
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable long-term lithium replenishment for enhancing energy density and cycle life of lithium-ion batteries†
Ganxiong
Liu‡
 ab,
Wang
Wan‡
a,
Quan
Nie
a,
Can
Zhang
a,
Xinlong
Chen
a,
Weihuang
Lin
c,
Xuezhe
Wei
b,
Yunhui
Huang
d,
Ju
Li
*e and
Chao
Wang
*a
ab,
Wang
Wan‡
a,
Quan
Nie
a,
Can
Zhang
a,
Xinlong
Chen
a,
Weihuang
Lin
c,
Xuezhe
Wei
b,
Yunhui
Huang
d,
Ju
Li
*e and
Chao
Wang
*a
aSchool of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: chaow@tongji.edu.cn
bSchool of Automotive Studies, Tongji University, Shanghai, 201804, China
cSchool of Chemical and Environmental Engineering, China University of Mining and Technology-Beijing, Beijing 100083, China
dState Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
eDepartment of Materials Science and Engineering and Department of Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: liju@mit.edu
First published on 28th December 2023
Abstract
A persistent challenge plaguing lithium-ion batteries (LIBs) is the consumption of active lithium with the formation of SEI. This leads to an irreversible lithium loss in the initial cycle and a gradual further exhaustion of active lithium in subsequent cycles. While prelithiation has been proven effective in compensating for this loss by introducing additional active lithium into batteries, prior studies have predominantly concentrated on offsetting the initial lithium loss, often overlooking the continuous lithium consumption that occurs throughout cycling. To address this challenge, we employed a sustained in situ lithium replenishment strategy that involves the systematic release of additional lithium inventory through precise capacity control during long-term cycling. Our method utilizes a lithium replenishment separator (LRS) coated with dilithium squarate-carbon nanotube (Li2C4O4–CNT) as the lithium compensation reagent. Placing Li2C4O4 on the separator rather than within the cathode significantly reduces disruptions in conduction pathways and inhibits catalytic reactions with LiFePO4, preventing the formation of carbon residues. When implemented in the LiFePO4||graphite battery system, our approach resulted in an impressive 12.9% capacity improvement in the initial cycle and a remarkable 97.2% capacity retention over 700 cycles, surpassing the comparison group, which exhibited 80% capacity retention after 426 cycles.
Broader contextThe development of a solid electrolyte interphase (SEI) in lithium-ion batteries (LIBs) inevitably consumes active lithium, resulting in a significant reduction of the reversible capacity and a shortened lifespan. Over the past decade, prelithiation has emerged as an effective strategy to counteract initial lithium loss. However, most efforts have focused solely on compensating for the initial lithium loss, neglecting the gradual depletion of lithium during cycling. This study introduces a controllable lithium replenishment strategy to achieve long-term capacity recovery within the battery. An air-stable Li2C4O4–CNT composite, characterized by its uniform spherical structure and excellent conductivity, was employed as a sacrificial reagent to provide additional lithium from the cathode side. By controlling the oxidation degree of Li2C4O4, the lithium replenishment process can be divided into several stages and precisely regulated. Consequently, we achieved higher energy density and significantly improved cycle life. |
Introduction
To meet the ever-growing energy demands, developing lithium-ion batteries (LIBs) with high energy density and prolonged cycle life has become a critical objective.1,2 However, due to unavoidable parasitic reactions such as the formation of solid electrolyte interface (SEI), the prevailing LIBs always suffer from active lithium loss (ALL), which is the primary cause of performance deterioration.3 While lithium metal batteries could offer a solution by providing excess lithium through Li metal anodes, their practical use is limited due to lithium metal's air instability and fragility.4,5 In contrast, in conventional LIBs where the cathode is the sole source of Li, ALL directly reduces the pool of reversibly transferable Li+ ions between cathode and anode, resulting in decreased energy density and cycle life. Consider that when all the active lithium in a cell comes from cathode-active materials like LiCoO2 or LiFePO4, every Li-ion that becomes inactive leaves an entire unit of CoO2 (13 times heavier than Li) or FePO4 (22 times heavier than Li) stranded on the cathode, functioning as inert “dead weight” forever. Thus, it would be more advantageous to employ a lighter and more cost-effective sacrificial Li source to compensate for ALL. Additionally, a controlled-release approach, similar to drug delivery mechanisms, would be preferable. Since ALL does not occur all at once, administering scheduled doses of full re-lithiation throughout a battery cell's entire lifespan, which can span decades, would yield significant advantages.When considering LIBs with a stable cathode material such as LiFePO4 (LFP), the degradation of the cathode structure leading to lithium consumption can be considered negligible.6 In this scenario, the ALL predominantly takes place on the anode side.7 Using prevailing commercial LIBs that employ graphite (Gr) or silicon-based (Si–C) anodes as an example, the ALL can be primarily attributed to the following two parts (Fig. 1):
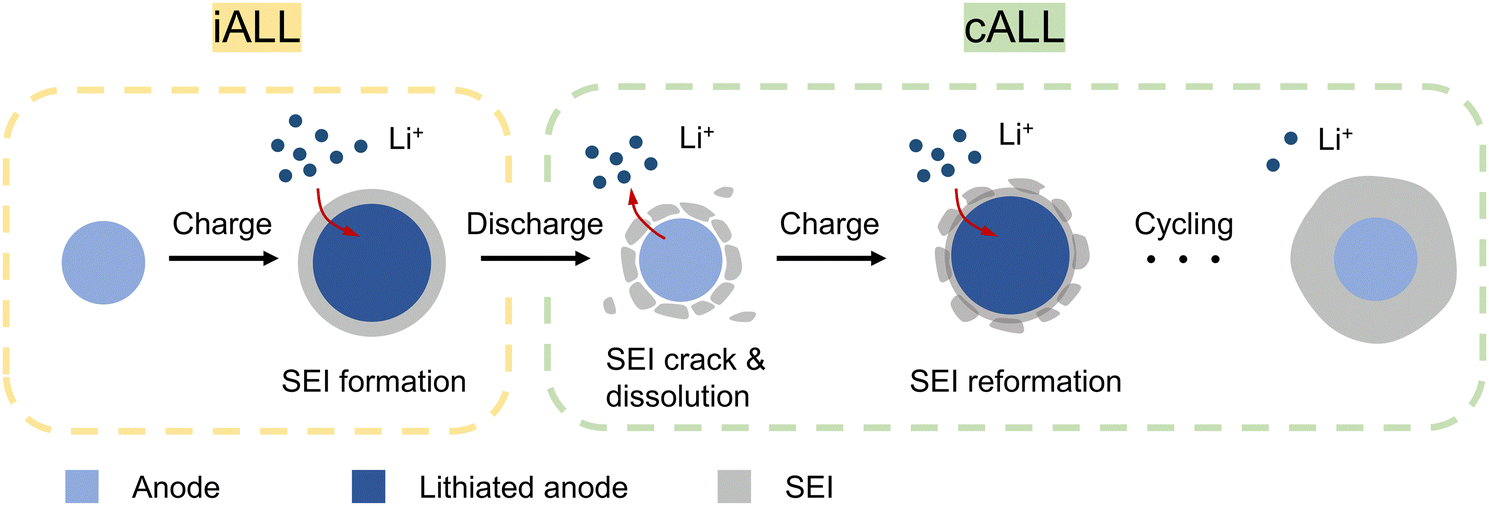 | ||
| Fig. 1 Schematic of the iALL and cALL that occurs during battery cycling. | ||
(I) Initial active lithium loss (iALL) due to the initial formation of SEI:
During the initial charging process, the organic electrolyte is electrochemically reduced and decomposed on the anode to form a SEI layer. This irreversible process leads to a significant iALL and a consequent low initial Coulombic efficiency (ICE).8 Graphite anodes typically experience an iALL of approximately 8–15%, while high-capacity alloy anodes (e.g., Si, Si–C, Sn, or SiOx) suffer even higher levels of iALL, ranging from 20–50%.9,10 Consequently, this phenomenon leads to reduced ICE and energy density of LIBs due to the cathode “dead weight” problem.
(II) Continuous active lithium loss (cALL) due to SEI-related side reactions:
During subsequent cycles, the anode materials gradually deplete active lithium ions due to ongoing side reactions that can thicken the SEI. This issue is particularly prominent in anodes with high volume change, leading to repeated breakage and growth of the SEI. As a result, active lithium ions are continuously consumed, resulting in a Coulombic efficiency (CE) lower than 100% during cycling.11 Assuming that all the decrease in CE is attributed to ALL at the anode, the capacity retention can be described by the equation: capacity retention = (CE)n, where n represents the cycle number.12 A low average CE signifies a substantial accumulation of lithium loss after prolonged cycling. However, the issue of cALL has often been disregarded, and limited research has been conducted on this matter.
To mitigate the ALL (ALL = iALL + cALL) issue and improve the energy density of current LIBs, a promising approach is through the implementation of a lithium replenishment strategy by storing an extra amount of lightweight active-lithium carriers in the battery system (which are expected to charge once, but not charged and discharged multiple times reversibly like the cathode-active materials).3,7,13 At present, lithium replenishment can be conducted as a separate pre-process before battery cycling, which is also known as prelithiation. For example, directly contacting or electrochemically reacting the anode with Li metal,14,15 or chemically reacting the anode with Li-containing reductants (e.g., Li-polycyclic aromatic hydrocarbons (Li-PAHs)),16,17 has been proposed to solve the ALL problems. Alternatively, lithium replenishment can also be achieved by incorporating high-capacity Li-containing additives directly into the cathode or anode materials during electrode processing.18–22 For example, previous studies have explored the use of sacrificial lithium salts (e.g., Li2O, Li2S, Li5FeO4, Co/Li2O, etc.) to compensate for the iALL in LIBs.18,23–28 However, these Li-rich additives usually exhibit strong alkalinity and poor air stability.29 Their decomposition can lead to the formation of poorly-conductive residues or voids, which can impede charge transfer and cause degradation in the cathode's structure.
Organic lithium salts like lithium oxalate (Li2C2O4) and dilithium squarate (Li2C4O4) are known for their stability.30–32 However, these sacrificial lithium salts can generate significant amounts of gas, potentially causing severe damage to the electrode structure and thereby degrading the battery's electrochemical performance. If, on the other hand, all the decomposed gas can be expelled from the cell, the specific capacity can be exceptionally high, reaching up to 3845 mA h g−1 (equivalent to that of pure lithium). Recently, Guo et al. introduced novel functional separators coated with Li2C2O4 for waste electrode recovery and coated with Li5FeO4 to prevent cathode slurry gelation.29,33 These functional separators can also prevent damage to the pristine electrode.
While significant efforts have been devoted to developing novel lithiation reagents and methods to compensate for iALL, the continuous and long-term capacity loss (cALL) that occurs throughout the entire cycle life of a battery has often been overlooked. To address both iALL and cALL, we propose a novel lithium replenishment separator (LRS). Unlike conventional lithium replenishment strategies that deplete the entire lithium inventory in the initial cycle to counteract iALL, our approach reserves an additional amount of active lithium inventory within the LRS. This reserve can be gradually released in subsequent cycles by precisely controlling the charge cutoff voltage and capacity. In this study, we utilized a well-formed Li2C4O4–CNT composite produced via spray drying as the sacrificial salt. Li2C4O4 was selected for its favorable characteristics, especially its moderate oxidation potential (Fig. 2a). Furthermore, we observed the catalysis of LFP to Li2C4O4. Coating Li2C4O4 on the separator reduces the contact area between Li2C4O4 and LFP, effectively inhibiting the catalytic decomposition of Li2C4O4 to CO2. This results in reduced carbon residue and leads to improved battery performance.
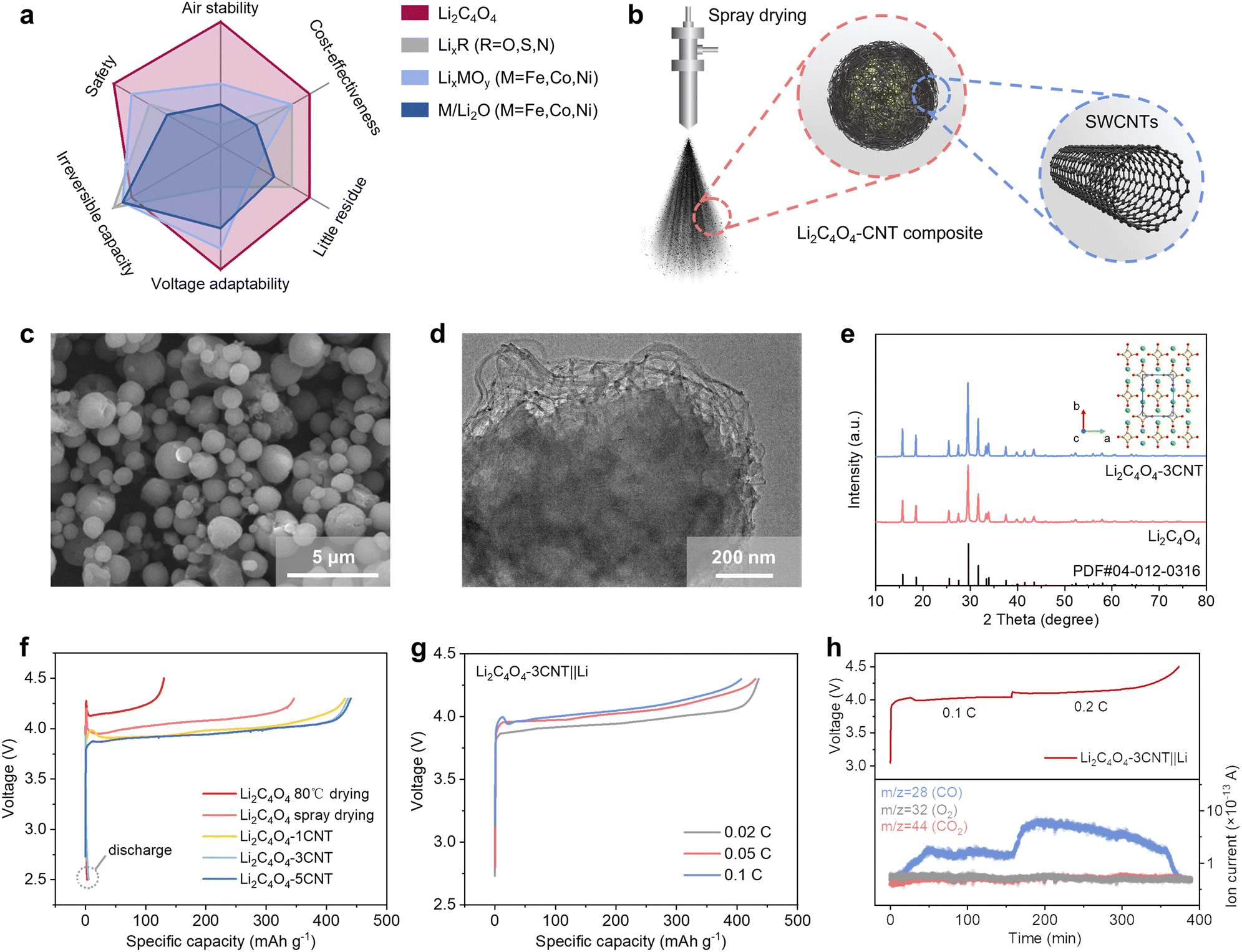 | ||
| Fig. 2 Synthesis and characterization of Li2C4O4–3CNT composite. (a) Figure of merits on Li2C4O4 with other representative prelithiation agents. (b) Schematic of the fabrication process of the Li2C4O4–CNT composites by spray drying method. (c) SEM and (d) TEM images of the as-prepared Li2C4O4–CNT composites. (e) XRD pattern of Li2C4O4 and Li2C4O4–CNT (inset is its corresponding crystal structure). (f) Galvanostatic charge–discharge profiles of Li2C4O4 at 0.02C. (g) Initial charge profiles of Li2C4O4–3CNT||Li half cells at different C-rates. (h) In situ differential electrochemical mass spectrometry (DEMS) test of the Li2C4O4–3CNT||Li half cells. | ||
Our innovative long-term lithium replenishment method ensures a sustained and controlled release of lithium ions throughout the battery's lifespan, effectively mitigating both the capacity loss arising from iALL and the capacity degradation associated with cALL, thus significantly extending the cycle life of LIBs. When applied to LFP||Gr full cells, our battery system exhibits exceptional electrochemical performance, demonstrating 12.9% capacity improvement in the initial cycle and excellent capacity retention of 97.2% after 700 cycles. While this capacity-controlled long-term lithium replenishment requires an automatic exhaust valve to degas in commercial cells, we have also developed an alternative automatic anode-supported long-term lithium replenishment strategy with the excess lithium from the first cycle stored in the graphite anode. This approach avoids subsequent degassing and is more compatible with existing battery manufacturing.
Results and discussion
Characterization of the Li2C4O4–CNT composite
While Li2C4O4 demonstrates outstanding combination properties compared with other relevant prelithiation materials, the electronic insulating nature of Li2C4O4 poses a challenge as it hinders the complete electrochemical decomposition of the material and leads to increased overpotential. To overcome these limitations, we employed a spray drying method to reduce the particle size of Li2C4O4 and incorporated CNT to form Li2C4O4–CNT composites (as shown in Fig. 2b). The scanning electron microscope (SEM) image of the Li2C4O4–3CNT composite is shown in Fig. 2c. A comparison with the bulk Li2C4O4 obtained through vacuum drying at 80 °C (Fig. S1, ESI†) reveals distinct differences in particle size and sphericity. In contrast to the bulk material, the particles produced via the spray drying method exhibit a more uniform size distribution, with a reduced size of approximately 1–2 μm and enhanced sphericity. These observations underscore the effectiveness of the spray drying technique in achieving a more controlled and homogeneous particle morphology for the Li2C4O4–CNT composite. The transmission electron microscope (TEM) result displayed in Fig. 2d reveals the uniform dispersion of Li2C4O4 particles within the conductive network formed by CNT. This nanoscale interplay facilitates efficient electronic and Li+ ion transport, which could enhance conductivity and electrochemical performance. Electronic conductivity tests reveal a significant enhancement in conductivity when 3 wt% CNT is incorporated into the composite, with a measured value of 1.09 × 10−3 S cm−1 (Fig. S2, ESI†). In contrast, the bare Li2C4O4 is an insulator, falling below the measurable range of the equipment.
