
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green ethylene production in the UK by 2035: a techno-economic assessment†
Andreas H.
Nyhus‡
,
Maria
Yliruka‡
 *,
Nilay
Shah
and
Benoît
Chachuat
*
*,
Nilay
Shah
and
Benoît
Chachuat
*
The Sargent Centre for Process Systems Engineering, Department of Chemical Engineering, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: m.yliruka@imperial.ac.uk; b.chachuat@imperial.ac.uk
First published on 18th January 2024
Abstract
Olefins production in the UK is the most emission-intensive sector of the chemical industry. Bringing thermocatalytic and electrocatalytic processes together, this paper compares nine process routes for green ethylene production from air-captured CO2 and off-shore wind electricity in order to displace fossil-based ethylene, with a particular focus on technology readiness for near-future deployment. The methanol-mediated thermocatalytic route has the lowest projected levelised cost at £2900 per ton of ethylene by 2035, closely followed by direct and tandem CO2 electroreduction routes in the range £2900–3200. The price of green ethylene at three times or more its current market price is confirmed through a sensitivity analysis varying the levelised cost of electricity, stack cost, and market price of propylene or oxygen simultaneously. While these green ethylene production processes would be carbon negative from a cradle-to-gate viewpoint, displacing a conventional ethane cracker with annual production capacity of 800 kt could consume as much as 46–66 TW h of renewable electricity, which is a major barrier to deployment.
Broader contextEthylene is the most widely produced organic compound globally. For decades, steam cracking has been the dominant method to produce ethylene from fossil fuels but its production is one of the largest CO2-emitting processes in the chemical industry, calling for the development of more sustainable production routes. Of the alternatives to defossilise ethylene, is the use of CO2 captured from ambient air as feedstock and renewable electricity from offshore wind in order for the produced ethylene to be fully compliant with the green chemical standard. Candidate technologies for producing such green ethylene at scale include a range of conventional and unconventional, thermocatalytic and electrocatalytic processes, which have to date been assessed separately and often using process boundaries that fail to comply with the green production standard. The techno-economic assessment here fills in this gap and seeks to predict a lower bound on future market price of green ethylene by 2035, with a particular focus on the UK context. |
1 Introduction
Light olefins, comprising ethylene and propylene, are fundamental building blocks in the chemical industry for the production of plastics, synthetic fibers and synthetic rubbers.1,2 In Europe light olefins are mostly produced through naphtha steam-cracking, while in the United Kingdom (UK) and the United States (US) alkane steam crackers are more common.3–5 A typical ethane cracker produces about 50% ethylene, 35% ethane, 5% C3+, and additional light hydrocarbons.2 In the UK alone, the annual olefins production amounts to 2.75 Mt and emits 3.1 Mt of CO2, making it the most emission-intensive process across the UK's chemical sector (share of 17%).6,7 It contributed about 0.5% of the UK's total greenhouse gas (GHG) emissions in 2019.8,9 Defossilising olefins production is particularly challenging given that fossil fuels are not only used to meet the utility demands, but also as the carbon feedstock itself.10Biomass, polymers and CO2 are all candidate feedstocks for defossilising light olefins and olefin-derived products.11–17 Since sustainably-sourced lignocellulosic biomass is in limited supply, its use may rather be prioritised for negative emission technologies such as bioenergy for power plants coupled with carbon capture and long-term geological storage.18 Challenges with polymer recycling, either mechanically or chemically, arise due to feedstock heterogeneity and availability.19 The number of times a polymer can undergo mechanical recycling is furthermore limited due to the degradation of its mechanical properties,20 while additives and impurities can cause catalyst deactivation in chemical recycling processes.21,22 In contrast to biomass and plastic waste, CO2 captured from industrial processes or extracted from the atmosphere can be purified to meet process specification for its utilisation. The UK has large CO2 storage capacities and aims to become a global leader in carbon capture, utilisation, and storage.23 The industrial clusters around Merseyside and on the East coast are expected to capture, respectively, 10 and 27 MtCO2 annually from industrial point sources from 2030.24–26 Large quantities of CO2 could therefore become available for CO2 utilisation projects, although no such development is yet planned in the UK.27 While CO2 from industrial processes and power plants remains ultimately linked to fossil fuel consumption, direct air capture (DAC) already has a negative carbon footprint for the current UK electricity mix (0.18 kgCO2 kW h−1 in 2022).28,29 Following the terminology applied to hydrogen production processes,30 only a production route based on atmospheric carbon and renewable energy may indeed qualify as green.
Two pathways for the utilisation of CO2 using renewable energy include: (i) thermocatalytic processes reliant on green hydrogen, and (ii) electroreduction of CO2 (CO2RR) powered by renewable electricity. The former can benefit from the economy of scale for large production volumes, minimize utility demands via heat integration, and produce valuable hydrocarbon co-products. Although at a lower technology readiness level (TRL) than thermocatalytic processes, CO2RR avoids losses incurred in converting electricity to hydrogen and will benefit from the large technological learning rates projected for hydrogen electrolysers in the near future (e.g., from $880 kW−1 today down to $400 kW−1 by 2030 for 10 MW PEM electrolysers).31 CO2RR may be used to directly produce ethylene or alternatively produce an intermediate that is subsequently converted to ethylene. Nevertheless, low-margins force the chemical industry to be risk-averse, causing a preference for thermocatalytic technologies which have greater maturity.32 Additionally, the delay induced by waiting for a low-TRL technology to become mature comes at the risk of missing emission reduction targets.33 There is a need, therefore, for comparing the available routes from CO2 and renewable energy to ethylene in a systematic manner, through taking current performance into account to decide whether to prioritise investment into scale-up or further research-and-development. Investment into the defossilisation of ethylene would have a significant impact across the chemical sector, as its embedded emissions are currently a major contributor to the cradle-to-gate emissions of numerous downstream products, including low- and high-density polyethylene (LDPE, HDPE), polyethylene terephthalate (PET), and polyvinyl chloride (PVC).34,35
1.1 Techno-economic review
Multiple techno-economic assessments of CO2 electroreduction processes have set out to determine which products could become competitive with conventional production routes, most commonly, formic acid, ethanol, carbon monoxide and ethylene.36–39 Orella et al.37 further included methane, Verma et al.38 methanol, and Jouny et al.39 methanol and n-propanol. Having the highest product value per electron, formic acid and carbon monoxide were the only products with positive end-of-life net present values under current market prices. Ethylene produced via CO2RR could be competitive with naphtha cracking, although not with ethane cracking.36 None of these studies focused on green ethylene production but instead assumed processes powered with grid electricity in a price range of $30–60 MW h−1 and fed with CO2 captured from large point sources at a price of $16.5–70 tCO2−1 to minimise production cost. Disregarding the alkaline conditions commonly used in CO2RR experiments, idealised assumptions were made for CO2 electrolysers with proton-exchange membrane (PEM) in terms of their faradaic efficiency, current density, and single-pass conversion. Additionally, liquid and gas separation costs were taken proportional to the product flowrates rather than based on the actual product composition.Going beyond the CO2 electroreduction processes, existing comparisons between thermocatalytic and electrocatalytic processes for producing methanol,40,41 methane42 and syngas43 from CO2 concluded that the former would be more profitable. It was also found that the production cost of these chemicals is typically driven by operating costs, accounting for more than 60% of the total cost. The losses incurred in the conversion of renewable electricity to H2 in any of the thermocatalytic routes could be afforded because water electrolysers presently enjoy higher energy efficiencies (around 70%) than CO2 electrolysers (dependent on target product but typically below 50%).40,42,43 A limitation in all of these studies is that the levelised cost of renewable electricity was set equal to the price of electricity, thereby neglecting the cost of balancing both daily and seasonal intermittency. Kim et al.41 even assumed a plant operation using free, surplus renewable electricity. Again, idealised assumptions on the performance of CO2 electrolysers were made,40,41 and only Welch et al.42 and Moreno-Gonzalez et al.43 considered a CO2 feedstock from DAC, which is necessary for the produced chemicals to qualify as green.
Regarding the complete route from CO2 to ethylene using renewable electricity, the thermocatalytic conversion of CO2 to methanol then to olefins (MS+MTO) was compared to CO2 methanation followed by oxidative coupling of methane (CTM+OCM)44 and to direct Fischer–Tropsch synthesis from CO2 (D-FT).45 Based on the energy content of the products, ethylene from D-FT could cost eight times as much as from MS+MTO due to the large utility requirement for the compression and pre-heating of its recycle stream. Ethylene from CTM+OCM could also be twice as costly as from MS+MTO,44 although the corresponding assessment did not account for differences in the product stream composition. In separate comparisons,46,47 both the MS and MTO stages were found to be cheaper than CTM and OCM, respectively. The cost advantage of MS over CTM was driven by the lower H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratio while MTO outcompeted OCM by generating a larger revenue from selling propylene compared to ethylene in the US market. Focusing only on electrocatalytic processes, Ramdin et al.48 compared the direct CO2RR to ethylene (D-eC2H4) to a two-stage electrolysis process converting CO2 to CO either at high- or low-temperature, followed by CO electroreduction (CORR) to ethylene. The high-temperature two-step route was predicted to be cheaper, while the total cost of both low-temperature routes were comparable. Modelling the separation processes in more detail than other studies, Ramdin et al.48 also confirmed the dominant costs to be the capital and operating costs of the electrolysers, contributing over 75% of the total system cost. Bringing together both types of processes, Ioannou et al.49 compared the electrocatalytic D-eC2H4 route to the thermocatalytic MS+MTO route. The latter was found to be the cheaper alternative since it had lower capital costs and could benefit from the higher energy efficiency of H2 electrolysers (65%) compared to CO2 electrolysers (34%). For scenarios in which electricity was generated by nuclear reactors or onshore wind turbines, Ioannou et al.49 predicted negative global warming potentials for both routes, yet production costs two to three times larger than naphtha cracking.
:CO2 ratio while MTO outcompeted OCM by generating a larger revenue from selling propylene compared to ethylene in the US market. Focusing only on electrocatalytic processes, Ramdin et al.48 compared the direct CO2RR to ethylene (D-eC2H4) to a two-stage electrolysis process converting CO2 to CO either at high- or low-temperature, followed by CO electroreduction (CORR) to ethylene. The high-temperature two-step route was predicted to be cheaper, while the total cost of both low-temperature routes were comparable. Modelling the separation processes in more detail than other studies, Ramdin et al.48 also confirmed the dominant costs to be the capital and operating costs of the electrolysers, contributing over 75% of the total system cost. Bringing together both types of processes, Ioannou et al.49 compared the electrocatalytic D-eC2H4 route to the thermocatalytic MS+MTO route. The latter was found to be the cheaper alternative since it had lower capital costs and could benefit from the higher energy efficiency of H2 electrolysers (65%) compared to CO2 electrolysers (34%). For scenarios in which electricity was generated by nuclear reactors or onshore wind turbines, Ioannou et al.49 predicted negative global warming potentials for both routes, yet production costs two to three times larger than naphtha cracking.
1.2 Contributions and outline
The techno-economic review above identifies the methanol-mediated thermocatalytic route as a promising candidate for low-cost green ethylene production from CO2 and renewable energy. While combined electrocatalytic and thermocatalytic processes via syngas and methanol have been reviewed,50 none of these combinations have been systematically compared through techno-economic analysis. Disparate system boundaries and costing methodologies in existing, often pairwise assessments make it difficult to draw meaningful conclusions about their overall cost-competitiveness.To supply either CO2 utilisation pathway with green electricity and hydrogen, the UK's renewable power generation capacity would need to be expanded.51 A whole systems perspective is much needed to account for the limitations on renewable generation capacity due to land availability, cost and public acceptance.51 However, current scenarios for the UK's future energy system fail to reflect these additional demands,52–54 due in part to the difficulty of integrating chemical production processes into whole-system energy modelling. Deciding early on a decarbonisation strategy across different sectors is however paramount to reducing the overall system costs associated with the transition to net-zero by 2050.55
This paper presents a comprehensive techno-economic analysis of nine process routes for green ethylene production from air-captured CO2 and renewable electricity from a dedicated offshore wind farm, by bringing thermocatalytic and electrocatalytic processes together. Using consistent system boundaries, process inventories are derived from best-in-class, heat-integrated thermocatalytic processes and both low- and high-temperature CO2 or CO electrolysers. The candidate routes are ranked according to the levelised cost of ethylene and a sensitivity analysis is conducted to confirm that the conclusions are robust towards changes in the levelised cost of electricity, stack cost, and market price of co-products. While the focus of this work lies in the comparison of defossilised ethylene production pathways on a process level, the process inventories computed herein could be used as part of any energy system model too.
The rest of the paper is structured as follows: Section 2 starts with reviewing the technology readiness and process conditions for selected production routes to green ethylene, then details the process modelling and levelised cost calculation methodology. Section 3 discusses the predicted process inventories and levelised costs of ethylene production for each route, before analysing the sensitivity of the top two routes, and examining systemic consequences beyond the process boundaries. Finally, Section 4 concludes the paper.
2 Methodology
2.1 Technology selection and modelling
A common system boundary is set around each process so that only renewable electricity, green hydrogen, CO2, air and water can enter, while only ethylene, hydrocarbon co-products, oxygen, residual CO2 and wastewater can leave. All hydrocarbon mixtures are separated in terms of their ethylene, ethane, propylene, propane, methane or C4+ constituents. Residual emissions from purge streams and carbon-capture processes and wastewater from the separation and purification are also accounted for as outputs.The processes of interest (Fig. 1) convert CO2 into ethylene either directly or via a single one-carbon intermediate (CO, CH4 and CH3OH). The conversion of CO2 to an intermediate is referred to as stage-1 and the subsequent conversion of the intermediate to ethylene as stage-2. For those processes needing syngas in stage-2, in situ produced CO is combined with green H2 in the relevant stoichiometry ratio. The ethylene production rate from stage-2 governs the production rate of the intermediate from stage-1, and both stages are integrated. Any unconverted CO2 or H2 from stage-2 is recycled to stage-1. Similarly, the heat and electricity utility demands of one stage may be met by excess heat or electricity from the other stage. A more systematic process integration using optimisation was beyond the scope of this work.
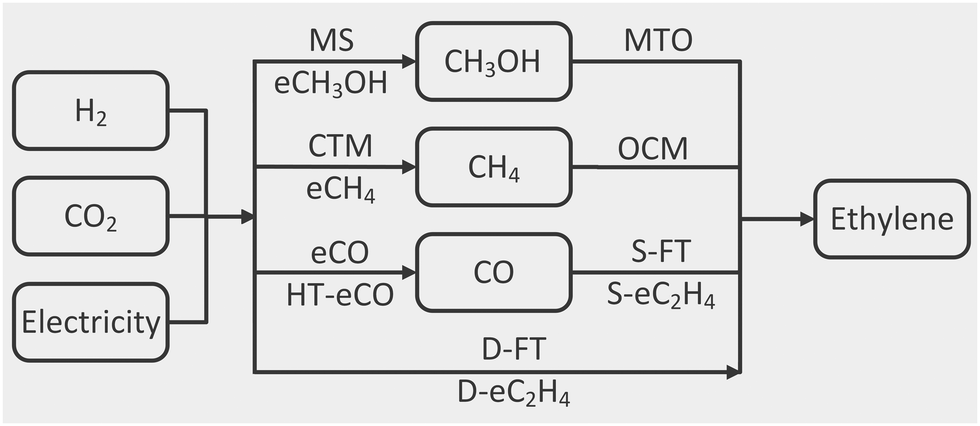 | ||
| Fig. 1 Overview of the processes and intermediates in the conversion of CO2 to ethylene. Abbreviations: MS: methanol synthesis; eCH3OH: CO2 electroreduction to methanol; MTO: methanol to olefin; CTM: CO2 methanation; eCH4: CO2 electroreduction to methane; OCM: oxidative coupling of methane; eCO: CO2 electroreduction to CO; HT-eCO: high-temperature CO2 electroreduction to CO; S-FT: syngas Fischer–Tropsch; S-eC2H4: CO electroreduction to ethylene; D-FT: direct Fischer–Tropsch; D-eC2H4: CO2 electroreduction to ethylene. Refer to Section A of the Appendix for details. | ||
A key outcome of the modelling conducted herein is linear input-output models for each (single- or two-stage) process derived from the detailed mass and energy process inventories and normalised to the production of 1 tonne of ethylene in the manner of a functional unit in life-cycle assessment (LCA). Such linear models are particularly suitable for integration into energy system models as part of future research.
The reference process are adapted to the new system boundary for green ethylene production by excluding any process unit generating an intermediate from fossil resources and including an air separation unit where a pure oxygen feed stream is required.56 Where applicable, the cryogenic distillation train by Liu et al.57 is added for the separation of hydrocarbon mixtures.
The mass and energy balances are subsequently updated, including the recycling of concentrated CO2 and H2 streams and any purge and flue gas streams. The utility demands are met by first integrating any excess heat and electricity. Where a net heat surplus is available after integration, a condensing steam turbine is designed to produce electricity.58 And where a net import of hot utility is required, the system is extended to include a hydrogen boiler. Finally, capital expenses from the reference processes are scaled according to the six-tenth rule58 and converted to 2019 prices using the Chemical Engineering Plant Cost Index (CEPCI).59
Refer to Section A of the appendix and Section S1 of the ESI,† for further details about the reference processes, including any adjustments made.
The electrochemical model describes the relationship between the electrical power and CO2 (or CO) feed rate with the flow rate of product. The electric current is set by the flow rate and the faradaic efficiency (ηF) of the product, while the required electrode area (Acell) follows from the current density (j). The cell voltage (Ecell) is calculated based on the half-cell potentials of the CO2RR (Ered) and oxygen evolution reaction (OER), which depart from the standard cell potentials due to both activation potentials and ohmic losses. While the half-cell potential at the anode could be reduced by oxidising organic molecules instead of water,61,62 this option would not be compliant with the set system boundary around renewable electricity, H2, CO2, air and water (cf. Section 2.1). The flow rates of the co-products and reactants are estimated from the product flow rate, the electrochemical reaction's stoichiometry (cf. Section A of the Appendix) and the single-pass CO2 or CO conversion (X). The parameter values for the electrocatalytic processes of interest are summarised in Table 1. The faradaic efficiencies, current densities, and cell voltages were all measured experimentally. The set single-pass CO2 or CO conversions of 40% were not measured, but deemed achievable by 2035 based on 10–40% conversion values reported in recent alkaline zero-gap electrolysers,63–66 largely limited by carbon losses due to carbonate formation.67,68
For the scale up of CO2 and CO electrolysers, a zero-gap cell design is preferred to a flow cell design as it reduces the overall cell resistance and increases performance, particularly at high current densities.66,73 The cell design and the balance of plant (BOP) layout of anion-exchange membrane (AEM) electrolysers are analogous to those of PEM electrolysers.74 A similar expression therefore is used for their capital cost, building on a specific stack cost estimate around £280 kW−1 by 2035.75 For 1 MW PEM electrolysers, the stack cost contributes about 35% of the total system cost;76 the BOP makes the other 65% in terms of power supply, deionised water circulation system, gas processing and cooling equipment, all expected to be manufactured at scale for electrolyer systems by 2035. Whereas the cost of the BOP scales with the power consumption of the electrolyser, the stack cost correlates with the electrode area.
To separate ethylene from the mixed gas products of the electrolysers, vacuum pressure swing adsorption (VPSA) processes are used,48,77 and any unconverted CO2 is recovered and recycled at a 97% capture rate by mixed-amine CO2 absorption.78 The separation of the liquid co-products (acetic acid, alcohols) is also considered when their concentration in the electrolyte exceeds 20 wt%. The utility demands of the separation train are met by hydrogen boilers and renewable electricity imports because they cannot be heat-integrated with the low-temperature electrolysers. Both the capital and utility costs of the separation processes are scaled up according to the total product flow rate. The total invested capital (TIC) comprises the fixed capital investments for the CO2 electrolyser and its associated separation train, as well as additional working capital, start-up and contingency costs.79
Refer to Section A of the Appendix for additional details about the reference CO2 electrolysers and to Section S2 of the ESI,† for further details about the electrochemical model and the costing methodology for the low-temperature CO2 electrolysers and their separation units.
Based on a scenario in which the cumulative capacity continues to grow at the same rate as in the past, a specific cost of £600 kW−1 is assumed for the SOEC system by 2035.84 This represents a >70% reduction from the present cost of 1200–2000 kW−1.84–88 The future capital cost of SOEC systems are perceived to be more uncertain than that of alkaline or PEM electrolysers,89 given the limited number of installations today84 and the anticipated technological improvements.90 The assumption that learning from high-temperature water electrolysis systems can be transferred to high-temperature CO2 electrolysis is consistent with the low-temperature CO2 electrolysers above.
Refer to Section A of the Appendix for additional details about the reference SOEC electrolysers and to Section S2 of the ESI,† for further details about the SOEC performance and costing.
2.2 Techno-economic analysis
The candidate process routes are compared in terms of the levelised cost of ethylene (LCOEt, eqn (1)), which accounts for the resource consumption, the revenues from co-products, and the annualised capital costs of the plant at scale. The cash-flow calculation assumes that the plant is erected during the first year, then operated at full capacity from the second year onward. The return on investment is neglected by setting the net present value of the project to zero.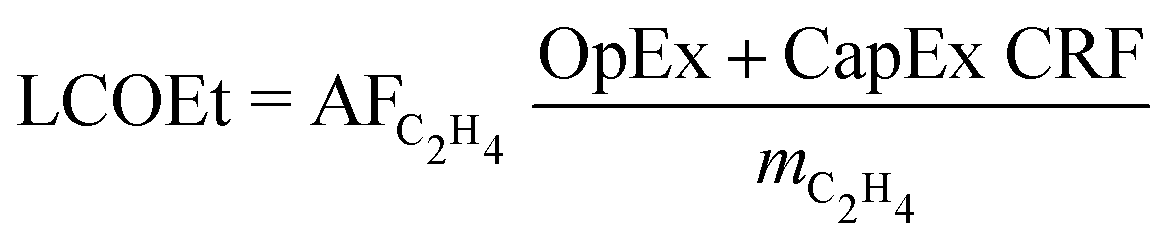 | (1) |
The capital expenditures (CapEx) are annualised using a uniform capital recovery factor (CRF). The operational expenditures (OpEx) comprise the variable utility costs and the fixed operating expenses for salaries, maintenance and overheads.
The economic allocation factors (AFj, eqn (2)) apportion the total annualised cost of a process among all the co-products (j ∈ {CH4, C2H4, C2H6, C3H8, C3H6, C4+, O2}), according to their share on the overall revenue.
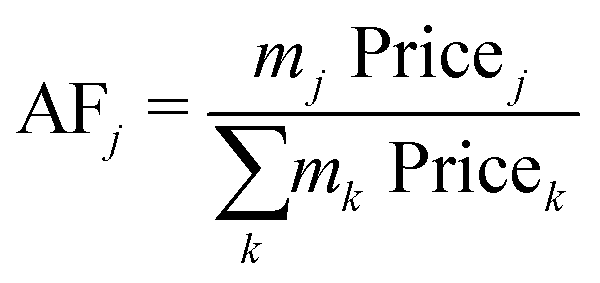 | (2) |
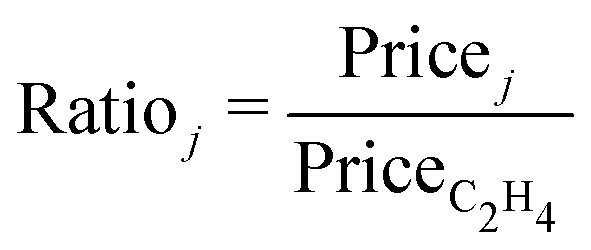 | (3) |
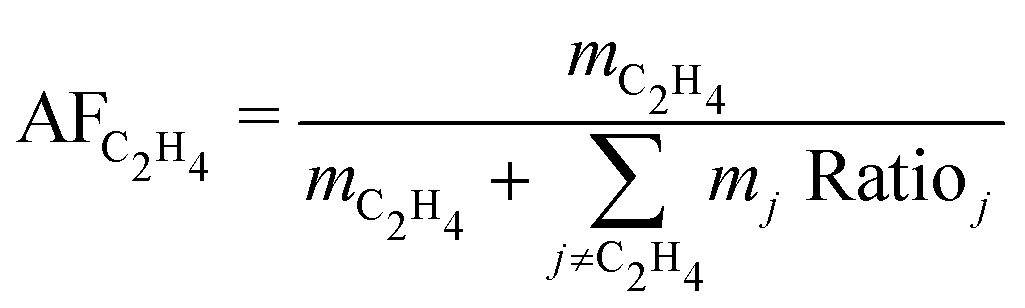 | (4) |
Refer to Sections S4 and S5 of the ESI,† for further details on economic calculations.
2.3 Case study and sensitivity analysis
The ethane cracker in Wilton (North Yorkshire, UK), with an annual production capacity of 800 kt, is chosen as the reference herein.5,91 A plant lifetime of 20 years is assumed, with a utilisation factor (UF) of 90% and discount rate of 6%.92,93 To study a transition to green ethylene in the near future, cost assumptions for 2035 are used where available, with all costs expressed in £2019 for consistency. The economic input parameters are summarised in Table 2 and discussed below.| Parameter | Unit | Nominal | Range |
|---|---|---|---|
| LT | year | 20 | |
| r | — | 0.06 | |
| UF | — | 0.9 | |
| CEPCI2019 | — | 607.3 | |
| m C2H4 | kt year−1 | 800 | |
| Costtax | £ tCO2−1 | 301.4 | |
| Costtww | £ tww−1 | 0.52 | |
| Costel | £ MW h−1 | 54.6 | 49.9–76.7 |
| CostH2 | £ MW h−1 | 74.3 | 67.9–104.3 |
| CostCO2 | £ tCO2−1 | 312.3 | 301.5–363.1 |
| Coststack | £ kW−1 | 283.5 | 235–782 |
| RatioCH4 | — | 0.13 | |
| RatioC2H6 | — | 0.15 | |
| RatioC3H6 | — | 0.87 | 0.70–1.05 |
| RatioC3H8 | — | 0.39 | |
| RatioC4+ | — | 0.8 | |
| RatioO2 | — | 0.13 | 0.00–0.13 |
While such large-scale production capacity is instrumental to contextualise the electricity consumption of green ethylene (cf. Section 3.4 below), some of the emerging technologies may not reach kilotonne annual production levels in the near future. It is worth reiterating that the process inventories and LCOEt for each route are calculated for smaller (relevant) process scales, as dictated by their reference processes (cf. Section A of the Appendix). The assessment methodology therefore relies both on technology scale-up and technology scale-out.
The cost of non-intermittent electricity (Costel), renewable hydrogen (CostH2) and air-captured CO2 (CostCO2) are based on a common value for the levelised cost of electricity (LCOE) of offshore wind in the UK.75 Balancing intermittency using both short- and long-term storage for the uninterrupted supply of renewable electricity is assumed to add 30% on top of the LCOE.94 The price of domestically produced green H2 is based on the LCOE using PEM electrolysers with an efficiency of 1.3 MW hel MW hH2,HHV−1, and adding an extra 36% for capital and operating costs of the electrolysers.75 Given the proximity of underground salt caverns to the Wilton production site95 and existing plans to build a hydrogen pipeline network in the North East of England by 2030,96 additional charges for transport and storage are henceforth neglected.97,98 For the ethylene product to be considered green, only CO2 from DAC is deemed a suitable feedstock. A cost of £312.3 tCO2−1 is estimated on the assumption that solid sorbent-based DAC is deployed at scale by 2035, including automated manufacturing of the CO2 collectors and based on the average relative humidity and temperatures in the UK,99 which is six-times higher than the cost for CO2 capture from cement.100,101 Air and water are assumed to be free of charge. The cost of residual process emissions (Costtax) is based on the 2035 UK carbon price of £301.4 tCO2−1,102 while a generic cost of £0.52 per ton of treated wastewater (Costww) is assumed.103
A further assumption in the economic comparison is that all the hydrocarbon co-products can be sold separately, although they are often integrated with downstream processing or combusted in existing plants.2 The market prices of hydrocarbon co-products (Pricej) are based on average European market prices in 2019,104 with the market price of ethylene itself (PriceC2H4) at £1000 tC2H4−1. The price of O2 co-product (PriceO2) is taken at £130 tO2−1.105 All of the relative prices (Ratioj, eqn (3)) are assumed to remain constant throughout the plant's lifetime. Notice, in particular, that ethylene has the highest market price amongst the co-products.
To determine the extent to which the process ranking is subject to key modelling assumptions, a sensitivity analysis is conducted in terms of the LCOE, the stack cost (Coststack), and the relative market prices of propylene (RatioC3H6) and oxygen (RatioO2) with respect to the ethylene price. A Monte Carlo simulation is conducted based on low-discrepancy Sobol sampling with 1024 scenarios, leading to a confidence interval smaller than 1.5% around the mean LCOEt value. All four uncertain factors are modelled with triangular probability distributions (cf.Table 2). The costs of uninterrupted renewable electricity (Costel), green H2 (CostH2), and CO2 feedstock (CostCO2) are assumed to be correlated with the LCOE of offshore wind, whose value ranges between current costs (£59.0 MW h−1) and projections for 2050 (£38.4 MW h−1).75,107 CostH2 is taken directly proportional to Costel since electricity is the main driver of green H2 price, while the variation range for CostCO2 accounts for the fact that electricity contributes around 40% of the cost of the solid sorbent-based DAC process.99 Since public data on propylene prices are limited, a range of ±20% is assumed.58 The upper and lower bounds for O2 price are so chosen to describe a scenario whereby the market price would decrease as a result of market saturation.49 The stack cost is varied between the capital cost of PEM electrolyser systems in 202075 and projected capital cost of a large-scale plant by 2030.31
3 Results and discussion
3.1 Process inventories
Process inventories for the nine selected routes at scale are shown in Fig. 2, with more details about the mass and energy balances available in Section S6 of the ESI.† All these inventories are scaled for 1 tonne of ethylene produced. Differentiated by colour, the processes correspond to three fully thermocatalytic routes (MS+MTO, CTM+OCM, D-FT), three fully electrocatalytic routes (eCO+S-eC2H4, HT-eCO+S-eC2H4, D-eC2H4), and three hybrid routes (eCO+S-FT, HT-eCO+S-FT, eCH4+OCM). The input-output inventory streams comprise electricity (subscript el), carbon dioxide (subscript CO2), hydrogen (subscripts H2,f and H2,h), ethylene (subscript C2H4), other hydrocarbons (subscript hc), oxygen (subscript O2), wastewater (subscript ww), and residual emissions (subscript CO2-eq).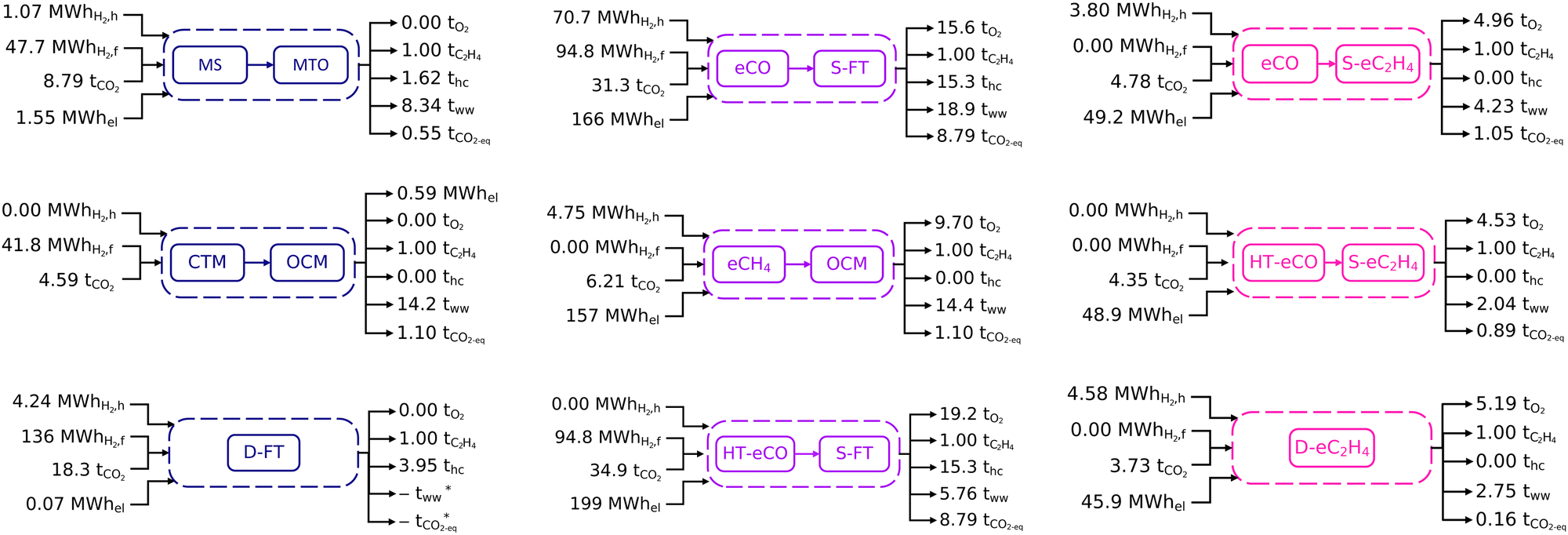 | ||
| Fig. 2 Process inventories for the production of 1 t of ethylene using fully thermocatalytic routes (blue), fully electrocatalytic routes (pink) and hybrid routes (purple). The hydrogen inputs distinguish heat (H2,h) and feedstock (H2,f). The hydrocarbon by-products (hc) include methane, ethane, propylene, propane and C4+. The waste streams consist of wastewater (ww) by the separation processes and residual GHG emissions (CO2-eq). *Not quantified. | ||
Since CO2 provides the carbon feedstock, the CO2 inventories closely correlate with the total hydrocarbon production (sum of C2H4 and hc). Most of the unconverted CO2 and intermediates are recycled within the process boundaries, therefore the net CO2 feedstock requirement is largely independent of the overall conversion rates. Despite the reference processes for D-FT and S-FT being tailored towards light olefins, their ethylene yields remain low and large amounts of CO2 are consumed in side reactions to co-products such as propylene and C4+. As a result, D-FT, eCO+S-FT and HT-eCO+S-FT have higher CO2 feedstock requirements than other routes. Likewise, the MS+MTO route has a large CO2 requirement due to significant propylene and C4+ side-production. Conversely, the electrocatalytic processes eCO+S-eC2H4, HT-eCO+S-eC2H4 and D-eC2H4 require less CO2 feedstock since ethylene is the single product. Even though ethylene is also the sole product of OCM, the CO2 requirement of the CTM+OCM and eCH4+OCM routes is intermediate as part of the unconverted methane is combusted in a turbine for electricity production, thus venting CO2 with the exhaust gas.
Hydrogen is either consumed as reaction feedstock (H2,f) or used as heating utility (H2,h). Neither of the electrocatalytic processes (eCO+S-eC2H4, HT-eCO+S-eC2H4, D-eC2H4) nor the methane-mediated route (eCH4+OCM) require hydrogen as feedstock since hydrogen is generated in situ from the humidified CO2 feed. However, as all low-temperature electrocatalytic processes have low single-pass conversion rates, they require hydrogen to provide heat for the regeneration of the amine sorbent used in the separation of unreacted CO2 before recycling. By contrast, the thermocatalytic processes consume hydrogen mainly as a feedstock. Given that the reactions are typically exothermic, the hydrogen feedstock is also indirectly used to meet part of the heat and electricity demands through process heat integration. However, additional hydrogen as heat utility is required for the D-FT process to preheat its large recycle stream and for the MS+MTO process despite using excess heat to preheat the reactor feed (cf. Section S6 of the ESI†). All of the thermocatalytic processes furthermore include turbines for converting excess reaction heat into electricity. But only for the integrated CTM+OCM process is the excess heat high enough for an electricity surplus to be generated and sold to the grid. The consumption pattern of the hybrid process eCO+S-FT reflects the characteristics of thermo- and electrocatalytic routes because significant amounts of hydrogen are consumed both as feedstock to achieve the necessary syngas composition and as heating fuel in the carbon capture process due to the low single-pass conversion of the eCO stage.
The electricity demands of the electrocatalytic and hybrid processes correlate with their cell voltages and faradaic efficiencies. Despite the high faradaic efficiency of the eCO and HT-eCO stages and the excess electricity generated in the S-FT stage (15.9 MW hel per tCO), eCO+S-FT and HT-eCO+S-FT have the highest electricity demand due to their large intermediate syngas stream (33.6 tCO per tC2H4); eCH4+OCM comes in third position as the eCH4 stage has by far the highest cell voltage. In contrast, the fully electrocatalytic routes eCO+S-eC2H4, HT-eCO+S-eC2H4 and D-eC2H4 consume 3 to 4 times less electricity than the hybrid routes. And although eCO+S-eC2H4, HT-eCO+S-eC2H4 require 10e− per molC2H4 compared to 12e− per molC2H4 in D-eC2H4, all three have similar electricity demands. For those processes having electrocatalytic conversion stages, the quantity of the oxygen by-product is commensurate to the electricity demand given the OER is the complementary half-cell reaction of the CO2/CO reduction reaction.
A significant amount of water is formed as by-product in both thermocatalytic and electrocatalytic processes. These wastewater streams either contain traces of hydrocarbons and alcohols in the case of thermocatalytic processes or correspond to an alkaline solution in the case of electrocatalytic processes, and they require treatment before crossing the process boundaries. Although the costs for an external wastewater treatment service were included in the TEA, on-site wastewater regeneration and recycling could also be implemented to lower the water footprint of the processes. The amount of wastewater produced by thermocatalytic processes generally correlates with the amount of hydrocarbons formed. The CTM+OCM process uses large amounts of steam to dilute the OCM reactor feed and therefore generates more wastewater than the MS+MTO process. Recall also that the wastewater stream of the D-FT process could not be quantified due to incomplete mass balances (cf. Section A.4). But as dewatering at the S-FT reactor outlet alone produces 18.81 tH2O per tC2H4, wastewater production is expected to be comparable for D-FT.
Direct (gross) emissions from the thermocatalytic processes are caused by purge streams, which are translated into CO2-equivalents on Fig. 2; while in electrocatalytic processes, the direct emissions correspond to any residual CO2 not captured by the mixed-amine absorption column. These direct emissions are lower than those of conventional ethane crackers (c. 0.74 tCO2 tC2H4−1)108 for both the direct electrochemical route D-eC2H4 and the thermochemical route MS+MTO, and slightly higher for the two-stage electrochemical routes eCO+S-eC2H4 and HT-eCO+S-eC2H4. By contrast, the hybrid processes eCO+S-FT and HT-eCO+S-FT present much larger direct emissions due to the Fischer–Tropsch synthesis having a small ethylene yield and forming a range of saleable co-products. Nevertheless, an economic allocation among these co-products (cf.Fig. 4 below) suggests that the direct emissions associated with ethylene would remain comparable to that of ethane cracking. Recall also that the purge stream and hence the emissions of the D-FT process could not be quantified due to incomplete mass balances (cf. Section A.4). Lastly, the carbon intensity of both the CTM+OCM and eCH4+OCM processes exceeds that of ethane cracking because significant CO2 emissions are caused by the combustion of unconverted methane from the OCM stage to generate electricity. Although not implemented here, capturing and recycling CO2 from this flue gas might be economically competitive given the high cost of DAC. It is also worth reiterating that the CO2 feedstock is much larger than the direct (gross) emissions in all of the green ethylene routes and so, unlike ethane crackers, the production processes are still net CO2 consumers. To draw final conclusions on the environmental impact, a thorough life-cycle assessment should be performed in future research.
3.2 Techno-economic comparison
The levelised costs of ethylene (LCOEt) from DAC and renewable electricity for the nine production pathways are compared in Fig. 3. The breakdown differentiates capital, fixed and variable operating costs, the latter being further differentiated into the contributions of CO2 feedstock, renewable energy (electricity, hydrogen), wastewater treatment, and carbon tax.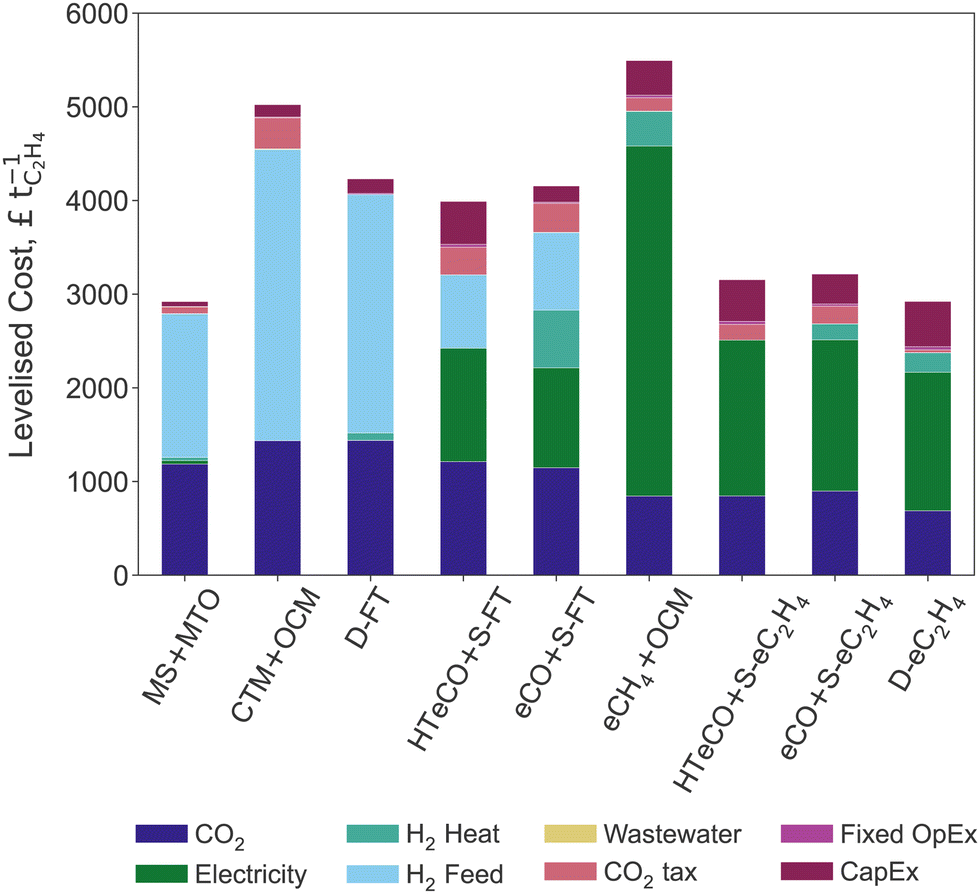 | ||
| Fig. 3 Levelised cost of ethylene and corresponding breakdown for the nine candidate routes. | ||
The predicted LCOEt ranges between £2920–5280 tC2H4−1. Remarkably, both the thermocatalytic process MS+MTO and the direct electroreduction process D-eC2H4 provide the lowest-cost options at around £2920 tC2H4−1. The most expensive routes are the methane-mediated hybrids with OCM, eCH4+OCM and CTM+OCM, both above £5000 tC2H4−1. The Fischer–Tropsch routes D-FT, eCO+S-FT and HT-eCO+S-FT have a comparable LCOEt in the range of £3930–4230 tC2H4−1. Given that the LCOEt of D-FT omits the carbon tax for direct emissions and wastewater treatment cost, its LCOEt is however underestimated. The three elecytrocatalytic routes D-eC2H4, eCO+S-eC2H4 and HT-eCO+S-eC2H4 also perform similarly with an LCOEt between £2920–3220 tC2H4−1.
All of these levelised costs can be interpreted as lower bounds on green ethylene market prices since they omit any profit margins. Even compared to a high market price of £1400 tC2H4−1 (recorded in April 2022)109 and after taxing the CO2 emissions of a conventional ethane cracker (c. £300 tC2H4−1 at 0.97 tCO2 tC2H4−1), the predicted green ethylene prices are far from being competitive with fossil-based ethylene.
The routes involving an electrocatalytic conversion step generally have a higher CapEx than the thermocatalytic processes (see Table 3). The eCO+S-eC2H4 process achieves the lowest CapEx among the electrocatalytic and hybrid routes because it requires a relatively small electrode area given its high current density (cf. Section 2.1.2). It is nevertheless 4 times more expensive than MS+MTO or CTM+OCM. Despite being fully thermocatalytic, D-FT requires larger unit and equipment sizes to make up for its low ethylene yield, which is why its CapEx is comparable to that of eCO+S-eC2H4 and 3.5 times greater to that of S-FT. The CapEx of the electrocatalytic and hybrid processes are dominated by the CO2/CO electrolyser cost, while the separation equipment and thermocatalytic stage contribute less than 6% and 12%, respectively.
| Route | CapEx, £2019Bn |
|---|---|
| MS+MTO | 1.17 |
| CTM+OCM | 1.21 |
| D-FT | 5.72 |
| HT-eCO+S-FT | 37.81 |
| eCO+S-FT | 13.56 |
| eCH4+OCM | 7.80 |
| HT-eCO+S-eC2H4 | 6.54 |
| eCO+S-eC2H4 | 4.88 |
| D-eC2H4 | 7.54 |
Despite the large differences observed in Table 3, the annualised CapEx only represents a minor share of the LCOEt for any of the processes (see breakdown in Fig. 3). The contributions of the fixed OpEx are also minor as they correspond to a small percentage of the CapEx (cf. Section 2.2). This conclusion holds insofar as the manufacturing of CO2/CO electrolysers can benefit from the learning rates that are predicted for PEM and SOEC electrolysers. It is also worth recalling that the assumed 5-year lifetime of the electrolyser stack by 2035 (cf. Section S2 of the ESI†) far exceeds current stability reports for low-temperature CO2/CO electrolyser cells.71,72,110,111 For instance, reducing the life-time of the D-eC2H4 electrolyser stack to 4 years is predicted to increase its CapEx by 19%. Therefore, extending the lifetime of the electrolyser stack is key towards improving the cost-competitiveness of electrocatalytic routes.112
It is clear from Fig. 3 that the cost of renewable energy—both renewable electricity and green H2—makes up the largest share of the LCOEt across all nine routes. For the fully thermocatalytic routes (MS+MTO, CTM+OCM, D-FT), green H2 contributes over 50% of the LCOEt. Instead, renewable electricity contributes about 50% of the LCOEt for the fully electrocatalytic routes (eCO+S-eC2H4, HT-eCO+S-eC2H4, D-eC2H4). The LCOEt of the hybrid route eCH4+OCM is also dominated by the electrochemical stage eCH4 due to its high cell voltage (cf.Table 1), while the procurement of hydrogen and electricity both contribute significantly to the LCOEt of eCO+S-FT and HT-eCO+S-FT. Except for eCO+S-FT (15%), the contribution of hydrogen as a heating utility to the LCOEt remains below 7%.
In all nine candidate routes, the CO2 feedstock has a notable contribution on the LCOEt because of the high cost of DAC. Substituting CO2 from DAC for fossil-based CO2 could reduce the share of CO2 feedstock to 3–10% of the LCOEt, thereby making its contribution significantly smaller than the cost of green electricity or hydrogen. Conversely, the residual emissions make a minor contribution to the LCOEt in any of the selected routes, not only for a carbon tax of £301.4 tCO2−1 (Table 2) but also in the high price scenario of BEIS with £504 tCO2−1 by 2035.102 Making MS+MTO cost-competitive with ethylene from a conventional ethane cracker (c. 0.74 tCO2 tC2H4−1) would entail a carbon price of £640 tCO2−1 or higher.
While the processes differ in conversion and ethylene yield, the LCOEt is ultimately governed by its economic allocation factor (AFC2H4, eqn (4)), which reflects the relative market prices of the co-products. A breakdown of the economic allocation factor for all nine routes is shown on Fig. 4. CTM+OCM is the only selected route with ethylene as the single saleable product (AFC2H4 = 100%). All the electrocatalytic routes produce oxygen as a saleable by-product alongside the desired carbon monoxide or hydrocarbons, leading to AFC2H4 values between 45–65%. Propylene and C4+ are the dominant co-products of the thermochemical routes MS+MTO and D-FT as well as the hybrid route eCO+S-FT. While MS+MTO has an AFC2H4 value over 40%, those of the Fischer–Tropsch syntheses are much lower due to low ethylene yields.
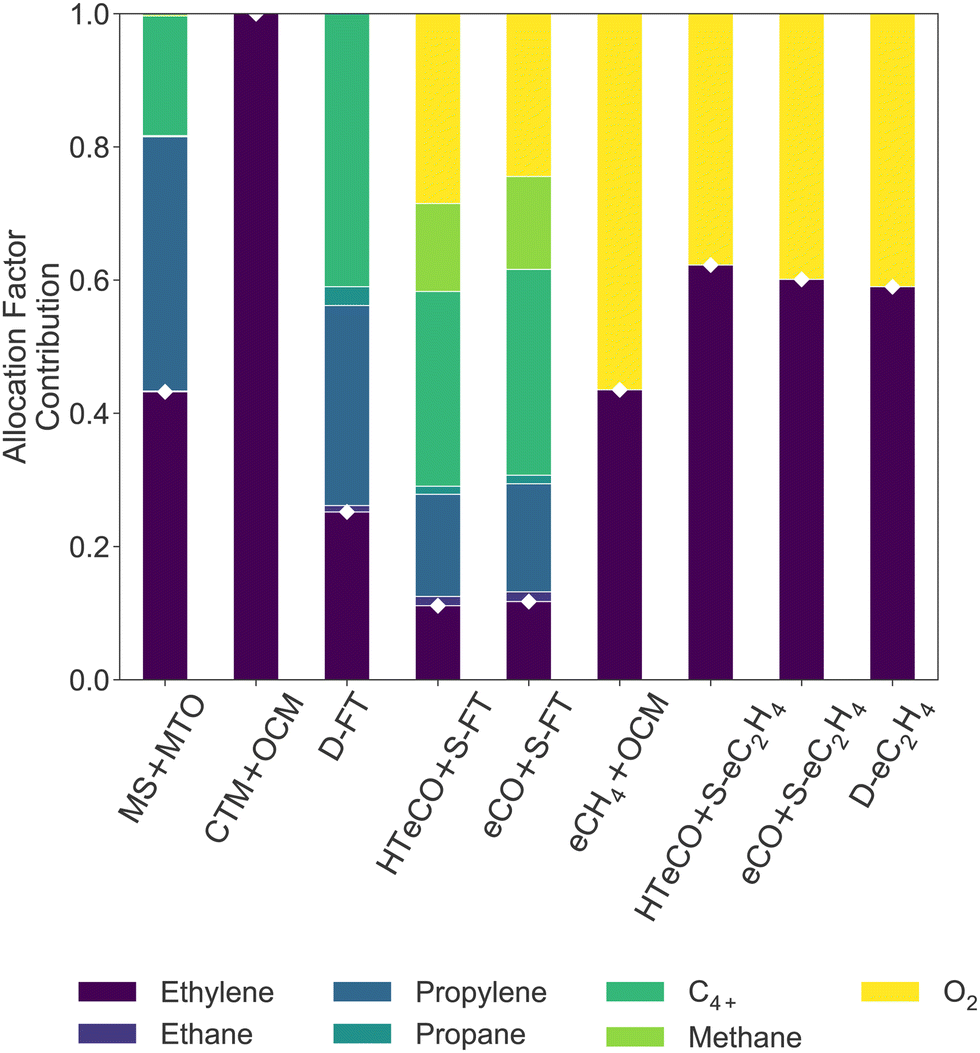 | ||
| Fig. 4 Economic allocation factors AFj (eqn (2)) of the saleable co-products in all nine candidate routes. The allocation factor of ethylene (AFC2H4, white diamonds) determines the LCOEt (eqn (1)). | ||
Based on this techno-economic comparison, it is unlikely for green ethylene production involving a Fischer–Tropsch synthesis (D-FT, eCO+S-FT, HT-eCO+S-FT) to be cost-competitive with other thermocatalytic processes, unless the value of its co-products or its conversion rate were to increase significantly. Neither of the OCM routes (CTM+OCM, eCH4+OCM) is furthermore competitive with MS+MTO given that OCM does not produce any other valuable hydrocarbon products than ethylene. The cost advantage of MS+MTO over CTM+OCM, eCH4+OCM or D-FT is consistent with previous techno-economic assessments.44,45
Finally, the LCOEt of all three electrocatalytic routes (eCO+S-eC2H4, HT-eCO+S-eC2H4, D-eC2H4) is within 10% of that of MS+MTO. This gap would increase significantly if the oxygen by-product from the electrolysers could no longer be valorised or if the life-time of the electrolyser stack did not extend beyond 3 or 4 years, as already noted. A single-pass conversion lower than the assumed 40% in AEM electrolysers (cf.Table 1) would also be detrimental to the LCOEt, due to the combined effect of increasing H2 consumption to recycle unconverted CO2, CO2 feed and CO2 emissions. For instance, a 30% single-pass conversion would already increase the LCOEt of both D-eC2H4 and eCO+S-eC2H4 by 5%. The overall similarity in LCOEt between eCO+S-eC2H4, HT-eCO+S-eC2H4 and D-eC2H4 is in agreement with recent techno-economic assessments, albeit conducted under different system boundaries.48,113 If it were not for the high electrolyte molarity (7 M KOH) in the selected D-eC2H4 reference (cf. Section A.4 of the Appendix),72 D-eC2H4 might as well rank behind the two tandem routes eCO+S-eC2H4 and HT-eCO+S-eC2H4. For instance, in a scenario with 1 M KOH electrolyte and target cell voltage of 3.5 V,114,115 the LCOEt of D-eC2H4 would already increase by 17% (£3600 tC2H4−1).
3.3 Sensitivity analysis
Results from the Monte Carlo analysis results for the two routes with the lowest LCOEt (MS+MTO and D-eC2H4) are summarised in Fig. 5, where uncertainties in the LCOE, stack cost, and market prices of propylene and oxygen relative to the ethylene price are varied simultaneously (cf. Section 2.3). The parity plot confirms that the production cost of the MS+MTO process remains the lowest across the range of sampled uncertainty scenarios. Only in scenarios with low propylene price, low stack cost, high LCOE and high oxygen price, does the LCOEt of D-eC2H4 become comparable to that of MS+MTO. This scenario analysis also reveals that the production cost of green ethylene at three times the current market price of (fossil-based) ethylene corresponds to a rather favourable scenario, due in large part to the skewed distributions of LCOE, stack cost and oxygen price.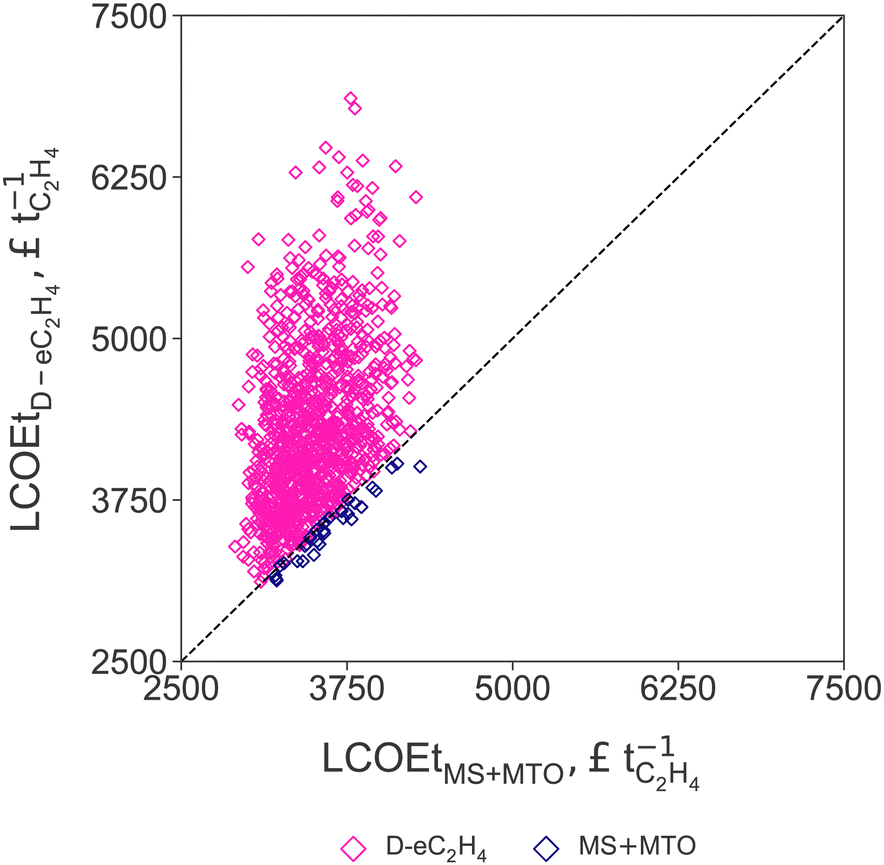 | ||
| Fig. 5 Parity plot comparing the levelised cost of ethylene (LCOEt) for the thermocatalytic production of ethylene via methanol (MS+MTO, on x-axis) and direct CO2 electroreduction to ethylene (D-eC2H4, on y-axis) for each uncertainty scenario. The levelised cost of electricity (LCOE), AEM stack cost, and market prices of propylene and oxygen are varied simultaneously (cf.Table 2). | ||
Recall that varying the LCOE of offshore wind between current costs (£59.0 MW h−1, +40%) and projections for 2050 (£38.4 MW h−1, −9%) changes the cost of non-intermittent renewable electricity, green hydrogen and air-captured CO2 proportionally (cf. Section 2.3). The LCOEt of both MS+MTO and D-eC2H4 processes correlate linearly with the LCOE (Fig. 6A). In particular, the ranking between MS+MTO and D-eC2H4 is preserved over the whole LCOE range. D-eC2H4 could become the cheapest option if the faradaic efficiency were to reach 80% (from the nominal 66% efficiency). But if PEMs were used instead of AEMs, the cell potential would increase by 25%, and the LCOEt gap between the two processes would widen; as it would if the electrolyte molarity was reduced to 1 M KOH (from the nominal 7 M KOH, cf. Section 3.2). Nevertheless, a more detailed modelling would be required to quantify the potential benefits afforded by adapting the operation of D-eC2H4 to fluctuating electricity prices.
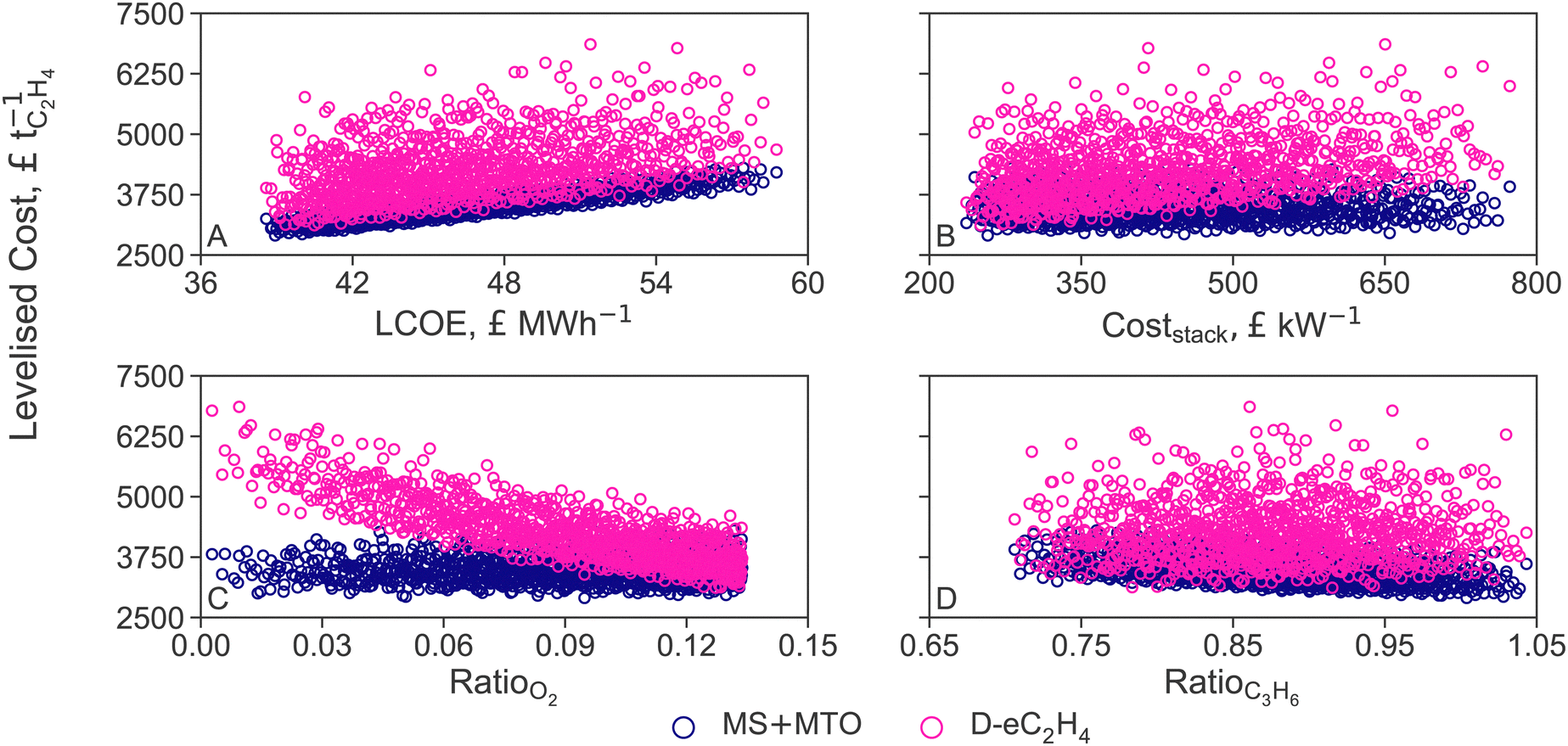 | ||
| Fig. 6 Sensitivity analysis of the levelised cost of ethylene (LCOEt) to variations in the levelised cost of electricity (LCOE, A), AEM stack cost (B) and market prices of oxygen (C) and propylene (D) for the thermocatalytic production of ethylene via methanol (MS+MTO) and direct CO2 electroreduction to ethylene (D-eC2H4). The costs of hydrogen and CO2 feedstock are varied simultaneously with LCOE (cf.Table 2). | ||
Reducing the stack cost from current prices to costs projected for 2030 would help close the LCOEt gap, but still not enough for D-eC2H4 to overtake MS+MTO (Fig. 6B). For D-eC2H4 to gain a competitive advantage over MS+MTO, reductions by over 12% in electricity consumption would be necessary, through improvement in faradaic efficiency and cell voltage. Given that the annualised CapEx contributes only 17% to the LCOEt (Fig. 3), the LCOEt of D-eC2H4 is however more sensitive to variations in the relative market price of oxygen than it is to the stack cost (compare Fig. 6B and D).
The large amount of oxygen by-product (5.2 tO2 tC2H4−1) makes its market price decisive for the economic allocation factor of D-eC2H4 (cf. Section 3.2). If the oxygen price were to drop as a consequence of market saturation, the LOCEt of D-eC2H4 would increase, even causing the LCOEt to double in scenarios where the oxygen price is null (Fig. 6C). Similarly, if the market price of oxygen were to remain at today's level (£130 tO2−1) while the market price of green ethylene tripled (£3000 tC2H4−1), the LCOEt of D-eC2H4 would be double that of MS+MTO.
Propylene is the main saleable co-product of the MS+MTO route and thereby influences the economic allocation factor. A negative correlation between the price ratio and the LCOEt is observed in Fig. 6D. But with about 1 tC3H6 produced for each tC2H4, propylene price variation exerts a smaller effect on MS+MTO than the oxygen price does on D-eC2H4 (compare Fig. 6C Even for a propylene price on par with ethylene, as observed for instance in April 2022,116 the LCOEt from MS+MTO would only decrease by a mere 2.3%.
Our finding that MS+MTO remains the lowest-cost process for green ethylene production across the majority of simulated scenarios suggests that scaling up of the current best available CO2 electrolysers may not be enough for producing low-cost green ethylene and significant performance improvement is needed for electrocatalytic routes to become competitive with MS+MTO. National green hydrogen production targets carry an additional risk to the competitiveness of D-eC2H4 as oxygen is also a by-product of water electrolysis. If the UK's ambitious target of 10 GW of low-carbon hydrogen production were to be produced in the form of green hydrogen, an extra 2 MtO2 per year would be added to the market by 2030. Without a concurrent increase in demand, e.g. for oxy-combustion117 or in wastewater treatment applications,118 the oxygen market price could drop. Meanwhile, the prices of green ethylene produced by MS+MTO could drop if the propylene price were to rise. Since ethylene and propylene are predominantly co-produced in naphtha crackers, their prices in the European market are currently highly correlated. But this interdependence could weaken in the future if the defossilisation of ethylene and propylene relied on separate feedstock. As indicated by the cost breakdown in Fig. 3 though, the LCOE from renewable power generators remains the most sensitive factor for competitive green ethylene prices by and large.
3.4 Beyond the process boundaries
At the time this paper is written, all three ethane steam crackers in the UK are reliant on shale gas imports from the US.119,120 CO2 utilisation for ethylene production therefore would promote the use of local resources such as offshore wind power. But a major impediment to the deployment of green ethylene lies with the huge demand for renewable electricity. The modelling conducted in the previous sections predicts that MS+MTO and D-eC2H4 would consume, respectively, 52 TW hel and 41 TW hel—assuming an efficiency of 1.3 MW hel MW hH2,HHV−1 for PEM electrolysers—to displace a single cracker with an annual capacity of 800 kt. In particular, these demands correspond to 13–16% of the UK's total electricity generation in 2022 (325 TW hel).121 On top of this, powering the DAC plant for MS+MTO and D-eC2H4 would require an additional 14 TW hel and 6 TW hel of renewable electricity, respectively, even with efficient heat pump systems and assuming an average electricity requirement of 2 MW hel tCO2−1.99 These figures also need to be contrasted with our reference ethane cracker in Wilton (UK) consuming 11.5 TW hel per year in total,6 a much smaller share (c. 3.5%) of the UK's annual electricity consumption.Alongside the huge demand for renewable energy to power green ethylene comes the physical footprint of a dedicated offshore wind farm. Assuming an average power per unit area of 3 W m−2 in the UK,122 the required offshore area would be 2800 km2 for MS+MTO and 2000 km2 for D-eC2H4, corresponding to 1.2–1.7 times the area of Greater London.123 Such figures put a clear pressure on the chemical industry to take a strategic decision on securing its supply of low-cost, low-carbon electricity. Delaying this decision could put the industry at risk of missing out on the best offshore wind farm leases, as the seabed areas in the UK exclusive economic zone (EEZ) are continuously auctioned. In the context of low-carbon ethylene production, the chemical industry might want to reconsider the idea of co-locating future chemical plants with nuclear plants that could supply both heat and power.124
Both geographic and geological characteristics govern the cost of green ethylene production. Like the UK, Northern Germany and Denmark have large offshore wind potentials in close proximity to underground salt caverns for low-cost, large-capacity hydrogen storage.125 Therefore, a LCOEt similar to our case study (Fig. 3) could be expected for production sites in those countries. While salt caverns are also available offshore in the North Sea, higher cost for offshore solution mining, injection wells and pipelines would exacerbate the cost of hydrogen storage. For production sites where geological hydrogen storage is unavailable, the use of pressure tanks could increase the hydrogen cost by 20%,98 in turn increasing the LCOEt of MS+MTO by 10%.
At the global level, the production cost of green ethylene will be dependent on the availability of renewables, climatic conditions, and financing conditions all together. Outside Europe, the Western US might offer suitable conditions for a similar LCOEt as in the UK (Fig. 3), but given that fossil-based ethylene in the US is less than half the price on the European market,126 green ethylene would be even less competitive on the US market. Comparable LCOE and LCOH to the UK could also be achieved in other parts of the World, such as the West Coast of Canada, the Arabic Peninsula or Australia.127,128 Nevertheless, the climatic conditions there would make CO2 capture using solid sorbent-based DAC 10–25% more expensive.99 A more detailed and systematic assessment is necessary to identify favourable locations for green ethylene production.
When deciding between D-eC2H4 and MS+MTO for the production of green ethylene, not only the cost but also the supply security during dark doldrums, periods in which little to no electricity is generated by offshore wind farms, should be considered. Renewable electricity is a localised resource that may not be transported over long-distances and for which supply and demand need to be matched at any point in time. Instead, local production of green hydrogen could be supplemented by imports from countries with similar or lower predicted production costs, such as Algeria and Australia.128,129 Even though hydrogen imports could also be reconverted to electricity for D-eC2H4, the associated losses would make it more cost-effective to directly use hydrogen in the MS+MTO process. For a future in which the chemical industry is mainly reliant on renewable power, a higher diversity of supply could bring greater price stability, as the price of commodity chemicals such as ethylene are otherwise at risk of becoming weather-dependent.
Finally, the main motivation behind displacing fossil-based ethylene for green ethylene lies in the prospect of producing carbon-negative ethylene and co-products (use phase excluded). To calculate the net CO2 emissions, the CO2 inlet streams are subtracted from the sum of scope-1 residual emissions and scope-2 emissions associated with electricity that is consumed either directly or for the production of H2 and CO2. Assuming an average carbon intensity of 7.5 kgCO2-eq MW hel−1,130 the allocated net emissions per tonne of ethylene produced amount to −3.3 tCO2-eq for MS+MTO and −1.9 tCO2-eq for D-eC2H4. Both thermocatalytic and electrocatalytic routes would thus present a negative carbon balance from a cradle-to-gate point of view. On the path to net-zero, this would allow for a dramatic reduction over conventional ethane steam crackers, whose cradle-to-gate emissions are estimated to exceed 3 tCO2-eq per tonne of ethylene with ethane from shale gas.108 A cradle-to-grave assessment encompassing the ethylene use phase in consumer products and any downstream emissions is nevertheless required to conclude on the negative emission potential of green ethylene as a whole.131
4 Conclusions
A techno-economic comparison between mature thermocatalytic and novel electrocatalytic processes was conducted for the production of green ethylene from air-captured CO2 and offshore wind electricity, with a view to displacing a conventional ethane cracker in the UK at the horizon of 2035. Similar to fossil-based processes today, the production cost of green ethylene would be dominated by the cost of energy and carbon feedstock, regardless of the process alternative. The fully-thermocatalytic route converting CO2 to methanol then to olefins would achieve the lowest cost (£2900 tC2H4−1), followed by fully-electrocatalytic routes (£2900–3200 tC2H4−1). Process routes relying on Fischer–Tropsch synthesis or oxidative coupling of methane would furthermore be non-competitive. Although considering a UK case study herein, similar levelised cost of green ethylene are expected in Northern Germany or Denmark.Apart from the levelised cost of electricity, the market price of the oxygen by-product and electrolyser stack cost are identified as key sensitive factors to the cost-competitiveness of the electrocatalytic processes. However, the methanol-mediated thermocatalytic route is predicted to out-compete the direct electroreduction of CO2 to ethylene, even for scenarios of high oxygen prices and low stack cost. From a cradle-to-gate perspective, both processes have the potential to reduce the carbon emissions of ethylene production significantly and achieve net-negative emissions between −3.3 to −1.9 tCO2-eq. But a major barrier to green ethylene deployment lies in its energy-intense nature, making it highly reliant on a large-scale supply of low-cost renewable electricity. Displacing a single cracker with an annual production capacity of 800 ktC2H4 would consume the equivalent of 14–20% of the UK's current power generation and require an offshore wind farm larger than the area of Greater London. Finally, transitioning from fossil fuels to intermittent renewable electricity requires a rethinking of the supply security of chemical processes, especially during dark doldrums. The process inventories developed herein could feed into decision-support models for analysing the wider consequences of ethylene defossilisation for the UK's future energy system. As an interim solution, ethylene production will continue to rely on fossil feedstock but could be retrofitted with CCS or H2 as fuel to lower its emission intensity.
Nomenclature
Acronyms
| CO2RR | Electroreduction of CO2 |
| CORR | Electroreduction of CO |
| AEM | Alkaline electrolyte membrane |
| AF | Allocation factor |
| UF | Utilisation factor |
| ASU | Air separation unit |
| BEIS | Department for Business, Energy & Industrial Strategy |
| BOP | Balance of plant |
| CapEx | Capital expenditures |
| CCS | Carbon capture and storage |
| CEPCI | Chemical engineering plant cost index |
| CRF | Capital recovery factor |
| DAC | Direct air capture |
| EEZ | Exclusive economic zone |
| FT | Fischer–Tropsch |
| FE | Faradaic efficiency |
| GHG | Greenhouse gas |
| HHV | Higher heating value |
| LCA | Life-cycle assessment |
| LCOE | Levelized cost of electricity |
| LCOEt | Levelized cost of ethylene |
| LCOH | Levelized cost of hydrogen |
| OER | Oxygen evolution reaction |
| OpEx | Operational expenditures |
| PEM | Proton exchange membrane |
| PTFE | Polytetrafluoroethylene |
| SOEC | Solid oxide electrolysis cell |
| TIC | Total invested capital |
| TRL | Technology readiness level |
| UK | United Kingdom |
| US | United States |
| VPSA | Vacuum pressure swing adsorption |
Subscripts
| el | Electricity |
| eq | Equivalents |
| f | Feedstock |
| h | Heating |
| hc | Hydrocarbons other than ethylene |
| stack | Electrolyser stack |
| tax | Carbon tax |
| ww | Wastewater |
Model parameters and variables
| η F | Faradaic efficiency |
| LT | Lifetime |
| E cell | Cell voltage |
| E red | Reduction potential |
| f O&M | Share of fixed operating costs on CapEx |
| j | Current density |
| m j | Production rate of product j |
| r | Interest rate |
| X | Conversion |
| AFj | Allocation factor of co-product j |
| A cell | Area of the electrode |
| Costj | Unit cost of process input/emission j |
| Pricej | Market price of co-product j |
| Ratioj | Ratio of market price of co-product j to ethylene |
Process technologies
| CTM | CO2 methanation |
| D-eC2H4 | Direct electroreduction of CO2 to ethylene |
| D-FT | Direct Fischer–Tropsch synthesis |
| eCH3OH | Electroreduction of CO2 to methanol |
| eCH4 | Electroreduction of CO2 to methane |
| eCO | Electroreduction of CO2 to CO |
| HT-eCO | High-temperature electroreduction of CO2 to CO |
| MS | Methanol synthesis |
| MTO | Methanol to olefins |
| OCM | Oxidative coupling of methane |
| S-eC2H4 | Electroreduction of CO to ethylene |
| S-FT | Syngas Fischer–Tropsch synthesis |
Author contributions
Andreas H. Nyhus: data curation, formal analysis, methodology, validation, software, investigation, visualisation, writing – original draft preparation. Maria Yliruka: conceptualisation, supervision, data curation, methodology, investigation, validation, writing – original draft preparation, writing – reviewing and editing. Nilay Shah: funding acquisition, writing – reviewing and editing. Benoît Chachuat: supervision, validation, writing-reviewing and editing.Data availability
Data is available upon request.Conflicts of interest
There are no conflicts to declare.Appendix
A Green ethylene production routes
This appendix provides further details about the selected reference processes in the conversion of CO2 to green ethylene (cf.Fig. 1). TRLs of these processes are summarised in Table 4.| Process | TRL |
|---|---|
| MS | 9 |
| eCH3OH | 4 |
| MTO | 9 |
| CTM | 9 |
| eCH4 | 4 |
| OCM | 6 |
| eCO | 4 |
| HT-eCO | 8 |
| S-FT | 4 |
| S-eC2H4 | 4 |
| D-FT | 4 |
| D-eC2H4 | 4 |
Methanol synthesis (MS). Production of methanol via CO2 hydrogenation (reactions (5) and (6)) has reached TRL 9, with commercial plants already operating in China.132 CO2 and H2 flow over a Cu/ZnO/Al2O3 catalyst in a fixed bed reactor under 76 bar and 210 °C to form methanol at a purity of 99.9 wt%.93
| CO2 + 3H2 ⇌ CH3OH + H2O | (5) |
| CO2 + H2 ⇌ CO + H2O | (6) |
CO2RR to methanol (eCH3OH). Electroreduction of CO2 to methanol (half-cell reactions (7) and (8)) over a Cu catalyst135 is most effective with a proton-exchange membrane (PEM) and alkaline electrolyte in a flowing cell set-up (TRL 4).136–139
| CO2 + 6H+ + 6e− → CH3OH + H2O | (7) |
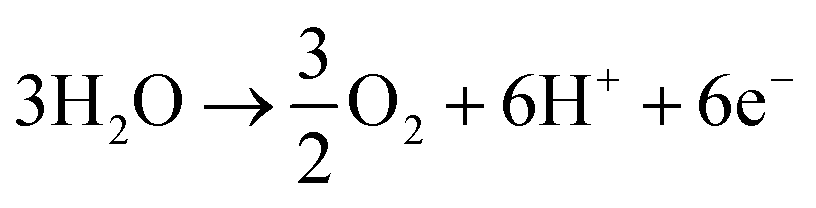 | (8) |
Methanol to olefins (MTO). Production of light olefins from methanol is at TRL 9, with commercial plants in operation in China.141 A SAPO-34 catalyst142 enables the conversion of methanol and steam at 3 bar and 400 °C to light olefins, achieving close to 100% conversion and 80% selectivity to ethylene and propylene at a product purity of 99.5 wt%.143 The hydrocarbon products [CH2] are formed via the dimethylether intermediate CH3OCH3 according to a complex exothermic reaction scheme,142 as summarised in reaction (9).
