
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards automated inclusion of autoxidation chemistry in models: from precursors to atmospheric implications†
Lukas
Pichelstorfer
 *abc,
Pontus
Roldin
d,
Matti
Rissanen
ef,
Noora
Hyttinen
g,
Olga
Garmash
eh,
Carlton
Xavier
cdi,
Putian
Zhou
c,
Petri
Clusius
c,
Benjamin
Foreback
cj,
Thomas
Golin Almeida
cf,
Chenjuan
Deng
k,
Metin
Baykara
cjl,
Theo
Kurten
f and
Michael
Boy
cjm
*abc,
Pontus
Roldin
d,
Matti
Rissanen
ef,
Noora
Hyttinen
g,
Olga
Garmash
eh,
Carlton
Xavier
cdi,
Putian
Zhou
c,
Petri
Clusius
c,
Benjamin
Foreback
cj,
Thomas
Golin Almeida
cf,
Chenjuan
Deng
k,
Metin
Baykara
cjl,
Theo
Kurten
f and
Michael
Boy
cjm
api-numerics, Neumarkt amW., 5202, Austria. E-mail: office@pi-numerics.com
bChemistry and Physics of Materials, University of Salzburg, A-5020, Austria
cInstitute for Atmospheric and Earth System Research/Physics, University of Helsinki, 00560 Helsinki, Finland
dDivision of Nuclear Physics, Department of Physics, Lund University, P. O. Box 118, 221 00 Lund, Sweden
eAerosol Physics Laboratory, Tampere University, 33720 Tampere, Finland
fDepartment of Chemistry, University of Helsinki, 00014 Helsinki, Finland
gDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, FI-40014 Jyväskylä, Finland
hDepartment of Atmospheric Sciences, University of Washington, Seattle, WA, USA
iSMHI/Swedish Meteorological and Hydrological Institute Research Department, Unit of Meteorology/Environment and Climate, SE-601 76 Norrköping, Sweden
jAtmospheric Modelling Centre Lahti, Niemenkatu 73, Lahti University Campus, 15140 Lahti, Finland
kState Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, 100084 Beijing, China
lClimate and Marine Sciences Department, Eurasia Institute of Earth Sciences, Istanbul Technical University, Maslak, Istanbul 34469, Turkey
mSchool of Engineering Science, Lappeenranta-Lahti University of Technology, 53851 Lappeenranta, Finland
First published on 9th July 2024
Abstract
In the last few decades, atmospheric formation of secondary organic aerosols (SOA) has gained increasing attention due to their impact on air quality and climate. However, methods to predict their abundance are mainly empirical and may fail under real atmospheric conditions. In this work, a close-to-mechanistic approach allowing SOA quantification is presented, with a focus on a chain-like chemical reaction called “autoxidation”. A novel framework is employed to (a) describe the gas-phase chemistry, (b) predict the products' molecular structures and (c) explore the contribution of autoxidation chemistry on SOA formation under various conditions. As a proof of concept, the method is applied to benzene, an important anthropogenic SOA precursor. Our results suggest autoxidation to explain up to 100% of the benzene-SOA formed under low-NOx laboratory conditions. Under atmospheric-like day-time conditions, the calculated benzene-aerosol mass continuously forms, as expected based on prior work. Additionally, a prompt increase, driven by the NO3 radical, is predicted by the model at dawn. This increase has not yet been explored experimentally and stresses the potential for atmospheric SOA formation via secondary oxidation of benzene by O3 and NO3.
Environmental significanceSecondary organic aerosols (SOA) are an important factor in predicting air quality and climate forcing by seeding clouds. During SOA formation, organic oxidation products of sufficiently low volatility partition to the particle phase to form airborne mass. A molecular level mechanistic description of this process seems out of reach for the time being: after the oxidation initiation, the chemistry quickly becomes complex and results in a vast variety of progressively oxygenated reaction intermediates and products, which are impossible to selectively follow by any current experimental methodologies. Similarly, the huge number of potential reaction paths render high-level computational predictions too expensive to derive. We propose an alternative methodology, in which highly sensitive chemical ionisation mass-spectrometry (CIMS) detection is utilized for semi-empirical autoxidation model generation. However, CIMS misses information about the isomeric oxidation pathways. In the first step to overcome this, a collective behaviour of isomeric radical species of unknown structures is considered. This assumption allows us to semi-empirically create lumped chemical schemes. |
1 Introduction
Adverse effects resulting from poor air quality nowadays represent one of the largest risks to health, causing several millions of premature deaths every year.1 This makes air pollution the top environmental mortality risk, posing a much higher risk than e.g. polluted water.2 In 2019, more than 99% of the global population lived in areas with outdoor PM2.5 (i.e. inhalable particles 2.5 μm or less in aerodynamic diameter3) levels not meeting WHO guidelines.4 In addition, while not being relevant in terms of total mass exposure, ultrafine particles (i.e., PM0.1) pose a severe health risk. After entering the body mainly through the lungs, ultrafine particles can relocate within the body to accumulate in all organs, including the brain.5 Typical symptoms include systemic inflammation and can be severe.6 The metric to quantify the hazard of inhalable aerosols is, despite being studied intensely, not fully illuminated.7 Airborne particle mass, number, and surface, as well as oxidative potential, are considered to be drivers for chronic and acute effects. Recently, Daellenbach et al. have suggested secondary organic aerosols (SOA; i.e. aerosols formed in the air from gaseous precursors8) to play a major role in contributing to aerosol mass burden and, in particular, to its oxidative potential at the European level.9 Further, they found that ambient, respirable particulate pollutants' toxicity is dominated by SOA from precursors emitted by anthropogenic activities. While primary aerosol particles impact air quality more locally and formation can be identified comparatively simply, the sources and formation pathways of SOA are complex and transport of precursor molecules can occur over large distances.10,11In the last decade, it has been shown, mainly by means of experiment, that highly oxygenated organic molecules (HOM, as defined by Bianchi et al.12) can form quickly upon oxidation of precursor volatile organic compounds (VOC) under atmospheric conditions.12,13 These, often short-lived, molecules deviate strongly from their parent VOC regarding their physical and chemical properties.14 This particularly impacts their SOA forming potential, as the saturation vapor pressure (psat) which governs the partition to the particle phase, drops significantly with advancing functionalization of the molecules.15 The formation of HOM involves a key chemical process called autoxidation. It describes a chain-like process of intra-molecular H-abstraction and O2 addition.16 Besides the advances in experimental approaches, findings based on theoretical considerations are increasingly available.13,17–19 Yet, there are few studies on deriving a mechanistic concept to predict SOA quantitatively based on the description of autoxidation chemistry under atmospheric conditions and covered by both theory and experiment.20,21 The approach by Donahue et al.,20 due to its formal simplicity, serves well to be applied in large scale models with substantial success. However, as the method does not aim to involve individual species but rather computes average parameters describing the evolution of gas phase chemistry and the partition of condensable vapors to the particle phase, the authors consider it less appropriate to approach a mechanistic understanding of the oxidation process. Thus, in the present work, we follow the approach by Roldin et al.,21 aiming for a close-to-mechanistic description of the autoxidation chemistry governing the process of molecular rearrangements and oxygen enrichment. We introduce the novel automated alkoxy/peroxy radical autoxidation mechanism framework (“autoAPRAM-fw” – see Methods Section 2.1 The autoAPRAM-fw for details), capable of setting up autoxidation chemistry schemes for any VOC system. Rate coefficients, if available, can be provided as an input. Otherwise they need to be derived from data. In this work, the framework is applied to benzene, a highly abundant and structurally prototypical aromatic molecule, related mainly to anthropogenic activities.11,22–25 Lately, there have also been reports of the importance of the biogenic emission of benzene and other BTEX species (i.e., toluene, ethylbenzene and xylenes).26,27 In this work, we constrain the rate coefficients of reactions related to benzene autoxidation chemistry. The approach is tested by reproducing pure gas-phase experiments and experiments investigating the SOA formation potential under high and low NOx conditions. Further, atmospheric implications are predicted by carrying out a parameterized study computing SOA mass yields for a range of benzene and NOx levels. Finally, atmospheric trajectory simulations are conducted to investigate the potential contribution of autoxidation chemistry and its impact on the benzene SOA evolution in the atmosphere.
2 Methods
In a first step, chemical reaction types describing benzene autoxidation chemistry are gathered from the literature (Section 2.2). Alongside28,29 a method describing the basic degradation of VOC in the atmosphere, we apply the autoAPRAM-fw to add, to data missing, the depiction of benzene autoxidation chemistry (Section 2.1). Simulation of pure gas-phase experiments allows missing information on reaction rate coefficients to be constrained (Section 2.3). In the next step, we assign potential molecular structures to the gas phase species formed (Section 2.4). This is done in order to compute their saturation vapor pressures by exploring several approaches such as state-of-the-art quantum chemistry calculations or group contribution methods (Section 2.5). In order to test the potential of the approach in predicting SOA formation, we replicate chamber experiments of benzene–OH oxidation in the presence of seed aerosol applying a detailed micro-physics and chemistry box model (Section 2.6). Potential atmospheric implications are illustrated by studying SOA yield under idealized atmospheric conditions (Section 2.6.4). This contrasts with findings from close-to-realistic Lagrangian-type atmospheric transport model calculations (Section 2.6.3).2.1 The autoAPRAM-fw
The automated alkoxy/peroxy radical autoxidation mechanism framework (autoAPRAM-fw) serves to generate a model describing autoxidation chemistry of VOCs in the gas phase. The chemistry scheme is based on MCM v3.3.1 describing the degradation of VOCs in the atmosphere.28,29 The autoAPRAM-fw consists of two modules: (a) autoReactions, which is applied to generate the differential equations describing autoxidation chemistry. It also defines product species names and creates a computer readable chemistry model. Further, it can be used to describe functional groups of product molecules; (b) autoSMILES, which creates potential structures of reaction products described by the SMILES convention.30 A graphical description of the framework is shown in Fig. 1.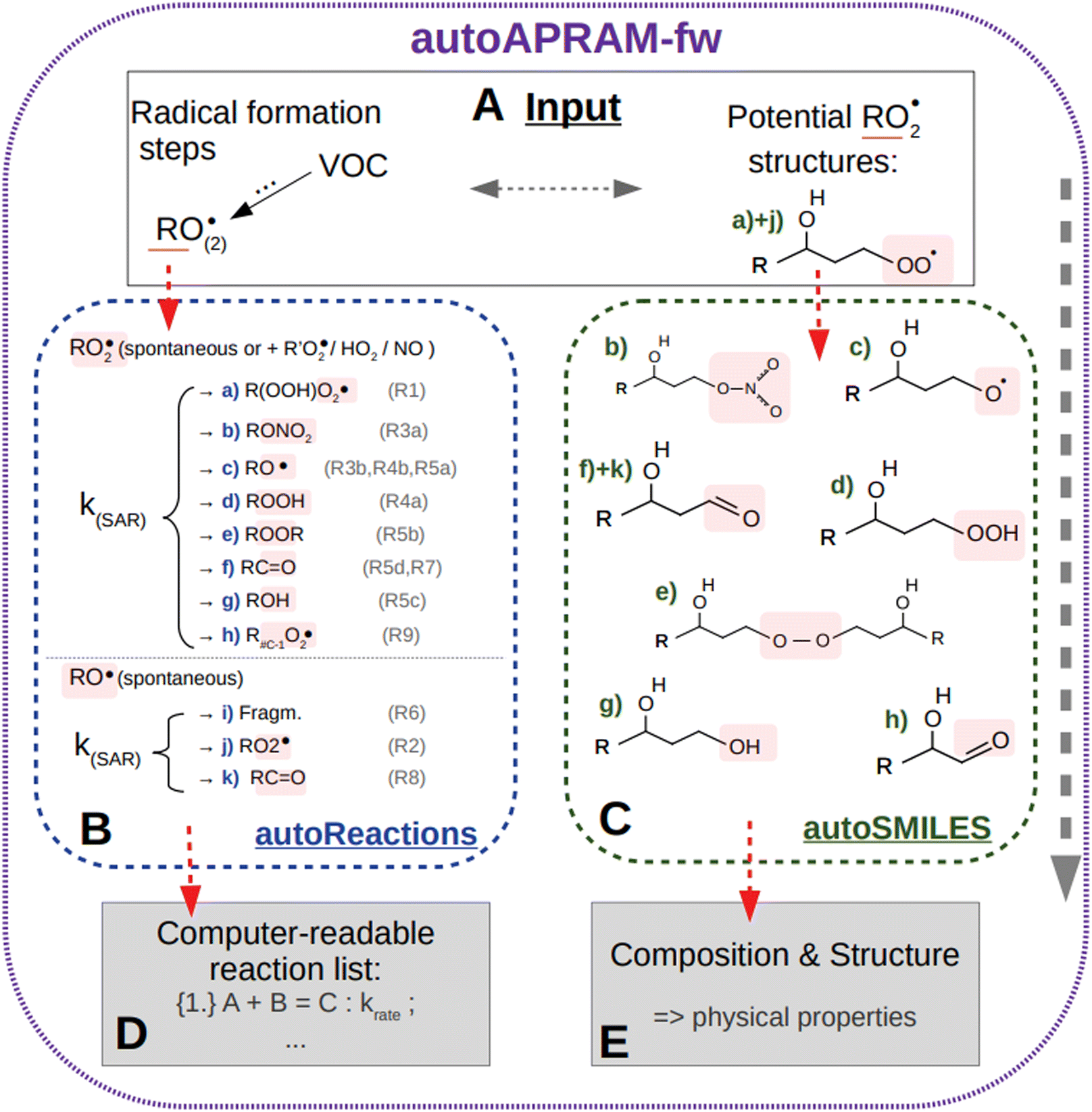 | ||
| Fig. 1 Overview of the autoAPRAM-fw. Sector (A) represents the input to submodules autoReactions (B) and autoSMILES (C), respectively. Outputs comprise a Fortran readable chemistry module from autoReactions (D), and molecular information on the species formed in autoSMILES (E). Note that the submodules may be run individually. | ||
As an input autoAPRAM-fw takes peroxy radical (RO2) names formed in MCM together with their atomic composition. Further, the RO2 composition of species formed by autoxidation chemistry which are not described in MCM needs to be provided by the user. They are not created by autoSMILES. After specification of the reaction types considered, autoReactions sets up a chemistry module. Reaction rate constants can be based on theory if available (e.g. structure–activity relationships – SAR) or can be set manually. Physical properties of the species formed can be determined from their probable structures, computed by autoSMILES, for the reaction types specified. In the present work we investigated the physical properties of product species using vapor pressure prediction methods “EVAPORATION”,31 “NANNOOLAL”32 and “MYRDAL/YALKOWSKI”33 for all species, and the selected species have been investigated using a high performance quantum chemistry method (COSMO-RS34–36).
The two modules autoReactions and autoSMILES can be run individually. The first one can be applied repeatedly to constrain the gas phase chemistry. Note that although the model can be used to produce close to explicit gas phase chemistry based on theory, there is still need for tuning the reaction rate constants such as branching ratios. AutoSMILES may be rerun to investigate the effect of varying structures of input-RO2 isomers. The framework itself is written in a way to describe any type of VOC undergoing autoxidation.
We are currently exploring the possibility of applying machine learning in the following way: a chemical scheme created by the autoAPRAM-fw is used together with MCM chemistry to represent a VOC system. Next, we optimise the newly developed chemistry schemes by performing an in-depth Bayesian analysis of the VOC system. To sample all model parameters, we use methods like Hamiltonian Monte Carlo with automatic differentiation and Markov Chain Monte Carlo (MCMC).37,38 Both techniques provide efficient solutions to high-dimensional problems. The goal is to sample and quantify model parameters to increase accuracy and reliability in chemical schemes.
2.2 Setting up the gas phase chemistry
The gas phase chemistry scheme generated is based on the master chemical mechanism (MCMv3.3.1), describing tropospheric degradation of hydrocarbons. However, it does not yet include autoxidation chemistry which is added by deploying the reaction types below: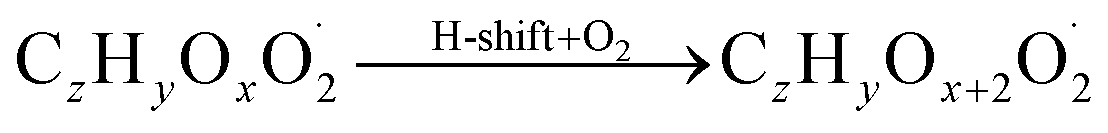 | (R1*) |
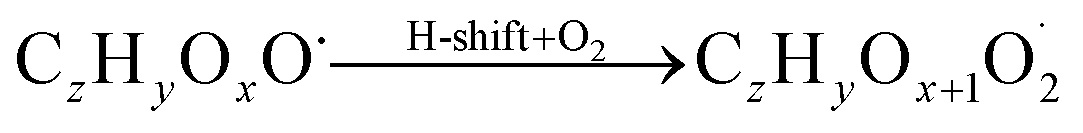 | (R2*) |
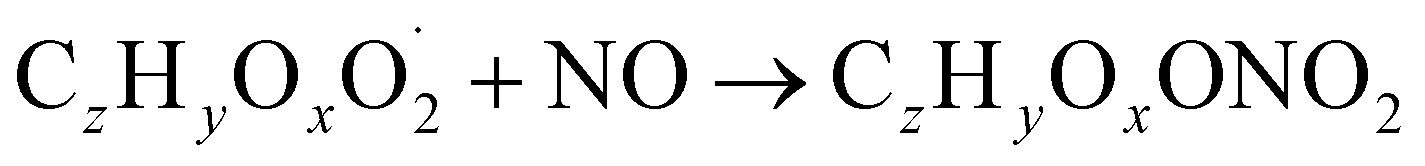 | (R3a) |
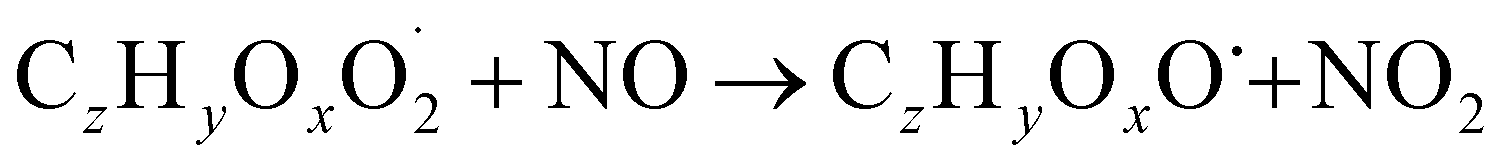 | (R3b) |
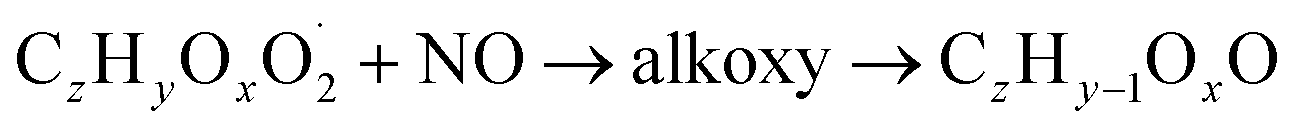 | (R3c) |
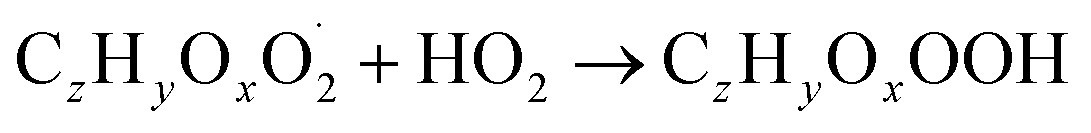 | (R4a) |
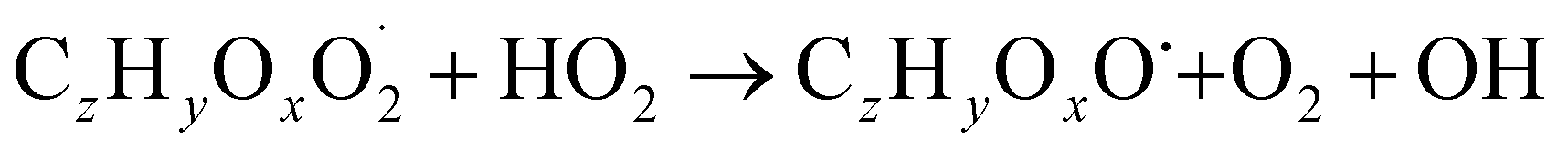 | (R4b) |
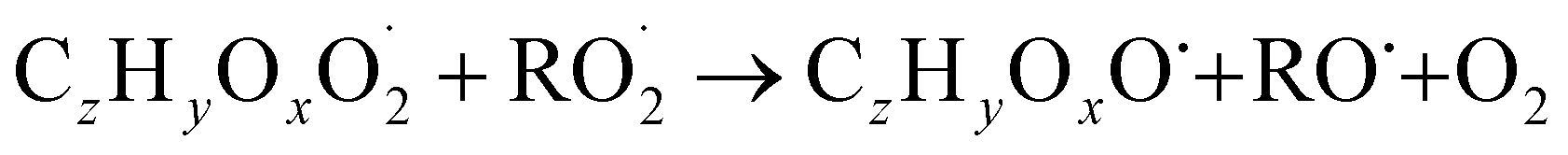 | (R5a) |
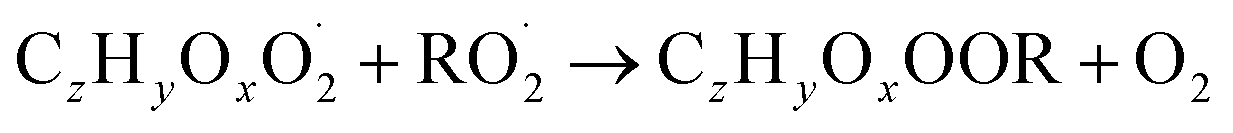 | (R5b) |
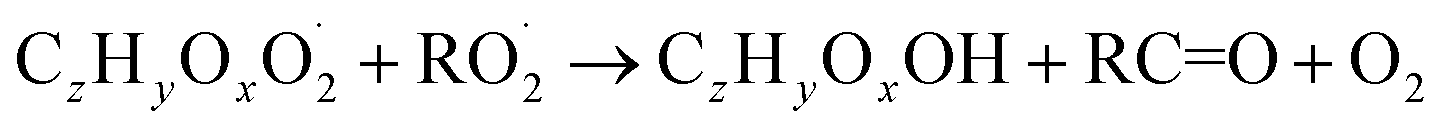 | (R5c) |
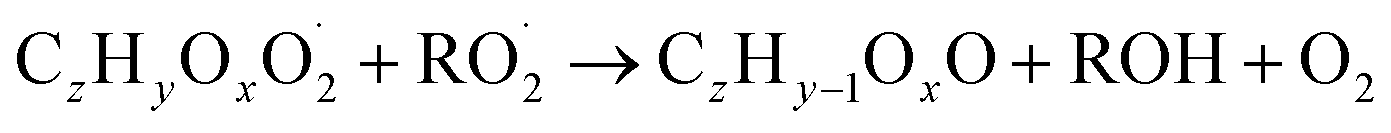 | (R5d) |
| CzHyOxO˙ → fragmentation | (R6) |
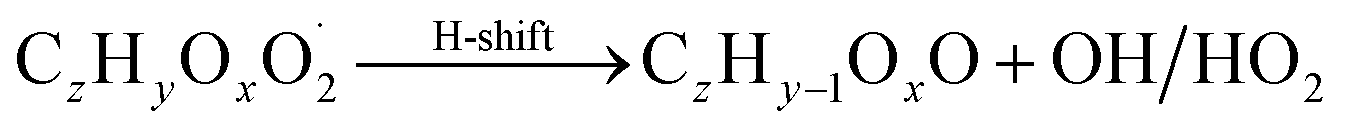 | (R7*) |
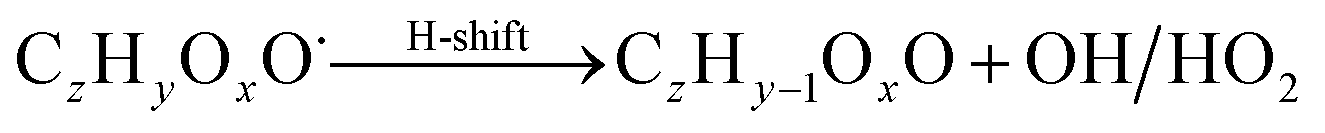 | (R8*) |
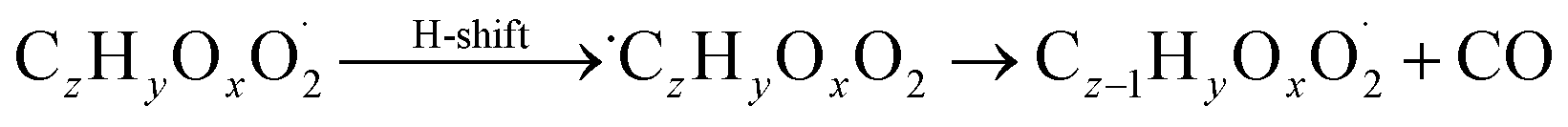 | (R9*) |
The considered reaction types are described in Bianchi et al.12 (reactions (R1*) and (R2*)), Orlando & Tyndall39 (reactions (R3)–(R8*)) and Crounse et al.40 (reaction (R9*)). In the current version, reactions marked with a “*” require constraining of the overall rate constants, while others require the distribution between product channels and consider a default overall rate (derived from MCM).
Upon choice of the reaction types considered, a computer readable chemistry model is created automatically by the autoAPRAM-fw. This allows new chemical systems to be quickly set up, various atmospherically relevant SARs to be considered or different RO2 isomers to be tested in a convenient way. The autoxidation chemistry connects to the MCM chemistry scheme mainly by using peroxy radicals formed in MCM.
2.3 Reaction rate coefficients
Constraining the reaction rate coefficients, and branching ratios in case of several product pathways, is neither trivial nor fully deterministic. Accordingly, potential solutions are to be expected from this method rather than the only “right answer”. In the present work, the following method has proven successful. It is based on the comparison of simulated mass spectra and experimentally determined high resolution mass spectra from nitrate (NO3−) chemical ionization mass spectrometry (CIMS) measurements. Note that these spectra need to be interpreted with care as discussed in the ESI section “Interpreting ion count data from CIMS measurements”.† Ideally, the comparison is done for an evolving chemistry. Experimental data should be taken from well-defined experimental setups (i.e., negligible wall effects; well-defined precursor consumption; well-known chemical systems). In the present work, we deployed flow tube experimental data, where available, for this task. The individual steps described below refer to a single specified point in time of simulation and experiment for which the chemistry code is constrained.Step 1 is for adjusting the hydrogen shift rates in order to reproduce observed peroxy radical levels (reaction (R1*)). This procedure has to be repeated after each of the other steps as H-shift rates interfere with the RO2 concentrations. Peroxy radicals with low oxygen number are not detected at full efficiency in NO3−-CIMS.41 Thus, their simulated concentration may well exceed the measured value.
In step 2, the rate coefficients determining dimer formation are constrained as the dimers are likely to be detected with high efficiency. Note that the least oxidised dimers may, similar to the monomers, be detected with reduced efficiency. Further, the branching ratios for RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (R5d) and ROH (R5c) formation are assigned. The formation of an alkoxy radical is calculated based on the difference between the overall rate coefficient describing RO2 + RO2 and the individual closed shell forming rate coefficients determined before (reactions (R5b)–(R5d)). Computing these numbers can be done best in case the RO2 + RO2 reactions are dominating compared to RO2 + HO2 or RO2 + NO.
O (R5d) and ROH (R5c) formation are assigned. The formation of an alkoxy radical is calculated based on the difference between the overall rate coefficient describing RO2 + RO2 and the individual closed shell forming rate coefficients determined before (reactions (R5b)–(R5d)). Computing these numbers can be done best in case the RO2 + RO2 reactions are dominating compared to RO2 + HO2 or RO2 + NO.
Step 3 comprises the determination of the branching ratio RO2 + HO2 → (a) ROOH or (b) RO + OH + O2 described in reactions (R4a) and (R4b). The overall rate coefficient for RO2 and HO2 is obtained from MCM chemistry for the given peroxy radical. Experimental conditions featuring a dominating HO2 sink for RO2 are favorable for taking this step. Otherwise, it is not possible to distinguish between species formed from RO2 + RO2 and RO2 + HO2 or RO2 + NO.
Step 4 requires the determination of isomeric closed shell species formation by intramolecular hydrogen abstraction from α-hydro(pero)xyl functional groups by O2. As a result a carbonyl functional group is formed (reaction (R7*)). This can be an important pathway under clean conditions, when RO2, HO2 and NO are not the dominating sinks of RO2.
Step 5 is the determination of RONO2 formation under high NOx conditions (reactions (R3a) and (R3b)). Here, the NOx level (relative to HO2 and RO2) is not too important as the nitrated products are distinctive (i.e., RONO2 cannot be confused with ROOH or ROH). This step also includes formation of alkoxy radicals that can either fragment (reaction (R6)), undergo RO-autoxidation to form RO2 species (reaction (R2*)) or form closed shell species (reaction (R8*)). Under high NO conditions, the formation of RO2via RO seems to be an important path to reproduce the observed peroxy radical levels.
Elimination of CO after hydrogen abstraction from an aldehyde group is considered in step 6 (reaction (R9*)). Resulting peroxy radicals have one less carbon atom in the chain compared to their parent VOC. For those species, steps 2–5 have to be repeated.
In the present work, steps 1 to 6 (except for steps 4 and 5) have been carried out for data from a flow tube setup. Step 5 was done for data gathered from a steady state type chamber experiment applying high NOx levels. Due to lack of experimental data from chamber experiments mimicking clean conditions rate coefficients to be constrained in step 4 were estimated (i.e. low VOC reacted, low NOx). Further, higher generation RO2 species are considered. Note that rate coefficients constrained from CIMS data to a great extent coincide with predictions from SARs for RO2 + RO2/HO2/NO:18,42
(1) ROOR: based on observed benzene ROOR formation and RO2 concentrations,43 we found the following relationship between the molecular mass of the RO2 species and the rate coefficient:
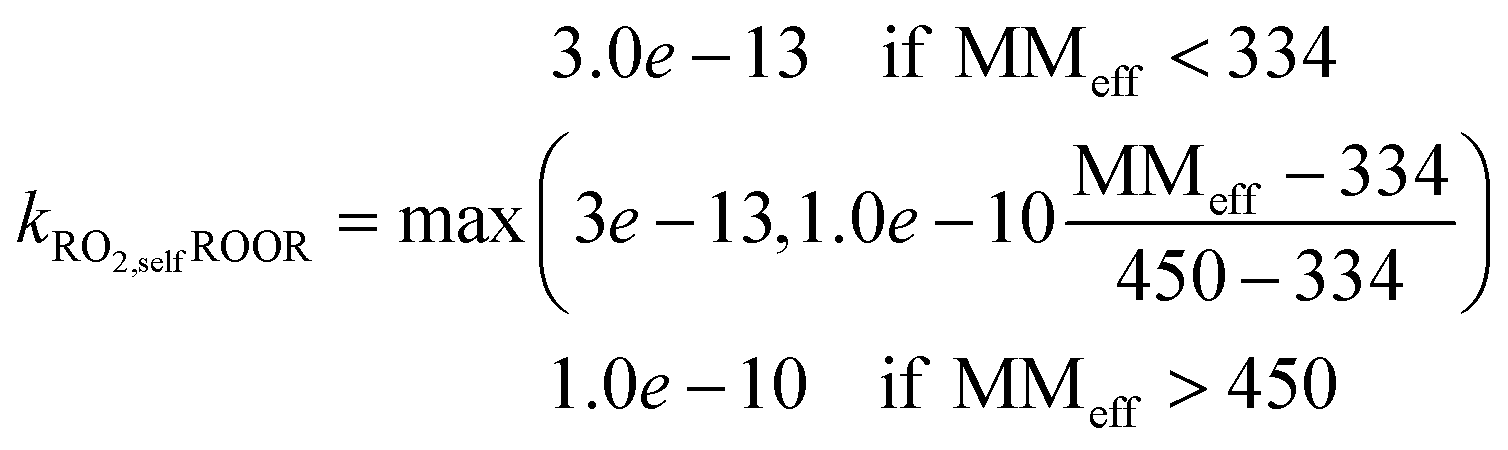 | (1) |
(2) ROOH yield: while the overall rate coefficient for RO2 + HO2 species is adopted from MCM, the branching ratios (see reactions (R4a) and (R4b)) were constrained against experimental data. To reproduce the experimental findings (by means of modelling), ROOH yields between 0.5% and 60%, with an increasing yield-trend towards higher oxidized species, are applied. For comparison, the bicyclic peroxy radical (named BZBIPERO2 in the present work), features an estimated ROOH yield of roughly 1%.42
(3) RONO2 yield: similar to reaction (R4), for the reaction RO2 + NO (reaction (R3)), overall rate coefficients are obtained from MCM generic rate coefficients and branching ratios towards RONO2 and RO are constrained against experimental data. Again, the alkoxy branch is dominating. RONO2 yields range from 0.2% to 4% which is in line with rate coefficients reported for similar molecular structures (0.3–2%).18,42
2.4 The molecular structures
The autoSMILES sub-module of the autoAPRAM-fw relies on SMILES-format inputs of potential peroxy radical structures in order to compute closed shell monomer and dimer structures. However, this information is currently not available for the vast majority of the considered RO2 species. As a consequence, an approach mainly based on theory is applied. For a few decades already a specific structure, the bicyclic peroxy radical (see ESI Fig. S11†) has been suggested to be a key RO2 structure that forms upon a single OH attack on benzene followed by O2 addition.23,25,42,46 This is in line with CIMS data showing C6H7O5 as the peroxy radical with the smallest oxygen number (note: an odd oxygen number and containing six carbon atoms; e.g. Molteni et al.43 or Wang et al.25). Accordingly, we base our considerations on this structure. To predict potential structures of other RO2 species, we considered unimolecular rearrangements, bimolecular reactions and multiple OH attacks. The aim is to provide at least one potential structure and its formation via plausible reaction paths for each peroxy radical composition detected by the mass spectrometer. The resulting formation scheme is shown in the ESI (Fig. S12†). Note that the focus of this scheme is to predict potential peroxy radical structures rather than suggesting an explicit reaction scheme. Further, radical termination pathways have been ignored in this illustration as the focus is put on radical retaining reactions. The resulting list of peroxy radical structures most likely represents only a fraction of RO2 species involved in the autoxidation chemistry.47We assume that the absence of information on RO2 structures, from either experiment or theoretical considerations, will most likely not change in the near future. However, due to the fact that theoretical considerations are applicable for suggesting structures at least for early-stage reaction products and beyond, while new technologies in mass spectrometry, such as versatile, structure specific charging techniques, are emerging,41,48 we expect that this gap in knowledge will continuously decrease.
2.5 Deriving the saturation vapor pressure
The saturation vapor pressure is an important quantity to derive the partition of a species between the vapor and condensed phase.49 Ideally, it is determined experimentally. However, as the present work involves hundreds of different species, this approach is not feasible. The vapor phase molecules formed upon oxidation of precursor VOC are characterized by their potential structures described by the SMILES convention. Corresponding psat are computed by means of two different approaches: for all species, psat is derived by applying group contribution methods, namely EVAPORATION,31 NANNOOLAL32 and MYRDAL/YALKOWSKI.33 ESI Fig. S13† graphically provides an overview of the volatility distribution. More information on the methods and how they compare to high-level quantum chemical calculations is provided in the ESI text.†All SOA simulations in this work are employing the group contribution methods to describe the species' vapor pressures. Variation of the SOA is spanned by methods NANNOOLAL and MYRDAL/YALKOWSKI, while EVAPORATION forms the center-line of the results. In case a single SOA result is reported (i.e. for atmospheric simulations), the psat is based on EVAPORATION.31 However, note that there is no evidence on which method computes best the psat of the unknown variety of structures.
