
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparison of molecular iodine evolution on the chemistry of lead and tin perovskites
Thomas
Webb
and
Saif A.
Haque
 *
*
Department of Chemistry, Molecular Sciences Research Hub, White city, London W12 0BZ, UK. E-mail: s.a.haque@imperial.ac.uk
First published on 24th April 2024
Abstract
The development of perovskite solar cells (PSCs) has gone from strength to strength over the last decade, enabling low-cost, flexible and high-efficiency photovoltaic devices. However, the significance of molecular iodine (I2) evolution within the perovskite layer on device longevity is only recently becoming realised. In lead-based perovskites, the formation of I2 is determinantal to both the photovoltaic performance of PSCs and the long-term stability. Likewise, I2 formation within tin perovskite is highly destructive; rapidly breaking down the composition of the perovskite layer, and severely limiting the shelf-life of photovoltaic devices. In both cases, the formation of I2 has a significant additional impact on the stability and function of all other elements of the PSC structure including the conductive metal oxide, metal electrode and charge transport layers (CTLs). In this perspective, we highlight the key role of iodine in dictating the performance and stability of lead and tin perovskite materials. In doing so we compare the similarities and differences between the formation mechanisms of molecular I2 in the lead and tin analogues while also considering its effect on the performance of PSCs through consideration of the various elements of the PSC structure. In discussing this challenge, we look to identify new emerging ways in which volatile iodine has been captured within other scientific fields and discuss the applicability, modification and utilisation of these strategies within PSCs. Finally, through consideration of the fundamental chemistry in these systems, we summarise the all-important role of iodine in PSCs, discuss efforts being made to mitigate the damage of I2 evolution, manage the redox chemistry, and provide design criteria for developing iodine-resilient PSCs.
 Thomas Webb | Thomas Webb is a postgraduate researcher in the Department of Chemistry at Imperial College London. He holds masters degrees in both Chemistry and Electronics and Electrical Engineering from Imperial College London and the University of Surrey, respectively. He is currently conducting his PhD doctoral studies at Imperial College London investigating the use of novel reductants and developing new strategies to stabilise tin perovskites under the supervision of Prof Saif A. Haque. |
 Saif A. Haque | Saif A. Haque is a Professor of Chemistry at Imperial College London. He is a chemist with a particular interest in renewable energy technologies, nanomaterials, functional electronic materials, photochemistry and solar energy conversion devices. His research is currently addressing the development and functional characterization of solar cell absorbers and devices based upon solution processable hybrid inorganic–organic semiconducting materials, inorganic metal chalcogenides, quantum dots and perovskites. Prior to becoming a professor, he was a Royal Society University Research Fellow at Imperial between 2005 and 2013. |
Broader contextIn recent years, perovskite materials which utilise an ABX3 crystal motif have been integrated into a wide range of optoelectronic and thermoelectric applications. Amongst these, perovskite solar cells (PSCs) and perovskite light-emitting diodes (PeLEDs) have emerged as forerunners to revolutionise their respective fields. Boasting solution processability and flexibility in the final products, PSCs and PeLEDs offer new opportunities in customisable and bespoke electronics while retaining remarkably high performance. However, solution processing the constituents of inorganic–organic halide salts into ionic lattice structure comes with the drawback of moisture instability and unwanted redox chemistry of reactive halides, specifically iodine. The incorporation of a corrosive triiodide/iodine redox couple in the earliest days of PSCs had a devastating impact on the shelf-life, often only taking minutes for the perovskite to break down. A paradigm shift to full solid-state architectures sought to rectify this issue, dramatically improving the shelf life to the scale of months. Nevertheless, while no longer implemented intentionally as an electrolyte, the evolution of iodine in situ during operation remains an obstacle to the commercialisation of perovskite optoelectronics with practical shelf lives. Indeed, the evolution of corrosive iodine and triiodide formation continues to place limitations on the long-term stability of perovskite optoelectronics, with far-reaching effects adversely impacting all the components of the device. It is therefore essential that the mechanisms of iodine generation within the device are identified to enable the development of suitable neutralisation strategies. The dynamic nature of these destabilising processes encourages the identification of dynamic solutions where iodine redox chemistry is managed sustainably. Such an approach avoids the depletion of iodide from the perovskite lattice by preventing its migration through the device architecture coupled with suitable redox chemistry to reform and replenish iodide. While newly emergent in the field of perovskite materials, we note significant advancements have been made in the field of iodine management in the fields of radioiodine disposal, medicine, and electrochemistry. These studies provide a head start in the integration and development of new strategies within the field of perovskite optoelectronics. Indeed, only if the effect of iodine release on long-term stability is understood and addressed effectively can perovskite optoelectronics achieve the longevity required for commercial technologies. |
Introduction
In just over a decade metal halide perovskite photovoltaics have grabbed the attention of the photovoltaic community. A wealth of research has enabled power conversion efficiencies (PCEs) to soar from 3.8% when first demonstrated, to beyond 26%.1–3 This rapid advancement in device performance has been made possible through a combination of breakthroughs in device architecture, defect passivation and compositional engineering.2 Indeed, the ability to tune the composition of the ABX3 lattice motif has enabled the production of light-harvesting layers with idealised spectral responses for capturing solar radiation.4–6 Furthermore, the high degree of control over the band structure has seen perovskites integrated into a range of applications including perovskite LEDs (Pe-LEDs), transistors and X-ray detectors, to name but a few.7–12 Nevertheless, the use of a solution processable ionic lattice also represents arguably the biggest drawback to their application - poor intrinsic stability.Unlike silicon and III–V semiconductors (GaAs, InGaAs) which comprise largely covalent structures, perovskite materials exist as a highly ionic lattice with an ABX3 motif.13–19 More recently, the definition of perovskite has been expanded beyond the classical ABX3 motif to include a range of structural variants including 2D and semi-2D crystal structures. These structures utilise either a monovalent (A′) or divalent (A′′) organic spacer cation to produce Ruddlesden–Popper phases (A2′An−1BnX3n+1) and Dion–Jacobsen phase (A′′An−1BnX3n+1) respectively.2,20,21 However, universal to all structures is the typical use of an organic (MA+, FA+,…) or inorganic (Cs+, Rb+,…) cation on the A-site, a metal cation, typically, Pb2+, Sn2+,Ge2+ on the B site and an anion, most often a halide, (Cl−, Br−, I−…) on the X-site.2,22–24 The ionic nature of the ABX3 lattice imparts several routes for instability in the PSCs including the tendency of the A-site organic cation to undergo solvation from the structure, unwanted redox chemistry of the constituent ions and the presence of mobile corrosive halide anions.25–29
In the earliest demonstration of perovskite devices, PSCs were prepared by adapting the pre-existing dye-sensitized solar cell (DSSC) architecture. In this configuration, the perovskite acts as a sensitizer on a titania scaffold, which in turn acts to collect electrons. The circuit is completed using an iodide/triiodide (I−/I3−) liquid electrolyte redox couple.1,30 As per the conventional DSSC devices, the use of iodine and triiodide proved to be a major source of instability, owing to its corrosive nature. Consequently, the shelf life of early PSCs was limited to mere minutes.1,30 Replacing the triiodide liquid electrolyte in favour of a fully solid-state hole transport layer unlocked significant improvements in both the device performance and stability.4,31,32 To this end, the removal of triiodide electrolytes, combined with a range of other optimisations, has enabled lead-perovskite devices prepared with carbon electrodes to exhibit stability under ambient conditions exceeding one year.33 Nevertheless, the evolution of I2 and the subsequent degradation reactions with I− ions to form I3− continue to haunt fully solid-state perovskites.34–36 Consequently, as per the early days of perovskite research, the generation I2 and I3− continue to limit the stability of perovskite, albeit on a longer timescale. Furthermore, the true extent and impact of the I2-induced degradation on the charge transport layers (CTLs) and electrodes is becoming clearer. As such the formation of I2 and I3− still hinders both the performance and longevity of PSCs, over a decade after being first identified as a problem.
Developments in compositional engineering have enabled the production of lead-free perovskites.37–40 These perovskites offer a lower-toxicity alternative to the use of lead, promising a lower bioavailability as well as further tunability over the band gap.24,41–43 Amongst these alternatives, tin-based perovskites have stood out as a leading candidate exhibiting an ideal bandgap of around 1.3 eV and PCEs exceeding 14%.37,39,44 These features make tin-perovskites a promising candidate for third-generation multijunction tandem applications.45 Nevertheless, tin perovskites suffer from a tendency to undergo facile oxidation when exposed to oxygen. In comparison, the oxidation of the B-site metal cation does not occur in the lead analogues owing to the increased inert pair effect down group IV. As such lead-based PSCs benefit from a larger energetic barrier to the formation of sp3 hybrid orbitals by virtue of its position below tin. The ability of tin perovskite to undergo oxidation to the Sn4+ state imparts significant instability on the thin films, facilitating rapid degradation of the material, increased trapping and self-p-type doping.26,46 This instability has recently been demonstrated to be exacerbated by the ability of SnI4 to further decompose into I2; where I2 is a strong oxidizer itself.47 As such, the stability of tin PSCs is limited to mere hours in ambient air with stability measurements most often collected under storage in inert conditions.48–50
The role of iodine and its equilibrium with iodide anions to form triiodide has been a long-running problem in developing stable-perovskite solar cells.1,30,32 While the transition to fully solid-state architectures undoubtedly had a significant impact on stability, the presence of iodine and triiodide within solid-state architectures has since been largely overlooked. Indeed, when compared to more established technologies such as silicon photovoltaics, the stability of both tin and lead PSCs continues to fall considerably short.28,51 A key contributor to this instability is the presence of mobile halides in PSCs and their tendency to oxidise into corrosive halogens such as I2 which severely reduces the device longevity. This effect is most easily observed upon a comparison of the choice of electrodes used when preparing PSCs designed for longevity where metals vulnerable to oxidation are frequently substituted with more inert carbon.33,52 As such, further work is needed to manage the impact of corrosive iodine formation as a failure mechanisms for PSCs.
The very recent identification of I2 formation in tin perovskite has been an interesting and potentially pivotal point in the search for stable tin PSCs.38,49,53,54 This discovery is made more interesting by the unique formation mechanisms of iodine within tin PSCs by virtue of the additional Sn4+ oxidation state. In turn, this creates a point of interest in deciphering which mechanisms are unique for tin PSCs and which are likely to be universal to both materials but may not yet have been discussed within the context of tin PSCs. From this comparison, lessons from the more established I2 chemistry of lead-perovskite can be compared and evaluated as risk factors to the stability of tin perovskites guiding future PSC design. Undoubtedly, universal to both cases is the effect of I2 formation on the other components of the device architecture including the metal oxide, CTLs and metal electrodes. After reviewing the chemistry of iodine formation pathways within lead and tin perovskites, we present a discussion detailing a range of strategies for its mitigation, highlighting opportunities to learn from cross-discipline approaches. In discussing these strategies, we highlight potential design criteria for fabricating PSCs that minimize iodine formation and propose regenerative redox pathways that can be implemented to improve chemical stability. Finally, we provide an outlook on the design of new beneficial molecules, polymers and redox couples which allow devices to operate sustainably and mitigate irreversible chemical changes.
Iodine in lead perovskites
To date, several mechanisms by which iodine can be generated within lead-based perovskites have been reported.34,35,55 Perhaps most frequently reported is the photolysis of PbI2 under exposure to illumination with photon energies exceeding 2.51 eV (494 nm) (eqn (1)).56–59 Localised domains of PbI2 are often reported within the perovskite layer, forming via several pathways. The most common of these include the thermal or moisture-induced loss of the A-site cation,60 and the inhomogeneous conversion of PbI2 in a two-step fabrication process.61,62 Alternatively, PbI2 may be deliberately added in excess within the precursor, reported to benefit the performance of devices.58,63,64 The photolysis of PbI2 into I2 within the perovskite also leads to the creation of metallic lead (Pb0), often observed as a shoulder in the perovskite Pb 4f orbital using X-ray photoelectron spectroscopy (XPS) (Fig. 1a).59,65–68 This phenomenon is amplified within PSCs prepared using metal oxide transport layers such as TiO2 and SnO2 which can behave as photocatalysts for iodide oxidation and Pb2+ reduction (eqn (1)) when exposed to UV light (Fig. 1b).69,70 The effect of photolysis on lead halide salts can be reduced by exchanging the halide with bromide or chloride anions which have stronger lead-halide bonds and therefore higher dissociation energies of 3.06 and 3.19 eV, respectively.56 As a consequence, the fraction of solar radiation with sufficient energy to reduce the lead in these cases is dramatically lowered. The replacement of iodide anions with bromide and chloride also affords a more compact lattice unit cell and stronger interactions between the inorganic framework and cation.71 Indeed, the reduced lattice parameters facilitate hydrogen bonding between CH3-X where X = Cl and Br, decreasing the halide mobility and reducing the available pathways for halide oxidation to occur.71,72 A similar reduction of halide mobility can be achieved through the incorporation of additives capable of forming halogen–halogen bonds.73,74 | ||
| Fig. 1 Mechanisms of I2 formation in Pb-based perovskite solar cells. (a) Ratio of lead as MAPbI3, PbI2 and metallic Pb0 from XPS. The fraction of Pb0 increases under illumination owing to PbI2 photolysis. Reprinted with permission.68 Copyright 2019, Royal Society of Chemistry. (b) Mechanism of UV-light driven metal–oxide photocatalysis of iodide anions to iodine. Reprinted with permission.69 Copyright 2019, Elsevier (c) energy diagram showing the formation of superoxide formation via the quenching of MAPbI3 excited states. (d) Change in fluorescence intensity of superoxide probe indicating the formation of superoxide on the scale of minutes from the perovskite when illuminated and exposed to oxygen. Reprinted with permission.81 Copyright 2015, Wiley. (e) Schematic of electrochemical oxidation of mixed MAPbBr1.5I1.5, changing the surface stoichiometry. Reprinted with permission.87 Copyright 2019, American Chemical Society. (f) Photograph of formamidinium iodide powder and solution before and after ageing in oxygen, where oxidation of iodide ions forms iodine changing the colour. Reprinted with permission.55 Copyright 2021, AAAS. | ||
In addition to light-driven photolysis and photocatalytic reactions, PbI2 is also sensitive to displacement with oxygen and moisture, degrading into PbO and releasing I2 vapour when exposed to ambient conditions (eqn (2)).75–77 Popov et al. observed that PbI2 films left exposed to ambient oxygen undergo discolouration from the release of iodine vapour, noting that small PbI2 grains are the most susceptible to this method of degradation.75 A second key mechanism by which oxygen can liberate I2 from perovskite is via the generation of reactive superoxide (O2−) when under illumination. In this mechanism, the photoexcited states of the perovskite are quenched through interaction with oxygen forming a highly aggressive O2− species (Fig. 1c and d). The highly reactive formed O2− quickly degrades pristine perovskite, liberating iodine (eqn (3)).78–83 Notably, the interaction of superoxide with PbI2 is also reported to yield I2 and PbO.78 Aristidou et al. revealed the formation of superoxide is dependent on the surface iodide vacancy defect density, which provides a low-energy site for the reduction of O2 to occur.78,84 The yield of iodine formation can thus be lowered via the removal of such defect sites through passivation or the production of films with large grain sizes. Superoxide yields can also be reduced via fast extraction of the photoexcited electrons on a time scale shorter than oxygen reduction.2,78,81 Alternatively, the generation of superoxide can be substantially suppressed via the substitution of iodide anions with bromide as demonstrated by Aziz et al.71
| PbI2 + hv → Pb0 + I2 | (1) |
| 2PbI2 + O2 → 2PbO + 2I2 | (2) |
| 4APbI3 + O−2 → 4PbI2 + 2I2 + H2O + 4A | (3) |
Perhaps least discussed within the literature, the formation of I2 can also occur within perovskite devices via the electrochemical oxidation of iodide anions at the electrodes under operation.35,36,85–87 This effect can be observed under relatively low electric fields, reported as low as 0.2 V μm−1.85 Such voltages are quite reasonable within operational PSCs and thus likely to generate I2 during operation, especially over the extended operation. We note that the electrochemical redox chemistry is likely to contribute significantly to instability during maximum power point (MPP) tests when measured under inert conditions.88 The electrochemical oxidation of I− anions is worsened by the mobile nature of the halide anions.29,89 This can lead to the depletion of iodine at the electrode interface, an effect shown experimentally using XPS.67 Similarly, Samu et al. found that the I−/Br− ratio in MAPbBr1.5I1.5 films varies with charge injection into the device.87 In contrast to iodide-rich phases, bromide phases exhibit greater resistance to electrochemical oxidation owing to a higher lattice energy; a result of greater electronegativity of the bromide ion and a stronger interaction with the lattice unit cell as compared to iodide.71,90 This leads to mixed halide perovskite films becoming increasingly bromide-rich/iodide deficient upon application of a voltage (Fig. 1e), giving the film an orange colour and damaging the film morphology.35,87,91 The link between composition and stability to electrochemical oxidation has to date received little attention but raises important questions concerning both the performance and stability of mixed halide PSCs. As such, management of the local halide composition around the electrodes, for the prevention of iodide permeation of the transport layers, is crucial for preventing the electrochemical oxidation of iodide.
In addition to iodine evolution from PbI2, the organic cations can also contribute to the evolution of iodine within lead-perovskites. Iodide salts are unstable when exposed to oxygen and moisture owing to their mild reducing nature (Table 1).92 A well-known example of this instability to oxidation is hydroiodic acid (HI), where stabilizers such as hypophosphoric acid are necessitated to stabilise the iodide ions and prevent I2 vapour forming.38,93,94 This chemistry extends to the organic cations used in perovskite precursors as reported by Chen et al., who reported that following exposure to air, formamidinium iodide (FAI) generates I2via the oxidation of I− (Fig. 1f).55 This effect can be visually identified as a yellowing of the FAI powder where I2 quickly reacts with I−, forming I3− and is often observed when FAI is stored improperly with exposure to oxygen.95 This oxidation of I− occurs via the FAI equivalence with FA and HI; the latter of which undergoes favourable oxidation with oxygen as discussed. This oxidation occurs on short timescales in FAI, possibly owing to the formation of sym-triazine as a product.96 To this end, the incorporation of poor-quality FAI, containing I2/I3−, dramatically reduces the stability and performance of PSCs.55,95 Alkylammonium bromides and chlorides offer greater stability to oxidation under ambient owing to better stabilisation of the negative charge on the bromide and chloride ions, yielding higher redox potentials (E°) (Table 1). Consequently, in mixed cation-halide systems, it may prove beneficial to use bromide and chloride formamidinium salts where possible, incorporating iodine via more stable methylammonium, alkali metal or lead salts.
| Redox couple | Potential E° (V)97 |
|---|---|
| Cl2 + 2e− → 2Cl− | +1.358 |
| Br2 + 2e− → 2Br− | +1.066 |
| I2 + 2e− → 2I− | +0.535 |
| O2 + 4H+ + 4e− → 2H2O | +1.229 |
Once formed, molecular iodine has a dramatic impact on the operational device stability of PSCs. I2 interacts rapidly with surface I− anions forming a highly corrosive I3− species.2,30,34,55 As might be expected, the formation of strong and corrosive oxidisers such as iodine and triiodide, play significant albeit rarely discussed roles in governing the stability of lead-perovskite solar cells. Indeed, the use of corrosive I3− electrolytes can be attributed to the shortcomings and failure of early DSSC-based PSCs, rapidly dissolving the perovskite into its constituent salts.1,2 Wang et al. observed that in dark conditions, I2 exposure to perovskite films leads to the formation of I3−via the combination of I2 and I− (Fig. 2a).34 The highly corrosive I3− then breaks down the perovskite structure into its constituents (PbI2, CH3NH2) and I2/HI which can reform I3−; where the triiodide continues the breakdown of the perovskite (Fig. 2a). In contrast, under illumination, degradation of the perovskite can occur via a photogenerated radical mechanism where either I2 undergoes photolysis to iodine radicals (I˙). This is made possible by the notably lower halide–halide bond strength of 151 kJ mol−1 allowing photolysis to happen at relatively low energies within the solar spectrum (>2.32 eV).98,99 Like I2, the formed I˙ radicals proceed to react with the accessible surface I− ions forming a reactive diiodide radical I2˙− species (eqn (9)). Alternatively, the reactive diiodide radical can be formed via the disproportionation of I3− (eqn (10)), helping rationalise the light-dependent mechanisms.34,99 The I2˙− radical that is generated then proceeds to break down the perovskite structure, similar to I3−.34 Removal of the illumination source reforms I3− and I− from the disproportionation of two equivalents I2˙−. It is likely that both with and without illumination, the degradation is accelerated by the mobility of I− species and the presence of surface defects, which contribute to the initial I2 concentration and the formation of I3−/I2˙−.
 | ||
| Fig. 2 Effects of I2 formation on PSC performance. (a) Mechanism of iodine accelerated degradation of MAPbI3 under illumination and darkness, both leading to the release of gaseous iodine. Adapted with permission.120 Copyright 2016, Springer Nature. (b) DFT simulated energy pathway from cubic α-FAPbI3, where iodine interstitials lower the activation energy to hexagonal δ-FAPbI3 formation following iodine exposure. (c) DFT simulated geometries of phase change from α to δ FAPbI3, where interstitial iodine provides lower energy transition states to the hexagonal phase. Adapted with permission.100 Copyright 2020, Elsevier. (d) Work-function of MAPbI3 pre and post I2 exposure (N2 used as carrier gas). Reprinted with permission.112 Copyright 2017, American Chemical Society. (e) Thermal admittance spectroscopy measured on MA0.7FA0.3PbI3 prepared with fresh perovskite solution, aged precursor shown to contain iodine and with BHC, where BHC is an iodine reductant. Reprinted with permission.55 Copyright 2021, AAAS. (f) Total and projected density of states (DOS) of MaPbI3 films without (above) and with (below) interstitial iodine, indicating the presence of trap states close to the valence band of MAPbI3. Reprinted with permission.116 Copyright 2017, American Chemical Society. (g) Simulated JV characteristics of PSCs modelled with isoenergetic carrier transfer into transport layers (carrier mobility μ = 1 × 10−4) and a blocking potential of 0.9 eV towards minority carriers. The simulation includes no permeation of iodine into the transport layers (solid) and the effect of iodine permeation (dashed) with no hole trapping (red) and an iodine-induced hole-trapping rate of 1 × 10−11 (green). Reprinted with permission.36 Copyright 2023, Royal Society of Chemistry. | ||
While MAPbI3 is a useful archetypal composition for studying the fundamental chemistry of perovskite systems we note the presence of additional, cation-dependent iodine degradation pathways, relevant for state-of-the-art PSCs. Indeed, FAPbI3 undergoes cation-specific degradation mechanisms when compared to MAPbI3via destabilising the cubic α-FAPbI3 photoactive phase. Tan et al. demonstrated that following exposure to I2 vapour, the barrier to the formation of hexagonal δ-FAPbI3 was lowered from 689 to 354 meV facilitated by the presence of interstitial iodine (Fig. 2b and c).100 Combining the accelerated I2-induced phase instability of α-FAPbI3 with the tendency of FAI to undergo halide oxidation presents a strong indicator that great care must be taken when storing and preparing high-performing perovskite compositions under ambient conditions.100
Perhaps less discussed is the influence of I2 within the precursor solution on the formation of iodoplumbates and the resulting morphology of perovskite films. Hu et al. proposed that during the perovskite crystallization process, polyiodide lead clusters, aggregate to form edge-sharing octahedral PbI64− iodoplumbates. These iodoplumbates lead to cuboctahedral voids which are subsequently occupied with an organic or inorganic cation yielding the perovsite crystal structure.101,102 The formation of these iodoplumbate colloids is often controlled within the literature via molecules with Lewis basicity, which displace I− as ligands on the Pb2+ centres.103,104 To this extent, dimethylsulfoxide (DMSO) has become widely prevalent as a co-solvent within the literature owing to its high Gutmann donor number, consequently reducing the number of higher-order polyiodides and slowing crystallisation.104–106 However, in the presence of excess iodine, the formation of higher coordination lead complexes becomes favoured reducing the efficacy of Lewis base additives.107,108 As such the presence of iodine in the precursor can strongly influence the perovskite morphology, which in turn can influence the stability.103,105,108,109
In the previous section, we discussed the different pathways to the formation of molecular iodine and subsequent degradation. We next look at the effect of the iodine, once formed, on the performance of PSCs. The effect of I2 on PSC performance is often overlooked; however recent studies have shown a significant impact on the intrinsic material properties of the perovskite when exposed to the halogen vapour. Kim et al. suggested that exposure to iodine p-type dopes MAPbI3, enhances electronic conductivity and reduces ionic conductivity.110,111 This occurs from the filling of intrinsic iodide vacancies in the film, preventing the trapping of carriers while also reducing the mobility of iodide vacancies as an ionic conduction mechanism.111 Likewise, work by Zohar et al. also studying the properties of the interaction between iodine and MAPbI3, reported a 150 meV deepening of the work function of MAPbI3, consistent with p-type doping (Fig. 2d).112 In this mechanism, iodine disproportionates on the perovskite surface, either neutralising a surface iodide vacancy (eqn (4)) or forming iodine interstitials (eqn (5)).112 In both cases, the perovskite becomes increasingly p-type doped. Perovskite materials are particularly susceptible to such p-type doping owing to their deep valence band energies and proximity to the iodine reduction potential around −5.7 eV.99,113 It has also been reported that iodine disproportionates on the perovskite surface to form a pair of oppositely charged interstitials 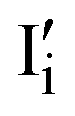 and
and 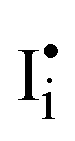 .34,114,115 We note that this mechanism is equivalent to those discussed through eqn (7) and (8), and thus can be used interchangeably to rationalise both p-type doping and electrochemical oxidation. As a result of iodine-induced p-type doping, the MAPbI3 films prepared by Zohar et al. showed an imbalance in the carrier diffusion lengths (D) changing from De: Dh = 590
.34,114,115 We note that this mechanism is equivalent to those discussed through eqn (7) and (8), and thus can be used interchangeably to rationalise both p-type doping and electrochemical oxidation. As a result of iodine-induced p-type doping, the MAPbI3 films prepared by Zohar et al. showed an imbalance in the carrier diffusion lengths (D) changing from De: Dh = 590![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :690 nm to De = 200:860 nm.112 We also note reactions (7) and (8) give an indicator of the ability of iodine to trap electrons from the conduction band. This is due to the energetics of the I2 and I3− redox potential when compared to vacuum which can be deeper than the conduction band of many perovskite materials.36,99
:690 nm to De = 200:860 nm.112 We also note reactions (7) and (8) give an indicator of the ability of iodine to trap electrons from the conduction band. This is due to the energetics of the I2 and I3− redox potential when compared to vacuum which can be deeper than the conduction band of many perovskite materials.36,99
 | (4) |
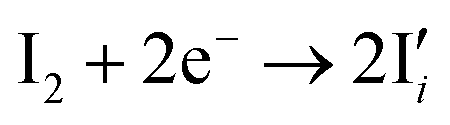 | (5) |
 | (6) |
 | (7) |
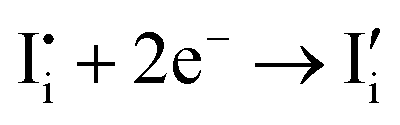 | (8) |
 | (9) |
 | (10) |
Given the mechanisms discussed above, it is not surprising that the exposure of PSCs to I2 has a significant impact on trapping and subsequent performance loss. Indeed, thermal admittance spectroscopy on perovskite films prepared with deliberate I2 inclusion at the precursor stage revealed significantly higher densities of shallow traps associated with the formation of I3− (Fig. 2e).55 Interestingly, in the case of fresh perovskites, the density of trap states could be further reduced through the addition of the reductant suggesting that iodine may be unavoidable within fresh perovskite precursors unless treated with a reductant. This could be a consequence of a necessitated equilibrium quantity of I2, something discussed later in sections of this review. The presence of shallow traps was further discussed by Li et al. who, using computational methods, related the shallow traps to interstitial iodide atoms which form through the dissociation of iodine on the perovskite surface (Fig. 2f).116 Liang et al. further correlated the shorter carrier lifetimes with the presence of I2 within perovskite films via encouraging photodecomposition of a prescribed PbI2 excess into I2 and Pb0 using illumination.66 The decrease in time-resolved photoluminescence (TRPL) lifetime and photoluminescence (PL) intensity is consistent with the formation of I3− shallow trap states and p-type doping, whereby increased intrinsic hole densities increase the likelihood Shockley–Read–Hall (SRH) recombination.112 More recently, Bitton et al. used computational modelling to relate the iodine chemistry active within the PSC architecture to the device physics.36 The study showed that both p-type doping via interstitial iodine formation and the reverse reaction, hole trapping from interstitial iodide anions, can dictate device performance. Indeed, the regeneration of neutral iodine species via the trapping of holes contributes significantly to losses in all photovoltaic parameters, an effect worsened when the transport layers are permeable to ions (Fig. 2g). Consequently, the evidence presents a strong argument for the prevention of iodine and iodide migration outside the active layer, an approach discussed in greater depth in later sections. In summary, the formation of I2 reduces the photovoltaic performance of PSCs through: (i) the formation of trap states and mobile iodide interstitials (mobile recombination centres), (ii) inadvertent p-type doping creating an imbalance in the diffusion length of free carriers, and (iii) breakdown of pristine perovskite via the formation of corrosive triiodide species. As such, the effect of I2 formation has been shown to directly contribute to losses in short-circuit current density (Jsc) and open-circuit voltage (VOC).55,66,112,117–119
In addition to the perovskite active layer, iodine also modifies the intrinsic properties and stability of the other layers which constitute the PSC architecture, such as the electrodes and charge transport layers. We will first consider the effect of iodine on the electrodes. The use of metals as electrodes in the presence of oxidising iodine is particularly problematic in the pursuit of long-term stable PSCs. This is particularly prevalent in metals such as silver, aluminium and copper, which undergo fast oxidation to form AgI, Al2I6 and CuI/CuI2 respectively (Fig. 3a–c).97,121–124 Li et al. measured the resistance across the silver electrodes on PSCs during degradation noting significant oxidation of the Ag under application of bias within the first 10 hours.125 Further studies by Wijesekara et al. used energy dispersive X-ray (EDX) coupled transmission electron microscopy (TEM) techniques to reveal that highly volatile iodine released from degrading perovskite films was sufficient to oxidise silver even in the absence of direct contact (Fig. 3d and e).126 Somewhat surprisingly, Shlenskaya et al., demonstrated that even gold, traditionally considered a stable metal when in the solid state, is not exempt from iodine-induced oxidation when used as an electrode.127 The damage to the gold electrode is triggered by an interaction between iodine with MA forming a highly reactive polyiodide species.128 The polyiodide melts, with chemical formula MAI-nI2, oxidize the surface of the gold forming a new tetragonal phase with the formula MA2Au2I6.127 More recently, similar reactions with Cu electrodes have been identified leading to the formation of MACu2I3.121 As such, the use of metals presents a significant challenge when used in iodine-based PSCs. This challenge may be overcome by using new inert electrodes such as carbon or carbon nanotube electrodes (CNTs), which have shown excellent longevity and can even exhibit enhanced properties following iodine doping.33,129 However, significant performance improvements are still required in devices prepared using oxidation-resistant electrodes to reach the state-of-the-art.130 Interestingly, the effect of I2 also extends to the transparent conductive metal oxide electrode, typically indium or fluorine-doped tin oxide (ITO/FTO). Ultraviolet photoelectron spectroscopy (UPS) and X-ray photoelectron (XPS) studies by Sun et al. report that exposure of I2 or Br2 to ITO modified the work function of the electrode, increasing significantly as a function of I2 concentration.131
 | ||
| Fig. 3 Effect of I2 on other components of PSC. (a) Oxidation of iodide to iodine and subsequent oxidation of silver electrode to AgI. Reprinted with permission.125 Copyright 2023, Elsevier. (b) and (c) Photographs of (b) fresh (Ag) and (c) aged PSCs with high quantities of AgI. (d) degradation of a perovskite film in proximity to Ag strips to measure AgI formation. (e) EDX-coupled TEM showing Iodide distribution across the silver strips demonstrating oxidation of the electrode occurs readily during perovskite degradation. Adapted with permission.126 Copyright 2021, Wiley. (f) Plot of spiro-OMeTAD HOMO deepening from exposure to I2 vapour. (g) JV curve data of PSCs prepared before and after exposure of the HTL to I2 vapour. Reprinted with permission.132 Copyright 2020, Wiley. (h) and (i) Schematic of current measurements across organic conductors exposed to I2 and (i) resulting change in current as a function of HOMO energy for a range of HTLs. (j) Schematic of I2 permeability from the perovskite through the HTL. (k) Plot of I2 permeability as a function of the energy of HOMO for a range of organic HTLs. Reprinted with permission.135 Copyright 2021, American Chemical Society. (l) Schematic of the influence of I2 on the deterioration of perovskite and NiO/PTAA interfaces and the associated effect on total PSC stability. Reprinted with permission.115 Copyright 2021, American Chemical Society. | ||
Like the electrodes and perovskite active layer, the charge transport layers can undergo both strong p-type doping and degradation following exposure to I2. 2,2′,7,7′-Tetrakis[N,N-Di(4-methoxyphenyl)amino]-9,9′-spirobifluorene (Spiro-OMeTAD), a common HTL, undergoes p-type doping with I2 leading to deepening of the highest occupied molecular orbital (HOMO) and the formation of a barrier to efficient hole extraction (Fig. 3f).91,132 The effect of this barrier can be observed in device JV characteristics presenting a characteristic “knee” feature and significantly worsened device performance (Fig. 3g).132 Similarly, deepening of the energy levels as a result of iodine doping has also been reported in poly(3-hexylthiophene)(P3HT), (6,6)-phenyl C60 butyric acid methyl ester (PCBM), C60 and a range of other small organic semiconductors upon exposure to I2.133–135 Kerner et al. related the extent to which p-type doping occurs to the first ionisation potential, referred to as the HOMO level energy in organic semiconductors.135 In the work, the authors report organic transport layers with a shallower HOMO, are most readily oxidised and therefore undergo the most significant doping. Experimentally, the authors find transport layers with shallower HOMOs yield the largest change in current following I2 exposure (Fig. 3h and i). Likewise, a similar relationship is reported between the HOMO energy and the permeability of I2 through the transport materials (Fig. 3j and k).135 It should also be noted the permeability of iodine with organic materials can be amplified by virtue of ionic interdiffusion with dopants such as Li+ and Co2+, necessitated in many organic HTLs, most notably spiro-OMeTAD.136 The choice of HTL is also essential for minimising the concentration of free holes and thus reducing the oxidation of free iodide ions (eqn (6)). The choice of the transport layer and its impact stability was recently discussed by Khadka et al.115 In the work the authors highlight the importance of maintaining an efficient hole-extracting interface for preventing both I2 evolution and the formation of destructive voids within the perovskite layer (Fig. 3l). Consequently, PSCs prepared using NiO, an inorganic HTL, exhibit better stability compared to PTAA, an organic HTL. This was attributed to the faster degradation of the organic component with I2 and the associated loss of the hole-collecting interface.115 Indeed, when compared to inorganic HTLs, organic polymers and molecules are susceptible to the slow iodination of unsaturated alkenes and aromatic rings with I2.135,137–140 Recently, it was suggested that P3HT can undergo crosslinking when exposed to I2, changing the material properties and likely the interface.135 In contrast, NiOx undergoes a surface anion exchange to form a thin NiI2 layer, but crucially will largely retain a hole-extracting interface.96,115 As such, choosing HTLs that are tolerant to I2 and undergo minimal p-type doping is crucial for reducing the formation of voids and improving the stability of PSCs.36
Iodine is a strong oxidizer and is formed via several mechanisms in lead-based PSCs, including from the precursors, superoxide formation and exposure to light or electric fields. Once formed, I2 dopes the perovskite active layer, forming shallow traps and reducing the performance of devices. Furthermore, the challenge of I2 and its associated forms I3− and I2˙−, the species responsible for the downfall of DSSC architectures, continues to hinder the longevity of PSCs. In this sense, the presence of mobile and reactive halide ions in a dynamic solid-state ion conductor remains a key cause of the instability associated with lead-based PSCs. This can be seen clearly when comparing lead-PSCs to previous generous inorganic technologies and represents a barrier to the long-term commercial success of lead-perovskite technologies. Equally significant is the effect of iodine on the other components required to make functioning PSCs. Like the active layer, iodine both dopes and degrades the charge transport layers and transparent metal oxides, hindering the extraction of carriers out of the cell. This in turn contributes further to the oxidation of iodide ions and further evolution of molecular iodine. As such, further work is needed in both identifying and remedying the mechanisms through which iodine can form and act on the perovskite in high-efficiency PSCs.
Iodine in tin perovskite
In the previous section, we discussed how I2 could compromise the medium and long-term stability of lead PSCs. The effect of iodine is significantly amplified in tin-perovskites owing to a predisposition to undergo oxidation from Sn2+ to an Sn4+ oxidation state. As such, when exposed to oxygen, tin perovskites undergo rapid and significant decomposition on the scale of minutes to hours (Fig. 4a). Very recently, I2 evolution has been identified as an additional key oxidiser responsible for the poor stability of tin perovskites. Interestingly, the ability of Sn nuclei to undergo sp3 hybridisation, forming an Sn(IV) oxidation state, creates new distinct iodine degradation pathways, not present within the lead systems previously discussed. As such the formation of the SnI4 oxidation product, the lead (Pb) analogue of which cannot form, creates new chemistry within the tin perovskite system by which iodine can be formed and oxidize pristine perovskite. The effect of this can be easily seen by comparing the reported stabilities of tin and lead PSCs.33,38,48,130 Furthermore, the presence of SnI4 is known to contribute to the trapping of carriers reducing the performance of PSCs.38 To make this challenge worse, the presence of small quantities of SnI4 is largely unavoidable within commercial precursors, often making batch-to-batch reproducibility difficult. | ||
| Fig. 4 Degradation mechanisms of ASnX3 perovskite. (a) Schematic of ASnX3 oxidation to SnI4 and SnO2 from exposure to O2. (b) Schematic of I2-mediated degradation of tin perovskite.49 (c) Inert storage stability of FASnI3 perovskite devices with and without NaBH4 as a reductant. Reprinted with permission.49 Copyright 2022, Elsevier. (d) Normalized UV-vis absorbance demonstrating the importance of combined oxygen and moisture on the stability of tin perovskite films. Reprinted with permission.38 Copyright 2021, Springer Nature. | ||
Once oxidised, the facile and rapid decomposition of SnI4 into I2 has been reported to occur from one of two mechanisms (Fig. 4b); (i) light-driven ligand metal charge transfer (LMCT),20 and (ii) the disproportionation of SnI4 with water and oxygen under atmospheric conditions.38 Considering first the former mechanism, when exposed to visible light, SnI4 undergoes a photoreduction forming SnI2, and releasing I2. Once the external light stimulus is removed, the formed I2 oxidizes the pristine perovskite back into SnI4 leading to degradation and the breakdown of the perovskite crystal structure. Through this mechanism, tin-PSCs can incur degradation when stored under illumination (light) even in an inert atmosphere (Fig. 4c).20 While yet to be discussed within the context of tin-PSCs it is likely that iodine radical mechanisms discussed in the previous section will also contribute to this degradation pathway, with the formation of iodine radicals unlikely to be exclusive to Pb-based systems. To date, the activity of iodine photoradicals has yet to be discussed within the tin-perovskite system. The degradation is further accelerated when exposed to ambient (moisture and oxygen) conditions. Lanzetta et al. revealed that when under ambient conditions, SnI4 within the film undergoes fast hydration to form HI and SnO2.38 As discussed previously, the HI then undergoes subsequent oxidation with oxygen yielding I2 and water. Once formed, the iodine rapidly oxidises perovskite to form SnI4, thus continuing the cycle. Crucially, with each iteration of the cycle two equivalents of iodine are formed leading to an exponential degradation mechanism which accelerates with each generation (Fig. 4d).38,42 Interestingly, the Sn2+ states undergo such fast and favourable oxidation when exposed to I2 that Sn2+ states in the form of SnS aerogels have previously been exploited to capture scavenge gaseous I2, forming SnI4 and SnI4(S8)2.141–143 It is therefore of fundamental importance to improve the tolerance of Sn2+ to oxidation to slow the evolution of I2.
In addition to the decomposition of SnI4, I2 has also been reported to form via the reaction of dimethyl sulfoxide (DMSO) and HI during the perovskite annealing process.53,54 While an interesting observation, and one which highlights an important route for I2 formation, the concentration of HI is likely to be low during the fabrication process when DMSO is present unless added intentionally as a means to modify grain size. I2 evolution from HI during the fabrication is therefore unlikely to contribute significantly to the long-term stability of PSCs, provided the precursors have been properly stored. Nevertheless, the formation of HI post-fabrication remains problematic, owing to oxidation with oxygen and potentially other chemical pathways such as interaction with transport layers.38 Very recently it has been suggested that as per the lead system, reactive superoxide species can be generated from tin perovskites.144,145 However, further work is needed to confirm this observation via directly observing the superoxide formation rather than measuring the consumption of a probe, which itself may be prone to instability. Furthermore, the probe used within the study, 1,3-diphenylisobenzofuran (DPBF), is sensitive to singlet oxygen O12 and great care must be taken to differentiate between the photogenerated oxygen compounds.146
In addressing the problem of iodine evolution within tin perovskites a two-part strategy is required, focusing simultaneously on both prevention and neutralisation. Firstly, great effort must be made to produce perovskites with high intrinsic stability. This can be achieved by ensuring low starting concentrations of both I2 and SnI4 which both contribute strongly to the instability as discussed. Secondly, an active iodine management strategy is required to neutralise the oxidising species as they form. Such strategies are required for both lead and tin perovskites and are therefore compared together later in the discussion sections of this review. Focusing on specific strategies to improve the intrinsic stability of tin perovskites, purification of the SnI2 precursor has become a well-established step for producing high-performing tin PSCs.38 This purification can take several forms and includes the use of a reductant such as metallic Sn nanoparticles to convert SnI4 to SnI2 within the precursor or performing a thermal treatment.147–150 Alternatively, high-quality SnI2 can be synthesised reproducibly in a laboratory environment through the reaction of tin metal and concentrated HCl followed by displacement of Cl with I2 and purification with Sn metal.37,151,152 Likewise, it is also important to note the potential for the cations to oxidize and release iodine if stored improperly. Furthermore, it is also likely that the cations can contribute HI to the precursor if improperly stored, which could encourage the formation of iodine during annealing.53,54
The intrinsic stability and tolerance of tin perovskite films to iodine can be improved via structural and compositional tuning. This can be achieved most directly via the substitution of iodide anions in favour of bromide (Fig. 5a). Perovskites prepared in which iodide ions are substituted in favour of bromide, exhibit improved intrinsic stability, forming stronger bonds with the metal centre, and undergoing photolysis at significantly higher dissociation energies.56,67,97,152,153 Furthermore, solar cells prepared with perovskite films comprising sub-stoichiometric bromide fractions have been shown to exhibit improved device performance, owing to greater structural order and reduced trapping.24,48,152,153 However, as per the lead analogues, the fraction of iodide suitable for replacement with bromide remains low to maintain good spectral overlap with solar irradiation. Consequently, halide exchange can only constitute a minor role in tin stabilizing strategies.
 | ||
| Fig. 5 Preventing I2 formation via structural tuning. (a) Normalized absorbance (500 nm) of FASn(I1−xBrx)3 showing improved stability of the perovskite film when substituting 25% I− with Br−. Reprinted with permission.153 Copyright 2018, American Chemical Society. (b) and (c) Photographs of the contact angle between substrate and water droplet on (b) a 3D perovskite surface, and (c) a 2D (neo-pentylbutylammonium iodide) capped 3D perovskite. Reprinted with permission.154 Copyright 2022, Wiley. (d) Normalized diffraction intensity of the 100 plane of FA1−xCsxSnI3 with varying concentrations of Cs cations. Reprinted with permission.26 Copyright 2017, American Chemical Society. (e) Relationship between incorporation of small cations and associated lattice strain on the carrier mobility of FA0.75MA0.25SnI3. Reprinted with permission.155 Copyright 2019, American Chemical Society. (f) and (g) 119Sn NMR spectra of (f) SnF2, SnI2, SnF4 and SnI4 reference positions and (g) combination of SnF2 and SnI4 showing peaks corresponding to SnI2 and SnF4. Reprinted with permission.156 Copyright 2021, Wiley. (h) Absorbance spectrum of pure tin (FASnI3) and mixed tin–lead (FASn0.5Pb0.5I3) perovskite, revealing near negligible SnI4 generation upon substitution of 50% Sn with Pb. Reprinted with permission.26 Copyright 2017, American Chemical Society. | ||
The cation choice (A-site) also contributes significantly to dictating the intrinsic stability of tin-perovskite thin films. Firstly, varying the hydrophobicity of the organic cation affords opportunities to prevent the interaction between the perovskite with moisture, essential for preventing SnI4 hydration, the first step of SnI4 chemical decomposition into I2 (Fig. 4b).38 This is of high importance owing to the tendency of the small organic cations such as MA+ and FA+ to undergo hydrogen bonding with water, prompting the hydrolysation of the tin iodide.157–159 The use of large hydrophobic cations is a promising approach to tackle this challenge by forming 2D or quasi-2D perovskites as hydrophobic capping layers.2,25,27,154 Alternatively, the interaction of the surface can be tuned via the application of thin protective interlayers, reducing the surface interaction with moisture and oxygen (Fig. 5b and c).160,161 Leijtens et al. demonstrated that substituting the FA cation with up to 20% Cs could improve the overall stability of the film, retaining its crystal structure for longer periods with increasing Cs fraction (Fig. 5d).26 The reduction in unit cell dimensions upon the addition of Cs is suggested to shorten and strengthen the Sn–I bond, increasing the energetic barrier to SnI4 formation. In addition to slowing oxidation, the reduction of lattice strain has also been demonstrated as beneficial to carrier mobility increasing from 27.07 cm2 V−1 s−1 in a pure MAFA-based perovskite up to 43.96 cm2 V−1 s−1 upon substituting 10% of the A site with Cs+ (Fig. 5e).155 When combined with a more hydrophobic capping layer, the two approaches offer a promising solution to surmounting the joint problems of oxidation and hydrolysation of SnI4 at the perovskite surface.
In the previous section, we discussed the mechanisms by which SnI4 readily generates I2via either illumination (LCMT) or displacement with oxygen. As such, it is vitally important to stabilise Sn2+ to avoid oxidation into Sn4+ and slow the cycle of degradation. Great success in this regard has been achieved via the addition of small fractions of SnF2 or SnCl2 to create a stoichiometric excess of Sn within the perovskite film. SnF2 as an additive was first identified as promising in perovskite solar cells over a decade ago, whereupon significant improvements in PSC photocurrents could be achieved upon inclusion of the additive.40 Similar observations were made by Kumar et al. who went on to attribute this photocurrent increase to a reduction in the hole carrier density, affording a more intrinsic perovskite semiconductor.162 The origin of the p-type doping for which SnF2 remedies was attributed to the formation of tin vacancies which form upon Sn2+ expulsion from the perovskite lattice.163,164 As discussed previously, within the lead-perovskites, reducing the intrinsic hole density is highly beneficial for avoiding the oxidation of iodide anions to iodine within the film. Furthermore, the addition of the SnF2 has been also shown to improve the morphology of the resultant film, creating a more compact interface and improving the moisture and oxygen resilience. Unsurprisingly, the addition of SnF2 has become amongst the most widely adopted tin-PSC additives within the literature. Recently, Zillner et al. suggested that the F− anions undergo accumulation at the interface with poly(3,4-ethylene dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS), suggesting the formation of a possible SnSx interlayer.165 However, the exact origin of the sulfur remains unclear within this work. Nevertheless, the results within the work suggest that an interaction between the HTL and SnF2 may occur. More recently, Pascual et al. offered mechanistic insights as to the ability of SnF2 to promote the reduction of SnI4 by observing that the combination of both species leads to the formation of SnF4 and SnI2 from 119Sn NMR.156 The driving force of this chemistry stems from the higher reacitivty of the fluoride ion relative to the iodide and therefore greater stability of SnF4 in the Sn4+ state relative to SnI2, promoting a ligand exchange between the two tin compounds. The resulting reduction in SnI4 will directly slow the formation of iodine and associated iodine-induced degradation.38
The stability of tin-based PSCs can also be improved via the partial substitution of tin cations in favour of lead, producing a tin–lead binary structure.166–168 When exposed to oxygen, two adjacent Sn2+ sites are required to form SnI4 and SnO2. Partially removing Sn2+ sites in favour of Pb2+ which cannot undergo the same chemistry discussed above. Moreover, this prevents the conventional mechanism of oxygen-induced degradation occurring in the immediately surrounding area (Fig. 5).38 Consequently, when well dispersed, each Pb2+ cation can prevent the oxidation of surrounding tin cations. As such, even at 50% Sn substitution in mixed tin–lead systems, dramatic reductions in SnI4 formation have been observed (Fig. 5h).26 The prevention of SnI4 formation has a dramatic effect on the suppression of I2 evolution slowing degradation. Consequently, tin–lead binary perovskites can achieve superior stability and device performance to that of pure tin.
While several studies have directly probed the effect of iodine on the device performance of lead PSCs, little attention has been dedicated to the consequences of I2 exposure on tin PSCs. This is owed to a combination of how recently I2 was discovered within tin perovskite and the significantly lower tolerance of tin perovskites to oxidizers, most notably oxygen. It therefore is useful to observe the doping characteristics and mechanisms of iodine within other semiconducting materials to speculate on the tin system. It is well documented that the addition of I2 to various semiconductors leads to p-type doping via the removal of an electron to form iodide interstitials (Ii−).34,112,115 This phenomenon has been demonstrated to occur across a wide range of materials including metal oxides, organic semiconductors and lead perovskites, suggesting similar doping phenomena likely to occur within the tin perovskite.111,112,132,133 Tin perovskite is well known to have p-type characteristics, a phenomenon previously linked to the presence of Sn2+ vacancies (Vsn).153,162,164 Furthermore, as tin perovskite degrades the p-type doping increases via the formation of more tin vacancies.38,153 It is therefore unclear to what extent the interaction of iodine with the tin perovskite may be contributing to the p-type doping observed within tin PSCs and, more significantly, to what extent the progressive doping may be circumvented through the neutralisation of iodine towards a more intrinsic semiconductor. In addition to doping tin-PSCs, the interaction of iodine with the perovskite leads to the formation of SnI4 and tin vacancies both previously reported to facilitate recombination.38,164,169,170 Lanzetta et al. revealed that the addition of +2 mol% of SnI4 to the precursor was sufficient to significantly lower the current to nearly a third while also reducing the voltage.38 As such improving the stability will inevitably enhance the performance and vice versa.
Discussion
In the previous sections, the significance of iodine formation on both the stability and performance of lead and tin PSCs was discussed. We next discuss strategies which look to mediate both the short and long-term impact of iodine formation on device performance. The most obvious approach to this end is the substitution of I− with other anions such as Br− or Cl−. These compositional changes improve the chemical stability via forming stronger bonds to the B-site metal centres while also having higher oxidation potentials. However, as discussed, changes to the band gap in addition to the formation of iodine-rich phases place practical limitations on the fraction of iodine which can be replaced.171 Consequently, new strategies and understanding are required to improve the stability of the highest performing iodine-rich PSCs which target (i) minimising the starting quantities of I2 and preventing I− oxidation during fabrication, (ii) developing dynamic processes which reduce the I2 as it is formed and (iii) minimising the evolution of iodine when subject to operational stresses. Consequently, the optimum strategy looks to produce PSCs with a low intrinsic concentration of I2, where oxidation is thermodynamically disfavoured. In this strategy, I2 formation from operational stresses is minimised to allow regenerative reducing redox reactions to compete and maintain a steady state of operation.Achieving a low intrinsic concentration of I2 within the perovskite precursor is essential in managing iodine which can catalyse degradation in both tin and lead-perovskites. As early as 2015 the merits of reductant incorporation within the precursor on I2 evolution, morphology and performance have been known. Zhang et al. identified that hypophosphorous acid (HPA), used commercially to stabilise hydroiodic acid, may be beneficial in reducing I2 and stabilising I−. This, in turn, leads to a reduction in the number of higher oxidation polyiodides within the precursor, enlarging grain size.117 Furthermore, when used in tin perovskite, HPA can reduce the tendency of Sn2+ to undergo oxidation.172,173 Recently, Chen et al. demonstrated a benzyl-substituted hydrazine salt, benzyl hydrazine hydrochloride (BHC), as a highly effective Iodine reductant within lead PSCs. After generating I2 within their MA0.7FA0.3PbI3 perovskite precursors, the authors incorporated a BHC additive, improving the average PCE of devices prepared from control and I2-containing solutions from 21.8 and 19.2% respectively, up to 23.3%. Crucially, the BHC-treated PSCs exhibited significantly improved stability with the BHC containing perovskite retaining over double the PCE of the control after 1000 hours under illumination.55 More recently, the same group incorporated the same BHC additive within all-perovskite tandems achieving an impressive large area (14.3 cm2) efficiency of 21.6%.174 Recently, 4-fluorobenzothiohydrazine was shown to stabilise all-inorganic CsPbI3 enabling a PCE of 21.41%, amongst the highest reported values for fully inorganic PSCs.175 Likewise, Zhang et al. used hydrazine sulfonate to stabilise narrow band-gap (≈1.24 eV) Pb–Sn binary PSCs enabling high PCEs of 23.17%.176 To this end, several hydrazine derivatives have been incorporated within PSCs owing to high oxidation potentials (Fig. 6) and the formation of inert N2 oxidation products.55,115,177–179 Nevertheless, there remains a gap in knowledge concerning design criteria for hydrazine additives. The recent success of benzylated hydrazine derivatives, suggests a milder and less aggressive reductant version of hydrazine is beneficial. Hydrazine can also be used as a reducing vapour to minimise the fraction of iodide anions which undergo oxidation during the deposition and annealing processes. Song et al. demonstrated that preparing tin-iodide PSCs under a hydrazine atmosphere was highly beneficial for reducing SnI4 during processing and subsequent filling of Sn2+ vacancies.180 However, it is important to note that while effective, hydrazine has several significant health and safety hazards including toxicity and a tendency to form a flammable vapour.181 As such the practicalities of using hydrazine as a reducing vapour during fabrication must also be considered, particularly when considering scalability.
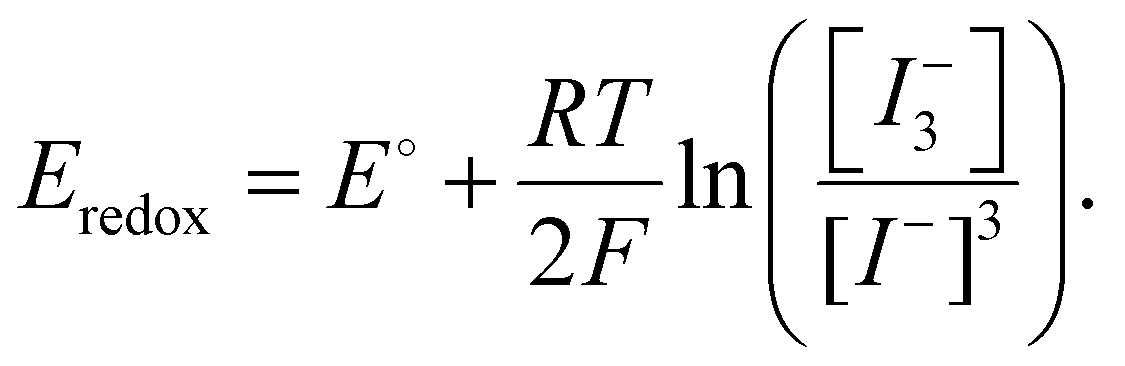 | (11) |
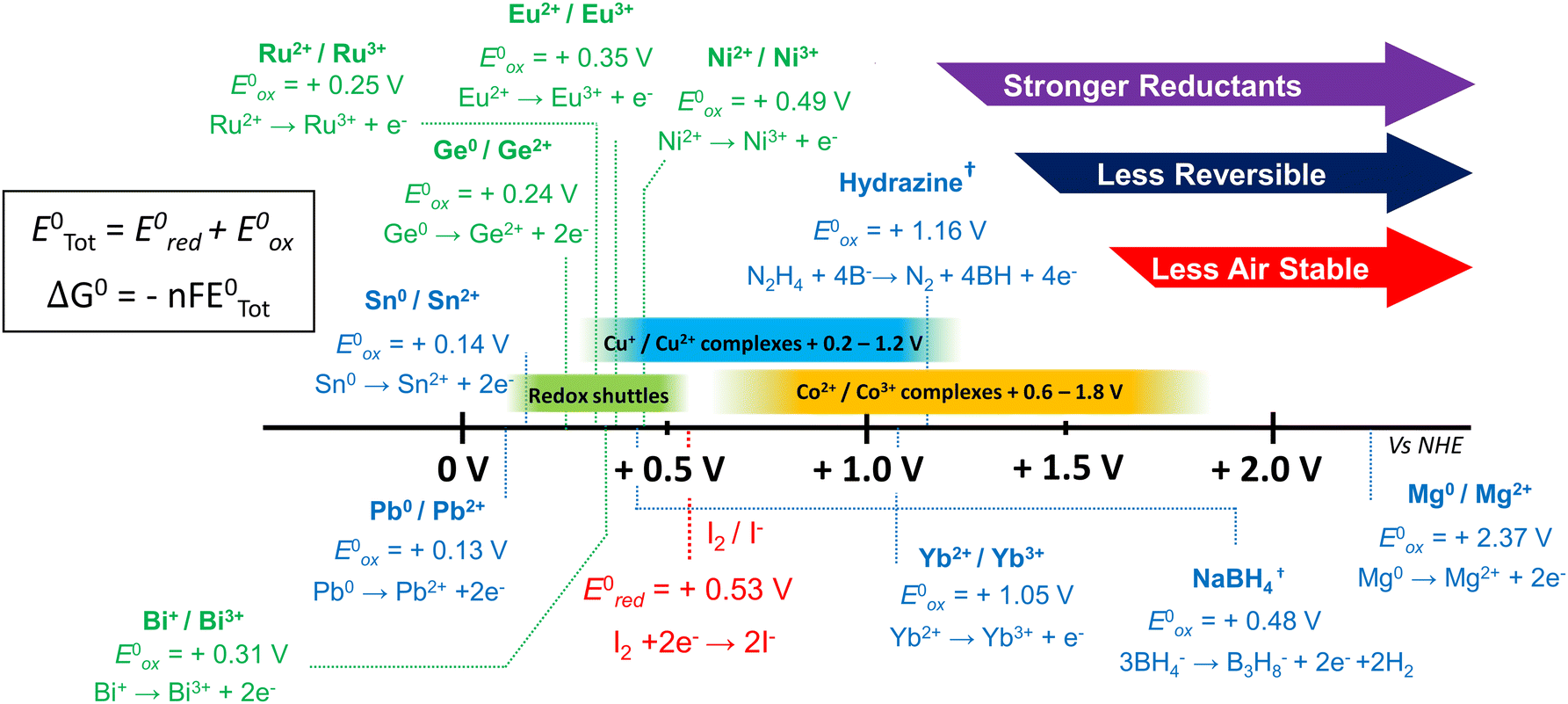 | ||
| Fig. 6 Oxidation potentials of various reductants. Plot of the oxidation potentials of different reductants vs. normal hydrogen electrode (NHE) under standard conditions. These values are provided as a rough estimate to aid the identification of useful redox couples. The iodine/triiodide reduction potential is given in red. Redox couples between the oxidation of Pb0/Pb2+ and I2/2I− (green) can undergo disproportionation to act as a redox shuttle for the formation of PbI2. Spontaneous redox chemistry occurs when ΔG < 0.†97,191–194 | ||
While undoubtedly beneficial, simply reducing the starting concentration of iodine within the precursor and during fabrication is insufficient to ensure long-term operational stability. This is a consequence of the propensity of iodine to form under operation. Furthermore, the oxidation of iodide to iodine and subsequent reduction back to iodide are subject to a dynamic equilibrium.100 As such the redox potential is likely influenced by the Nernst potential (eqn (11)), a function of the concentration ratio of the oxidised and reduced species.99 This makes achieving completely iodine-free PSCs thermodynamically difficult. For this reason, we speculate that a low equilibrium concentration of iodine may be unavoidable and thus management within the active layer inevitably becomes necessary. This can be achieved via the application of dynamic reduction processes or by influencing the iodine–iodide–triiodide equilibrium constant to discourage the oxidised form.100 Sanchez-Diaz et al. utilized a mild active reductant (Fig. 6) namely, sodium borohydride (NaBH4), to purify the precursor while also dynamically reducing I2 during operation under illumination. The strategy was shown to be effective at improving the stability of tin perovskite devices, leading to a 130% increase in the initial PCE after 500 hours under illumination in an inert atmosphere. In comparison, the control devices (no NaBH4) failed after 200 h.49 However, like hydrazine-based compounds, exposure of the NaBH4 to air leads to the oxidation of the reductant, and is thus most effective when used in encapsulated devices. As such, finding effective iodine reductants which are stable to ambient conditions presents a difficult challenge. A potential solution presented by Liu et al. details how I2 evolution can be managed during processing via the addition of methylamine to the precursor solution. The authors of this study suggest that the addition of methylamine as a base results in an increase in precursor basicity leading to a disproportionation reaction of iodine into I− and iodate anions (IO3−) mediated by trace water.109,182 Further investigation into the mechanism revealed a correlation between the pka and the efficacy of disproportionation, with strong Arrhenius bases such as NaOH and KOH most effective at preventing I2 evolution.183 Such strategies represent an effective way to influence the iodine–iodide–triiodide equilibrium processes. Very recently, Sun et al. reported the use of potassium formate (HCOO−K+) as an effective and oxygen-stable iodine reductant.95 The study demonstrates that iodine formation within the precursor solution can be mitigated via the addition of formate anions yielding CO2 and both I− and H+ ions. These products are benign to the function of the PSCs enabling a high performance of 23.8% and good accelerated stability in ambient conditions.95
Despite recent advances in device PCE and stability, the reliance on a finite concentration of a reductant additive which may become increasingly depleted during operation is sub-optimal. An alternative strategy involves the design and integration of new redox mediator compounds within the perovskite active layer (Fig. 7a). These redox shuttles are designed to undergo disproportionation, facilitating I2 reduction before regenerating to the initial state via undergoing redox chemistry with other ions. Such regenerative approaches which are not consumed during operation hold great promise for asserting greater control over the redox processes within PSCs. This approach was well demonstrated by Wang et al. who opted for the use of the Lanthanide metal salt europium iodide (Eu3+/Eu2+) redox couple to reduce I2 formed within the perovskite into more benign I−.118 The Eu3+ could then be regenerated via the oxidation of metallic Pb0 into Pb2+ (Fig. 7b), producing a beneficial regenerative cycle which, crucially, can function perpetually without depleting the additive. By removing I2 and Pb0-based traps, PSCs prepared with Eu2+ exhibited improved PCE, increasing from 18.5% to 20.7%. The devices incorporating the additional regenerative redox pathways also exhibited far superior stability retaining a 93% initial PCE when stored under illumination and 91% when stored at 85 °C, in an inert atmosphere over a 1500-hour test period. The desirable redox potential of the Eu2+/Eu3 redox couple has since seen the iodide salt (EuI2) replaced in favour of various alternative salts with a range of counter anions.184–186 Nevertheless, the benefits of redox pathways to reduce I2 have yet to be fully realised. Indeed, several transition and lanthanide metals have suitable redox potentials to encourage redox chemistry between Pb0 and I2 as shown in Fig. 6. Furthermore, the oxidation potentials of many metals can be tuned by the choice of ligand. In this regard, the Cu+/Cu2+ redox pair is particularly promising owing to a library of reported complexes with oxidation potentials between the lead and iodine redox reactions.187,188 We also highlight the excellent oxidation potentials of Ni2+ and Ru2+ complexes to act as redox shuttles (Fig. 6).189,190 Additionally, milder regenerative reductants are less reactive towards oxygen and ambient conditions, improving the effectiveness of the approach compared to the use of stronger reductants.
 | ||
| Fig. 7 Strategies which can be implemented within the PSC to improve the I2 stability. (a) Schematic of PSC structure highlighting modification of the active layer. (b) Eu2+/3+ redox shuttle to facilitate I2 reduction and Pb0 oxidation. Reprinted with permission.118 Copyright 2019, AAAS. (c) Structure of β-cyclodextrin and schematic of I2 capture around the perovskite grain. Adapted with permission.195 Copyright 2023, American Chemical Society. (d) Thermal stability of PSCs prepared with and without poly(2-vinylpyridine) (P2VP) with the perovskite layer. Reprinted with permission.196 Copyright 2023, American Chemical Society. (e) Schematic of PSC structure highlighting the incorporation of iodine-blocking interlayers. (f) Structure of Cu(II)/Zn(II) porphyrins used as an interlayer to block/capture I2 and protect spiro-OMeTAD. (g) DFT calculated stable equilibrium structures of iodine-coordinated Cu(II)porphyrin structure. (h) Improvement in PSC photostability upon incorporation of a Cu(II) porphyrin. Adapted with permission.207 Copyright 2021, Chinese Chemical Society. (i) Schematic of PSC structure highlighting strategies which involve the transport layers. (j) and (k) Trend in charge extraction capability of various HTLs with PEA0.2FA0.8SnI3 perovskite and (k) corresponding stability of the perovskites. Reprinted with permission.38 Copyright 2021, Springer Nature. (l) Mechanism of down-conversion of high-energy UV-light via TiO2 absorption, subsequent energy transfer to Eu3+ and re-emission in the visible spectrum. Reprinted with permission.223 Copyright 2018, American Chemical Society. | ||
In addition to being thermodynamically favourable, it is also important to consider the kinetics of reduction pathways. Indeed, the chosen reductant must be in proximity to I2, rather than allowing the volatile halogen to escape into the wider device stack or atmosphere. Li et al. proposed the use of β-cyclodextrin (β-CD) an oligosaccharide cage formed of glucose sub-units; β-CD captures iodine molecules within the perovskite layer, providing opportunities for redox chemistry between I2 and Pb0 to compete kinetically with I2 escape (Fig. 7c). Consequently, by delaying and preventing the escape of I2, redox reactions between metallic Pb0 and captured I2 species become competitive.195 As such even after 200 h of aging, the FA1−xCsxPbI3 prepared with β-CD exhibited little structural decomposition and no Pb0 peaks within the XRD pattern. Crucially, MPP tracking at elevated temperatures and illumination, yielded improved device stability when prepared with β-CD, retaining an impressive 88.2% PCE after 1000 h. Similarly, polyvinylpyrrolidone (PVP), a polymeric iodophor widely used as an antiseptic, has also gained recent attention through its ability to form a complex with and act as a reservoir for I2 capture within perovskites.196–201 Interestingly, conflicting reports have so far been provided as to the exact optimum application with Kang et al. choosing to add the iodine-bound form (PVP-I) whereas Yang et al. opted for poly(2-vinylpyridine) (P2VP) with no starting bound-iodine.196,198 In the former approach, the PVP-I provided a reservoir to replace iodine, preventing irreversible loss and depletion. While early DFT studies have warned about the impact of irreversible iodine depletion from the ABX3, little research has been offered as to how stoichiometry may be replenished dynamically and sustainably; this being essential for device longevity.36 In the approach taken by Yang et al. the non-iodized form is incorporated within the PSC, whereby captured iodine generated from iodide oxidation is used effectively to oxidise metallic Pb0 traps and reform perovskite. Nevertheless, Both PVP-I and P2VP-containing devices attained high efficiencies approaching 23%. Furthermore, devices prepared using P2VP as an iodine-blocking layer strategy exhibited impressive operational stability (under inert N2 conditions) retaining 90% PCE after 1000 h MPP tracking under illumination and 90.9% after 750 h heating at 85 °C (Fig. 7d). In contrast, for PVP-I, only dark low humidity stability measurements are provided leaving considerable question marks over unwanted iodine release when exposed to operational stimuli such as temperature, illumination and sustained electrical bias.36
Developing strategies which confine iodine within the active layer is a powerful approach that provides opportunities to encourage favourable dynamic redox chemistry. Such chemistry provides beneficial restorative pathways to compete with degradation occurring by chemical instability and operational stimulus. To this end, many lessons in the design of effective iodine capture can be learned from the rapidly developing field of radioiodine I127 capture, medical polymeric iodophores and iodine retention in electrochemical cells. The use of organic molecules or polymers which can be tuned and spin-coated into the PSC structure (Fig. 7e) presents a promising approach. Studies investigating the uptake of I2 within these compounds have reported that the ability to bind to iodine can be improved via the presence of an extended π-system or the addition of Lewis-base functionality such as amines and thiols.202–205 To this end, the use of D–π–A porphyrin derivatives has been shown to enhance stability by mitigating both Pb0 and iodine when integrated into PSCs.206 Luo et al. demonstrated the use of small bipyridine cages (BPy-cage) which bind to I2via an I–N Lewis-base interaction. The Bpy-cage showed remarkable I2 loading capacities of up to 3230 mg g−1. Likewise, extended conjugated systems were recently used by Wang et al. to form a range of polymeric aromatic framework (PAF) iodine scavengers with adsorption coefficients of up to 289 mg g−1.203 Interestingly, the PAF utilises a triphenylamine core, similar to that used in organic HTLs such as poly(triaryl amine) (PTAA) and poly(N,N′-bis-4-butylphenyl-N,N′-bisphenyl)benzidine (Poly-TPD). This structural similarity to pre-existing PSC materials is promising for further development and integration into PSCs if the PAFs can be spin-coated sufficiently thinly as an interlayer. The recent functionalisation of triphenylamines for iodine capture is also highly promising for the development of new polymeric organic semiconductor transport layers which allow for carrier extraction while also preventing iodine depletion of the active layer. Early work on developing iodine confinement layers has already shown promising results with copper(II), Zinc(II) and cobalt(II) porphyrin layers improving hole extraction while significantly improving light and thermal stability via means of coordination to I2 (Fig. 7f–h).207–209 These studies demonstrate the opportunities afforded by both the use of transition metal complexes and iodine-capturing interlayers to encapsulate the PSC architecture and beyond.
The integration of iodine sorbents as interlayers to encapsulate the perovskite active layer offers an another alternative strategy to managing iodine chemistry within the device. To this end, several classes of materials have been developed for iodine capture including those based on zeolites,210 chalcogels (chalcogenide aerogels),141 and metal/covalent-organic frameworks (MOFs/COFs).142,211–213 Through drawing small quantities of iodine out of the perovskite cell, before encouraging redox chemistry and reintroduction of I− the build-up of corrosive quantities of I2 within the active layer can be managed.38,42,49 This effect was recently discussed by Bitton et al. who noted that the gradual loss of I2 leads to an irreversible degradation of the device on the time scale of months to years while concentrated I2 build-up within the active layer could lead to degradation on the time scale of days.36 In such cases, it may be preferable to neutralise iodine at the interfaces via design of an interlayer rather than within the bulk through incorporation as an additive species. This is most prevalent in the case of tin-perovskites where Sn2+ sites are highly sensitive to I2-induced oxidation leading to rapid degradation into SnI4. As an alternative approach, low-permeability metal oxide materials can be used to form protective barriers which can bind to iodine outside the active layer to protect the device constituents and minimise electrochemical oxidation. Al2O3, a well-established insulating scaffold material in PSCs is known to undergo surface physisorption to I2.214,215 This imparts promising iodine-blocking properties while physisorption to the interface rather than chemisorption, combined with good steric access to bound I2, allows for the design of reductive processes.215,216 It has also been suggested that Al2O3, MgO and La2O3 may also reduce physisorbed I2 to I−, possibly via under-coordinated surface O2− anions acting as electron donors.214,215 Indeed, the development of transition metal oxides for oxygen reduction reaction purposes could be noteworthy for the development of I2-confinement layers suitable for use in perovskite optoelectronics.217 Recent studies using Al2O3 have demonstrated impressive efficiencies reaching 25.5% and demonstrating improved operational stability when using Al2O3 compared to using self-assembling monolayers (SAMs) alone, demonstrating their active stabilising role.218
The inclusion of reducing additives or regenerative redox pathways within the PSC stack imparts clear measurable improvements in both device stability and photovoltaic performance. Nevertheless, to reduce the strain on processes which seek to remove and subsequently reduce iodine it is essential to minimise the generation of iodine at source. Specifically, only when I2-generated processes can be sufficiently minimised and managed can a balance be achieved where the oxidation of iodide is matched by the reduction of iodine enabling devices to operate sustainably. As such, maintaining a steady state where the reduction of I2 can match the oxidation of I− is essential for cells that can theoretically operate perpetually. The most significant unavoidable operational stimuli for iodine generation are the presence of electrical bias and illumination. The evolution of I2via the electrochemical oxidation of iodide ions under operation has been reported as a significant degradation pathway in Pb-based PSCs. It is noteworthy that similar electrochemical oxidation is also likely to occur within tin perovskite owing to the low threshold voltage (0.2 V μm−1) for such redox chemistry.85 The formation of I2 within tin-perovskites under electrical bias may also contribute to the instability of tin PSCs under operation in inert atmospheres. The effect of electrochemical oxidation is amplified within light-emitting devices, where perovskites have been hailed as promising tunable emitters, capable of high external quantum efficiencies. In this application, the applied voltage bias and current are higher than that of PSCs, increasing the rate of iodine generation as a function of applied bias.87 One solution to this issue may be to reduce the mobility of I− ions, preventing migration to the electrodes where oxidation occurs. Bai et al. reported that the placing of 2D layers at the perovskite interfaces can block the path of mobile I− preventing the accumulation of I− at the perovskite interface.2,219,220 Another solution involves the use of additives such as fluorinated aromatics that encourage halogen-halogen interactions.221 Likewise, the formation of localized regions of Br-rich 3D perovskite close to the interface via a secondary spin coating step can help produce a perovskite interface more resilient to oxidation (Table 1).2,85 This approach has the additional benefit of reducing the impact of oxygen and moisture on the active layer and potentially preventing back-recombination across the interface.2 Zhao et al. demonstrated impressive stability by utilizing a chloride-rich 2D-perovskite layer (Cs2PbI2Cl2) on top of CsPbI3 perovskite to mitigate iodine migration into the CuSCN hole transport layer.222 The authors found that preventing iodide anions from entering the HTL, even after a period of 2000 hours, enabled impressive improvements in stability under a range of temperature and humidity conditions. Furthermore, by producing a secondary perovskite layer, excess surface PbI2 can be converted into a perovskite phase, preventing photolysis to iodine. This was demonstrated by Liang et al., who prepared PSCs with a 5% PbI2 excess and showed an improvement in stability upon treatment with n-Butylammonium bromide (nBABr).66
Similarly, the presence of mobile iodide ions combined with the intrinsic free hole population contributes to the formation of I2via the activation of Ii+/Ii− interstitial pairs. Khadka et al. reported that reducing the concentration of free holes through an efficient HTL interface leads to lower yields of I2 formation.115 Consequently, the choice of transport layers has a significant influence on the rate of I− oxidation (Fig. 7i). Furthermore, the need to dope many organic semiconductors such as Spiro-OMeTAD can further exacerbate oxidation through ionic interdiffusion drawing halide anions within the HTL.136 This is particularly damaging owing to the high hole density within the HTL, with simulations revealing significantly poorer device performance when iodide is permitted to permeate within the HTL.36 Recently, we demonstrated that the choice of HTL has a significant impact on the stability of tin PSCs.38 This study highlighted a link between efficient hole extraction and improved stability of the tin perovskite layer. Better hole extraction at the perovskite/HTL interface likely leads to fewer free holes available to oxidise iodide (Fig. 7j and k). For this reason, it is essential that the HTL interface has good intrinsic stability and maintains efficient extraction of holes through an energetically aligned interface to produce devices with better longevity.115 In this regard, inorganic HTLs which are less reactive to halogens than organics, are likely to improve resilience to I2 for long-term device stability. The choice of transport layer can also serve in a secondary capacity as a UV filter for the perovskite, reducing superoxide formation and photolysis of metal–halide bonds. While metal oxides such as TiO2 and SnO2 can absorb within this range, modification is required to prevent a photocatalysis effect encouraging halide oxidation. Hang et al. successfully mitigated the photocatalytic effects of SnO2 by designing a localized chloride-rich perovskite phase at the interface. The strong Pb–Cl bond in the chloride-rich phase offered protection against photocatalytic iodine formation and ultimately improved device stability, retaining >80% PCE following 500 h under UV irradiation (100 mW cm−2, 365 nm).69 Alternatively, low-cost filtering of UV light can be achieved by treating metal oxides with photocurable fluoropolymers and silanes/siloxanes.224–226 Similarly, down-converters may offer effective protection against high-energy solar radiation by promoting energy transfer from the metal oxide to a secondary visible emitter (Fig. 7l).227
Essential to developing new strategies which combat I2 formation is the need for accurate characterisation techniques. In the case of iodine identification, this is made challenging by (i) the very low vapour pressure of I2, (ii) the presence of I− in the perovskite structure, and (iii) the lack of symmetry allowed vibrational modes.228 UV-visible absorbance spectroscopy has proven a popular approach, particularly when investigating the precursor solution chemistry, due to the high-molar extinction coefficient and distinctive red/pink colouration.26,38,55,229 This approach can be extended to solid films via means of submerging perovskite film(s) within an anhydrous insoluble solvent, often toluene, under inert conditions.26,38,55 If done with sufficient care, the concentration of extracted iodine can then be quantified via the Beer–Lambert law. X-ray photoelectron spectroscopy can also be used to complement more quantitative absorbance techniques, offering additional insights regarding the ratio of I−/I2 within solid-state films.49 Additionally, XPS can offer topographical spatial resolution on the distribution of I2 either via XPS mapping or in combination with etching techniques.67 Similarly, compositional analysis is also possible using mass spectrometry techniques. However, in the solid state, generating secondary ions from the solid-state film leads to a significant reduction of I2 to I−, making it difficult to distinguish between perovskite and I2. As such, mass spectrometry techniques are most effective when measuring the emission of volatile gases from a solid sample following some stimuli. It is noteworthy that Juarez-Perez et al. demonstrated that MAPbI3 inherently releases iodine when placed under vacuum.67 As such, when using vacuum-based techniques it is pertinent to note the presence of potential experimental artefacts by virtue of the low vapour pressure. This means that vacuum-based measurement techniques will produce an underestimate of the initial iodine concentration. Nevertheless, once degassed, XPS and Mass spectrometry techniques under a vacuum are highly effective tools for identifying I2 evolution. Such techniques have previously been used to identify the formation of I2 under stimuli such as light, temperature and electronic bias.67,114,120 Another approach to monitor iodine generation under operation is by measuring the resistance across a metal electrode of a known distance. This approach was effectively used by Li et al. to give insights into the conversion of conductive Ag into insulating AgI and presents a useful test method to investigate the formation and confinement of volatile iodine as a function of time.125 Such measurements provide useful metrics in stabilising high-performance metal electrodes, a critical point of failure in high-performing perovskite device longevity.230
Conclusion
The self-assembly of a semiconducting material from the combination of ionic salts offers many benefits including solution processability, low production cost, high-throughput commercialisation and compositional tunability. Nevertheless, producing an ionic semiconductor from a lattice of charged halide and organic constituents inevitably raises challenges associated with reactivity and chemical stability. This can readily be seen via comparison with pre-existing silicon and III–V technologies which use covalent bonding, enabling shelf-lives measurable in decades.231 To this end, numerous studies have detailed the chemical instability associated with the perovskite ionic lattice, of note, the organic cation stability,222,232,233 phase instabilities,234–236 and high aqueous solubility.237 As summarised within this review, the oxidation of I− to corrosive molecular iodine species can occur via several means and has far-reaching implications within the PSC stack. Furthermore, the low thermodynamic barrier of Sn(II) and Ge(II) to oxidation, creates additional challenges in the development of lead-free perovskites and all perovskite tandem devices.Addressing the formation of iodine is a complex issue which requires a deeper understanding of the thermodynamic driving force of the iodide–iodine–triiodide equilibrium. This includes understanding the effect of acidity, mobility and reductants on the equilibrium constant and the effects of thermodynamic quantities of iodine. Such studies will likely lead the way in developing new approaches to tackle the challenge of iodine perhaps through stabilising the iodide X-site, further thermodynamically disfavouring the side of the oxidation products. This could be achieved through new advances in halogen bonding and stabilisation of terminal X-halide sites. We anticipate that such strategies are most effective when used to encourage, kinetically or otherwise, redox chemistry between Pb0 and I2. Indeed, by virtue of the I−/I3−/I2 dynamic equilibrium, careful design of iodine sorbents is required such that iodide is not leeched from the perovskite lattice either directly via the sorbent or influencing the iodide–iodine–triiodide equilibrium towards the side of oxidation as per Le Chatelier's principle.100 In this regard, the use of easy-to-disperse, small molecules with a low iodine capacity such as crown ethers, porphyrins or oligosaccharides is desirable, preventing the accumulation of large quantities of iodine. Furthermore, the use of small sorbents can better sterically facilitate redox chemistry with Pb0 or other redox shuttles and reductants helping establish an active restorative pathway to compete with degradation. The search for new redox shuttles to regenerate Pb2+ and I− of the active layer draws an interesting parallel to the previous work conducted within DSSC technologies for the replacement of iodine electrolytes. These studies, combined with a wealth of interdisciplinary studies on the design of transition metal complexes may provide a useful starting point for controlling the redox chemistry within PSCs.189,238
However, the approach discussed above may be unsuitable, in its current state, for use in tin-perovskites owing to the absence of a species to undergo oxidation (Pb0) and severe adverse chemistry to I2 within the active layer. Nevertheless, in the case of non-lead B-site cations (Sn, Ge), exploration of alternative regenerative redox pathways could be developed to facilitate sustainable disproportionation reactions to oppose degradation. An example of this could be using weak reductants with redox potentials sensitive to the chemical environment such as acidity, where HI is formed during oxidation. Alternatively, in such instances, the use of irreversible reductants may be effective if the formation mechanisms of I2 are sufficiently slowed. To date, modified-hydrazine derivatives have stood out as particularly promising. Another approach is to disfavour oxidation, this can be done by management of HI formed during degradation. Likewise, stabilisation of reactive halide defects and under passivation of under-coordination is crucial to thermodynamically stabilise the reduced form of I−.38 It is also pertinent to note, that in tin-based perovskites iodine acts catalytically in a self-sustaining degradation loop which regenerates greater quantities of iodine with each generation.38,42 Consequently, the removal of small quantities of iodine will yield dramatic improvements in perovskites with B2+ sites prone to oxidation.
In recent years, steady progress has been in minimising iodide migration, a key process in the formation of iodine. Similarly, the modification of robust inorganic transport layers to provide simple, and low-cost UV protection is highly promising. We note that the identification of new high-performing electrode materials has been a particularly difficult hurdle to stable high-performing PSCs. Conventional electrode materials such as gold, silver and copper are highly sensitive to oxidation with iodine vapours, undergoing visible discolouration and limiting operational stability.239,240 Conversely, while more stable, carbon alternatives currently are yet to match the performance of metal-based electrodes. This failure mechanism could be managed using iodine-blocking layers at the interface or strategies which aim to confine I2 within the active layer with molecular sorbents such as MOFs or transition metal porphyrins to prevent the distribution of volatile and corrosive halogen vapour.36 To this end, we note the rapid recent development of large covalent organic frameworks as highly effective iodine sorbents.241,242 This class of I2 scavenger offers useful tunability over pore size and functional groups to improve distribution close to grains, tune capacity and maintain pores large enough to sterically allow reduction within the cage. Very recently, Xie et al. demonstrated a new COF named COF-TAPT capable of storing 2380 mg g−1 of I2 when stored at 25 °C under dry conditions.242 Such large loading capacities may enable the integration of monolayers to encapsulate the perovskite layer and confine iodine from mobilising within the PSC stack while the high porosity enables the presence of redox chemistry to occur. When coupled with an implemented reduction pathway such as a redox shuttle, irreversible reductant or reaction with Pb0, the use of iodine confinement strategies could circumvent the need for lower-efficiency inert electrodes.
Drawing inspiration from the fields of radioiodine sequestration, tuneable COFs, iodine retention within electrochemical cells and polymeric iodophors has already seen promising initial success at addressing the sensitive redox chemistry present in PSCs.195,198 Equally, in recent years studies identifying new mechanisms of I2 generation both within lead and tin have enabled a better understanding of the destructive redox chemistry that takes place within ionic perovskite solar cells under operation. Of particular interest is the effect of bias voltage on electrochemical oxidation within tin PSCs, yet to be addressed within the field and a possible contributor to low reported MPP stability even under inert atmospheres.49 Similarly, the effect of illumination on iodine radical formation has yet to be fully explored and requires greater understanding. Perhaps most important is gaining a greater understanding of the equilibria, and its dependencies, between the oxidised and reduced forms of iodine within PSCs. Indeed, only if I2-generating processes can be sufficiently minimised can a regenerative balance of oxidation and reduction be designed to facilitate sustainable control of the redox chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. H gratefully acknowledges funding from the Engineering and Physical Sciences Research Council (EPSRC, EP/X012344/1).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- T. Webb, S. J. Sweeney and W. Zhang, Device Architecture Engineering: Progress toward Next Generation Perovskite Solar Cells, Adv. Funct. Mater., 2021, 31, 2103121 CrossRef CAS.
- J. Park, et al., Controlled growth of perovskite layers with volatile alkylammonium chlorides, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Efficient Hybrid Solar Cells Based on Meso-Superstructured Organometal Halide Perovskites, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- A. Walsh, Principles of Chemical Bonding and Band Gap Engineering in Hybrid Organic–Inorganic Halide Perovskites, J. Phys. Chem. C, 2015, 119, 5755–5760 CrossRef CAS PubMed.
- B. Park, et al., Chemical engineering of methylammonium lead iodide/bromide perovskites: tuning of opto-electronic properties and photovoltaic performance, J. Mater. Chem. A, 2015, 3, 21760–21771 RSC.
- J. Cao, et al., Preparation of Lead-free Two-Dimensional-Layered (C8H17NH3)2SnBr4 Perovskite Scintillators and Their Application in X-ray Imaging, ACS Appl. Mater. Interfaces, 2020, 12, 19797–19804 CrossRef CAS PubMed.
- L. Pan, S. Shrestha, N. Taylor, W. Nie and L. R. Cao, Determination of X-ray detection limit and applications in perovskite X-ray detectors, Nat. Commun., 2021, 12, 5258 CrossRef CAS PubMed.
- H. Wei and J. Huang, Halide lead perovskites for ionizing radiation detection, Nat. Commun., 2019, 10, 1066 CrossRef PubMed.
- X.-K. Liu, et al., Metal halide perovskites for light-emitting diodes, Nat. Mater., 2021, 20, 10–21 CrossRef CAS PubMed.
- Q. Van Le, H. W. Jang and S. Y. Kim, Recent Advances toward High-Efficiency Halide Perovskite Light-Emitting Diodes: Review and Perspective, Small Methods, 2018, 2, 1700419 CrossRef.
- A. Liu, et al., High-performance inorganic metal halide perovskite transistors, Nat. Electron., 2022, 5, 78–83 CrossRef CAS.
- C. K. Møller, Crystal Structure and Photoconductivity of Cæsium Plumbohalides, Nature, 1958, 182, 1436 CrossRef.
- D. Weber, CH3NH3PbX3, ein Pb(II)-System mit kubischer Perowskitstruktur/CH3NH3PbX3, a Pb(II)-System with Cubic Perovskite, Structure, 1978, 33, 1443–1445 Search PubMed.
- O. Knop, R. E. Wasylishen, M. A. White, T. S. Cameron and M. J. M. V. Oort, Alkylammonium lead halides. Part 2. CH3NH3PbX3 (X = Cl, Br, I) perovskites: cuboctahedral halide cages with isotropic cation reorientation, Can. J. Chem., 1990, 68, 412–422 CrossRef CAS.
- D. Kumar and M. Johari, Characteristics of silicon crystal, its covalent bonding and their structure, electrical properties, uses, AIP Conf. Proc., 2020, 2220, 040037 CrossRef CAS.
- G. Ackland, Semiempirical model of covalent bonding in silicon, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 10351–10355 CrossRef CAS PubMed.
- Y. Kim, et al., III–V colloidal nanocrystals: control of covalent surfaces, Chem. Sci., 2020, 11, 913–922 RSC.
- R. M. Feenstra and J. A. Stroscio, 5.3. Gallium Arsenide, in Methods in Experimental Physics, ed. J. A. Stroscio & W. J. Kaiser, Academic Press, 1993, vol. 27, pp. 251–276 Search PubMed.
- P. Li, et al., Phase Pure 2D Perovskite for High-Performance 2D–3D Heterostructured Perovskite Solar Cells, Adv. Mater., 2018, 30, 1805323 CrossRef PubMed.
- E.-B. Kim, M. S. Akhtar, H.-S. Shin, S. Ameen and M. K. Nazeeruddin, A review on two-dimensional (2D) and 2D-3D multidimensional perovskite solar cells: Perovskites structures, stability, and photovoltaic performances, J. Photochem. Photobiol., C, 2021, 48, 100405 CrossRef CAS.
- N. J. Jeon, et al., Compositional engineering of perovskite materials for high-performance solar cells, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, The emergence of perovskite solar cells, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Lead-free solid-state organic–inorganic halide perovskite solar cells, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS.
- Z. Zhu, et al., Interaction of Organic Cation with Water Molecule in Perovskite MAPbI 3: From Dynamic Orientational Disorder to Hydrogen Bonding, Chem. Mater., 2016, 28, 7385–7393 CrossRef CAS.
- T. Leijtens, R. Prasanna, A. Gold-Parker, M. F. Toney and M. D. McGehee, Mechanism of Tin Oxidation and Stabilization by Lead Substitution in Tin Halide Perovskites, ACS Energy Lett., 2017, 2, 2159–2165 CrossRef CAS.
- J. Ye, et al., Enhanced Moisture Stability of Perovskite Solar Cells With Mixed-Dimensional and Mixed-Compositional Light-Absorbing Materials, Sol. RRL, 2017, 1, 1700125 CrossRef.
- M. V. Khenkin, et al., Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures, Nat. Energy, 2020, 5, 35–49 CrossRef.
- S. Kim, et al., Relationship between ion migration and interfacial degradation of CH3NH3PbI3 perovskite solar cells under thermal conditions, Sci. Rep., 2017, 7, 1200 CrossRef PubMed.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, 6.5% efficient perovskite quantum-dot-sensitized solar cell, Nanoscale, 2011, 3, 4088 RSC.
- S. D. Stranks, et al., Electron–Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- H.-S. Kim, et al., Lead Iodide Perovskite Sensitized All-Solid-State Submicron Thin Film Mesoscopic Solar Cell with Efficiency Exceeding 9%, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- G. Grancini, et al., One-Year stable perovskite solar cells by 2D/3D interface engineering, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour, Nat. Energy, 2016, 2, 1–8 Search PubMed.
- J. T. DuBose, P. S. Mathew, J. Cho, M. Kuno and P. V. Kamat, Modulation of Photoinduced Iodine Expulsion in Mixed Halide Perovskites with Electrochemical Bias, J. Phys. Chem. Lett., 2021, 12, 2615–2621 CrossRef CAS PubMed.
- S. Bitton and N. Tessler, Perovskite ionics – elucidating degradation mechanisms in perovskite solar cells via device modelling and iodine chemistry, Energy Environ. Sci., 2023, 16, 2621–2628 RSC.
- X. Jiang, et al., One-Step Synthesis of SnI2 (DMSO)x Adducts for High-Performance Tin Perovskite Solar Cells, J. Am. Chem. Soc., 2021, 143, 10970–10976 CrossRef CAS PubMed.
- L. Lanzetta, et al., Degradation mechanism of hybrid tin-based perovskite solar cells and the critical role of tin (IV) iodide, Nat. Commun., 2021, 12, 2853 CrossRef CAS PubMed.
- G. Li, et al., Ionic Liquid Stabilizing High-Efficiency Tin Halide Perovskite Solar Cells, Adv. Energy Mater., 2021, 11, 2101539 CrossRef CAS.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, All-solid-state dye-sensitized solar cells with high efficiency, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- N. K. Noel, et al., Lead-free organic–inorganic tin halide perovskites for photovoltaic applications, Energy Environ. Sci., 2014, 7, 3061–3068 RSC.
- L. Lanzetta, T. Webb, J. M. Marin-Beloqui, T. J. Macdonald and S. A. Haque, Halide Chemistry in Tin Perovskite Optoelectronics: Bottlenecks and Opportunities, Angew. Chem., Int. Ed., 2023, 62, e202213966 CrossRef CAS PubMed.
- Y. Gao, Y. Hu, C. Yao and S. Zhang, Recent Advances in Lead-Safe Perovskite Solar Cells, Adv. Funct. Mater., 2022, 32(52), 2208225 CrossRef CAS.
- J. Zhou, et al., Chemo-thermal surface dedoping for high-performance tin perovskite solar cells, Matter, 2022, 5, 683–693 CrossRef CAS.
- S. Gu, et al., Tin and Mixed Lead–Tin Halide Perovskite Solar Cells: Progress and their Application in Tandem Solar Cells, Adv. Mater., 2020, 32, 1907392 CrossRef CAS PubMed.
- N. Sun, et al., Architecture of p-i-n Sn-Based Perovskite Solar Cells: Characteristics, Advances, and Perspectives, ACS Energy Lett., 2021, 6, 2863–2875 CrossRef CAS.
- T. Dohi and Y. Kita, Oxidizing Agents, Iodine Chemistry and Applications, John Wiley & Sons, Ltd, 2014, pp. 277–301 DOI:10.1002/9781118909911.ch16.
- H. Yao, et al., Strategies for Improving the Stability of Tin-Based Perovskite (ASnX3) Solar Cells, Adv. Sci., 2020, 7, 1903540 CrossRef CAS PubMed.
- J. Sanchez-Diaz, et al., Tin perovskite solar cells with >1,300 h of operational stability in N2 through a synergistic chemical engineering approach, Joule, 2022, 6(4), 861–883 CrossRef CAS PubMed.
- B. Li, et al., Efficient and Stable Tin Perovskite Solar Cells by Pyridine-Functionalized Fullerene with Reduced Interfacial Energy Loss, Adv. Funct. Mater., 2022, 32, 2205870 CrossRef CAS.
- D. L. Staebler, R. S. Crandall and R. Williams, Stability of n-i-p amorphous silicon solar cells, Appl. Phys. Lett., 1981, 39, 733–735 CrossRef CAS.
- Y. Hu, et al., Stable Large-Area (10 × 10 cm2) Printable Mesoscopic Perovskite Module Exceeding 10% Efficiency, Sol. RRL, 2017, 1, 1600019 CrossRef.
- J. Pascual, et al., Origin of Sn(ii) oxidation in tin halide perovskites, Mater. Adv., 2020, 1, 1066–1070 RSC.
- M. I. Saidaminov, et al., Conventional Solvent Oxidizes Sn(II) in Perovskite Inks, ACS Energy Lett., 2020, 5, 1153–1155 CrossRef CAS.
- S. Chen, X. Xiao, H. Gu and J. Huang, Iodine reduction for reproducible and high-performance perovskite solar cells and modules, Sci. Adv., 2021, 7(10), eabe8130 CrossRef CAS PubMed.
- M. Benavides-Garcia and K. Balasubramanian, Bond energies, ionization potentials, and the singlet–triplet energy separations of SnCl2, SnBr2, SnI2, PbCl2, PbBr2, PbI2, and their positive ions, J. Chem. Phys., 1994, 100, 2821–2830 CrossRef CAS.
- G. Tumen-Ulzii, et al., Detrimental Effect of Unreacted PbI2 on the Long-Term Stability of Perovskite Solar Cells, Adv. Mater., 2020, 32, 1905035 CrossRef CAS PubMed.
- F. Liu, et al., Is Excess PbI2 Beneficial for Perovskite Solar Cell Performance?, Adv. Energy Mater., 2016, 6, 1502206 CrossRef.
- A. F. Akbulatov, et al., Temperature Dynamics of MAPbI3 and PbI2 Photolysis: Revealing the Interplay between Light and Heat, Two Enemies of Perovskite Photovoltaics, J. Phys. Chem. Lett., 2021, 12, 4362–4367 CrossRef CAS PubMed.
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Impact of H2O on organic–inorganic hybrid perovskite solar cells, Energy Environ. Sci., 2017, 10, 2284–2311 RSC.
- M. Wang, Y. Feng, J. Bian, H. Liu and Y. Shi, A comparative study of one-step and two-step approaches for MAPbI3 perovskite layer and its influence on the performance of mesoscopic perovskite solar cell, Chem. Phys. Lett., 2018, 692, 44–49 CrossRef CAS.
- M. Yavari, et al., A synergistic Cs2CO3 ETL treatment to incorporate Cs cation into perovskite solar cells via two-step scalable fabrication, J. Mater. Chem. C, 2021, 9, 4367–4377 RSC.
- A. Marchioro, et al., Unravelling the mechanism of photoinduced charge transfer processes in lead iodide perovskite solar cells, Nat. Photonics, 2014, 8, 250–255 CrossRef CAS.
- Q. Chen, et al., Controllable Self-Induced Passivation of Hybrid Lead Iodide Perovskites toward High Performance Solar Cells, Nano Lett., 2014, 14, 4158–4163 CrossRef CAS PubMed.
- J. F. Verwey, Time and intensity dependence of the photolysis of lead halides, J. Phys. Chem. Solids, 1970, 31, 163–168 CrossRef CAS.
- J. Liang, et al., Origins and influences of metallic lead in perovskite solar cells, Joule, 2022, 6, 816–833 CrossRef CAS.
- E. J. Juarez-Perez, et al., Photodecomposition and thermal decomposition in methylammonium halide lead perovskites and inferred design principles to increase photovoltaic device stability, J. Mater. Chem. A, 2018, 6, 9604–9612 RSC.
- J. D. McGettrick, et al., Sources of Pb(0) artefacts during XPS analysis of lead halide perovskites, Mater. Lett., 2019, 251, 98–101 CrossRef CAS.
- P. Hang, et al., An Interlayer with Strong Pb-Cl Bond Delivers Ultraviolet-Filter-Free, Efficient, and Photostable Perovskite Solar Cells, iScience, 2019, 21, 217–227 CrossRef CAS PubMed.
- S. Ito, S. Tanaka, K. Manabe and H. Nishino, Effects of Surface Blocking Layer of Sb2S3 on Nanocrystalline TiO2 for CH3NH3PbI3 Perovskite Solar Cells, J. Phys. Chem. C, 2014, 118, 16995–17000 CrossRef CAS.
- A. Aziz, et al., Understanding the Enhanced Stability of Bromide Substitution in Lead Iodide Perovskites, Chem. Mater., 2020, 32, 400–409 CrossRef CAS.
- L. McGovern, M. H. Futscher, L. A. Muscarella and B. Ehrler, Understanding the Stability of MAPbBr3 versus MAPbI3: Suppression of Methylammonium Migration and Reduction of Halide Migration, J. Phys. Chem. Lett., 2020, 11, 7127–7132 CrossRef CAS PubMed.
- X. Fu, et al., Halogen-halogen bonds enable improved long-term operational stability of mixed-halide perovskite photovoltaics, Chem, 2021, 7, 3131–3143 CAS.
- M. L. Ball, J. V. Milić and Y.-L. Loo, The Emerging Role of Halogen Bonding in Hybrid Perovskite Photovoltaics, Chem. Mater., 2022, 34, 2495–2502 CrossRef CAS.
- G. Popov, et al., Atomic Layer Deposition of PbI2 Thin Films, Chem. Mater., 2019, 31, 1101–1109 CrossRef CAS.
- C. Mortan, et al., Preparation of methylammonium lead iodide (CH3NH3PbI3) thin film perovskite solar cells by chemical vapor deposition using methylamine gas (CH3NH2) and hydrogen iodide gas, Energy Sci. Eng., 2020, 8, e734 Search PubMed.
- A. Senocrate, G. Y. Kim, M. Grätzel and J. Maier, Thermochemical Stability of Hybrid Halide Perovskites, ACS Energy Lett., 2019, 4, 2859–2870 CrossRef CAS.
- N. Aristidou, et al., Fast oxygen diffusion and iodide defects mediate oxygen-induced degradation of perovskite solar cells, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- Y. Ouyang, et al., Photo-oxidative degradation of methylammonium lead iodide perovskite: mechanism and protection, J. Mater. Chem. A, 2019, 7, 2275–2282 RSC.
- X. Feng, et al., Photon-generated carriers excite superoxide species inducing long-term photoluminescence enhancement of MAPbI3 perovskite single crystals, J. Mater. Chem. A, 2017, 5, 12048–12053 RSC.
- N. Aristidou, et al., The Role of Oxygen in the Degradation of Methylammonium Lead Trihalide Perovskite Photoactive Layers, Angew. Chem., 2015, 127, 8326–8330 CrossRef.
- N. Aristidou, C. Eames, M. S. Islam and S. A. Haque, Insights into the increased degradation rate of CH3NH3PbI3 solar cells in combined water and O2 environments, J. Mater. Chem. A, 2017, 5, 25469–25475 RSC.
- D. Bryant, et al., Light and oxygen induced degradation limits the operational stability of methylammonium lead triiodide perovskite solar cells, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- L. Lanzetta, N. Aristidou and S. A. Haque, Stability of Lead and Tin Halide Perovskites: The Link between Defects and Degradation, J. Phys. Chem. Lett., 2020, 11, 574–585 CrossRef CAS PubMed.
- Y. Yuan, et al., Electric-Field-Driven Reversible Conversion Between Methylammonium Lead Triiodide Perovskites and Lead Iodide at Elevated Temperatures, Adv. Energy Mater., 2016, 6, 1501803 CrossRef.
- L. A. Frolova, N. N. Dremova and P. A. Troshin, The chemical origin of the p-type and n-type doping effects in the hybrid methylammonium–lead iodide (MAPbI3) perovskite solar cells, Chem. Commun., 2015, 51, 14917–14920 RSC.
- G. F. Samu, et al., Electrochemical Hole Injection Selectively Expels Iodide from Mixed Halide Perovskite Films, J. Am. Chem. Soc., 2019, 141, 10812–10820 CrossRef CAS PubMed.
- L. K. Ono, Y. Qi and S. (Frank) Liu, Progress toward Stable Lead Halide Perovskite Solar Cells, Joule, 2018, 2, 1961–1990 CrossRef CAS.
- Z. Li, et al., Extrinsic ion migration in perovskite solar cells, Energy Environ. Sci., 2017, 10, 1234–1242 RSC.
- D. D. Wagman, The NBS Tables of Chemical and Thermodynamic Properties: Selected Values for Inorganic and C1 and C2 Organic Substances in SI Units, National Bureau of Standards, Washington, DC, 1982, vol. 11 Search PubMed.
- Z. Xu, R. A. Kerner, J. J. Berry and B. P. Rand, Iodine Electrochemistry Dictates Voltage-Induced Halide Segregation Thresholds in Mixed-Halide Perovskite Devices, Adv. Funct. Mater., 2022, 32, 2203432 CrossRef CAS.
- J. Paquette and B. L. Ford, Iodine chemistry in the +1 oxidation state. I. The electronic spectra of OI−, HOI, and H2OI+, Can. J. Chem., 1985, 63, 2444–2448 CrossRef CAS.
- W. J. Husa, The preparation of diluted hydriodic acid and syrup of hydriodic acid, J. Am. Pharm. Assoc., 1931, 20, 759–762 CAS.
- J. M. Robinson, P. T. Herndon, P. L. Holland and L. D. Marrufo, Regeneration and Recovery of Hydriodic Acid after Reduction of Polyols to Fuels1, Org. Process Res. Dev., 1999, 3, 352–356 CrossRef CAS.
- D. Sun, et al., Chemical Reduction of Iodine Impurities and Defects with Potassium Formate for Efficient and Stable Perovskite Solar Cells, Adv. Funct. Mater., 2023, 2303225 CrossRef CAS.
- S. Thampy, B. Zhang, J.-G. Park, K.-H. Hong and J. W. P. Hsu, Bulk and interfacial decomposition of formamidinium iodide (HC(NH2)2I) in contact with metal oxide, Mater. Adv., 2020, 1, 3349–3357 RSC.
- W. Haynes, D. Lide and B. Thomas, CRC Handbook of Chemistry and Physics: A Ready Reference Book of Chemical and Physical Data, CRC Press, 2016, vol. 1, p. 2017 Search PubMed.
- A. Saiz-Lopez, R. W. Saunders, D. M. Joseph, S. H. Ashworth and J. M. C. Plane, Absolute absorption cross-section and photolysis rate of I2, Atmos. Chem. Phys., 2004, 4, 1443–1450 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Characteristics of the Iodide/Triiodide Redox Mediator in Dye-Sensitized Solar Cells, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- S. Tan, et al., Shallow Iodine Defects Accelerate the Degradation of α-Phase Formamidinium Perovskite, Joule, 2020, 4, 2426–2442 CrossRef CAS.
- Q. Hu, et al., In situ dynamic observations of perovskite crystallisation and microstructure evolution intermediated from [PbI6]4− cage nanoparticles, Nat. Commun., 2017, 8, 15688 CrossRef CAS PubMed.
- B. Li, Q. Dai, S. Yun and J. Tian, Insights into iodoplumbate complex evolution of precursor solutions for perovskite solar cells: from aging to degradation, J. Mater. Chem. A, 2021, 9, 6732–6748 RSC.
- I. Hwang, Challenges in Controlling the Crystallization Pathways and Kinetics for Highly Reproducible Solution-Processing of Metal Halide Perovskites, J. Phys. Chem. C, 2023, 127, 24011–24026 CrossRef CAS.
- J. C. Jr Hamill, J. Schwartz and Y.-L. Loo, Influence of Solvent Coordination on Hybrid Organic–Inorganic Perovskite Formation, ACS Energy Lett., 2018, 3, 92–97 CrossRef CAS.
- G. S. Shin, S.-G. Kim, Y. Zhang and N.-G. Park, A Correlation between Iodoplumbate and Photovoltaic Performance of Perovskite Solar Cells Observed by Precursor Solution Aging, Small Methods, 2020, 4, 1900398 CrossRef CAS.
- O. Shargaieva, et al., Hybrid perovskite crystallization from binary solvent mixtures: interplay of evaporation rate and binding strength of solvents, Mater. Adv., 2020, 1, 3314–3321 RSC.
- J. Kim, et al., Synthesis of stable iodoplumbate and perovskite for efficient annealing-free device and long-term storage, SusMat, 2023, 3, 821–833 CrossRef CAS.
- J. Kim, et al., Unveiling the Relationship between the Perovskite Precursor Solution and the Resulting Device Performance, J. Am. Chem. Soc., 2020, 142, 6251–6260 CrossRef CAS PubMed.
- Z. Liu, et al., Chemical Reduction of Intrinsic Defects in Thicker Heterojunction Planar Perovskite Solar Cells, Adv. Mater., 2017, 29, 1606774 CrossRef PubMed.
- A. Senocrate and J. Maier, Solid-State Ionics of Hybrid Halide Perovskites, J. Am. Chem. Soc., 2019, 141, 8382–8396 CrossRef CAS PubMed.
- G. Y. Kim, et al., Large tunable photoeffect on ion conduction in halide perovskites and implications for photodecomposition, Nat. Mater., 2018, 17, 445–449 CrossRef CAS PubMed.
- A. Zohar, et al., What Is the Mechanism of MAPbI3 p-Doping by I2? Insights from Optoelectronic Properties, ACS Energy Lett., 2017, 2, 2408–2414 CrossRef CAS.
- S. Meloni, G. Palermo, N. Ashari-Astani, M. Grätzel and U. Rothlisberger, Valence and conduction band tuning in halide perovskites for solar cell applications, J. Mater. Chem. A, 2016, 4, 15997–16002 RSC.
- F. Fu, et al., I 2 vapor-induced degradation of formamidinium lead iodide based perovskite solar cells under heat–light soaking conditions, Energy Environ. Sci., 2019, 12, 3074–3088 RSC.
- D. B. Khadka, Y. Shirai, M. Yanagida and K. Miyano, Insights into Accelerated Degradation of Perovskite Solar Cells under Continuous Illumination Driven by Thermal Stress and Interfacial Junction, ACS Appl. Energy Mater., 2021, 4, 11121–11132 CrossRef CAS.
- W. Li, J. Liu, F.-Q. Bai, H.-X. Zhang and O. V. Prezhdo, Hole Trapping by Iodine Interstitial Defects Decreases Free Carrier Losses in Perovskite Solar Cells: A Time-Domain Ab Initio Study, ACS Energy Lett., 2017, 2, 1270–1278 CrossRef CAS.
- W. Zhang, et al., Enhanced optoelectronic quality of perovskite thin films with hypophosphorous acid for planar heterojunction solar cells, Nat. Commun., 2015, 6, 10030 CrossRef CAS PubMed.
- L. Wang, et al., A Eu3+-Eu2+ ion redox shuttle imparts operational durability to Pb-I perovskite solar cells, Science, 2019, 363, 265–270 CrossRef CAS PubMed.
- S. Bitton and N. Tessler, Perovskite ionics – elucidating degradation mechanisms in perovskite solar cells via device modelling and iodine chemistry, Energy Environ. Sci., 2023, 16, 2621–2628 RSC.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour, Nat. Energy, 2016, 2, 1–8 Search PubMed.
- N. N. Udalova, et al., New Aspects of Copper Electrode Metamorphosis in Perovskite Solar Cells, J. Phys. Chem. C, 2020, 124, 24601–24607 CrossRef CAS.
- G. Jones and B. B. Kaplan, The Iodide, Iodine, Tri-Iodide Equilibrium And The Free Energy Of Formation Of Silver Iodide, J. Am. Chem. Soc., 1928, 50, 1845–1864 CrossRef CAS.
- B. J. Riley, S. Chong and C. L. Beck, Iodine Vapor Reactions with Pure Metal Wires at Temperatures of 100–139 °C in Air, Ind. Eng. Chem. Res., 2021, 60, 17162–17173 CrossRef CAS.
- S. I. Troyanov, T. Krahl and E. Kemnitz, Crystal structures of GaX3 (X = Cl, Br, I) and AlI3, Z. Kristallogr. - Cryst. Mater., 2004, 219, 88–92 CrossRef CAS.
- H. Li, et al., Probing the stability of perovskite solar cell under working condition through an ultra-thin silver electrode: Beyond the halide ion diffusion and metal diffusion, Chem. Eng. J., 2023, 458, 141405 CrossRef CAS.
- A. Wijesekara, et al., Enhanced Stability of Tin Halide Perovskite Photovoltaics Using a Bathocuproine—Copper Top Electrode, Adv. Energy Mater., 2021, 11, 2102766 CrossRef CAS.
- N. N. Shlenskaya, N. A. Belich, M. Grätzel, E. A. Goodilin and A. B. Tarasov, Light-induced reactivity of gold and hybrid perovskite as a new possible degradation mechanism in perovskite solar cells, J. Mater. Chem. A, 2018, 6, 1780–1786 RSC.
- A. A. Petrov, et al., A new formation strategy of hybrid perovskites via room temperature reactive polyiodide melts, Mater. Horiz., 2017, 4, 625–632 RSC.
- Y. Zhao, J. Wei, R. Vajtai, P. M. Ajayan and E. V. Barrera, Iodine doped carbon nanotube cables exceeding specific electrical conductivity of metals, Sci. Rep., 2011, 1, 83 CrossRef PubMed.
- L. Fagiolari and F. Bella, Carbon-based materials for stable, cheaper and large-scale processable perovskite solar cells, Energy Environ. Sci., 2019, 12, 3437–3472 RSC.
- X. H. Sun, et al., Photoelectron spectroscopic study of iodine- and bromine-treated indium tin oxides and their interfaces with organic films, Chem. Phys. Lett., 2003, 370, 425–430 CrossRef CAS.
- G. Tumen-Ulzii, et al., Understanding the Degradation of Spiro-OMeTAD-Based Perovskite Solar Cells at High Temperature, Sol. RRL, 2020, 4, 2000305 CrossRef CAS.
- Z. Zhuo, et al., Efficiency improvement of polymer solar cells by iodine doping, Solid-State Electron., 2011, 63, 83–88 CAS.
- T. R. Ohno, G. H. Kroll, J. H. Weaver, L. P. F. Chibante and R. E. Smalley, Doping of C60 with iodine, Nature, 1992, 355, 401 CrossRef.
- R. A. Kerner, et al., Organic Hole Transport Material Ionization Potential Dictates Diffusion Kinetics of Iodine Species in Halide Perovskite Devices, ACS Energy Lett., 2021, 6, 501–508 CrossRef CAS.
- L. Yuan, et al., A conformally bonded molecular interface retarded iodine migration for durable perovskite solar cells, Energy Environ. Sci., 2023, 16, 1597–1609 RSC.
- J. H. Hildebrand and H. A. Benesi, Interaction of Iodine with Aromatic Hydrocarbons, Nature, 1949, 164, 963 CrossRef.
- S. Stavber, M. Jereb and M. Zupan, Electrophilic Iodination of Organic Compounds Using Elemental Iodine or Iodides, Synthesis, 2008, 1487–1513 CrossRef CAS.
- G. Sumrell, B. M. Wyman, R. G. Howell and M. C. Harvey, Reaction Of Lower Olefins And Iodine In A Liquid Phase: Novel Preparation Of Alkene Iodohydrins, Can. J. Chem., 1964, 42, 2710–2712 CrossRef CAS.
- K. Čebular and S. Stavber, Molecular iodine as a mild catalyst for cross-coupling of alkenes and alcohols, Pure Appl. Chem., 2018, 90, 377–386 CrossRef.
- K. S. Subrahmanyam, et al., Chalcogenide Aerogels as Sorbents for Radioactive Iodine, Chem. Mater., 2015, 27, 2619–2626 CrossRef CAS.
- B. J. Riley, D. A. Pierce and J. Chun, Efforts to Consolidate Chalcogels with Adsorbed Iodine, 2013, PNNL-22678, 1097940, DOI:10.2172/1097940 , https://www.osti.gov/servlets/purl/1097940/.
- R. Pénélope, L. Campayo, M. Fournier, A. Gossard and A. Grandjean, Solid sorbents for gaseous iodine capture and their conversion into stable waste forms, J. Nucl. Mater., 2022, 563, 153635 CrossRef.
- Z. Zhang, et al., Revealing Superoxide-Induced Degradation in Lead-free Tin Perovskite Solar Cells, Energy Environ. Sci., 2022, 15(12), 5274–5283 RSC.
- Y. Zhang, et al., Highly Efficient Tin Perovskite Solar Cells via Suppressing Superoxide Generation, Sol. RRL, 2023, 7, 2200997 CrossRef CAS.
- X.-F. Zhang and X. Li, The photostability and fluorescence properties of diphenylisobenzofuran, J. Lumin., 2011, 131, 2263–2266 CrossRef CAS.
- T. Nakamura, et al., Sn(IV)-free tin perovskite films realized by in situ Sn(0) nanoparticle treatment of the precursor solution, Nat. Commun., 2020, 11, 3008 CrossRef CAS PubMed.
- M. Ozaki, et al., Solvent-Coordinated Tin Halide Complexes as Purified Precursors for Tin-Based Perovskites, ACS Omega, 2017, 2, 7016–7021 CrossRef CAS PubMed.
- T. Rath, et al., Photovoltaic properties of a triple cation methylammonium/formamidinium/phenylethylammonium tin iodide perovskite, J. Mater. Chem. A, 2019, 7, 9523–9529 RSC.
- F. Gu, et al., Improving Performance of Lead-Free Formamidinium Tin Triiodide Perovskite Solar Cells by Tin Source Purification, Sol. RRL, 2018, 2, 1800136 CrossRef.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- B.-B. Yu, et al., Oriented Crystallization of Mixed-Cation Tin Halides for Highly Efficient and Stable Lead-Free Perovskite Solar Cells, Adv. Funct. Mater., 2020, 30, 2002230 CrossRef CAS.
- S. J. Lee, et al., Reducing Carrier Density in Formamidinium Tin Perovskites and Its Beneficial Effects on Stability and Efficiency of Perovskite Solar Cells, ACS Energy Lett., 2018, 3, 46–53 CrossRef CAS.
- X. Liu, et al., Influence of Halide Choice on Formation of Low-Dimensional Perovskite Interlayer in Efficient Perovskite Solar Cells, Energy Environ. Mater., 2022, 5, 670–682 CrossRef.
- K. Nishimura, et al., Relationship between Lattice Strain and Efficiency for Sn-Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2019, 11, 31105–31110 CrossRef CAS PubMed.
- J. Pascual, et al., Fluoride Chemistry in Tin Halide Perovskites, Angew. Chem., Int. Ed., 2021, 60, 21583–21591 CrossRef CAS PubMed.
- F. Wang, et al., Organic Cation-Dependent Degradation Mechanism of Organotin Halide Perovskites, Adv. Funct. Mater., 2016, 26, 3417–3423 CrossRef CAS.
- K. L. Svane, et al., How Strong Is the Hydrogen Bond in Hybrid Perovskites?, J. Phys. Chem. Lett., 2017, 8, 6154–6159 CrossRef CAS PubMed.
- C. Kamal, et al., Coupling Methylammonium and Formamidinium Cations with Halide Anions: Hybrid Orbitals, Hydrogen Bonding, and the Role of Dynamics, J. Phys. Chem. C, 2021, 125, 25917–25926 CrossRef CAS PubMed.
- T. Webb, et al., A Multifaceted Ferrocene Interlayer for Highly Stable and Efficient Lithium Doped Spiro-OMeTAD-based Perovskite Solar Cells, Adv. Energy Mater., 2022, 12(26), 2200666 CrossRef CAS.
- M. Li, et al., Hydrophobic Polystyrene Passivation Layer for Simultaneously Improved Efficiency and Stability in Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2018, 10, 18787–18795 CrossRef CAS PubMed.
- M. H. Kumar, et al., Lead-Free Halide Perovskite Solar Cells with High Photocurrents Realized Through Vacancy Modulation, Adv. Mater., 2014, 26, 7122–7127 CrossRef CAS PubMed.
- S. Gupta, D. Cahen and G. Hodes, How SnF2 Impacts the Material Properties of Lead-Free Tin Perovskites, J. Phys. Chem. C, 2018, 122, 13926–13936 CrossRef CAS.
- D. Ricciarelli, D. Meggiolaro, F. Ambrosio and F. De Angelis, Instability of Tin Iodide Perovskites: Bulk p-Doping versus Surface Tin Oxidation, ACS Energy Lett., 2020, 5, 2787–2795 CrossRef CAS.
- J. Zillner, et al., The Role of SnF2 Additive on Interface Formation in All Lead-Free FASnI3 Perovskite Solar Cells, Adv. Funct. Mater., 2022, 32, 2109649 CrossRef CAS.
- C. Li, et al., Low-bandgap mixed tin–lead iodide perovskites with reduced methylammonium for simultaneous enhancement of solar cell efficiency and stability, Nat. Energy, 2020, 5, 768–776 CrossRef CAS.
- Z. Yang, X. Zhang, W. Yang, G. E. Eperon and D. S. Ginger, Tin–Lead Alloying for Efficient and Stable All-Inorganic Perovskite Solar Cells, Chem. Mater., 2020, 32, 2782–2794 CrossRef CAS.
- R. Prasanna, et al., Design of low bandgap tin–lead halide perovskite solar cells to achieve thermal, atmospheric and operational stability, Nat. Energy, 2019, 4, 939–947 CrossRef CAS.
- S. Shao, et al., Field-Effect Transistors Based on Formamidinium Tin Triiodide Perovskite, Adv. Funct. Mater., 2021, 31, 2008478 CrossRef CAS.
- Q. Tai, J. Cao, T. Wang and F. Yan, Recent advances toward efficient and stable tin-based perovskite solar cells, EcoMat, 2019, 1, e12004 CrossRef.
- R. A. Kerner, Z. Xu, B. W. Larson and B. P. Rand, The role of halide oxidation in perovskite halide phase separation, Joule, 2021, 5, 2273–2295 CrossRef CAS.
- W. Li, et al., Addictive-assisted construction of all-inorganic CsSnIBr2 mesoscopic perovskite solar cells with superior thermal stability up to 473 K, J. Mater. Chem. A, 2016, 4, 17104–17110 RSC.
- T. J. Macdonald, L. Lanzetta, X. Liang, D. Ding and S. A. Haque, Engineering Stable Lead-Free Tin Halide Perovskite Solar Cells: Lessons from Materials Chemistry, Adv. Mater., 2023, 35, 2206684 CrossRef CAS PubMed.
- X. Dai, et al., Efficient monolithic all-perovskite tandem solar modules with small cell-to-module derate, Nat. Energy, 2022, 7, 923–931 CrossRef CAS.
- Y. Duan, et al., 21.41%-Efficiency CsPbI3 Perovskite Solar Cells Enabled by an Effective Redox Strategy with 4-Fluorobenzothiohydrazide in Precursor Solution, Adv. Funct. Mater., 2024, 34(10), 2312638 CrossRef CAS.
- W. Zhang, et al., Component Distribution Regulation in Sn-Pb Perovskite Solar Cells through Selective Molecular Interaction, Adv. Mater., 2023, 35, 2303674 CrossRef CAS PubMed.
- L. Wang, et al., Favorable grain growth of thermally stable formamidinium-methylammonium perovskite solar cells by hydrazine chloride, Chem. Eng. J., 2022, 430, 132730 CrossRef CAS.
- W. Chen, et al., Improving the Efficiency of Hole-Conductor-Free Carbon-Based Planar Perovskite Solar Cells with Long-Term Stability by Using the Hydrazine Acetate Additive via the One-Step Method, ACS Appl. Electron. Mater., 2021, 3, 5211–5218 CrossRef CAS.
- Y. Che, et al., Hydrazide Derivatives for Defect Passivation in Pure CsPbI3 Perovskite Solar Cells, Angew. Chem., Int. Ed., 2022, 61, e202205012 CrossRef CAS PubMed.
- T.-B. Song, et al., Importance of Reducing Vapor Atmosphere in the Fabrication of Tin-Based Perovskite Solar Cells, J. Am. Chem. Soc., 2017, 139, 836–842 CrossRef CAS PubMed.
- J. K. Niemeier and D. P. Kjell, Hydrazine and Aqueous Hydrazine Solutions: Evaluating Safety in Chemical Processes, Org. Process Res. Dev., 2013, 17, 1580–1590 CrossRef CAS.
- Z. Liu, et al., Gas-solid reaction based over one-micrometer thick stable perovskite films for efficient solar cells and modules, Nat. Commun., 2018, 9, 3880 CrossRef PubMed.
- Y. Chen, et al., Impacts of alkaline on the defects property and crystallization kinetics in perovskite solar cells, Nat. Commun., 2019, 10, 1112 CrossRef PubMed.
- X. Feng, X. Lv, J. Cao and Y. Tang, Continuous Modification of Perovskite Film by a Eu Complex to Fabricate the Thermal and UV-Light-Stable Solar Cells, ACS Appl. Mater. Interfaces, 2022, 14, 55538–55547 CrossRef CAS PubMed.
- Y. Chen, et al., Dual Passivation of Perovskite and SnO2 for High-Efficiency MAPbI3 Perovskite Solar Cells, Adv. Sci., 2021, 8, 2001466 CrossRef CAS PubMed.
- D. Cortecchia, et al., Layered Perovskite Doping with Eu3+ and β-diketonate Eu3+ Complex, Chem. Mater., 2021, 33, 2289–2297 CrossRef CAS PubMed.
- E. A. Ambundo, et al., Influence of Coordination Geometry upon Copper(II/I) Redox Potentials. Physical Parameters for Twelve Copper Tripodal Ligand Complexes, Inorg. Chem., 1999, 38, 4233–4242 CrossRef CAS.
- J. D. Cope, et al., Tuning the copper(II)/copper(I) redox potential for more robust copper-catalyzed C—N bond forming reactions, Eur. J. Inorg. Chem., 2020, 1278–1285 CrossRef CAS PubMed.
- Q. D. Lin, Gregory; Diao, Tianning. Experimental Electrochemical Potentials of Nickel Complexes, Synlett, 2021, 1606–1620 CAS.
- E. Eskelinen, S. Luukkanen, M. Haukka, M. Ahlgrén and T. A. Pakkanen, Redox and photochemical behaviour of ruthenium(II) complexes with H2dcbpy ligand (H2dcbpy = 2,2′-bipyridine-4,4′-dicarboxylic acid), J. Chem. Soc., Dalton Trans., 2000, 2745–2752, 10.1039/B004751L.
- W. J. Evans, Perspectives in reductive lanthanide chemistry, Coord. Chem. Rev., 2000, 206–207, 263–283 CrossRef CAS.
- T. Wang, Y. Wang, J. Lei, K.-J. Chen and H. Wang, Electrochemically induced surface reconstruction of Ni-Co oxide nanosheet arrays for hybrid supercapacitors, Exploration, 2021, 1, 20210178 CrossRef PubMed.
- D. van der Westhuizen, K. G. von Eschwege and J. Conradie, Electrochemistry and spectroscopy of substituted [Ru(phen)3]2+ and [Ru(bpy)3]2+ complexes, Electrochim. Acta, 2019, 320, 134540 CrossRef CAS.
- D. Aurbach, et al., Prototype systems for rechargeable magnesium batteries, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- X. Li, et al., Iodine-trapping strategy for light-heat stable inverted perovskite solar cells under ISOS protocols, Energy Environ. Sci., 2023, 16, 6071–6077 RSC.
- H. Yang, et al., Enhancing the Stability of Perovskite Solar Cells through an Iodine Confining Strategy, ACS Energy Lett., 2023, 8, 3793–3799 CrossRef CAS.
- D. N. Makhayeva, G. S. Irmukhametova and V. V. Khutoryanskiy, Polymeric Iodophors: Preparation, Properties, and Biomedical Applications, Rev. J. Chem., 2020, 10, 40–57 CrossRef CAS PubMed.
- D.-H. Kang, C. Ma and N.-G. Park, Antiseptic Povidone–Iodine Heals the Grain Boundary of Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2022, 14, 8984–8991 CrossRef CAS PubMed.
- Z. Dai, et al., Perovskite Films Treated with Polyvinyl Pyrrolidone for High-Performance Inverted Perovskite Solar Cells, ACS Appl. Energy Mater., 2022, 5, 4448–4460 CrossRef CAS.
- Y. Niu, et al., Improved crystallinity and self-healing effects in perovskite solar cells via functional incorporation of polyvinylpyrrolidone, J. Energy Chem., 2022, 68, 12–18 CrossRef CAS.
- C. M. Lee, et al., Improved device efficiency and lifetime of perovskite light-emitting diodes by size-controlled polyvinylpyrrolidone-capped gold nanoparticles with dipole formation, Sci. Rep., 2022, 12, 2300 CrossRef CAS PubMed.
- X. Qian, et al., Capture and Reversible Storage of Volatile Iodine by Novel Conjugated Microporous Polymers Containing Thiophene Units, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- J. Wang, et al., Synthesis of N-containing porous aromatic frameworks via Scholl reaction for reversible iodine capture, Microporous Mesoporous Mater., 2021, 310, 110596 CrossRef CAS.
- V. Safarifard and A. Morsali, Influence of an amine group on the highly efficient reversible adsorption of iodine in two novel isoreticular interpenetrated pillared-layer microporous metal–organic frameworks, CrystEngComm, 2014, 16, 8660–8663 RSC.
- A. S. Munn, et al., Iodine sequestration by thiol-modified MIL-53(Al), CrystEngComm, 2016, 18, 8108–8114 RSC.
- C.-L. Mai, et al., Donor–π–Acceptor Type Porphyrin Derivatives Assisted Defect Passivation for Efficient Hybrid Perovskite Solar Cells, Adv. Funct. Mater., 2021, 31, 2007762 CrossRef CAS.
- G.-B. Xiao, et al., Lead and Iodide Fixation by Thiol Copper(II) Porphyrin for Stable and Environmental-Friendly Perovskite Solar Cells, CCS Chem., 2021, 3, 25–36 CrossRef CAS.
- G.-B. Xiao, Z.-F. Yu, J. Cao and Y. Tang, Encapsulation and Regeneration of Perovskite Film by in Situ Forming Cobalt Porphyrin Polymer for Efficient Photovoltaics, CCS Chem., 2020, 2, 488–494 CrossRef CAS.
- P. Yan, et al., Chemical encapsulation of perovskite film by tetra-thiol copper(II) porphyrin for stable and clean photovoltaics, Org. Electron., 2021, 93, 106158 CrossRef CAS.
- T. C. T. Pham, et al., Capture of iodine and organic iodides using silica zeolites and the semiconductor behaviour of iodine in a silica zeolite, Energy Environ. Sci., 2016, 9, 1050–1062 RSC.
- J. Huve, et al., Porous sorbents for the capture of radioactive iodine compounds: a review, RSC Adv., 2018, 8, 29248–29273 RSC.
- D. Luo, Y. He, J. Tian, J. L. Sessler and X. Chi, Reversible Iodine Capture by Nonporous Adaptive Crystals of a Bipyridine Cage, J. Am. Chem. Soc., 2022, 144, 113–117 CrossRef CAS PubMed.
- B. J. Riley, J. D. Vienna, D. M. Strachan, J. S. McCloy and J. L. Jerden, Materials and processes for the effective capture and immobilization of radioiodine: A review, J. Nucl. Mater., 2016, 470, 307–326 CrossRef CAS.
- M. Che, C. Naccache and B. Imelik, Electron spin resonance studies on titanium dioxide and magnesium oxide—Electron donor properties, J. Catal., 1972, 24, 328–335 CrossRef CAS.
- M. Gliński and U. Ulkowska, Reaction of iodine with metal oxides, Can. J. Chem., 2011, 89, 1370–1374 CrossRef.
- K. K. Miller, A. Rezende, A. J. A. de, Aquino, D. Tunega and M. L. Pantoya, Adsorption and exchange reactions of iodine molecules at the alumina surface: modelling alumina-iodine reaction mechanisms, Phys. Chem. Chem. Phys., 2022, 24, 11501–11509 RSC.
- D. A. Kuznetsov, et al., Tuning Redox Transitions via Inductive Effect in Metal Oxides and Complexes, and Implications in Oxygen Electrocatalysis, Joule, 2018, 2, 225–244 CrossRef CAS.
- W. Peng, et al., Reducing nonradiative recombination in perovskite solar cells with a porous insulator contact, Science, 2023, 379, 683–690 CrossRef CAS PubMed.
- Y. Bai, et al., Dimensional Engineering of a Graded 3D–2D Halide Perovskite Interface Enables Ultrahigh Voc Enhanced Stability in the p-i-n Photovoltaics, Adv. Energy Mater., 2017, 7, 1701038 CrossRef.
- Y. Lin, et al., Enhanced Thermal Stability in Perovskite Solar Cells by Assembling 2D/3D Stacking Structures, J. Phys. Chem. Lett., 2018, 9, 654–658 CrossRef CAS PubMed.
- M. A. Ruiz-Preciado, et al., Supramolecular Modulation of Hybrid Perovskite Solar Cells via Bifunctional Halogen Bonding Revealed by Two-Dimensional 19F Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 2020, 142, 1645–1654 CrossRef CAS PubMed.
- X. Zhao, et al., Accelerated aging of all-inorganic, interface-stabilized perovskite solar cells, Science, 2022, 377, 307–310 CrossRef CAS PubMed.
- L. Jiang, et al., High-Performance Perovskite Solar Cells with a Weak Covalent TiO2:Eu3+ Mesoporous Structure, ACS Appl. Energy Mater., 2018, 1, 93–102 CrossRef CAS.
- H. Lee, et al., UV-Blocking and Transparent Polydimethylsiloxane Film for Improving Stability of Perovskite Photovoltaics, ACS Appl. Opt. Mater., 2023, 1, 1208–1216 CrossRef CAS.
- Y. Sun, et al., Enhanced UV-light stability of organometal halide perovskite solar cells with interface modification and a UV absorption layer, J. Mater. Chem. C, 2017, 5, 8682–8687 RSC.
- R. Keshavarzi, N. Molabahrami, N. Afzali and M. Omrani, Improving Efficiency and Stability of Carbon-Based Perovskite Solar Cells by a Multifunctional Triple-Layer System: Antireflective, UV-Protective, Superhydrophobic, and Self-Cleaning, Sol. RRL, 2020, 4, 2000491 CrossRef CAS.
- R. Datt, et al., Downconversion Materials for Perovskite Solar Cells, Sol. RRL, 2022, 6, 2200266 CrossRef CAS.
- G. P. Baxter, C. H. Hickey and W. C. Holmes, The vapor pressure of iodine, J. Am. Chem. Soc., 1907, 29, 127–136 CrossRef.
- Y. Zhou, et al., How Photogenerated I2 Induces I-Rich Phase Formation in Lead Mixed Halide Perovskites, Adv. Mater., 2024, 36(1), 2305567 CrossRef CAS PubMed.
- H. Wang, H. Liu, W. Li, L. Zhu and H. Chen, Inorganic perovskite solar cells based on carbon electrodes, Nano Energy, 2020, 77, 105160 CrossRef CAS.
- J. Pastuszak and P. Węgierek, Photovoltaic Cell Generations and Current Research Directions for Their Development, Materials, 2022, 15(16), 5542 CrossRef CAS PubMed.
- T. Zhang, et al., Profiling the organic cation-dependent degradation of organolead halide perovskite solar cells, J. Mater. Chem. A, 2017, 5, 1103–1111 RSC.
- K. Liu, et al., Covalent bonding strategy to enable non-volatile organic cation perovskite for highly stable and efficient solar cells, Joule, 2023, 7, 1033–1050 CrossRef CAS.
- Z. Qiu, N. Li, Z. Huang, Q. Chen and H. Zhou, Recent Advances in Improving Phase Stability of Perovskite Solar Cells, Small Methods, 2020, 4, 1900877 CrossRef CAS.
- S. J. Yoon, M. Kuno and P. V. Kamat, Shift Happens. How Halide Ion Defects Influence Photoinduced Segregation in Mixed Halide Perovskites, ACS Energy Lett., 2017, 2, 1507–1514 CrossRef CAS.
- B. Park and S. I. Seok, Intrinsic Instability of Inorganic–Organic Hybrid Halide Perovskite Materials, Adv. Mater., 2019, 31, 1805337 CrossRef PubMed.
- F. Li and M. Liu, Recent efficient strategies for improving the moisture stability of perovskite solar cells, J. Mater. Chem. A, 2017, 5, 15447–15459 RSC.
- T. J. Collins, Designing Ligands for Oxidizing Complexes, Acc. Chem. Res., 1994, 27, 279–285 CrossRef CAS.
- E. M. Sanehira, et al., Influence of Electrode Interfaces on the Stability of Perovskite Solar Cells: Reduced Degradation Using MoOx/Al for Hole Collection, ACS Energy Lett., 2016, 1, 38–45 CrossRef CAS.
- Y. Han, et al., Degradation observations of encapsulated planar CH3NH3PbI3 perovskite solar cells at high temperatures and humidity, J. Mater. Chem. A, 2015, 3, 8139–8147 RSC.
- J. Chang, et al., Tetrathiafulvalene-based covalent organic frameworks for ultrahigh iodine capture, Chem. Sci., 2021, 12, 8452–8457 RSC.
- Y. Xie, et al., Efficient and simultaneous capture of iodine and methyl iodide achieved by a covalent organic framework, Nat. Commun., 2022, 13, 2878 CrossRef CAS PubMed.
Footnote |
| † The oxidation potential of hydrazine and NaBH4 are pH-dependent varying significantly with oxidation product. |
| This journal is © The Royal Society of Chemistry 2024 |