Fig. 2e showcases the X-ray diffraction (XRD) pattern of Li2C4O4, demonstrating its excellent agreement with the dilithium squarate phase (PDF #04-012-0316). The inserted image in Fig. 2e illustrates the crystal structure of Li2C4O4, revealing the presence of a squarate anion bonded with two lithium ions. The Fourier-transform infrared (FTIR) spectra of Li2C4O4 exhibit two absorption peaks at 1515 cm−1 and 1100 cm−1 (Fig. S3, ESI†), corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and C–C stretching vibration, respectively. The absence of C–O bonds in the FT-IR spectra indicates that Li2C4O4 is composed of two Li+ ions and a C4O42− ion with four CO bonds and C–C bonds. The thermogravimetric (TG) profile presented in Fig. S4 (ESI†) indicates that Li2C4O4 exhibits good thermal stability, with an onset thermal decomposition temperature close to 400 °C. When exposed to humid air for 7 days, the material exhibited water absorption of 15 wt% without any signs of chemical deterioration, demonstrating its excellent stability towards moisture. Furthermore, an air-exposure experiment was conducted to assess the air stability of Li2C4O4. After being stored in the air for over 180 days, the XRD pattern showed minimal differences compared to the freshly prepared sample (Fig. S5a, ESI†). No peaks associated with OH− or CO32− were observed in both the FTIR spectra and the Nuclear Magnetic Resonance (NMR) spectra, indicating the absence of LiOH or Li2CO3 formation (Fig. S5b and c, ESI†). The charge profile of the long-term air-exposed Li2C4O4–3CNT also remained unaffected (Fig. S5d, ESI†), further confirming the excellent air stability of Li2C4O4.
O stretching vibration and C–C stretching vibration, respectively. The absence of C–O bonds in the FT-IR spectra indicates that Li2C4O4 is composed of two Li+ ions and a C4O42− ion with four CO bonds and C–C bonds. The thermogravimetric (TG) profile presented in Fig. S4 (ESI†) indicates that Li2C4O4 exhibits good thermal stability, with an onset thermal decomposition temperature close to 400 °C. When exposed to humid air for 7 days, the material exhibited water absorption of 15 wt% without any signs of chemical deterioration, demonstrating its excellent stability towards moisture. Furthermore, an air-exposure experiment was conducted to assess the air stability of Li2C4O4. After being stored in the air for over 180 days, the XRD pattern showed minimal differences compared to the freshly prepared sample (Fig. S5a, ESI†). No peaks associated with OH− or CO32− were observed in both the FTIR spectra and the Nuclear Magnetic Resonance (NMR) spectra, indicating the absence of LiOH or Li2CO3 formation (Fig. S5b and c, ESI†). The charge profile of the long-term air-exposed Li2C4O4–3CNT also remained unaffected (Fig. S5d, ESI†), further confirming the excellent air stability of Li2C4O4.
The electrochemical performance of the as-prepared sample was evaluated using Li–metal half cells. Fig. 2f displays the charge and discharge curves of the pure Li2C4O4 and Li2C4O4–CNT composite within the voltage range of 2.5 to 4.3 V at a charging rate of 0.02C. The Li2C4O4 cathode derived from vacuum drying exhibits an oxidation potential of approximately 4.15 V, with a capacity of 130 mA h g−1. However, this capacity falls significantly short of the theoretical value of 425 mA h g−1, indicating an insufficient decomposition process. In contrast, the addition of CNT and the utilization of the spray drying method significantly enhance the electrochemical activity of the materials by a reduced decomposition potential and improved capacity utilization. Specifically, the pure spray-dried Li2C4O4 displays an oxidation potential above 4.0 V, with an initial activation energy barrier of 4.2 V. In contrast, the inclusion of 1 wt% CNT results in a decreased oxidation potential of 3.9 V and a reduced initial activation energy barrier of 4.0 V. Further increasing the CNT content to 3 wt% eliminates the initial activation energy barrier, with an oxidation potential at 3.9 V and an improved charging capacity of 435 mA h g−1. However, the addition of 5 wt% CNT does not yield any additional enhancements in electrochemical performance. Therefore, 3 wt% was chosen as the final CNT content. The initial discharge curves indicate a negligible capacity, affirming the viability of Li2C4O4 as a sacrificial salt for compensating for ALL. Additionally, the CV curve (Fig. S6, ESI†) reveals an oxidation peak at 3.86 V with no observed reduction peaks, further confirming the complete irreversibility of lithium extraction from Li2C4O4. The rate performance of the Li2C4O4–3CNT is shown in Fig. 2g. As the charging rate increased from 0.02 to 0.1C, a slight increase in the decomposition potential was observed due to larger polarization. However, the irreversible capacity remained largely unaffected. These findings provide valuable insights for determining the optimal current during the formation stage of full cells.
In previous studies on Li2C4O4, there was speculation regarding its decomposition into CO2 and C.31,32,34 However, no direct evidence was presented to support this hypothesis. Hu et al. conducted an ex situ gas analysis and assumed that the decomposition reaction is Li2C4O4 → 2CO2 + 2C + 2Li+ + 2e−. However, they also detected CO in the gas analysis, which they considered to be in an intermediate state.30 Herein, we employed in situ differential electrochemical mass spectrometry (DEMS) measurements to investigate the gas evolution during the decomposition of Li2C4O4–3CNT. As depicted in Fig. 2h, only CO (m/z = 28) was detected during the decomposition of Li2C4O4, and its signal increased proportionally as the current was raised. There were no detections of other gases such as CO2 (m/z = 44) or O2 (m/z = 32). This result indicates that the decomposition mechanism of Li2C4O4 can be represented as follows:
| Li2C4O4 → 4CO + 2Li+ + 2e− | (1) |
This process involves full deposition without residual components, liberating two active Li+ ions. Importantly, the generated CO gas can be efficiently eliminated by degassing before battery cycling, without adding to the overall weight of the battery. Consequently, the effective capacity of Li2C4O4 can potentially reach as high as 3845 mA h g−1 (theoretical capacity of Li) when the mass of the produced gas is not considered.
Comparison of cathode and separator lithium replenishment for LFP||Gr full cells
The decomposition of Li2C4O4 can produce gases and generate pores in batteries when it is utilized as a lithium replenishing agent. When Li2C4O4 is incorporated into the cathode, it can impact the structure of the pristine electrode, potentially resulting in substantial changes in electrochemical performance.34 To address these potential side effects, we have chosen to place Li2C4O4 on the separator through a simple blade coating method. The resulting LRS exhibited a bi-layered structure, with one side featuring an evenly distributed coating layer, while the other side retained the original morphology of the separator (Fig. S7, ESI†). No penetration of the slurry is observed, confirming the maintained electronic insulation properties of the separator. The coating layers demonstrated excellent adhesion to the PP separator, maintaining their integrity without any powder detachment even after undergoing repetitive folding and recovery cycles (Fig. S8, ESI†). With the coating side facing the cathode, the Li2C4O4–3CNT composite can be oxidized during the charging process and act as an additional reservoir of Li to compensate for ALL (Fig. S9, ESI†). Herein, we have conducted a detailed comparison between cathode and separator lithium replenishment (CLR and SLR) strategies based on Li2C4O4.DEMS analyses were conducted on both the CLR and SLR to examine the decomposition behavior of Li2C4O4 in half cells. For CLR, the additive amounts to 5 wt%, while for SLR, the thickness of the coating layer is 4 μm. The mass of Li2C4O4 was kept consistent between the two cases to enable a direct comparison of gas production. As shown in Fig. 3a and b, the decomposition process resulted in the generation of both CO and CO2, which is different from the findings in Fig. 2h, where only CO was detected during Li2C4O4 decomposition. Moreover, the quantity of CO2 generated in SLR was notably lower than that in CLR. This suggests that the charged state of LFP (FePO4) may act as a catalyst for the decomposition of Li2C4O4 to CO2 and C. The proposed reaction is expressed below:
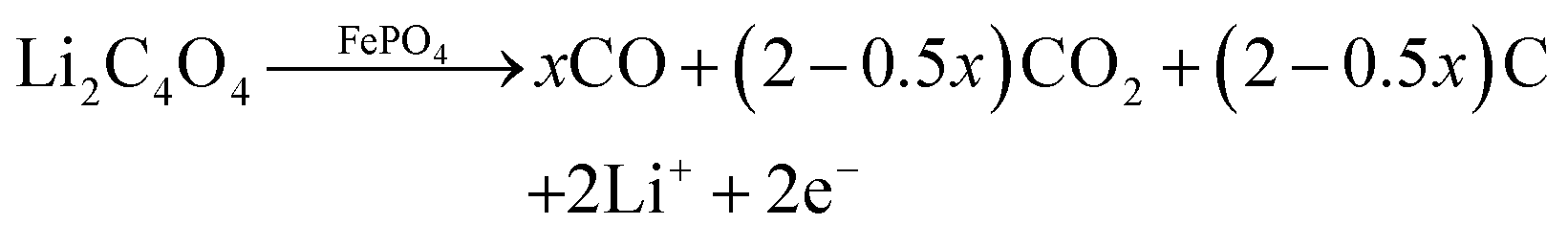 | (2) |
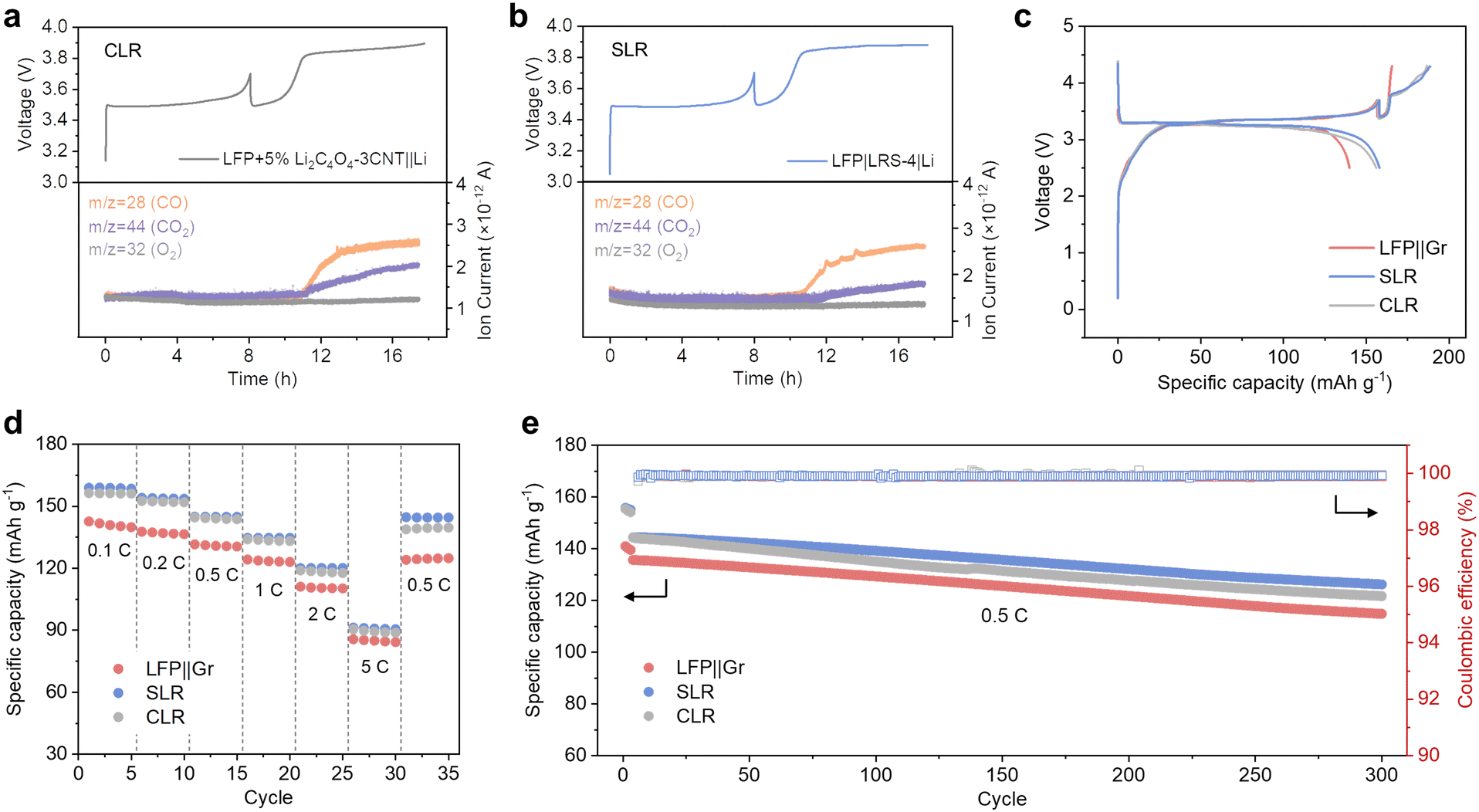 | ||
| Fig. 3 Comparison of the electrochemical performance of LFP||Gr full cells with CLR (LFP + 5% Li2C4O4–3CNT||Gr) and SLR (LFP|LRS-4|Gr) strategies. (a) and (b) In situ DEMS tests of the LFP||Li half cells. (c) Initial charge–discharge profiles of LFP||Gr full cells. (d) Rate performance of LFP||Gr full cells. (e) Comparison of the cycling stability of the full cells at 0.5C. | ||
When Li2C4O4 is situated in the cathode, it has a broader interaction with LFP, potentially accounting for the increased levels of CO2 observed compared to when Li2C4O4 is placed in the separator, where only the interface comes into contact with LFP. For further verification, we introduced a porous 5 μm-thick CNT film between the cathode and LRS. This film can effectively isolate LFP and LRS and serves as an excellent electron and Li+ conductor. The DEMS result with this configuration demonstrates a significant reduction in the quantity of CO2 (Fig. S10, ESI†), aligning well with our hypothesis regarding the LFP-catalyzed decomposition of Li2C4O4.
The electrochemical performance of both CLR and SLR was further investigated. In Fig. 3c, the initial charge and discharge profiles of the full-cell configurations with CLR and SLR are presented. Notably, both CLR and SLR cells demonstrated increased initial charge and discharge capacities when compared to the pristine cell (LFP||Gr). In LFP||Gr full cell, a lithium loss of 14.5% was observed, resulting in a discharge capacity of 140 mA h g−1. In contrast, SLR and CLR cells exhibited significantly improved performance, with a charge capacity of 188 and 186 mA h g−1, and a discharge capacity of 158 and 156 mA h g−1. These values closely align with those achieved in an LFP||Li half cell. Furthermore, the oxidation potential of Li2C4O4 for CLR and SLR remained similar, at approximately 3.8 V.
The rate and long-term cycling performance of the LFP||Gr, SLR, and CLR cells are illustrated in Fig. 3d and e. Both the SLR and CLR cells exhibit superior rate performance compared to the comparison cell. However, after several cycles at 5C, the capacity of the CLR cell is difficult to recover to the initial level, while the SLR cell shows a good capacity recovery once the rate decreases to 0.5C. Concerning long-term cycling performance, the CLR cell exhibits a faster rate of capacity degradation compared to the SLR cell (Fig. 3e). This difference could potentially be attributed to the presence of gas pores, residual carbon, and incompletely decomposed Li2C4O4 residing in the electrodes in the case of CLR. These factors may lead to the partial isolation of cathode particles and structural damage to the cathode, consequently negatively impacting the overall performance of the cell. As shown in Fig. S11 (ESI†), the cycled LFP electrode with the CLR method exhibits some uniform gas pores. Attributed to the spray drying method, the Li2C4O4–CNT particles exhibit uniform size and shape, resulting in well-defined pore characteristics. This minimizes the adverse effects of CLR compared to prior literature.34 In contrast, SLR effectively separates the prelithiation reagents from the cathode materials, thereby mitigating potential integrity issues of the electrode after lithium replenishment. On the one hand, the improved contact between Li2C4O4 and carbon black, which is diluted by LFP in the case of CLR, enhances the complete decomposition of Li2C4O4. On the other hand, after eliminating the direct formation of pores in the cathode, the LFP electrode with SLR maintains a good structure after Li2C4O4–3CNT decomposition. These observations further support the notion that SLR can effectively preserve the cathode's structural integrity, contributing to its superior electrochemical performance.
The electrochemical impedance of CLR, SLR, and the control cell is depicted in Fig. S12 (ESI†). Before cycling, the charge transfer resistance (Rct) of CLR is slightly higher than that of the pristine cell. This difference is likely due to the low electronic conductivity of the additive, which could impede electron transfer (Fig. S13b, ESI†). In contrast, the SLR cell exhibits lower Rct and internal resistance (Re) compared to the pristine cell. This improvement can be attributed to enhanced electron transfer in SLR cells, where the conductive separator acts as a secondary current collector (Fig. S13c, ESI†).35,36 This facilitates electron transfer and enhances the utilization of active material, resulting in reduced resistance and improved rate performance with the SLR strategy. After the first cycle, impedance increases in all cases, but SLR consistently exhibits the lowest impedance, while CLR is slightly lower than the control cell (Fig. S14, ESI†). The generation of pores in the cathode and separator after Li2C4O4–3CNT decomposition may enhance electrolyte accessibility, thereby contributing to the reduced impedance.37 SEM images of the LRS after the first charge are shown in Fig. S15 (ESI†), depicting a uniform distribution of pores generated in the separator. Such a separator can effectively transport both electrons and lithium ions, which likely contributes to the good electrochemical performance of the SLR strategy.
Performance of the lithium replenishment separator
To thoroughly assess the performance of the LRS, we conducted a comprehensive investigation of several key properties. These properties include an electrochemical performance with various loadings of Li2C4O4–3CNT, electrolyte uptake capacity, ionic conductivity, thermal stability, and so on. By controlling the slurry coating process, we were able to precisely tailor the thickness of the coating layer on the LRS to accommodate the diverse lithium compensation needs of various battery systems. Cross-sectional images of the LRS in Fig. 4a showcased varying thicknesses of the prelithiation layer, ranging from 5 μm to 15 μm. As the thickness increases, the areal mass loading of Li2C4O4–3CNT exhibits a linear increase from 0.32 to 0.98 mg cm−2, corresponding to a capacity range of approximately 0.13 to 0.42 mA h cm−2. The decomposition efficiency of Li2C4O4–3CNT remains consistently at around 100% (Fig. 4b), and the oxidation plateaus stay below 4 V at different loadings (Fig. S16, ESI†). This consistency ensures a high lithium utilization ratio.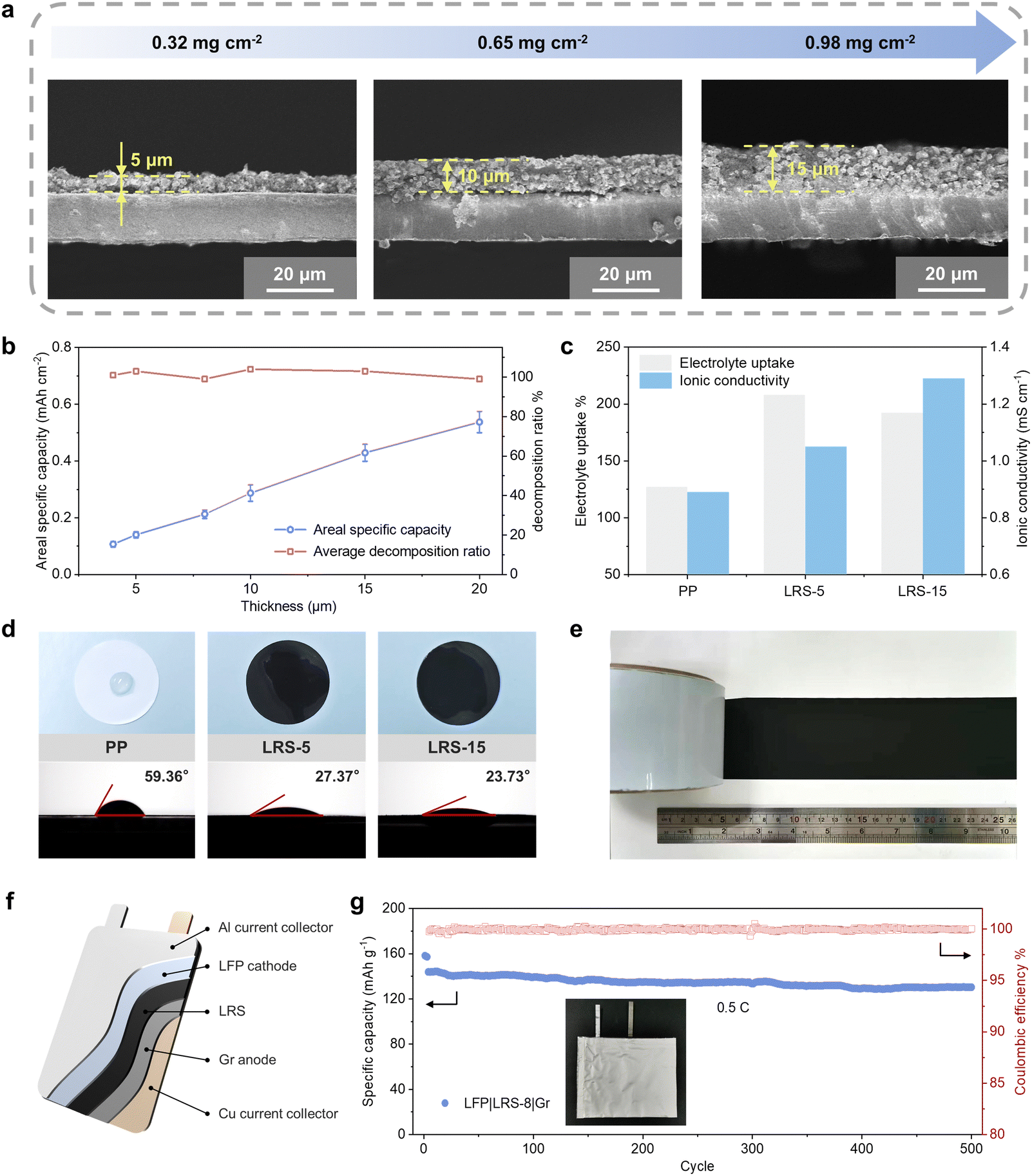 | ||
| Fig. 4 Characterization of the lithium replenishment separator. (a) Cross-sectional morphology and thickness of the Li2C4O4–CNT coating layer. (b) Summarized relationship between area capacity and the thickness of the coating layer. (c) The electrolyte uptake capacity and ionic conductivity of the PP separator and LRS. (d) The wetting properties of various separators and their corresponding contact angles with carbonate electrolyte. (e) The prepared LRS wound on a reel. (f) Schematic diagram of an LFP||Gr pouch cell with LRS. (g) Cycling performance of the LFP|LRS-8|Gr pouch cell at 0.5C (inset is the optical image of the pouch cell). | ||
It is worth noting that the “second current collector” effect prevents the increase in electrochemical impedance with the growing thickness of the separator. As shown in Fig. S17a (ESI†), when employing an LRS with a 15-μm-thick coating layer, the LFP|LRS-15|Gr cell exhibits a low initial Rct of 16.23 Ω. In contrast, when the amount of Li2C4O4–3CNT in the CLR method is increased to 15 wt%, undecomposed Li2C4O4 can result in poor interparticle contact and significant polarization due to reduced electrical conductivity. Consequently, the Rct of the LFP + 15% Li2C4O4–3CNT||Gr cell reaches 28.15 Ω. Even if some Li2C4O4 on the separator undergoes partial decomposition in the first cycle, the remaining portion has little impact on the charge transfer properties of the LFP cathode (Fig. S17b, ESI†). This allows for the preservation of a portion of Li2C4O4 as a lithium reservoir to compensate for cumulative active lithium loss (cALL) in subsequent cycles.
Fig. 4c shows the electrolyte absorption ability and ionic conductivity of LRS. After full immersion in the carbonate-based electrolyte, the LRS-5 and LRS-15 samples demonstrated enhanced electrolyte uptake percentages of 208% and 192%, respectively, compared to 127% for the PP separator. This increased electrolyte absorption is beneficial for facilitating ion transport and reducing interface impedance. Consequently, the LRS exhibited higher ionic conductivity, with values of 1.05 cm−1 for LRS-5 and 1.29 mS cm−1 for LRS-15, in comparison to 0.89 mS cm−1 for the PP separator. The modified separators also exhibited enhanced wettability, as evidenced by the instant spreading of the liquid electrolyte on the prelithiation side (Fig. 4d). LRS-5 and LRS-15 demonstrated contact angles of 27.37° and 23.73°, respectively, while the uncoated separator maintained a larger contact angle of 59.36°. These improvements effectively reduce the internal polarization of the cell, complementing the explanation of the enhanced rate performance brought by SLR and further illustrating its superiority over CLR. Additionally, the thermal stability of the separator was improved. As shown in Fig. S18 (ESI†), the PP separator experienced significant structural deformation when stored at 150 °C for 10 minutes, while the LRS-15 exhibited only slight shrinkage.
Fig. 4e demonstrates the fabrication of the LRS wound on a reel at a meter scale, highlighting its potential for industrial manufacturing. To evaluate the performance of LRS at a larger scale, an LFP|LRS-8|Gr pouch cell was assembled (Fig. 4f). The cycling performance of the pouch cell at 0.5C is shown in Fig. 4g. After 500 cycles, the cell maintains a discharge capacity of 130.2 mA h g−1, with a high capacity retention of 90.49%. These results indicate the promising potential of our lithium replenishment method for energy storage applications.
In situ long-term lithium replenishment
The situation regarding iALL and cALL at different cycles is illustrated in Fig. 5a (assuming that all capacity fading originates from ALL in an LFP||Gr system). In the initial cycle, the LFP||Gr full cells experience an iALL of 14.5%. Subsequently, after 50 cycles, the accumulated cALL reaches 2.5%, and it rises to 10.5% after 200 cycles. After 500 cycles, the cALL becomes 1.6 times that of iALL, highlighting the significant need for compensation for cALL during cycling. It is important to note that these findings are based on coin-cell test. In commercial pouch cells, the rate of cALL increase is expected to be lower. However, for extended cycles, such as thousands of cycles, the cALL can still be significant.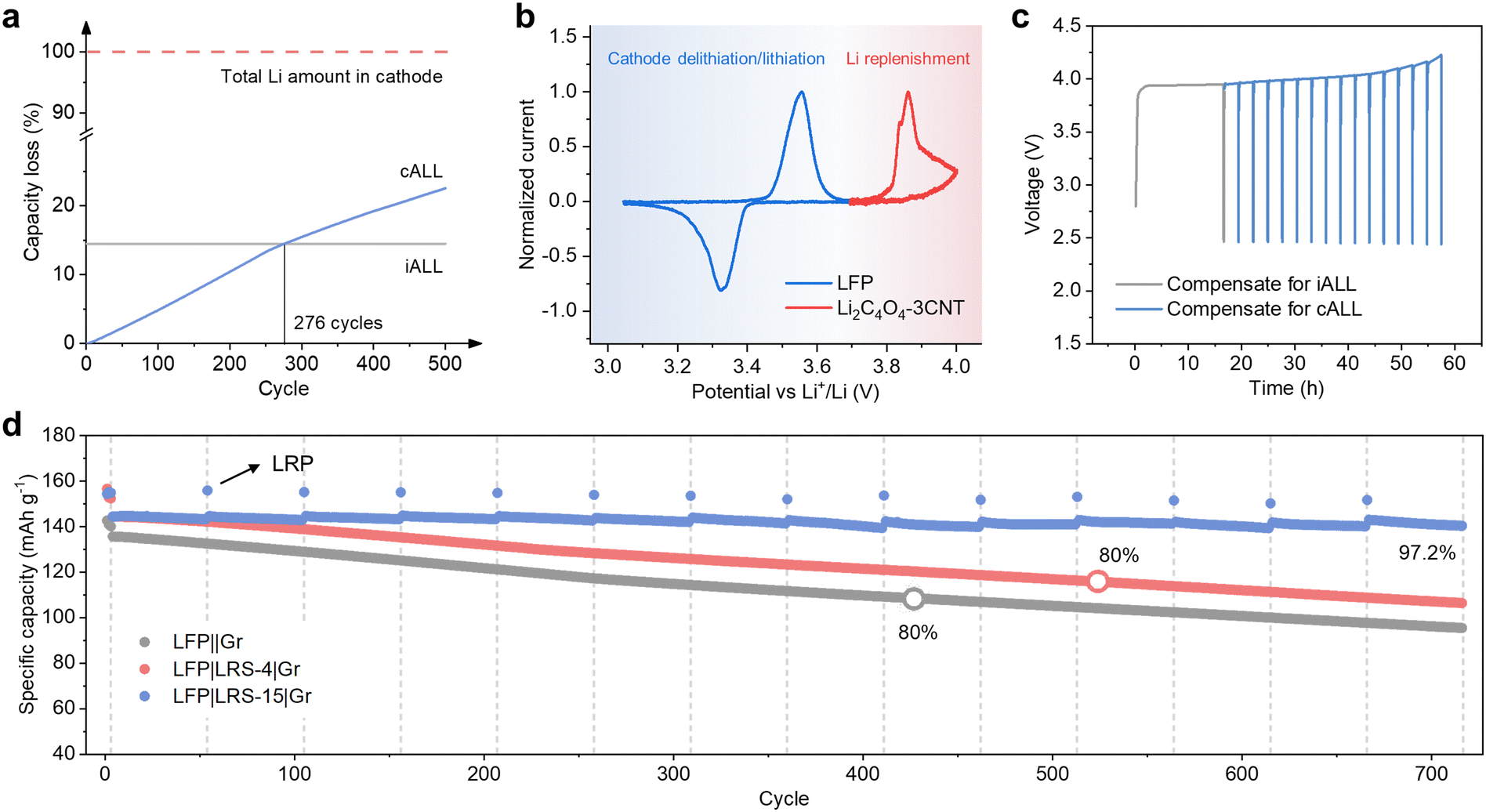 | ||
| Fig. 5 Electrochemical evaluation of the long-term lithium replenishment strategy (a) the ratio of iALL and cALL after different cycles. (b) CV curves of LFP and Li2C4O4–3CNT. (c) The controlled-release process verified by LRS-15||Li half cell. (d) Cycling performance of LFP||Gr full cells at 0.5C with long-term lithium replenishment strategy. | ||
When lithium replenishment is precisely calibrated to compensate solely for iALL, the battery lacks excess lithium to counterbalance cALL in subsequent cycles. Consequently, it exhibits a capacity degradation rate similar to that of a battery without lithium replenishment. To address long-term capacity degradation resulting from cALL, we propose a lithium replenishment strategy designed to enhance the cycling performance of lithium-ion batteries (LIBs) throughout their entire lifecycle. In this approach, we introduce the concept of the “lithium replenishment degree” (LRD) to quantitatively measure the surplus amount of active lithium ions available for compensation. The LRD is calculated as the ratio of the capacity of the sacrificial lithium reservoir to the capacity of the cathode:
| LRD ≡ sacrificial lithium salt capacity/cathode capacity |
When the pre-loaded lithium reservoir is precisely adjusted to offset the loss of iALL, the LRD is equivalent to ICE. To enable lithium compensation throughout the entire cycle life of the batteries, it is necessary to introduce a higher LRD into the batteries, with the surplus LRD serving as a reservoir of lithium gradually released during extended cycling.
In addition to increasing the LRD, another critical aspect is ensuring that the oxidation of the lithium replenishment agent occurs after the oxidation of the cathode and that its potential is lower than the voltage threshold that the cathode can tolerate. This arrangement allows for the controlled release of lithium through the charging process. The normalized CV curves of LFP and Li2C4O4–3CNT are shown in Fig. 5b, clearly illustrating that the oxidation potential of Li2C4O4 is 0.3–0.4 V higher than that of LFP. Furthermore, Li2C4O4–3CNT can be fully decomposed below 4.3 V, thus avoiding high voltage damage to the LFP cathode. By adjusting the charging capacity and charge cutoff voltage, we can precisely control the release of active lithium from the Li2C4O4 decomposition. When the battery has undergone a certain number of cycles or its capacity has decreased to a certain level, we establish a lithium replenishment point (LRP) for the battery to restore its capacity.
The controlled release process of LRS-15 (LRD ≈ 69%) was simulated in a half cell and is presented in Fig. 5c. In the first cycle, roughly 30% of the Li2C4O4 decomposed to compensate for iALL, and 15 LRPs were established. Fig. S19 (ESI†) displays the charge–discharge curves for the 9th lithium replenishment and the subsequent charge and discharge curves during the 1st and 50th cycles, all with the same current cycling LFP in a full cell (2.5–3.7 V). At each LRP, approximately 0.02 mA h cm−2 of active lithium was released. The oxidation potential of Li2C4O4 is approximately 4 V at the 9th LRP, and no capacity is observed within the voltage range of 2.5–3.7 V. These results demonstrate the precise control over the release of active lithium from LRS.
The proposed long-term lithium replenishment strategy was further validated in a full-cell configuration. After every 50 cycles, a LRP was introduced to release 4.7% of the stored lithium inventory. As shown in Fig. 5d, the cycling performance of the full cell exhibited a significant improvement. The battery's capacity was replenished after each LRP before the complete depletion of the Li2C4O4 on the LRS. With the long-term lithium replenishment strategy, the specific capacity of LFP remained at 140.4 mA h g−1 after 716 cycles at 0.5C. This outperformed the cells that only compensated for the iALL (106.4 mA h g−1), and the cells without lithium compensation (95.5 mA h g−1). The capacity retention of the long-term lithium replenishment reached 97.2% after 716 cycles, making a substantial increase compared to the cell with iALL compensation alone (73.8%) and the one without lithium replenishment (70.3%).
The corresponding charge and discharge curves with long-term lithium replenishment at the 10th, 100th, 300th, and 700th cycles are shown in Fig. S20 (ESI†), respectively. It can be observed that the charge and discharge profiles maintain a good overlap throughout the long cycling, indicating stable electrochemical performance. Moreover, the polarization remains unchanged during the extended cycling process, indicating the preservation of excellent electrode kinetics. After the delithiation of LFP, the oxidation plateau of Li2C4O4 gradually increased with the consumption of Li2C4O4 (Fig. S21, ESI†). The dynamic balance between active lithium loss and replenishment eliminates the issues of excessive lithium plating. Importantly, when the LFP||Gr cell was charged using the same test procedure as the LFP|LRS-15|Gr cell, the capacity exhibited no significant difference compared to the cell charged under normal conditions (voltage range of 2.5–3.7 V) (Fig. S22, ESI†). This proves that the capacity recovery is not due to the high charging cutoff voltage and low charging rate at each LRP.
The configuration of the LRP can be adjusted to suit specific requirements. For example, the LRP can also be set to occur after every 100 cycles, as shown in Fig. S23 (ESI†). To further optimize the strategy, the battery management system (BMS) can be implemented to monitor the state of health (SOH) of the batteries. The SOH can serve as the trigger condition for initiating the lithium replenishment process. When the SOH deteriorates to a predetermined value, the charging conditions can be adjusted to facilitate the release of active lithium ions.
It is important to note that the lithium compensation process achieved by Li2C4O4 is accompanied by gas production. To make this approach practical, it is necessary to design batteries equipped with automatic exhaust valves capable of releasing gas when pressure levels reach a certain threshold. However, the integration of such an exhaust system into battery manufacturing processes remains relatively uncommon and may incur additional production costs. To address this issue, we have developed an alternative anode-supported automatic long-term lithium replenishment strategy. In this approach, a higher LRD is also required, but the fundamental distinction lies in releasing all the active lithium during the initial cycle and increasing the N/P ratio to store excess lithium within the graphite anode as LiCx. By adopting this approach, no additional gas is generated during cycling, and any gas produced during the first cycle can be released through the normal degassing process. This ensures the safe and efficient operation of the battery while minimizing the impact of gas generation on its overall performance.
The initial charge and discharge curves of the full cells with varying LRDs, specifically 25.86%, 34.96%, and 41.71%, are shown in Fig. 6a–c. To prevent lithium–metal plating, the corresponding N/P ratios are increased to 1.4, 1.5, and 1.6, respectively. The increased N/P ratio leads to a slight decrease in ICE (Fig. S24a, ESI†), but the cycling performance is not affected (Fig. S24b, ESI†). As depicted in Fig. 6d, the continuous replenishment of active Li+ from LiCx enabled the capacity to remain stable for the first 178, 292, and 390 cycles, respectively. Once the lithium inventory was depleted, the capacity began to decline at a similar rate to the control group. With an LRD of 41.71%, the specific capacity remains at 139.7 mA h g−1 after 450 cycles, whereas for LRD values of 25.86% and 34.96%, the capacities are 127.5 mA h g−1 and 136.1 mA h g−1, respectively. The corresponding capacity retentions were 96.6%, 98.3%, and 88.7%, signifying a substantial improvement in electrochemical performance compared to LRD values that solely compensated for iALL. This approach offers greater flexibility in the selection of prelithiation agents or methods. However, its drawback lies in the limited lithium storage capacity of graphite, resulting in a constrained enhancement of the battery's energy density. Nevertheless, when considering the anode mass ratio at the pack level, the increase in the N/P ratio is negligible in comparison to the overall enhancement in total energy density and cycle life. A comparative analysis of various prelithiation methods is presented in Fig. 6e, highlighting the advantages of our lithium replenishment approaches over other recently reported prelithiation methods.
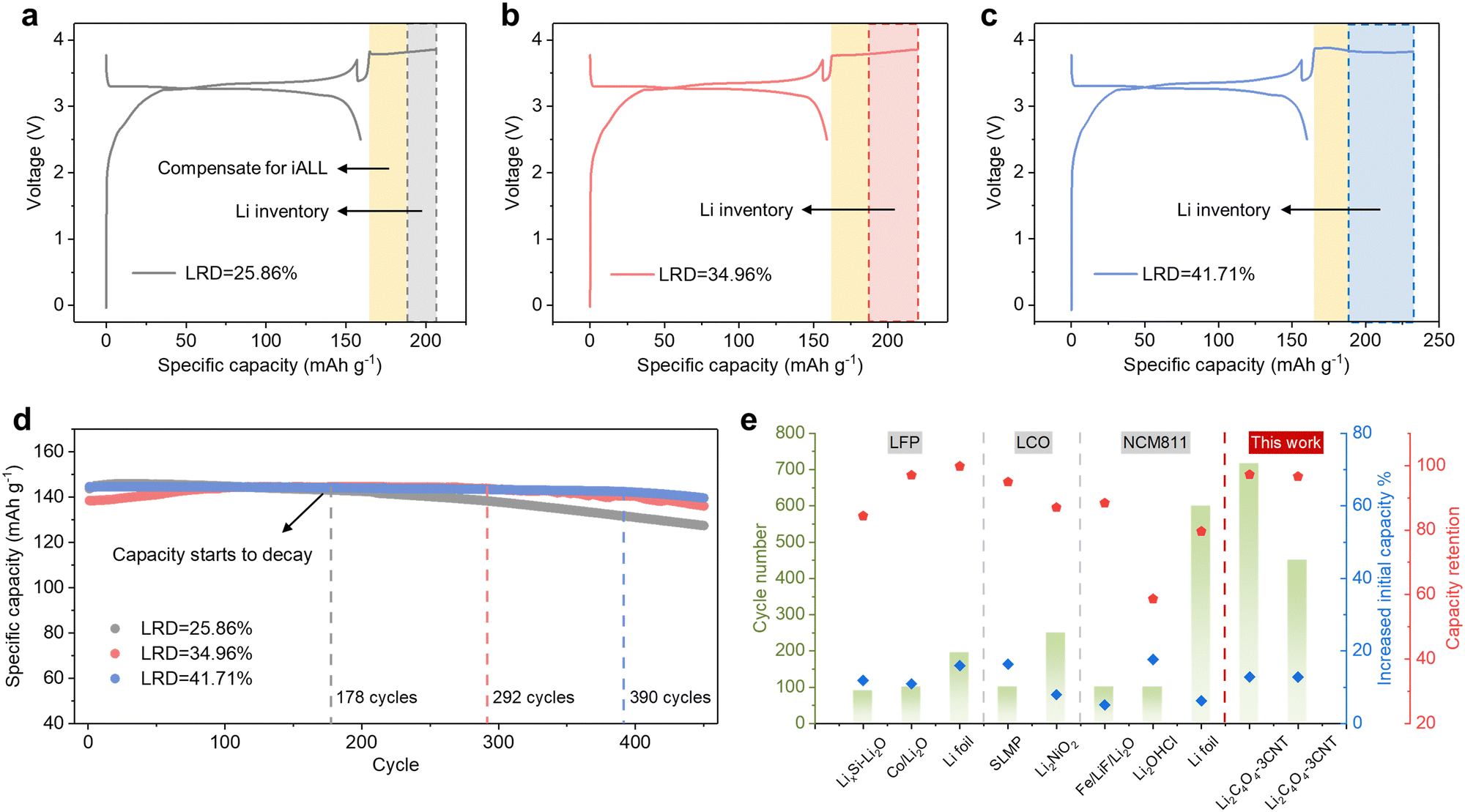 | ||
| Fig. 6 Anode-supported long-term lithium replenishment strategy (a)–(c) Initial charge–discharge profiles of LFP||Gr full cells with different LRD. (d) Cycling performance of LFP||Gr full cells at 0.5C with additional Li inventory stored in the anode. (e) Comparison of the full cells electrochemical performance with various prelithiation methods.15,18,21,22,25,38,39 | ||
Conclusions
In summary, we have developed an innovative strategy to enhance the energy density and cycle life of LIBs by employing a lithium replenishment separator (LRS). Our approach involves the use of Li2C4O4–CNT composite as a capacity-controlled sacrificial agent, which delivers a significant irreversible capacity of 425 mA h g−1, and as high as 3845 mA h g−1 if the decomposition product, are fully degassed from the cell. Compared to the conventional cathode lithium replenishment (CLR) method, our separator lithium replenishment (SLR) method offers substantial advantages. It effectively inhibits the catalytic decomposition of Li2C4O4 to CO2 and C by LFP particles, reducing carbon residue in the battery and producing higher energy density. The generated gas may be released by an automatic exhaust valve when the pressure level reaches a certain threshold. The LRS also acts as a secondary current collector, reducing impedance, and integrates seamlessly with existing LIB manufacturing processes. The LRS areal capacity can be precisely tailored by adjusting the Li2C4O4–3CNT coating layer thickness to fulfill various lithium compensation needs. When implemented in LFP||Gr full cells, the iALL was effectively compensated.Building upon this, we further developed an innovative long-term lithium replenishment strategy to address continuous active lithium loss (cALL) as well. By optimizing the lithium replenishment degree (LRD), additional doses of Li2C4O4 are dispensed, which can be systematically released by regulating the charging capacity and cutoff voltage during long-term cycling. This results in continuous replenishment of active lithium throughout the entire lifecycle of LIBs, leading to full capacity and prolonged cycle life. With the incorporation of a battery management system (BMS) to monitor the battery's state-of-health (SOH), future research in this field could optimize the settings for a more efficient and intelligent full-cycle-life lithium compensation. Additionally, we propose a strategy of storing additional lithium inventory in the graphite anode to eliminate gassing during cycling. This ensures spontaneous and sustainable lithium replenishment throughout the battery's lifespan. These results provide fresh insights into achieving in situ lithium replenishment and effectively mitigating lithium loss during cycling, promoting the advancement of high-energy and long-lasting LIBs.
Experimental
Materials synthesis
1.14 g squaric acid (Aladdin, 99.0% purity) and 0.74 g lithium carbonate (Adamas, 99.9% purity) were dissolved in 50 mL deionized water (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The resulting mixture was stirred for 2 hours at room temperature to form the Li2C4O4 solution. CNT dispersion (Ocsial, 0.4 wt% in H2O, diameter: 1.6 ± 0.6 nm, length ≥ 5 μm) was initially diluted to 0.1 wt% with deionized water and then added to the solution. Subsequently, the prepared Li2C4O4–CNT solution was introduced into a spray dryer at a rate of 15 mL min−1 at 180 °C, to obtain the Li2C4O4–CNT composite. Different CNT contents were prepared, and the quantity of CNT added was calculated based on the total mass of the composite.
:1). The resulting mixture was stirred for 2 hours at room temperature to form the Li2C4O4 solution. CNT dispersion (Ocsial, 0.4 wt% in H2O, diameter: 1.6 ± 0.6 nm, length ≥ 5 μm) was initially diluted to 0.1 wt% with deionized water and then added to the solution. Subsequently, the prepared Li2C4O4–CNT solution was introduced into a spray dryer at a rate of 15 mL min−1 at 180 °C, to obtain the Li2C4O4–CNT composite. Different CNT contents were prepared, and the quantity of CNT added was calculated based on the total mass of the composite.
Preparation of the LRS and cell assembly
The LRS was prepared by mixing 80 wt% of Li2C4O4–3CNT, 10 wt% Super P carbon, and 10 wt% polyvinylidenefluoride (PVDF) in N-methyl-2-pyrrolidone (NMP). The obtained slurry was cast onto a PP separator (Celgard A273) using a line coater and dried in a vacuum oven overnight at 60 °C. The thickness of the coating layer can be adjusted by using different models of the line coaters. The LFP and Gr electrodes were prepared by mixing active materials, Super P carbon, and PVDF in a weight ratio of 8:1:1 and 9:0.5:0.5 in NMP. The resulting slurry was coated onto Al/Cu foil and dried overnight at 80 °C. The active materials mass loading of LFP and Gr electrodes was approximately 7 and 3.6 mg cm−2, and the diameter of the electrodes was both 12 mm. And the N/P ratio was around 1.1. The electrolyte used was 1 M LiPF6 in a 3:7 vol/vol mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) with 2 wt% vinylene carbonate (VC). 60 μL electrolyte was used for each coin cell. All the coin cells (CR2025) were assembled in a glove box filled with Ar, with moisture and oxygen content less than 0.1 ppm. The lithium replenishment separator was used with the coating side face to the cathode. In the CLR method, the Li2C4O4–3CNT composite was directly added to the LFP slurry. Specific capacity calculations for the full cells were based on the mass of LFP.
Material characterization
The morphologies of the Li2C4O4–CNT composite were analyzed using scanning electron microscopy (SEM, FEI, QUANTA 250FEG) and transmission electron microscopy (TEM, Tecnai F30). The crystal structure was evaluated by X-ray diffractometer with Cu Kα radiation (Bruker D8 Advance) using a Bragg–Brentano geometry between 10° and 90° at a step size of 0.02° s−1. Fourier transform infrared (FTIR) spectra were recorded using a Bruker VERTEX 70 FTIR spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker-500 MHz NMR Spectrometer. The thermal stability of Li2C4O4 was investigated by thermogravimetric analysis (TGA, Q600 SDT) under airflow at a heating rate of 5 °C min−1, ranging from room temperature to 800 °C. The evolving gases were analyzed by in situ differential electrochemical mass spectrometry (DEMS, Shanghai LingLu Co., Ltd). For ex situ material characterizations, cells were dissembled in an Ar-filled glove box, and the cycled electrodes or separators were thoroughly washed with diethyl carbonate (DEC) to remove impurities.Electrochemical characterization
Electrochemical tests were conducted using the Neware test system (CT4008, Neware) at 25 °C. The half cells were cycled in the voltage range of 2.5–4.3 V (vs. Li+/Li). The LiFePO4||Gr full cells with LRS were firstly charged to 3.7 V under 0.1C (1C ≡ 160 mA h g−1), then the batteries were further charged to 4.3 V with a lower current density of 0.02C for Li2C4O4 (1C = 425 mA h g−1). The charging capacity was monitored to control the degree of Li compensation based on demand. For batteries with a long-term lithium replenishment strategy, a portion of Li2C4O4 is used to compensate for the initial lithium loss, while the remainder serves as lithium inventory, slowly released in subsequent cycles. At each LRP, the same test procedure (0.1C to 3.7 V, then switched to a lower current and charged to a higher voltage, terminated when charging to a certain capacity) was used to control the release of active lithium. For batteries with extra lithium sources stored in anodes, larger N/P ratios were used to prevent lithium plating. CV and EIS experiments were conducted on the electrochemical workstation (GAMRY INTERFACE 1010E), the scan rate of CV is 0.03 mV s−1, and the frequency range of EIS is 100 kHz to 0.1 Hz with a voltage amplitude of 10 mV.Author contributions
G. L. and W. W. contributed equally to this work. G. L., Y. H., and C. W. conceived the research idea. G. L. and C. W. designed the experiments. G. L. and W. W. carried out materials synthesis and electrochemical characterization. W. W., Q. N., C. Z., and X. C. assisted with material characterization. W. L. contributed to conducting the DEMS measurements, and X. W. assisted in revising the paper. All authors discussed the results and contributed to the data analysis. G. L., W. W., and J. L. wrote the paper with contributions from all authors. C. W. supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3803400), the Shanghai Pujiang Program (22PJ1413400), and the Fundamental Research Funds for Central Universities.Notes and references
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J.-M. T. M. Armand, Nature, 2008, 451, 652–657 CrossRef PubMed.
- R. Zhan, X. Wang, Z. Chen, Z. W. Seh, L. Wang and Y. Sun, Adv. Energy Mater., 2021, 11, 2101565 CrossRef CAS.
- W. Wu, W. Luo and Y. Huang, Chem. Soc. Rev., 2023, 52, 2553–2572 RSC.
- Y.-Y. Wang, X.-Q. Zhang, M.-Y. Zhou and J.-Q. Huang, Nano Res. Energy, 2023, 2, e9120046 CrossRef.
- L.-X. Yuan, Z.-H. Wang, W.-X. Zhang, X.-L. Hu, J.-T. Chen, Y.-H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 RSC.
- J. Sun, L. Huang, G. Xu, S. Dong, C. Wang and G. Cui, Mater. Today, 2022, 58, 110–118 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- J. C. B. A. J. Smith, S. Trussler and J. R. Dahn, J. Electrochem. Soc., 2010, 157, A196–A202 CrossRef.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2020, 31, 2005581 CrossRef.
- H. Xu, S. Li, C. Zhang, X. Chen, W. Liu, Y. Zheng, Y. Xie, Y. Huang and J. Li, Energy Environ. Sci., 2019, 12, 2991–3000 RSC.
- C. Yang, H. Ma, R. Yuan, K. Wang, K. Liu, Y. Long, F. Xu, L. Li, H. Zhang, Y. Zhang, X. Li and H. Wu, Nat. Energy, 2023, 8, 703–713 CrossRef CAS.
- J. Jang, I. Kang, J. Choi, H. Jeong, K. W. Yi, J. Hong and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 14473–14480 CrossRef CAS PubMed.
- J. Choi, H. Jeong, J. Jang, A. R. Jeon, I. Kang, M. Kwon, J. Hong and M. Lee, J. Am. Chem. Soc., 2021, 143, 9169–9176 CrossRef CAS PubMed.
- Y. Sun, H.-W. Lee, Z. W. Seh, N. Liu, J. Sun, Y. Li and Y. Cui, Nat. Energy, 2016, 1, 15008 CrossRef CAS.
- J. Zhao, J. Sun, A. Pei, G. M. Zhou, K. Yan, Y. Y. Liu, D. C. Lin and Y. Cui, Energy Storage Mater., 2018, 10, 275–281 CrossRef.
- P. Jezowski, O. Crosnier, E. Deunf, P. Poizot, F. Beguin and T. Brousse, Nat. Mater., 2018, 17, 167–173 CrossRef CAS PubMed.
- J. Zhao, H. W. Lee, J. Sun, K. Yan, Y. Liu, W. Liu, Z. Lu, D. Lin, G. Zhou and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7408–7413 CrossRef CAS PubMed.
- L. Guo, C. Xin, J. Gao, J. Zhu, Y. Hu, Y. Zhang, J. Li, X. Fan, Y. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2021, 60, 13013–13020 CrossRef CAS PubMed.
- Y. Sun, H.-W. Lee, Z. W. Seh, G. Zheng, J. Sun, Y. Li and Y. Cui, Adv. Energy Mater., 2016, 6, 1600154 CrossRef.
- K. Park, B.-C. Yu and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502534 CrossRef.
- J. M. Du, W. Y. Wang, A. Y. S. Eng, X. X. Liu, M. T. Wan, Z. W. Seh and Y. M. Sun, Nano Lett., 2020, 20, 546–552 CrossRef CAS PubMed.
- X. Su, C. Lin, X. Wang, V. A. Maroni, Y. Ren, C. S. Johnson and W. Lu, J. Power Sources, 2016, 324, 150–157 CrossRef CAS.
- H. Park, T. Yoon, Y.-U. Kim, J. H. Ryu and S. M. Oh, Electrochim. Acta, 2013, 108, 591–595 CrossRef CAS.
- Y. Zhan, H. Yu, L. Ben, B. Liu, Y. Chen, Y. Wu, H. Li, W. Zhao and X. Huang, J. Mater. Chem. A, 2018, 6, 6206–6211 RSC.
- Q. Meng, M. Fan, X. Chang, H. Li, W. P. Wang, Y. H. Zhu, J. Wan, Y. Zhao, F. Wang, R. Wen, S. Xin and Y. G. Guo, Adv. Energy Mater., 2023, 13, 2300507 CrossRef CAS.
- B. Shen, B. Sarkodie, L. zhang, H. Jiang, C. Li and Y. Hu, Energy Storage Mater., 2022, 45, 687–695 CrossRef.
- M. Arnaiz, D. Shanmukaraj, D. Carriazo, D. Bhattacharjya, A. Villaverde, M. Armand and J. Ajuria, Energy Environ. Sci., 2020, 13, 2441–2449 RSC.
- D. Shanmukaraj, S. Grugeon, S. Laruelle, G. Douglade, J.-M. Tarascon and M. Armand, Electrochem. Commun., 2010, 12, 1344–1347 CrossRef CAS.
- M. Fan, Q. Meng, X. Chang, C. F. Gu, X. H. Meng, Y. X. Yin, H. Li, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2022, 12, 2103630 CrossRef CAS.
- A. Gomez-Martin, M. M. Gnutzmann, E. Adhitama, L. Frankenstein, B. Heidrich, M. Winter and T. Placke, Adv. Sci., 2022, 9, e2201742 CrossRef PubMed.
- H. Shao, F. Ai, W. Wang, H. Zhang, A. Wang, W. Feng and Y. Huang, J. Mater. Chem. A, 2017, 5, 19892–19900 RSC.
- J. Zhu, Y. Ge, D. Kim, Y. Lu, C. Chen, M. Jiang and X. Zhang, Nano Energy, 2016, 20, 176–184 CrossRef CAS.
- X. Guo, C. Wang, W. Wang, Q. Zhou, W. Xu, P. Zhang, S. Wei, Y. Cao, K. Zhu, Z. Liu, X. Yang, Y. Wang, X. Wu, L. Song, S. Chen and X. Liu, Nano Res. Energy, 2022, 1, e9120026 CrossRef.
- X. Li, Y. Li, Y. Tang, L. Zhang and J. Huang, J. Power Sources, 2021, 496, 229868 CrossRef CAS.
- M. G. Kim and J. Cho, J. Mater. Chem., 2008, 18, 5880–5887 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03740a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |