Highly stable manganese oxide cathode material enabled by Grotthuss topochemistry for aqueous zinc ion batteries†
Received
2nd December 2023
, Accepted 15th January 2024
First published on 16th January 2024
Abstract
The design and synthesis of manganese oxide-based materials with high-rate performance and long cycle life is a major challenge for aqueous zinc-ion batteries (AZIBs). This research reports the presence of a synergistic collaboration between vacancies, lattice water and nickel ions on enhancing the hydrated protons hopping via the Grotthuss mechanism for high-performance zinc ion batteries. The Grotthuss mechanism allows for the efficient transfer of a proton charge without the actual movement of the molecule over long distances, resulting in high ionic conductivity. NiMn3O7·3H2O achieves a capacity of 318 mA h g−1 under 200 mA g−1 and 121 mA h g−1 under 5 A g−1 with a retention of 91% after 4000 cycles. The relationship between the remarkable performance and Grotthuss topochemistry is investigated using techniques including synchrotron X-ray absorption spectroscopy and density functional theory. Protons prefer to bond with O2− ions on the Mn–O layer, and proton transfer is favoured in the presence of vacancies. The continuous hopping of protons within the host material induces periodic, temporary local structural changes in the lattice. This dynamic behaviour alters the energy barriers for ions intercalation and deintercalation. Nickel ions facilitate the ongoing mobility of hydrated protons via Grotthuss hopping by preserving the system's electrical neutrality, which counterbalances the dynamic changes caused by proton migration. This study provides insight into the Grotthuss conduction mechanism for the development of high-performance cathode materials in AZIBs.
Broader context
As an electrochemical energy storage system, zinc-ion batteries have attracted significant attention, benefiting from the low cost and abundant reserves of zinc. However, the practical utility of zinc-ion batteries is restricted by the limited performance of the cathode materials. Manganese oxide cathode materials have been extensively researched because of their comparatively high discharge plateau and satisfactory theoretical capacity. Although significant efforts have been undertaken to enhance the performance (lifespan and capacity) of manganese oxides, understanding the energy storage mechanism remains constrained. Notably, the hydrated proton collaborates with the zinc cation to complete the intercalation/deintercalation process, hence enhancing the battery's capacity. However, the mechanism of hydrated protons remains insufficiently understood. Here, we uncover the presence of a synergistic collaboration between vacancies and lattice water on the Grotthuss mechanism of hydrated protons. Experiments and computational studies have revealed that vacancies in the lattice serve as a driving force for proton hopping, while water molecules act as conduits for proton hopping during the Grotthuss process. Hydrated protons exhibit a tendency to migrate via vacancy sites with reduced barriers. This would enable the hydrated proton to move through the periodic variation of vacancies. Moreover, the stability of the cathode material is improved because nickel cations preserve the system's electrical neutrality, which counterbalances the dynamic changes caused by charge carriers’ migration in the lattice. We use a controllable hydrothermal approach to generate abundant vacancies and improve the diffusion mechanism. As a result, the cathode material NiMn3O7 that has been developed demonstrates an improved energy storage mechanism.
|
Introduction
Driven by the practical need for safety and cost-effectiveness, aqueous zinc-ion batteries (AZIBs) have recently gained significant attention. MnO2 has attracted extensive research due to its relatively high discharge plateau and lifespan. However, the development of manganese oxide cathode materials is hindered by their unsatisfactory lifespan at low current densities, which is attributed to the bivalency of the Zn2+ cation, i.e., the strong electrostatic interaction between the Zn2+ cations and the host frameworks slows down its transport. The high desolvation energy of Zn2+ at the electrode–electrolyte interface, compared to its monovalent counterpart, imposes an extra energy penalty on its intercalation in manganese oxide. The relatively strong electrostatic repulsion between the charge carrier and manganese oxide during intercalation and deintercalation also limits ion transfer in AZIBs. Thus, enhancing the kinetics of charge carriers within manganese oxide materials is a crucial aspect of AZIBs cathode research.1,2
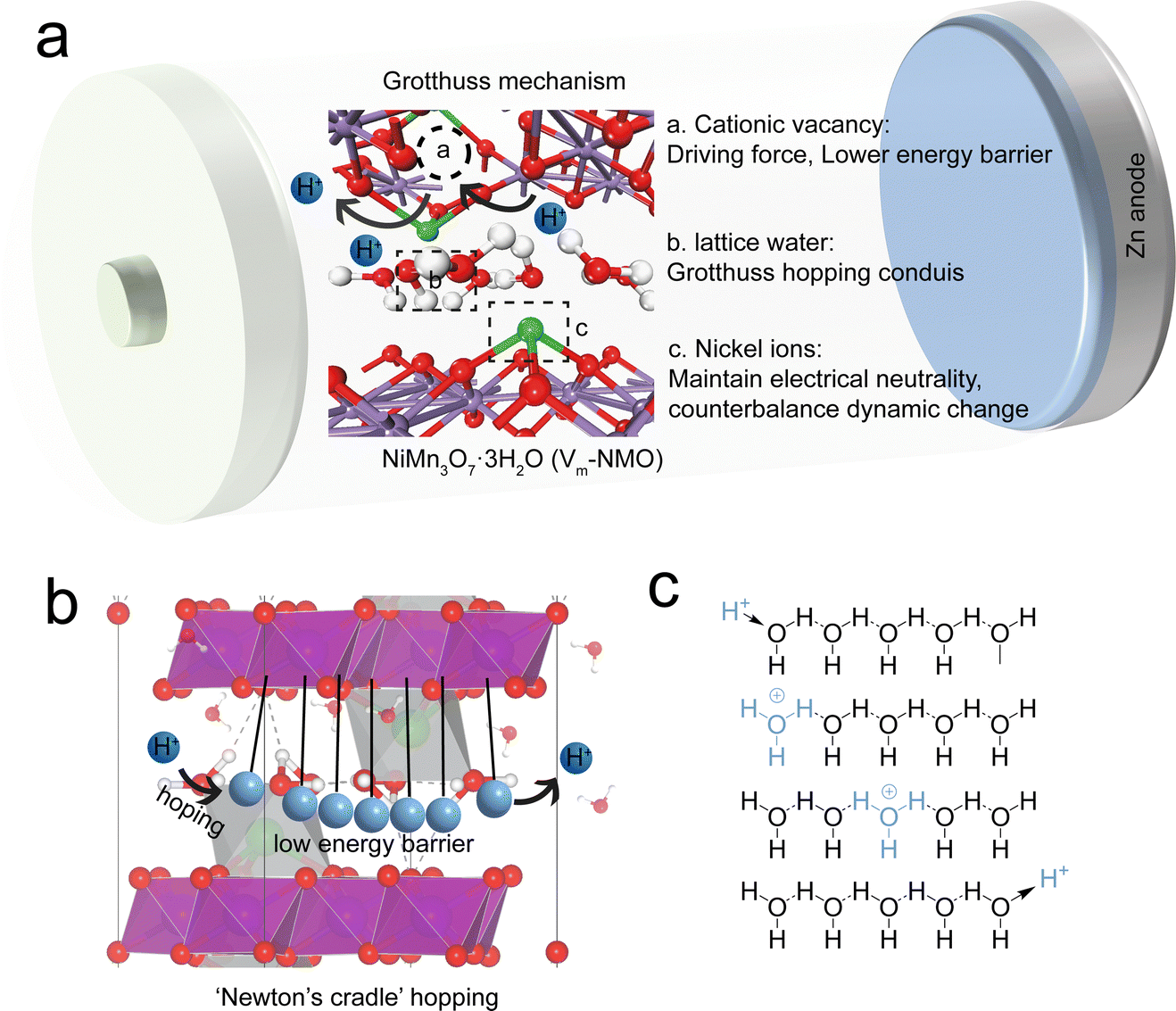
According to several prior studies, the Grotthuss mechanism is considered to be an efficient conductivity process because it implies the collective chain-like proton transfer, where the net charge, in principle, can be transported faster than a proton itself.3–7 The Grotthuss mechanism provides more efficient proton transport because it does not require the mobility of entire molecules. The synergy between defect engineering and the Grotthuss mechanism could enhance the charge carrier kinetics (Fig. 1(a)). A recent study indicates that Grotthuss conduction can occur in defective Prussian blue analogues.8 In this mechanism, a hydrogen atom hops between two hydrogen-bonded water molecules, displacing one of the molecule's existing H atoms and causing a cascade of identical displacements along the hydrogen-bonding network.9 This motion is akin to Newton's cradle (Fig. 1(b)), with correlated local displacements leading to long-range transport of protons.10,11 This allows for the fast, efficient transfer of protons without the need for a proton to travel the entire structure solely. The continuous hopping of protons across the host material can induce periodic but temporary, local structural changes in the lattice, simultaneously changing the energy barriers for zinc ion intercalation and deintercalation in the lattice, which optimise the diffusion pathways for zinc ions. Furthermore, the presence of manganese vacancies in the lattice not only serves as the low energy barrier pathways for proton hopping but also can serve as additional sites for both proton and zinc ion intercalation and increase the capacity of the battery. Defect engineering has been proven to be a facile strategy to modify the electronic structures of host materials and alter the energy barrier for charge carrier transfer.12–16 The most prevalent cation vacancy in manganese oxides is Mn vacancy. By comparing the density of states of manganese oxide with and without vacancies, it has been proposed that introducing Mn vacancies into the manganese oxide framework can lead to an increase in the charge density at the Fermi level and can result in a lower surface energy barrier.17 Furthermore, unlike the homogeneous charge distribution in typical manganese oxide, the electrons in defective manganese oxide congregate near the Mn vacancy. Defect-induced local structural changes and charge redistribution can facilitate charge transfer and accommodate the volume changes associated with ion intercalation.
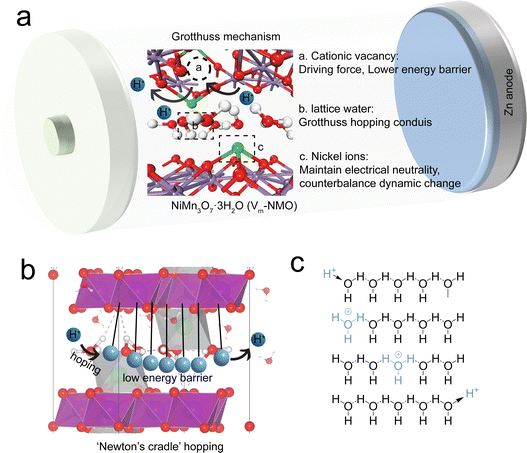 |
| | Fig. 1 (a) Hopping Grotthuss mechanism for proton conductivity. (b) Newton's cradle illustration of proton hopping Grotthuss mechanism in NiMn3O7 lattice. (c) Illustration of hydrated proton hopping via Grotthuss mechanism. | |
A strategic approach needs to be developed for achieving the Grotthuss mechanism in manganese oxide-based AZIBs. This approach should focus on two key aspects: (i) constructing a continuous hydrogen bond network in lattice to facilitate the proton migration from the electrolyte to cathode material interlayer space and (ii) mitigating the energy barrier for charge transfer within the host cathode material via defect engineering.18–20 By effectively addressing these challenges, the efficient operation of AZIBs through the Grotthuss conduction mechanism can be realised, leading to improved energy storage performances.
This study presents a unique strategy to enhance the rate performance and long-term cycling stability of aqueous zinc ion batteries by utilising the Grotthuss mechanism in manganese oxide. A facile hydrothermal synthesis process was employed to prepare Ernie nickelite NiMn3O7. We found that the specific peak intensity and lattice structure in NiMn3O7 could be tailored by altering the synthesis conditions. The optimisation led to the formation of vacancies in the lattice. For convenience, the resulting samples are ascribed as Vm-NMO (with vacancy) and NMO (without vacancy). It was demonstrated that Vm-NMO can be used as highly reversible and high-capacity cathode materials for aqueous zinc ion batteries. The prepared cathode material Vm-NMO, containing abundant cation vacancies, exhibits an enhanced ion transfer and a high capacity of 318 mA h g−1 at a current density of 200 mA g−1, along with a stable capacity of 121 mA h g−1 at 5 A g−1 and a capacity retention of 91% over 4000 cycles was achieved. Furthermore, the energy storage mechanism of the Vm-NMO cathode material was systematically investigated by combining synchrotron X-ray absorption spectroscopy with density functional theory (DFT) simulations. In aqueous electrolytes, protons react with water molecules, forming hydronium ions (H3O+). The numerous lattice water molecules within the host material create a continuous hydrogen bond network, which opens corridors for the migration of abundant water molecules from the electrolyte to the interlayer spaces in the layered manganese oxides. Our results indicate that protons prefer to bond with O2− ions on the Mn–O layer rather than staying with water molecules. Additionally, energy barrier calculations for proton transfer with and without Mn4+ vacancies indicate that proton transfer is favoured in the presence of defects. Mn4+ vacancies are the driving force for proton hopping in the Vm-NMO systems. The continuous hopping of protons within the host material induces periodic, temporary local structural changes in the lattice. This dynamic behaviour alters the energy barriers for ion intercalation and deintercalation, optimising the diffusion pathways for zinc ions. Moreover, the presence of manganese vacancies in the lattice serves as low-energy barrier pathways for proton hopping and additional sites for proton and zinc ion intercalation, resulting in an increased battery capacity. This dynamic behaviour raises an interesting point regarding the Grotthuss-like mechanism on the Mn–O layer in Vm-NMO.
Results and discussion
Characterisation of the Vm-NMO and NMO cathode material structure
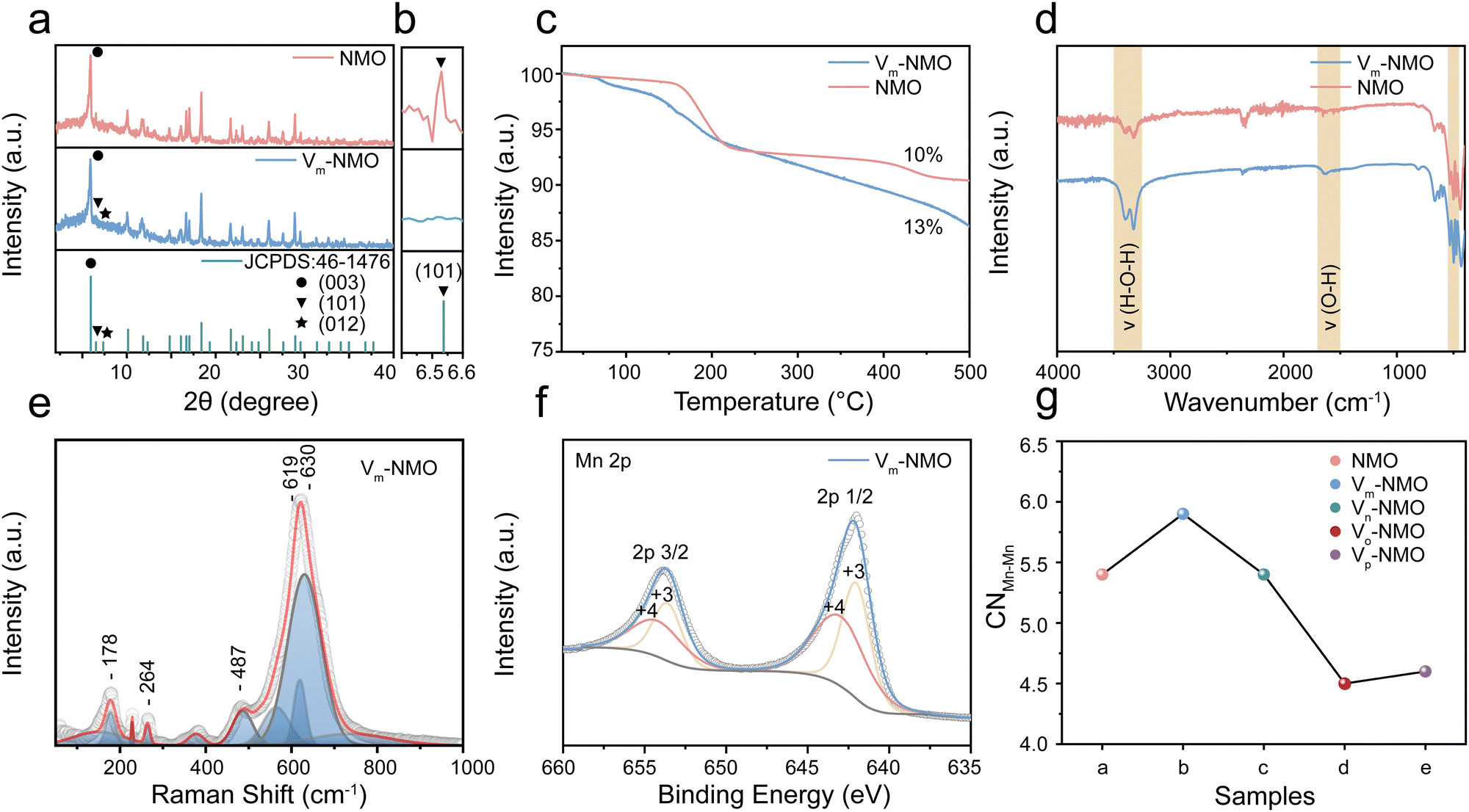
NiMn3O7 nanoparticles were synthesised using a facile hydrothermal synthesis method, and the Vm-NMO nanoparticle with vacancies was synthesised by optimising the synthesis parameter.21 Detailed synthesis process is included in ESI.† To examine the impact of the synthesis parameter on the crystal structure. We conducted a series of experiments to investigate by varying the amount of synthesis precursors and temperature. The effects of different NH4Cl adding amounts were examined in ESI,† Fig. S1. The synthesis experiment indicates that the optimal NH4Cl concentration for promoting NiMn3O7 formation is 0.04 mol L−1. Concentrations exceeding 0.4 mol L−1 result in a phase transition from NiMn3O7 to alpha-MnO2. The impact of varied synthesis temperatures was studied in ESI,† Fig. S2. The XRD results show that the formation of vacancies in NiMn3O7 is highly sensitive to the temperature. The optimized NiMn3O7 synthesis temperature is 200 °C, lower temperature than 180 °C can lead to mixed phase products while temperature higher than 260 °C can lead to the product phase transition from NiMn3O7 to NiMn2O4. The X-ray diffraction (XRD) pattern of the as-prepared Vm-NMO and NMO (Fig. 2(a) and (b)) matched well with Ernie nickelite NiMn3O7·3H2O (JCPDS: 46-1476), which belongs to hexagonal R*(148) space group and has corresponding lattice constants of a = 7.5293(4) Å and c = 20.752(1) Å.22 Defects and vacancies in the crystal structure of the material can alter the XRD pattern by modifying the spacing between the crystal planes. The main difference between Vm-NMO and NMO lies in (101), (012) and (003) crystal planes. The significant absence of (101) and (012) in the Vm-NMO lattice indicates lattice changes in the crystal structure, which can be due to preferred orientation or preferred crystal growth. Additionally, the intensity of the (003) lattice plane is significantly reduced, indicating disrupted long-range order of Vm-NMO and increased disorder of the lattice. The Mn atoms, which are octahedrally coordinated by six oxygen atoms, and form MnO6 octahedra that connect along common edges to create a two-dimensional layer. The Mn3O7 layer is formed by periodically removing 1/7 of the Mn atoms in a triangular lattice, resulting in a maple-leaf-lattice (MLL) structure consisting of Mn4+ ions. The Mn3O7 layers are separated by a nonmagnetic block layer composed of Ni2+ ions located above and below the Mn-vacant sites of the Mn3O7 layer, as well as lattice water molecules. The Mn–Mn distance between the layers (6.8 Å) is much greater than the layer thickness (2.8 Å), providing ample room for ion diffusion within the crystal lattice and helping to accommodate the volume changes by reducing the strain that occurs during ion intercalation and extraction process.23 The spacious layered framework also permits the insertion of water molecules from the electrolyte into the interlayer space.24 The presence of crystal water in the lattice was confirmed by thermogravimetric analysis measurement (TGA), which showed that Vm-NMO contains 13% lattice water (Fig. 2(c)), consistent with its chemical formula. In comparison, NMO lost 10% of weight during TGA testing. Previous research confirmed that the lattice water molecules are located at the layered manganese oxide interlayer spaces.25–28 TGA results indicate that Vm-NMO has a higher crystal water content, which could contribute to building a continuous hydrogen bond network and screening the electrostatic interaction between charge carriers and the host framework.29 The chemical vibrational modes in the as-prepared materials were confirmed by Fourier transform infrared spectroscopy (FTIR). A series of absorption peaks in Fig. 2(d) could be assigned to different metal oxide vibration modes of Mn–O (530–570 cm−1, 600–620 cm−1), Ni–O (480–490 cm−1) and the vibration of O–H, H–O–H (3200–3400 cm−1) in crystal water, respectively, which are in line with previous research.30–32 The intensity of the water vibration signals in Vm-NMO is stronger than that in NMO, indicating that Vm-NMO contains more lattice water. The contact angle measurement of the Vm-NMO material is 83°, which is smaller than the contact angle of NMO (122°). The difference in contact angle suggests that the Vm-NMO material has better wettability (ESI,† Fig. S3). Raman spectroscopy was utilised to examine the intricate structural distinctions between Vm-NMO and NMO, employing an excitation wavelength of 532 nm. The Vm-NMO Raman profile showed three distinct bands in the range of 450 to 700 cm−1 (Fig. 2(e)): two obvious Raman features at ∼490 and ∼620–640 cm−1 assigned to out-of-plane Mn–O vibrations perpendicular to the layers and the band around 570 cm−1 correspond to in-plane Mn–O stretching vibration along with the octahedral layers in Vm-NMO. Raman features of NMO with different vibrational modes were compared to Vm-NMO (ESI,† Fig. S4). A dominant in-plane Mn–O stretching vibration was observed at 571 cm−1, and an out-of-plane Mn–O vibration was found at 621 cm−1. These in-plane and out-of-plane bands are sensitive markers related to the changes in the coordination states of octahedral Mn cations and structural distortions of the related Mn–O environments.20,33,34 The valence of the as-prepared cathode materials was investigated by X-ray photoelectron spectroscopy (XPS). The Mn 2p XPS profiles shown in Fig. 2(f) and ESI,† Fig. S5 exhibit two distinct peaks in both Vm-NMO and NMO materials, illustrating the co-existence of Mn4+ and Mn3+. We found that the ratio of Ni/Mn in Vm-NMO is higher than that of the NMO material. As shown in ESI,† Fig. S6 and S7, the normalised ratios of Ni, Mn, and O in Vm-NMO is about 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13:33 while the ratio in NMO is 1:13:34. This difference can be attributed to the presence of cation vacancies in the Vm-NMO.35,36 In the crystal structure, cation vacancies create local geometrical distortions and changes in the coordination of neighbouring atoms, leading to an altered distribution of cations within the material. Ni and Mn have similar ionic radii, making Ni a suitable substitute for Mn in the crystal lattice.37,38 The charge neutrality of the material is maintained by the presence of cation vacancies, which can accommodate the substitution of Ni ions without significantly altering the overall charge balance.18 The comprehensive analysis of the structural differences between Vm-NMO and NMO was investigated by X-ray absorption fine structure analysis. The Mn–Mn coordination number of Vm-NMO depends on the specific arrangement and concentration of the vacancies, which is an important structural parameter that can influence various material properties, such as electronic structure, transport properties, and reactivity.39 NMO, Vm-NMO, Vn-NMO, Vo-NMO and Vp-NMO are synthesised by varying the addition of NH4Cl from 0 mol L−1 to 1 mol L−1 while keeping other conditions the same. In Fig. 2(g), an increase in Mn–Mn coordination number was observed in Vm-NMO. This increase can be due to the presence of lattice vacancies, inducing structural rearrangements and facilitating the formation of additional coordination bonds between neighbouring manganese atoms to share their electronic states.19 With a higher coordination number, there is an expanded overlap of atomic orbitals between adjacent manganese atoms. The overlapping orbitals increase the delocalisation of electronic states and reduce the energy separation between the occupied and unoccupied orbitals. This increased connectivity between manganese atoms lowers the energy barriers that charges need to overcome to move between adjacent sites. Charge carriers, including proton and zinc ions, can more easily transfer from one manganese atom to another, enabling more efficient charge storage.40–42
:13:33 while the ratio in NMO is 1:13:34. This difference can be attributed to the presence of cation vacancies in the Vm-NMO.35,36 In the crystal structure, cation vacancies create local geometrical distortions and changes in the coordination of neighbouring atoms, leading to an altered distribution of cations within the material. Ni and Mn have similar ionic radii, making Ni a suitable substitute for Mn in the crystal lattice.37,38 The charge neutrality of the material is maintained by the presence of cation vacancies, which can accommodate the substitution of Ni ions without significantly altering the overall charge balance.18 The comprehensive analysis of the structural differences between Vm-NMO and NMO was investigated by X-ray absorption fine structure analysis. The Mn–Mn coordination number of Vm-NMO depends on the specific arrangement and concentration of the vacancies, which is an important structural parameter that can influence various material properties, such as electronic structure, transport properties, and reactivity.39 NMO, Vm-NMO, Vn-NMO, Vo-NMO and Vp-NMO are synthesised by varying the addition of NH4Cl from 0 mol L−1 to 1 mol L−1 while keeping other conditions the same. In Fig. 2(g), an increase in Mn–Mn coordination number was observed in Vm-NMO. This increase can be due to the presence of lattice vacancies, inducing structural rearrangements and facilitating the formation of additional coordination bonds between neighbouring manganese atoms to share their electronic states.19 With a higher coordination number, there is an expanded overlap of atomic orbitals between adjacent manganese atoms. The overlapping orbitals increase the delocalisation of electronic states and reduce the energy separation between the occupied and unoccupied orbitals. This increased connectivity between manganese atoms lowers the energy barriers that charges need to overcome to move between adjacent sites. Charge carriers, including proton and zinc ions, can more easily transfer from one manganese atom to another, enabling more efficient charge storage.40–42
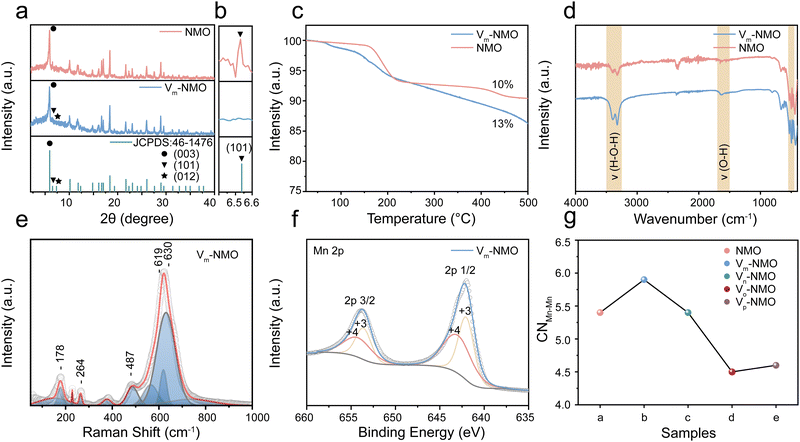 |
| | Fig. 2 Material characterisation of as-prepared Vm-NMO and NMO (a) XRD profile. (b) Enlarged XRD profile. (c) TGA profile. (d) FTIR profile. (e) Raman profile. (f) XPS Mn 2p profile. (g) XAFS fitting on the Mn–Mn coordination numbers. | |
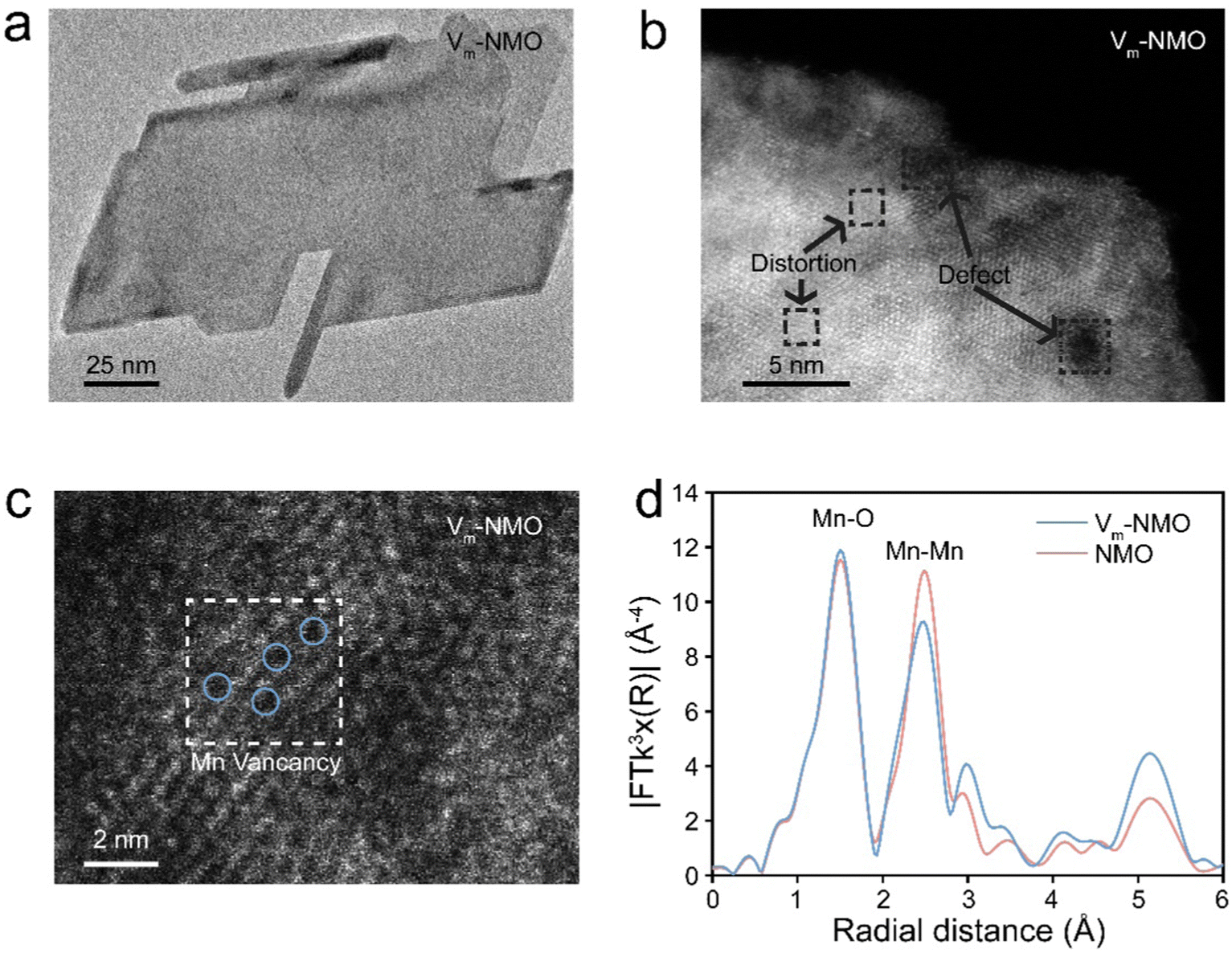
Multiple techniques were employed to thoroughly examine the morphology and understand the role of vacancies in as-prepared materials. Scanning electron microscope (SEM) was used to analyse the morphology of Vm-NMO, revealing a homogeneous three-dimensional flower-like particle composed of nanoplates, as shown in ESI,† Fig. S8. The structural symmetry and crystallinity of as-prepared Vm-NMO materials were studied using transmission electron microscopy (Fig. 3(a)), which showed that an average length of Vm-NMO nanoplate is 150 nm. An inter-layer distance of 0.68 nm in the Vm-NMO was illustrated by high-resolution transmission electron microscopy (HRTEM) (ESI,† Fig. S9), which corresponds to the (003) lattice plane in Vm-NMO. This lattice space result is consistent with relevant XRD findings. The elemental mapping results demonstrated that Ni, Mn, and O are uniformly distributed across Vm-NMO nanoplates and NMO nanorods (ESI,† Fig. S10–S14). Scanning transmission electron microscopy (STEM) images provide atomic-level insights into lattice distortion and defects within Vm-NMO lattice (Fig. 3(b)). Atomic-resolution STEM images of Vm-NMO show direct evidence of atomically scattered Mn vacancies throughout the Vm-NMO nanoplate (Fig. 3(c)). Lattice vacancies can facilitate hydrated proton transfer by lowering the energy barrier, which enables Grotthuss topochemistry. The k3-weighted Fourier transform of the extended X-ray absorption fine structure (EXAFS) can provide information about the local atomic environment surrounding Mn atoms.43 The Mn K-edge EXAFS spectrum of the as-prepared materials showed two peaks located at 1.5 Å and 2.5 Å, corresponding to the Mn–O (the six coordinated oxygen atoms of the nearest neighbouring atom around the manganese atom) and Mn–Mn (six coordinated manganese atom edge sharing through oxygen atom within [MnO6] octahedral slab) shells, respectively. The position and intensity of these peaks provide information about the Mn–O and Mn–Mn bond length and coordination number in the material. A reduction in Mn–Mn peak intensity was observed in the Vm-NMO sample, suggesting a reduction in the coordination number of Mn cations in the Vm-NMO material. It is demonstrated that the local structure of the Vm-NMO material possesses defects in the crystal lattice. This reduction further supports the presence of Mn vacancies in Vm-NMO44,45
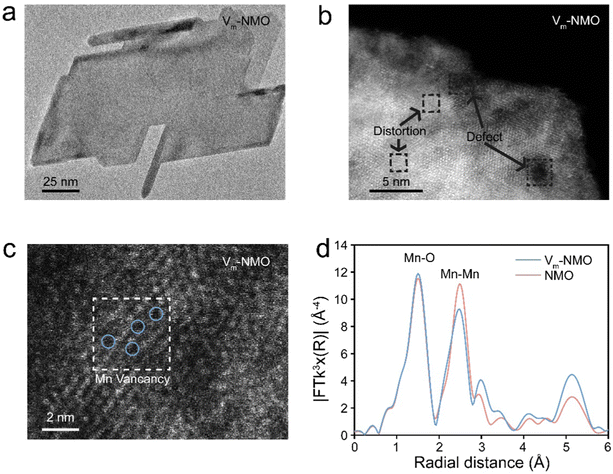 |
| | Fig. 3 Material characterisation of as-prepared NiMn3O7·3H2O (a) TEM profile. (b) HRTEM profile. (c) Atomic resolution STEM profile. (d) k3-weighted Fourier transform of the extended X-ray absorption fine structure data for Vm-NMO and NMO. | |
Computational simulation on proton migration
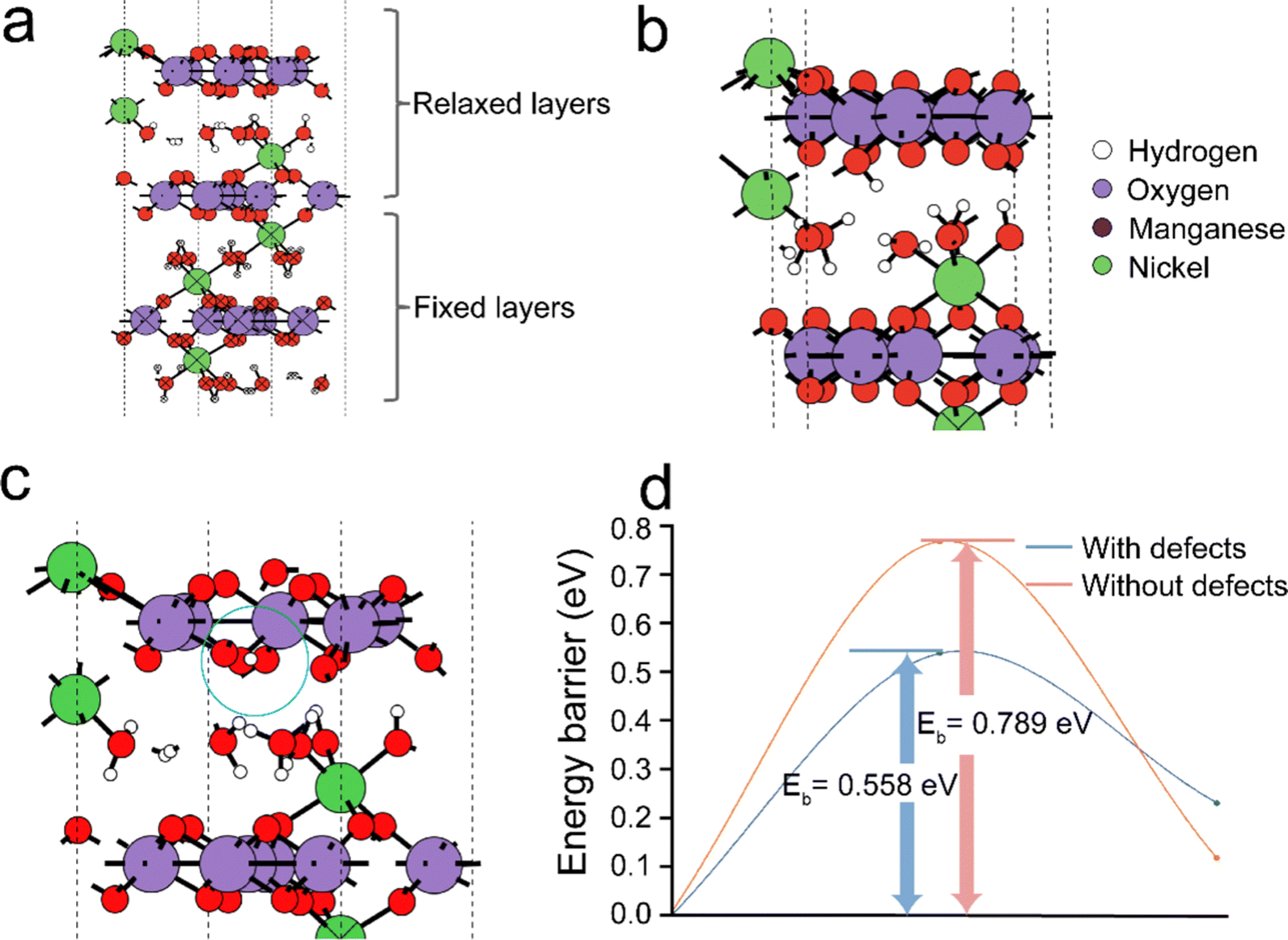
To further gain insight into the proton hopping mechanism with and without cationic (Mn4+) vacancies in the NiMn3O7 (001) system, density functional theory (DFT) based quantum chemical calculations were performed using the Vienna Ab initio Simulation Package (VASP).41,46 We note that while our experimental results indicated the dominance of the (003) facet of NiMn3O7, we chose to focus on the (001) facet for the sake of simplicity in our calculations, allowing meaningful comparisons and benchmarking with previous theoretical studies.17,47–49 At first, the stable sites for proton adsorption on the NiMn3O7 (001) surface were evaluated (Fig. 4(a)). For these calculations, three models were considered, i.e., (i) proton in the proximity of the crystal water molecules between the NiMn3O7 layers, (ii) proton bonded to an Mn4+ ion and (iii) proton bonded to an O2− ion on the NiMn3O7 layers. The fully relaxed geometries revealed that in (i) and (ii) above, the protons were transferred to a nearby O2− ion and, in the third case, remained adsorbed on the O2− ion (ESI,† Fig. S15). The adsorption of protons is favoured on the O2− ions of the NiMn3O7 layers. Previously, it was reported that the transition metal vacancies (Mn, Cd, Zn) could contribute to easy proton absorption/desorption and enable exceptionally high proton conduction.50–55 Our experiments also evidenced the presence of Mn4+ vacancies in the NiMn3O7 system.
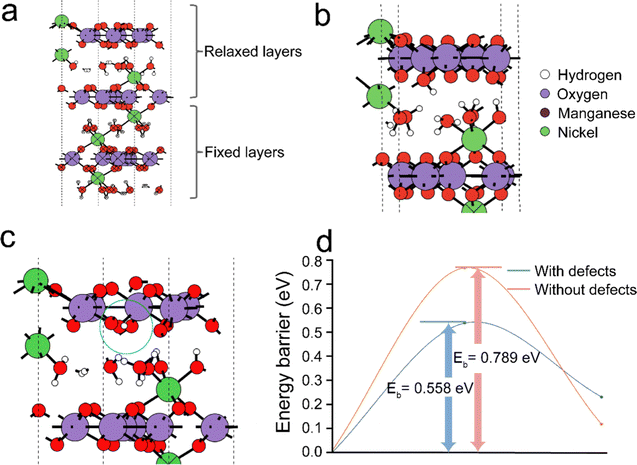 |
| | Fig. 4 (a) Fully relaxed pristine NiMn3O7 (001) model showing the positions of different ions and interlayer water molecules. To properly reproduce the majority of the structure, the position of the bottom two layers of NiMn3O7 and water crystal molecules are held constant. Fully relaxed geometries of adsorbed protons in the NiMn3O7 (001) surface (b) without and (c) with Mn4+ defects and (d) the energy barriers of proton hopping with and without Mn4+ defects. | |
Therefore, in the next step, the adsorption of a proton was carried out around an Mn4+ defect, i.e., (iv) on an O2− ion next to the defect site, (v) on another O2− ion away from the defect site and (vi) on the defect site. The relaxed structures showed that in the fourth and fifth cases, the proton remained bonded to the O2− ions, but in the sixth case, as expected, the proton migrated to a nearby O2− ion close to the defect site (ESI,† Fig. S16). The calculated adsorption energies of the protons in the pristine models without Mn4+ vacancies are in the range of −2.97 eV to −3.07 eV, while in the models with Mn4+ vacancies, they are in the range of −4.03 eV to −5.07 eV (ESI,† Fig. S17). This indicates higher stability of protons in the Vm-NMO systems with cationic vacancies. The Bader charges of −1.13 eV and −1.21 eV on the O2−, respectively for NiMn3O7 systems without and with Mn4+ respectively indicate that this may be due to relatively larger charge localisation in the defective model. Additionally, within the NiMn3O7 system with Mn4+ defect, we found that when protons are adsorbed on the O2− ions close to the defect site, they are ∼−1.04 eV more stable than when they are slightly further away. This difference indicates that the protons could easily hop to a site closer to the defect site. As shown in Fig. 4(d), the calculated energy barriers for proton hopping between two consecutive O2− ions in the absence (i.e., 0.79 eV) and presence (i.e., 0.56 eV) further support these findings. Therefore, from the DFT calculations, we conclude that Mn4+ vacancies are the driving force for proton hopping in NiMn3O7.
Electrochemical performance of Vm-NMO and NMO materials
To systematically study the electrochemical performance of the defective material for AZIBs, we prepared cathode materials and investigated them using cyclic voltammetry (CV) curves in Swagelok cells with zinc metals as the anode. The CV curves of NMO reveal two pairs of redox peaks, corresponding to proton intercalation/deintercalation at ∼1.12 V/1.63 V and zinc ion intercalation/deintercalation at ∼1.35 V/1.57 V. In contrast, Vm-NMO cathode material exhibits reduced polarisation of redox reactions (187 mV vs. 210 mV), implying enhanced ion transport.56 The presence of Mn4+ vacancies in Vm-NMO and its lattice water network can create pathways for Grotthuss conduction, enabling the electrode to be charged and discharged close to theoretical capacity and maintain stability for thousands of cycles. Moreover, as shown in ESI,† Fig. S18, the CV curves of Vm-NMO remain unchanged after first cycle activation, demonstrating the high reversibility. The galvanostatic intermittent titration technique (GITT) was employed to estimate the diffusion coefficient of Zn2+ (DZn2+) in Vm-NMO material with manganese vacancies. Fig. S19 (ESI†) illustrates that the average DZn2+ values at the charge and discharge plateaus are approximately 10−10 to 10−11 cm2 s−1, respectively. This Zn diffusivity is comparable to the Li diffusion coefficient (typically 10−10 cm s−1) reported in the literature. In contrast, the NMO electrode exhibits a lower DZn2+ (10−11 to 10−12 cm2 s−1) and a larger overpotential, indicating the sluggish charge/discharge mobility. The electrochemical impedance spectroscopy (EIS) reveals that Vm-NMO defective cathode material with a hydrogen-bonding network promotes charge transfer. The charge transfer resistance of Vm-NMO (43 Ω) is significantly lower than NMO (75 Ω), illustrating that manganese vacancies with a continuous hydrogen bond network within the Vm-NMO lattice could enhance charge transfer in the system. The galvanostatic charge–discharge curves of Vm-NMO and NMO cathode materials are illustrated in Fig. 5(c), and these were obtained under various current densities, ranging from 0.2 to 5 A g−1, with eight cycles at each current density. Noticeable charge and discharge plateaus could be observed, with a high specific capacity of 318 mA h g−1 achieved at 0.2 A g−1. The Vm-NMO cathode material possesses substantial cycling performance (91% over one hundred cycles). Moreover, the specific capacity retains 38% after increasing the current density 25-fold to 5 A g−1. The detailed cycling and rate performance are illustrated in Fig. 5(d). As the current density increases stepwise, the corresponding specific capacity is retained at 254, 215, 173 and 121 mA h g−1 at 0.5, 1, 2 and 5 A g−1, respectively. After the current density returns to 0.2 A g−1, the specific capacity recovers, demonstrating stable charge/discharge performances under different current densities. The remarkable rate performance enables Vm-NMO to maintain stably under high current densities, with a capacity retention of 91% over 4000 cycles (Fig. 5(f)). In contrast, the rate performance of NMO under identical conditions is lower than that of Vm-NMO, indicating superior ion transport in the Vm-NMO cathode material.
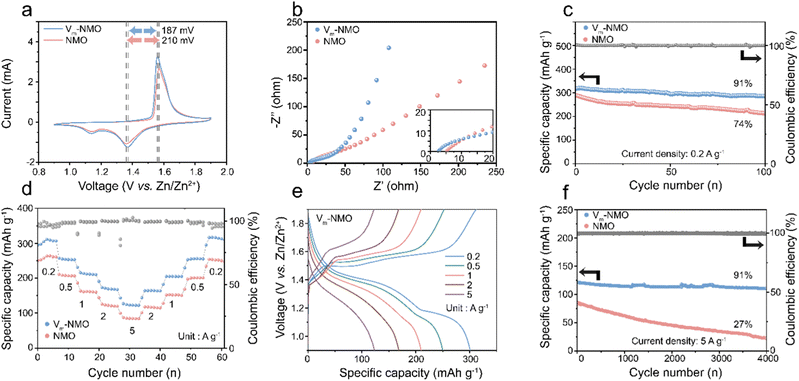 |
| | Fig. 5 Electrochemical performances of as-prepared Zn||Vm-NMO batteries. (a) CV curves of as-prepared materials at different scan rates. (b) EIS profiles of Vm-NMO and NMO. (c) Cycling performance of Vm-NMO and NMO at the current density of 0.2 A g−1. (d) Rate performance. (e) Galvanostatic charge–discharge profiles of Vm-NMO cathodes at different current densities; (f) cycling performance of Zn||Vm-NMO and Zn||NMO batteries at the current density of 5 A g−1. | |
Kinetic evaluation on the Vm-NMO and NMO cathode materials
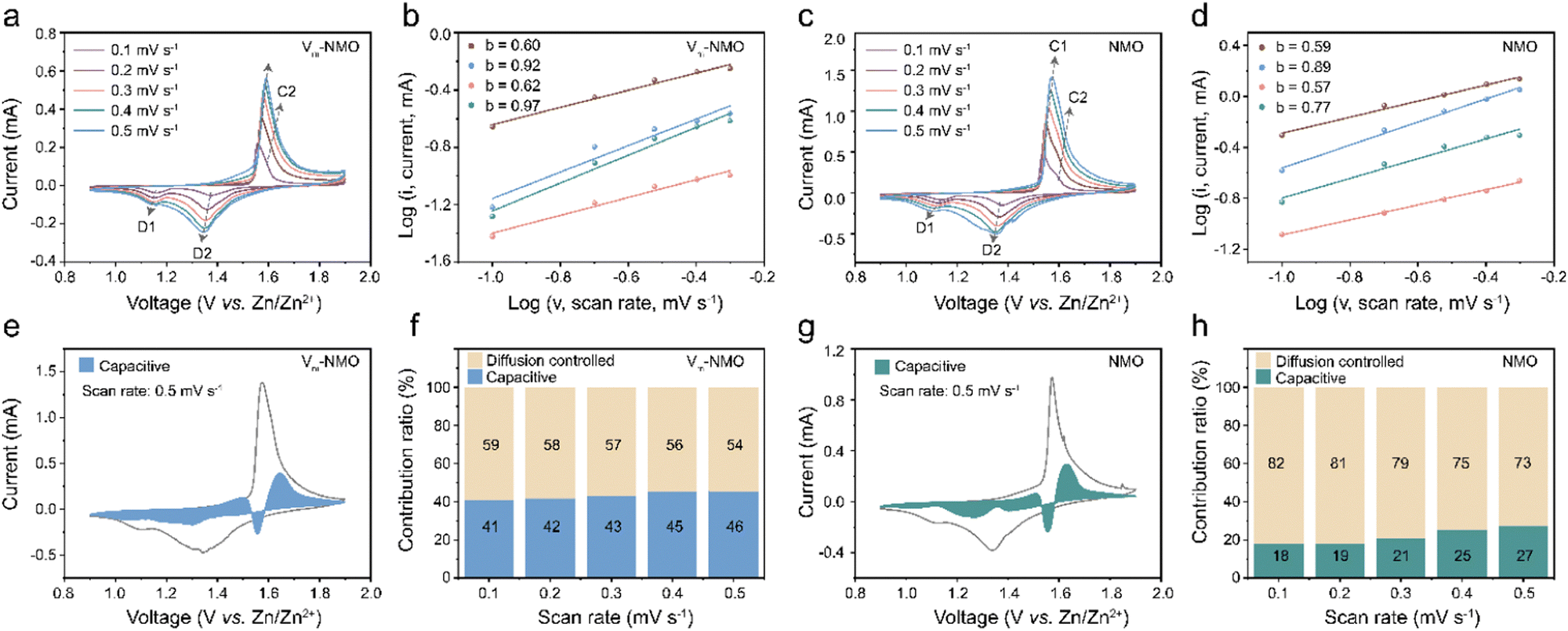
To further investigate the influence of the Grotthuss mechanism on the intercalation process in AZIBs, we analysed the electrochemical processes of Vm-NMO and NMO using CV tests at multiple scan rates. The b value, capacitive contributions, and capacitive ratios of Vm-NMO and NMO were compared in Fig. 6. As shown in Fig. 6(a), two pairs of redox peaks (C1/D1 and C2/D2) appeared in the CV curve of Vm-NMO, and these redox peak positions shifted with increasing scan rate. To investigate the diffusion behaviour in Vm-NMO, the corresponding processes of charge storage were calculated according to the linear relationship between logi versus logv using the equation below:In this equation, the peak current i and sweep rate v were directly obtained, and a, b are parameters that change accordingly. The value of b within a range of 0.5 to 1 implies different charge storage behaviour with respect to capacitive (b = 1) and diffusion-controlled (b = 0.5) behaviours. Accordingly, the linear fitting b values result from the CV curves of both Vm-NMO and NMO were between 0.5 to 1, indicating that the ion storage behaviour comprises both diffusion-controlled and capacitive behaviours. In Fig. 6(b), the b values of Vm-NMO are 0.60, 0.92, 0.62, and 0.97, indicating an enhanced capacitive behaviour that could contribute to faster ion storage kinetics. In comparison, the b values of NMO (Fig. 6(d)) are obviously lower than that of Vm-NMO, indicating a sluggish ion storage process. These results indicate that an improved ion transfer ability is due to the fast displacive hydrated proton hopping mechanism. To further illustrate the storage behaviour of Vm-NMO, capacitive contribution ratios were revealed under different scan rates (Fig. 6(e)–(h)). The capacitive behaviour contributes to 41% of overall storage at a scan rate of 0.1 mV s−1, and this ratio gradually increases to 46% as the scan rate rises to 0.5 mV s−1. In contrast, NMO exhibits only 18% of capacitive behaviour under a scan rate of 0.1 mV s−1. A similar trend is observed at different scan rates, indicating that capacitive behaviour is enhanced (ESI,† Fig. S20 and S21), which aligns with the b value results. These results indicate the enhancement in charge transfer can be attributed to the facilitated displacive hydrated proton hopping, which creates pathways for efficient ion migration.
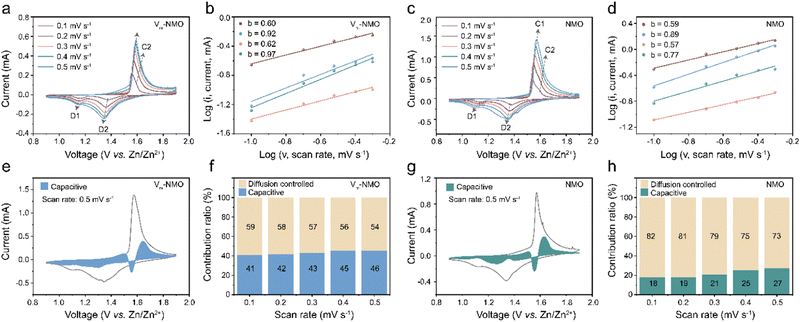 |
| | Fig. 6 Electrochemical characterisation of as-prepared NiMn3O7·3H2O. (a) CV plots of Vm-NMO at different scan rates. (b) b-value of Vm-NMO. (c) CV plots of NMO in different scan rates. (d) b-value of NMO. (e) Capacitive contribution of Vm-NMO at the scan rate of 0.5 mV s−1. (f) Capacitive ratio of Vm-NMO in different scan rates. (g) Capacitive contribution of NMO at the scan rate of 0.5 mV s−1. (h) Capacitive ratio of NMO under different scan rates. | |
Electrochemical insertion/deinsertion reaction mechanism
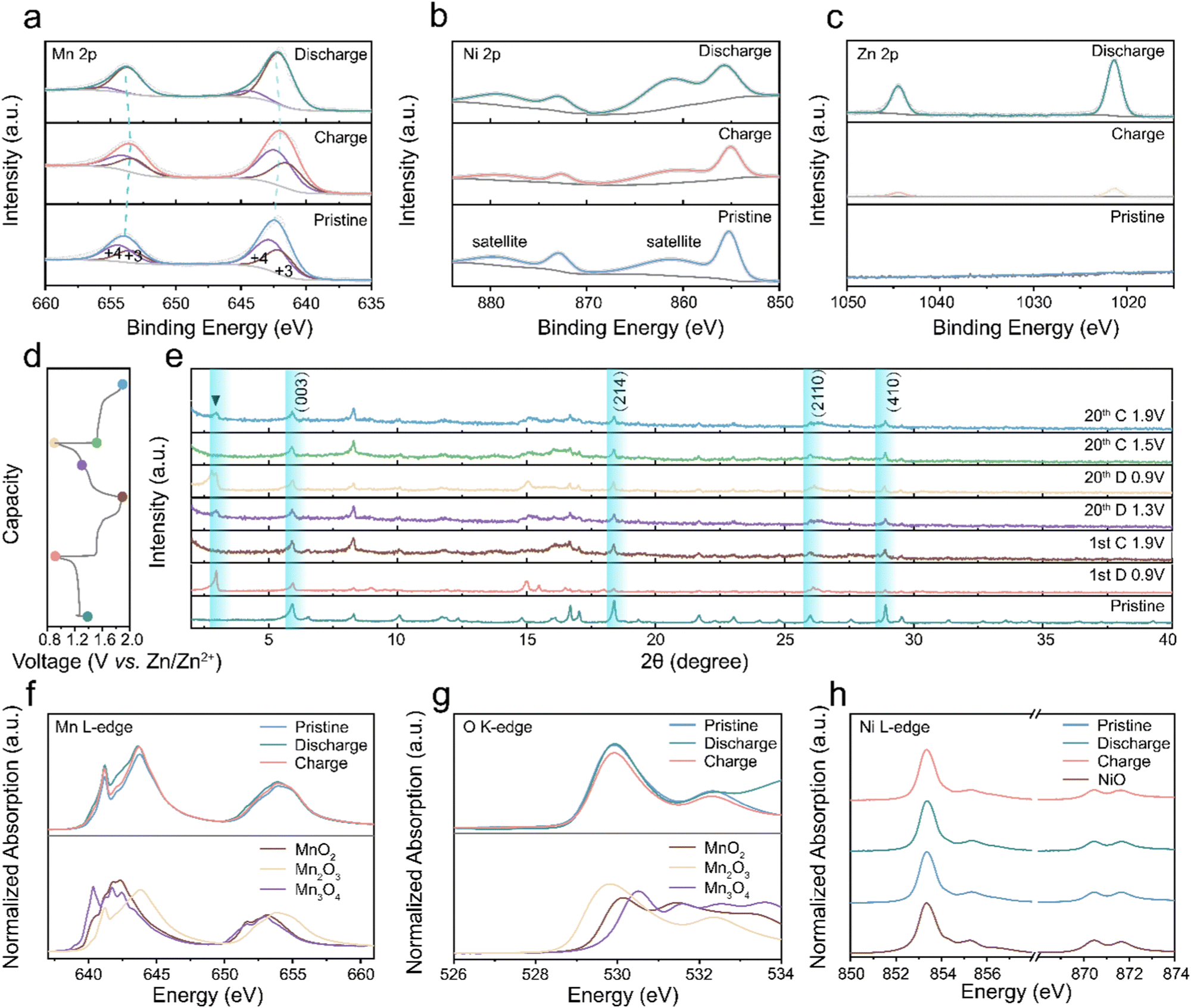
Ex situ XPS tests were conducted to uncover the valence changes during the charging and discharging.57Fig. 7(a) illustrates the classic manganese valence change in AZIBs. The Mn 2p spectra fitting results, featuring two deconvoluted peaks, indicate a mixture of multiple Mn valence states.58 In the discharging state, the manganese valence peak position undergoes a negative shift, signifying the reduction of manganese oxide compared to the pristine state due to charge carrier intercalation. During the charging process, Mn 2p peaks shift positively toward the pristine position, indicating reversible valence changes throughout charge and discharge cycles.59–63Fig. 7(b) and Fig. S22 (ESI†) demonstrate a persistent Ni 2p signal in various states, confirming the stability of nickel atoms in the cathode material, where it balances the charge induced by vacancies. The nickel atoms in Vm-NMO remain inactive during charging and discharging cycles, with interlayer nickel ions compensating for the charge imbalance caused by manganese vacancy and contributing to cycling stability. In contrast, the Ni 2p XPS intensity of cycled NMO electrode decreased, indicating a partial deintercalation of nickel in NMO electrodes upon cycling. Fig. 7(d) and (e) show ex situ XRD results that reveal characteristic peaks associated with the discharging process of the Vm-NMO electrode. The reversible peak appearance and disappearance of the zinc hydroxide trifluoromethyl sulfonate peak at the low degree indicates the proton intercalation during the discharging process.64,65 Consistent with numerous manganese-based materials studies, zinc hydroxide trifluoromethyl sulfonate forms through the integration of OH−, Zn(CF3SO3)2, and H2O in the electrolyte following proton transfer to the Vm-NMO electrode.66,67 The characteristic peaks disappear in the subsequent charging phase, implying a reversible reaction. The formation and disappearance of XRD characteristic peaks highlight the reversible proton transfer during the discharging process.
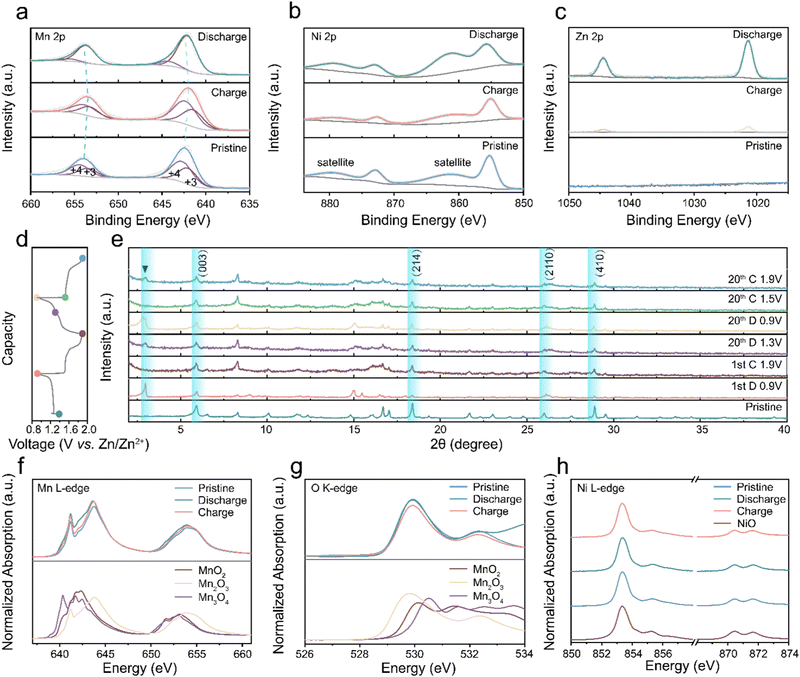 |
| | Fig. 7 The charge storage mechanism of Vm-NMO batteries. Ex situ XPS profiles of Vm-NMO at different states: (a) Mn 2p. (b) Ni 2p. (c) Zn 2p. (d) and (e) Ex situ XRD profiles of Vm-NMO at different states. (f) Mn L3,2-edge NEXAFS profiles at different states. (g) O K-edge NEXAFS profiles of cathode materials in different states. (h) NEXAFS profiles of Ni L-edge of cathode materials in different states. | |
Mn L-edge Near Edge X-ray Absorption Fine Structure (NEXAFS) spectra provide insights into the local bonding environment of manganese atoms in the cathode material. The pristine state spectrum exhibits multiple peaks corresponding to different electronic transitions and variations in coordination environment with oxygen atoms, namely A (638–639 eV, 2p–3d, octahedral), B (641–642 eV, 2p–3d, tetrahedral), C (643–644 eV, 2p–3d, mixed octahedral and tetrahedral sites).68 This indicates that the Mn ions in Vm-NMO can occupy both octahedral and tetrahedral sites, particularly Mn ions situated at the edge or corner sites of MnO6 octahedral units. The crystal structure of Vm-NMO comprises edge-sharing MnO6 octahedra layers interconnected by sharing corners with other octahedra. Fig. 7(f) of Mn L3,2-edge NEXAFS reveals that the shape of Vm-NMO in the charging and pristine state resembles MnO2(IV), verifying that pristine valence is close to +4 and Mn valence can be reduced to pristine in the charging state. The systematic shift of the L3 leading edge position and gravity centre of the L3/L2 edge towards lower energies is observable in the fully discharging state, indicating that the Mn oxidation state in the discharging state is lower than in the pristine state and the fully charging state. Additionally, the Mn L-edge intensity between 641–642 eV increased in the discharging state, the similarity in the peak shape to Mn3+, indicating Mn atoms were reduced to a valence close to Mn(III) in the discharged state after zinc ion intercalation. Moreover, O K-edge NEXAFS spectra, determined by the hybridisation of O 2p and Mn 3d states, lead to electron transitions from ligand O 1s orbitals to unoccupied transition metal 3d orbitals. In Fig. 7(g), the O K-edge spectra include several distinct peaks around 529 eV and 532 eV. In the pristine and fully charging state, two absorption peaks can be identified due to the Mn(IV) with a 3d3 (t32ge0g) configuration, while a lower energy peak belongs to spin-down t2g and spin-up eg transitions, and a higher energy peak originate from the spin-down eg transitions. In the fully discharged state, the peak intensity increases with a broad absorption peak identifiable between 530–531 eV, caused by the Mn(III) content with a 3d4 (t32ge1g) configuration. The octahedral layers in NiMn3O7 are separated by tetrahedral voids occupied by nickel cations.23,69 In the charging state, Ni L-edge NEXAFS spectroscopy provides valuable information about the oxidation state of Ni and the coordination environment of Ni atoms in the material. The NEXAFS spectrum offers insight into the redox activity and stability of nickel ions in the material. The peak A (853 eV) and peak B (855 eV) are attributed to transitions from Ni 2p3/2 core level to the unoccupied 3d orbitals and unoccupied 4p orbitals, respectively. The intensity of these two peaks depends on the oxidation state of the Ni ions and is sensitive to changes in the local coordination environment of the Ni atoms. The multiple peaks in Fig. 7(h) illustrate the high spin Ni2+ electronic structure and its octahedral coordination structure. The intensity and peak position remain stable in the charging and discharging states, indicating that Ni2+ is electrochemically stable in the Vm-NMO lattice. Nickel ions facilitate the ongoing mobility of hydrated protons via Grotthuss hopping by preserving the system's electrical neutrality, which counterbalances the dynamic changes caused by proton migration.
Conclusion
In summary, our research demonstrates the synergistic collaboration between vacancies, lattice water and nickel ions on enhancing the hydrated protons hopping via the Grotthuss mechanism for high-performance zinc ion battery cathodes. Energy barrier calculations for proton transfer with and without Mn4+ vacancies indicate that proton transfer is favoured in the presence of defects. Mn4+ vacancies act as the driving force for proton hopping in the Vm-NMO systems. Additionally, the presence of lattice water molecules within the host material facilitates the migration of hydrated protons from the electrolyte to the interlayer spaces in the lattice. The simulation results indicate that protons prefer to bond with O2− ions on the Mn–O layer rather than staying with water molecules. The continuous hopping of protons within the host material induces periodic, temporary local structural changes in the lattice. This dynamic behaviour alters the energy barriers for ion intercalation and deintercalation, thereby optimising their diffusion pathways. Moreover, the presence of manganese vacancies in the lattice serve as additional sites for hydrated proton and zinc ion intercalation, resulting in an increased capacity. The nickel ions increase the stability by preserving the system's electrical neutrality. The as-prepared Vm-NMO cathode material exhibits enhanced ion transfer and a high specific capacity of 318 mA h g−1 at 200 mA g−1. Furthermore, it demonstrates excellent rate performances, and remarkable cycling stability, with a capacity retention of 91% over 4000 cycles. Overall, this study provides insights into the utilisation of the Grotthuss mechanism and manganese oxide's distinct characteristics for the advancement of energy storage technologies. These findings highlight the potential of utilising the Grotthuss mechanism to develop high-performance energy storage materials.
Author contributions
F. Z.: conceptualisation, methodology, software, data acquisition, data curation, formal analysis, investigation, validation, visualisation, writing – original draft, and writing – review and editing. J. L.: conceptualisation, methodology, formal analysis, and writing – review and editing. A. C.: conceptualisation, data acquisition, formal analysis, software, and writing – review and editing. L. L., L. K., H. D., X. G., and Y. T.: data acquisition, software, formal analysis. F. L., T. L.: methodology, data acquisition, and writing – review and editing. I. P.: funding acquisition, supervision, and writing – review and editing. G. H.: conceptualisation, methodology, funding acquisition, supervision, and writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council (EPSRC, EP/V027433/3), EPSRC Centre for Doctoral Training in Molecular Modelling and Materials Science (EP/L015862/1), UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding (101077226; EP/Y008707/1), Open Foundation of the State Key Laboratory of Silicate Materials for Architectures at WUT (no. SYSJJ2020-04), Shanghai Scientific and Technological Innovation Project (22520710100) and the Royal Society (RGS/R1/211080; IEC/NSFC/201261) for funding support. AC acknowledges the use of the Sulis Tier 2 HPC platform at the University of Warwick funded by EPSRC (EP/T022108/1) and ARCHER2 via the membership of the UK's HEC Materials Chemistry Consortium funded by EPSRC (EP/R029431). We acknowledge the B18 beamline (proposal no. SP32905), B07-B beamline (proposal no. SI29340), and electron Physical Science Imaging Centre instruments E01 and E02 (proposal no. MG32058, MG32035) at Diamond Light Source for the allocated experiment sessions. We thank Dr Iuliia Mikulska, Dr Veronica Celorrio, and Dr Diego Gianolio for their support during our B18 beamtime session, and Dr Pilar Ferrer and Dr David C. Grinter for their support during our B07-B beamtime session, and Dr David G. Hopkinsonc and Dr Christopher S. Allen for their support during our STEM session. We thank Prof. K. Holt for the discussion on FTIR spectrum.
References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3334 RSC.
- R. H. Tunuguntla, F. I. Allen, K. Kim, A. Belliveau and A. Noy, Nat. Nanotechnol., 2016, 11, 639–644 CrossRef CAS PubMed.
- I. Popov, Z. Zhu, A. R. Young-Gonzales, R. L. Sacci, E. Mamontov, C. Gainaru, S. J. Paddison and A. P. Sokolov, Commun. Chem., 2023, 6, 77 CrossRef CAS PubMed.
- C. Dellago, M. M. Naor and G. Hummer, Phys. Rev. Lett., 2003, 90, 105902 CrossRef PubMed.
- Z. Cao, Y. Peng, T. Yan, S. Li, A. Li and G. A. Voth, J. Am. Chem. Soc., 2010, 132, 11395–11397 CrossRef CAS PubMed.
- C. Dellago and G. Hummer, Phys. Rev. Lett., 2006, 97, 245901 CrossRef PubMed.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123–130 CrossRef CAS.
- T. Miyake and M. Rolandi, J. Phys.: Condens. Matter, 2016, 28, 023001 CrossRef PubMed.
- H. Zhang, W. Wu, Q. Liu, F. Yang, X. Shi, X. Liu, M. Yu and X. Lu, Angew. Chem., Int. Ed., 2021, 60, 896–903 CrossRef CAS PubMed.
- W. Sun, Z. Xu, C. Qiao, B. Lv, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Sci., 2022, 9, e2201679 CrossRef PubMed.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y. W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- Y. Fu, Q. Wei, G. Zhang, X. Wang, J. Zhang, Y. Hu, D. Wang, L. Zuin, T. Zhou, Y. Wu and S. Sun, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- J. Li, N. Luo, L. Kang, F. Zhao, Y. Jiao, T. J. Macdonald, M. Wang, I. P. Parkin, P. R. Shearing, D. J. L. Brett, G. Chai and G. He, Adv. Energy Mater., 2022, 12, 2201840 CrossRef CAS.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- Z. Ma, X. M. Shi, S. I. Nishimura, S. Ko, M. Okubo and A. Yamadas, Adv. Mater., 2022, 34, e2203335 CrossRef PubMed.
- S.-i Ohkoshi, K. Nakagawa, K. Tomono, K. Imoto, Y. Tsunobuchi and H. Tokoro, J. Am. Chem. Soc., 2010, 132, 6620–6621 CrossRef CAS PubMed.
- D. Chen, D. Ding, X. Li, G. H. Waller, X. Xiong, M. A. El-Sayed and M. Liu, Chem. Mater., 2015, 27, 6608–6619 CrossRef CAS.
- Y. Zhang, H. Zhang, L. Fang, J. Deng and Y. Wang, Electrochim. Acta, 2017, 245, 32–40 CrossRef CAS.
- F. Putzolu, G. Balassone, M. Boni, M. Maczurad, N. Mondillo, J. Najorka and F. Pirajno, Ore Geol. Rev., 2018, 97, 21–34 CrossRef.
- Y. Haraguchi, A. Matsuo, K. Kindo and Z. Hiroi, Phys. Rev. B, 2018, 98, 064412 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- D. Yuan, X. Li, H. Yao, Y. Li, X. Zhu, J. Zhao, H. Zhang, Y. Zhang, E. T. J. Jie, Y. Cai and M. Srinivasan, Adv. Sci., 2023, 10, 2206469 CrossRef CAS PubMed.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- L. Kang, M. Zhang, Z.-H. Liu and K. Ooi, Spectrochim. Acta, 2007, 67, 864–869 CrossRef PubMed.
- X. Xie, J. Li, Z. Xing, B. Lu, S. Liang and J. Zhou, Nat. Sci. Rev., 2023, 10, nwac281 CrossRef CAS PubMed.
- R. Yi, X. Shi, Y. Tang, Y. Yang, P. Zhou, B. Lu and J. Zhou, Small Struct., 2023, 4, 2300020 CrossRef CAS.
- Z. M. Chan, D. A. Kitchaev, J. N. Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5261–E5268 CAS.
- Y.-K. Hsu, Y.-C. Chen, Y.-G. Lin, L.-C. Chen and K.-H. Chen, Chem. Commun., 2011, 47, 1252–1254 RSC.
- Y. Li, Y. Lu, X. Jiang, L. Lu, J. Qin, D. Yang, J.-L. Chen, L. Zhang, D. Wang and A. Lei, Energy Storage Mater., 2023, 54, 553–562 CrossRef.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2023, 33, 2209698 CrossRef CAS.
- S. Mazen, N. Abu-Elsaad and A. Nawara, Phys. Solid State, 2020, 62, 1183–1194 CrossRef CAS.
- C. Liang, F. Kong, R. C. Longo, C. Zhang, Y. Nie, Y. Zheng and K. Cho, J. Mater. Chem., 2017, 5, 25303–25313 RSC.
- J. Schmalhorst, A. Thomas, S. Kämmerer, O. Schebaum, D. Ebke, M. Sacher, G. Reiss, A. Hütten, A. Turchanin and A. Gölzhäuser, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 014403 CrossRef.
- J. H. Lee, G. Ali, D. H. Kim and K. Y. Chung, Adv. Energy Mater., 2017, 7, 1601491 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. Li, Z. Liu, S. Han, P. Zhou, B. Lu, J. Zhou, Z. Zeng, Z. Chen and J. Zhou, Nano-Micro Lett., 2023, 15, 237 CrossRef CAS PubMed.
- P. Bhobe, K. Priolkar and P. Sarode, Phys. Rev. B, 2006, 74, 224425 CrossRef.
- T. Ressler, S. L. Brock, J. Wong and S. L. Suib, J. Phys. Chem. B, 1999, 103, 6407–6420 CrossRef CAS.
- F. Wang, J. Deng, S. Impeng, Y. Shen, T. Yan, G. Chen, L. Shi and D. Zhang, Chem. Eng. J., 2020, 396, 125192 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- W. Yang, Y. Zhu, F. You, L. Yan, Y. Ma, C. Lu, P. Gao, Q. Hao and W. Li, Appl. Catal., B, 2018, 233, 184–193 CrossRef CAS.
- C. Y. Ouyang, Ž. Šljivančanin and A. Baldereschi, J. Chem. Phys., 2010, 133, 204701 CrossRef CAS PubMed.
- J. A. Dawson, H. Chen and I. Tanaka, ACS Appl. Mater. Interfaces, 2015, 7, 1726–1734 CrossRef CAS PubMed.
- H. Lind, B. Wickman, J. Halim, G. Montserrat-Sisó, A. Hellman and J. Rosen, Adv. Sustain. Syst., 2021, 5, 2000158 CrossRef CAS.
- L. Li, X. Feng, Y. Nie, S. Chen, F. Shi, K. Xiong, W. Ding, X. Qi, J. Hu, Z. Wei, L.-J. Wan and M. Xia, ACS Catal., 2015, 5, 4825–4832 CrossRef CAS.
- B. Ammundsen, D. J. Jones, J. Roziere and G. R. Burns, Chem. Mater., 1995, 7, 2151–2160 CrossRef CAS.
- W. Zhao, X. Feng, W. Tan, F. Liu and S. Ding, J. Environ. Sci., 2009, 21, 520–526 CrossRef CAS PubMed.
- E. Silvester, A. Manceau and V. A. Drits, Am. Mineral., 1997, 82, 962–978 CAS.
- X. Qian, L. Chen, L. Yin, Z. Liu, S. Pei, F. Li, G. Hou, S. Chen, L. Song, K. H. Thebo, H.-M. Cheng and W. Ren, Science, 2020, 370, 596–600 CrossRef CAS PubMed.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 RSC.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- M. Harada, F. Kotegawa and M. Kuwa, ACS Appl. Energy Mater., 2022, 5, 278–294 CrossRef CAS.
- Z. Zhong, J. Li, L. Li, X. Xi, Z. Luo, G. Fang, S. Liang and X. Wang, Energy Storage Mater., 2022, 46, 165–174 CrossRef.
- D. Zhang, J. Cao, X. Zhang, N. Insin, S. Wang, J. Han, Y. Zhao, J. Qin and Y. Huang, Adv. Funct. Mater., 2021, 31, 2009412 CrossRef CAS.
- Y. Liu, Z. Qin, X. Yang and X. Sun, Adv. Funct. Mater., 2022, 32, 2106994 CrossRef CAS.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- Y. Ma, Y. Ma, T. Diemant, K. Cao, X. Liu, U. Kaiser, R. J. Behm, A. Varzi and S. Passerini, Adv. Energy Mater., 2021, 11, 2100962 CrossRef CAS.
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, L. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219136 CrossRef CAS PubMed.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 CrossRef CAS PubMed.
- P. Oberholzer, E. Tervoort, A. Bouzid, A. Pasquarello and D. Kundu, ACS Appl. Mater. Interfaces, 2019, 11, 674–682 CrossRef CAS PubMed.
- L. Wang, K.-W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
-
K. D. Kwon and G. Sposito, in Advances in the Environmental Biogeochemistry of Manganese Oxides, ed. X. Feng, W. Li, M. Zhu and D. L. Sparks, American Chemical Society, Washington, DC, 1st edn, 2015, ch. 4, vol. 1197, pp. 51–64.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d3ee04161a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jianwei
Li
b,
Arunabhiram
Chutia
a,
Jianwei
Li
b,
Arunabhiram
Chutia