2.6 Numerical simulations: ADCHAM & ADCHEM applications
The aerosol dynamics gas and particle phase chemistry model for laboratory CHAMber studies (ADCHAM50) and the trajectory model for aerosol dynamics, gas and particle phase CHEMistry and radiative transfer (ADCHEM21,51) are deployed in the present work. ADCHAM serves to compute phase change of inorganics (H2SO4, NH3, HNO3) and organics (MCM and autoAPRAM species) by considering Brownian coagulation, condensation, evaporation and dissolution. To ease computation, only organic species with a psat lower than 1 Pa (which corresponds to a saturation mass concentration C* or roughly 0.1 g m−3) are considered to potentially partition to the particle phase. Diffusion in the carrier gas is described by Fuller's method. Detailed model inputs can be found in the ESI (section “Model input specifications”).†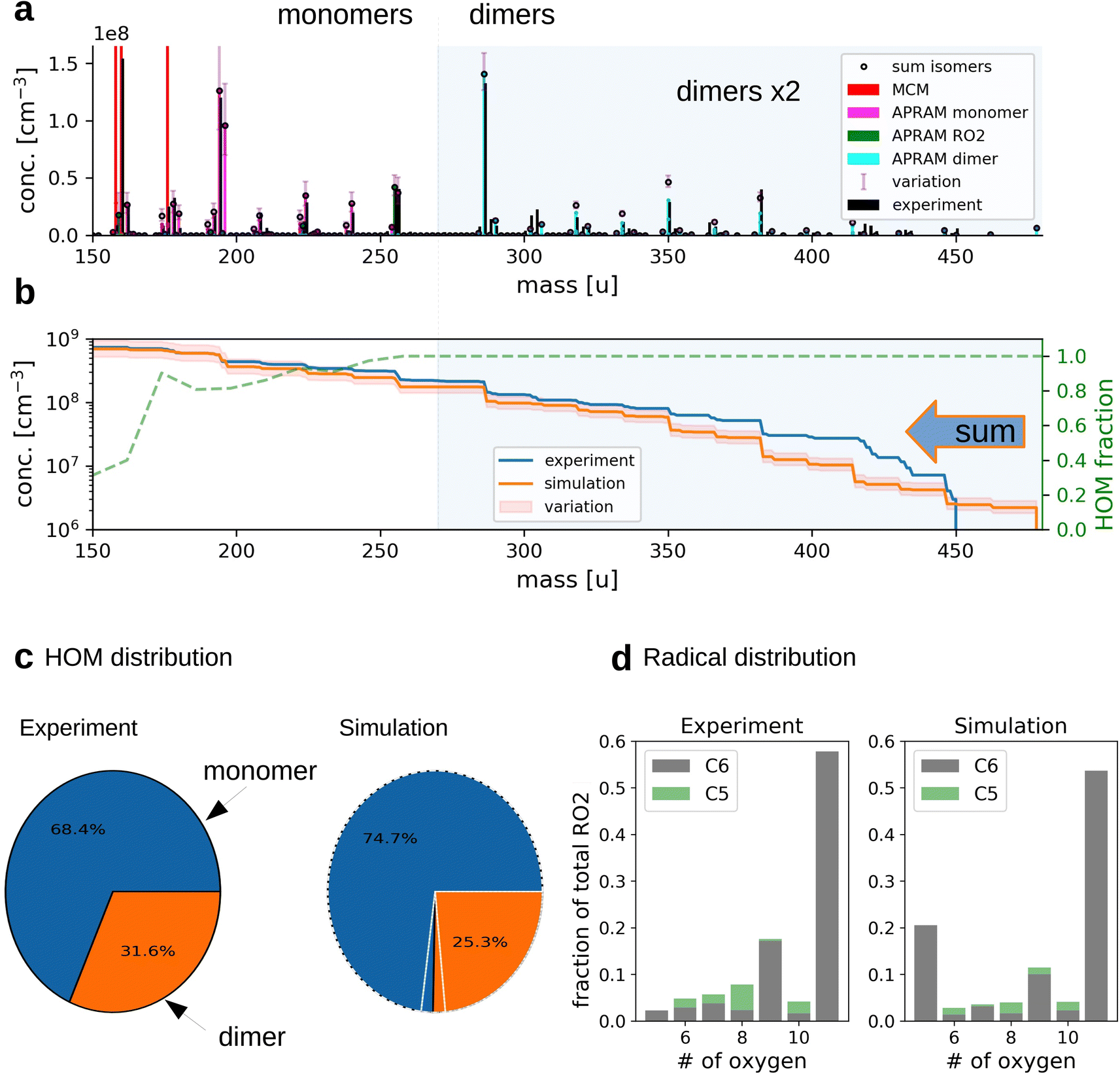 | ||
| Fig. 2 The simulation of OH oxidation of benzene in the presence of UV light. Panel (a) depicts the gas-phase molecular mass distribution from flow tube experiment43 (black bars) and the simulation results (colored bars show different subsets of species from one simulation; “sum isomers” represents the sum of simulated isomers; note that dimer concentrations are raised by a factor of 2 to increase readability). Panel (b) shows cumulative representation of the model results, starting at the upper observed molecular mass limit. The secondary axis indicates the computed, unitless HOM fraction. Panel (c) depicts the distribution of HOM species between the monomers and dimers (variation due to wall loss assumptions is indicated by white lines for simulated data). Panel (d) illustrates the frequency of peroxy radicals of different oxygenation states and carbon number. The insets indicate radicals with 6 (“C6”) and 5 (“C5”) carbon atoms. | ||
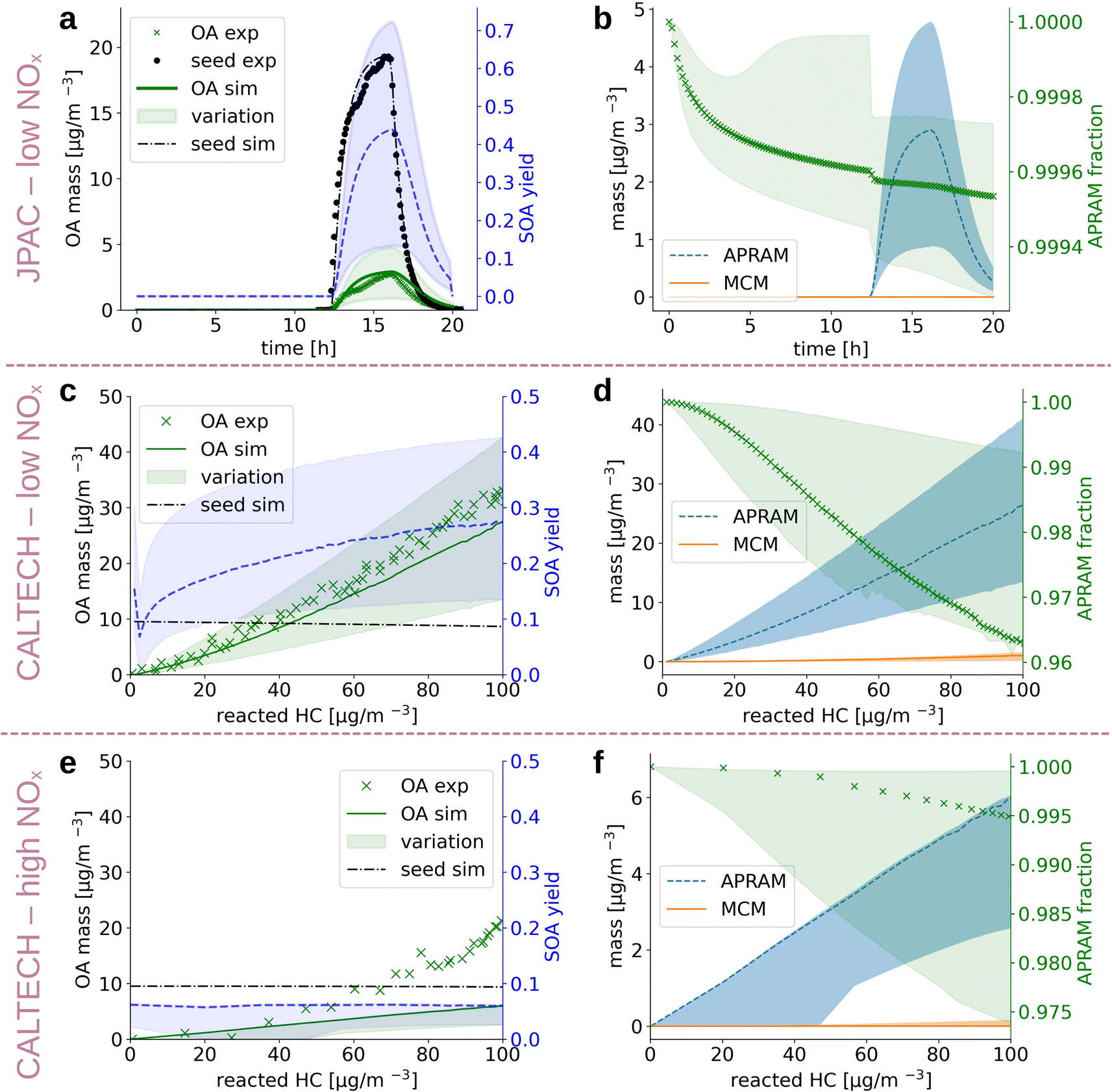 | ||
| Fig. 3 The simulation results when reproducing experimental chamber runs (panels (a and b) and (c–f) refer to the JPAC chamber and CALTECH chamber, respectively). Panels on the left hand side (a, c and e) depict the organic aerosol formed (simulation: “OA sim”; experiment: “OA exp”) as well as the seed aerosol present (simulation: “seed sim”; experiment: “seed exp”), respectively, shown on the left hand ordinate. The unitless, simulated SOA yield (dashed line; shaded area denotes the variation) is illustrated by the right hand ordinate. Results are shown as a function of reaction time (panels (a and b)) and reacted hydrocarbon (panels (c–f)). Panels (b, d, and f) contain computed information on the distribution of mass formed by MCM chemistry and by the autoxidation chemistry (APRAM), respectively. The variation of modeled data results from the different psat-determination methods applied. | ||
In this work, the third case, “11 am”, is discussed in detail. Plots showing respective results for the cases “9 am” and “10 am” can be found in the ESI.† Data are discussed from 1 day upwind to 3 days downwind Malmö, except for the airmass trajectories (full trajectories over the 10 day simulation period are given in ESI Fig. S1†). The simulation days 7 to 1 day upwind are applied to allow a build-up of the gas-phase species.
Detailed information on the approach can be found in the ESI section “Description of parametric yield calculations”.† ESI Table ST2† provides an overview of the inputs.
3 Results
3.1 Pure gas-phase simulations
The applied chemistry scheme is set up by coupling the basic description of tropospheric benzene degradation, obtained from the master chemical mechanism (MCM; see the Methods section for details), to the autoxidation chemistry scheme created by the autoAPRAM-fw. Missing information on reaction rate coefficients is constrained based on flow tube and chamber experiments. Abridged, MCM describes the degradation of VOC species including the formation of peroxy radicals which produce oxygenated closed shell species by unimolecular or bimolecular reactions. Multiple generations of oxidations are considered. Thermodynamically more stable fragmentation products build up via the alkoxy radical paths, ultimately forming CO and CO2.28,29 Formally, the autoAPRAM-fw adds the formation of high oxygen containing species by considering autoxidation chemistry in addition to the MCM chemistry scheme. Besides various closed shell product forming reactions, autoxidation (consisting of an intramolecular hydrogen abstraction followed by the addition of O2) of peroxy-(RO2) and alkoxy-(RO) radicals is the key process. Implementing the combined chemistry scheme into the aerosol dynamics model ADCHAM50 enables computational reproduction of pure gas-phase experiments focusing on autoxidation and competing reactions.Computer simulations of a flow tube setup employed by Molteni et al.43 show the applicability of the approach (Fig. 2, panels (a)–(d); dimer concentrations were raised by a factor of 2 for better visibility). Note that monomers refer to reaction products having the same or smaller carbon number compared to the VOC precursor. Dimers form by the accretion reaction of monomer-radicals. Variation of the results originates from diffusive losses and is described in detail in the Methods section (see Section 2.6). As depicted by panel (a), the model is able to reproduce the atomic mass distribution measured using a nitrate chemical ionization mass spectrometer (NO3−-CIMS). Further, the monomer to dimer ratio and the radical distribution are reproduced (Fig. 2, panels (c) and (d)). Deviation of modeled and experimental concentrations is observed for the least oxygenated peroxy radicals and their closed shell derivatives (i.e., species with molecular mass below 180 amu; without reagent ions), excluding dimers (panel (a)). This, however, is not surprising as the detection efficiency of these species is lowered due to their structural specifications.41
Slight underestimation of the formation of dimers in the range above 400 amu is observed (see Fig. 2b). However, no sound explanation is available yet as the precursor molecules, highly oxidized RO2, are assumed to be detected with the highest efficiency and the formation of their dimeric products is considered to proceed close to the kinetic limit (∼10−10 cm3 s−1). Potentially, the underestimation of simulated dimer formation featuring high atomic mass might originate from ignored oxidation of closed shell dimers (of lower oxygen content), followed by autoxidation steps or by general underrepresentation of RO2 concentration.
Since no flow tube data is available for benzene under elevated NOx ([NOx] > 1 ppbv) conditions, we used results from a steady state chamber setup to constrain the model.66 Note that we consider the retrieval of potential rate coefficients from the flow tube setup to be more accurate. The reason is the degree of complexity in mixing reactors featuring comparatively long residence times (processes to consider: chemical reactions, mixing, wall partitioning, nucleation, aerosol formation, in- and out-flux). In comparison, the flow tube represents a “simple” setup (processes to consider: chemical reactions and limited wall interaction). Accordingly, we consider the chamber-derived rate coefficients to be less accurate. The only rate coefficients derived from the elevated-NOx experiments are reaction types (R3) and (R8*) (see Section 2.2 “Setting up the gas-phase chemistry” for more information on the reaction types).
While the monomer distribution is largely reproduced with regard to nitrate species formation, the dimer concentrations are underestimated by the model due to the scavenging of their precursor species, the peroxy radicals, by NO (see ESI Fig. S15†). The approach of stable closed shell species (i.e., closed shell products are not further oxidised) formation in the autoAPRAM-fw produces, compared to experiment, dimers of similar mass and composition. However, their abundance is considerably underestimated (∼90%) under high NOx conditions. Note that, in contrast to low NOx conditions, the simulated HOM dimer fraction is much lower (dimer fraction ∼ 2%). Detailed results under high NOx conditions are discussed in the ESI (section “High NOx conditions”).†
3.2 Partition to the particle phase
Alkoxy and peroxy radicals form upon oxidation of the parent VOC. The radicals can undergo autoxidation and, as a consequence, isomerize to multi-functionalized molecules. The properties of these species deviate substantially from the parent VOCs. In order to compute their properties, we derived the potential molecular structures. Due to the fact that these molecules are often short lived, diverse (with regard to chemical composition and, most likely, isomeric variation), and only comprise a small fraction of the pool of reaction products, their structures have not yet been approached experimentally, besides a few exceptions.67 Thus, we base the structural suggestions on theoretical work and available knowledge on similar reaction classes (see Sections 2.2 and 2.4). The autoSMILES sub-module of the autoAPRAM-fw computes likely structures for all closed shell species. Potential structures of the peroxy radicals serve as an input. The applied approach is discussed in detail in the Methods section. Partitioning of oxidation products between the gaseous and the condensed phases, in the presence of a condensation sink, is strongly affected by the species saturation vapor pressure. The psat is obtained by applying group contribution methods (e.g., O'Meara et al.68) to all structures. A quantum chemistry based statistical thermodynamics method, conductor-like screening model for real solvents (COSMO-RS34–36), is applied exclusively to a few species as it requires detailed analysis and extensive computational resources (see Section 2.5 Deriving the saturation vapor pressure).Despite being an intensely studied molecule only a few benzene oxidation experiments conducted under well defined, atmospherically relevant conditions which report SOA information can be found in the literature.64,66 In the present work we set up the ADCHAM model for the CALTECH54 and JPAC53 chambers to simulate seeded OH oxidation in the presence of UV light. Fig. 3 depicts simulation results and experimental data. Panels (a), (c) and (e) compare reported64,66 and simulated aerosol mass formed and show calculated SOA mass yield based on the simulated data. Note that SOA mass yield is defined as SOA formed divided by benzene mass reacted. Values for aerosol mass formed agree well with the observations for panels (a) and (c), representing low NOx conditions ([NO] and [NO2] < 1 ppt). Accordingly, the mass yields reported in the literature were reproduced under reported conditions. Garmash et al. derived a maximum SOA mass yield of 40%, while we found 42% (12% to 71%), and Ng et al. observed a maximum mass yield of 37% which compares to 28% (14% to 42%) in the simulation in the CALTECH chamber. For the high NOx case shown in panel (e), the SOA formation potential is underestimated (simulated mass yield of roughly 7% (3% to 7%) vs. experimentally observed 26%). Ranges in simulated SOA yield result from different psat-estimation methods applied (see Section 2.5 Deriving the saturation vapor pressure). Fig. 3, panels (b), (d) and (f) represent the contribution of MCM and autoAPRAM species, respectively, to the organic aerosol formed. In all three cases, the model suggests clear dominance (>94%) of the species formed via autoxidation. In the steady state experiment from the JPAC chamber, the fraction of autoAPRAM species is highest (>99%). Notably, the heterogeneous nucleation at the seed particle surface is initiated by species formed by autoxidation in all simulations (i.e., the autoAPRAM fraction approaches 100% at t → 0 s after seed aerosol addition which corresponds to low reacted hydrocarbon in the CALTECH case). Further, note a conceptional difference, “steady state” vs. “evolving chemistry” between experimental setups related to Fig. 3 panels (a) and (b) (i.e., resembling the JPAC chamber) and (c)–(f) (i.e., resembling the CALTECH chamber), respectively, which is highlighted by the evolution of the seed aerosol: while panel (a) shows an increase in seed aerosol upon production, followed by its decay, panels (c) and (e) represent an almost constant seed aerosol concentration present in the chamber.
3.3 Atmospheric implications
VOC concentrations have been reported almost all around the globe. Benzene is a prototypical aromatic molecule whose emission is related to mainly anthropogenic activities and is detected at high levels in many environments.26,27,69 Besides benzene being toxic itself, its basic risk to health is via the oxidation in the atmosphere to contribute to the formation of nitrogen-containing species and secondary organic aerosols.70 If emitted by a combustion process, benzene is mixed with NOx. While NOx, and in particular NO, concentrations decrease relatively quickly upon transport away from its source, benzene has an atmospheric lifetime of roughly 12 days.71 As a consequence, benzene is oxidized in the presence of varying NOx mixing ratios. To demonstrate this process, we selected airmass trajectories (a) originating from the clean air above the Arctic Ocean between Greenland and Svalbard; (b) traveling over an urban European region (Malmö/Copenhagen); and (c) are characterized by changing NOx conditions during benzene-SOA formation.To showcase the effects of autoxidation chemistry on benzene atmospheric oxidation and SOA formation, the Lagrangian atmospheric chemistry transport and aerosol dynamics model ADCHEM was employed.51 A more detailed description of the model setup is provided in the Methods section (2.6 Numerical simulations: ADCHAM & ADCHEM applications) and references therein. It was configured to simulate three air-mass trajectories arriving at Malmö, southern Sweden in the end of April, 2021. The simulations are named by the time the air-masses pass the city of Malmö: “9 am”, “10 am” and “11am” (UTC + 1). For the selected cases, air masses from the Arctic Ocean between Greenland and Svalbard are transported over Scandinavia and the Baltic Sea before they arrive in Malmö 7 days later. Downwind Malmö the air masses are transported over Copenhagen and spend ∼48 hours over the North Sea before they arrive near Brussels (see ESI Fig. S1†). Results are analyzed in detail from one day upwind Malmö to the end of the simulation (Fig. 4). Based on emission inventories, the benzene concentrations remain low (<0.1 ppbv) until the calculated trajectories move over southern Sweden, and peak over Copenhagen at ∼0.7 ppbv (Fig. 4, panel (b); see also Fig. S2 and S3†). Oxidant evolution, summarized in panel (a), shows a typical diurnal behaviour: OH level rises during the day. NO and NO2, if emission is low, are converted to the NO3 radical by O3. This process is suppressed during the daytime by photo-reactions: 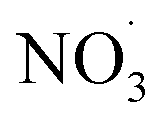 and NO2 are photolyzed to NO2 and NO, respectively, which explains the source-independent increase in NO during daytime.
and NO2 are photolyzed to NO2 and NO, respectively, which explains the source-independent increase in NO during daytime.
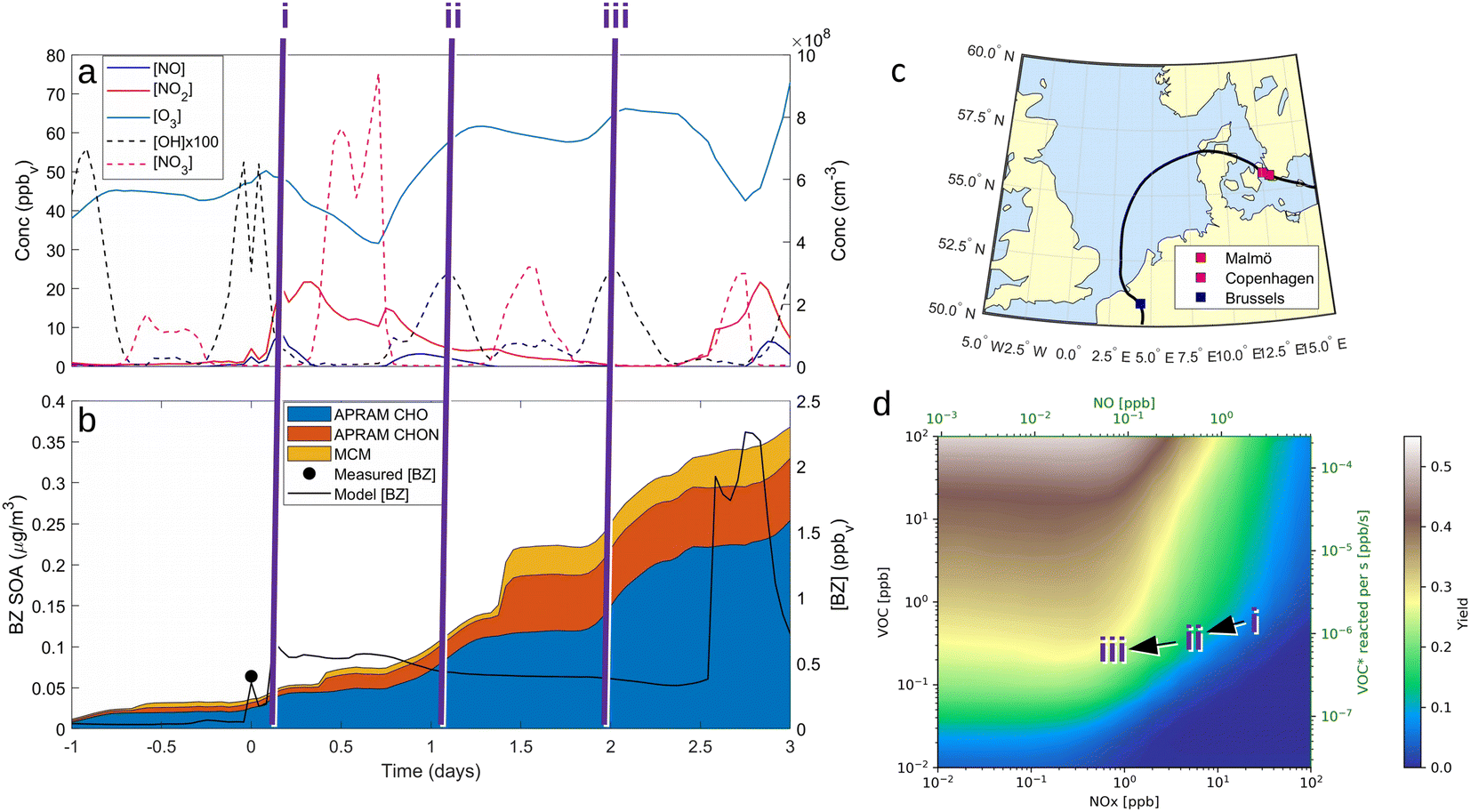 | ||
Fig. 4 ADCHEM results along the case 3 (“11 am”) air mass trajectory. Panel (a) shows the modelled gas-phase concentrations of NO, NO2, O3, OH and 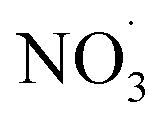 from 1 day upwind (−1 day) to 3 days downwind Malmö. Note that OH concentration is scaled up by a factor of 100 to increase readability. Panel (b) shows the modelled benzene gas-phase concentrations (model [BZ]) and the modelled benzene SOA mass concentrations of non-nitrate APRAM species (APRAM CHO – species of atomic composition CxHyOz), APRAM organonitrates (APRAM CHON – nitrogen-containing species) and MCM species. The modelled benzene is also compared with the observed benzene concentrations at the measurement station Dalaplan in Malmö (measured [BZ]). The air mass trajectory path is displayed in panel (c). Panel (d) illustrates the computed SOA mass yield (unitless) from OH oxidation of benzene in the presence of different levels of NOx and VOC. Bottom and top abscissas depict NOx and NO concentrations. VOC mixing ratio and VOC turnover are shown by the left and the right hand ordinate, respectively. The color code indicates the mass yield value. Note the highlighted points in chemical space (“i”, “ii” & “iii”) in all panels. from 1 day upwind (−1 day) to 3 days downwind Malmö. Note that OH concentration is scaled up by a factor of 100 to increase readability. Panel (b) shows the modelled benzene gas-phase concentrations (model [BZ]) and the modelled benzene SOA mass concentrations of non-nitrate APRAM species (APRAM CHO – species of atomic composition CxHyOz), APRAM organonitrates (APRAM CHON – nitrogen-containing species) and MCM species. The modelled benzene is also compared with the observed benzene concentrations at the measurement station Dalaplan in Malmö (measured [BZ]). The air mass trajectory path is displayed in panel (c). Panel (d) illustrates the computed SOA mass yield (unitless) from OH oxidation of benzene in the presence of different levels of NOx and VOC. Bottom and top abscissas depict NOx and NO concentrations. VOC mixing ratio and VOC turnover are shown by the left and the right hand ordinate, respectively. The color code indicates the mass yield value. Note the highlighted points in chemical space (“i”, “ii” & “iii”) in all panels. | ||
Benzene, due to its aromatic structure, is oxidized only by OH at a considerable rate.42 The resulting peroxy RO2 species drives the SOA formation during daytime via autoxidation chemistry (see “APRAM CHO” in Fig. 4b). Over Malmö and Copenhagen, the high NO concentrations partially suppress the autoxidation of peroxy radicals formed by OH oxidation of benzene, which results in less highly oxygenated organic molecules. The impact on aerosol mass yield and SOA composition is shown in the ESI (Fig. S4–S6).† As the air masses are transported away from the source (emissions of benzene and NOx), the chemical system shifts to less NOx-mediated autoxidation chemistry. The effect on SOA mass formed can be seen when comparing the situations “i”, “ii” and “iii” in Fig. 4, panel (b). Further, this is graphically illustrated in panel (d) which aims to depict the dependence of SOA mass yield on VOC (i.e., benzene), OH and NOx.
There is a distinct increase in modeled SOA mass after sunset which appears low as benzene levels are low; however, the ΔSOA gets more apparent after the air parcels have passed over the Oresund region (i.e. t = 0 days in Fig. 4, panel (b)). During the first few hours after the sunset during days 1 and 2 downwind Malmö/Copenhagen, rapid 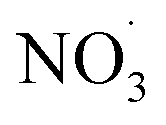 oxidation of the intermediate MCM benzene oxidation product 4-nitrocatechol (C6H5O4N), which is formed and accumulates in the gas-phase during the daytime, contributes to the formation of a highly oxygenated di-nitrate RO2 species (C6H5O11N2). This results in substantial organonitrate SOA mass formation, composed mainly of organonitrate dimers (“CHON” Fig. 4, panel (b) and ESI Fig. S2 and S3, panel (b)†). Those species, although highly oxygenated, are not considered HOM (according to definition by Bianchi et al.12) as the formation does not include autoxidation steps. Note that this formation pathway remains to be investigated experimentally. Yet, it is entirely based on modeling results.
oxidation of the intermediate MCM benzene oxidation product 4-nitrocatechol (C6H5O4N), which is formed and accumulates in the gas-phase during the daytime, contributes to the formation of a highly oxygenated di-nitrate RO2 species (C6H5O11N2). This results in substantial organonitrate SOA mass formation, composed mainly of organonitrate dimers (“CHON” Fig. 4, panel (b) and ESI Fig. S2 and S3, panel (b)†). Those species, although highly oxygenated, are not considered HOM (according to definition by Bianchi et al.12) as the formation does not include autoxidation steps. Note that this formation pathway remains to be investigated experimentally. Yet, it is entirely based on modeling results.
The trajectory model runs are contrasted by the more general picture of daytime benzene-SOA formation potential (see Fig. 4, panel (d)). The SOA mass yield is calculated applying a box model (ADCHAM, see Section 2.6 Numerical simulations: ADCHAM & ADCHEM applications for details) mimicking the daytime atmospheric conditions and a range of VOC and NOx mixing ratios. Generally, predicted yields vary between less than 1% for high NOx and low VOC conditions and up to 50% under extreme conditions (high VOC and low NOx). The computed mass yields show a slight increase with increasing NO as long as NO is low ([NO] < 0.1 ppbv). Above roughly 0.1 ppbv, the mass yield shows a distinct negative correlation with increasing NO. On the other hand, there is a positive correlation between the benzene turnover and yield. Note that these computed yields are highly dependent on OH concentration, despite being relatively insensitive regarding variations in seed aerosol properties, sunlight (neglecting the strong indirect effect via OH formation), methane concentration or O3 (ESI Fig. S7†). Further, note that the OH and O3 concentrations were set to constant values in these simulations in order to allow investigation of mass yields under various VOC/NOx concentrations at a typical daytime OH level. This reflects the assumption that the VOC of interest is not dominating the OH sink. A strong variation in oxidants due to different VOC or NOx levels would complicate the interpretation of individual data points as well as the comparability of mass yield results.
4 Discussion
4.1 Limitations of the mechanism development
The presented methodology aims to investigate the potential impact of autoxidation chemistry on (a) the gas-phase chemistry, (b) SOA formation in seeded flow reactor experiments, and (c) atmospheric SOA formation. Since the approach gathers information and data from various sources, caution is in order. In the following, the main limiting factors are discussed together with the arising consequences for the presented results.Another consequence of the underrepresentation of isomeric variability is the fact that some reactions suggested may not be possible with regard to their chemical structure.
Limitations regarding the saturation vapor pressures assigned can be found in the ESI.†
4.2 Limitations of the numerical simulations
In this section focus is put on the limitations of SOA simulations arising in context with the chemical scheme proposed. General limitations of the numerical simulations of aerosol dynamics using ADCHAM or ADCHEM are discussed elsewhere.21,50,51Limited knowledge on the partitioning of molecules to the chamber walls is a challenge. Since the benzene structures are covered by the autoAPRAM-fw feature 5 and more oxygen atoms, a first order loss term was applied (see Section 2.6.2). The wall partitioning is of great relevance in chamber simulations as it directly affects the production rates inferred: the higher the wall loss rate is, the higher is the reaction rate coefficient forming the depositing species in order to explain the observed mass-peak hight.
In the atmospheric simulations, the main limitations related to autoxidation chemistry are:
- The reaction type (R5) in which the peroxy radical of interest reacts with the pool of peroxy radicals. Rate coefficients for this reaction have been derived in the flow tube experiment which comprises “hotter” (i.e., concentrations of OH and HO2 are much higher than atmospheric ones) chemistry than the atmosphere and very different timescales (i.e., slow reactions underrepresented compared to the atmosphere).
- While the reaction type (R5) may be overrepresented in the atmospheric simulations, the reaction types (R7*)–(R9*) may gain more importance in the atmosphere compared to the chamber.
Another limitation is the temperature dependence, particularly of the RO2 autoxidation reaction, which is considered exponential. In this work the temperature dependence is neglected due to lack of data.
4.3 Utility of the autoAPRAM-fw
Despite the limitations listed in previous sections, there is broad utility of the approach drafted. The autoAPRAM-fw may be used to:(1) Produce atomic peak lists for a set of predefined (autoxidation) reactions, e.g., to be used in the analysis of CIMS spectra. The automated nature of the framework allows large numbers of peaks to be provided quickly. The product description allows isomers and reaction pathways to be considered.
(2) Create, within seconds, sets of chemical reactions governing autoxidation based on selection of the parent VOC and reaction types chosen. Rate coefficients need to be provided (pure guess or based on SAR).
(3) Assign potential rate coefficients to the schemes created based on experimental data as described in Section 2.3. This process requires substantial working time. However, this limiting factor will be overcome soon as we are currently evaluating two approaches to automatically assign potential solutions for rate coefficients from experimental data: (a) a random-forest based method, run on a computer cluster and (b) an analytically-based approach providing roughly 103 rate coefficients per second during evaluation of the chemical scheme against CIMS data.
(4) Characterise the products formed in the autoxidation scheme by means of: (a) atomic composition, (b) functional groups and (c) likely SMILE-structures. This allows various methods to be considered to derive saturation vapor pressures.
(5) Set up an autoxidation chemistry scheme for any parent VOC. The approach is not limited to benzene.
(6) Implement new reaction types to consider new theoretical/experimental findings.
(7) Consider an increased isomeric variety of input radicals. In the benzene example, a single peroxy radical species represents all peroxy radical isomers (i.e., the RO2 with 7 oxygen atoms: Bzo_RO2_O7 describes RO2 isomers of composition C6H7O7).
(8) Couple autoxidation chemistry schemes to MCM and solve them with an aerosol-dynamics and chemistry model. The resulting model setup is capable of investigating the mechanistic behaviour of the chemistry in detail. A similar degree of detail is not met in any other approach aiming to be implemented in aerosol-dynamics and chemistry models. Still, the suggested method is much more lumped than other approaches aiming to cover the full autoxidation chemistry.72
To summarize, the presented approach does not aim to be mechanistic or predictive with regard to structure-dependent reaction pathways. Further, the reaction rate coefficients applied are not determined from structure–activity relationships. The autoAPRAM-fw serves to create reaction schemes to be related to experimental data in order to quantify the rate coefficients of reaction types that explain the observed rearrangement of the CIMS peaks. SARs are considered when comparing the rate coefficients found to previous results from similar systems. This is done in order to check the plausibility of their magnitude. Thus, the proposed autoxidation scheme for benzene represents a single, potential solution. It allows observed CIMS-data evolution as well as SOA formation to be reproduced while proposing realistic (according to the literature) reaction parameters.
5 Conclusion
Autoxidation chemistry is a key for understanding the atmospheric source of many multiply functionalized molecules and their potential partition to the condensed phase.14 Applying reaction-rules of alkoxy- and peroxy radical chemistry enables sets of chemical reaction equations to be compiled in an almost automated fashion using the autoAPRAM-fw.Currently, the fitting of reaction rate coefficients to allow for reproduction of the experimentally obtained gas phase species mass spectrum is a limiting factor: it requires considerable manpower (of the order of weeks for a system like benzene). To overcome this, we are currently exploring two approaches: (1) a randomized search for good fits of rate coefficients; and (2) an analytical solution to obtain rate coefficients from stationary and steady state-like experimental conditions. Approach (1) requires substantial computational effort while the analytical method can be run on a personal computer within seconds.
In the present work we show that these reactions and their rate coefficients may be constrained against experimental data from the flow tube and chamber to reproduce the part of the mass spectrum covered by HOM species within the limits of calculated error (typically less than 10% for relative concentrations of HOM species), including the distribution of peroxy radicals (see Fig. 2). Thereby, we found a potential relationship (see eqn (1)) between the rate coefficient and the molecular mass for the ROOR formation via reaction (R5b). Predicting the species vapor pressure by group contribution methods proves successful to reproduce observed mass yields in seeded experiments found in the low NO regime (experimental: 40%66 and 37%64vs. simulated: 42% (range: 12% to 71%) and 28% (range: 14% to 42%)), regardless of the experiment type. In contrast, formation of condensed phase species is under-predicted under high NO conditions (experimental: 26% vs. simulated: 7%). Potential reasons for this discrepancy are diverse: (a) the present approach neglects the oxidation of closed shell autoAPRAM species. Thus, if terminating reactions are dominating, the propagation of radical chemistry may be under-represented. (b) The prediction of the nitrate species' vapor pressure in the group contribution methods, generally, is considered less accurate compared to non-nitrated species as the experimental data is scarce.68 Reasons for scarce experimental data are, besides others, their high reactivity which makes the handling difficult. (c) The high reactivity of NOx species affects oxidation experiments in chambers and flow tubes as well. Accordingly, availability of high resolution CIMS data for nitrate species during VOC oxidation experiments is very limited which exacerbates model constraint. (d) Formation of highly functionalized organic species in the presence of high NO mixing ratios likely does not proceed via RO2 autoxidation as NO strongly limits the peroxy radical lifetime (ESI Fig. S8†). Thus, more fundamental work on nitrated species autoxidation and properties of the respective products will help to improve the model representation. Further note that reactive uptake of the oxidation products at the surfaces of the condensed phase is not modeled explicitly. Accordingly, the calculated SOA mass may be underrepresented in case reactive uptake is of importance, as could be the case for the product organonitrates. Another species group potentially contributing to reactive uptake is epoxides: they may form via secondary oxidation of aromatic species. At the aerosol surface they transform into less volatile species via acid-catalyzed ring-opening reactions.73
Clearly, the molecular structures and their isomeric distribution are subject to speculation. Likely, this situation will remain for an unforeseen time. Similarly, the exact retrieval of saturation vapor pressures for given molecule structures is difficult. However, based on the set of potential peroxy radical isomers (see Fig. S12†) and the methods applied to derive the saturation vapor pressure (see Section 2.5 “Deriving the saturation vapor pressure”), we found that the description of autoxidation and its effect on the molecular structures is still crucial for modeling SOA formation from benzene. This is valid, independent of the choice of underlying RO2 structures and independent of the choice of psat-derivation method.
The presence of high mixing ratios of NO and HO2 poses additional challenges as their nitrate and ROOH yield by NO and H addition, respectively, is suggested to be very low for peroxy radicals attached to a C-ring.42 The yields of closed shell species during the RO2 reaction with NO and HO2 are predicted to be, besides the latter being highly uncertain, of the order of 0.1% and 1% respectively.42 Thus, the species allow for efficient NOx and, likely but less certain, HOx cycling. However, the molecular rearrangements of resulting alkoxy species are hard to predict.74,75 In the present chemistry scheme, nitrate and ROOH yields in the range of 0.2% to 4% and 0.5% to 60% are applied. While the yields of RONO2 species can be determined from CIMS data, the ROOH yields are less distinct as they can't be distinguished from species formed via other bi- or uni-molecular reactions, featuring the same composition.
Clearly, the model predicts potential, high mass yields of up to 55% from benzene oxidation under atmospheric-like daytime conditions (i.e. low 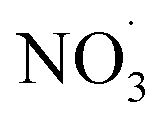 concentration; see Fig. 4, panel (d)). Additional SOA formation is predicted from
concentration; see Fig. 4, panel (d)). Additional SOA formation is predicted from 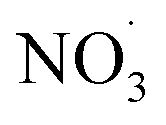 oxidation of benzene–OH oxidation products shortly after sunset, resulting in the formation of highly oxygenated organonitrates. The modeled SOA mass yield positively correlates with increasing benzene turnover. This finding is supported by recent work showing a positive correlation between benzene turnover and HOM formation.66 The effect of NO on autoxidation is less uniform: at NO concentrations less than about 0.1 ppbv, the yield positively correlates with NO, while at higher nitrogen oxide levels, there is a negative correlation. A low NO concentration, due to high yield of alkoxy radicals, may speed up autoxidation.76 Note that RO H-shift rates are suggested to be orders of magnitude faster (about 103 to 107 s−1)74 compared to rate coefficients for peroxy radical H-shifts which are reportedly below 1 s−1 with few faster exceptions.16,19,77 H-shift rates for peroxy radicals in the range from 0.01 s−1 to 1.5 s−1 are applied in the current benzene-autoxidation scheme (see ESI section “AutoAPRAM – benzene scheme”†). If NO concentration exceeds the level of 0.1 ppbv, it starts to scavenge the RO2 pool which forms the basis for peroxy radical autoxidation reactions and low-volatility dimer formation (see ESI Fig. S8†). For both RO and RO2 species, H-shift rates strongly depend on the substitution and span of the H-shift.78
oxidation of benzene–OH oxidation products shortly after sunset, resulting in the formation of highly oxygenated organonitrates. The modeled SOA mass yield positively correlates with increasing benzene turnover. This finding is supported by recent work showing a positive correlation between benzene turnover and HOM formation.66 The effect of NO on autoxidation is less uniform: at NO concentrations less than about 0.1 ppbv, the yield positively correlates with NO, while at higher nitrogen oxide levels, there is a negative correlation. A low NO concentration, due to high yield of alkoxy radicals, may speed up autoxidation.76 Note that RO H-shift rates are suggested to be orders of magnitude faster (about 103 to 107 s−1)74 compared to rate coefficients for peroxy radical H-shifts which are reportedly below 1 s−1 with few faster exceptions.16,19,77 H-shift rates for peroxy radicals in the range from 0.01 s−1 to 1.5 s−1 are applied in the current benzene-autoxidation scheme (see ESI section “AutoAPRAM – benzene scheme”†). If NO concentration exceeds the level of 0.1 ppbv, it starts to scavenge the RO2 pool which forms the basis for peroxy radical autoxidation reactions and low-volatility dimer formation (see ESI Fig. S8†). For both RO and RO2 species, H-shift rates strongly depend on the substitution and span of the H-shift.78
A close to realistic atmospheric transport simulation, applying the complete chemistry scheme developed, suggests that condensable species formed by benzene autoxidation chemistry (added on top of the MCM chemistry scheme by applying the autoAPRAM-fw) can have a significant share of the anthropogenic contribution to ambient organic aerosol mass. This fraction (up to 20%), likely, is over-predicted, as autoxidation chemistry, in the model, is still limited to a few VOC species: benzene, which is the focus of the work, as well as α-pinene, β-pinene, limonene and carene.21 Additionally, the simulations for Malmö, Sweden, cover a period in April with cold Arctic air masses, which naturally limits the contribution of biogenic species.
Apparent mass yields from daytime autoxidation chemistry (determined from ADCHEM simulations) are similar to parametric simulations under comparable conditions (yields of about 20–52%; ESI Fig. S4–S6† and <55%, respectively). There are differences between apparent mass yields in the atmospheric runs and mass yield from the parametric simulations: the parametric runs consider daytime conditions only. As a consequence the 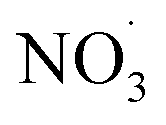 radical levels remain low and OH is the dominating oxidant. In the atmospheric simulations, OH oxidation-products of benzene accumulate during the day and are oxidized by
radical levels remain low and OH is the dominating oxidant. In the atmospheric simulations, OH oxidation-products of benzene accumulate during the day and are oxidized by 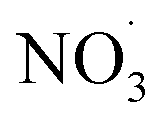 radicals at dawn (see ESI Fig. S9†). Since OH is low while new SOA forms, the mass yield (ΔSOA/Δbenzene) can get very high as benzene is only oxidized by OH (Xu et al., 2020). As a result, apparent mass yields of up to 1000% are modeled for short periods of time. The simulated
radicals at dawn (see ESI Fig. S9†). Since OH is low while new SOA forms, the mass yield (ΔSOA/Δbenzene) can get very high as benzene is only oxidized by OH (Xu et al., 2020). As a result, apparent mass yields of up to 1000% are modeled for short periods of time. The simulated 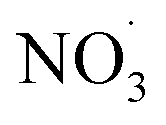 oxidation of benzene-products seems to question the use of a general benzene mass yield based on OH oxidation experiments. Note that the benzene-SOA formation initialized by nitrate radicals is a model prediction and remains to be examined experimentally.
oxidation of benzene-products seems to question the use of a general benzene mass yield based on OH oxidation experiments. Note that the benzene-SOA formation initialized by nitrate radicals is a model prediction and remains to be examined experimentally.
Direct comparison between atmospheric (simulation) data and mass yield simulations is generally difficult as characterizing parameters don't fully match (e.g., temperature, relative humidity). However, clearly, benzene and its intermediate oxidation products are oxidized under changing NOx, OH˙ and 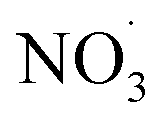 levels which on the one hand affects the formation of HOM species (in particular the dimer formation) and on the other hand impacts on the nitrogen-containing species' contribution to SOA. As the air masses are transported away from hotspots of anthropogenic activity (i.e. emission sources of benzene and NOx), the conversion of benzene to SOA via OH oxidation becomes more effective due to a shift in the chemical regime towards less NOx mediated chemistry (see Fig. 4). The formation of SOA, during daytime, is as expected based on experimental findings.64,66 In contrast, the increase of
levels which on the one hand affects the formation of HOM species (in particular the dimer formation) and on the other hand impacts on the nitrogen-containing species' contribution to SOA. As the air masses are transported away from hotspots of anthropogenic activity (i.e. emission sources of benzene and NOx), the conversion of benzene to SOA via OH oxidation becomes more effective due to a shift in the chemical regime towards less NOx mediated chemistry (see Fig. 4). The formation of SOA, during daytime, is as expected based on experimental findings.64,66 In contrast, the increase of 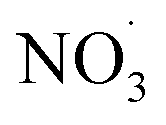 at dusk results in a distinct and timely limited increase in modeled SOA formation. This effect has not yet been investigated experimentally.
at dusk results in a distinct and timely limited increase in modeled SOA formation. This effect has not yet been investigated experimentally.
The authors assume that considering autoxidation chemistry for additional VOC species will most likely increase the formation of SOA and reduce the share of individual components. However, bear in mind that mixtures of VOC species may decrease the individual SOA yield via scavenging the oxidants or by forming higher volatility RO2 + RO2 reaction products as shown for the α-pinene–isoprene system.65 On the other hand, it has been shown recently that VOC mixtures may also increase mass yields significantly: Faiola et al. found augmentation of the aerosol formation by roughly 50–130% from real monoterpene–sesquiterpene mixtures emitted by stressed Scots pine trees.79 Consequently, we think that a mechanistic representation of autoxidation chemistry, to some extent, is essential to improve the prognostic capacity in experiments and under atmospheric conditions. This includes the construction of models able to capture various aspects observed experimentally. Further, upon successful reproduction of experiments by means of modelling, reduction of the code to a reduced formal extent is inevitable to allow application in large scale models.80 A mechanistic model-representation of key observations from experiments will most likely be successful with regard to predictions in future changing atmospheres, whereas empirically based approaches may fail as a result of prevailing conditions not resembling those in their empirical foundation.
Data availability
Data from the flow tube, JPAC chamber (high/low NOx) and CALTECH chamber (high/low NOx) can be accessed online (https://doi.org/10.5281/zenodo.8087267). Data from ADCHEM atmospheric simulations are available upon request (E-mail: office@pi-numerics.com).Author contributions
LP, PR, MR, TK and MiB designed the research; LP, PR, MR, NH, CD, OG, CX, PZ, PC, BF, TGA, MeB, TK and MiB conducted the research; LP, PR, MR, MeB, MiB, NH, and OG analyzed the data; LP and PR developed the models; LP, PR, MR and MiB wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Austrian Science Funds (FWF), grant no. J-4241 Schrödinger Programme. Swedish Research Councils FORMAS and VR (FORMAS project no. 2018-01745; VR project no. 2019-05006). Crafoord foundation (project no. 20210969). Research Council of Finland, grant no. 338171, 331207 and 336531. Research Council of Finland (Center of Excellence, grant number 346369). Research Council of Finland (ACCC Flagship, 337549). Jane and Aatos Erkko Foundation (JAES). EU H2020 project FORCeS (821205). European Commission Horizon Europe project FOCI (101056783). This project has received funding from the European Research Council under the European Union's Horizon 2020 Research and Innovation Programme under grant no. 101002728. University of Helsinki and Stockholm University (Autumn 2020 Arctic Avenue). Computational resources were provided by the CSC-IT Center for Science, Finland. We would like to thank the Environmental Department City of Malmö for kindly providing benzene observations from the Dalaplan station in Malmö.Notes and references
- WHO Global Air Quality Guidelines, Particulate Matter (PM2.5 and PM10), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide, Wold Health Organization, Geneva, 2021 Search PubMed.
- R. Fuller, P. J. Landrigan, K. Balakrishnan, G. Bathan, S. Bose-O'Reilly, M. Brauer, J. Caravanos, T. Chiles, A. Cohen, L. Corra, M. Cropper, G. Ferraro, J. Hanna, D. Hanrahan, H. Hu, D. Hunter, G. Janata, R. Kupka, B. Lanphear, M. Lichtveld, K. Martin, A. Mustapha, E. Sanchez-Triana, K. Sandilya, L. Schaefli, J. Shaw, J. Seddon, W. Suk, M. M. Téllez-Rojo and C. Yan, Pollution and health: a progress update, Lancet Planet. Health, 2022, 6, e535–e547 CrossRef PubMed.
- Particulate Matter (PM) Pollution, https://www.epa.gov/pm-pollution, accessed, 8 November, 2022 Search PubMed.
- Ambient (Outdoor) Air Pollution, https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health, accessed, 8 November, 2022 Search PubMed.
- D. E. Schraufnagel, The health effects of ultrafine particles, Exp. Mol. Med., 2020, 52, 311–317 CrossRef CAS PubMed.
- M. W. Frampton, Systemic and cardiovascular effects of airway injury and inflammation: ultrafine particle exposure in humans, Environ. Health Perspect., 2001, 109, 529–532 CAS.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Pöschl, S. Takahama, A. Takami, H. Tong, B. Weber, A. Yoshino and K. Sato, Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- W. C. Hinds and Y. Zhu, Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles, Wiley, Hoboken, NJ, 3rd edn, 2022 Search PubMed.
- K. R. Daellenbach, G. Uzu, J. Jiang, L.-E. Cassagnes, Z. Leni, A. Vlachou, G. Stefenelli, F. Canonaco, S. Weber, A. Segers, J. J. P. Kuenen, M. Schaap, O. Favez, A. Albinet, S. Aksoyoglu, J. Dommen, U. Baltensperger, M. Geiser, I. El Haddad, J.-L. Jaffrezo and A. S. H. Prévôt, Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 2020, 587, 414–419 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- N. Riemer, A. P. Ault, M. West, R. L. Craig and J. H. Curtis, Aerosol Mixing State: Measurements, Modeling, and Impacts, Rev. Geophys., 2019, 57, 187–249 CrossRef.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- S. Iyer, A. Kumar, A. Savolainen, S. Barua, C. Daub, L. Pichelstorfer, P. Roldin, O. Garmash, P. Seal, T. Kurtén and M. Rissanen, Molecular rearrangement of bicyclic peroxy radicals is a key route to aerosol from aromatics, Nat. Commun., 2023, 14, 4984 CrossRef CAS PubMed.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- L. Vereecken, B. Aumont, I. Barnes, J. W. Bozzelli, M. J. Goldman, W. H. Green, S. Madronich, M. R. Mcgillen, A. Mellouki, J. J. Orlando, B. Picquet-Varrault, A. R. Rickard, W. R. Stockwell, T. J. Wallington and W. P. L. Carter, Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry: Perspective on Gas-Phase Atmospheric Chemical Kinetic Mechanism, Int. J. Chem. Kinet., 2018, 50, 435–469 CrossRef CAS.
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS.
- L. Vereecken and B. Nozière, H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 2011, 11, 3303–3318 CrossRef CAS.
- P. Roldin, M. Ehn, T. Kurtén, T. Olenius, M. P. Rissanen, N. Sarnela, J. Elm, P. Rantala, L. Hao, N. Hyttinen, L. Heikkinen, D. R. Worsnop, L. Pichelstorfer, C. Xavier, P. Clusius, E. Öström, T. Petäjä, M. Kulmala, H. Vehkamäki, A. Virtanen, I. Riipinen and M. Boy, The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 2019, 10, 4370 CrossRef CAS PubMed.
- T. Han, Z. Ma, W. Xu, L. Qiao, Y. Li, D. He and Y. Wang, Characteristics and source implications of aromatic hydrocarbons at urban and background areas in Beijing, China, Sci. Total Environ., 2020, 707, 136083 CrossRef CAS PubMed.
- D. R. Glowacki, L. Wang and M. J. Pilling, Evidence of Formation of Bicyclic Species in the Early Stages of Atmospheric Benzene Oxidation, J. Phys. Chem. A, 2009, 113, 5385–5396 CrossRef CAS PubMed.
- A. W. Birdsall, J. F. Andreoni and M. J. Elrod, Investigation of the Role of Bicyclic Peroxy Radicals in the Oxidation Mechanism of Toluene, J. Phys. Chem. A, 2010, 114, 10655–10663 CrossRef CAS PubMed.
- S. Wang, R. Wu, T. Berndt, M. Ehn and L. Wang, Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 2017, 51, 8442–8449 CrossRef CAS PubMed.
- M. Rocco, E. Dunne, M. Peltola, N. Barr, J. Williams, A. Colomb, K. Safi, A. Saint-Macary, A. Marriner, S. Deppeler, J. Harnwell, C. Law and K. Sellegri, Oceanic phytoplankton are a potentially important source of benzenoids to the remote marine atmosphere, Commun. Earth Environ., 2021, 2, 175 CrossRef.
- P. K. Misztal, C. N. Hewitt, J. Wildt, J. D. Blande, A. S. D. Eller, S. Fares, D. R. Gentner, J. B. Gilman, M. Graus, J. Greenberg, A. B. Guenther, A. Hansel, P. Harley, M. Huang, K. Jardine, T. Karl, L. Kaser, F. N. Keutsch, A. Kiendler-Scharr, E. Kleist, B. M. Lerner, T. Li, J. Mak, A. C. Nölscher, R. Schnitzhofer, V. Sinha, B. Thornton, C. Warneke, F. Wegener, C. Werner, J. Williams, D. R. Worton, N. Yassaa and A. H. Goldstein, Atmospheric benzenoid emissions from plants rival those from fossil fuels, Sci. Rep., 2015, 5, 12064 CrossRef CAS PubMed.
- C. Bloss, V. Wagner, M. E. Jenkin, R. Volkamer, W. J. Bloss, J. D. Lee, D. E. Heard, K. Wirtz, M. Martin-Reviejo, G. Rea, J. C. Wenger and M. J. Pilling, Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 2005, 5, 641–664 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders, V. Wagner and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 181–193 CrossRef CAS.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Model., 1988, 28, 31–36 CrossRef CAS.
- S. Compernolle, K. Ceulemans and J.-F. Müller, EVAPORATION: a new vapour pressure estimation method for organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 2011, 11, 9431–9450 CrossRef CAS.
- Y. Nannoolal, J. Rarey and D. Ramjugernath, Estimation of pure component properties, Fluid Phase Equilib., 2008, 269, 117–133 CrossRef CAS.
- P. B. Myrdal and S. H. Yalkowsky, Estimating Pure Component Vapor Pressures of Complex Organic Molecules, Ind. Eng. Chem. Res., 1997, 36, 2494–2499 CrossRef CAS.
- A. Klamt, Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, Refinement and Parametrization of COSMO-RS, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- F. Eckert and A. Klamt, Fast solvent screening via quantum chemistry: COSMO-RS approach, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- H. Haario, M. Laine, A. Mira and E. Saksman, DRAM: efficient adaptive MCMC, Stat. Comput., 2006, 16, 339–354 CrossRef.
- H. Haario, E. Saksman and J. Tamminen, An adaptive Metropolis algorithm, Bernoulli, 2001, 7, 223–242 CrossRef.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294 RSC.
- J. D. Crounse, H. C. Knap, K. B. Ørnsø, S. Jørgensen, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 2012, 116, 5756–5762 CrossRef CAS PubMed.
- N. Hyttinen, R. V. Otkjær, S. Iyer, H. G. Kjaergaard, M. P. Rissanen, P. O. Wennberg and T. Kurtén, Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds, J. Phys. Chem. A, 2018, 122, 269–279 CrossRef CAS PubMed.
- L. Xu, K. H. Møller, J. D. Crounse, H. G. Kjaergaard and P. O. Wennberg, New Insights into the Radical Chemistry and Product Distribution in the OH-Initiated Oxidation of Benzene, Environ. Sci. Technol., 2020, 54, 13467–13477 CrossRef CAS PubMed.
- U. Molteni, F. Bianchi, F. Klein, I. El Haddad, C. Frege, M. J. Rossi, J. Dommen and U. Baltensperger, Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 2018, 18, 1909–1921 CrossRef CAS.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Accretion Product Formation from Self- and Cross-Reactions of RO 2 Radicals in the Atmosphere, Angew. Chem., Int. Ed., 2018, 57, 3820–3824 CrossRef CAS PubMed.
- U. Molteni, M. Simon, M. Heinritzi, C. R. Hoyle, A.-K. Bernhammer, F. Bianchi, M. Breitenlechner, S. Brilke, A. Dias, J. Duplissy, C. Frege, H. Gordon, C. Heyn, T. Jokinen, A. Kürten, K. Lehtipalo, V. Makhmutov, T. Petäjä, S. M. Pieber, A. P. Praplan, S. Schobesberger, G. Steiner, Y. Stozhkov, A. Tomé, J. Tröstl, A. C. Wagner, R. Wagner, C. Williamson, C. Yan, U. Baltensperger, J. Curtius, N. M. Donahue, A. Hansel, J. Kirkby, M. Kulmala, D. R. Worsnop and J. Dommen, Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 2019, 3, 873–883 CrossRef CAS.
- R. Atkinson, W. P. L. Carter, K. R. Darnall, A. M. Winer and J. N. Pitts, A smog chamber and modeling study of the gas phase NOx-air photooxidation of toluene and the cresols, Int. J. Chem. Kinet., 1980, 12, 779–836 CrossRef CAS.
- A. H. Goldstein and I. E. Galbally, Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- M. P. Rissanen, J. Mikkilä, S. Iyer and J. Hakala, Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 2019, 12, 6635–6646 CrossRef CAS.
- T. Mattila, M. Kulmala and T. Vesala, On the condensational growth of a multicomponent droplet, J. Aerosol Sci., 1997, 28, 553–564 CrossRef CAS.
- P. Roldin, A. C. Eriksson, E. Z. Nordin, E. Hermansson, D. Mogensen, A. Rusanen, M. Boy, E. Swietlicki, B. Svenningsson, A. Zelenyuk and J. Pagels, Modelling non-equilibrium secondary organic aerosol formation and evaporation with the aerosol dynamics, gas- and particle-phase chemistry kinetic multilayer model ADCHAM, Atmos. Chem. Phys., 2014, 14, 7953–7993 CrossRef.
- P. Roldin, E. Swietlicki, G. Schurgers, A. Arneth, K. E. J. Lehtinen, M. Boy and M. Kulmala, Development and evaluation of the aerosol dynamics and gas phase chemistry model ADCHEM, Atmos. Chem. Phys., 2011, 11, 5867–5896 CrossRef CAS.
- D. B. Ingham, Diffusion of aerosols from a stream flowing through a cylindrical tube, J. Aerosol Sci., 1975, 6, 125–132 CrossRef.
- T. F. Mentel, J. Wildt, A. Kiendler-Scharr, E. Kleist, R. Tillmann, M. Dal Maso, R. Fisseha, T. Hohaus, H. Spahn, R. Uerlings, R. Wegener, P. T. Griffiths, E. Dinar, Y. Rudich and A. Wahner, Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 2009, 9, 4387–4406 CrossRef CAS.
- M. D. Keywood, V. Varutbangkul, R. Bahreini, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol Formation from the Ozonolysis of Cycloalkenes and Related Compounds, Environ. Sci. Technol., 2004, 38, 4157–4164 CrossRef CAS PubMed.
- X. Zhang, C. D. Cappa, S. H. Jathar, R. C. McVay, J. J. Ensberg, M. J. Kleeman and J. H. Seinfeld, Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5802–5807 CrossRef CAS PubMed.
- G. Rolph, A. Stein and B. Stunder, Real-time Environmental Applications and Display System: Ready, Environ. Model. Software, 2017, 95, 210–228 CrossRef.
- C. Xavier, M. Baykara, R. Wollesen de Jonge, B. Altstädter, P. Clusius, V. Vakkari, R. Thakur, L. Beck, S. Becagli, M. Severi, R. Traversi, R. Krejci, P. Tunved, M. Mazzola, B. Wehner, M. Sipilä, M. Kulmala, M. Boy and P. Roldin, Secondary aerosol formation in marine Arctic environments: a model measurement comparison at Ny-Ålesund, Atmos. Chem. Phys., 2022, 22, 10023–10043 CrossRef CAS.
- C. Granier, S. Darras, H. Denier van der Gon, J. Doubalova, N. Elguindi, B. Galle, M. Gauss, M. Guevara, J.-P. Jalkanen, J. Kuenen, C. Liousse, B. Quack, D. Simpson and K. Sindelarova, The Copernicus Atmosphere Monitoring Service global and regional emissions (April 2019 version), DOI:10.24380/D0BN-KX16.
- M. Sofiev, J. Soares, M. Prank, G. de Leeuw and J. Kukkonen, A regional-to-global model of emission and transport of sea salt particles in the atmosphere, J. Geophys. Res., 2011, 116, D21302 CrossRef.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- M. E. Jenkin, J. C. Young and A. R. Rickard, The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- R. W. de Jonge, J. Elm, B. Rosati, S. Christiansen, N. Hyttinen, D. Lüdemann, M. Bilde and P. Roldin, Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 2021, 21, 9955–9976 CrossRef.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 2007, 7, 3909–3922 CrossRef CAS.
- G. McFiggans, T. F. Mentel, J. Wildt, I. Pullinen, S. Kang, E. Kleist, S. Schmitt, M. Springer, R. Tillmann, C. Wu, D. Zhao, M. Hallquist, C. Faxon, M. Le Breton, Å. M. Hallquist, D. Simpson, R. Bergström, M. E. Jenkin, M. Ehn, J. A. Thornton, M. R. Alfarra, T. J. Bannan, C. J. Percival, M. Priestley, D. Topping and A. Kiendler-Scharr, Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 2019, 565, 587–593 CrossRef CAS PubMed.
- O. Garmash, M. P. Rissanen, I. Pullinen, S. Schmitt, O. Kausiala, R. Tillmann, D. Zhao, C. Percival, T. J. Bannan, M. Priestley, Å. M. Hallquist, E. Kleist, A. Kiendler-Scharr, M. Hallquist, T. Berndt, G. McFiggans, J. Wildt, T. F. Mentel and M. Ehn, Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 2020, 20, 515–537 CrossRef CAS.
- S. Tomaz, D. Wang, N. Zabalegui, D. Li, H. Lamkaddam, F. Bachmeier, A. Vogel, M. E. Monge, S. Perrier, U. Baltensperger, C. George, M. Rissanen, M. Ehn, I. El Haddad and M. Riva, Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry, Nat. Commun., 2021, 12, 300 CrossRef CAS PubMed.
- S. O'Meara, A. M. Booth, M. H. Barley, D. Topping and G. McFiggans, An assessment of vapour pressure estimation methods, Phys. Chem. Chem. Phys., 2014, 16, 19453–19469 RSC.
- A. Sekar, G. K. Varghese and M. K. R. Varma, Analysis of benzene air quality standards, monitoring methods and concentrations in indoor and outdoor environment, Heliyon, 2019, 5, e02918 CrossRef PubMed.
- World Health Organization, WHO Guidelines for Indoor Air Quality: Selected Pollutants, WHO, Copenhagen, 2010 Search PubMed.
- N. J. Blake and D. R. Blake, in Encyclopedia of Atmospheric Sciences, Elsevier, 2003, pp. 2438–2446 Search PubMed.
- B. Aumont, S. Szopa and S. Madronich, Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 2005, 5, 2497–2517 CrossRef CAS.
- Z. Fu, F. Ma, Y. Liu, C. Yan, D. Huang, J. Chen, J. Elm, Y. Li, A. Ding, L. Pichelstorfer, H.-B. Xie, W. Nie, J. S. Francisco and P. Zhou, An overlooked oxidation mechanism of toluene: computational predictions and experimental validations, Chem. Sci., 2023, 14, 13050–13059 RSC.
- L. Vereecken and J. Peeters, A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 2010, 12, 12608 RSC.
- L. Vereecken and J. Peeters, Decomposition of substituted alkoxy radicals—part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 2009, 11, 9062 RSC.
- W. Nie, C. Yan, L. Yang, P. Roldin, Y. Liu, A. L. Vogel, U. Molteni, D. Stolzenburg, H. Finkenzeller, A. Amorim, F. Bianchi, J. Curtius, L. Dada, D. C. Draper, J. Duplissy, A. Hansel, X.-C. He, V. Hofbauer, T. Jokinen, C. Kim, K. Lehtipalo, L. Nichman, R. L. Mauldin, V. Makhmutov, B. Mentler, A. Mizelli-Ojdanic, T. Petäjä, L. L. J. Quéléver, S. Schallhart, M. Simon, C. Tauber, A. Tomé, R. Volkamer, A. C. Wagner, R. Wagner, M. Wang, P. Ye, H. Li, W. Huang, X. Qi, S. Lou, T. Liu, X. Chi, J. Dommen, U. Baltensperger, I. El Haddad, J. Kirkby, D. Worsnop, M. Kulmala, N. M. Donahue, M. Ehn and A. Ding, NO at low concentration can enhance the formation of highly oxygenated biogenic molecules in the atmosphere, Nat. Commun., 2023, 14, 3347 CrossRef CAS PubMed.
- E. Praske, R. V. Otkjær, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, Atmospheric autoxidation is increasingly important in urban and suburban North America, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 64–69 CrossRef CAS PubMed.
- R. V. Otkjær, H. H. Jakobsen, C. M. Tram and H. G. Kjaergaard, Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 2018, 122, 8665–8673 CrossRef PubMed.
- C. L. Faiola, A. Buchholz, E. Kari, P. Yli-Pirilä, J. K. Holopainen, M. Kivimäenpää, P. Miettinen, D. R. Worsnop, K. E. J. Lehtinen, A. B. Guenther and A. Virtanen, Terpene Composition Complexity Controls Secondary Organic Aerosol Yields from Scots Pine Volatile Emissions, Sci. Rep., 2018, 8, 3053 CrossRef CAS PubMed.
- A. Kaduwela, D. Luecken, W. Carter and R. Derwent, New directions: atmospheric chemical mechanisms for the future, Atmos. Environ., 2015, 122, 609–610 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00054d |
| This journal is © The Royal Society of Chemistry 2024 |