| 2CH3OH + H2O ⇌ CH3OCH3 ⇌ [CH2] + H2O | (9) |
CO2 methanation (CTM). Methanation of CO2 has been commercially operated in Germany since 2013,145 thus considered TRL 9. Methane is formed by reacting H2 and CO2 in a stoichiometric ratio of 4 (reactions (6), (10) and (11)) over a Ni/MgAl2O4 catalyst in an adiabatic fixed-bed reactor at 10 bar and at 350 °C.92
| CO2 + 4H2 ⇌ CH4 + 2H2O | (10) |
| CO + 3H2 ⇌ CH4 + H2O | (11) |
CO2 electroreduction to methane (eCH4). Electroreduction of CO2 to methane (half-cell reactions (12) and (13)) appears as the most immature technology of all the considered CO2RR routes (TRL 4). Recent research has been more focused on other CO2RR products such as CO or C2H4 than CH4.147
| CO2 + 6H2O + 8e− → CH4 + 8OH− | (12) |
| 8OH− → 2O2 + 4H2O + 8e− | (13) |
Oxidative coupling of methane (OCM). This process was demonstrated at pilot scale (TRL 6) by Siluria Technologies in the US.149 Ethylene is produced by reacting CH4 and O2 (reactions (14)–(16)) over a Na2WO4-Mn/SiO2(W-Mn/SiO2) catalyst in a cooled reactor with inlet at 350 °C and outlet at 305 °C under 9 bar.150
| 2CH4 + ½O2 ⇌ C2H6 + H2O | (14) |
| C2H6 + ½O2 ⇌ C2H4 + H2O | (15) |
| C2H6 ⇌ C2H4 + H2 | (16) |
Low-temperature CO2 electroreduction to CO (eCO). Common amongst experimental studies on CO2 electroreduction to CO (half-cell reactions (17) and (18); TRL 4) is the use of a flowing alkaline electrolyte,152–154 sometimes paired with an ionic liquid.155,156 Candidate transition metal catalysts include Zn,153 Ni,154 Ag,155 Co,69 and Au.157
| CO2 + H2O + 2e− → CO + 2OH− | (17) |
| 2OH− → ½O2 + H2O + 2e− | (18) |
High-temperature CO2 electroreduction to CO (HT-eCO). SOEC technology relies heavily on decades-long research and development effort that has gone into commercialising solid oxide fuel cells (SOFC). It has reached TRL 8 for dry electrolysis of CO2 to CO (half-cell reactions (19) and (20)), for instance with Haldor Topsoe's patented carbon monoxide generator, eCOs™ for small-scale applications.83,158 This high-temperature electrolysis operates at 600–850 °C where it benefits from a reduced theoretical minimum cell voltage and faster reaction kinetics.80 In contrast to low-temperature CO2 electrolysers which require a liquid (aqueous) electrolyte, the oxide ions migrate through a ceramic electrolyte in a SOEC, most commonly Y2O3-stabilised ZrO2 (YSZ). The CO2 electrode needed to conduct both ions and electrons is therefore a ceramic-metal material, usually Ni-YSZ.159 Often separated by gadolinium-doped cerium oxide layer,160 a layer of lanthanum strontium manganite (LSM) perovskite can serve as the anode.161
| CO2 + 2e− → CO + O2− | (19) |
| O2− → ½O2 + 2e− | (20) |
Syngas Fischer–Tropsch synthesis (S-FT). While FT synthesis is already used at commercial scale for fuel production, its application in olefins production is still at the research stage (TRL 4).162,163 The exothermic conversion of syngas to low olefins (reaction (21)) over a Fe–Mn–Cu–K catalyst has been reported under 10 bar and 330 °C.57
| nCO + nH2 → [CH2]n + nH2O | (21) |
CO electroreduction to ethylene (S-eC2H4). Cu has emerged as the catalyst of choice for CO reduction to ethylene under alkaline conditions (half-cell reactions (22) and (23)) in lab-scale electrolysers (TRL 4).71,165–167
| 2CO + 4H2O + 8e− → C2H4 + 8OH− | (22) |
| 8OH− → 2O2 + 4H2O + 8e− | (23) |
Direct Fischer–Tropsch synthesis (D-FT). Similar to FT synthesis of low olefins from syngas (S-FT), the development of a bifunctional catalyst for direct FT synthesis from CO2 (reactions (6) and (21)) remains at the research stage (TRL 4).168–170 Direct conversion of CO2 and H2 to ethylene through FT was also reported using a Fe-K/γ-Al2O3 catalyst under 10 bar and 300 °C.171 Do and Kim172 recently designed a heat-integrated process using this Fe-K/γ-Al2O3 catalyst, which was selected as reference. Nevertheless the mass balances provided by Do and Kim172 are incomplete in that they do not allow quantifying the purge and wastewater streams from D-FT.
CO2 electroreduction to ethylene (D-eC2H4). Similar to CO electroreduction to ethylene, research on direct electroreduction of CO2 to ethylene (half-cell reactions (24) and (25)) has converged on Cu as the catalyst of choice, with operation under flowing alkaline conditions (TRL 4).110,173
| 2CO2 + 8H2O + 12e− → C2H4 + 12OH− | (24) |
| 12OH− → 3O2 + 6H2O + 12e− | (25) |
Acknowledgements
Maria Yliruka is funded by the UK Engineering and Physical Sciences Research Council (EPSRC) under grant EP/R045518/1. Benoît Chachuat and Nilay Shah gratefully acknowledge funding by the EPSRC under grants EP/V011863/1 and EP/V042432/1. The authors would like to thank Ana Somoza-Tornos and Mahinder Ramdin (TU Delft) for the fruitful discussions on CO2RR; David Danaci (Imperial College London) for his guidance on the separation of C2H4/CO mixtures and his feedback on the manuscript; Marwan Sendi (Imperial College London) providing detailed insights into his solid-sorbent DAC process model; and the anonymous reviewers for their constructive suggestions on the manuscript.Notes and references
- International Energy Agency, The Future of Petrochemicals towards More Sustainable Plastics and Fertilisers Together Secure Sustainable, International Energy Agency technical report, 2018.
- H. Zimmermann and R. Walzl, in Ullmann's Encyclopedia of Industrial Chemistry, ed. G. Bellussi, M. Bohnet, J. Bus, K. Drauz, H. Greim, K.-P. Jackel, U. Karst, A. Kleemann, G. Kreysa, T. Laird, W. Meier, E. Ottow, M. Roper, J. Scholtz, K. Sundmacher, R. Ulber and U. Wietelmann, Wiley, 7th edn, 2011, ch. Ethylene, vol. 40, pp. 1–58 Search PubMed.
- ExxonMobil, https://www.exxonmobil.co.uk/company/overview/uk-operations/fife-operations/our-operations.
- Ineos, https://www.ineos.com/businesses/ineos-olefins-polymers-europe/products/.
- SABIC UK, Teesside – Wilton Site, 2021.
- Technical report prepared by the UK Department of Energy and Climate Change and Department for Business, Innovation and Skills, Industrial Decarbonisation and Energy Efficiency Roadmaps to 2050 Chemicals, 2015.
- P. W. Griffin, G. P. Hammond and J. B. Norman, Appl. Energy, 2018, 227, 587–602 CrossRef CAS.
- Department for Business, Energy & Industrial Strategy and National Statistics, 2019 UK Greenhouse Gas Emissions, Final Figures, 2021.
- Element Energy Ltd, Deep-Decarbonisation Pathways for UK Industry, Climate Change Committee technical report, 2020.
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- M. Yang, X. Tian and F. You, Ind. Eng. Chem. Res., 2018, 57, 5980–5998 CrossRef CAS.
- J. van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 CrossRef CAS.
- D. S. Mallapragada, Y. Dvorkin, M. A. Modestino, D. V. Esposito, W. A. Smith, B.-M. Hodge, M. P. Harold, V. M. Donnelly, A. Nuz, C. Bloomquist, K. Baker, L. C. Grabow, Y. Yan, N. N. Rajput, R. L. Hartman, E. J. Biddinger, E. S. Aydil and A. D. Taylor, Joule, 2023, 7, 23–41 CrossRef CAS.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B.-M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. Van Steenberge, D. R. D'hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef.
- J.-P. Lange, Energy Environ. Sci., 2021, 14, 4358–4376 RSC.
- BEIS, Net Zero Strategy: Build Back Greener, Department of Business, Energy and Industrial Strategy technical report, 2021.
- SusChem, Cefic, PlasticsEurope, EuPC and ECP4, Sustainable Plastics Strategy, 2020.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- BEIS, Carbon Capture, Usage and Storage, Department of Business, Energy and Industrial Strategy technical report, 2022.
- I. Esau and R. Watts, Upstream, 2021.
- GE Gas Power, Technip Energies and GE Gas Power Awarded FEED Study for Teesside Power, Carbon Capture and Compression Project in the UK, 2021.
- BP p.l.c, BP Selects Johnson Matthey's LCH Technology for Its First Low Carbon (Blue) Hydrogen Project, 2023.
- HM Government, CCUS Net Zero Investment Roadmap. Capturing Carbon and a Global Opportunity., 2023.
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- National Grid Electricity System Operator, Historic Generation Mix & Carbon Intensity, 2020.
- A. Velazquez Abad and P. E. Dodds, Energy Policy, 2020, 138, 111300 CrossRef.
- A. H. Reksten, M. S. Thomassen, S. Møller-Holst and K. Sundseth, Int. J. Hydrogen Energy, 2022, 47, 38106–38113 CrossRef CAS.
- C. Tsay, R. C. Pattison, M. R. Piana and M. Baldea, Comput. Chem. Eng., 2018, 112, 180–189 CrossRef CAS.
- C. F. Heuberger, I. Staffell, N. Shah and N. Mac Dowell, Nat. Energy, 2018, 3, 634–640 CrossRef.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- T. Rommens, A. Tenhunen, L. Mortensen and I. Vanderreydt, Greenhouse Gas Emissions and Natural Capital Implications of Plastics (Including Biobased Plastics), European Environment Agency – European Topic Centre on Waste and Materials in a Green Economy technical report, 2021.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- M. J. Orella, S. M. Brown, M. E. Leonard, Y. Román-Leshkov and F. R. Brushett, Energy Technol., 2020, 8, 1–12 Search PubMed.
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- M. A. Adnan and M. G. Kibria, Appl. Energy, 2020, 278, 1–12 CrossRef.
- H. Kim, B. Lee, D. Lim, C. Choe and H. Lim, Green Chem., 2021, 23, 7635–7645 RSC.
- A. J. Welch, I. A. Digdaya, R. Kent, P. Ghougassian, H. A. Atwater and C. Xiang, ACS Energy Lett., 2021, 6, 1540–1549 CrossRef CAS.
- M. Moreno-Gonzalez, A. Berger, T. Borsboom-Hanson and W. Mérida, Energy Convers. Manage., 2021, 244, 1–17 CrossRef.
- Z. Zhao, J. Jiang and F. Wang, J. Energy Chem., 2021, 56, 193–202 CrossRef CAS.
- T. N. Do, C. You and J. Kim, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- W. Hoppe, S. Bringezu and N. Wachter, J. CO2 Util., 2018, 27, 170–178 CrossRef CAS.
- A. P. Ortiz-Espinoza, M. M. Noureldin, M. M. El-Halwagi and A. Jiménez-Gutiérrez, Comput. Chem. Eng., 2017, 107, 237–246 CrossRef CAS.
- M. Ramdin, B. De Mot, A. R. Morrison, T. Breugelmans, L. J. Van Den Broeke, J. P. Trusler, R. Kortlever, W. De Jong, O. A. Moultos, P. Xiao, P. A. Webley and T. J. Vlugt, Ind. Eng. Chem. Res., 2021, 60, 17862–17880 CrossRef CAS PubMed.
- I. Ioannou, S. C. D'Angelo, A. J. Martín, J. Pérez-Ramírez and G. Guillén-Gosálbez, ChemSusChem, 2020, 13, 6370–6380 CrossRef CAS PubMed.
- L. Berkelaar, J. van der Linde, J. Peper, A. Rajhans, D. Tiemessen, L. van der Ham and H. van den Berg, Chem. Eng. Res. Des., 2022, 182, 194–206 CrossRef CAS.
- A. Kaetelhoen, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 166, 11187–11194 CrossRef PubMed.
- B. Fais, I. Keppo, M. Zeyringer, W. Usher and H. Daly, Energy Strategy Rev., 2016, 13–14, 154–168 CrossRef.
- National Grid ESO, Future Energy Scenarios 2022, 2022.
- HM Government, Industrial Decarbonisation Strategy, Department of Business, Energy and Industrial Strategy technical report, 2021.
- M. Victoria, K. Zhu, T. Brown, G. B. Andresen and M. Greiner, Nat. Commun., 2020, 11, 6223 CrossRef CAS PubMed.
- M. Gazzani, D. M. Turi and G. Manzolini, Int. J. Greenhouse Gas Control, 2014, 20, 293–309 CrossRef CAS.
- Y. Liu, H. Kamata, H. Ohara, Y. Izumi, D. S. W. Ong, J. Chang, C. K. Poh, L. Chen and A. Borgna, Ind. Eng. Chem. Res., 2020, 59, 8728–8739 CrossRef CAS.
- G. Towler and R. Sinnot, Chemical Engineering Design – Principles, Practice and Economics of Plant and Process Design, Elsevier, 3rd edn, 2022 Search PubMed.
- S. Jenkins, Chemical Engineering Plant Cost Index: 2019 Annual Value, 2019.
- S. Hernandez-Aldave and E. Andreoli, Catalysts, 2020, 10(6), 713 CrossRef CAS.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS PubMed.
- M. Duarte, J. Hereijgers, N. Daems, S. Van Daele and T. Breugelmans, J. Environ. Chem. Eng., 2022, 10, 107836 CrossRef CAS.
- Q. Xu, A. Xu, S. Garg, A. B. Moss, I. Chorkendorff, T. Bligaard and B. Seger, Angew. Chem., Int. Ed., 2023, 62, e202214383 CrossRef CAS PubMed.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. OBrien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- B. Endrödi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Shi and S. Holdcroft, J. Phys. Chem. C, 2021, 125, 25446–25454 CrossRef CAS.
- C. Wang, Y. Liu, H. Ren, Q. Guan, S. Chou and W. Li, ACS Catal., 2022, 12, 2513–2521 CrossRef CAS.
- X. Y. Zhang, W. J. Li, X. F. Wu, Y. W. Liu, J. Chen, M. Zhu, H. Y. Yuan, S. Dai, H. F. Wang, Z. Jiang, P. F. Liu and H. G. Yang, Energy Environ. Sci., 2022, 15, 234–243 RSC.
- Y. Ji, C. Yang, L. Qian, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2021, 600, 847–853 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- Green Hydrogen Cost Reduction. Scaling up Electrolysers to Meet the 1.5 C Climate Goal, International Renewable Energy Agency technical report, 2020.
- BEIS, Hydrogen Production Costs 2021, Department of Business, Energy and Industrial Strategy technical report, 2021.
- A. Mayyas, M. Ruth, B. Pivovar, G. Bender and K. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, NREL technical report, 2018.
- J. E. Bachman, D. A. Reed, M. T. Kapelewski, G. Chachra, D. Jonnavittula, G. Radaelli and J. R. Long, Energy Environ. Sci., 2018, 11, 2423–2431 RSC.
- D. A. Jones, Technoeconomic Evaluation of MEA versus Mixed Amines and a Catalyst System for CO2 Removal at Near-Commercial Scale at Duke Energy Gibson 3 Plant and Duke Energy Buck NGCC Plant, Lawrence Livermore National Laboratory technical report, 2018.
- G. A. Buchner, A. W. Zimmermann, A. E. Hohgräve and R. Schomäcker, Ind. Eng. Chem. Res., 2018, 57, 8502–8517 CrossRef CAS.
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- C. Mittal, C. Hadsbjerg and P. Blennow, Chem. Eng. World, 2017, 52, 44–46 Search PubMed.
- N. B. Jakobsson, C. F. Pedersen and J. B. Hansen, Process for Producing CO from CO2 in a Solid Oxide Electrolysis Cell, Haldor Topsoe A/S, US Pat., US 10494728 B2, 2019 Search PubMed.
- G. Glenk, P. Holler and S. Reichelstein, Energy Environ. Sci., 2023, 16, 6058–6070 RSC.
- H. Böhm, A. Zauner, D. C. Rosenfeld and R. Tichler, Appl. Energy, 2020, 264, 114780 CrossRef.
- Clean Hydrogen Joint Undertaking, Strategic Research and Innovation Agenda 2021–2027, 2022.
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 2, 17110 CrossRef.
- H. Nami, O. B. Rizvandi, C. Chatzichristodoulou, P. V. Hendriksen and H. L. Frandsen, Energy Convers. Manage., 2022, 269, 116162 CrossRef CAS.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- T. Smolinka, N. Wiebe, P. Sterchele, A. Palzer, F. Lehner, M. Jansen, S. Kiemel, R. Miehe, S. Wahren and F. Zimmermann, Studie IndWEDe. Industrialisierung Der Wasserelektrolyse in Deutschland: Chancen Und Herausforderungen Für Nachhaltigen Wasserstoff Für Verkehr, Strom Und Wärme, Bundesministerium für Verkehr und digitale Infrastruktur (BMVI) technical report, 2018.
- Environment Agency, Wilton Olefins Installation. Permit Number EPR/BS3590IE, Environment Agency technical report, 2016.
- R. Chauvy, D. Verdonck, L. Dubois, D. Thomas and G. De Weireld, J. CO2 Util., 2021, 47, 1–10 Search PubMed.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- J. Price, I. Keppo and P. E. Dodds, Energy, 2023, 262, 125450 CrossRef CAS.
- J. D. Williams, J. Williamson, D. Parkes, D. J. Evans, K. L. Kirk, N. Sunny, E. Hough, H. Vosper and M. C. Akhurst, J. Energy Storage, 2022, 53, 105109 CrossRef.
- East Coast Hydrogen, East Coast Hydrogen Feasibility Report, 2021.
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583–3602 RSC.
- D. S. Mallapragada, E. Gençer, P. Insinger, D. W. Keith and F. M. O'Sullivan, Cell Rep. Phys. Sci., 2020, 1, 100174 CrossRef.
- M. Sendi, M. Bui, N. Mac Dowell and P. Fennell, One Earth, 2022, 5, 1153–1164 CrossRef.
- IEAGHG, Deployment of CCS in the Cement Industry, 2013.
- S. Hughes and A. Zoelle, Cost of Capturing CO2from Industrial Sources, National Energy Technology Laboratory technical report, 2022.
- BEIS, Valuation of Greenhouse Gas Emissions: For Policy Appraisal and Evaluation, 2020.
- United Utilities, Wholesale Sewerage, United Utilities technical report, 2019.
- ICIS, ICIS Chemical Business, ICIS technical report, 2019.
- M. Rivarolo, D. Bellotti, A. Mendieta and A. Massardo, Energy Convers. Manage., 2014, 79, 74–84 CrossRef CAS.
- J. G. Speight, Rules of Thumb for Petroleum Engineers, John Wiley & Sons, 2017 Search PubMed.
- R. Wiser, J. Rand, J. Seel, P. Beiter, E. Baker, E. Lantz and P. Gilman, Nat. Energy, 2021, 6, 555–565 CrossRef.
- F. Keller, R. P. Lee and B. Meyer, J. Cleaner Prod., 2020, 250, 119484 CrossRef CAS.
- M. Hurley, European Ethylene Contract Reference Price in April Set at 1665 €/t for April, ICIS News, 2022.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C. T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- M. Goldman, A. Prajapati, E. Duoss, S. Baker and C. Hahn, Curr. Opin. Electrochem., 2023, 39, 101248 CrossRef CAS.
- G. Leonzio, B. Chachuat and N. Shah, Sustain. Prod. Consum., 2023, 43, 124–139 CrossRef.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2019, 577, 509–513 CrossRef PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- ICIS News, 2022.
- T. Kato, M. Kubota, N. Kobayashi and Y. Suzuoki, Energy, 2005, 30, 2580–2595 CrossRef CAS.
- H. Mohammadpour, R. Cord-Ruwisch, A. Pivrikas and G. Ho, Chem. Eng. Sci., 2021, 246, 117008 CrossRef CAS.
- A. Torchia and R. Shamseddine, Saudi's SABIC Signs Deal to Use U.S. Shale Gas at British Plant, 2015.
- INEOS Group, INEOS Insight Docks at Grangemouth Bringing US Shale Gas to the UK for the First Time, 2016.
- H. Ritchie, M. Roser and P. Rosado, Our World in Data, Energy, 2023, https:// ourworldindata.org/energy.
- D. J. C. MacKay, Sustainable Energy–Without the Hot Air, 2009 Search PubMed.
- G. L. Authority, Land Area and Population Density, Ward and Borough, 2018 Search PubMed.
- A. Kadak, The Chemical Engineer, 2022, June 972, https://www.thechemicalengineer.com/features/energy-bringing-nuclear-back/ Search PubMed.
- D. G. Caglayan, N. Weber, H. U. Heinrichs, J. Linßen, M. Robinius, P. A. Kukla and D. Stolten, Int. J. Hydrogen Energy, 2020, 45, 6793–6805 CrossRef CAS.
- K. Allen, G. Baida, B. Balboa, L. Berton, B. Mk, B. Brooks, F. Dobashi, K. Hays, et al., Global Petrochemical Outlook H2 2019, Petrochemicals Special Report, S&P Global Platts technical report, 2019.
- X. Kan, L. Reichenberg, F. Hedenus and D. Daniels, arXiv, 2022, preprint, arXiv.2202.02257 DOI:10.48550/arXiv.2202.02257.
- D. Freire Ordóñez, C. Ganzer, T. Halfdanarson, A. González Garay, P. Patrizio, A. Bardow, G. Guillén-Gosálbez, N. Shah and N. Mac Dowell, Energy Adv., 2023, 2, 2042–2054 RSC.
- R. Taylor, E. Raphael, C. Lewis, R. Berridge and J. Howes, Expansion of Hydrogen Production Pathways Analysis – Import Chains, Department of Business, Energy & Industrial Strategy technical report, 2022.
- Royal HaskoningDHV, Norfolk Boreas, Greenhouse Gas Footprint Assessment, ExA.AS-3.D14.V1, 2020.
- S. E. Tanzer and A. Ramírez, Energy Environ. Sci., 2019, 12, 1210–1218 RSC.
- Carbon Recycling International, The Shunli CO2-to-Methanol Plant: Commercial Scale Production in China.
- M. J. Matzen, M. H. Alhajji and Y. Demirel, J. Adv. Chem. Eng., 2015, 5, 1–13 Search PubMed.
- I. L. Wiesberg, J. L. de Medeiros, R. M. Alves, P. L. Coutinho and O. Q. Araújo, Energy Convers. Manage., 2016, 125, 320–335 CrossRef CAS.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- A. P. Periasamy, R. Ravindranath, S. M. Senthil Kumar, W. P. Wu, T. R. Jian and H. T. Chang, Nanoscale, 2018, 10, 11869–11880 RSC.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 130, 14345–14349 CrossRef.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- M. Ye, H. Li, Y. Zhao, T. Zhang and Z. Liu, in Mesoscale Modeling in Chemical Engineering Part II, ed. G. B. Marin and J. Li, Academic Press, 2015, ch. 5, vol. 47 of Advances in Chemical Engineering, pp. 279–335 Search PubMed.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- Y. H. Chen, W. Hsieh, H. Chang and C. D. Ho, J. Taiwan Inst. Chem. Eng., 2022, 130, 103893 CrossRef CAS.
- S. Jasper and M. M. El-Halwagi, Processes, 2015, 3, 684–698 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- G. Iaquaniello, S. Setini, A. Salladini and M. De Falco, Int. J. Hydrogen Energy, 2018, 43, 17069–17081 CrossRef CAS.
- B. Li, L. Liu, M. Yue, Q. Niu, M. Li, T. Zhang, W. Xie and Q. Wang, Green Chem., 2024, 26, 103–121 RSC.
- H. Pan and C. J. Barile, Energy Environ. Sci., 2020, 13, 3567–3578 RSC.
- J. Liu, J. Yue, M. Lv, F. Wang, Y. Cui, Z. Zhang and G. Xu, Carbon Resour. Convers., 2022, 5, 1–14 CrossRef CAS.
- V. Spallina, I. C. Velarde, J. A. M. Jimenez, H. R. Godini, F. Gallucci and M. Van Sint Annaland, Energy Convers. Manage., 2017, 154, 244–261 CrossRef CAS.
- L. S. Layritz, I. Dolganova, M. Finkbeiner, G. Luderer, A. T. Penteado, F. Ueckerdt and J. U. Repke, Appl. Energy, 2021, 296, 117049 CrossRef CAS.
- H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu and C. He, Nano Energy, 2020, 70, 104454 CrossRef CAS.
- K. Liu, J. Wang, M. Shi, J. Yan, Q. Jiang, K. H. Liu, M. M. Shi, J. M. Yan, Q. Jiang and J. Z. Wang, Adv. Energy Mater., 2019, 9, 1900276 CrossRef.
- H. B. Yang, S. F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H. R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- Haldor Topsoe, https://www.topsoe.com/processes/carbon-monoxide.
- Y. Song, Z. Zhou, X. Zhang, Y. Zhou, H. Gong, H. Lv, Q. Liu, G. Wang and X. Bao, J. Mater. Chem. A, 2018, 6, 13661–13667 RSC.
- A. Hauch, K. Brodersen, M. Chen, C. Graves, S. H. Jensen, P. S. Jørgensen, P. V. Hendriksen, M. B. Mogensen, S. Ovtar and X. Sun, ECS Trans., 2017, 75, 3 CrossRef CAS.
- A. Nechache and S. Hody, Renewable Sustainable Energy Rev., 2021, 149, 111322 CrossRef CAS.
- Y. Ni, Y. Liu, Z. Chen, M. Yang, H. Liu, Y. He, Y. Fu, W. Zhu and Z. Liu, ACS Catal., 2019, 9, 1026–1032 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- D. Xiang, S. Yang and Y. Qian, Energy Convers. Manage., 2016, 110, 33–41 CrossRef CAS.
- H. P. Duong, N.-H. Tran, G. Rousse, S. Zanna, M. W. Schreiber and M. Fontecave, ACS Catal., 2022, 12, 10285–10293 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- N. S. Romero Cuellar, C. Scherer, B. Kaçkar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef CAS.
- G. Centi, A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- J. Gao, C. Jia and B. Liu, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- M. A. Marvast, M. Sohrabi, S. Zarrinpashne and G. Baghmisheh, Chem. Eng. Technol., 2005, 28, 78–86 CrossRef CAS.
- S. Najari, G. Gróf, S. Saeidi and F. Gallucci, Int. J. Hydrogen Energy, 2019, 44, 4630–4649 CrossRef CAS.
- T. N. Do and J. Kim, Energy Convers. Manage., 2020, 214, 112866 CrossRef CAS.
- H. Q. Liang, S. Zhao, X. M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03064d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |