
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrical-energy storage into chemical-energy carriers by combining or integrating electrochemistry and biology
Largus T.
Angenent
 *abcde,
Isabella
Casini
a,
Uwe
Schröder
f,
Falk
Harnisch
g and
Bastian
Molitor
ae
*abcde,
Isabella
Casini
a,
Uwe
Schröder
f,
Falk
Harnisch
g and
Bastian
Molitor
ae
aEnvironmental Biotechnology Group, Department of Geosciences, University of Tübingen, Schnarrenbergstr. 94-96, 72076 Tübingen, Germany. E-mail: l.angenent@uni-tuebingen.de
bAG Angenent, Max Planck Institute for Biology, Max Planck Ring 5, 72076 Tübingen, Germany
cDepartment of Biological and Chemical Engineering, Aarhus University, Gustav Wieds vej 10D, 8000 Aarhus C, Denmark
dThe Novo Nordisk Foundation CO2 Research Center (CORC), Aarhus University, Gustav Wieds vej 10C, 8000 Aarhus C, Denmark
eCluster of Excellence – Controlling Microbes to Fight Infections, University of Tübingen, Auf der Morgenstelle 28, 72076 Tübingen, Germany
fInstitute for Biochemistry, University of Greifswald, Felix-Hausdorff-Straße 4, 17487 Greifswald, Germany
gUFZ-Helmholtz Centre for Environmental Research, Department Microbial Biotechnology, Permoserstrasse 15, 04318, Leipzig, Germany
First published on 27th March 2024
Abstract
Our societies must reconsider current industrial practices and find carbon-neutral alternatives to avoid the detrimental environmental effects that come with the release of greenhouse gases from fossil-energy carriers. Using renewable sources, such as solar and wind, allows us to circumvent the burning of fossil energy carriers to produce electrical energy. However, this leads to a spatial-temporal discrepancy between production and demand, necessitating the ability to store vast amounts of electrical energy. Physical storage of electrical energy, such as hydropower and underground pressure storage, as well as the conversion of electrical energy into chemical energy, such as with batteries, can offer vast storage capacities. Another route of storing electrical energy at a massive scale is its conversion into chemical-energy carriers by combining or integrating electrochemistry with biology. Here, we will give an overview of the potential of these biological-storage technologies. Based on the order in which they combine or integrate biological and electrochemical steps, we will discuss the current state of research on these technologies in three distinct sections: (1) electrochemistry followed by biology; (2) biology followed by electrochemistry; and (3) integrated electrochemistry and biology. We will discuss research needs and opportunities in an outlook section at the end.
Broader contextOne of the most critical aspects of a low-carbon future is to store electrical energy for up to months to manage the electric load during imbalances between production and consumption. Many opportunities exist, including hydropower, underground pressure storage, and different kinds of batteries. Another opportunity is to use microbiological processes to convert electrical energy into chemical-energy carriers or directly store electrical energy. For both, biology needs to be combined with electrochemistry. Demonstration plants already exist that place water electrolysis before fermentation to produce and store methane within the natural gas infrastructure (power-to-gas with biomethanation). The opposite order is also possible – fermentation followed by electrolysis such as Kolbe electrolysis. This order is attractive because electrochemical conversions are much faster than biological conversions, which lends it to switch it on only during short peak production periods to manage the load. Finally, integrating biology into electrochemistry enables directly storing electrical energy in chemical-energy carriers, for example, in rechargeable microbial electrochemical systems. |
Introduction
The collective societies of the world need to produce an increasing amount of electrical energy and chemical-energy carriers to face an ever-growing population and economic development. At the same time, greenhouse-gas emissions must be rapidly lowered to prevent environmental collapse from climate change, making fossil sources passé. Fortunately, technologies to generate ample electrical energy from renewable sources, such as wind and solar, are already well established and are becoming economically compatible with fossil-fuel sources.1,2 As a result, several countries have seriously increased the share of renewable electrical energy in the grid and aim at generating fossil-free electrical energy in the near future.3 Here, we use the term electrical energy to include a time aspect (i.e., power × time), and refer to power to describe the availability at a given instant (i.e., current × voltage), or for consistency with an existing terminology (e.g., power-to-gas [PtG]).A major limitation of renewable energy sources is their intermittency, which leads to a discrepancy between short-term (e.g., day/night), long-term (e.g., seasonal), and local availabilities of sufficient electrical energy.4 Implementing intelligent electric grids and extending grid infrastructure can partly overcome the temporal and local distribution discrepancy, but societal and political issues must be considered.5 Regardless, long-term storage is required in the form of physical energy and chemical-energy carriers through underground pressure storage, hydropower, and various kinds of batteries (e.g., flow batteries, lithium-ion batteries).6 However, some of these abiotic-storage technologies are limited by geographical constraints or low energy densities,7 while batteries have a high environmental cost in cradle-to-grave assessments.8 Therefore, the solution of electrical-energy storage would likely need to come from a mixture of several technologies, depending on the geographical location. Developing such technologies and building them to store enough electrical energy should be priorities of research and development activities and the societal imperative right now.
Among the storage technologies, those that combine electrochemistry with biology (biological storage) should also be considered. They could require a lower infrastructure investment when compared to, for example, hydropower by utilizing the already available natural gas grid and its massive storage capacity. In Germany alone, the capacity for natural gas storage is 24.6 billion m3, which is enough to provide natural gas for 80 days in wintery conditions.9 While some biological-storage technologies are already in an advanced state,10,11 others need considerable research and development before they can be implemented at a vast scale, if ever possible. It has been evaluated that advanced biological-storage technologies can be cost-competitive compared to fossil fuels when specific benchmarks of electrical-energy pricing, carbon dioxide gas (referred to here as carbon dioxide) availability, and process efficiency are met.4
Here, we review biological-storage technologies that convert electrical energy into chemical-energy carriers by combining electrochemistry and biology either in a combined system with several process steps in series or integrated into one single process step. Based on how the electrochemical and biological steps are combined together in these technologies, we structured the review in three sections: (1) electrochemistry followed by biology; (2) biology followed by electrochemistry; and (3) integrated electrochemistry and biology (Fig. 1). The review is not comprehensive but includes pertinent examples from the literature for each of the three sections. We discuss the advantages/disadvantages, the state-of-the-art, and the future potential of these concepts, and we end this review with an outlook to highlight research needs and opportunities. For policymakers, the message is clear: enable building as many wind turbines and photo-voltaic systems as possible within a short time frame. This is because science & technology plus entrepreneurship within a stimulating political climate will rapidly find ways to store electrical energy during periods of excess power at low or even negative electrical-energy prices and to balance the electrical grid that needs re-building to serve these needs.12
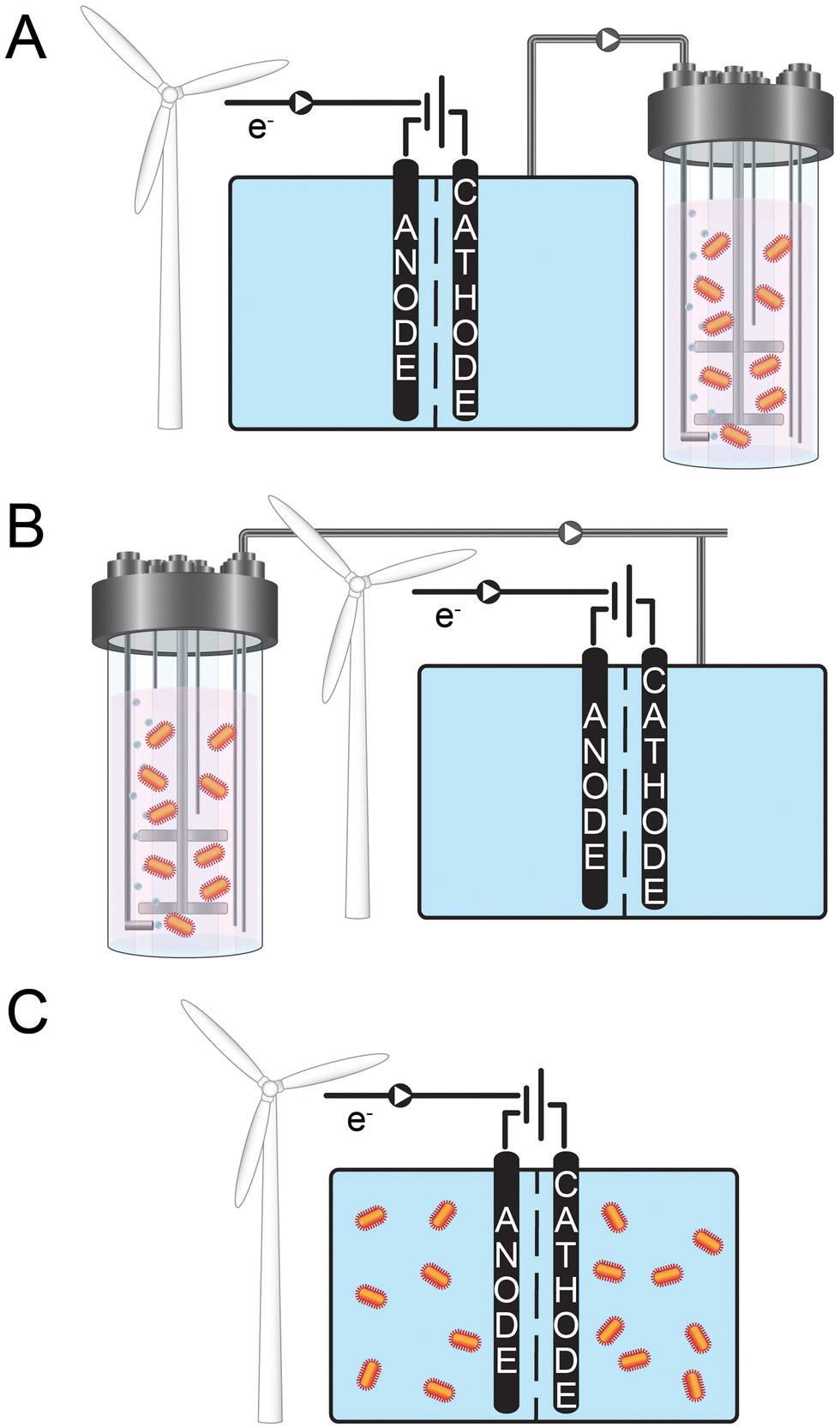 | ||
| Fig. 1 This thematic figure summarizes the concepts of the three sections of this review. All three panels depict renewably produced electrical energy (represented by the wind turbine) that is then converted via an electrochemical system. However, the placement of the biological system varies: (A) the biological step is placed after the electrochemical system; (B) the biological step occurs before the electrochemical system; and (C) the biology is integrated within the electrochemical system. | ||
Conventional electrolysis without biology
Water is split into molecular hydrogen and dioxygen gas, hereafter referred to as hydrogen and oxygen, in the conventional abiotic electrochemical system (i.e., water electrolysis cell or electrolyzer) that is placed before the biological step (Fig. 1A). Multiple reviews detail technological and economic assessments of water electrolysis, especially concerning storing electrical energy.11,13,14 Therefore, only a summary is provided here. Water electrolysis connects a reduction reaction at the cathode to produce hydrogen and an oxidation reaction at the anode to produce oxygen (eqn (1) [neutral or alkaline] and eqn (2) [acidic], respectively). For any electrochemical system, the minimum required cell potential consists of the theoretical cell potential plus the overpotentials at the anode and cathode. Thus, the overpotentials must be minimized to only several hundred mV to maintain high energy efficiencies (and reduce power consumption). For conventional water electrolysis, the overpotentials at the anode (oxygen evolution reaction) and cathode (hydrogen evolution reaction) have been lowered to acceptable levels throughout a long development period. Four methods of electrolysis are commonly described: (1) alkaline electrolysis (AE), which uses a liquid alkaline electrolyte; (2) polymer electrolyte membrane (PEM) electrolysis, which uses a proton exchange membrane; (3) anion exchange membrane (AEM) electrolysis, which uses an AEM and which is also known as alkaline PEM; and (4) solid-oxide electrolysis (SOEC), which operates at high temperatures with a solid-oxide electrolyte.13,15 In addition, membrane-less electrolyzers may become standard due to cost reduction16in tandem with the ability to separate the gases via a cryogenic system.In Europe, primarily, the first two technologies are used at the demonstration- or industrial-scale.17 When it comes to the choice of an electrolyzer for electrical-energy storage, the following factors should be considered: (i) energy efficiency; (ii) ability to exhibit a dynamic behavior (i.e., ability to be switched off and on); (iii) tolerance to low minimal loads; (iv) ability to produce hydrogen at high pressures; (v) lifetime; and (vi) capital and operating expenditures (OPEX).15 Finally, depending on the geographical location, the requirement for fresh water may need to be evaluated, while the development of electrolysis with seawater is still ongoing.18–20 The required ∼15 L of fresh water per 1 kg of hydrogen is not critical in, for example, Germany,21 but it may be elsewhere. In addition, a large percentage (∼50%) of water may be recoverable in the biological step such as with biomethanation (eqn (3)).
| 2H2O + 2e− → H2 + 2OH− | (1) |
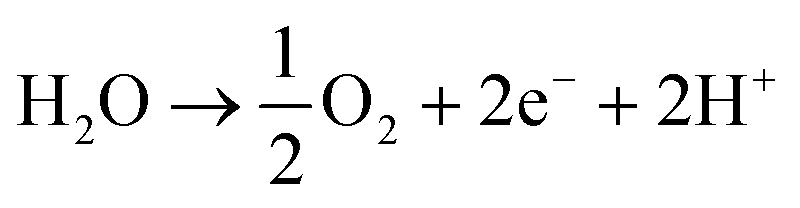 | (2) |
| 4H2 + CO2 → CH4 + 2H2O | (3) |
To compare different biological storage technologies, to decide the order in which unit operations are placed within a technology, or whether to plan integrating electrochemistry and biology, we should consider the energy requirements and capital expenditures (CAPEX) for post-processing needs for electrolyzers, which include drying and handling the pressure of the product gas hydrogen. For hydrogen alone, it was estimated that drying and handling pressure account for ∼15% of the CAPEX for a 1-MW PEM electrolyzer at a production capacity of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per year at an uninstalled cost of $258/kW.22 Therefore, if instead, an integrated approach is used to produce, for example, a soluble chemical, such as acetate, with a microbe inside the electrochemical cell, the gas drying and pressure handling equipment of an electrolyzer would be superfluous, reducing the CAPEX considerably. On the other hand, any soluble chemical product would need purification, adding considerable energy requirements and CAPEX.23 Thus, a detailed techno-economic analysis is necessary to account for all the complexities, including the treatment of organic wastes that possibly would be generated by growing microbial cells.
000 units per year at an uninstalled cost of $258/kW.22 Therefore, if instead, an integrated approach is used to produce, for example, a soluble chemical, such as acetate, with a microbe inside the electrochemical cell, the gas drying and pressure handling equipment of an electrolyzer would be superfluous, reducing the CAPEX considerably. On the other hand, any soluble chemical product would need purification, adding considerable energy requirements and CAPEX.23 Thus, a detailed techno-economic analysis is necessary to account for all the complexities, including the treatment of organic wastes that possibly would be generated by growing microbial cells.
Section I: electrochemistry followed by biology
Power-to-gas with biomethanation
Power-to-gas (PtG) is the overall terminology for technology that converts electrical energy into hydrogen or methane gas (referred to here as methane) for storage as chemical-energy carriers.7,15 An electrolysis step to split water into hydrogen and oxygen is utilized, which is followed by a methanation step when methane instead of hydrogen is the desired gas. The methanation step can be abiotic or biological (i.e., biomethanation via gas fermentation), with the latter being our focus in this review (Fig. 1A). Initially, the nomenclature power-to-gas was not used. A previous review had traced the concept of storage of electrical energy as methane to the work of Hashimoto24 during the early 1990s in Japan.13 Hashimoto proposed to use photovoltaic technology in the desert to meet global energy demands sustainably. However, this solution would require storing and transporting the generated electrical energy. Therefore, as a first step, he proposed electrolyzing seawater into hydrogen and oxygen on-site, converting the electrical energy into chemical-energy carriers – stored in hydrogen bonds. Even 30 years later, however, the use of seawater still needs to be further developed as mentioned above.To simultaneously help address climate change, which is mainly caused by rising levels of carbon dioxide in the atmosphere due to anthropogenic activity,25 Hashimoto24 suggested utilizing carbon dioxide and reacting it with hydrogen to form methane in an abiotic methanation step (eqn (3)). This enables recycling carbon without further removing sequestered carbon from fossil fuels.26 A recent life-cycle analysis showed that including carbon-capturing during electrical-energy storage would, indeed, considerably improve its environmental benefits.7 Finally, Hashimoto24 proposed to convert the produced methane into a liquid to be stored and transported as liquefied natural gas (LNG).
However, a more simple and pertinent solution in many societies is to store methane as renewable natural gas in the existing natural-gas grid with its vast storage capacities.6,9 For such an application, converting hydrogen to methane is advantageous because regulations limit the hydrogen content within the natural-gas grid.13 In addition, hydrogen has a substantially higher diffusive nature and a considerably lower energy density than methane,5 and leads to the embrittlement of metal piping. Consequently, its use as a direct electrical carrier would require rebuilding the natural gas grid, which is unnecessary when methane is used as the energy carrier. During an interim period, we may need both hydrogen and methane storage and transport,27 with the possibility of laying a new hydrogen pipeline next to the existing natural gas pipeline to minimize regulatory delays. Regardless of the type of gas to store energy, both leaked hydrogen and methane would contribute to radiative forcing.28 Thus, any new storage and grid system would need to prevent fugitive emissions. Even though this is harder to do for hydrogen than methane, hydrogen would always have a lower greenhouse gas potential than methane with a current leakage rate of up to 2% for natural gas in the US.29 In addition, the generation of methane requires a reliable source of carbon dioxide. Furthermore, this carbon dioxide is then released again into the atmosphere upon methane oxidation. Thus, the electrical-energy storage technology with methane is not a carbon capture and sequestration (CCS) technology, but it does offset fossil-fuel-based natural gas and promotes a circular carbon economy.
The abiotic method of converting hydrogen and carbon dioxide into methane (eqn (3)), which Hashimoto24 alluded to in the early nineties, is the thermochemical Sabatier process that utilizes a metal catalyst (most commonly nickel or ruthenium).11,13,21 The Sabatier process efficiently converts carbon dioxide into methane (83–90%).30 Still, several aspects of biomethanation are advantageous, which is why we focus on this route here (PtG-biomethanation in Table 1). First, the abiotic process requires high temperatures and pressures between 200–550 °C and 100–10000 kPa, respectively. This makes the Sabatier process less user-friendly in decentralized locations than biological systems that operate between 35–65 °C and have an overpressure of only 100–200 kPa.
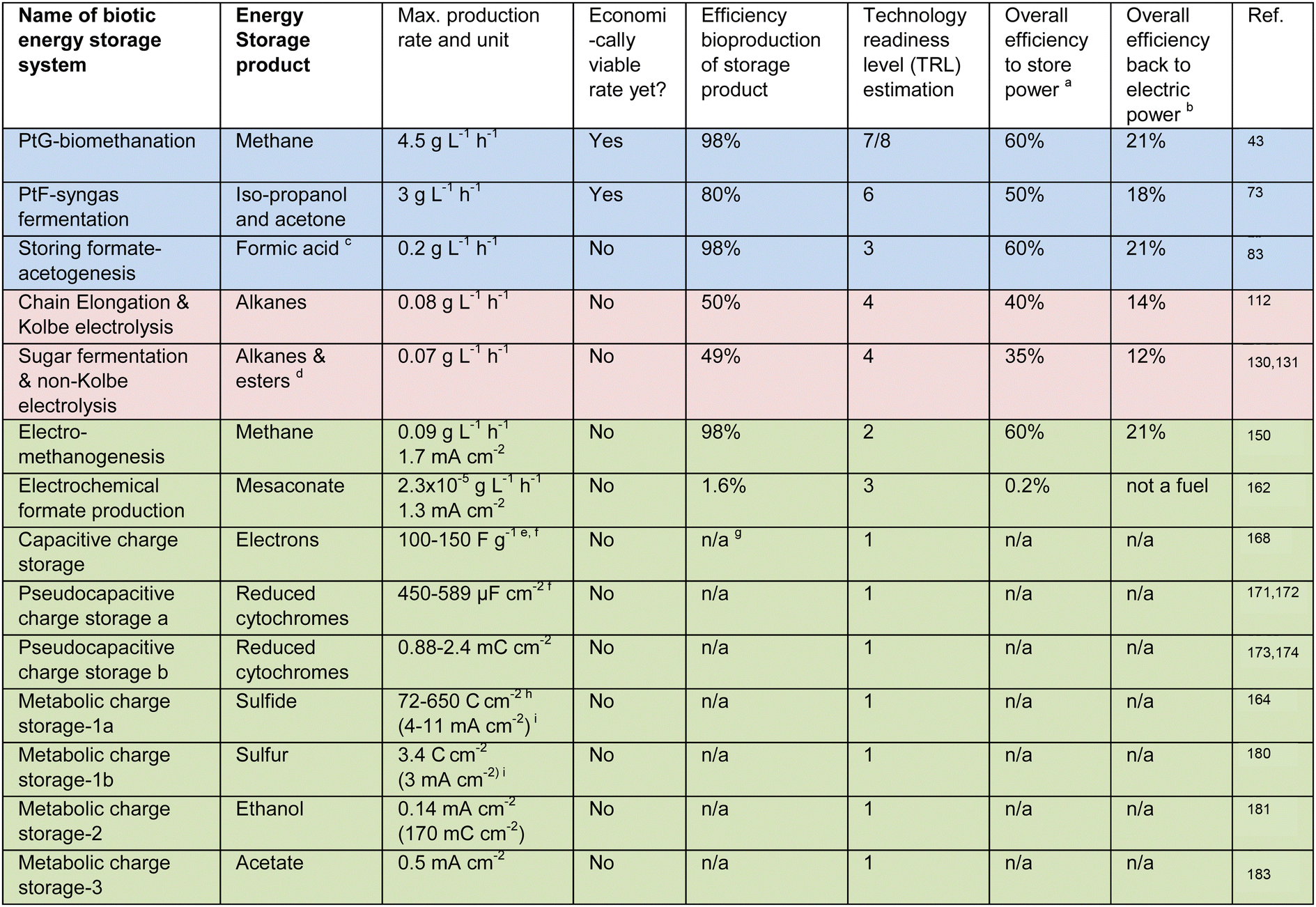
|
a Electrolyzer efficiency of 60%.
b Combustion conversion 35% for all fuels.
c Based on a specific activity of 3 mmol gcell protein−1 h−1 with 1 mg mL−1 cell protein in a 1.5-L bioreactor.
d Based on a specific activity of 0.012 gHAA gcell dry weight−1 h−1 with 5.7 gcelldryweight L−1.
e High surface area electrode: 2000 m2 g−1 of specific surface area.
f Charge capacity (C cm−2) is not available in/cannot be derived from the respective publication.
g Not available; the information is not available in/cannot be derived from the respective publication.
h Porous, three-dimensional electrode.
i Transient (exponentially decaying) currents due to exhaustive conversion of electrode associated storage compounds.
|
|---|
|
Second, metal catalysts in the Sabatier process are prone to deactivation (e.g., fouling, poisoning, and thermal degradation) due to contaminants in the gas stream such as sulfur compounds.30 Depending on the source of carbon dioxide, the gas stream may need to be extensively purified, which adds costs and complexity to the storage system, very likely excluding decentralized power-to-gas conversion systems. A list of maximum impurities in the gas stream toward metal catalysts is shown in Table 4.2 of Lehner et al.31 The volatile sulfur or other compounds that are generated during, for example, anaerobic digestion of wastes to biogas do not compromise the biomethanation system.32,33 On the contrary, sulfur compounds and trace metals act as growth nutrients and would, therefore, be advantageous for the applied microbial culture, which are methane-producing microbes (methanogens).
Third, the Sabatier process is a non-dynamic process that cannot be quickly tuned or switched off and restarted during a temporary lull in renewable power supply. On the contrary, thermophilic biomethanation is known to be a dynamic process, which was shown with a non-pure culture of methane-producing microbes (methanogens) in a trickle bed bioreactor.34 Thermophilic methanogens (∼65 °C) pause their metabolism when at suboptimal thermal growing conditions (∼25 °C). Therefore, biomethanation can be switched off and on by cooling and heating the fermentation broth, respectively, to manage the requirement for a dynamic process.34 As a result, a smaller-capacity hydrogen storage system can be installed upstream of the bioreactors to make biomethanation a dynamic process, which may result in considerably lower CAPEX. For the non-dynamic Sabatier process, the CAPEX for hydrogen storage may comprise 15–25% of the total CAPEX.35
The biomethanation step utilizes methanogens as microbial catalysts in bioreactors.32,36,37 All isolated methanogens are obligate anaerobes with a growth requirement to produce methane38 and belong to the domain of Archaea.39 Hydrogenotrophic methanogens use carbon dioxide as the carbon source and electron acceptor, and hydrogen as the electron donor, and in biotechnology can be utilized at high rates for gas fermentation.40 The carbon dioxide is reduced by using hydrogen to produce methane and water (eqn (3)) by going through one round of the Wolfe cycle.41 During the Wolfe cycle, several enzymes utilize the following coenzymes as C1-carriers and redox cofactors: methanofuran, tetrahydromethanopterin, coenzyme M, coenzyme B, F420, and ferredoxin.41
Besides the advantageous dynamic behavior of thermophilic methanogens, it was already known in the 1980s and 1990s that thermophilic hydrogenotrophic methanogenesis with pure cultures of Methanothermobacter thermautotrophicus achieved very high kinetic rates.42,43 For example, Schill et al.43,44 attained a volume of methane production per reactor volume per day (VVD) of 163 L L−1 day−1 (4.5 g L−1 h−1 at standard temperature and pressure) based on the wet volume (Table 1). Recently, a genetic system for M. thermautotrophicus was developed to possibly optimize the system and broaden the substrate and product spectrum.45 Other thermophilic and hyperthermophilic (>80 °C) methanogenic strains were compared specifically for their characteristics as potential biocatalysts in power-to-gas applications.46
The previous studies with M. thermautotrophicus achieved these high VVDs despite a low solubility of hydrogen into the liquid solution, and thus a sluggish hydrogen transfer rate, which is the limiting factor of biomethanation. The solubility of hydrogen at thermophilic conditions is lower than for mesophilic conditions, which is a disadvantage, but this is overcome by the higher conversion rates and the higher diffusibility for hydrogen at thermophilic conditions. Regardless, the bench-scale bioreactor systems utilized vigorous mixing in completely stirred tank reactors (energy intensive),42–44 and by increasing the pressure.32,47 Scale-up with a mixed bubble column during demonstration-scale projects maintained the high methane production rates of the bench-scale systems at much-reduced mixing intensities (conserving energy) due to the innate increase of: (1) the gas residence time; and (2) pressure from the added height of the liquid column compared to the bench-scale bioreactor.48 This resulted in higher VVD rates while maintaining a 98% methane content in the gas phase.48,49 Based on these promising results, several companies are developing their first industrial-scale projects based on biomethanation.11,48,50
Three other features besides the high VVDs make thermophilic power-to-gas applications more favorable compared to mesophilic (∼35 °C) conditions. First, there is a lower risk of microbial contamination under thermophilic conditions when pure cultures of methanogens are utilized. Second, there is a lower cooling requirement for thermophilic conditions than for mesophilic conditions because hydrogenotrophic methanogenesis is an exothermic (i.e., heat-generating) process.37 Third, as a consequence of the exothermic metabolism, M. thermautotrophicus experiences an entropy-retarded growth.43,51 The considerable decrease in the entropy (conversion of 5 moles of gases into 1 mole of gas and 2 moles of liquid water) is overcompensated by enormous heat production (vast decrease in enthalpy) by all hydrogenotrophic methanogens.52 This means that growth is inefficient, while methane production is efficient, which is advantageous for PtG processes.32 Entropy-retarded growth was more pronounced for thermophilic (60 °C) compared to mesophilic methanogens (39 °C), because the heat produced to only compensate for the loss of entropy was 80% vs. 50%, respectively.53 Indeed, Guneratnam54 found a higher efficiency in methane production at 65 °C than at 55 °C with an open culture of microbial consortia (i.e., reactor microbiome).
Power-to-fuel with syngas fermentation
Liquid energy carriers of chemical energy (i.e., fuels), such as ethanol, can also be produced by placing the bioprocess after an electrochemical system in a power-to-fuel (PtF) system. It is possible to utilize two different types of electrochemical systems to produce: (1) hydrogen at the cathode of a conventional electrolysis system plus augmenting carbon dioxide; and (2) carbon monoxide and hydrogen at a cathode of a carbon dioxide electrolysis system plus leftover carbon dioxide.55,56 Carbon monoxide is one of the main constituents of synthesis gas (syngas), with hydrogen and carbon dioxide as the other pertinent gases. In addition to electrochemical methods to produce syngas from carbon dioxide and water, other sources are the: (1) production of basic oxygen furnace (BOF) gas during steelmaking; and (2) thermochemical production of syngas through solid-waste conversion. Regardless of whether carbon dioxide electrolysis becomes mainstream, a vast volume of syngas will be available with high concentrations of carbon monoxide (PtF-syngas fermentation in Table 1). A syngas fermentation industry is already maturing to produce fuels and chemicals, such as ethanol, from syngas.26,57Due to thermodynamical reasons, 66% of the carbon leaves syngas fermentation as carbon dioxide when syngas only contains carbon monoxide.26 The carbon loss is decreased and becomes even negligible when increasing quantities of hydrogen are added to the syngas mixture.26 For syngas fermentation, the ratios of hydrogen, carbon monoxide, and carbon dioxide can be variable.26,58 That is why electrical-energy storage by supplying syngas with external hydrogen from conventional electrolysis during syngas fermentation is very lucrative – the carbon atoms in the syngas can all end up in the fuel with lower local carbon dioxide emissions as a result. The fuel from the power-to-fuel system can be used again for electrical energy generation as part of a true electrical-energy storage system. However, the overall energy efficiency is disappointing (Table 1). The fuel (the stored chemical-energy carrier) can also be utilized for transportation or heat production. Regardless, the carbon in the fuel will ultimately be oxidized again and released as carbon dioxide. Thus, similarly to the power-to-gas system with biomethanation, energy-storage requirements and offsetting fossil fuels are met (circular economy), but CCS requirements are not.
Similar to power-to-gas, both abiotic and biological processes are available to produce liquid fuels from syngas. For power-to-fuel, the Fischer–Tropsch process is analogous to the Sabatier process from the previous section. In comparison, the biological syngas fermentation process exhibits the same three first advantages than biomethanation: (1) a lower operating temperature and pressure; (2) a lower sensitivity to deactivation by gas contaminants; and (3) a more dynamic behavior. Furthermore, syngas fermentation has higher product specificities than the abiotic Fischer–Tropsch process. However, its volumetric production rates are considerably lower, which is a clear disadvantage of the biological process.
Acetogens are utilized as the biocatalyst for syngas fermentation. These acetogens employ the Wood–Ljungdahl pathway to reduce carbon dioxide as part of autotrophic growth.59 In the linear Wood–Ljungdahl pathway, carbon dioxide is reduced into acetyl-coenzyme A through the methyl and carbonyl branches. The electrons for this reduction are either derived from hydrogen or carbon monoxide oxidation. Carbon monoxide can enter the pathway directly in the carbonyl branch. Acetogens have this in common with hydrogenotrophic methanogens; however, the utilized coenzymes and enzymatic reactions are distinctively different between methanogens and acetogens. The carbon from the carbon dioxide is transferred and reduced by the following coenzymes in acetogens: tetrahydrofolate, NAD/NADP, and ferredoxin. Acetyl-coenzyme A is then further converted into acetate and other fermentation products.60
Wild-type acetogens in pure culture can produce ethanol, which can serve as a fuel.61,62 In addition, when combined with another wild-type bacterium in a co-culture, they can produce different fuels such as n-butanol and n-hexanol.56,63,64 However, for all these systems, fuels must be separated from the fermentation broth by, for example, distillation, stripping, or pervaporation,65 and the necessary energetic cost would need to be taken into consideration when evaluating the overall energy efficiency for electrical-energy storage.
Genetic engineering of acetogens is performed in academic and commercial settings to generate other fuels or optimize gas fermentation further. Heterologous pathways for novel fuels from hydrogen and carbon dioxide or syngas are introduced into the acetogen, and unwanted by-products can be avoided or reduced by gene deletions.58,66 Researchers have developed and optimized powerful genetic tools, such as different transformation protocols, gene deletions and regulation, and gene editing with CRISPR/Cas technology, for Clostridium ljungdahlii and Clostridium autoethanogenum.67–70
Some examples of the utilization of these genetic tools to broaden and optimize the power-to-fuel platform are: (1) increasing ethanol production by the deletion of the bifunctional alcohol dehydrogenase in C. autoethanogenum;71 (2) improving autotrophic growth by inactivation of one carbon monoxide dehydrogenase in C. autoethanogenum;72 (3) implementing vitamin prototrophy as an alternative for selection of engineered strains in large-scale processes;67 and (4) producing n-butanol, acetone, or iso-propanol as alternative fuels.61,73 In an extensive team effort to optimize pathways, strains, and bioprocessing, Liew et al.73 achieved iso-propanol and acetone production rates of ∼ 3 g L−1 h−1 at selectivities >90% at the pilot-scale (Table 1).
Even when attaining the mature performance results of Liew et al.,73 the products of syngas fermentation consist of small molecules with a carbon-chain length of only three carbon atoms (C3). The reason for producing small molecules is that acetogens grow under limited energy availability, which is a characteristic of their anaerobic growth conditions. Therefore, strategies need to be developed to boost the available energy for the production of fuel that is longer in chain length.74 One of these strategies may be to provide nitrate as an additional electron acceptor instead of ammonia as the nitrogen source75,76 because it results in higher ATP production and biomass growth. However, biochemical research is necessary to understand the exact mechanisms. The study by Liew et al.73 shows that the research around syngas fermentation is rapidly maturing. Indeed, LanzaTech (Skokie, IL, USA), who was a partner in that study, is already operating syngas fermentation at industrial scales.
Other electrical-energy storage routes with acetogens
Acetogens are flexible when it comes to the gas mixture they can be fed with, as explained above, but this does impact the product specificities. Acetogens grow well with hydrogen and carbon dioxide but do not produce as much fuel as when carbon monoxide is present.77 Instead, the predominant product from carbon dioxide and hydrogen is acetate, which, as an intermediate, can still be utilized to generate chemical-energy carriers. Acetogenic bacteria can produce acetate in a first anaerobic bioreactor, which then can be converted by, for example, genetically modified yeast in a second aerobic bioreactor into lipids78 or other fuels,79 such as alkanes.80 This may not necessarily lead to extraordinarily more energy consumption for aeration because pure oxygen is already available in excess from the conventional electrolyzer when co-located. With such a system, renewable electrical energy can even be stored as human food as part of a power-to-protein strategy.81 In fact, acetogens efficiently reduces carbon dioxide into acetate as an intermediate product towards carbon fixation with aerobic yeast or fungal cells.82Another direct option to store renewable electrical energy in chemical-energy carriers with acetogens is the accumulation of formate by Acetobacterium woodii within the gas-fermentation broth by an instrinsic enzymatic reaction with hydrogen and carbon dioxide or syngas (Storing formate-acetogenesis in Table 1). This can occur under specific growth-limiting conditions at a volumetric production rate of 0.2 g L−1 h−1 (Table 1),83 by, for example, adding monensin, which is an ionophore antibiotic, or by genetic modification.84 The enzymatic reaction is reversible,83 which is ideal for storage because hydrogen and carbon dioxide would be freed quickly from the gas-fermentation broth again by consuming soluble formate in the same bioreactor.84 Researchers found that the hydrogen-dependent carbon dioxide reductase (HDCR) from A. woodii or Thermoanaerobacter kivui was responsible for formate formation at superior efficiencies compared to metal catalysts.84 This enzyme is membrane-bound.85 Formate was formed and oxidized again at a maximum concentration of 330 mM in an A. woodii culture,84 while E. coli accumulated a formate concentration of 500 mM.86
Rather than an enzymatic reversal to hydrogen and carbon dioxide within a single fermentation tank with A. woodii or T. kivui, dilute formate can be utilized as a substrate. For example, the same acetogens can be utilized for chemical production after changing the growth conditions, further realizing the dynamic adeptness of this biological process. Thus, when renewable electrical energy is plentiful, formate can be quickly stored within the fermentation broth. Then, a slower process of subsequent bioconversion into chemicals (i.e., acetate) and cellular growth can be carried out by the same acetogens, albeit a large part of the formate-bound carbon will be rereleased as carbon dioxide due to thermodynamic reasons, requiring the addition of hydrogen to prevent such rerelease during the bioconversion step.
Section II: biology followed by electrochemistry
The superior rates for electrochemistry represent an advantage
Placing the bioprocess before the abiotic electrochemical system is another possible order to store electrical energy87 (Fig. 1B). The bioprocess would first generate an intermediate product that is then upgraded electrochemically into fuel with energy-storing chemical bonds (Fig. 2). We first discuss the advantages of this order for an example of chemical production (not a fuel). Then, we discuss the first fuel-production strategy: coupling microbial chain elongation with Kolbe electrolysis. Finally, we will discuss the second fuel-production strategy: aerobic fermentation followed by non-Kolbe electrolysis. Other strategies for fuel production exist, which we will not further discuss because this review is not exhaustive. These are, for example: (1) the fermentation to oleic acid followed by an electrochemical decarboxylation;88 and (2) the electrochemical upgrading of acetone into a drop-in fuel.89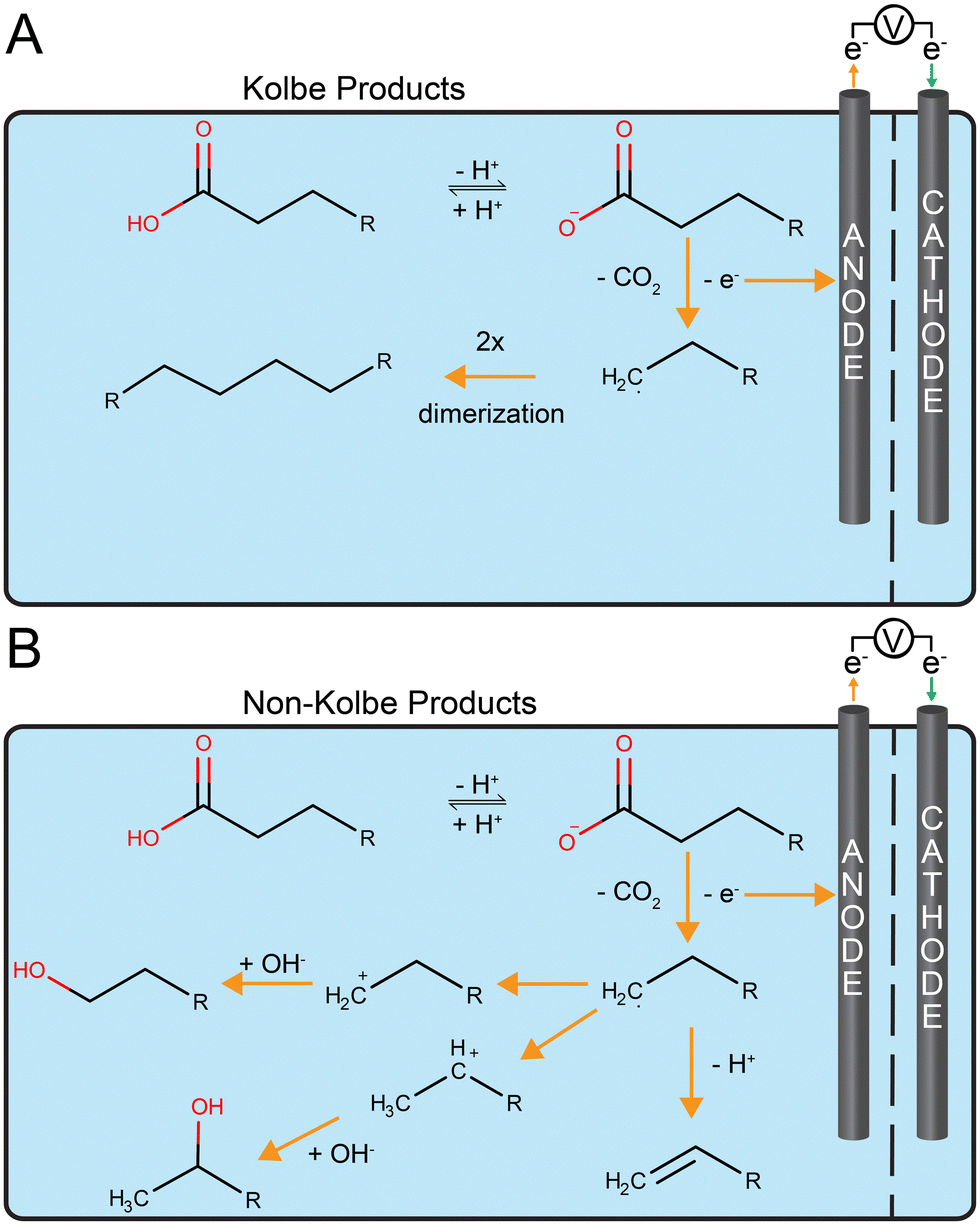 | ||
| Fig. 2 Examples for the biological step followed by electrochemistry: (A) medium-chain carboxylates from microbial chain elongation are electrochemically converted using Kolbe electrolysis (decarboxylation and dimerization) that yield hydrocarbons as drop-in fuels; and (B) long-chain carboxylates from fermentation by Pseudomonas taiwanensis VLB120 pSB01 with xylose as the substrate are electrochemically converted using non-Kolbe electrolysis (decarboxylation without dimerization), which yielded hydrocarbons that can be utilized as drop-in fuel additives. The orange arrows depict reactions and electron flow at the anode (discharging of Kolbe and non-Kolbe reactions). | ||
Generally, the production rates for biological systems are relatively slow, with production rates of at least 1 g L−1 h−1 for economically viable systems.90,91 This results in a relatively large fermentation volume with relatively low parasitic electrical-energy losses of approximately 3%.92,93 Therefore, the bioprocess would operate continuously, even when the supply/demand ratio of renewable electrical energy is low (i.e., high electrical-energy pricing). On the contrary, electrochemical systems with much faster rates (100 mA cm−2 of electrode area for product formation from carbon dioxide94), but with an innate high electrical-energy consumption, can be operated intermittently and only when the supply/demand ratio of renewable electrical energy is high at low or even negative electrical-energy pricing.
For commodity chemical production of polyamides and polyesters (not for fuel production), the order of biology followed by electrochemistry has already been studied.91 Suastegui et al.95 found that the fermentation of glucose to muconate (i.e., a 6-carbon dicarboxylate) was 96× slower than the electrochemical hydrogenation of muconate into 3-hexenedioate (i.e., a precursor for nylon-6,6), albeit both unit processes were not optimized. Because of this considerable difference in the production rate, this biology/electrochemical order represents a unique opportunity to take advantage of the dynamic behavior of renewable electrical-energy supply. For example, the biological system would continuously produce muconate, whereas the electrochemical system would only need to be powered 1% of the time to match the rate of biological muconate production during low or negative electric pricing. In case such a very low-capacity utilization percentage is unfeasible, a system can be optimized with a continuously operated large biological system and a relatively small electrochemical system that would operate most of the time but not at periods of high electric pricing.
Microbial chain elongation and Kolbe electrolysis
Microbial chain elongation with reactor microbiomes in non-axenic conditions can treat and upgrade a plethora of different organic wastes or wastewaters to produce medium-chain carboxylates, including n-caproate (C6), n-heptanoate (C7), and n-caprylate (C8).96,97 Reactor microbiomes are well equipped to deal with the complexity and variability of organic compounds because primary fermentation converts all polysaccharides, proteins, and lipids into a pool of similar intermediates. They are short-chain carboxylates, including acetate (C2), propionate (C3), lactate (C3), and n-butyrate (C4), and some ethanol (C2), with hydrogen and carbon dioxide as the intermediate gases.98 A diverse community of microbes can perform this primary fermentation step that includes hydrolysis and acidogenesis as part of the carboxylate platform, which is similar to anaerobic digestion of wastes to generate biogas.98The difference between anaerobic digestion and chain elongation are the final steps in the anaerobic food web of the open culture. These final steps are part of secondary fermentation.98 For chain elongation to occur, acetoclastic methanogenesis (eqn (4)) from anaerobic digestion should be entirely suppressed by operating the bioreactor at: (1) mildly-acidic conditions (pH ∼ 5.5);99 (2) relatively low hydraulic retention times;100 or (3) conditions of by-product inhibition of n-caprylate.101 Some hydrogenotrophic methanogenesis (eqn (3)) will still be active at mildly acidic pH levels, which reduces the hydrogen partial pressure. This is beneficial as long as the hydrogen partial pressure does not drop below 1 kPa to prevent ethanol oxidation to acetate that is not coupled to the reverse β-oxidation pathway (i.e., excessive ethanol oxidation).102
The reverse β-oxidation pathway replaces part of acetogenesis and methanogenesis from anaerobic digestion by elongating short-chain carboxylates to medium-chain carboxylates. This pathway utilizes ethanol (C2), lactate (C3), or monomers of sugars (C5 or C6) as electron donors and carbon/energy sources. Adding external electron donors to organic wastes is not always necessary, which is a clear economic advantage.103 Below, the electron donor ethanol and the electron acceptor n-butyrate to produce n-caproate are used in example reaction (eqn (5)).
| CH3COOH → CH4 + CO2 | (4) |
| 6C2H5OH + 5CH3CH2CH2CHOOH → 5CH3(CH2)4COOH + CH3COOH + 2H2 + 4H2O | (5) |
Thiolase is an essential enzyme in the circular reverse β-oxidation pathway, which condenses two acyl-CoA molecules.104 Energy for bacterial growth is partly conserved through substrate-level phosphorylation via oxidation of one of the six moles of ethanol to acetate (eqn (5)). Additional energy for bacterial growth comes from transport-coupled phosphorylation through the Rnf complex and ATPases.96,105 A phylogenetic-diverse group of bacteria has the genetic capabilities to perform reverse β-oxidation, with Clostridium kluyveri as the first representative that was studied.106 Other pathways that are being investigated as potential routes to n-caproate include the reversed tricarboxylic acid (TCA) combined with the reverse β-oxidation pathway and the fatty acid biosynthesis pathway.107
Depending on the substrate composition, there might be the need to add electron donors. For example, short-chain carboxylates from the primary fermentation of food waste have been converted into medium-chain carboxylates by adding external ethanol.108 This process has now been scaled up as a demonstration plant in the Netherlands with plans for an industrial-scale plant.109 However, when compounds in the organic waste can be first converted into lactate through homofermentation,110 the external addition of electron donors may no longer be necessary, which circumvents procuring ethanol.103 Similar to anaerobic digestion, chain elongation can reach a high product selectivity of >50%.103,111
The mildly acidic conditions to prevent acetoclastic methanogenesis have proven to be ideal for the in-line extraction of the medium-chain carboxylate products by membrane-based liquid–liquid extraction (i.e., pertraction).99 Eventually, the carboxylates are extracted into an alkaline extraction solution of pH ∼ 9, which has been directly coupled to Kolbe electrolysis to store electrical energy in the chemical bonds of alkanes, which are drop-in fuels (Fig. 2A).112 The extraction solution from pertraction has shown to be a good connection point between the relatively sluggish biological system (0.07 g L−1 h−1) and the fast electrochemical system (Chain elongation and Kolbe electrolysis in Table 1). The natural product n-caproate can be accumulated in the extraction solution until electrical-energy prices drop. Periodically switching on Kolbe electrolysis then reduces the n-caproate concentration by its conversion into a mixture of hydrocarbons (mainly alkanes), which is innately removed by phase separation. Kolbe electrolysis of n-caproate also produces 1.8 × 10−2 g hydrogen per g of n-decane (1.3 mol hydrogen per mol n-decane). For the calculation, we used the experimentally validated 70% and 90% Coulombic efficiency for Kolbe electrolysis at the anode and the hydrogen evolution reaction at the cathode, respectively.113,114 Considering a carbon efficiency of 50% (mol C mol C−1) for the overall conversion of waste substrate into Kolbe product by biological and electrochemical conversions,112 the system generates 1.1 × 10−2 g hydrogen per gram of carbon in the waste substrate (6.4 × 10−2 mol hydrogen per mol carbon in the substrate). The produced hydrogen can be utilized as described above.
The Kolbe reaction (Fig. 2A) during Kolbe electrolysis specifically converts medium-chain carboxylates into alkanes by electrochemical decarboxylation at the anode and dimerization of the formed alkyl-radicals (eqn in Fig. 2A). Thereby, one carbon atom per carboxylate is oxidized to carbon dioxide, and is not included in the product. The carbon dioxide in the off-gas could be recovered by utilizing the hydrogen that is also generated. Interestingly, the Kolbe reaction was the first known electro-organic reaction and can be performed at ambient temperatures and in aqueous solutions, making it environmentally benign.115 Research on Kolbe reactions became sporadic until recently.116 In the 1980s, chain elongation had been coupled to Kolbe electrolysis for the first time.117,118 Such possible coupling to produce fuels from organic wastes has recently made a renaissance.112,119,120
At the current state of technology development, the alkaline extraction solution from chain elongation achieves a total medium-chain carboxylate concentration of ∼1 mol L−1. During proof-of-concept research with Kolbe electrolysis, the concentration of the medium-chain carboxylates in this extraction solution was decreased to ∼0.5 mol L−1 within 5.5 h at a carboxylate conversion rate of 2.1 × 10−3 mol cm−2 h−1 (per electrode area) after which the alkaline extraction can be reused. A Coulombic efficiency for the Kolbe reaction of up to 80% was achieved under these conditions,112 which is the efficiency of electrons from the electrical energy that ends up in the fuel. This represented an energy input for the medium-chain carboxylate-to-fuel conversion of 0.1 kW h mol−1 at a >5 V potential difference and a current density of 72 mA cm−2. The total electron efficiency (expressed in chemical oxygen demand [COD] equivalents) of the entire coupled process – from ethanol-rich corn beer to a hydrocarbon mixture (drop-in fuel) – was roughly 0.5 g CODfuel g CODcorn beer−1, which is promising (Table 1).112
Low alkane accumulation during earlier preliminary work in our groups and the work by others121 taught us that the existing knowledge on the Kolbe reaction was not sufficient. The influence of the environmental conditions on the anolyte, such as the pH, had to be carefully assessed and optimized.122 More importantly, the formation of agglomerates and micelle-like structures hindered electrochemical conversion due to electrode blockage.123 In contrast, short-chain carboxylates do not form the agglomerates and micelle-like structures at the electrode surface, explaining why mixtures of carboxylic acids, including ones with shorter chains, were advantageous.113,118 Kolbe electrolysis may require further alterations in the configuration of the electrochemical system, such as flow-reactors or sonoelectrochemistry, which utilizes ultrasound in electrochemistry.124 In the past, Kolbe electrolysis primarily utilized platinum (Pt) electrodes. To reduce the CAPEX, platinized titanium (Ti) instead of Pt was used for Kolbe electrolysis and found to be highly efficient.125 A recent breakthrough was made through the use of rapid alternating polarity to limit pH gradients at electrodes, which made Pt superfluous. The Kolbe reaction occurred for diverse substrates with only amorphous carbon such as reticulated vitreous carbon. However, acetone is needed as a reaction solvent, and medium-chain carboxylates as a reactant have not been tested yet.126 In addition, the overpotentials were high, resulting in a required potential difference of 10 V,126 which would lead to unsustainably high power consumption rates and inferior energy efficiencies. However, this potential difference could be lowered considerably by, for example, improving the electrolytic conductivity and by making the process continuous.127
Sugar-to-hydroxy acid bioconversion and non-Kolbe electrolysis
For the other type of reaction, which is referred to as a non-Kolbe reaction (Fig. 2B), an acid moiety undergoes an electrochemical decarboxylation at the anode followed by radical formation, which is similar to the Kolbe reaction. However, no subsequent dimerization occurs, but rather a further loss of a proton, yielding a carbocation (i.e., a positively charged carbon atom on a carbon chain). This also forms hydrocarbons, including alkanes and alcohols (eqn in Fig. 2), albeit with shorter chain lengths than the Kolbe reaction. Depending on the reaction conditions, ethers and esters can also be formed.128 When the dimerization generates lengthy hydrocarbon products that may block the electrode, the non-Kolbe reaction can be the preferred reaction compared to the Kolbe reaction. Even with non-Kolbe reactions, carboxylic acids from plant oils can only be converted when using, for example, sonoelectrochemistry forming emulsions to allow sufficient contact with the anode surface for the reaction to occur and to remove the hydrocarbons from the electrodes.129An example of electrical-energy storage combining a biological step followed by non-Kolbe electrolysis was the conversion of a glucose-rich medium (as a proxy for a sugar-waste stream) to produce a drop-in diesel fuel (Sugar fermentation and non-Kolbe electrolysis in Table 1).130 The intermediate products from the biological step were 3-hydroxy decanoic acid and low amounts of 3-(3-hydroxy-alkanoyloxy)alkanoates from fermentation with genetically modified Pseudomonas taiwanensis VLB120 pSB01130 at a volumetric production rate of 0.07 g L−1 h−1 (Table 1).131 The non-Kolbe reaction had to be performed in methanol as an organic solvent under alkaline conditions to assure: (1) a sufficient solubility of the reactant; and (2) a high selectivity for the final product.130 The final product mixture contained more than 90% C9 oxygenates (e.g., 2-nonanone, nonanal, 1-nonanol, nonanal-dimethyl-acetal, and methyl-nonanoate), possessing drop-in fuel-like qualities such as a cetane number of 63.
Noteworthy, a complete conversion for non-Kolbe electrolysis was possible after 30 min with a selectivity of up to 95% and 60% for a synthetic solution and a real fermentation solution with 3-hydroxydecanoic acid (3-HDA), respectively. However, this result was obtained with relatively low Coulombic efficiencies of, for example, ∼25% for the synthetic solution with 3-HDA. With a shorter reaction time of 10 min rather than 30 min, higher Coulombic efficiencies of 71% were reported for a synthetic solution. However, this resulted in a residual substrate in the synthetic solution. Thus, work on a mechanistic understanding, for example, by using online mass-spectrometry,132 and subsequent electrochemical engineering at biologically compatible conditions would be necessary to optimize the non-Kolbe reaction further.
Section III: integration of biology into electrochemistry
Electrochemical hydrogen production and integrated biomethanation
Conventional water electrolysis is the unit process with the highest CAPEX for the overall power-to-gas system that includes biomethanation in a separate bioreactor,133 even though the cost of electrolysis is decreasing.11 This is why academic institutions and companies are working on integrating methanogenic microbes within the catholyte of microbial electrochemical systems to combine biology and electrochemistry into one system. It would circumvent drying and handling of pressured hydrogen of conventional electrolysis altogether, as discussed above, and possibly reduce the CAPEX. The field of study is called microbial electrochemistry, and when carbon dioxide is reduced at the cathode, it is called microbial electrosynthesis.134,135Initially, integrating methanogenesis with the cathode of electrochemical systems was thought to be a process with direct-electron transfer between the electrode and the methanogenic microbes,136 which was referred to as electromethanogenesis (Table 1). However, a mechanistic understanding of this direct-electron transfer was elusive. Early work by Villano et al.137 showed that methane production at the cathode started immediately when hydrogenotrophic methanogens from a conventional fermenter without electrodes were introduced to the cathode. The absence of a lag phase suggested that specific membrane-located proteins to wire electrons through the non-conductive membrane, which would be necessary for direct-electron transfer, were either: (1) already expressed; or (2) not necessary. If assumed that such membrane-bound proteins would be energetically unfavorable for such microbes when not used, the absence of a lag phase would indicate that another electron-transfer mechanism was active (or at least active at higher transfer rates) than direct electron transfer.
Therefore, a more likely electron-transfer mechanism for electromethanogenesis is mediated-electron transfer with soluble hydrogen or formate as intermediates. Hydrogen and formate can be produced at the cathode and can be immediately taken up and converted by methanogens to methane. This results in very low concentrations of these intermediates, masking this mediated electron-transfer mechanism. Hydrogen can theoretically be produced at pH 7 at a standard redox potential of −0.414 V vs. a standard hydrogen electrode (SHE). However, overpotentials push the potential of hydrogen evolution down to <−0.69 V vs. SHE. Certain enzymes such as hydrogenases and formate dehydrogenases, which are located at the membranes of methanogens, can reduce the overpotential. This opens the possibility of hydrogen production at less negative potentials,138 explaining why methane was produced at the cathode with a potential of −0.65 V vs. SHE.137
Deutzmann et al.139 showed that these enzymes play a pertinent role in electromethanogenesis by knocking out the encoding genes in Methanococcus maripaludis. Without these (de)hydrogenases, the mutant strain could not provide the electrochemical advantage provided by the wild type. More recently, Boto et al.140 provided similar evidence for the acetogen C. ljungdahlii. They observed that a planktonic culture performed better than a biofilm culture in a microbial electrochemical system and further proving that hydrogen is the mediator for electron transfer for microbial electrosynthesis. These important results showed that the functional mechanism of electromethanogenesis or microbial electrosynthesis is mediated-electron transfer with hydrogen as the most likely intermediate to guarantee faster rates. The results do not dispute the existence of direct electron transfer for certain groups of methanogens. In fact, in very specific natural environments, methanogens could take up electrons from conductive materials at relatively low rates.141
Even though hydrogenases and formate dehydrogenases provide a valid electrochemical advantage at the cathode,139 Kracke et al.142 electro-plated biocompatible metals, such as nickel molybdenum (NiMo), at the cathode to further reduce the overpotential. In a follow-up paper, they achieved a VVD of 1.4 L L−1 day−1 and a Coulombic efficiency of 90% with NiMo and M. maripaludis in a microbial electrochemical system,143 which was further improved to 2.2 L L−1 day−1 and a Coulombic efficiency of 99% with 3-D printed electrodes.144 This translated to an electrode-surface-area-corrected current density of 0.72 mA cm−2 and is in the range for which hydrogen bubble formation was circumvented.
However, beyond 1 mA cm−2, which is necessary for an economically viable scale-up, hydrogen bubble formation would prevent biofilm formation and a high Coulombic efficiency.144 Increased overpotentials and many other problems due to bubble formation at electrodes are not only a limitation for biological electrochemical systems but for all abiotic electrochemical systems and are not always fully acknowledged.145 On the other hand, when controlled and understood well in abiotic electrochemical systems, bubble formation has led to benefits such as a superior transport of reactants.145 Kempler et al.,146 for example, were able to reduce overpotentials by changing the high-surface-area three-dimensional structures of electrodes, leading to the well-distributed formation of small oxygen bubbles. Thus, bubble formation may create very specific problems and opportunities around biofilm formation in microbial electrochemical systems. Modeling has shown that active biofilms can, in principle, achieve high current densities for microbial electrosynthesis.147 In that case, bubble formation should be prevented to maintain hydrogen in the soluble form and to maintain a proper biofilm (it will be sloughed off during bubble formation).144
A superior electrochemical configuration for electromethanogenesis, which is called a zero-gap cell design,148 was utilized by Geppert et al.149 They set a current density of 3.5 mA cm−2 with Nickel as a cathode catalyst, and achieved a VVD of 12.5 L L−1 day−1 at a 64% Coulombic efficiency, but only for a day. Baek et al.150 included a vapor-fed anode system to the zero-gap cell design to limit the pH imparity across the membrane and reached a VVD of 2.9 L L−1 day−1 (0.09 g L−1 h−1 in Table 1) for several weeks at a current density of 1.7 mA cm−2 with a Pt catalyst.150 A 1.5–3.5 mA cm−2 applied current density was, until recently, a relatively high current density for a microbial electrochemical system, but it is very low compared to an abiotic carbon dioxide electrolyzer, which may be operated at 250 mA cm−2. However, for an industrial-size commercial translation, a current density of 50–100 mA cm−2 may be necessary.151
Fortunately, a much higher current of 30 mA cm−2 was applied with a hybrid zero-gap cell design, reaching 2.2 V, with a pentlandite-type cathode catalyst (Fe3Ni3Co3S8).152 It is doubtful that this hybrid system is a true microbial electrochemical cell because the cathode is shielded from the methanogenic culture broth (identified as the catholyte in the paper) by a hydrogen-diffusing Teflon layer. Regardless, the VVD was 2.4 L L−1 day−1 at an estimated Coulombic efficiency of ∼70% for ∼200 days. With the Teflon layer, the principle is reminiscent of a vapor-fed cathode system by Rossi et al.153 in an abiotic zero-gap cell: (1) maintaining a wet cathode (with water from the anolyte crossing the membrane); (2) getting rid of the gas product (with hydrogen diffusing through a Teflon layer); and (3) relinquishing a catholyte (by shielding the fermentation broth with Teflon). It is important to understand the advantage of this hybrid system152 compared to placing water electrolysis before fermentation or whether the miniaturization effort for the hybrid zero-gap cell design is an advantage at the bench only. Possibly, not having to dry hydrogen and deal with pressurized hydrogen could be the advantage (as mentioned above).
Electrochemical formate production and integrated biological conversion
Similar to power-to-gas with biomethanation, the separate fermentation after a carbon dioxide electrolysis system could also be integrated within the electrochemical system as part of the power-to-fuel system, which was mentioned earlier for carbon monoxide.4,56 Carbon monoxide and hydrogen could also be produced at the cathode of a microbial electrochemical system after which a biological conversion within the catholyte would occur. A first proof-of-concept has now been published around this concept.154 An abiotic carbon dioxide electrolysis system can, in addition to carbon monoxide, produce formate, methanol, acetate, ethanol, and ethylene.4Of these products, the electrochemical reduction of carbon dioxide to formate at the cathode (eqn (6)) has been integrated with biology in different ways (Electrochemical formate production in Table 1).155 Li et al.156 pioneered the use of an electrochemical system with an indium cathode to produce formate and introduced Ralstonia eutropha H16 as the production host to obtain isobutanol and 3-methyl-1-butanol as the fuels. Indium possesses a relatively high selectivity for formate.157,158 However, the electrochemical reduction of carbon dioxide into formate at the indium cathode needed to be further optimized to occur under biocompatible conditions (i.e., ambient temperature, ambient pressure, and neutral pH).159,160
| CO2 + 2H+ + 2e− → HCOOH | (6) |
Under biocompatible conditions, but without the presence of microbes, Hegner et al.160 demonstrated that a formate production rate of ∼0.25 mmol cm−2 h−1 was achieved with an electrical-energy consumption of ∼0.142 kW h per mole of formate, a Coulombic efficiency of 83%, and a current density of 16 mA cm−2. For a different study, the formate production rate and the current density decreased from ∼0.06 to ∼0.04 mmol cm−2 h−1 and from 3.5 to 2.8 mA cm−2, respectively, during the scale-up from 50 mL to 1 L.161
Finally, the team integrated the optimized electrochemical reduction of carbon dioxide to formate and a biological catalyst, namely, the modified Methylobacterium extorquens AM-1 strain, to produce the C5-polymer precursors mesaconate and 2S-methyl succinate in one pot (1-L wet volume) but during sequential production periods. This resulted in a maximum formate production rate and current density of ∼2.8 μmol cm−2 h−1 and 1.3 mA cm−2 (Table 1), respectively, at a Coulombic efficiency of 13% due to ample hydrogen production (87%), which is 6.7 mol hydrogen per mole formate produced (0.29 g hydrogen per g formate produced). The conversion rate of formate during the biological period equaled the formation rate of formate during the electrochemical period, resulting in a mesaconate production rate of 0.179 μmol L−1 h−1 (2.3×10−5 g L−1 h−1 in Table 1) and a 2S-methyl succinate production rate of 0.129 μmol L−1 h−1 (2.1×10−5 g L−1 h−1).162 Similar to integrated biomethanation, current densities are considerably lower for the integrated systems than the 50–100 mA cm−2 that would be necessary for an economically viable technology.
Energy storage in rechargeable microbial electrochemical systems
Rather than converting electrical energy in one direction into chemical bonds for energy storage with integrated systems, it is also possible to reversibly store electrical energy into biofilm electrodes using electrochemically active bacteria.163 A very early study worked on this storage concept in the 1990s.164 First, the system was charged during which microbial sulfate reducers converted soluble sulfate into sulfide, which precipitated the sulfide on a metal-oxide-coated, three-dimensional electrode as an insoluble transition metal sulfide. This metal sulfide acted as the electric-energy storage compound because it was re-oxidized to sulfate after connecting the anode with an oxygen cathode, rereleasing the electrical energy in a microbial fuel cell.164 After this first example, other rechargeable microbial electrochemical systems were developed, which we distinguish here in: (1) capacitive-; (2) pseudocapacitive-; and (3) metabolic-charge storage, based on their physical basis.Capacitive-charge storage (first charge storage) is based on the reorientation of the electrochemical double layer at the electrode-solution interface upon a polarity change of the electrode and does not involve Faradaic (i.e., redox) reactions (Fig. 3A and Table 1). As an abiotic mechanism, it does not rely on and does not require a biological activity for its functioning. As a first example, fungal cells, such as the mycelium of the grey-oyster fungus Pleurotus ostreatus on wet wood shavings, increased the capacitance from 57 to 189 pF compared to the wood shavings alone with a 10-mm electrode spacing.165 Using these values, in combination with the applied charge/discharge voltage of 50 V, a corresponding charge capacity increase from 2.9 to 9.5 nC can be calculated. However, due to missing data on electrode dimensions, no surface area values can be derived. A second example is the utilization of open cultures of electrochemically active bacteria in biofilms of microbial electrochemical systems.166 However, we do not report quantitative numbers here from this work because the authors only provided cumulative capacities recorded over a series of discharges. Due to the relatively minor charge density (i.e., current density × time) of the electrochemical double layer, capacitive electrodes require a large electrochemically active surface area, realized by, for example, an activated-carbon coating.167 Thus, carbon granules of a specific surface area of 2000 m2 g−1 have a specific double-layer capacitance in the order of 100–150 F g−1 (Capacitive charge storage in Table 1).168
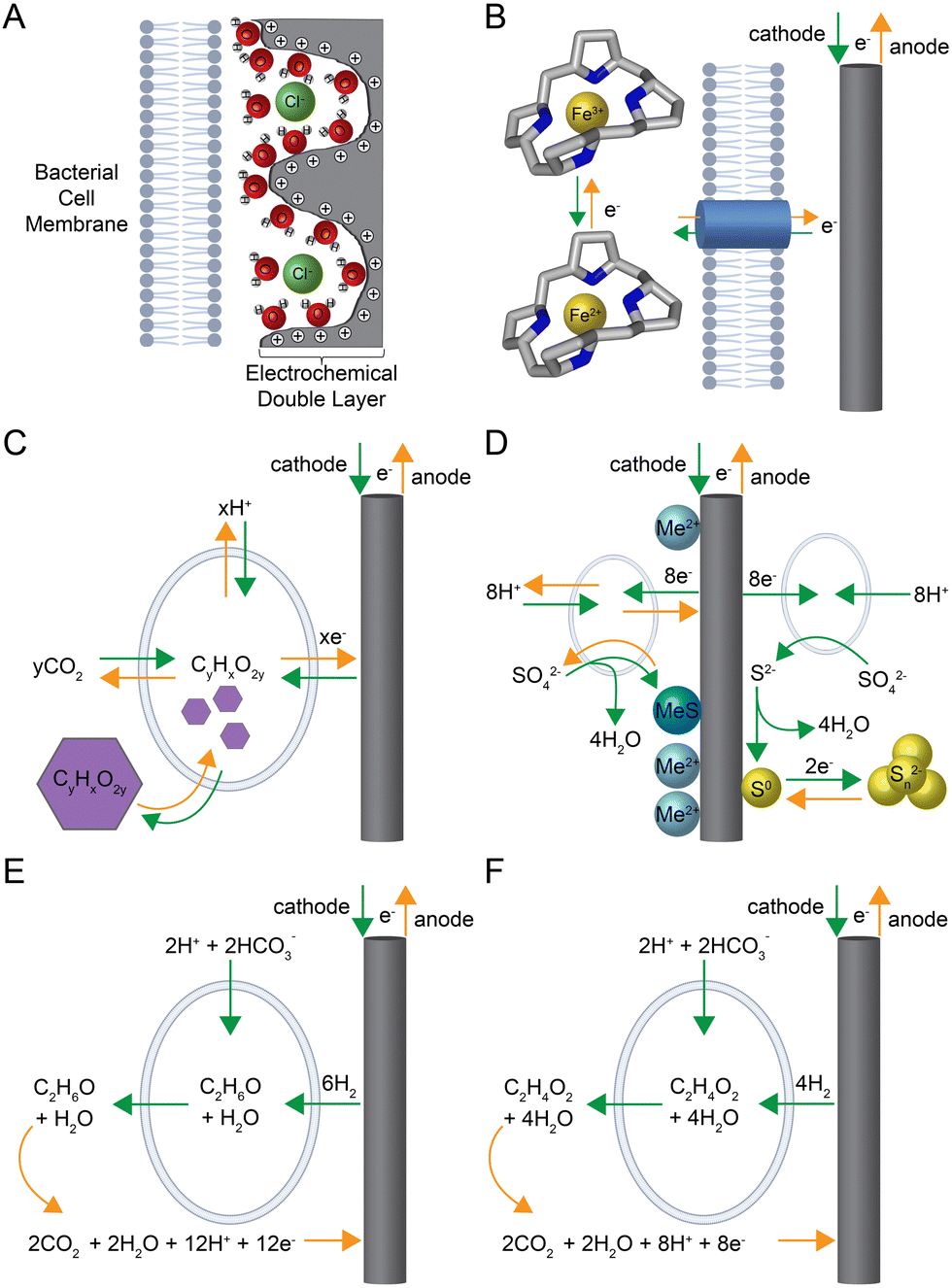 | ||
| Fig. 3 Modes of charge storage in biofilm electrodes: (A) capacitive-charge storage upon reorientation of the electrochemical double layer at the electrode–electrolyte (biofilm) interface; (B) pseudocapacitive-charge storage via reduction and oxidation of reversible electron carriers, such as the cytochrome pool, within electrochemically active bacteria and their biofilms; (C) metabolic-charge storage within microbial cells by producing and consuming a polymer; (D) metabolic-charge storage outside of the microbial cell with sulfur compounds either with a metal hydroxide complex (left side of electrode) or with polysulfide (right side of electrode); (E) metabolic-charge storage outside of the microbial cell with ethanol in a biofilm; and (F) metabolic-charge storage outside of the microbial cell with acetate in a biofilm. Green arrows depict processes and reactions during charging, while orange arrow depict processes and reactions during discharging. The cell membranes were created with BioRender.com. | ||
The concept of capacitive-charge storage could find an application in microbial electrochemical systems for which the fluidized bed (i.e., moving bed) capacitive granules, which are made of activated carbon, are not in permanent contact with the current collector. Therefore, charge storage becomes essential.169 This fluidized-bed system was limited by the electrical resistance rather than the ionic resistance of counter ions at the current collector.170
Faradaic processes form the basis for pseudocapacitive-charge storage (second charge storage), which is based on the reduction and oxidation of surface-confined redox species (Fig. 3B and Table 1). For an electrochemically active biofilm, pseudocapacitance is caused by a reversible charge and discharge of microbial redox compounds such as extracellular, outer-membrane, and periplasmatic cytochromes or adsorbed flavins.163 For example, Malvankar et al.171 proposed using Geobacter sulfurreducens biofilms, which contain relatively high concentrations of c-type cytochromes, as the basis for bioelectrochemical supercapacitors. Of note is that the term supercapacitor generally describes electrochemical elements exploiting both capacitive and pseudocapacitive processes. They achieved a biofilm capacitance of 589 μF cm−2.171 Subsequently, a similar value of 450 μF cm−2 (Pseudocapacitive charge a in Table 1) was also achieved by ter Heijne et al.172 The charge density of such biofilms can be derived via integration of the oxidation and reduction signals of the respective non-turnover cyclic voltammograms, yielding values between approximately 0.88–2.4 mC cm−2 (Pseudocapacitive charge b in Table 1).173,174 The advantage of capacitors and pseudocapacitors lies in the fast charge and discharge process and the high degree of electrochemical reversibility (low overpotentials). Unfortunately, the ability to store charge is limited because of the restricted number of cytochromes in the biofilm.
Metabolic-charge storage (third charge storage) overcomes this limitation by exploiting a reversible microbial-metabolism trait with electrochemically active bacteria (Fig. 3C–F). We will first discuss the reversible charge storage within bacterial cells and then outside the cell. A magnetosome-battery hypothesis had been developed during evolutionary studies, with the postulation that reversible biomineralization of magnetite or greigite within the bacterial cells could produce energy for these bacteria.175 However, it is not yet clear whether the internal reduction and oxidation of biominerals can be used for electrical-energy storage.176 Such cellular energy storage within bacterial cells is possible with organic polymers such as polyhydroxyalkanoates.177–179 As a proof-of-concept, a sugar (i.e., glucose and fructose) was first converted to an organic polymer, which is oxidized, releasing electrons and carbon dioxide at the anode. For this to work as storage, however, instead of a sugar, carbon dioxide would need to be first reduced with electrical energy into a polymer, which would then need to be oxidized again, freeing the stored electrical energy (Fig. 3C).
Three different approaches for metabolic charge-storage elements with compounds outside of the bacteria cell have been published to store energy in: (1) an insoluble precipitate, which we exemplified above for the first rechargeable microbial electrochemical system;164,180 (2) a bacterial biofilm;181,182 or (3) the adjacent medium solution.183,184 For the first approach, the central biochemical step is the formation of sulfide by microbial sulfate reduction before sulfur-rich compounds are stored. Two different sulfur-rich storage compounds have been tried (Metabolic charge storage 1a-b in Table 1): (i) Habermann and Pommer164 utilized metal hydroxide-coated electrodes to precipitate and accumulate metal sulfide into a three-dimensional electrode as the charging step, with a subsequent re-oxidation to sulfate as the discharging step (left side of Fig. 3D). At pH 7, the charge storage capacity was 72 C cm−2 (and up to 650 C cm−2 at pH 9) at a three-dimensional electrode with an estimated current density of 4–11 mA cm−2 during a transient discharge curve (Metabolic charge storage 1a in Table 1);164 (ii) Izadi et al.180 utilized the precipitation of elemental sulfur at the electrode surface via abiotic anodic sulfide oxidation. As a solid-state-electrochemical process, this sulfur was then reduced to polysulfide (charging) and oxidized back to sulfur (discharging) (right side of Fig. 3D). The experimental results show a charge-storage capacity of 3.4 C cm−2 and an estimated current density of 3 mA cm−2 at a planar electrode (Metabolic charge storage 1b in Table 1).
For the second approach, Yates et al.181 applied a switchable electrochemically active biofilm, which was isolated from the anode of a marine-sediment microbial fuel cell and capable of microbial electrosynthesis to store carbon dioxide and after polarity switching, to rerelease carbon dioxide (Metabolic charge storage 2 in Table 1). Energy storage in such biofilm resembles the concept of a classical rechargeable battery. They referred to this biofilm as a Cathode–Anode (CANode) biofilm with a Desulfobulbaceae member as a functionally active microbe to take up electrons as hydrogen at the cathode to produce ethanol from bicarbonate and to get rid of electrons by ethanol oxidation at the anode (Fig. 3E).182 Regardless of the mechanism, steady-state charging and discharging current densities of 0.14 mA cm−2 and estimated storage capacities of 170 mC cm−2 were achieved (Table 1).181,182
For the third approach, Molenaar et al.183 developed a storage technology that was composed of two hydraulically connected microbial electrochemical systems. The approach resembles a reversible fuel cell or a so-called redox-flow battery (Metabolic charge storage 3 in Table 1). For charging, an electrochemically active biofilm at the cathode in the first system reduces carbon dioxide from bicarbonate to acetate, which increases in concentration in the microbial electrolyte solution.184 For discharging, the acetate is re-oxidized from the microbial electrolyte solution by an electrochemically active biofilm at the anode of the hydraulically connected second system (Fig. 3F). At the counter-electrode side of both systems, a ferri-/ferrocyanide electrolyte solution was used, allowing an electrochemically reversible redox process. The microbial electrolyte and the ferri-/ferrocyanide electrolyte solutions can circulate between the two microbial electrochemical systems. A later study from the same team developed a three-compartment microbial electrochemical system with the ferri-/ferrocyanide electrolyte in the middle to circumvent the circulation of this electrolyte solution to achieve a current density of 0.5 mA cm−2 (Table 1).183
Outlook
Efficient storage technologies for electrical energy are essential for the transition of our societies to a renewable-based and circular economy because the load of the electric grid has to be managed. Biology can play an important role. In this outlook section, we will follow the organization of the main text and Table 1 to discuss research needs and possibilities to make the combination or integration of electrochemistry and biology into a viable energy storage option. Electrolysis of water is in an advanced stage; however, for implementation, post-processing steps, such as drying the gas, must be taken into consideration, as mentioned in the text. Combining and placing a water electrolysis unit before a bioreactor within a power-to-gas process for biomethanation already offers a way of storing electrical energy into the chemical-energy carrier methane, with enormous storage capacities in existing natural gas grids. This advanced technology can already be implemented on a large scale with an estimated technology readiness level (TRL) of 7/8 (Table 1). Still, research areas that include genetic modification of the pure methanogenic culture could lead to either side product formation with methane as the main product for ATP production or other reduced-product formation without methane production. Synthetic and systems biology techniques are needed for the latter to make sure that enough ATP is produced.One of the main advantages of methane production is that no product extraction is necessary because methane freely bubbles out of the fermentation broth. This is different when a different product, such as isopropanol, is produced with syngas fermentation. For any of the liquid fuels, extraction and separation technology as part of an elaborate post-processing system is necessary, which is often one of the most sensitive parameters in a techno-economic analysis. Therefore, research projects on product extraction and separation technology are as important as optimizing fermentation and should be performed simultaneously during development.
Kolbe electrolysis can play a role in post-processing as well because the electrochemical conversion into a more hydrophobic product leads to easier extraction from the fermentation broth. This is what occurs when a medium-chain carboxylate is converted into a longer alkane, resulting in innate phase separation. Research on Kolbe and non-Kolbe electrolysis is, therefore, important, especially because this has been an underrepresented research area and a similar intensive research trajectory is necessary as it was for water electrolysis to lower the overpotentials for acceptable energy efficiencies. Another area of research is to find out why Kolbe electrolysis often works better with synthetic substrates than real substrates. Low concentrations of fermentation side products may prevent a proper Kolbe reaction. Finally, even though Kolbe electrolysis after fermentation could be advantageous due to the possibility of intermittent power utilization based on electric power pricing, Kolbe products (fuels) are considerably cheaper than medium-chain carboxylates. This destruction in value currently prevents scale-up efforts and further development. Possibly an advantageous and cheaper extraction with Kolbe electrolysis could partly overcome the destruction in product value.
Electromethanogenesis, within the area of microbial electrosynthesis, has seen considerable improvements in current densities and methane production rates in recent years. The transfer of the zero-gap cell design from abiotic electrochemistry to microbial electrochemistry has driven this. A recent solution to completely protect the cathode catalyst from biology by introducing a hydrogen-diffusing Teflon layer within the zero-gas cell design has even further increased the current densities to 30 mA cm−2.152 However, this raises the question of whether this is even a microbial electrochemical cell and why not just combine electrochemistry followed by biology (power-to-gas with biomethanation). Thus, researchers in the field of microbial electrochemistry now need to considerably improve the production rates of microbial electrochemical systems to be on par with abiotic electrochemistry, while electrochemistry is still truly integrated with biology.
Specifically for microbial electrochemistry, there seems to currently be a discrepancy between a maximum current density of 1 mA cm−2 to circumvent bubble formation of hydrogen at the cathode144 and a required current density of 50–100 mA cm−2 to make the technology economically viable,151 as described earlier. To close this gap, we can envision two different research directions for microbial electrochemistry, which is in agreement with Jourdin and Burdyny.147 The first direction promotes active biofilms by developing high-surface-area, three-dimensional structures of cathodes, which reduce the needed specific current density per actual surface area, and which would evenly distribute only small bubbles if they are even formed. The second direction prevents biofilm formation all together, possibly with new catalyst layers, and bubble formation could aid in preventing biofilm growth. Without biofilms, we can simply recirculate planktonic methanogens from a bioreactor as the catholyte to take up the soluble hydrogen. The short contact time between planktonic cells and cathodes could reduce the stress that the microbes would encounter, however, care should be taken that all hydrogen is taken up to maintain high Coulombic efficiencies (and thus energy efficiencies) with increasing current densities. Staying within the area of microbial electrosynthesis, electrochemical formate production that is integrated into a bioreactor shows limited current densities and a severe incompatibility problem. Here, detailed research on the (detrimental) effect of media components on performance is required, but without pertinent breakthroughs, it is hard to imagine as a promising energy storage.
The current densities for charge storage systems are also too low. In fact, energy storage in rechargeable microbial electrochemical systems is still in its infancy. Due to the underlying physical restrictions and limited biofilm thicknesses, capacitive and pseudocapacitive energy storage in biofilm electrodes appears to be the most limited and can only be increased by using three-dimensional electrode structures. The use of these systems explicitly as energy storage systems appears to make little sense, particularly in comparison to conventional capacitors and rechargeable batteries. The higher charge densities have been achieved by exploiting inorganic compounds (e.g., elemental sulfur or sulfur compounds) that are being produced by microbial metabolism (Metabolic charge storage 1 in Table 1). Research efforts could, in the long-term, achieve storage capacities in the range of conventional rechargeable batteries. Yet, important issues, such as the improvement of electrochemical reversibility and chemical stability in a microbial environment, must be addressed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the VIP+ program of the Federal Ministry of Education and Research in Germany (03VP06911 and 03VP06912) to LTA and FH, the Deutsche Forschungsgemeinschaft (eBiotech, SPP2240 – project number 445506379) to LTA and FH, the Alexander von Humboldt Foundation in the framework of the Alexander von Humboldt Professorship to LTA, the Novo Nordisk Foundation CO2 Research Center with grant number NNF21SA0072700 to LTA, the Deutsche Forschungsgemeinschaft under Germany's Excellence Strategy (EXC 2124 – 390838134) to LTA and BM, and the Helmholtz Association within the integration platform at the UFZ to FH: Tapping nature's potential for sustainable production and a healthy environment. US acknowledges funding by the U.S. Office of Naval Research Global Program (Grant Nr. N62909-19-1-2025). We thank Dr James Byrne (University of Bristol, UK) for the helpful discussion around magnetosomes. Finally, we thank the three anonymous manuscript reviewers for making the review better.References
- R. Wiser, K. Jenni, J. Seel, E. Baker, M. Hand, E. Lantz and A. Smith, Nat. Energy, 2016, 1, 16135 CrossRef.
- S. Comello, S. Reichelstein and A. Sahoo, Renewable Sustainable Energy Rev., 2018, 92, 744–756 CrossRef.
- A. Das, L. Emele, F. Meinke-Hubeny, I. Moorkens, C. Nissen and M. Tomescu, Renewable energy in Europe 2018 recent growth and knock-on effects, Report EEA Report No 20/2018, European Environment Agency, Luxembourg, 2018.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- T. Persson, J. MurPhy, A.-K. Jannasch, E. Ahern, J. Liebetrau, M. Trommler and J. Toyama, A Perspective on the Potential Role of Biogas in Smart Energy Grids, IEA Bioenergy, 2014 Search PubMed.
- A. Ajanovic and R. Haas, Wiley Interdiscip. Rev.: Energy Environ., 2019, 8, e318 Search PubMed.
- A. Sternberg and A. Bardow, Energy Environ. Sci., 2015, 8, 389–400 RSC.
- A. R. Dehghani-Sanij, E. Tharumalingam, M. B. Dusseault and R. Fraser, Renewable Sustainable Energy Rev., 2019, 104, 192–208 CrossRef.
- German Federal Ministry for Economic Affairs and Energy, Conventional Energy Sources: Still indispensable for a reliable energy supply, https://www.bmwi.de/Redaktion/EN/Dossier/conventional-energy-sources.html, (accessed Jan. 21, 2020, 2020).
- M. Bailera, P. Lisbona, L. M. Romeo and S. Espatolero, Renewable Sustainable Energy Rev., 2017, 69, 292–312 CrossRef CAS.
- M. Thema, F. Bauer and M. Sterner, Renewable Sustainable Energy Rev., 2019, 112, 775–787 CrossRef CAS.
- F. Harnisch and R. Lehneis, Next Sustainability, 2023, 2, 100010 CrossRef.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- S. Schiebahn, T. Grube, M. Robinius, V. Tietze, B. Kumar and D. Stolten, Int. J. Hydrogen Energy, 2015, 40, 4285–4294 CrossRef CAS.
- G. F. Swiegers, A. L. Hoang, A. Hodges, G. Tsekouras, C.-Y. Lee, K. Wagner and G. Wallace, Curr. Opin. Electrochem., 2022, 32, 100881 CrossRef CAS.
- C. Wulf, J. Linßen and P. Zapp, Energy Procedia, 2018, 155, 367–378 CrossRef.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- F. Zhang, L. Yu, L. Wu, D. Luo and Z. Ren, Trends Chem., 2021, 3, 485–498 CrossRef CAS.
- L. Yu, J. Xiao, C. Huang, J. Zhou, M. Qiu, Y. Yu, Z. Ren, C.-W. Chu and J. C. Yu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202382119 CrossRef CAS PubMed.
- C. Schnuelle, J. Thoeming, T. Wassermann, P. Thier, A. von Gleich and S. Goessling-Reisemann, Energy Res. Soc. Sci., 2019, 51, 187–197 CrossRef.
- A. Mayyas, M. Ruth, B. Pivovar, G. Bender and K. Wipke, Manufacturing cost analysis for proton exchange membrane water electrolyzers, NREL/Division Report NREL/TP-6A20-72740, Institution, Golden, CO, 2018.
- C. I. Cabrera-Rodríguez, C. M. Cartin-Caballero, E. Platarou, F. A. de Weerd, L. A. M. van der Wielen and A. J. J. Straathof, Sep. Purif. Technol., 2018, 203, 56–65 CrossRef.
- K. Hashimoto, Mater. Sci. Eng., A, 1994, 179–180, 27–30 CrossRef.
- K. Hashimoto, M. Yamasaki, K. Fujimura, T. Matsui, K. Izumiya, M. Komori, A. A. El-Moneim, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Asami, Mater. Sci. Eng., A, 1999, 267, 200–206 CrossRef.
- B. Molitor, H. Richter, M. E. Martin, R. O. Jensen, A. Juminaga, C. Mihalcea and L. T. Angenent, Bioresour. Technol., 2016, 215, 386–396 CrossRef CAS PubMed.
- E. Zeyen, M. Victoria and T. Brown, Nat. Commun., 2023, 14, 3743 CrossRef CAS PubMed.
- N. Warwick, P. Griffiths, J. Keeble, A. Archibald, J. Pyle and K. Shine, Atmospheric implications of increased hydrogen use, U. Government/Division, Institution, 2022.
- R. A. Field and R. G. Derwent, Int. J. Hydrogen Energy, 2021, 46, 30190–30203 CrossRef CAS.
- M. Younas, L. Loong Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- M. Lehner, R. Tichler, H. Steinmüller and M. Koppe, Power-to-gas: Technology and business models, Springer, Cham, 2014 Search PubMed.
- M. R. Martin, J. J. Fornero, R. Stark, L. Mets and L. T. Angenent, Archaea, 2013, 157529 Search PubMed.
- L. Rachbauer, G. Voitl, G. Bochmann and W. Fuchs, Appl. Energy, 2016, 180, 483–490 CrossRef CAS.
- D. Strübing, A. B. Moeller, B. Mößnang, M. Lebuhn, J. E. Drewes and K. Koch, Appl. Energy, 2018, 232, 543–554 CrossRef.
- W. Liu, F. Wen and Y. Xue, J. Mod. Power Syst. Clean Energy, 2017, 5, 439–450 CrossRef.
- S. Rittmann, A. Seifert and C. Herwig, Biomass Bioenergy, 2012, 36, 293–301 CrossRef CAS.
- L. T. Angenent, J. G. Usack, J. Xu, D. Hafenbradl, R. Posmanik and J. W. Tester, Bioresour. Technol., 2018, 247, 1085–1094 CrossRef CAS PubMed.
- R. K. Thauer, A.-K. Kaster, H. Seedorf, W. Buckel and R. Hedderich, Nat. Rev. Microbiol., 2008, 6, 579–591 CrossRef CAS PubMed.
- N. R. Pace, Science, 1997, 276, 734–740 CrossRef CAS PubMed.
- K. C. Costa and J. A. Leigh, Curr. Opin. Biotechnol, 2014, 29, 70–75 CrossRef CAS PubMed.
- R. K. Thauer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15084 CrossRef CAS PubMed.
- P. Schönheit, J. Moll and R. K. Thauer, Arch. Microbiol., 1980, 127, 59–65 CrossRef.
- N. Schill and U. von Stockar, Thermochim. Acta, 1995, 251, 71–77 CrossRef CAS.
- N. Schill, W. M. van Gulik, D. Voisard and U. von Stockar, Biotechnol. Bioeng., 1996, 51, 645–658 CrossRef CAS PubMed.
- C. Fink, S. Beblawy, M. Enkerlin Andreas, L. Mühling, T. Angenent Largus, B. Molitor and M. Schleper Christa, mBio, 2021, 12, e02766–02721 CrossRef CAS PubMed.
- L.-M. Mauerhofer, S. Zwirtmayr, P. Pappenreiter, S. Bernacchi, A. H. Seifert, B. Reischl, T. Schmider, R.-S. Taubner, C. Paulik and S. K. M. R. Rittmann, Commun. Biol., 2021, 4, 289 CrossRef CAS PubMed.
- P. A. Pappenreiter, S. Zwirtmayr, L.-M. Mauerhofer, S. K.-M. R. Rittmann and C. Paulik, Eng. Life Sci., 2019, 19, 537–544 CrossRef CAS PubMed.
- D. Rusmanis, R. O’Shea, D. M. Wall and J. D. Murphy, Bioengineered, 2019, 10, 604–634 CrossRef CAS PubMed.
- D. Fontaine, P. Grima, M. Hoerl, L. Mets, M. Forstmeier and D. Hafenbradl, Commun. Agric. Appl. Biol. Sci., 2017, 81, 183–187 Search PubMed.
- M. Thema, T. Weidlich, M. Hörl, A. Bellack, F. Mörs, F. Hackl, M. Kohlmayer, J. Gleich, C. Stabenau, T. Trabold, M. Neubert, F. Ortloff, R. Brotsack, D. Schmack, H. Huber, D. Hafenbradl, J. Karl and M. Sterner, Energies, 2019, 12, 1670 CrossRef CAS.
- N. A. Schill, J. S. Liu and U. von Stockar, Biotechnol. Bioeng., 1999, 64, 74–81 CrossRef CAS PubMed.
- U. von Stockar and J. S. Liu, Biochim. Biophys. Acta Bioenerg., 1999, 1412, 191–211 CrossRef CAS PubMed.
- R. Muñoz-Tamayo, M. Popova, M. Tillier, D. P. Morgavi, J.-P. Morel, G. Fonty and N. Morel-Desrosiers, PLoS One, 2019, 14, e0226243 CrossRef PubMed.
- A. J. Guneratnam, E. Ahern, J. A. FitzGerald, S. A. Jackson, A. Xia, A. D. W. Dobson and J. D. Murphy, Bioresour. Technol., 2017, 225, 308–315 CrossRef CAS PubMed.
- F. Liew, M. E. Martin, R. C. Tappel, B. D. Heijstra, C. Mihalcea and M. Köpke, Front. Microbiol., 2016, 7, 694 Search PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- N. Fackler, B. D. Heijstra, B. J. Rasor, H. Brown, J. Martin, Z. Ni, K. M. Shebek, R. R. Rosin, S. D. Simpson, K. E. Tyo, R. J. Giannone, R. L. Hettich, T. J. Tschaplinski, C. Leang, S. D. Brown, M. C. Jewett and M. Köpke, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 439–470 CrossRef PubMed.
- F. R. Bengelsdorf and P. Dürre, Microb. Biotechnol., 2017, 10, 1167–1170 CrossRef PubMed.
- H. L. Drake, A. S. Gößner and S. L. Daniel, Ann. N. Y. Acad. Sci., 2008, 1125, 100–128 CrossRef CAS PubMed.
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, 2008, 1784, 1873–1898 CrossRef CAS PubMed.
- M. Köpke, C. Held, S. Hujer, H. Liesegang, A. Wiezer, A. Wollherr, A. Ehrenreich, W. Liebl, G. Gottschalk and P. Dürre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13087–13092 CrossRef PubMed.
- H. Richter, M. E. Martin and L. T. Angenent, Energies, 2013, 6, 3987–4000 CrossRef CAS.
- M. Diender, A. J. M. Stams and D. Z. Sousa, Biotechnol. Biofuels, 2016, 9, 82 CrossRef PubMed.
- H. Richter, B. Molitor, M. Diender, D. Z. Sousa and L. T. Angenent, Front. Microbiol., 2016, 7, 1773 Search PubMed.
- N. Qureshi, I. S. Maddox and A. Friedl, Biotechnol. Prog., 1992, 8, 382–390 CrossRef CAS.
- C. M. Humphreys and N. P. Minton, Curr. Opin. Biotechnol, 2018, 50, 174–181 CrossRef CAS PubMed.
- F. J. Annan, B. Al-Sinawi, C. M. Humphreys, R. Norman, K. Winzer, M. Köpke, S. D. Simpson, N. P. Minton and A. M. Henstra, Appl. Microbiol. Biotechnol., 2019, 103, 4633–4648 CrossRef CAS PubMed.
- R. Zhao, Y. Liu, H. Zhang, C. Chai, J. Wang, W. Jiang and Y. Gu, ACS Synth. Biol., 2019, 8, 2270–2279 CrossRef CAS PubMed.
- G. Philipps, S. de Vries and S. Jennewein, Biotechnol. Biofuels, 2019, 12, 112 CrossRef PubMed.
- P.-F. Xia, I. Casini, S. Schulz, C.-M. Klask, L. T. Angenent and B. Molitor, ACS Synth. Biol., 2020, 9, 2162–2171 CrossRef CAS PubMed.
- F. Liew, A. M. Henstra, M. Köpke, K. Winzer, S. D. Simpson and N. P. Minton, Metab. Eng., 2017, 40, 104–114 CrossRef CAS PubMed.
- F. Liew, A. M. Henstra, K. Winzer, M. Köpke, S. D. Simpson and N. P. Minton, mBio, 2016, 7, e00427–00416 CrossRef CAS PubMed.
- F. E. Liew, R. Nogle, T. Abdalla, B. J. Rasor, C. Canter, R. O. Jensen, L. Wang, J. Strutz, P. Chirania, S. De Tissera, A. P. Mueller, Z. Ruan, A. Gao, L. Tran, N. L. Engle, J. C. Bromley, J. Daniell, R. Conrado, T. J. Tschaplinski, R. J. Giannone, R. L. Hettich, A. S. Karim, S. D. Simpson, S. D. Brown, C. Leang, M. C. Jewett and M. Köpke, Nat. Biotechnol., 2022, 40, 335–344 CrossRef CAS PubMed.
- B. Molitor, E. Marcellin and L. T. Angenent, Curr. Opin. Chem. Biol., 2017, 41, 84–92 CrossRef CAS PubMed.
- D. F. Emerson, B. M. Woolston, N. Liu, M. Donnelly, D. H. Currie and G. Stephanopoulos, Biotechnol. Bioeng., 2019, 116, 294–306 CrossRef CAS PubMed.
- C.-M. Klask, N. Kliem-Kuster, B. Molitor and L. T. Angenent, Front. Microbiol., 2020, 11, 724 CrossRef PubMed.
- J. K. Heffernan, K. Valgepea, R. de Souza Pinto Lemgruber, I. Casini, M. Plan, R. Tappel, S. D. Simpson, M. Köpke, L. K. Nielsen and E. Marcellin, Front. Bioeng. Biotechnol., 2020, 8, 204 CrossRef PubMed.
- P. Hu, S. Chakraborty, A. Kumar, B. Woolston, H. Liu, D. Emerson and G. Stephanopoulos, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3773 CrossRef CAS PubMed.
- D. Kiefer, M. Merkel, L. Lilge, M. Henkel and R. Hausmann, Trends Biotechnol., 2021, 39, 397–411 CrossRef CAS PubMed.
- T. Lehtinen, H. Virtanen, S. Santala and V. Santala, Biotechnol. Biofuels, 2018, 11, 228 CrossRef PubMed.
- B. Molitor, A. Mishra and L. T. Angenent, Energy Environ. Sci., 2019, 12, 3515–3521 RSC.
- A. Mishra, J. N. Ntihuga, B. Molitor and L. T. Angenent, Joule, 2020, 4, 1142–1147 CrossRef.
- K. Schuchmann and V. Müller, Science, 2013, 342, 1382 CrossRef CAS PubMed.
- F. M. Schwarz, J. Moon, F. Oswald and V. Müller, Joule, 2022, 6, 1304–1319 CrossRef CAS.
- H. M. Dietrich, R. D. Righetto, A. Kumar, W. Wietrzynski, R. Trischler, S. K. Schuller, J. Wagner, F. M. Schwarz, B. D. Engel, V. Müller and J. M. Schuller, Nature, 2022, 607, 823–830 CrossRef CAS PubMed.
- M. Roger, F. Brown, W. Gabrielli and F. Sargent, Curr. Biol., 2018, 28, 140–145.e142 CrossRef CAS PubMed.
- F. Harnisch and C. Urban, Angew. Chem., Int. Ed., 2018, 57, 10016–10023 CrossRef CAS PubMed.
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22, 286–301 RSC.
- N. Shevchenko, J. Villafuerte, H. Ling, C. J. Walkling, D. D. Zhang, B. G. Harvey and M. Mascal, Sustainable Energy Fuels, 2023, 7, 569–573 RSC.
- U.S. DOE, Breaking the Biological Barriers to Cellulosic Ethanol: A Joint Research Agenda, U.S. Department of Energy Office of Science, 2006 Search PubMed.
- J. E. Matthiesen, M. Suástegui, Y. Wu, M. Viswanathan, Y. Qu, M. Cao, N. Rodriguez-Quiroz, A. Okerlund, G. Kraus, D. R. Raman, Z. Shao and J.-P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 7098–7109 CrossRef CAS.
- M. Kimming, C. Sundberg, Å. Nordberg, A. Baky, S. Bernesson, O. Norén and P. A. Hansson, Biomass Bioenergy, 2011, 35, 1572–1581 CrossRef CAS.
- M. Pöschl, S. Ward and P. Owende, Appl. Energy, 2010, 87, 3305–3321 CrossRef.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783 CrossRef CAS PubMed.
- M. Suastegui, E. Matthiesen John, M. Carraher Jack, N. Hernandez, N. Rodriguez Quiroz, A. Okerlund, W. Cochran Eric, Z. Shao and J.-P. Tessonnier, Angew. Chem., Int. Ed., 2016, 55, 2368–2373 CrossRef CAS PubMed.
- L. T. Angenent, H. Richter, W. Buckel, C. M. Spirito, K. J. J. Steinbusch, C. M. Plugge, D. P. B. T. B. Strik, T. I. M. Grootscholten, C. J. N. Buisman and H. V. M. Hamelers, Environ. Sci. Technol., 2016, 50, 2796–2810 CrossRef CAS PubMed.
- A. Duber, R. Zagrodnik, J. Chwialkowska, W. Juzwa and P. Oleskowicz-Popiel, Sci. Total Environ., 2020, 728, 138814 CrossRef CAS PubMed.
- M. T. Agler, B. A. Wrenn, S. H. Zinder and L. T. Angenent, Trends Biotechnol., 2011, 29, 70–78 CrossRef CAS PubMed.
- M. T. Agler, C. M. Spirito, J. G. Usack, J. J. Werner and L. T. Angenent, Energy Environ. Sci., 2012, 5, 8189–8192 RSC.
- T. I. M. Grootscholten, K. J. J. Steinbusch, H. V. M. Hamelers and C. J. N. Buisman, Bioresour. Technol., 2013, 136, 735–738 CrossRef CAS PubMed.
- R. Palomo-Briones, J. Xu, C. M. Spirito, J. G. Usack, L. H. Trondsen, J. J. L. Guzman and L. T. Angenent, Chem. Eng. J., 2022, 446, 137170 CrossRef CAS.
- M. Roghair, T. Hoogstad, D. P. B. T. B. Strik, C. M. Plugge, P. H. A. Timmers, R. A. Weusthuis, M. E. Bruins and C. J. N. Buisman, Environ. Sci. Technol., 2018, 52, 1496–1505 CrossRef CAS PubMed.
- J. Xu, J. Hao, J. J. L. Guzman, C. M. Spirito, L. A. Harroff and L. T. Angenent, Joule, 2018, 2, 280–295 CrossRef CAS.
- S. Esquivel-Elizondo, C. Bağcı, M. Temovska, B. S. Jeon, I. Bessarab, R. B. H. Williams, D. H. Huson and L. T. Angenent, Front. Microbiol., 2021, 11, 594524 CrossRef PubMed.
- W. Buckel and R. K. Thauer, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 94–113 CrossRef CAS PubMed.
- H. A. Barker, M. D. Kamen and B. T. Bornstein, Proc. Natl. Acad. Sci. U. S. A., 1945, 31, 373–381 CrossRef CAS PubMed.
- H. Kim, S. Kang and B.-I. Sang, Bioresour. Technol., 2022, 344, 126211 CrossRef CAS PubMed.
- T. I. M. Grootscholten, F. Kinsky dal Borgo, H. V. M. Hamelers and C. J. N. Buisman, Biomass Bioenergy, 2013, 48, 10–16 CrossRef CAS.
- P. Candry, J. M. Carvajal-Arroyo, S. Pratt, J. Sousa, Ç. Akyol, F. Fatone and R. Ganigué, in Resource Recovery from Water: Principles and Application, ed. I. Pikaar, J. Guest, R. Ganigué, P. Jensen, K. Rabaey, T. Seviour, J. Trimmer, O. van der Kolk, C. Vaneeckhaute and W. Verstraete, IWA Publishing, 2022 DOI:10.2166/9781780409566_0159.
- S. E. Daly, J. G. Usack, L. A. Harroff, J. G. Booth, M. P. Keleman and L. T. Angenent, ACS Sustainable Chem. Eng., 2020, 8, 13934–13944 CrossRef CAS.
- L. T. Angenent, D. Zheng, S. Sung and L. Raskin, Water Environ. Res., 2002, 74, 450–461 CrossRef CAS PubMed.
- C. Urban, J. Xu, H. Strauber, T. R. dos Santos Dantas, J. Muhlenberg, C. Hartig, L. T. Angenent and F. Harnisch, Energy Environ. Sci., 2017, 10, 2231–2244 RSC.
- K. Neubert, M. Hell, M. Chávez Morejón and F. Harnisch, ChemSusChem, 2022, 15, e202201426 CrossRef CAS PubMed.
- L. F. M. Rosa, K. Röhring and F. Harnisch, Fuel, 2024, 356, 129590 CrossRef CAS.
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- P. F. Levy, J. E. Sanderson, R. G. Kispert and D. L. Wise, Enzyme Microb. Technol., 1981, 3, 207–215 CrossRef CAS.
- P. F. Levy, J. E. Sanderson and L. K. Cheng, J. Electrochem. Soc., 1984, 131, 773–777 CrossRef CAS.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- D. Klüh, W. Waldmüller and M. Gaderer, Clean Technol., 2021, 3, 1–18 CrossRef.
- W. C. Khor, S. Andersen, H. Vervaeren and K. Rabaey, Biotechnol. Biofuels, 2017, 10, 180 CrossRef PubMed.
- C. Stang and F. Harnisch, ChemSusChem, 2016, 9, 50–60 CrossRef CAS PubMed.
- C. Urban and F. Harnisch, ChemElectroChem, 2017, 4, 1378–1389 CrossRef CAS.
- T. J. Mason, J. P. Lorimer and D. J. Walton, Ultrasonics, 1990, 28, 333–337 CrossRef CAS.
- K. Neubert, M. Schmidt and F. Harnisch, ChemSusChem, 2021, 14, 3097–3109 CrossRef CAS PubMed.
- Y. Hioki, M. Costantini, J. Griffin, K. C. Harper, M. P. Merini, B. Nissl, Y. Kawamata and P. S. Baran, Science, 2023, 380, 81–87 CrossRef CAS PubMed.
- P. Drögemüller, T. Stobbe and U. Schröder, ChemSusChem, 2024, 17, e202300973 CrossRef PubMed.
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schäfer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS.
- T. R. dos Santos, F. Harnisch, P. Nilges and U. Schröder, ChemSusChem, 2015, 8, 886–893 CrossRef CAS PubMed.
- J. Meyers, J. B. Mensah, F. J. Holzhäuser, A. Omari, C. C. Blesken, T. Tiso, S. Palkovits, L. M. Blank, S. Pischinger and R. Palkovits, Energy Environ. Sci., 2019, 12, 2406–2411 RSC.
- T. Tiso, R. Zauter, H. Tulke, B. Leuchtle, W.-J. Li, B. Behrens, A. Wittgens, F. Rosenau, H. Hayen and L. M. Blank, Microb. Cell Fact., 2017, 16, 225 CrossRef PubMed.
- J. Ranninger, P. Nikolaienko, K. J. J. Mayrhofer and B. B. Berkes, ChemSusChem, 2022, 15, e202102228 CrossRef CAS PubMed.
- N. Kassem, J. Hockey, C. Lopez, L. Lardon, L. T. Angenent and J. W. Tester, Sustainable Energy Fuels, 2020, 4, 4644–4661 RSC.
- U. Schröder, F. Harnisch and L. T. Angenent, Energy Environ. Sci., 2015, 8, 513–519 RSC.
- K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers and D. R. Lovley, mBio, 2010, 1, e00103-10 CrossRef PubMed.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS PubMed.
- M. Villano, F. Aulenta, C. Ciucci, T. Ferri, A. Giuliano and M. Majone, Bioresour. Technol., 2010, 101, 3085–3090 CrossRef CAS PubMed.
- M. Rosenbaum, F. Aulenta, M. Villano and L. T. Angenent, Bioresour. Technol., 2011, 102, 324–333 CrossRef CAS PubMed.
- J. S. Deutzmann, M. Sahin and A. M. Spormann, mBio, 2015, 6, e00496-15 CrossRef PubMed.
- S. T. Boto, B. Bardl, F. Harnisch and M. A. Rosenbaum, Green Chem., 2023, 25, 4375–4386 RSC.
- A.-E. Rotaru, N. R. Posth, C. R. Löscher, M. R. Miracle, E. Vicente, R. P. Cox, J. Thompson, S. W. Poulton and B. Thamdrup, Limnetica, 2019, 38, 21–40 Search PubMed.
- F. Kracke, A. B. Wong, K. Maegaard, J. S. Deutzmann, M. A. Hubert, C. Hahn, T. F. Jaramillo and A. M. Spormann, Commun. Chem., 2019, 2, 45 CrossRef.
- F. Kracke, J. S. Deutzmann, W. Gu and A. M. Spormann, Green Chem., 2020, 22, 6194–6203 RSC.
- F. Kracke, J. S. Deutzmann, B. S. Jayathilake, S. H. Pang, S. Chandrasekaran, S. E. Baker and A. M. Spormann, Front. Microbiol., 2021, 12, 696473 CrossRef PubMed.
- J. K. Lee and A. Bazylak, Joule, 2021, 5, 19–21 CrossRef.
- P. A. Kempler, Z. P. Ifkovits, W. Yu, A. I. Carim and N. S. Lewis, Energy Environ. Sci., 2021, 14, 414–423 RSC.
- L. Jourdin and T. Burdyny, Trends Biotechnol., 2021, 39, 359–369 CrossRef CAS PubMed.
- R. Phillips and C. W. Dunnill, RSC Adv., 2016, 6, 100643–100651 RSC.
- F. Geppert, D. Liu, E. Weidner and A. T. Heijne, Int. J. Hydrogen Energy, 2019, 44, 21464–21469 CrossRef CAS.
- G. Baek, R. Rossi, P. E. Saikaly and B. E. Logan, Water Res., 2022, 219, 118597 CrossRef CAS PubMed.
- A. Prévoteau, J. M. Carvajal-Arroyo, R. Ganigué and K. Rabaey, Curr. Opin. Biotechnol, 2020, 62, 48–57 CrossRef PubMed.
- R. Rad, T. Gehring, K. Pellumbi, D. Siegmund, E. Nettmann, M. Wichern and U.-P. Apfel, Cell Rep. Phys. Sci., 2023, 4, 101526 CrossRef CAS.
- R. Rossi, G. Baek and B. E. Logan, Environ. Sci. Technol., 2022, 56, 1211–1220 CrossRef CAS PubMed.
- I. Schwarz, A. Rieck, A. Mehmood, R. Bublitz, L. Bongers, D. Weuster-Botz and T.-P. Fellinger, ChemElectroChem, 2024, 11, e202300344 CrossRef CAS.
- P. Izadi and F. Harnisch, Joule, 2022, 6, 935–940 CrossRef.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596 CrossRef CAS PubMed.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electrochem. Soc., 1990, 137, 1772–1778 CrossRef CAS.
- Y. Pei, H. Zhong and F. Jin, Energy Sci. Eng., 2021, 9, 1012–1032 CrossRef CAS.
- C. Gimkiewicz, R. Hegner, M. F. Gutensohn, C. Koch and F. Harnisch, ChemSusChem, 2017, 10, 958–967 CrossRef CAS PubMed.
- R. Hegner, L. F. M. Rosa and F. Harnisch, Appl. Catal., B, 2018, 238, 546–556 CrossRef CAS.
- R. Hegner, K. Neubert, L. F. M. Rosa and F. Harnisch, ChemElectroChem, 2019, 6, 3731–3735 CrossRef CAS.
- R. Hegner, K. Neubert, C. Kroner, D. Holtmann and F. Harnisch, ChemSusChem, 2020, 13, 5295–5300 CrossRef CAS PubMed.
- A. ter Heijne, M. A. Pereira, J. Pereira and T. Sleutels, Trends Biotechnol., 2021, 39, 34–42 CrossRef CAS PubMed.
- W. Habermann and E. H. Pommer, Appl. Microbiol. Biotechnol., 1991, 35, 128–133 CrossRef CAS.
- A. E. Beasley, A. L. Powell and A. Adamatzky, arXiv, 2020, preprint, arXiv:2003.07816 DOI:10.48550/arXiv.2003.07816.
- A. Deeke, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2012, 46, 3554–3560 CrossRef CAS PubMed.
- A. Deeke, T. H. J. A. Sleutels, A. T. Heijne, H. V. M. Hamelers and C. J. N. Buisman, J. Power Sources, 2013, 243, 611–616 CrossRef CAS.
- L. Caizán-Juanarena, C. Borsje, T. Sleutels, D. Yntema, C. Santoro, I. Ieropoulos, F. Soavi and A. ter Heijne, Biotechnol. Adv., 2020, 39, 107456 CrossRef PubMed.
- A. Deeke, T. H. J. A. Sleutels, T. F. W. Donkers, H. V. M. Hamelers, C. J. N. Buisman and A. Ter Heijne, Environ. Sci. Technol., 2015, 49, 1929–1935 CrossRef CAS PubMed.
- C. Borsje, T. Sleutels, C. J. N. Buisman and A. ter Heijne, J. Environ. Chem. Eng., 2021, 9, 105556 CrossRef CAS.
- N. S. Malvankar, T. Mester, M. T. Tuominen and D. R. Lovley, Chem. Phys. Chem., 2012, 13, 463–468 CrossRef CAS PubMed.
- A. t Heijne, D. Liu, M. Sulonen, T. Sleutels and F. Fabregat-Santiago, J. Power Sources, 2018, 400, 533–538 CrossRef CAS.
- I. Schmidt, A. Pieper, H. Wichmann, B. Bunk, K. Huber, J. Overmann, P. J. Walla and U. Schröder, ChemElectroChem, 2017, 4, 2515–2519 CrossRef CAS.
- Y. Liu, F. Harnisch, K. Fricke, R. Sietmann and U. Schröder, Biosens. Bioelectron., 2008, 24, 1006–1011 CrossRef CAS PubMed.
- H. Vali and J. L. Kirschvink, in Iron Biominerals, ed. R. B. Frankel and R. P. Blakemore, Springer US, Boston, MA, 1991, pp. 97–115 DOI:10.1007/978-1-4615-3810-3_7.
- R. E. Kopp, C. Z. Nash, J. L. Kirschvink and J. R. Leadbetter, AGU Fall Meeting, 2004.
- S. Freguia, K. Rabaey, Z. Yuan and J. Keller, Environ. Sci. Technol., 2007, 41, 2915–2921 CrossRef CAS PubMed.
- K. Nishio, Y. Kimoto, J. Song, T. Konno, K. Ishihara, S. Kato, K. Hashimoto and S. Nakanishi, Environ. Sci. Technol. Lett., 2014, 1, 40–43 CrossRef CAS.
- F. Kubannek, U. Schröder and U. Krewer, Bioelectrochemistry, 2018, 121, 160–168 CrossRef CAS PubMed.
- P. Izadi, M. N. Gey, N. Schlüter and U. Schröder, iScience, 2021, 24, 102822 CrossRef CAS PubMed.
- M. D. Yates, L. Ma, J. Sack, J. P. Golden, S. M. Strycharz-Glaven, S. R. Yates and L. M. Tender, Environ. Sci. Technol. Lett., 2017, 4, 374–379 CrossRef CAS.
- R. L. Mickol, B. J. Eddie, A. P. Malanoski, M. D. Yates, L. M. Tender and S. M. Glaven, Appl. Environ. Microbiol., 2021, 87, e01676–01621 CrossRef CAS PubMed.
- S. D. Molenaar, M. Elzinga, S. G. Willemse, T. Sleutels, A. ter Heijne and C. J. N. Buisman, ChemElectroChem, 2019, 6, 2464–2473 CrossRef CAS.
- S. D. Molenaar, A. R. Mol, T. H. J. A. Sleutels, A. ter Heijne and C. J. N. Buisman, Environ. Sci. Technol. Lett., 2016, 3, 144–149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |