
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing highly durable reversal-tolerant anodes via integrating high-surface-area Ti4O7 supported Pt and Ir@IrOx for proton exchange membrane fuel cells†
Zheng
Li
 abc,
Yongbiao
Mu
bc,
Qing
Zhang
bc,
Haodong
Huang
bc,
Xianbin
Wei
d,
Lin
Yang
bc,
Guanxiong
Wang
e,
Tianshou
Zhao
*abc,
Gang
Wu
*f and
Lin
Zeng
*bc
abc,
Yongbiao
Mu
bc,
Qing
Zhang
bc,
Haodong
Huang
bc,
Xianbin
Wei
d,
Lin
Yang
bc,
Guanxiong
Wang
e,
Tianshou
Zhao
*abc,
Gang
Wu
*f and
Lin
Zeng
*bc
aDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: zhaots@sustech.edu.cn
bShenzhen Key Laboratory of Advanced Energy Storage, Department of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: zengl3@sustech.edu.cn
cSUSTech Energy Institute for Carbon Neutrality, Southern University of Science and Technology, Shenzhen 518055, China
dDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China
eShenzhen Academy of Aerospace Technology, Shenzhen 518057, China
fDepartment of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: gangwu@buffalo.edu
First published on 15th January 2024
Abstract
Fuel starvation during the fuel cell operation inevitably leads to high potential anodes, which causes carbon corrosion and catalyst layer collapse and poses a challenge to the durability of proton exchange membrane fuel cells. Herein, Ti4O7 with a high specific surface area was synthesized facilely and utilized to replace carbon as the anode catalyst support. Previously, the initial performance of the Ti4O7-supported catalyst was obstructed by the disadvantaged electrical conductivity of Ti4O7. This work obtains a comparable polarization performance after optimizing the Ti4O7-supported anode catalyst layer parameters by shortening the electron transfer pathway and increasing metal coverage. Meanwhile, reversal-tolerant anodes (RTAs) were fabricated by the traditional IrO2 addition and core–shell structured Ir@IrOx to validate the applicability of the Ti4O7 support. Typically, the Ir@IrOx/Pt/Ti4O7-fabricated RTA with low Ir loading displays an approximately ten times longer reversal time (6 hours) and two orders of magnitude lower degradation rate than a conventional carbon-supported counterpart. The degradation origin of the Ti4O7-supported anodes was also studied by postmortem characterizations, pointing to the Pt oxidation caused by the formation of TiOx thin layers on the Pt surface. The study aims to promote the development of carbon-free anodes and provide a bright perspective for practical high-performance, low-degradation, and cost-friendly RTA fabrication.
Broader contextDurability and cost are essential factors for the commercial application of proton exchange membrane fuel cells (PEMFCs). Anode cell reversal happens inevitably during the operation of PEMFCs and causes irreversible damage to the anode catalyst layer. Commercially, IrO2 or Ir black is utilized to alleviate the destruction of cell reversal even though it deteriorates the performance and increases the cost. Hence, an enhanced anode is highly desired for practical PEMFCs to address commercial issues. This work is dedicated to the development of a high-performance, low-degradation, and cost-friendly anode through integrating high-surface-area Ti4O7 supported Pt and core–shell structured Ir@IrOx to significantly improve the reversal tolerance while maintaining polarization performance with super-low degradation. Through postmortem studies, along with advanced characterization, the degradation mechanisms of carbon-supported and Ti4O7-supported catalysts are comprehensively compared. This work represents the most promising carbon-free support candidate and paves the way for future research of reversal tolerance anodes for PEMFCs. |
1. Introduction
Driven by the growing demand for carbon-free energy, the “hydrogen society” concept has been put forward toward the “carbon neutral” goal. Among all hydrogen energy utilization units, proton exchange membrane fuel cells (PEMFCs) are the most promising for commercialization, attracting intensive attention and investigation.1 However, durability and cost are the main hindrances to broader commercialization.2 Among all the problems, the cell reversal would damage the anode catalyst layer and further reduce durability. In particular, due to the local fuel starvation, the potential between the anode and cathode is narrowing or even reversing during the cell reversal. The carbon oxidation reaction (COR) and oxygen evolution reaction (OER) happen to maintain the external current and internal proton flow instead of the initially desirable hydrogen oxidation reaction (HOR), while the oxygen reduction reaction (ORR) remains unchanged in the cathode.3 Notably, the COR is more harmful than the OER because the irreversible corrosion of carbon leads to the detachment of Pt nanoparticles,4 increased internal resistance,5 and the collapse of the catalyst layer,6 significantly deteriorating performance.7| 2H2O → H+ + O2 + 4e− E0 = 1.23 V (vs. RHE) | (1) |
| C + 2H2O → CO2 + 4H+ + 4e− E0 = 0.21 V (vs. RHE) | (2) |
| C + H2O → CO + 2H+ + 4e− E0 = 0.52 V (vs. RHE) | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and 0.5 mgPt cm−2,16 which is far from the practical application. Alternatively, carbon is added into titanium oxide-supported catalyst layers to offset the unsatisfactory electronic conductivity.17–20 However, carbon corrosion during the cell reversal is inevitable once the carbon exists and thus causes performance decay.
15 and 0.5 mgPt cm−2,16 which is far from the practical application. Alternatively, carbon is added into titanium oxide-supported catalyst layers to offset the unsatisfactory electronic conductivity.17–20 However, carbon corrosion during the cell reversal is inevitable once the carbon exists and thus causes performance decay.
In this work, we synthesized a pure Ti4O7 anode with a high specific surface area by a facile hydrogen-free method. After the Pt deposition, the Pt/Ti4O7 anode at a 0.1 mgPt cm−2 loading exhibited comparable performance to a commercial Pt/C catalyst under different operating conditions. The key to improving Ti4O7-supported catalyst layer performance is shortening the electronic transfer pathway and increasing the metal coverage on the Ti4O7 surface, achieved by constructing a thin catalyst layer and high Pt content in the anode. Traditional IrO2 addition and novel Ir@IrOx core–shell Ir deposition were applied to fabricate reversal-tolerant anodes (RTAs) to boost OER activity. The results showed that the membrane electrode assembly (MEA) with the Ti4O7-supported catalyst effectively prolonged the first reversal time and decreased the performance degradation rate. The performance degradation was investigated, and the results indicate that the degradation originates from the Pt oxidation by forming a TiOx thin layer at the Pt surface, verified by postmortem EIS, XPS, SEM, and TEM characterization.
2. Results and discussion
2.1 Ti4O7 synthesis and optimization of catalyst layer parameters
The procedure applied to synthesize Ti4O7 is illustrated in Fig. 1a. The XRD patterns under different synthesis conditions are presented in Figure S1 to confirm the synthesis of Ti4O7. When the mass ratio of PEG 400 to TiOSO4 (based on TiO2) is 1, the diffraction peaks of the (110) and (211) facets of TiO2 appear at 27.4° and 54.3°, indicating over-oxidation during synthesis. The mass ratio of PEG400 to TiOSO4 (based on TiO2) was increased to 1.5 for a thicker PEG 400 coating, resulting in significantly attenuated TiO2 peak intensities. Furthermore, particles near the gas flow aggregated together and turned white due to uneven gas flow distribution and exposure to high temperatures. Therefore, the PEG@TiO2 powder was ground with an agate mortar and then placed in a sealed quartz crucible before heating. The XRD analysis revealed that the grinding and sealing processes effectively inhibit TiO2 formation and promote the desirable Ti4O7 phase. Due to the difference in properties between Ti4O7 and carbon, the Pt/Ti4O7 anode catalyst layer parameters were optimized, including Pt content, ionomer content, and Pt loading. First, the Pt content of Pt/Ti4O7 was controlled by the mass ratio of Pt precursor to Ti4O7 in the synthesis process. A higher Pt coverage on the Ti4O7 surface can reduce the Ti4O7 exposure and form a connected metal nanoparticle network, creating a potential electron transfer pathway and offsetting the disadvantaged conductivity. Based on the observations made during the synthesis process, the product yield is higher (over 90%) when the Pt content is 20% to 40%. However, if the Pt content is increased to 50%, the product yield drops significantly to 81.3%. This indicates a considerable mass loss for 50% Pt/Ti4O7 during the synthesis. Therefore, the complete surface loading of Ti4O7 is 40% Pt/Ti4O7, while for 50% Pt/Ti4O7, there is an over-Pt loading. The overloading of Pt causes the aggregation of Pt and decreases the Pt utilization, making it difficult for Pt deposition. The polarization curves of 20% to 50% Pt/Ti4O7 anodes are shown in Fig. 1b, further confirming the optimization of Pt content at 40%. The performance of Pt/Ti4O7 is also significantly impacted by the ionomer content. As demonstrated in Fig. 1c, the polarization performance improves noticeably when the ionomer content decreases from 25 wt% to 4 wt% due to the thinner ionomer layer. However, when the ionomer content decreases to 3 wt%, the performance declines, particularly in the large current density range. This can be attributed to the limited proton transfer ability caused by the low ionomer content at this stage. Subsequently, the Pt loading was investigated and optimized. As shown in Fig. 1d, the Pt/Ti4O7 anode with 0.1 mgPt cm−2 displays the highest polarization performance compared to 0.2 mgPt cm−2 and 0.05 mgPt cm−2. A similar outcome was observed in other titanium oxide-supported catalysts.15 The MEA with a Pt/C anode with different Pt loadings was also tested. As shown in Fig. S2 (ESI†), Pt/C anodes with 0.1 and 0.2 mgPt cm−2 exhibit similar polarization performance, indicating an anode loading of 0.1 mgPt cm−2 is sufficient, and the cathode limits the performance. However, when the Pt loading further decreases to 0.05 mgPt cm−2, the performance decay with more than 100 mV loss at 1000 mA cm−2 is observed due to the limited active site of the anode, implying 0.1 mgPt cm−2 is also the optimized loading of Pt/C in the anode catalyst layer. Eventually, the optimized parameters of the Pt/Ti4O7 anode catalyst layer are determined as a Pt content of 40 wt%, an ionomer content of 4 wt%, and a Pt loading of 0.1 mgPt cm−2. Under these conditions, the Pt/Ti4O7 anode catalyst layer showed similar polarization performance to commercial Pt/C under different flow rates. Specifically, at 1000 mA cm−2 under 0.5 L/1 L fixed flows, the Pt/Ti4O7 anode catalyst layer achieved a polarization voltage of 0.670 V compared to 0.673 V for the commercial Pt/C. Similarly, at 1000 mA cm−2 under 1.5/2.5 stoichiometry flows, the Pt/Ti4O7 anode catalyst layer achieved a polarization of 0.638 V compared to 0.639 V for the commercial Pt/C (Fig. 1e). The high specific surface area of Ti4O7 enables a high Pt content Pt/Ti4O7 anode and further results in a thinner catalyst layer at a given Pt loading, which is vital for comparable performance. The schematic illustration of the optimization principle is demonstrated in Fig. 1f. Meanwhile, as shown in Fig. S3 (ESI†), the reversal tests were also performed for Pt/C and Pt/Ti4O7 anodes without Ir by replacing the H2 with N2 while maintaining a constant current density (200 mA cm−2) to simulate the fuel starvation. The result shows that the MEA with Pt/Ti4O7 as an anode catalyst displays a reversal time double that of the MEA with Pt/C as the anode even though the time duration is limited without the OER catalyst (53 s vs. 135 s), suggesting the enhanced anti-oxidation ability for Ti4O7 under high potential. Further characterization has been performed to investigate the properties of high-surface area Ti4O7. The electronic conductivities of the XC-72R carbon support, Ti4O7, and TiO2 were measured between 1 and 30 MPa using the four-terminal method. As shown in Fig. S4 (ESI†), the electrical conductivity of Ti4O7 is only one order of magnitude lower than that of XC-72R at the same pressure. Specifically, the electrical conductivity of Ti4O7 is 2.37 S cm−1 under 30 MPa, while the value for XC-72R is 26.9 S cm−1. By contrast, the conductivity of TiO2 was also measured as a reference with 4.3 × 10−4 S cm−1, indicating a significant improvement by introducing an oxygen vacancy. | ||
| Fig. 1 (a) Schematic diagram of the synthesis procedure of Pt/Ti4O7. H2/air polarization curves of Pt/Ti4O7 anodes under different (b) Pt contents, (c) ionomer contents, and (d) Pt loadings. (e) Comparison of commercial Pt/C and Pt/Ti4O7 under stoichiometry flows (1.5/2.5) and fixed flows (0.5 L/1 L). (f) Summary of the Pt/Ti4O7 anode catalyst layer parameter optimization principle. | ||
2.2 Characterization of Ti4O7, Pt/Ti4O7 and Ir@IrOx/Pt/Ti4O7
The catalyst deposition is demonstrated in Fig. 2a. TEM was used to investigate the morphology of the as-prepared Ti4O7. As shown in Fig. 2b, the size of Ti4O7 is about 200–300 nm with a smooth surface, and a lattice space of 0.334 nm was acquired and attributed to Ti4O7 (020), as shown in Fig. 2c. EDS mapping results are also presented in Fig. 2c, and the relative atomic fraction is displayed in Fig. S5 (ESI†). The Ti and O elements are distributed evenly, and the atomic fraction analyzed by EDS mapping is roughly 61.15% for O and 38.85% for Ti. This is close to the theoretical atomic fraction of Ti4O7 (63.6% for O and 36.4% for Ti). The specific surface area of the as-prepared Ti4O7 was determined using the nitrogen adsorption isotherm. As shown in Fig. 2d, it has a specific surface area of 166 m2 g−1, close to the carbon support XC-72R (with a surface area of 215 m2 g−1). The morphology of 40% Pt/Ti4O7 was examined using HR-TEM, as shown in Fig. 2e, f and Fig. S6 (ESI†). The Ti4O7 support is coated with an intensive Pt nanoparticle shell. Pt nanoparticles of 3–4 nm diameter are fully distributed on the Ti4O7 support, displaying a high metal coverage and confirming the 40% Pt content optimization in Fig. 1b. As illustrated in Fig. 2f, the atomic TEM image demonstrates the deposition of Pt with a lattice space of 0.227 nm. Besides, the Pt nanoparticles connect mutually following the distribution of Pt nanoparticles, indicating the potential electron transfer pathway. Further EDS mapping validates the uniform dispersion of Ti and O elements and the outer distribution of Pt, as shown in Fig. S7 (ESI†). | ||
| Fig. 2 (a) Schematic diagram of the synthesis procedure of Ir@IrOx/Pt/Ti4O7. (b, c) TEM images of Ti4O7 and the corresponding EDS mapping. (d) N2 adsorption and desorption isotherms of XC-72R and Ti4O7. (e) and (f) HR-TEM images of 40% Pt/Ti4O7. (g) XRD patterns of carbon-supported and Ti4O7-supported catalysts. (h) and (i) TEM images of Ir@IrOx/Pt/Ti4O7 and (j) the corresponding EDS mapping. (k) XPS Pt 4f and Ir 4f spectra of Ir@IrOx/Pt/Ti4O7. (l) XPS depth profiles of Ir 4f for Ir@IrOx/Pt/Ti4O7 with various Ar sputtering times. | ||
Due to the sluggish OER activity of Pt, Ir species will be added to boost the OER activity and prolong the OER plateau, protecting the anode catalyst layer from carbon corrosion. Traditionally, the micro-size commercial IrO2 is utilized to physically mix with an anode catalyst to form an RTA. However, the large IrO2 particles reduce the expensive Ir utilization and disrupt the mass transfer pathway. An enhanced OER activity catalyst with higher Ir utilization is desired further to protect the anode catalyst layer under anode reversal. The nano-size core–shell Ir@IrOx/Pt/Ti4O7 directly deposits Ir species on the Pt/Ti4O7 surface, greatly improving the Ir utilization and minimizing the repercussion of mass transfer pathway distortion. The XRD patterns of Pt/C, Ir@IrOx/Pt/C, Pt/Ti4O7, Ir@IrOx/Pt/Ti4O7 and Ir@IrOx directly deposited on Ti4O7 (Ir@IrOx/Ti4O7) are shown in Fig. 2g. For carbon-supported catalysts, the diffraction peaks at 39.7°, 46.2°, 67.4°, 81.3°, and 85.7° emerged on the Pt/C and Ir@IrOx/Pt/C samples, which corresponded to the (111), (200), (220), (311), and (222) facets of metallic Pt, respectively. A strong peak appeared between 20° and 30°, which is attributed to the carbon support. For Ti4O7-supported catalysts, the diffraction peaks of Pt/Ti4O7 and Ir@IrOx/Pt/Ti4O7 samples are still well indexed to the characteristic diffraction peaks of Ti4O7, implying that successful deposition of Pt on the Ti4O7 support and the as-prepared high specific surface area Ti4O7 maintained the original crystalline structure even after 12 hours of reduction reaction and 4 hours of air exposure heating. The size of Pt nanoparticles is estimated based on the Pt (220) peak using the Scherrer equation.21,22 The calculation results are shown in Fig. 2g, and similar Pt sizes are obtained for all samples. No Ir or IrO2 peak is observed for Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 since the Ir particles are small, and the signal is covered by the strong Pt signal. Specifically, for better comparison and to examine the chemical state of Ir, the Ir content is increased to 50% for Ir@IrOx directly deposited on Ti4O7 (Ir@IrOx/Ti4O7) so that the Ir peak can be observed. As shown in Fig. 2g, Ir@IrOx/Ti4O7 presents a strong XRD peak of Ir@IrOx/Ti4O7 at 40.6°, matching the (111) facet of metallic Ir. Besides, no peak is found at 28.0° and 34.7°, characteristic peaks for the oxidation state of Ir. Similarly, the XRD pattern of Ir@IrOx directly deposited on carbon (Ir@IrOx/C) is shown in Fig. S8 (ESI†). The diffraction peaks at 40.6°, 69.1°, and 83.4° confirm the metallic state of Ir.
The TEM images of Ir@IrOx/Pt/Ti4O7 are displayed in Fig. 2h, i and Fig. S9 (ESI†). The size of the Ti4O7 support is again confirmed by measuring the particle size in Fig. 2h. The HR-TEM image is shown in Fig. 2i, and two typical-size nanoparticles deposit intensively on the Ti4O7 surface. The larger particles are confirmed as Pt by analyzing the lattice space of 0.227 nm, while the smaller particles are Ir with a lattice space of 0.222 nm. The corresponding EDS mapping was also conducted, as shown in Fig. 2j. The Pt and Ir elements are distributed thoroughly and evenly on the surface of Ti and O elements, indicating a metal-connected nanoparticle network. XPS was utilized to investigate the valence states of Pt and Ir in Ir@IrOx/Pt/Ti4O7, as shown in Fig. 2k. The binding energy positions of Pt(0) 4f are located at 71.0 eV and 74.3 eV, while the Pt(II) 4f peaks are present at 72.4 eV and 75.7 eV, and the Pt(IV) 4f peaks appear at 74.2 eV and 77.5 eV.23 Similarly, the binding energy positions of Ir(0) 4f lie at 60.8 eV and 63.8 eV, while the Ir(IV) 4f peaks present at 61.7 eV and 64.7 eV, and the Ir(III) 4f peaks appear at 62.6 eV and 65.6 eV.24 According to the deconvolution analysis of Pt 4f and Ir 4f spectral peaks, Pt(0) and Ir(IV) present a dominant percentage even though Pt(II), Pt(IV), Ir(0), and Ir(III) also exist. A similar test was conducted for Ir@IrOx/Pt/C to eliminate the Ir–O interaction between Ti4O7 and Ir. As shown in Fig. S10 (ESI†), the Ir(IV) is still dominant compared with Ir(II) and Ir(0), suggesting the existence of IrOx species. To further confirm the structure of the Ir species, XPS tests were performed at various sputtering depths to reveal the Ir valence state distribution, and the results are shown in Fig. 2l. The sputtering depth interval is 0.025 nm for each adjacent XPS Ir 4f spectrum, and the total sputtering depth is 0.25 nm. The Ir 4f7/2 and 4f5/2 peaks show negative core-level shifts during continuous etching when the sputtering depth is below 0.1 nm, displaying a higher Ir(0) 4f peak area ratio. The reason can be explained by the surface oxidation of Ir nanoparticles and the oxidation layer thickness is approximately 0.1 nm. When the sputtering depth keeps on increasing up to 0.25 nm, the Ir 4f peaks remain almost unchanged, implying the metallic Ir dominates inside, which is consistent with the XRD results. Considering the metallic Ir XRD diffraction peaks, metallic Ir lattice space observed in TEM images and surface Ar sputtering of XPS analysis, it can be concluded that the external Ir on the particle surface is oxidized, while the inner Ir remains in the metallic state, indicating the core–shell Ir@IrOx structure.
2.3 Electrochemical results of Ti4O7-supported catalysts
The electrochemical performance was evaluated by the half-cell tests and MEA full-cell tests. First, linear sweep voltammetry (LSV) was conducted to assess the HOR and OER activities. As shown in Fig. 3a, Pt/Ti4O7 exhibits satisfactory HOR activity similar to the commercial Pt/C, with diffusion-limited plateaus reached within 70 mV. Meanwhile, IrO2 can hardly oxidize hydrogen below 0.3 V (vs. RHE), indicating little contribution to the anode HOR activity. As presented in Fig. 3b, the OER activity test demonstrated that Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 share almost identical activity. Commercial IrO2 displayed a larger overpotential than Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 (390 mV vs. 360 mV@10 mA cm−2) due to the larger particle size and lower catalyst utilization. The OER activity of Pt/C is also sluggish below 1.7 V (vs. RHE), implying the majority of OER activity of the RTA comes from the Ir. For better comparison with Ti4O7 and carbon supports, the polarization performance tests at the stoichiometry flow (1.5/2.5) before and after the reversal tests were performed by traditionally adding commercial IrO2 to form Pt/C + IrO2 and Pt/Ti4O7 + IrO2 reversal tolerance anodes, as shown in Fig. 3d. Similarly, the polarization performances of Ir@IrOx/Ti4O7 and Ir@IrOx/Pt/C fabricated RTAs are presented in Fig. 3e. In addition, the reversal tests were performed, and the results are displayed in Fig. 3c. Initially, no significant performance difference is observed for Pt/C + IrO2 and Pt/Ti4O7 + IrO2 RTAs before the reversal tests with the maximum power density of 915 and 937 mW cm−2 (0.606 V vs. 0.613 V @ 1000 mA cm−2). The reversal tests were also conducted, and the Pt/Ti4O7 + IrO2 RTA can achieve a reversal time four times longer than the Pt/C + IrO2 RTA (112 min vs. 27 min). The period between 1.4 V and 2.0 V is prominently longer for the Pt/Ti4O7 + IrO2 RTA due to the enhanced anti-corrosion ability of Ti4O7. The polarization performance after the reversal test is also shown in Fig. 3d, and the results show that the Pt/C + IrO2 RTA loses approximately 38% performance in 27 min (from 915 to 573 mW cm−2 and from 0.606 V to 0.523 V @ 1000 mA cm−2). Meanwhile, the Pt/Ti4O7 + IrO2 RTA displays about 92% performance retention after a 112 min reversal time (from 937 to 860 mW cm−2 and from 0.613 V to 0.586 V @ 1000 mA cm−2), suggesting that using Ti4O7 as the catalyst support can effectively prolong the reversal time and protect the catalyst layer structure. Likewise, in terms of the Ir@IrOx/Pt/C and Ir@IrOx/Ti4O7 RTAs, similar initial performances are presented with a maximum power density of 1000 and 973 mW cm−2 (0.603 V vs. 0.609 V @ 1000 mA cm−2). For the Ir@IrOx/Pt/C RTA, the voltage reaches the terminal voltage in 39 min, while that of the Ir@IrOx/Pt/Ti4O7 RTA does not drop rapidly but is maintained up to 367 min, which is nearly ten times longer than that of the Ir@IrOx/Pt/C RTA. The postmortem performances were also tested to evaluate the degradation of Ir@IrOx/Pt/Ti4O7 and Ir@IrOx/Pt/C RTAs. Regarding the Ir@IrOx/Pt/C RTA, noticeable performance loss was observed even though the reversal time is significantly shorter than that of the Ir@IrOx/Pt/Ti4O7 RTA, as shown in Fig. 3e. The maximum power density of Ir@IrOx/Pt/C decreases apparently by 467 mW cm−2, from 1000 mW cm−2 to 533 mW cm−2 (from 0.603 V to 0.485 V @ 1000 mA cm−2), showing a 47% performance loss in half-hour of cell reversal duration. By contrast, the Ir@IrOx/Pt/Ti4O7 RTA presents a better performance retention ability, and the maximum power density reduces by 112 mW cm−2, from 973 mW cm−2 to 861 mW cm−2 (from 0.609 V to 0.593 V @ 1000 mA cm−2), with 88% performance retention after more than six hours of cell reversal test. The situation is similar when applying the fixed flow (0.5/1 L min−1), as shown in Fig. S11 (ESI†). Sufficient flows are provided to minimize the effect of catalyst layer thickness since Ti4O7 has a much larger mass weight when compared with carbon, which means a thinner catalyst layer thickness. Based on the similar initial performance, the Ir@IrOx/Pt/Ti4O7 and Pt/Ti4O7 + IrO2 RTAs can maintain 85% and 90% maximum power density after the reversal test. The first reversal times and degradation rates of the four anode MEAs are summarized in Fig. 3f. The Ti4O7-supported RTAs display a significantly longer reversal time and two orders of magnitude lower degradation rates than carbon-supported counterparts. Particularly, Ir@IrOx/Pt/Ti4O7 and Pt/Ti4O7 + IrO2 RTAs present 0.044 and 0.241 mV min−1 at 1000 mA cm−2 voltage loss, while the values for Ir@IrOx/Pt/C and Pt/C + IrO2 are 3.03 and 3.07 mV min−1 at 1000 mA cm−2, suggesting enhanced reversal tolerance and better durability for the Ti4O7-supported RTAs. The reversal test principle is illustrated in Fig. 3g for easier understanding. The reversal test was conducted by replacing the H2 with N2 while maintaining a constant current density (200 mA cm−2) to simulate fuel starvation.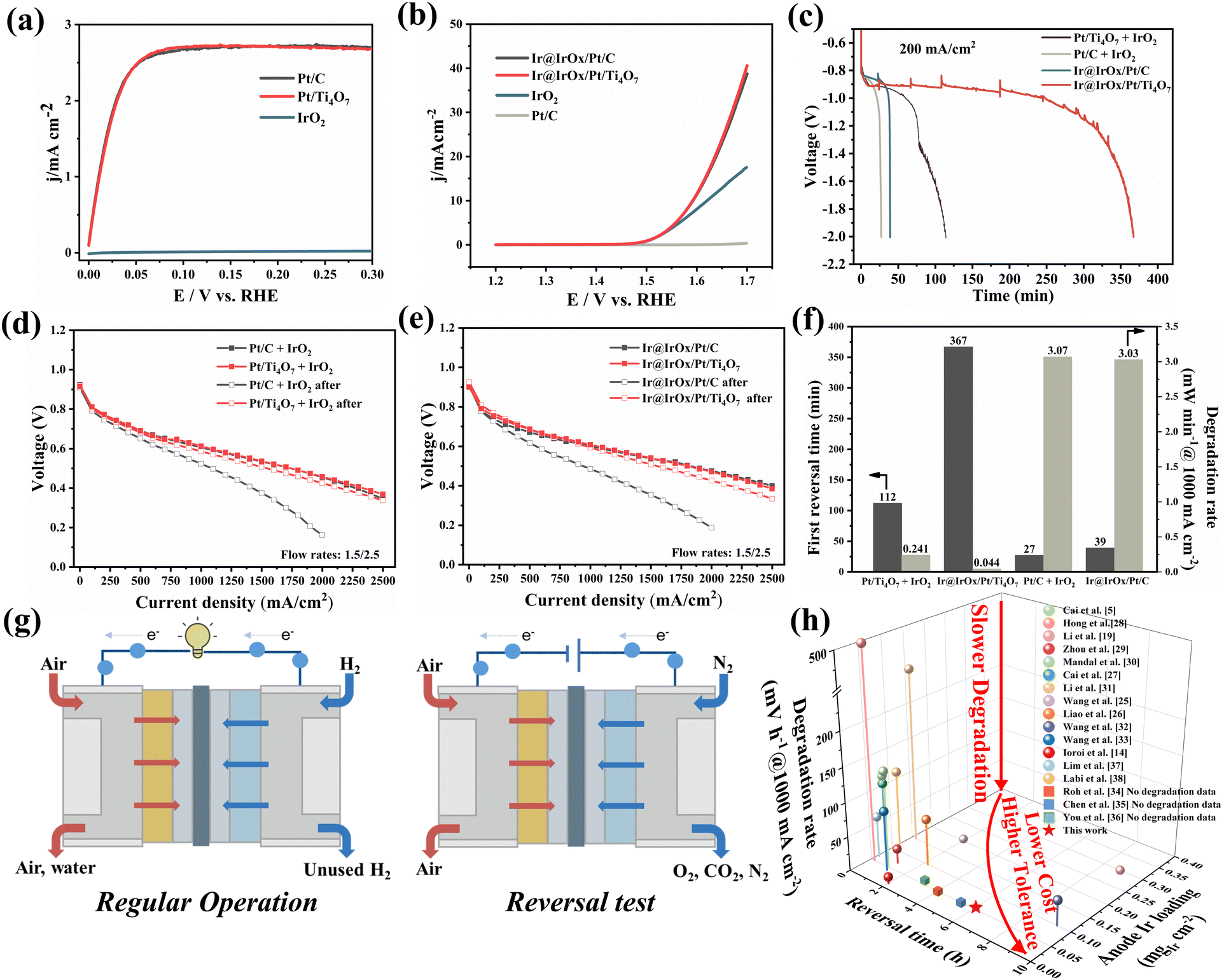 | ||
| Fig. 3 (a) HOR polarization curves of Pt/Ti4O7, the commercial Pt/C, and the commercial IrO2 in H2-saturated 0.1 M HClO4, (b) OER polarization curves of Ir@IrOx/Pt/C, Ir@IrOx/Pt/Ti4O7, commercial Pt/C and commercial IrO2 in N2-saturated 0.5 M H2SO4 at a scan rate of 5 mV s−1 and rotational speed of 1600 rpm. (c) Reversal tolerance test and H2/air polarization curves of (d) Pt/C + IrO2 and Pt/Ti4O7 + IrO2, (e) Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 RTAs under stoichiometry flows (1.5/2.5). (f) Comparison of reversal times and maximum power density degradation rates under fixed flows. (g) Schematic illustration of the anode reversal test. Components from left to right: cathode flow field plate, CGDL, CCL, PEM, ACL, AGDL, and anode flow field plate. (h) The comparison of anode Ir loading, first reversal time and degradation rate with recently reported literature. | ||
EIS at 200 mA cm−2 was also conducted to analyze the impedance change before and after the reversal tests. The impedances were fitted by the equivalent circuit model shown in Fig. S12 and S13 (ESI†), and the fitting results are summarized in Table S1 (ESI†). As shown in Fig. S12 (ESI†), the initial impedances are similar for Pt/C + IrO2 and Pt/Ti4O7 + IrO2 anode cells. Qualitatively, the initial ohmic resistances of the samples mentioned above, reflected by the x-left intercepts in the high-frequency region, are almost identical. Furthermore, the diameter of the semicircle represents the charge transfer resistance. In most cases, the charge transfer resistance of the HOR is considerably smaller than that of the ORR because of the faster kinetics.25,26 This makes isolating the HOR charge transfer resistance challenging unless the anode electron or proton transfer pathways are obstructed. The slight difference in the cathode charge resistance barely affects the polarization performance. After the reversal tests, the impedance of the Pt/C +IrO2 anode cell increases. At the same time, the Pt/Ti4O7 + IrO2 counterpart remains stable, as shown in the hollow point curves in Fig. S12 (ESI†). Specifically, concerning the Pt/C + IrO2 anode cell, the increased resistances lie on the ohmic and the anode charge transfer resistance.
In contrast, the Pt/Ti4O7 + IrO2 anode cell mainly concentrates on the anode charge transfer resistance, indicating the enhanced anode catalyst layer structure protection ability of the Ti4O7 support. When it comes to Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 anode cells, there is an analogous situation where the ohmic resistance and anode charge transfer resistance both increase significantly for the Ir@IrOx/Pt/C anode cell, as shown in Fig. S13 (ESI†). By comparison, the ohmic resistance for the Ir@IrOx/Pt/Ti4O7 anode cell barely changes, while the anode charge transfer resistance becomes more prominent. According to the EIS results, the degradation of the Ti4O7-supported anode catalyst layer is not caused by the catalyst layer structure damage, which is validated by SEM images, but due to the obstruction of electron and proton transfer pathways, indicating that the anode catalyst has undergone oxidation. Since the ionomer facilitates the transfer of protons, it is reasonable to infer and consider the resistance to electron transfer.
It is also vital to compare the performance of RTAs multidimensionally and simultaneously, including the Ir loading, first reversal time, and degradation rate. Since the Ir loading represents the cost of RTA fabrication, the reversal time reflects the reversal tolerance ability, and the degradation rate indicates the practical utilization potential. It is meaningless to chase a long reversal time but sacrifice the MEA performance or cease to function after the reversal test because the reversal time is relatively short in the actual operation. It is significant to keep balance among the three parameters. As shown in Fig. 3h and Table S2 (ESI†), the combination of low Ir loading, long reversal time, and slow performance degradation achieved by the Ir@IrOx/Pt/Ti4O7 is comparable to, and in some cases even surpasses, those reported for recent RTAs.5,14,19,25–38
2.4 Postmortem characterization and degradation origin
The in-plane and cross-section of anode catalyst layers were scanned using SEM to evaluate the anode catalyst layer destruction, as illustrated in Fig. 4a. For the commercial IrO2 addition samples, micro-size IrO2 is observed in the in-plane images before the reversal tests. The surfaces of the carbon-supported anode catalyst layers become rough and irregular after the reversal tests. At the same time, the darker area is attributed to the adhesive carbon fibers in the GDL, indicating severe carbon corrosion during the reversal test.6,39 The micro-size holes appear on the carbon-supported anode surface, further suggesting the destruction of catalyst layers. Notable thinner anode catalyst layer thickness is also found when the Pt/C + IrO2 anode decreases from 3.2 μm to 2.5 μm and the Ir@IrOx/Pt/C anode reduces from 2.6 μm to 2.1 μm. By contrast, the Ti4O7-supported anode catalyst layer displayed a smooth anode catalyst layer surface even though a much longer reversal time was experienced. Meanwhile, a significantly enhanced anode protection ability is found according to the consistent thickness of anode catalyst layers before and after the reversal tests (all approximately 1.5 μm). The SEM characterization further proves the better anode catalyst layer structure retention ability of the Ti4O7-supported catalyst, suggesting the boosted durability when using the Ti4O7 support. After the reversal tests, the TEM images of Pt/C +IrO2 and Pt/Ti4O7 +IrO2 are also shown in Fig. 4b–e and Fig. S14 (ESI†). The characterization focuses on Pt/C +IrO2 and Pt/Ti4O7 +IrO2 anodes since the Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 anodes follow a similar degradation phenomenon, but the Ir nanoparticles complicate the morphological observations. As shown in Fig. 4b, the structure of the carbon support becomes spiculate, and the Pt nanoparticles agglomerated with each other in certain areas of the Pt/C +IrO2 anode catalyst after the reversal test. Besides, irregular holes in the higher magnification TEM images appear inside the carbon support structure, as marked with dotted lines in Fig. 4c, suggesting severe carbon corrosion. Moreover, the lattice fringes of carbon intensively appear. In contrast, the amorphous carbon barely shows up, indicating the instability of amorphous carbon under high potential during the reversal test and preferential corrosion of amorphous carbon. In terms of the Pt/Ti4O7 + IrO2 anode, as shown in Fig. 4d, the Ti4O7 support maintains the original structure with a 200 to 300 nm diameter. Meanwhile, the Pt nanoparticles still deposit uniformly on the Ti4O7 surface, as proven by the EDS mapping (Fig. 4d and Fig. S15, ESI†). In addition, the encapsulation of a thin layer of TiOx covering the Pt particles is observed in the higher magnification TEM image due to the strong metal–support interaction (SMSI),40 which is indicated by white arrows in Fig. 4e. A multi-physics model was employed utilizing an aggregation model (Fig. S16, ESI†) to gain a more comprehensive understanding of carbon corrosion phenomena. As depicted in Fig. 4f, the CO2 concentration exhibits an increasing trend over time. For instance, the average CO2 concentration within the ionomer domain rises from 0 mol m−3 at 0 hours to 1.1 mol m−3 at 2 hours. Concurrently, the carbon particle undergoes gradual corrosion as time progresses. | ||
| Fig. 4 (a) SEM images of Pt/C +IrO2, Pt/Ti4O7 + IrO2, Ir@IrOx/Pt/C and Ir@IrOx/Pt/Ti4O7 before and after the reversal test. Each inset represents the in-plane anode surface. (b) and (c) TEM images of the Pt/C + IrO2 anode catalyst after the reversal test with different magnifications. (d) and (e) HAADF and TEM images of the Pt/Ti4O7 +IrO2 anode catalyst after the reversal test and the corresponding EDS mapping. (f) CO2 concentration distribution and carbon corrosion as a function of time for the carbon-supported anode catalyst. | ||
XPS was performed containing Pt 4f, Ti 2p, and O 1s core-level photoemission spectra before and after the reversal tests to compare and investigate the degradation origin of the Ti4O7-supported anode catalyst layer. As shown in Fig. 5, the relative concentrations of different valence states of Pt, Ti and O were calculated based on the spectral weights of the peaks after deconvolution. For Pt 4f, the peak position and deconvolution are presented before, and the content of Pt(0) significantly decreases from more than 60% to 50% after the reversal tests for both Pt/Ti4O7 + IrO2 and Ir@IrOx/Pt/Ti4O7 anodes. By contrast, the content of Pt(II) rises from approximately 20% to more than 30%. At the same time, Pt(IV) remains stable, suggesting that Pt(0) turns into Pt(II) and the Pt is oxidized during the reversal test. Furthermore, the peaks of Ti(III) 2p3/2 and Ti(IV) 2p3/2 lie at 457.1 eV and 458.5 eV, with a 5.7 eV splitting value variation.15,41 According to the Ti 2p spectral analysis, the contents of Ti remain steady throughout the reversal test as the Ti(III) and Ti(IV) contents fluctuate within 1%, which can be considered an experimental system error. The stable Ti(III) and Ti(IV) contents indicate that the Ti4O7 is hardly oxidized under high potential during the reversal test, consistent with previous research.14,42 Moreover, the O 1s spectra are also examined to distinguish the peaks of lattice oxygen (530.0 eV), adsorbed oxygen species in the oxygen vacancies (531.4 eV), surface hydroxyl (532.4 eV), and surface water (533.8 eV).15,43 The surface water peak emerges sharply after the reversal test, attributed to the water generated and adsorbed during the test. The oxygen adsorbed on the oxygen vacancy is still dominant even after the reversal tests, suggesting the prominent existence of Ti4O7. As previously reported,40,44,45 the Ti(III) can move to the Pt surface due to the SMSI and form a thin TiOx layer, which is permeable to proton and hydrogen and will not affect the HOR and Pt hydrogen under potential deposition (H-UPD) formation. According to Fig. S17 (ESI†), the CV tests reveal no significant difference in H-UPD before and after the reversal tests, suggesting normal hydrogen absorption and desorption. Based on these results, the decrease in performance observed after the reversal test is attributed to Pt oxidation. This oxidation occurs due to the formation of a thin layer of TiOx on the surface of the Pt, which impedes the electron transfer.
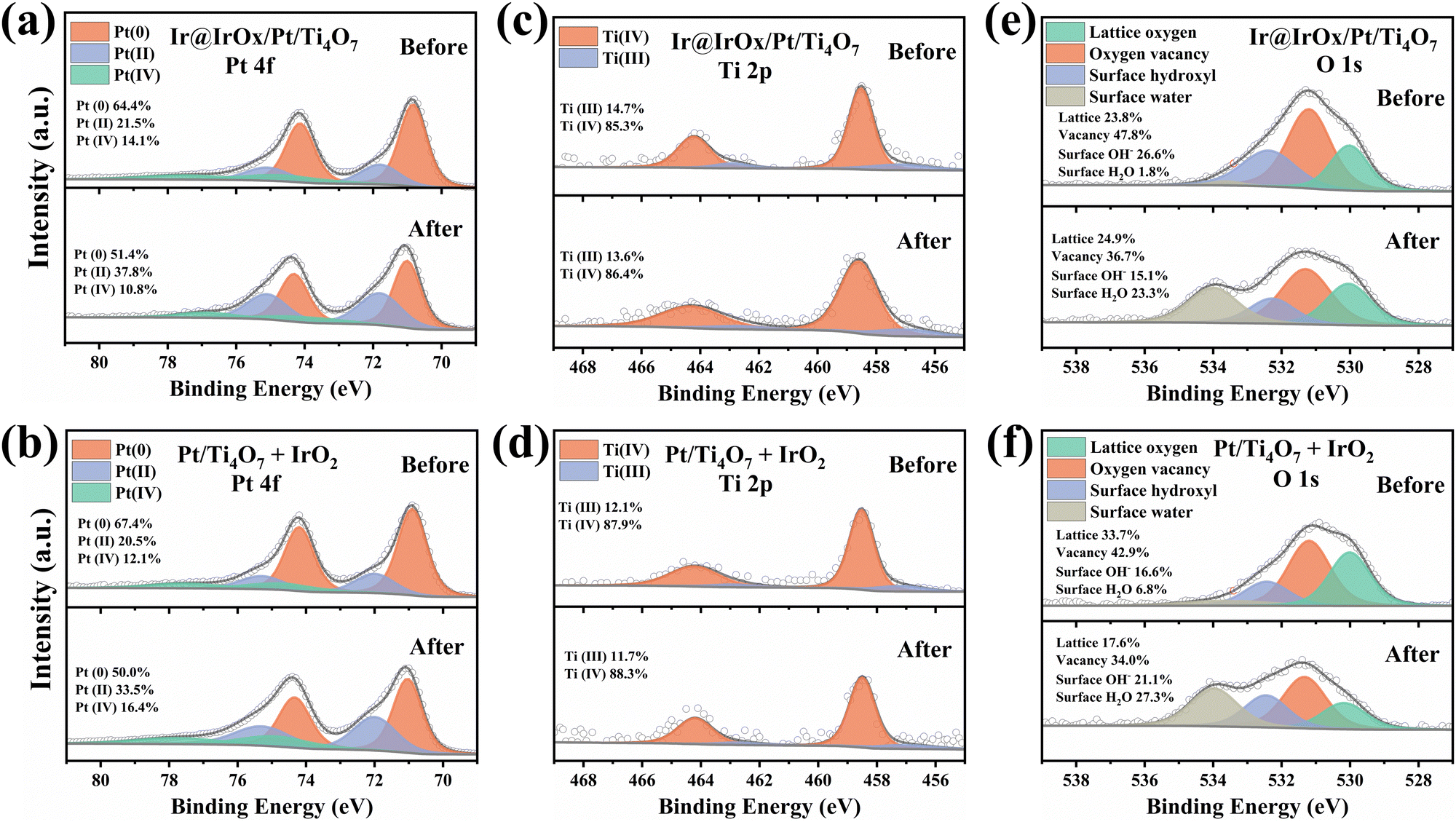 | ||
| Fig. 5 XPS (a) and (b) Pt 4f, (c) and (d) Ti 2p and (e) and (f) O 1s spectra of Ti4O7-supported catalysts (Ir@IrOx/Pt/Ti4O7 and Pt/Ti4O7 + IrO2) before and after the reversal test. | ||
3. Conclusions
In this contribution, we synthesized a high specific surface area Ti4O7 anode by a facile hydrogen-free method and utilized it as the anode catalyst support. The Ti4O7-supported anode catalyst layer parameters were optimized, displaying comparable performance with carbon-supported counterparts. Meanwhile, the screening of Ti4O7-supported anode catalyst layers showed enhanced reversal tolerance than a carbon-supported catalyst, whether in traditional IrO2 addition or novel Ir@IrOx/Pt/Ti4O7 core–shell Ir deposition. Specifically, the Ir@IrOx/Pt/Ti4O7 RTA presented a 367 min first reversal time and 0.044 mV min−1 at 1000 mA cm−2 voltage degradation rate, approximately ten times higher and two orders of magnitude lower than those of the Ir@IrOx/Pt/C RTA under a low Ir loading (0.05 mgIr cm−2), respectively. Even when applying the traditional IrO2 addition method, the Pt/Ti4O7 + IrO2 RTA still showed a four times longer reversal time and two orders of magnitude lower degradation rate than the Pt/C + IrO2 RTA, maintaining almost identical initial polarization performance. The origin of Ti4O7-supported catalyst degradation was also investigated and attributed to the Pt oxidation caused by the formation of TiOx thin layers at Pt surfaces, which was verified by postmortem EIS, XPS, SEM, and TEM characterizations. This study reveals a novel practical perspective for RTA design by replacing carbon supports. It paves the way for future research of carbon-free anodes for proton exchange membrane fuel cells.4. Experimental
4.1 Synthesis of the Ti4O7 support
Ti4O7 was prepared by a facile hydrogen-free method developed elsewhere.46 Typically, titanyl sulfate (TiOSO4) was applied as the Ti precursor and then hydrolyzed as TiO2 in water. Subsequently, PEG400 was added under continuous stirring so that TiO2 was coated with a uniform PEG400 layer. The mass ratio of PEG400 to TiO2 was kept at 1.5. After that, the water was removed at 80 °C, and the temperature increased to 150 °C for 5 hours. The as-prepared brown powder was ground using an agate mortar and transferred to a sealing quartz crucible. Then, the sample was heated to 967 °C under pure argon at 10 °C min−1. After the sample was cooled to room temperature, the powder was ground again, then the fine powder was collected.4.2 Synthesis of Pt/Ti4O7 and IrOx@Ir/Pt/Ti4O7
Pt/Ti4O7 was synthesized by a simple ethanol reduction method. Typically, 18 mg Ti4O7 was added into a 500 mL flask containing 200 mL ethanol and 200 mL DI water. After ultrasonication for 1 hour, H2PtCl6·xH2O (Sigma-Aldrich, 37.5% Pt content) aqueous solution with 12 mg Pt was added into the above dispersion, which was then stirred for 3 hours at room temperature to obtain a homogeneous solution. Subsequently, the solution was heated to 95 °C for 12 hours. When it cooled down to room temperature, the black powder was collected by vacuum filtration, followed by sufficient hot DI water washing. Lastly, Pt/Ti4O7 was obtained by drying at 60 °C overnight.IrOx@Ir/Pt/Ti4O7 was synthesized using the as-prepared Pt/Ti4O7. Initially, Pt/Ti4O7 was synthesized to support the Ir sol. The weight ratio of Ir to Pt was controlled at 1:2. The Ir sol was obtained using a hydrothermal method with aqueous IrCl3·3H2O solution (Macklin, 53% Ir content) containing 6 mg Ir and 68 mL of anhydrous ethanol. The solution was mixed and stirred in a 100 mL Teflon-lined stainless steel autoclave and then sealed and maintained at 150 °C for 4 hours. After cooling to room temperature, the prepared Pt/Ti4O7 was added to the Ir sol, and the Ir was adsorbed onto the Pt/Ti4O7 surface. The mixture was heated to 60 °C with continuous stirring to dry the solvent. It is important to note that nitrogen gas was purged into the mixture during the drying process to prevent the self-ignition of the nano-scale catalysts. The collected powders were annealed at 120 °C for 3 hours to strengthen the metal–support interactions. Finally, the powder was rinsed with deionized water and dried to obtain IrOx@Ir/Pt/Ti4O7. Similar treatment proceeded with commercial Pt/C to obtain the IrOx@Ir/Pt/C. For comparison, the IrOx@Ir/Ti4O7 was prepared by adding Ti4O7 into Ir sol and following the same subsequent processes.
4.3 Characterizations
The surface area of the catalyst was determined by BET analysis using nitrogen isotherms (Micromeritics ASAP 2460). X-ray diffraction (XRD) was performed on a 3 kW Rigaku SmartLab diffractometer with a Cu Kα radiation source at 40 kV and 30 mA at a scan rate of 8° min−1. The electronic conductivity was measured using an automatic powder resistivity tester (Suzhou Jingge Electronic Co. Ltd) with a pressure between 1 MPa and 30 MPa. X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Fisher ESCALAB Xi+ using monochromated Al Kα radiation. Scanning electron microscopy (SEM) images were acquired using a ZEISS Sigma 300 with an accelerating voltage of 5 kV. A Talos F200X G2 was used to obtain the transmission electron microscopy (TEM) images. The TEM images were acquired at 200 kV with the samples deposited on a holey carbon film-covered copper grid. High-resolution transmission electron microscopy (HR-TEM) was conducted using a Titan Themis G2 double spherical aberration-corrected transmission electron microscope with a 300 kV accelerating voltage.4.4 Electrode preparation and performance testing
All half-cell tests were conducted in a conventional three-electrode cell with an electrochemical workstation (Shanghai CH Instruments Inc.) on Pine Instruments under room temperature. For the HOR test, the catalyst ink was prepared using isopropyl alcohol, water, and Nafion D520 ionomer. The mass ratio of the ionomer to dry catalyst ink was kept at 30% for the carbon-supported catalyst and IrO2, and 4% for the Ti4O7-supported catalyst. Linear sweep voltammetry (LSV) was performed from 0 to 0.3 V vs. RHE at a scan rate of 5 mV s−1 and a rotational rate of 1600 rpm in H2-saturated 0.1 M HClO4. A glassy carbon electrode (GCE) with a diameter of 5 mm was employed as the working electrode, while a Pt mesh and a reversible hydrogen electrode (RHE) were selected as the counter electrode and reference electrode, respectively. The metal loading was controlled at 20 μg cm−2. A similar protocol was followed for the OER test. The electrolyte and the reference electrode were switched to 0.5 M H2SO4 and Ag/AgCl. Besides, the LSV scanning range was adjusted to 1.2 to 1.7 V vs. RHE. The Ir or Pt loading was controlled at 10 μg cm−2.Regarding the full-cell test, the anode catalyst ink was prepared by ultrasonicating the mixture of the catalyst, D520 Nafion ionomer, and 1:1 water/isopropanol solvent for 1.5 hours. The mass content of ionomer was kept at 30 wt% for Pt/C, Pt/C + IrO2 and IrOx@Ir/Pt/C, while the same parameter for Pt/Ti4O7, Pt/Ti4O7 + IrO2 and IrOx@Ir/Pt/Ti4O7 was 4 wt%. The cathode catalyst ink was treated similarly using the 47.1 wt% Pt/C (TEC10EA50E, Tanaka) catalyst. Then, the ink was sprayed onto both sides of the proton exchange membrane (Nafion 211, 25 μm, Chemours) with an effective area of 4 cm2. The Pt loading was controlled at 0.1 mgPt cm−2 and 0.4 mgPt cm−2 for the anode and cathode, respectively. The catalyst-coated membrane (CCM) and gas diffusion layers (MB30, AvCarb) were assembled to form the MEA. Two pieces of Teflon with 150 μm thickness were used as sealing gaskets to prevent gas leakage. The single-cell tests were all conducted on a Hephas HTS-125 fuel cell test station at 80 °C with 100% relative humidity (RH). Back pressures of hydrogen and air were 100 kPa. For the cell reversal test, 0.4 L min−1 nitrogen and 0.2 L min−1 air were supplied at the anode and cathode, during which the outlets of both the anode and cathode were exposed to ambient pressure. A constant current density of 0.2 A cm−2 was applied, and the cell voltage was recorded using an electrochemical workstation (Solartron EnergyLab XM). Then, the cell would shut down when the voltage reached −2.0 V.30,47 The polarization performance, cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS) tests were conducted before and after the cell reversal tests. The EIS was performed at 200 mA cm−2 under fully humidified H2/air, and the frequency was scanned from 100 kHz to 0.1 Hz. The CV was conducted in fully humidified N2/H2 with fixed flows and ambient pressure, scanning from 0.1 to 1.0 V at a scan rate of 50 mV s−1. To evaluate the polarization performance, two flow rate conditions were tested (fixed flows of 0.5/1 L min−1 and stoichiometry of 1.5/2.5) for comparison. The fixed flow test was carried out, ensuring ample provision of gases to address the mass transfer problem. Meanwhile, the stoichiometry flow test provided a practical condition to assess the actual performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 52276198 and 52293414), Shenzhen Key Laboratory of Advanced Energy Storage (No. ZDSYS20220401141000001) and the Fund of Shenzhen Municipal Central Government to Guide Local Science and Technology (2021Szvup084). The authors would also like to acknowledge the TEM resources of the Pico Center at SUSTech Core Research Facilities, which was supported by the Presidential Fund and the Development and Reform Commission of Shenzhen Municipality.References
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- Z. Li, Y. Wang, Y. Mu, B. Wu, Y. Jiang, L. Zeng and T. Zhao, Renewable Sustainable Energy Rev., 2023, 176, 113182 CrossRef CAS.
- P. Ren, P. Pei, Y. Li, Z. Wu, D. Chen and S. Huang, Prog. Energy Combust. Sci., 2020, 80, 100859 CrossRef.
- C. Cai, Y. Rao, J. Zhou, L. Zhang, W. Chen, Z. Wan, J. Tan and M. Pan, J. Power Sources, 2020, 473, 228542 CrossRef CAS.
- Y. Wang, C. Zhou, X. Xie, C. Yang, Q. Feng, J. Zou, X.-Z. Yuan, J. Fan, L. Zeng, H. Li and H. Wang, Int. J. Hydrogen Energy, 2020, 45, 996–1007 CrossRef CAS.
- Z. Li, H. Huang, Y. Wang, Y. Mu, Y. Jiang, Q. Zhang, T. Zhao and L. Zeng, Chem. Eng. J., 2023, 144189 CrossRef CAS.
- S. Ye, in PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications, ed. J. Zhang, Springer, London, 2008, pp. 835–860 Search PubMed.
- H. J. Liu, M. Q. Luo, L. X. Yang, C. L. Zeng and C. Fu, Ceram. Int., 2022, 48, 25538–25546 CrossRef CAS.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- J. E. Graves, D. Pletcher, R. L. Clarke and F. C. Walsh, J. Appl. Electrochem., 1991, 21, 848–857 CrossRef CAS.
- X. Sun, Z. Wang, W. Yan and C. Zhou, Catalysts, 2022, 12, 480 CrossRef CAS.
- J.-E. Won, D.-H. Kwak, S.-B. Han, H.-S. Park, J.-Y. Park, K.-B. Ma, D.-H. Kim and K.-W. Park, J. Catal., 2018, 358, 287–294 CrossRef CAS.
- T. Ioroi and K. Yasuda, J. Power Sources, 2020, 450, 227656 CrossRef CAS.
- S.-H. You, S.-M. Jung, K.-S. Kim, J. Lee, J. Park, H. Y. Jang, S. Shin, H. Lee, S. Back, J. Lee and Y.-T. Kim, ACS Energy Lett., 2023, 2201–2213 CrossRef CAS.
- S.-Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898–13899 CrossRef CAS PubMed.
- F. T. Angerasa, C.-Y. Chang, E. A. Moges, W.-H. Huang, K. Lakshmanan, Y. Nikodimos, J.-F. Lee, N. G. Habtu, M.-C. Tsai, W.-N. Su and B. J. Hwang, Mater. Today Energy, 2023, 34, 101312 CrossRef CAS.
- B. M. Stühmeier, A. M. Damjanović, K. Rodewald and H. A. Gasteiger, J. Power Sources, 2023, 558, 232572 CrossRef.
- Y. Li, W. Song, G. Jiang, Y. Yang, H. Yu, Z. Shao, F. Duan and Y. Yang, Front. Energy, 2022, 16, 852–861 CrossRef.
- Z. Wang, X. Jin, F. Chen, X. Kuang, J. Min, H. Duan, J. Li and J. Chen, J. Colloid Interface Sci., 2023, 650, 901–912 CrossRef CAS PubMed.
- C. Wang, Z. X. Liu, Z. Q. Mao, J. M. Xu and K. Y. Ge, Chem. Eng. J., 2005, 112, 87–91 CrossRef CAS.
- Y. Wang, J. Jin, S. Yang, G. Li and J. Qiao, Electrochim. Acta, 2015, 177, 181–189 CrossRef CAS.
- Z. Liu, L. M. Gan, L. Hong, W. Chen and J. Y. Lee, J. Power Sources, 2005, 139, 73–78 CrossRef CAS.
- C. E. Moore, F. Afsahi, A. P. Young and E. L. Gyenge, J. Phys. Chem. C, 2019, 123, 23361–23373 CrossRef CAS.
- Y. Wang, J. Liao, Z. Li, B. Wu, J. Lou, L. Zeng and T. Zhao, Int. J. Hydrogen Energy, 2022, 47, 13101–13111 CrossRef CAS.
- J. Liao, S. Zaman, Y. Wang, M. Yang, L. Yang, M. Chen and H. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 4092–4100 CrossRef CAS PubMed.
- C. Cai, Z. Wan, Y. Rao, W. Chen, J. Zhou, J. Tan and M. Pan, J. Power Sources, 2020, 455, 227952 CrossRef CAS.
- B. K. Hong, P. Mandal, J.-G. Oh and S. Litster, J. Power Sources, 2016, 328, 280–288 CrossRef CAS.
- X. Zhou, H. Ji, B. Li and C. Zhang, ACS Omega, 2020, 5, 10099–10105 CrossRef CAS PubMed.
- P. Mandal, B. K. Hong, J.-G. Oh and S. Litster, J. Power Sources, 2018, 397, 397–404 CrossRef CAS.
- Y. Li, L. Zhao, X. Du, W. Gao, C. Zhang, H. Chen, X. He, C. Wang and Z. Mao, Chem. Eng. J., 2023, 461, 141823 CrossRef CAS.
- Y. Wang, Y. Jiang, J. Liao, Z. Li, T. Zhao and L. Zeng, ACS Appl. Mater. Interfaces, 2022, 14, 56867–56876 CrossRef CAS PubMed.
- J. Wang, X. Zhou, B. Li, D. Yang, H. Lv, Q. Xiao, P. Ming, X. Wei and C. Zhang, Int. J. Hydrogen Energy, 2020, 45, 8930–8940 CrossRef CAS.
- C.-W. Roh, H.-E. Kim, J. Choi, J. Lim and H. Lee, J. Power Sources, 2019, 443, 227270 CrossRef CAS.
- W. Chen, C. Cai, S. Li, J. Tan and M. Pan, Int. J. Hydrogen Energy, 2021, 46, 8749–8757 CrossRef CAS.
- E. You, M. Min, S.-A. Jin, T. Kim and C. Pak, J. Electrochem. Soc., 2018, 165, F3094–F3099 CrossRef CAS.
- K. H. Lim, W. H. Lee, Y. Jeong and H. Kim, J. Electrochem. Soc., 2017, 164, F1580–F1586 CrossRef CAS.
- T. Labi, F. Van Schalkwyk, S. M. Andersen, P. Morgen, S. C. Ray and J. Chamier, J. Power Sources, 2021, 490, 229568 CrossRef CAS.
- Y. Wang, X. Xie, C. Zhou, Q. Feng, Y. Zhou, X.-Z. Yuan, J. Xu, J. Fan, L. Zeng, H. Li and H. Wang, J. Power Sources, 2020, 449, 227542 CrossRef CAS.
- B. M. Stühmeier, S. Selve, M. U. M. Patel, T. N. Geppert, H. A. Gasteiger and H. A. El-Sayed, ACS Appl. Energy Mater., 2019, 2, 5534–5539 CrossRef.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- T. Arai, O. Takashi, K. Amemiya and T. Takahashi, SAE Int. J. Alt. Power., 2017, 6, 145–150 CrossRef.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- T. N. Geppert, M. Bosund, M. Putkonen, B. M. Stühmeier, A. T. Pasanen, P. Heikkilä, H. A. Gasteiger and H. A. El-Sayed, J. Electrochem. Soc., 2020, 167, 084517 CrossRef CAS.
- B.-J. Hsieh, M.-C. Tsai, C.-J. Pan, W.-N. Su, J. Rick, H.-L. Chou, J.-F. Lee and B.-J. Hwang, Electrochim. Acta, 2017, 224, 452–459 CrossRef CAS.
- M. Chisaka, W. Nagano, B. Delgertsetseg and T. Takeguchi, Chem. Commun., 2021, 57, 12772–12775 RSC.
- X. Zhou, Y. Yang, B. Li and C. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 2455–2461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03921h |
| This journal is © The Royal Society of Chemistry 2024 |