosion. Linear sweep voltammetry (LSV) tests confirmed that IDE effectively inhibited parasitic reactions, reducing the response current density in the IDE20 electrolyte (Fig. 5j). The considerable reduction in cell thickness after IDE modification, even after about 2600 cycles, in comparison to the thickness after about 40 cycles of the unmodified cell, implies that IDE can effectively suppress the hydrogen evolution reaction (HER) (Fig. S29, ESI†). The reduction of the response current density in the IDE20 electrolyte in the LSV test also demonstrates the ability of IDE to inhibit oxygen evolution reaction (OER) (Fig. S30, ESI†). The inhibition of Zn corrosion and improved oxidative stability are attributed to the increased stability of water clusters, which is a result of stronger hydrogen bonding interactions formed between IDE and water. The stability of O2 adsorption corrosion was investigated by immersing Zn foils in the two electrolytes for 3 and 5 days.42,43 Optical photographs revealed that Zn electrodes immersed in the 2 M Zn(OTf)2 electrolyte exhibited petal-like corrosion products on their surfaces, whereas the surfaces of the Zn electrodes immersed in IDE20 remained smooth, indicating that IDE contributed to the inhibition of O2 adsorption corrosion (Fig. S31 and S32, ESI†).
To verify the practical application potential of the electrolyte with the addition of the IDE, full cells were assembled with VOH and ZVO as cathodes, respectively. As evidenced in Fig. 6a and b, the CV results reveal that during the initial 6 cycles, the intensity of the reduction peak and oxidation peak of the Zn|2 M Zn(OTf)2|VOH batteries changes obviously, indicating the gradual inactivation of VOH. For the Zn|IDE20|VOH batteries, all CV curves remain almost coincident, indicating the superior electrochemical stability and reversibility of VOH electrodes. The VOH and ZVO cathodes in IDE20 consistently deliver stable and higher capacities across a range of current densities, from 0.2 A g−1 to 10 A g−1. Moreover, they display highly reversible capacities when the current density returns to 0.5 A g−1. (Fig. 6c, f and Fig. S33, ESI†)
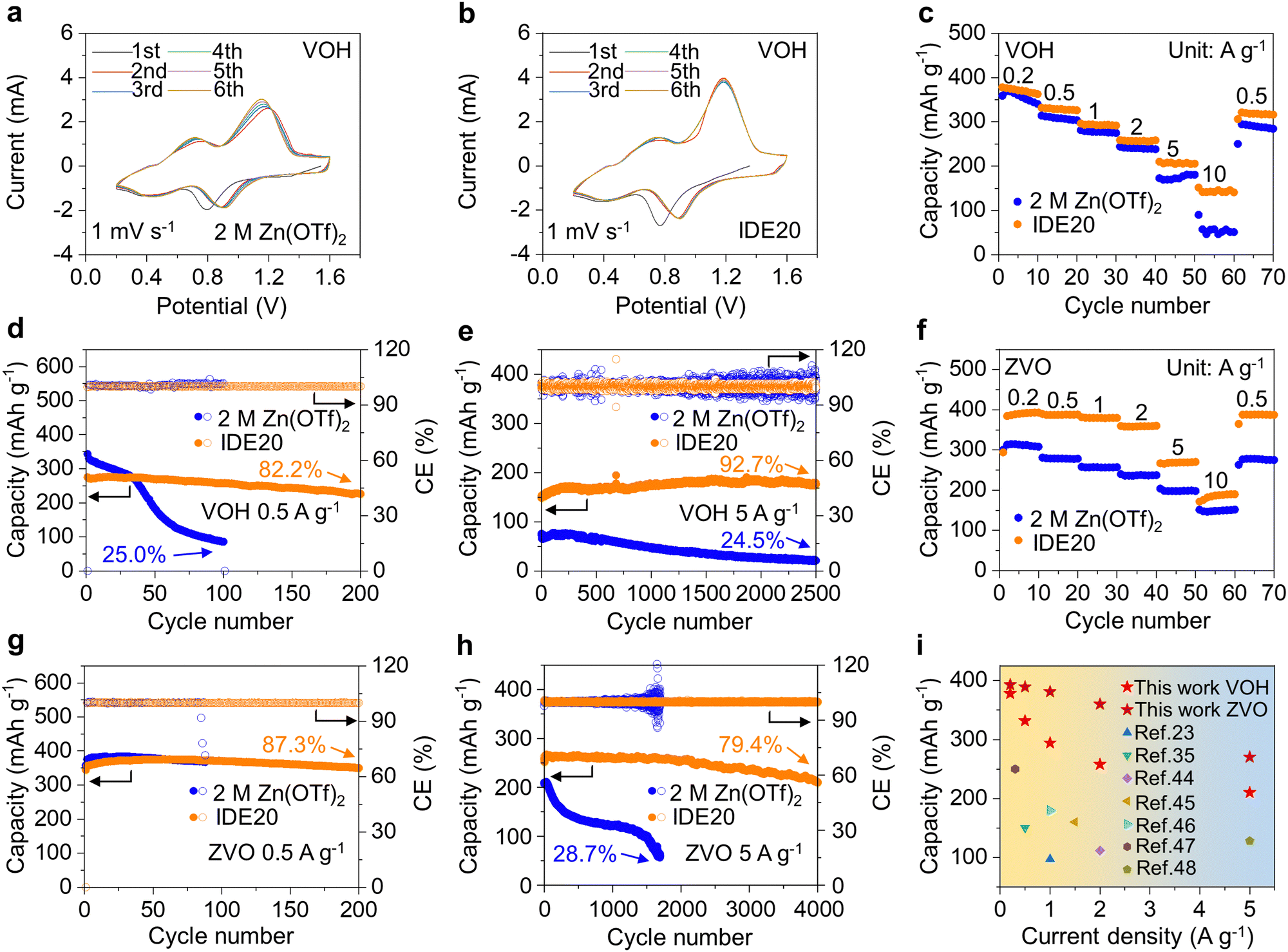 | ||
| Fig. 6 Electrochemical performance of Zn‖VOH (a)–(e) and Zn‖ZVO (f)–(h) cells. (a) and (b) The initial six CV cycles at a scan rate of 1 mV s−1. (c) and (f) Rate performance. (d) and (g) Cyclic stability and CE at a specific current of 0.5 A g−1. (e) and (h) Cyclic stability and CE at a specific current of 5 A g−1. (i) Comparison of the rate performance of Zn‖VOH and Zn‖ZVO full cells with previous reports. | ||
For the VOH electrode in IDE20 electrolyte, it offers a maximum capacity of 276.3 mA h g−1 at a current density of 0.5 A g−1 (Fig. 6d and Fig. S34, ESI†) and 190.3 mA h g−1 at a current density of 5 A g−1, lasting for more than 2500 cycles (Fig. 6e). Similarly, the ZVO electrode in IDE20 electrolyte exhibits a maximum capacity of 375.4 mA h g−1 at a current density of 0.5 A g−1 (Fig. 6g and Fig. S35, ESI†) and 266.6 mA h g−1 at a current density of 5 A g−1, lasting for more than 4000 cycles (Fig. 6h). When compared to previously published efforts on electrolyte alteration for AZIBs, this work stands out for obtaining improved rate performance (Fig. 6i and Table S2, ESI†).23,35,44–48 To meet commercial grade requirements, a VOH cathode with a mass loading of 19.39 mg cm−2 was synthesized, and cells using the IDE20 electrolyte exhibited excellent stability with CE >99.1% and slow capacity decay over 400 cycles (Fig. S36 and S37, ESI†).
It becomes evident that 2 M Zn(OTf)2 electrolytes have a detrimental impact on the electrochemical performance of V-based cathodes. These electrolytes lead to severe dissolution and irreversible structural changes, resulting in continuous loss of active material and structural disintegration. On the other hand, the IDE20 electrolyte functions through interfacial water masking. IDE molecules are preferentially adsorbed on the cathode surface to inhibit water penetration. Simultaneously, IDE reduces the presence of free water and bound water in the Zn2+ solvation structure by forming strong hydrogen bonds with water molecules. This unique combination of these two factors effectively suppresses water activity and, consequently, the cathode's dissolution, even at different current densities. As a result, the IDE20 electrolyte offers the promise of enabling long-lasting and durable AZIBs.
Conclusions
In conclusion, this study introduces an innovative approach for interfacial water masking aimed at preventing cathodic dissolution in AZIBs. The addition of the IWMA IDE disrupts the original network of hydrogen bonds between water molecules in the electrolyte. IDE molecules then form strong hydrogen bonds with water, reducing the presence of free water molecules in the electrolyte and decreasing the amount of bound water in the Zn2+ solvation structure. These combined effects effectively inhibit water activity within the system. Furthermore, IDE molecules not only reduce water activity but also adsorb onto the cathode surface. Together, these mechanisms create two protective barriers that prevent polar water molecules from damaging the crystal structure of the cathode materials. Meanwhile, substantial spatial resistance and symmetrical charge distribution of IDE prevent it from participating in the solvation structure of Zn2+, lowering the desolvation energy of hydrated Zn2+ at the cathode–electrolyte interface and improving the battery's rate performance.In terms of electrochemical performance, VOH stands out as the most promising cathode material among the different electrolyte formulations. It exhibits excellent capacity retention, retaining 82.2% of its capacity after 200 cycles at a low current density of 0.5 A g−1 and reaches a maximum capacity of 190.3 mA h g−1 at a current density of 5 A g−1, with an impressive 92.7% capacity retention after 2500 cycles. Similarly, the ZVO electrode in the IDE20 electrolyte demonstrates remarkable stability, with 87.3% capacity retention after 200 cycles at a current density of 0.5 A g−1, and up to 266.6 mA h g−1 at a current density of 5 A g−1, sustaining this performance for more than 4000 cycles. This IWMA method presents an innovative strategy for electrolyte design and holds significant promise for the development of long-lasting and durable AZIBs.
Author contributions
Conceptualization: W. Z., Z. S. and Y. L. Experimental design and investigation: W. Z. and Z. S. Data analyses: W. Z., Z. S., H. C., J. M., S. Z., Y. K. and Y. L. NMR characterization: W. Z. Calculation: J. M. and H. C. Writing – original draft: W. Z. Writing – review & editing: W. Z., Z. S., H. C., Y. K. and Y. L.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors extend their gratitude to Shiyanjia Lab for providing invaluable assistance with the XRD, ICP-OES and XPS analysis. We thank N. Zheng at State Key Laboratory of Chemical Engineering in Zhejiang University for performing SEM. This work is supported by the National Key R&D Program of China (2018YFA0209600), the National Natural Science Foundation of China (22022813, 21878268), the Zhejiang Provincial Natural Science Foundation of China (LQ24B030002), the China Postdoctoral Science Foundation (2022M722729, 2023T160571) and the Technology Project of Institute of Wenzhou (XMGL-CX-202204, XMGL-KJZX-202208).Notes and references
- W. Zhong, J. Zhang, Z. Li, Z. Shen, S. Zhang, X. Wang and Y. Lu, Green Chem. Eng., 2023, 4, 264–284 CrossRef.
- Z. Shen, J. Mao, G. Yu, W. Zhang, S. Mao, W. Zhong, H. Cheng, J. Guo, J. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218452 CrossRef CAS PubMed.
- Q. Yao, F. Xiao, C. Lin, P. Xiong, W. Lai, J. Zhang, H. Xue, X. Sun, M. Wei, Q. Qian, L. Zeng and Q. Chen, Battery Energy, 2023, 2, 20220065 CrossRef CAS.
- X. Wang, Z. Zhang, B. Xi, W. Chen, Y. Jia, J. Feng and S. Xiong, ACS Nano, 2021, 15, 9244–9272 CrossRef CAS PubMed.
- D.-S. Liu, Y. Zhang, S. Liu, L. Wei, S. You, D. Chen, M. Ye, Y. Yang, X. Rui, Y. Qin and C. C. Li, Adv. Funct. Mater., 2022, 32, 2111714 CrossRef CAS.
- W. Li, C. Han, Q. Gu, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2020, 10, 2001852 CrossRef CAS.
- B. Fei, Z. Liu, J. Fu, X. Guo, K. Li, C. Zhang, X. Yang, D. Cai, J. Liu and H. Zhan, Adv. Funct. Mater., 2023, 33, 2215170 CrossRef CAS.
- J. Yue, L. Lin, L. Jiang, Q. Zhang, Y. Tong, L. Suo, Y.-S. Hu, H. Li, X. Huang and L. Chen, Adv. Energy Mater., 2020, 10, 2000665 CrossRef CAS.
- S. Liu, J. He, D.-S. Liu, M. Ye, Y. Zhang, Y. Qin and C. C. Li, Energy Storage Mater., 2022, 49, 93–101 CrossRef.
- Z. Xing, G. Xu, X. Xie, M. Chen, B. Lu, J. Zhou and S. Liang, Nano Energy, 2021, 90, 106621 CrossRef CAS.
- L. Ding, L. Wang, J. Gao, T. Yan, H. Li, J. Mao, F. Song, S. Fedotov, L.-Y. Chang, N. Li, Y. Su, T. Liu and L. Zhang, Adv. Funct. Mater., 2023, 33, 2301648 CrossRef CAS.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- L. Zhang, J. Hu, B. Zhang, J. Liu, H. Wan, L. Miao and J. Jiang, J. Mater. Chem. A, 2021, 9, 7631–7639 RSC.
- W. Yang, Y. Yang, H. Yang and H. Zhou, ACS Energy Lett., 2022, 7, 2515–2530 CrossRef CAS.
- D. Xu, H. Wang, F. Li, Z. Guan, R. Wang, B. He, Y. Gong and X. Hu, Adv. Mater. Interfaces, 2019, 6, 1801506 CrossRef.
- J. Guo, J. Ming, Y. Lei, W. Zhang, C. Xia, Y. Cui and H. N. Alshareef, ACS Energy Lett., 2019, 4, 2776–2781 CrossRef CAS.
- B. Shuai, C. Zhou, Y. Pi and X. Xu, ACS Appl. Energy Mater., 2022, 5, 6139–6145 CrossRef CAS.
- L. Zhang, X. Qin, L. Wang, Z. Zhao, L. Mi and Q. Lu, Front. Chem. Sci. Eng., 2023, 17, 1244–1253 CrossRef CAS.
- H. Liu, L. Jiang, B. Cao, H. Du, H. Lu, Y. Ma, H. Wang, H. Guo, Q. Huang, B. Xu and S. Guo, ACS Nano, 2022, 16, 14539–14548 CrossRef CAS PubMed.
- D. Bin, Y. R. Wang, A. G. Tamirat, P. Zhu, B. B. Yang, J. Wang, J. H. Huang and Y. Y. Xia, ACS Sustainable Chem. Eng., 2021, 9, 3223–3231 CrossRef CAS.
- A. Clarisza, H. K. Bezabh, S. K. Jiang, C. J. Huang, B. W. Olbasa, S. H. Wu, W. N. Su and B. J. Hwang, ACS Appl. Mater. Interfaces, 2022, 14, 36644–36655 CrossRef CAS PubMed.
- Y. Zhu, J. Yin, X. Zheng, A.-H. Emwas, Y. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC.
- T. Li, Y. Lim, X. Li, S. Luo, C. Lin, D. Fang, S. Xia, Y. Wang and H. Yang, Adv. Energy Mater., 2022, 12, 2103231 CrossRef CAS.
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song, Y. Cui, J. Liu, Z. Wang, S. Li, Q. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- L. Miao, R. Wang, S. Di, Z. Qian, L. Zhang, W. Xin, M. Liu, Z. Zhu, S. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9678 CrossRef CAS PubMed.
- T. Zhang, J. Yang, H. Wang, H. Yu, Q. Li, L. Chen, Y. Chen and T. Wang, Energy Storage Mater., 2024, 65, 103085 CrossRef.
- M. Li, X. Feng, J. Yin, T. Cui, F. Li, J. Chen, Y. Lin, X. Xu, S. Ding and J. Wang, J. Mater. Chem. A, 2023, 11, 25545–25554 RSC.
- L. Zhang, B. Zhang, J. Hu, J. Liu, L. Miao and J. Jiang, Small Methods, 2021, 5, 2100094 CrossRef CAS PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- X. Zhang, F. Xue, X. Sun, T. Hou, Z. Xu, Y. Na, Q. An, Z. Chen, S. Cai and C. Zheng, Chem. Eng. J., 2022, 445, 136714 CrossRef CAS.
- Q. Zhang, K. Xia, Y. Ma, Y. Lu, L. Li, J. Liang, S. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS.
- X. Wang, S. Li, W. Zhang, D. Wang, Z. Shen, J. Zheng, H. L. Zhuang, Y. He and Y. Lu, Nano Energy, 2021, 89, 106353 CrossRef CAS.
- P. Lin, J. Cong, J. Li, M. Zhang, P. Lai, J. Zeng, Y. Yang and J. Zhao, Energy Storage Mater., 2022, 49, 172–180 CrossRef.
- S. Zhang, M. Ye, Y. Zhang, Y. Tang, X. Liu and C. C. Li, Adv. Funct. Mater., 2023, 33, 2208230 CrossRef CAS.
- Y. Mei, Y. Liu, W. Xu, M. Zhang, Y. Dong and J. Qiu, Chem. Eng. J., 2023, 452, 139574 CrossRef CAS.
- G. Yu, Y. Zhou, R. Yang, M. Wang, L. Shen, Y. Li, N. Xue, X. Guo, W. Ding and L. Peng, J. Phys. Chem. C, 2015, 119, 12325–12334 CrossRef CAS.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- C. Wang, X. Zeng, J. Qu, J. M. Cairney, Q. Meng, P. J. Cullen and Z. Pei, Matter, 2023, 6, 3993–4012 CrossRef CAS.
- T. Wei, H. Zhang, Y. Ren, L. E. Mo, Y. He, P. Tan, Y. Huang, Z. Li, D. Zhu and L. Hu, Adv. Funct. Mater., 2023, 2312506 CrossRef.
- Z. Huang, Z. Li, Y. Wang, J. Cong, X. Wu, X. Song, Y. Ma, H. Xiang and Y. Huang, ACS Energy Lett., 2023, 8, 372–380 CrossRef CAS.
- W. Zhuang, Q. Chen, Z. Hou, Z. Sun, T. Zhang, J. Wan and L. Huang, Small, 2023, 19, 2300274 CrossRef CAS PubMed.
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2022, 34, 2206963 CrossRef CAS PubMed.
- L. Su, L. Liu, B. Liu, J. Meng and X. Yan, iScience, 2020, 23, 100995 CrossRef CAS PubMed.
- J. Zhang, P. Li, Y. Wang, Z. Zhao and Z. Peng, Adv. Funct. Mater., 2023, 33, 2305804 CrossRef CAS.
- Y. Wang, T. Wang, S. Bu, J. Zhu, Y. Wang, R. Zhang, H. Hong, W. Zhang, J. Fan and C. Zhi, Nat. Commun., 2023, 14, 1828 CrossRef CAS PubMed.
- W. Zhang, M. Dong, K. Jiang, D. Yang, X. Tan, S. Zhai, R. Feng, N. Chen, G. King, H. Zhang, H. Zeng, H. Li, M. Antonietti and Z. Li, Nat. Commun., 2022, 13, 5348 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, W. K. Pang, W. Lie, J. A. Yuwono, G. Liang, S. Liu, A. M. D. Angelo, J. Deng, Y. Fan, K. Davey, B. Li and Z. Guo, Nat. Commun., 2023, 14, 2720 CrossRef CAS PubMed.
- H. Yu, D. Chen, Q. Li, C. Yan, Z. Jiang, L. Zhou, W. Wei, J. Ma, X. Ji, Y. Chen and L. Chen, Adv. Energy Mater., 2023, 13, 2300550 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04208a |
| ‡ The authors contribute to the work equally. |
| This journal is © The Royal Society of Chemistry 2024 |