
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-year, high-time resolution aerosol chemical composition and mass measurements from Fairbanks, Alaska†
Ellis S.
Robinson
 *a,
Michael
Battaglia, Jr‡
b,
James R.
Campbell
cd,
Meeta
Cesler-Maloney
cd,
William
Simpson
cd,
Jingqiu
Mao
cd,
Rodney J.
Weber
b and
Peter F.
DeCarlo
*a
*a,
Michael
Battaglia, Jr‡
b,
James R.
Campbell
cd,
Meeta
Cesler-Maloney
cd,
William
Simpson
cd,
Jingqiu
Mao
cd,
Rodney J.
Weber
b and
Peter F.
DeCarlo
*a
aEnvironmental Health & Engineering, Johns Hopkins University, Baltimore, Maryland, USA. E-mail: shipleyrobinson@gmail.com; pdecarl1@jhu.edu
bSchool of Earth & Atmospheric Sciences, Georgia Institute of Technology, Atlanta, Georgia, USA
cChemistry & Biochemistry, University of Alaska Fairbanks, Fairbanks, Alaska, USA
dGeophysical Institute, University of Alaska Fairbanks, Fairbanks, Alaska, USA
First published on 29th April 2024
Abstract
Fairbanks-North Star Borough, Alaska (FNSB) regularly experiences some of the worst wintertime air quality in the United States. Exceedances of the EPA's 24 h fine particulate matter (PM2.5) rule are common, and can last for weeks-long periods. Here we present sub-hourly measurements of chemically-speciated aerosol measurements over a 25 month span from an Aerosol Chemical Speciation Monitor (ACSM). This dataset includes measurements from all four seasons and over three separate winters (2020, 2021, 2022). It spans a long enough duration to provide an overview of typical seasonal and diurnal variations in aerosol concentrations, composition, and sources in Fairbanks. We observe consistent high PM2.5 concentrations in wintertime, which is dominated by organic aerosol (OA) and, to a lesser extent, sulfate (SO4). We perform factor analysis of the OA using Positive Matrix Factorization (PMF), which reveals three factors, two of which are attributable to primary sources. These primary OA factors are highest in concentration and fractional contribution during wintertime. We show that high concentration periods are correlated with cold temperatures, and enriched in those organic aerosol components related to primary emissions. High concentration periods are also enriched in SO4, though we show that some of the “SO4” measured by the ACSM is very likely organosulfur compounds, which are more prevalent at high concentrations. We also show that within winter, there are significantly different diurnal patterns in PM components depending on meteorological parameters. This analysis is important for understanding air quality patterns in Fairbanks, and as context for the 2022 ALPACA measurement campaign.
Environmental significanceMulti-year, high-time resolution aerosol chemical composition and mass measurements from Fairbanks, Alaska: Fairbanks, AK has some of the worst wintertime air quality in the US due to strong local sources and frequent periods of low dispersion. We present a unique dataset spanning three years (2020–2022), including three winters, that illustrates how particulate matter (PM) emissions sources and meteorology interact and lead to polluted conditions. We show how PM chemical composition changes across season, temperature, and pollution level. These findings are highly relevant to the population of Fairbanks and other similar areas by shedding light on what aspects of the pollution problem in Fairbanks may be addressable at the source. This also establishes important context for interpreting results from the 6-week intensive “ALPACA” field campaign during 2022. |
1 Introduction
Fairbanks-North Star Borough (FNSB), Alaska suffers from perennially poor air quality during wintertime. The area regularly exceeds the health-based PM2.5 threshold put forth by the U.S. Environmental Protection Agency (EPA) in the National Ambient Air Quality Standard (NAAQS) of 35 μg m−3 average over 24 hours, sometimes for multiple days at a time.1 FNSB has been a designated EPA non-attainment area for the 24 hours PM2.5 NAAQS since 2009, and was reclassified as a “serious” non-attainment area in 2017. The area regularly makes the American Lung Association's list of cities with the worst air quality, including as recently as 2021.1,2Poor air quality can be a common feature for urban areas at high latitude in winter, where temperatures are very low and there is very little sunlight; Fairbanks is an extreme example.3 The cause of the pollution is typically local emissions that become trapped near the surface under low-dispersion conditions. Winters in Fairbanks are characterized by extreme cold (reaching as low as −40 °C), very light winds, and strong surface-based inversions (frequently 0.5 °C m−1 or more in the first 10 m above ground level). Fairbanks has a mix of local sources, including industry and power generation, transportation, and residential heating.4 Oil and biomass combustion are the primary fuel sources for residential heating, with relatively little use of natural gas or electric.5 Per capita demand for heating is very high given the extreme cold.
Previous studies of FNSB's air quality problems show that organic aerosol (OA) dominates wintertime PM mass (55–75%).4 Source apportionment studies have found domestic wood burning to be the largest single source of PM pollution, representing between 40–80% of wintertime PM in Fairbanks.6–8 Sulfate (SO4) is the second-most prominent aerosol component, representing 15–20% of wintertime PM in the above studies.4,9,10 More recent PMF analysis from Ye and Wang showed decreasing woodsmoke and increasing SO4 as a fraction of total PM over time. Multiple studies12,13 have investigated the sources and mechanisms underlying the observed high SO4 levels, but have been unable to reproduce the high PM2.5 SO4 concentrations (>10 μg m−3) through modeling. Recent work from Moon et al.10 shows that a large fraction of wintertime SO4 are primary emissions, suggesting that the gap between measurements and models may be attributable to emission inventory inaccuracy for sulfur-containing fuels.
Despite the focus that air quality issues in FNSB have received, significant questions remain. Most of the PM speciation work in Fairbanks has been done using filter-sampling, which may have significant artifacts related to partitioning when sampling at ultra-cold temperatures. Additionally, filter sampling, typically done over the course of many hours or days, lacks the temporal resolution required to understand intraday changes and diurnal patterns of different aerosol species. Much of the above literature was published using data from ∼2010 or earlier, and so these studies may not accurately reflect the current air quality problem in FNSB, as PM levels have declined over the past decade, per long-running monitoring data from the Alaska Department of Environmental Conservation (ADEC).14 Despite these decreases, FNSB is still a non-attainment area with respect the EPA thresholds.
Here we present sub-hourly aerosol concentration and composition data from an Aerosol Chemical Speciation Monitor (ACSM) stationed in downtown Fairbanks from January 2020 through February 2022. The long duration of the deployment, the high time resolution, and the chemical composition information make this a novel dataset. Insights on air quality in this unique region may also apply to other urban areas that experience extreme cold and limited photochemistry associated with wintertime in high latitudes. These types of conditions have been under-studied.
In this paper we present our time series of speciated aerosol measurements, including source-resolved OA factors; we explore diurnal trends of aerosol species across seasons; and we shed light on the possible underlying reasons for the unexplained high particulate SO4 levels previously observed. Our findings are relevant to the air quality literature on northern latitude urban areas, of interest to the population of FNSB, and important for understanding the context for the 2022 winter intensive ALPACA field campaign (Alaskan Layered Pollution and Chemical Analysis).15
2 Methods
2.1 Measurement location & spatial context
The primary focus of this paper is data collected from an ACSM instrument, stationed in downtown Fairbanks, AK. Our measurement campaign took place from mid-January 2020 to the end of February 2022. Due to instrument downtime and repairs, we have a few noteworthy gaps in our measurements: during July 2020, and between April and October of 2021. Despite these missing data periods, we have coverage across all four seasons, and three winters. Winter is the main focus of this paper as Fairbanks' local air quality problems are largely a wintertime phenomenon.8,16,17 Fairbanks also can be subject to pollution episodes from regionally- and long-range transported wildfire smoke11 during summer, but the scope of that problem is not easily addressable through local action. No measurements were made during the above-mentioned gaps, but the dataset is otherwise continuous.The ACSM was installed at an existing air quality monitoring site operated by ADEC. The site is part of the US EPA's National Core (NCore) monitoring network, and is situated in downtown Fairbanks along the banks of the Chena River. Fig. 1a shows the location of the sampling site within the domain, and its proximity to major roadways and population density. We refer to the measurement site as the “NCore” site from herein.
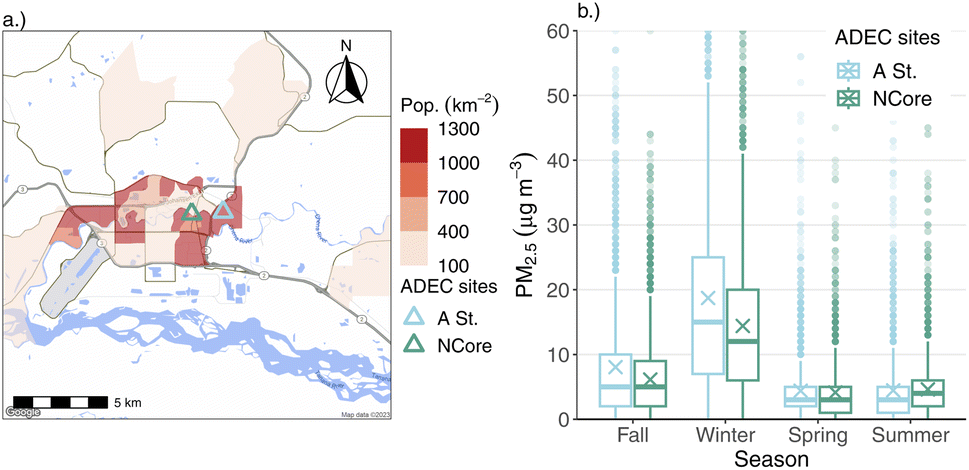 | ||
| Fig. 1 Land use context for the ACSM measurements, which was stationed at the Alaska Department of Environmental Conservation (ADEC) NCore field site. (a) Overview map of Fairbanks that shows reference monitor location sites relative to population density and major roads. Population density is shown at the level of census block groups per the 2020 U.S. Census. (b) Seasonal comparison of hourly PM2.5 concentration data at the ADEC NCore and A Street monitoring sites for the duration of the ACSM measurement time period. Mean concentrations are marked with an “×.” Note: the Y-axis is cut off at 60 μg m−3 to emphasize differences between median and mean concentrations between the two sites, which does exclude a few outlier points above this cut-off. | ||
Downtown Fairbanks has a history of impacts from traffic emissions.18 FNSB was designated as a non-attainment area for carbon monoxide (CO), largely due to automobile emissions, and the NCore site was established in 2011 on account of these legacy reasons. Another ADEC PM2.5 monitoring station was established in 2019 (“A Street”), about 1.7 km east of the NCore site, due to concerns over high PM2.5 levels in residential areas from woodsmoke emissions that weren't fully captured by measurements at NCore. The A Street monitoring site was created as part of the State Implementation Plan (SIP) established to address the perennial non-attainment of the area. Recent mobile monitoring work suggests that the downtown site is relatively more impacted by local traffic emissions and less by local biomass-burning emissions during winter in Fairbanks compared to nearby residential areas (e.g., the neighborhood containing the A Street site).19 The highest PM2.5 concentrations are typically found in North Pole, a neighboring town approximately 20.6 km southeast of downtown Fairbanks. Wintertime PM2.5 concentrations in North Pole are out of attainment, and have a larger relative contribution from woodsmoke to total PM.7 The focus of this paper, however, is air quality in downtown Fairbanks.
2.2 Aerosol chemical speciation monitor
For this study we used an ACSM featuring a quadrupole mass spectrometer with unit mass resolution, a PM2.5 aerodynamic lens, and a “capture vaporizer” (CV).22 The use of the PM2.5 lens allows for transmission of large particles unlike the standard AMS aerodynamic lens, to facilitate direct comparison with regulatory PM2.5 measurements.23 Similarly, the CV is meant to eliminate the artifact of particle bounce24 attributable to some particle phase states and compositions, which can result in undercounting of PM mass for AMS and ACSM instruments compared to other PM measurements using the standard vaporizer (SV). We assume collection efficiency (CE) is equal to 1 for all components measured by the ACSM because of the CV.25
We operate the ACSM at 30 minutes temporal resolution. Sub-hourly measurements of chemical composition, along with the long duration of these measurements, represent the most novel aspects of this study, as they take a large step forward in understanding the behavior of aerosol species, compared to one-in-three day, 24 h average speciated filter data from the NCore site. This allows us to explore diurnal patterns of aerosol constituents, including source-attributed PMF factors for OA, in a way that has not been done before in Fairbanks or other sub-arctic urban areas, especially across seasons. Typical ACSM operation requires alternating between measuring aerosols and gases versus gases-only, in order to correct the ion signal for contributions from background gases. For each 30 minutes run, the ASCM would sample aerosol particles and gases for 15 minutes followed by 15 minutes of sampling air through a HEPA filter to remove aerosol signal. The difference in mass spectral signal is then attributed to aerosol particles.26
Using the default relative ionization efficiency (RIE) for SO4 (RIE = 1.2) and the RF value from the previous instrument calibration made prior to deployment (RF = 3.95 × 10−11 amps μg−1 m−3), we choose a AB reference value that results in the best agreement between ACSM and NCore filter SO4. We define best agreement with be when the AB-corrected ACSM SO4 measurements the NCore filter SO4 have a slope equal to one in comparison with one another, as shown in Fig. 2a. We then apply the AB reference value determined from this SO4-only comparison to the other aerosol species measured by the ACSM, as is typical.
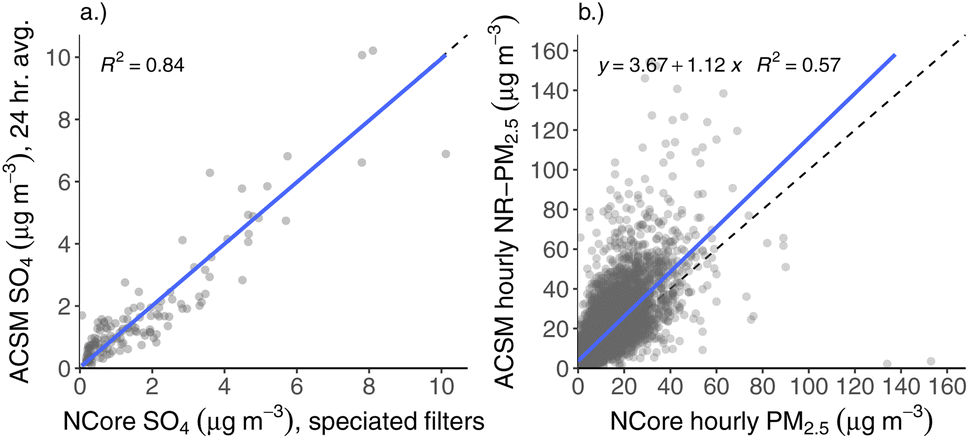 | ||
| Fig. 2 Correlation plots between ACSM and ADEC NCore data. (a) Comparison of ACSM SO4, averaged over the 24 h period of filter collection for the speciated, one-in-three-day filter data from NCore. This plot is used to pick a single airbeam reference value such that the slope between the two SO4 measurements is 1. (b) Comparison of ACSM total NR-PM2.5 with hourly PM2.5 from NCore, after choosing the optimized airbeam reference value based on the SO4 comparison. ACSM data are averaged to the same timeframe as the NCore data for both comparisons. | ||
2.3 PMF analysis
We conducted PMF analysis in order to perform source-apportionment for the OA fraction of NR-PM2.5. PMF has been described in detail elsewhere,29 including specifically for the application to AMS data.30 In brief, PMF is an algorithmic technique that deconvolves the OA matrix into a linear combination of static mass spectral “factors.” We followed the common approach for applying PMF to AMS and ACSM datasets of exploring the solution space for multiple factors (1- through 6-factor solutions) and rotations (fpeak values from −1 to 1).30 None of the factor mass spectra were constrained a priori, as is common with some approaches to PMF (e.g., SoFi31). In addition, we ran PMF on various seasonal subsets of the data, to compare solutions across seasons (e.g., summer-only vs. winter-only vs. full dataset). We used the ACSM analysis panel (v.1.6.1.6) to prepare both the OA MS and error matrices. We performed PMF using the PMF2.exe algorithm with the PMF Evaluation Tool (PET v.2.08) with unit mass ions up to m/z 149. For organic matrix and error matrix preparation we applied the following through the PET analysis panel: (1) removed NaNs and 0 columns, (2) removed “Bad” m/zs using a signal-to-noise ratio (SNR) <0.2 and downweighted “weak” m/zs with SNR <2.2.4 Diurnal pattern calculations
For diurnal pattern calculations, we have segmented our data in a few important ways. First, each quantity (e.g., aerosol species concentration, temperature, etc.) that we present diurnal patterns for we calculate the mean value within each hour of the day. For example, all data between 9:00 am and 10:00 am would be used to calculate the mean value for the 9 o'clock hour, within a given categorical filter (e.g., Spring + weekend). Weekend vs. weekday data filtering was done based on Monday–Friday being weekdays, and Saturday–Sunday being weekends, and all distinctions between days used a 12:00 am cut time. Additionally, we split our data into four seasons, which we operationally defined in the following way: “Winter” was all data from December, January, and February; “Spring” was all data from March, April, and May; “Summer” was all data from June, July, and August; and “Fall” was all data from September, October, and November. These same operationally-defined seasons are used by Ye and Wang in a previous study of Fairbanks' air quality. We include the previous year's December with January and February when considering the winter season (e.g., December 2020 and January–February 2021 comprise “Winter 2021”).2.5 Ancillary data sources
There were multiple data sources important for this study beyond the ACSM dataset. We gathered hourly meteorological data (wind direction, wind speed, and ground-level air temperature) collected at Fairbanks International Airport using the worldMet package for R, which scrapes the NOAA ISD (https://www.ncdc.noaa.gov/isd). These meteorological data are imperfect for use in this context, given that the airport is removed by roughly 4 miles from the NCore site downtown, and may reflect somewhat different wind patterns. Nonetheless, we use the airport meteorological data in the absence of other data and hope that they reflect wind and temperature behavior in downtown Fairbanks. Additionally, data from this weather station that have winds below 1.5 m s−1 are considered “calm”, and are given the value of 0 m s−1. Observations from this weather station are not always made at even intervals, however, and so we compute hourly means in order to standardize the time series, which results in some hourly-average wind speeds below 1.5 m s−1. For this reason, we display both median and mean wind speed values in diurnal plots (e.g., Fig. 7), where median wind speeds of zero indicate that winds are “calm” for the majority of the time.We also downloaded the one-in-three-day, 24 h speciated PM2.5 filter data from the EPA website (https://aqs.epa.gov/aqsweb/airdata/download_files.html), which was used for the “data calibration” procedure with ACSM SO4 described above (shown in Fig. 2a). These speciated filter data run through the duration of the ACSM dataset. We downloaded hourly BAM PM2.5 data measured at both aforementioned ADEC sites (A Street and NCore), which is used for Fig. 1b and 2b. Hourly ADEC PM2.5 data were retrieved from the EPA website for measurements taken prior to Jan. 1, 2022 (data up to Jan. 1, 2022 are considered finalized by ADEC). Hourly PM2.5 data from Jan. 1, 2022 to the end of the campaign (Feb. 25, 2022) were downloaded directly from the ADEC website (https://dec.alaska.gov/air/air-monitoring/alaska-air-quality-real-time-data). ADEC declares these data as preliminary, but we still use them for the comparison here and find no meaningful difference of the preliminary data with respect to the ACSM compared with the finalized data. Lastly, we downloaded gas criteria pollutant data (ozone (O3), sulfur dioxide (SO2), and CO) made at the NCore site from the EPA website.
2.6 Data analysis software used
We used multiple data analysis software tools for this study. We used the ACSM Local (v1.6.1.6) data processing package written for Igor Pro (v8) to process raw ACSM data files and to apply the ion transmission correction. To perform PMF analysis, we used the PMF Evaluation Tool (PET, v2.08A) for Igor Pro. Both ACSM Local and PET are created and maintained by Aerodyne Research, Inc. We performed all other data analysis and visualizations using R software (v3.6.3). In R, we used the worldMet, tidyverse, ggmap, scales, and lubridate libraries.3 Results & discussion
3.1 PM2.5 comparisons
3.2 PMF solution
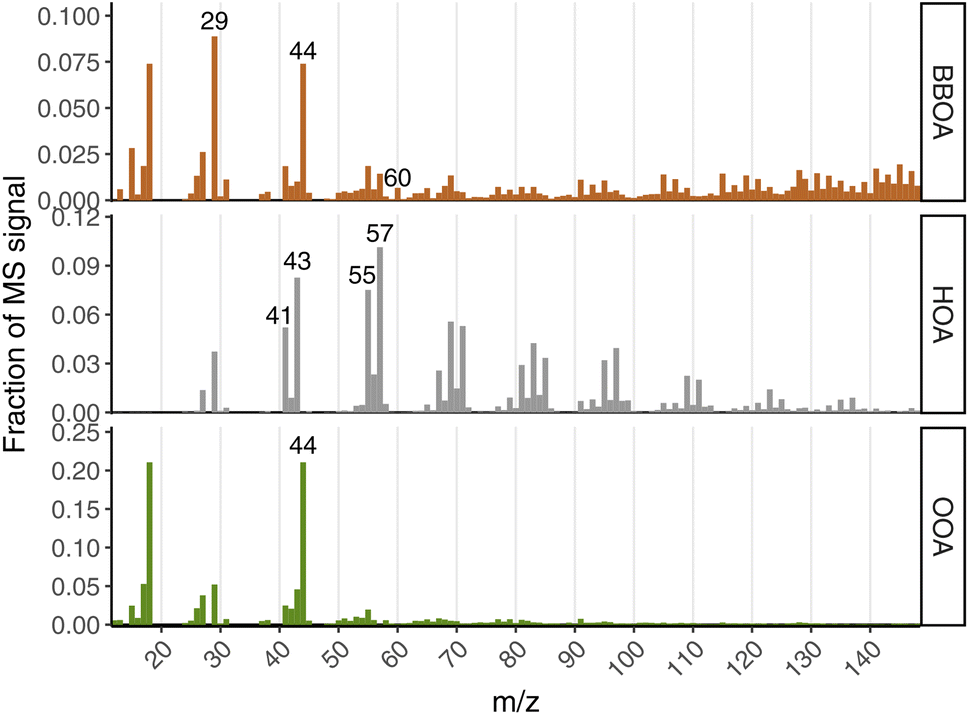 | ||
| Fig. 3 Normalized mass spectra of each PMF factor for the 3-factor solution. | ||
The HOA factor profile is highly similar to other previously published HOA profiles in the literature, and generally represents non-biomass fuel combustion emissions and/or lubricating oil emissions. In the Fairbanks context, this could possibly include OA from gasoline vehicle exhaust, diesel vehicle exhaust, and/or furnace heating oil emissions. The average cosine similarity of our HOA factor to three other published HOA factors was 0.920 (Zhang et al.:32 0.921, Ng et al.:33 0.925, and Crippa et al.:34 0.913), which is high in the context of comparing PMF factors across studies.30 It exhibits the common “picket fence” fragmentation pattern of reduced hydrocarbon molecules, and has prominent peaks at m/z 43, m/z 55, and m/z 57, which typically contain substantial contributions from reduced fragments C3H7+, C4H7+, and C4H9+, respectively. Without high-resolution mass spectra, we cannot definitively conclude that all (or nearly all) of the signal in the HOA factor derives from non-oxygenated CxHy fragments, but ratios of m/z 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m/z 55 and m/z 43:m/z 41 above 1 are both typical of canonical HOA mass spectra, and is true for our HOA factor as well. Based on the diurnal pattern (discussed in more depth below) it seems likely that our HOA factor is largely from vehicle emissions. We also see that HOA correlates most strongly (R2 = 0.5) with CO during wintertime compared to all other aerosol species and OA factors (see Fig. S2†).
:m/z 55 and m/z 43:m/z 41 above 1 are both typical of canonical HOA mass spectra, and is true for our HOA factor as well. Based on the diurnal pattern (discussed in more depth below) it seems likely that our HOA factor is largely from vehicle emissions. We also see that HOA correlates most strongly (R2 = 0.5) with CO during wintertime compared to all other aerosol species and OA factors (see Fig. S2†).
Our BBOA factor likely represents fresh wood-burning PM emissions. It contains almost all (92%) of the signal at m/z 60, which is typically uniquely comprised of the C2H4O2+ ion. This is the main unique ion fragment in AMS mass spectra of levoglucosan,35 which is a marker of fresh biomass burning aerosol emissions.36 The BBOA factor also contains a high fraction of mass at larger m/z (e.g., m/z > 70) compared to the OOA factor, which is a common feature of BBOA factor profiles in comparison to OOA from previous studies.33,34 Compared to three other BBOA factors reported in the literature, we computed an average cosine similarity of 0.767 (Ng et al.:33 0.734, Crippa et al.:34 0.770, and Crippa et al.:34 0.797). This similarity value is lower than that of our HOA factor compared against other HOA factors from the literature, but is expected to be lower given that biomass-burning tends to have more-variable composition,35 and the CV tends to fragment molecules to a greater degree compared to the library mass spectra from SV instruments. Indeed, we see this with m/z 60 in this instrument: despite our BBOA containing the large majority of the m/z 60 signal, and there being no shortage of actual biomass-burning PM in Fairbanks, we see that the fraction of this marker ion (f60) is substantially lower (f60 = 0.0065) than that of BBOA profiles from previously published studies using the SV (f60 ∼ 0.15–0.04). Similarly, the fraction of m/z 73 (another marker for BBOA emissions35) to total OA (f73 = 0.002) in the BBOA factor is also lower than in SV instruments, though we do see that m/z 60 and m/z 73 are relatively well-correlated (R2 = 0.85 during winter). Despite these differences with the reference spectra, interpreting this PMF factor as “BBOA” is reasonable based on its spectral characteristics in the context of CV vs. SV differences, as well as its diurnal pattern, which we discuss in a subsequent section.
OOA is the least straightforward factor of the three to interpret, despite being very spectrally similar to other previously-published OOA factor profiles. The average cosine similarity between our OOA profile and three other studies was 0.953 (Zhang et al.:32 0.918, Lanz et al.:37 0.987, and Ng et al.:33 0.954). The most prominent peak in the OOA MS is at m/z 44, which is dominated by the CO2+ ion, and comes from oxygen-containing groups such as e.g., carboxylic acids, alcohols, carbonyls. From mid-latitude studies using AMS PMF, OOA factors are often attributed to secondary OA (SOA). In Fairbanks, it is not clear that there should be much photochemically-formed SOA during wintertime, given the low solar insolation and low O3.17 Nonetheless, OOA is the dominant PMF factor, making up the majority of OA across all seasons. It comprises 53% of the OA in winter, and is even higher in all other seasons.
Despite the spectral similarity, we do not interpret our OOA factor as directly comparable to other OOA factors from the literature that represent SOA. It likely represents a mix of real biomass-burning OA and some kind of oxidized OA, which could also be biomass-burning OA just with more atmospheric aging. The three-factor solution identifies a “fresh” BBOA factor (discussed above), which contains all of m/z 60; this helps us establish a lower-bound on what the contribution of primary biomass-burning emissions is to total OA (∼31%) during wintertime. There are a few reasons why this is likely just a lower-bound, and that some real biomass-burning PM gets apportioned into the OOA factor, however. First, as discussed in more depth in Section 3.5, the diurnal patterns of OOA and BBOA are very similar, indicating that there is likely not any hard boundary between the two factors with respect to sources. Second, there is strong temporal correlation between OOA and BBOA in wintertime (R2 = 0.81), which also may indicate similar source(s) (see Fig. S1†). Third, we know that the limitations of our ACSM-due to the unit-mass resolution mass spectrometer and the CV-will tend to decrease the complexity of the mass spectrum, and thus homogenize spectral differences between factor profiles. Specifically, the CV tends to lead to more fragmentation,38 and Zheng et al.39 show that a CV-ACSM apportioned more mass to SOA versus POA using PMF for a side-by-side comparison with a SV-AMS. Fourth and lastly, we know from previous AMS studies that BBOA spectra tend to be more variable than other primary OA types e.g., HOA, which reflects how variable biomass burning emissions themselves are. It is reasonable to expect variable composition across the ensemble of biomass burning emissions in Fairbanks, given the likely variability in stove types, stove operation, stages of fire (e.g., smoldering vs. flaming), and fuels used,40–42 and thus a single PMF factor may not fully capture all biomass-burning emissions in this context. So, we label the PMF factor as “OOA” given its spectral similarity to previous results, but think it likely represents a mix of aerosol types that likely includes some biomass-burning PM during winter. Indeed, a previous study from Kotchenruther identified an “aged woodsmoke factor” in their source-attribution for PM in Fairbanks,6 which our OOA factor may reflect during wintertime.
OOA is the dominant PMF factor during summertime, comprising roughly 82% of OA. We do not see a strong correlation between OOA (or any other aerosol species or OA factor) and O3 during summertime (see Fig. S2†). This may be again due to the fact that our OOA factor represents a mix sources, which is summer may include biogenic SOA, aged woodsmoke from long-range transport, and/or other aged OA. Nonetheless, as mentioned by Campbell et al.,27 the large amount of OA, which is dominated by OOA and, to a lesser extent, BBOA, likely provides a large, mildly hygroscopic volume for aqueous-phase reactions to occur even in these cold and dark conditions.
The two-factor solution yields an HOA factor that is very spectrally similar to the HOA factor from the three-factor solution (cosine similarity of 0.999), as well as a non-HOA factor that has clear signatures of both biomass burning PM (e.g., contains all m/z 60) and oxidized aerosol (e.g., large peaks at m/z 44 and m/z 18). Cosine similarity of this non-HOA factor compared to the BBOA and OOA factors from the three-factor solution was high (0.920 and 0.989, respectively), which also indicates shared chemical characteristics. This mixed factor accounts for roughly 80% of the OA mass during wintertime, which is roughly the same as the sum of the OOA and BBOA factors (81%) from the three-factor solution. Thus, both the two- and three-factor solutions indicate that HOA contributes roughly 20% of the wintertime OA mass, with the other roughly 80% being some mix of fresh biomass-burning emissions and more-oxidized OA. The three-factor solution, however, offers more specificity and is more quantitative with respect to biomass burning emissions. For these reasons, we present the three-factor PMF solution as that which best describes the mix of OA in Fairbanks, as measured by our CV-ACSM.
3.3 Time series and seasonal trends
Fig. 4 shows the full ACSM time series for the duration of deployment. Panels show, from top to bottom, (a) the relative contribution of each aerosol species and OA factor to the total NR-PM2.5 mass (monthly average), (b) the monthly mean concentrations of each aerosol species with OA separated into component PMF factors, (c) ambient ground-level temperature, and (d) the raw ACSM time series of the major aerosol species, which is at 30 minutes temporal resolution.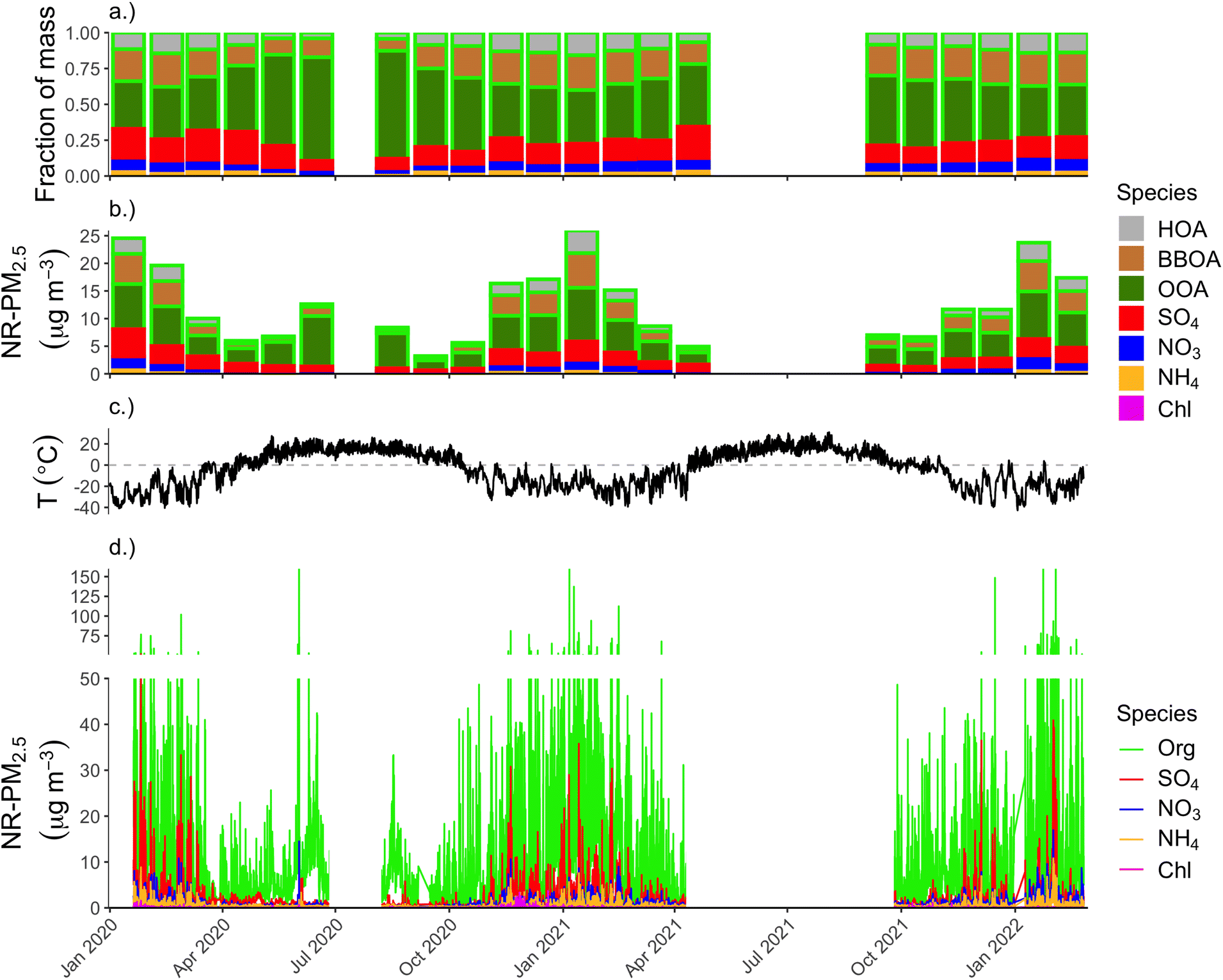 | ||
| Fig. 4 Time series plot of the full dataset (a) monthly-average fractional contribution of each aerosol species and OA PMF factor to total NR-PM2.5. (b) monthly-average mean concentration of all aerosol species, where the sum of all bars is the total NR-PM2.5 concentration. (c) Ambient temperature, measured at the Fairbanks International Airport. (d) Raw time series of 30 minutes average ACSM data for all major aerosol species. | ||
Multiple aspects of the time series panel in Fig. 4 are notable: first and most obviously, concentrations of all aerosol species are higher during the cold winter months compared to spring, summer, and fall months. During winter months, roughly 13% of our half-hourly measurements were above the 24 h PM2.5 NAAQS of 35 μg m−3, and roughly 61% of the measurements were higher than the annual average PM2.5 NAAQS of 12 μg m−3. Most of the high-concentration spikes in concentration above the 24 h NAAQS are driven by sharp increases in OA, but there are many instances in the winter months of SO4 “spikes” exceeding 10 μg m−3, and going as high as 52 μg m−3. This SO4 behavior is starkly different from mid-latitude urban areas where SO4 aerosol tends to be regional in spatial scale,43 and thus concentrations typically do not vary on the order of ∼10 μg m−3 on short, sub-hour-long timescales that we see in the winter in Fairbanks. This behavior in the SO4 time series implies either rapidly-changing boundary layer conditions and/or local emissions of SO4 or SO4-forming precursors that convert to SO4 on fast (sub-hour) timescales.
In the top two panels of Fig. 4 we summarize the seasonal patterns of NR-PM2.5 concentration and composition by showing month-long averages. The second-from-top panel (b) shows monthly average concentrations for each aerosol species, with the height of each bar representing species mass concentration and the sum of all bars representing the total NR-PM2.5 mass concentration. These data show that NR-PM2.5 mass in winter 2022 during the ALPACA campaign is roughly similar to the previous two winters. Average total NR-PM2.5 concentrations for January for 2020, 2021, and 2022 were 24.8, 26.0, and 23.8 μg m−3, respectively. Across the full winter (Dec.–Feb.), average concentrations for 2020, 2021, and 2022 were 19.3, 17.7, and 20.7 μg m−3, respectively.
Average concentrations during spring, summer, and fall periods were much lower than winter, at 7.5, 10.5, and 8.82 μg m−3, respectively. Composition varied between winter and these other seasons as well. OA had the highest fractional contribution in the summer (89%) and the lowest in the spring (70%), though OA dominates NR-PM2.5 mass across all seasons. Roughly 76% of NR-PM2.5 is OA in the wintertime.
Summer air quality in Fairbanks can be heavily impacted by transported wildfire smoke. Our measurements only provide information on one summer (summer 2020), and so may not be indicative of summer air quality in Fairbanks more generally. Total PM2.5 measured by ADEC at both A Street and NCore sites showed summer PM2.5 concentrations during summer 2021 being roughly double that of summer 2020. According to the Alaska Interagency Coordination Center,44 within Alaska there were roughly 180000 acres burned during 2020, the large majority of which happens during summer. In 2019 and 2022, by contrast, roughly 2.7 and 3.2 million acres were burned in Alaska, respectively. Obviously comparing total acres burned across Alaska does not translate directly to air quality impacts in Fairbanks, specifically, but we use this as a proxy that suggests that the summer data we report here are likely on the low end of wildfire-related air quality impacts compared to other summers.
The OOA factor is the largest contributor to OA across all seasons, though varies considerably over the course of the year. In summer, OOA represents 82% of all OA (and 73% of total NR-PM2.5). In winter, OOA is 53% of OA, while BBOA is 31% and HOA is 15%. If the HOA and BBOA factors represent the aerosol components that can be reduced due to behavior changes and/or locally-implemented emissions controls, roughly half of the OA in the wintertime could be addressable through modifiable factors. However, as mentioned in the previous section, we think it is likely that biomass burning emissions are represented in PMF space by both the BBOA and OOA factors, and thus BBOA's fractional contribution to OA of 31% represents a lower-bound of how much of the OA is attributable to biomass burning. Inclusion of some fraction of the OOA would increase the fraction of total OA attributable to biomass-burning emissions sources.
The second highest contributor to NR-PM2.5 is SO4, which makes up 13% of NR-PM2.5 during wintertime and only 6% during summer. SO4 fraction is highest in the spring (19%), though this is when absolute concentrations of NR-PM2.5 were lowest. During winter months, SO4 makes up fractionally more of the total NR-PM2.5 at high mass concentrations, as shown in Fig. 5a. Fig. 5b shows a histogram of wintertime total NR-PM2.5 concentration (bottom panel), binned by intervals of 10 μg m−3. The top panel of 5 shows the average fractional contribution in each concentration bin for each aerosol species and OA factors to the total. Thus, SO4 is a major driver of the highest concentrations we see in Fairbanks in the winter. Note, for each aerosol species, the absolute concentration also tends to increase for each concentration bin (e.g., there is more BBOA at higher concentrations than lower concentration, but its fractional contribution might be lower due to increases in other constituents, like SO4).
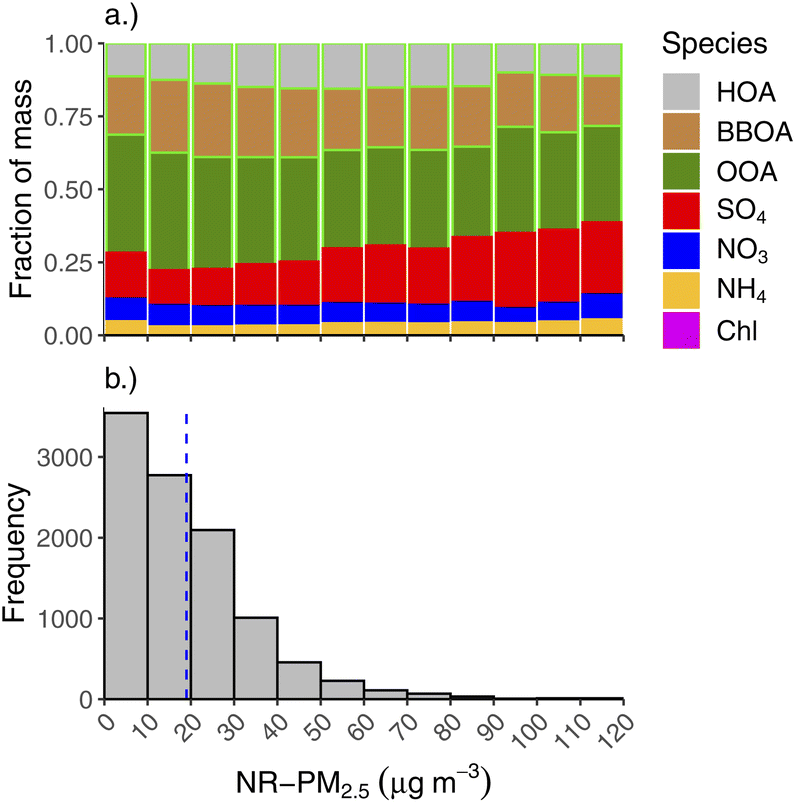 | ||
| Fig. 5 Aerosol composition as a function of total 30 minutes NR-PM2.5 concentration during wintertime. (a) Top panel shows the fractional contribution of each aerosol species and OA PMF factor to the total mass for 10 μg m−3 concentration bins. (b) Bottom panel is a histogram illustrating the frequency of NR-PM2.5 concentrations during winter. Mean wintertime concentration is marked with a blue line on the histogram. | ||
3.4 Aerosol concentrations and ambient temperature
There is a strong relationship between ambient temperature and PM concentration and composition in Fairbanks, illustrated in Fig. 6. Obviously temperature is highly correlated with season, and we have already shown that concentrations are higher in winter compared to summer. Nonetheless, important behavior is revealed by looking at PM concentrations vs. temperature across the entire dataset. Fig. 6 shows all ACSM measurements of total NR-PM2.5 concentration plotted against ambient temperature, and colored by the fraction of the sum of HOA and BBOA PMF factors to total OA. We overlay box plots of NR-PM2.5 concentration within temperature bins of 5 °C increments.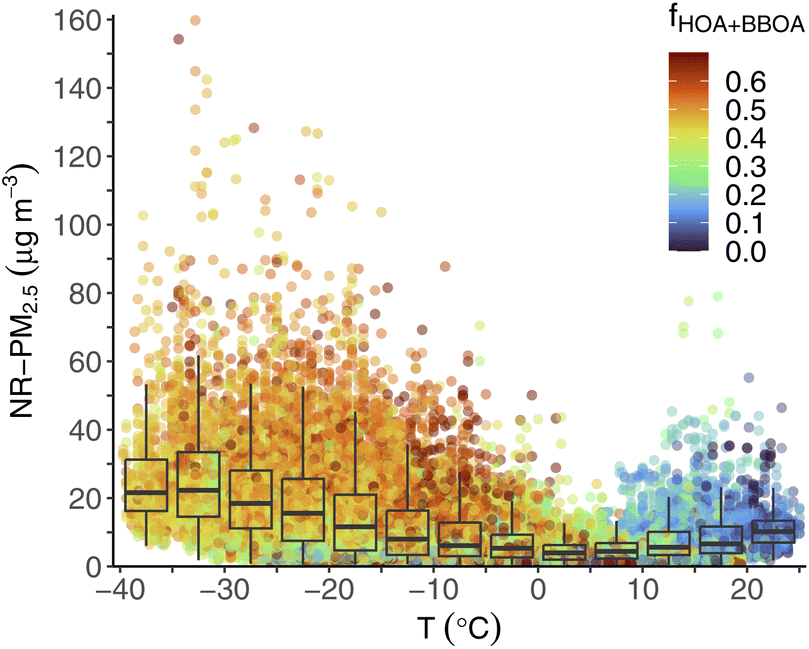 | ||
| Fig. 6 Total 30 minutes NR-PM2.5 concentration as a function of ambient temperature. Points are colored by the fraction of OA from the two primary PMF factors (HOA & BBOA), labeled fHOA+BBOA. Overlaid box plots indicate the distribution of NR-PM2.5 concentration within 5 °C temperature bins. | ||
The peak concentrations we measured were almost exclusively at low temperatures: for example, almost all (93%) NR-PM2.5 >60 μg m−3 were below −10 °C. Median concentrations within 5 °C bins also increased as temperatures decreased. The highest median 5 °C bin concentration was 22.3 μg m−3 at −35 to −30 °C. We observed similar patterns of increasing concentrations at colder temperature for other quantities (e.g., 75th percentile, minimum concentration, etc.).
PM composition is also correlated with temperature, as cold periods tend to be more impacted by primary source contributions than warm periods. We illustrate this by coloring the points on Fig. 6 by the fraction of OA from one of the two primary OA factors, HOA or BBOA, fHOA+BBOA. During warm periods, e.g., 15 °C and above, fHOA+BBOA tends to be low (<0.20), as indicated by green or blue-colored points. During colder periods, especially when concentrations are highest, most of the points are yellow/orange/red, indicating higher contributions from primary sources (fHOA+BBOA > 0.45). As others have documented,16,17,19 meteorological conditions (e.g., surface temperature inversions that lead to low dispersion) play a key role in trapping pollution in Fairbanks, leading to high PM concentrations. During these cold temperature periods, primary emissions make up a higher relative contribution to PM concentrations. This is consistent with high demand for activities that drive primary emissions (e.g., home heating using wood stoves, car idling for warming during startup) as well as gas-particle partitioning that heavily favors the condensed phase at ultra-low temperatures.
3.5 Diurnal behavior of aerosol species
The diurnal behavior of aerosol species varied by season as well, which we show in Fig. 7. This plot shows the mean hourly concentrations of total NR-PM2.5, each major aerosol species, and the three OA PMF factors. Each hourly mean species concentration is calculated for each season and day type (weekday vs. weekend). The panel groupings are meant to allow for quick visual assessments of: (1) the contribution of OA to total NR-PM2.5 as a function of hour of day across seasons, (2) comparisons of the concentrations and daily patterns between the three OA species, and (3) comparisons of the concentrations and daily patterns between each of the inorganic species.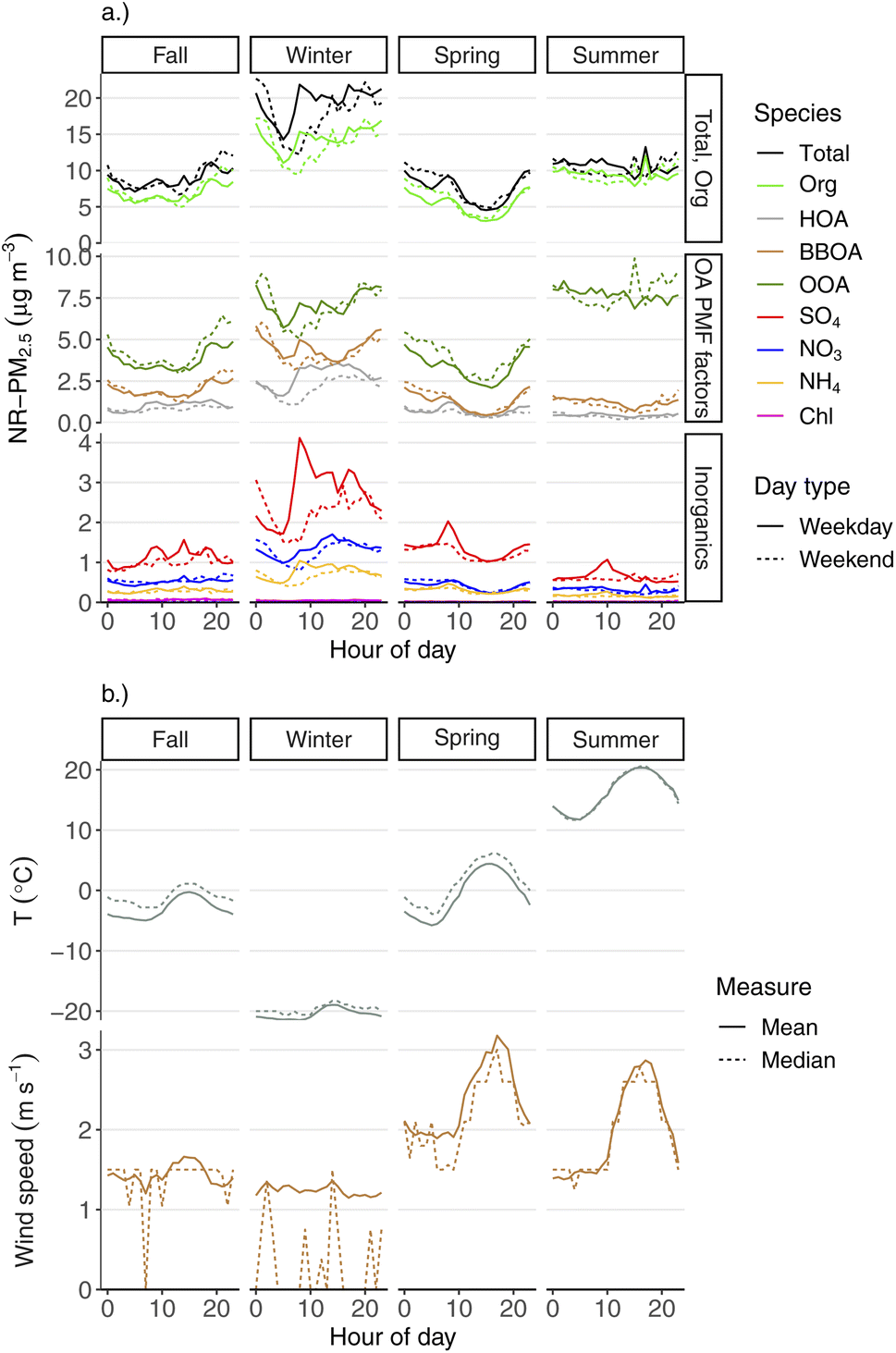 | ||
| Fig. 7 Seasonal diurnal plots of hourly (a) mean aerosol species and OA PMF factor concentrations, and (b) mean and median temperature and wind speed. Each diurnal pattern is calculated for each season (Fall, Winter, Spring, and Summer) and type of day (weekend vs. weekday). | ||
Across all seasons, we see that total NR-PM2.5 and OA follow the same broad diurnal trend as one another. This is unsurprising given that OA makes up the majority of aerosol mass in Fairbanks in all seasons. We also see that NR-PM2.5 and OA have distinct diurnal patterns depending on the season.
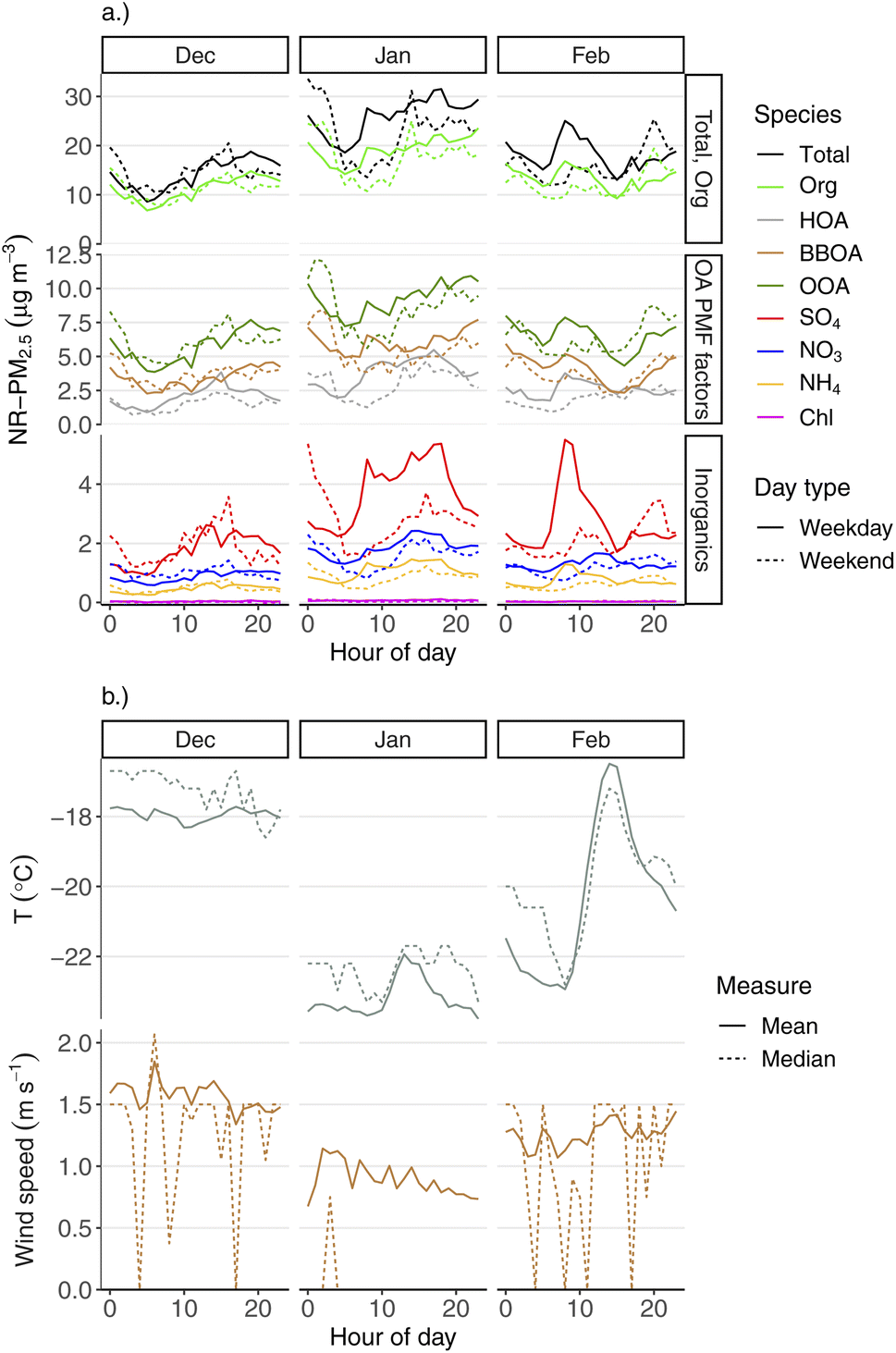 | ||
| Fig. 8 Monthly diurnal plots of hourly (a) mean aerosol species and OA PMF factor concentrations, and (b) mean and median temperature and wind speed. Each diurnal pattern is calculated for each wintertime month (December, January, and February) and type of day (weekend vs. weekday). | ||
Concentrations of the primary OA factors (HOA and BBOA) are at their lowest point during early mornings, with pronounced increases at other parts of the day. This also contributes to the overall wintertime diurnal pattern of total NR-PM2.5, in addition to the meteorological influence discussed above. Both HOA and BBOA have morning increases (around 8 am), suggesting higher activity from primary emitters during this time, which is in line with expectations of both morning commuting and, possibly, higher wood stove use following morning wake-up. The morning increase of HOA is shifted towards later hours in the day during weekends compared to weekdays, which is likely reflective of different weekend vs. weekday driving patterns. During weekdays, the diurnal HOA concentration peaks around typical evening commute time, which is consistent with its source being primary vehicle emissions. HOA concentrations tend to decline starting in the mid-to-late evening. If our HOA factor had a significant contribution from heating-oil combustion particles from residential heating, we might expect to see an increase in the evening, similar to BBOA. These diurnal and weekday/weekend patterns, and the mass spectral profile (see Fig. 3), are what leads us to deduce that this HOA factor is largely vehicular in origin. However, it seems likely that heating-oil combustion exhaust would have a very similar mass spectrum in this instrument to lubricating oil or diesel exhaust, and so we cannot completely rule out a contribution from this source to our HOA factor.
The diurnal behavior of BBOA during wintertime evenings is the opposite of HOA. BBOA concentrations increase in the mid-to-late evening, likely reflecting increased wood stove use as people are in their houses preparing for sleep. Wood stoves also tend to have higher emission factors during startup compared to steady-state use (due to e.g., cold fuels, cold catalyst, etc.),45 which may also explain the BBOA increase in the early evening compared to later in the evening and early morning. Conversely, overnight wood stove use (while people are sleeping) may tend to have lower emissions compared to evening hours, which could also help explain the decrease of BBOA signal in the early mornings, in addition to increased dispersion as discussed above. OOA follows a similar diurnal pattern to BBOA, which is one of the reasons why we assume that OOA also represents real biomass-burning PM. Early afternoon is when we see the daily minimum for BBOA, which likely reflects relatively reduced wood stove use due to both warmer mid-day temperatures and/or people being in their homes less.
Interestingly, SO4 shows similar patterns to HOA, both diurnally and with respect to weekday/weekend behavior. Like HOA, there is a strong morning increase, around typical morning work and school commute times. Similarly, it stays high throughout the day, and then decreases following evening commute times. The weekend/weekday differences for HOA and SO4 are similar as well, with both the morning increase and evening decrease being shifted later on weekends. The diurnal similarity to HOA suggests that some meaningful fraction of the sulfate could be linked to vehicle emissions, either through direct SO4 emission or precursor emissions followed by fast secondary chemistry.
Within the winter season, there are distinct diurnal patterns for aerosol species, wind speed, and temperatures for each month as well. These patterns are shown in the same way as the previous plot, but grouped by winter month (December, January, February) in Fig. 8. Most notably, the diurnal temperature pattern is quite different in February compared to December or January (Fig. 8b). December and January have relatively flat temperature profiles over the course of the day. February, however, shows a 5 °C temperature swing between early-morning minimum temperature and afternoon maximum. This reflects the lack of solar heating in the darkest part of the year (late December) compared to February. All aerosol species have an inverse concentration pattern in February compared to temperature, where early-morning concentrations tend to be at a maximum, but fall to an afternoon minimum, coincident with the temperature increase. This very likely reflects greater dispersion during this afternoon period, as a result of e.g., increased boundary layer height due to solar heating. Within each winter month, we still see very different diurnal behavior between PM constituents, including the primary pollutants (BBOA, HOA, SO4).
Fig. S4† shows the wintertime diurnal pattern for all aerosol species and wind speed and temperature for each of 2020, 2021, and 2022. These plots only use January and February data, as we do not have December 2019 data for the ACSM. We see similar patterns for each species by year, e.g., the morning increase in SO4, highest levels of BBOA factor being overnight, etc.
We also see an afternoon “clean out” in spring for all species, which reflects higher dispersion driven by temperature increases and an afternoon maximum in wind speed. We do not see a similar afternoon clean-out during summer for most aerosol species, despite wind speeds having a similar diurnal pattern across the two seasons. For example, there is little diurnal variation at all for NR-PM2.5, OA, or OOA during the summer. We speculate that this may be due to a combination of nearly round-the-clock daylight (providing opportunity for photochemical PM production) and the prevalence of more regional-scale PM sources (e.g., wildfire smoke and/or photochemical PM).
3.6 SO4 fragmentation pattern
Previous studies have shown that the SO4 signal in the AMS can contain contributions from organosulfur (OS) compounds. Those studies looking at AMS fragmentation patterns of OS compounds have included sodium hydroxymethanesulfonate (Na-HMS);46 organosulfates, sulfonates, and sulfonic acids;47 and sulfones.48 With the exception of methanesulfonic acid (MSA), these OS compounds contain very little HSO3+ or H2SO4+ in their mass spectra, and reduced contribution from SO3+ compared to the (NH4)2SO4 mass spectrum; MSA has some HSO3+, but no H2SO4+ or SO3+. Ammonium sulfate ((NH4)2SO4), by contrast, has contributions from each of SO3+, HSO3+, and H2SO4+ in its mass spectra.We probe the idea that some of the “SO4” signal in our dataset may come from OS compounds by examining the wintertime mass spectrum fragmentation pattern of SO4 as a function of total concentration, using the same binning approach from Fig. 5. Fig. 9 shows the ratio of each HySOx+ fragment relative to SO+ (m/z 48), which is one of the two largest SO4 ion signals in the AMS, the other being SO2+ (m/z 64). We choose to report these HySOx+/SO+ ratios, following the analysis of Hu et al.48 Since this is a unit-mass resolution instrument, our attribution of the SO4-related signal at m/z 48 as “SO+” is not measured directly, but rather estimated based on the default AMS frag table, but we use the chemical ion fragment names for clarity.
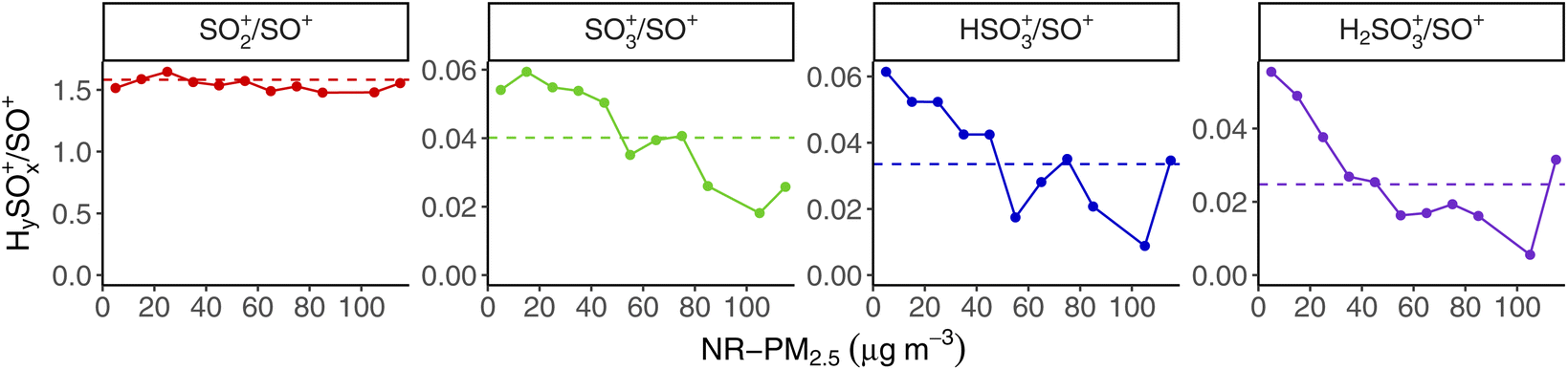 | ||
| Fig. 9 CV ACSM wintertime ratios of the major sulfur-containing fragment ions (SO2+, SO3+, HSO3+, and H2SO4+) to SO+ that make up AMS SO4 as a function of total NR-PM2.5 concentration. Ratios are determined from the slope of the regression fit for all data within each 10 μg m−3 NR-PM2.5 concentration bin. Average for all wintertime data is shown by the horizontal dotted line in each panel. | ||
SO2+/SO+ is relatively flat across all concentration bins in Fig. 9, with a total average value (marked with a dotted horizontal line) of 1.73. However, we see that each of the other HySOx+ ions decreases relative to SO+ with higher total NR-PM2.5 concentrations. This trend is consistent with OS compounds contributing relatively more to total SO4 signal in the AMS at higher concentrations.
Previous work in Fairbanks also shows that OS compounds make up a significant fraction of particulate sulfur especially at high concentrations. Campbell et al.27 estimated that 26–41% of particulate sulfur in Fairbanks is in the form of S(IV) during polluted episodes (where PM2.5 > 35 μg m−3), and that a majority of this S(IV) is expected to be hydroxymethanesulfonate (HMS). Moon et al.10 find that HMS contributes significantly (26%) to total PM sulfur during the most-polluted portion of the ALPACA field campaign. HMS forms via the aqueous phase reaction of SO2 and formaldehyde (HCHO). This formation pathway for particulate sulfur does not require high oxidant levels reliant on the availability of sunlight, unlike e.g., the gas phase oxidation of SO2 by OH or the aqueous phase oxidation of SO2 by H2O2 and O3.49 While HCHO can be formed through photochemical reactions with a variety of VOCs,50 HCHO is also a primary emission of vehicular combustion and biomass-burning,51 which likely dominate over photochemical sources in the cold, dark winter conditions of Fairbanks.52 Our data do not independently confirm that some of the AMS SO4 signal is HMS specifically, but the change we see in SO4 fragmentation patterns at higher NR-PM2.5 concentrations is consistent with a greater contribution from OS compounds like HMS.
Some of the ACSM SO4 signal being OS compounds formed from vehicular HCHO emissions could help explain the similarity between HOA and SO4 diurnal patterns. It also is consistent with the trend we see of ACSM SO4 contributing a larger fraction to total NR-PM2.5 at higher mass concentrations, where HOA and BBOA make up a larger fraction of total OA than at lower mass concentration. OS formation from HCHO would also imply that these combustion-related HCHO emissions are rate-limiting much of the time, as opposed to SO2, in the aqueous phase reaction between HCHO and SO2 that forms OS compounds (like HMS).
Because of the significantly reduced signal at SO3+, HSO3+, and H2SO4+ in CV instruments compared to SV instruments due to greater fragmentation, even when measuring (NH4)2SO4, there are much higher signal-to-noise issues when trying to quantify OS compounds. Preliminary analysis of ALPACA data from a high-resolution time-of-flight SV AMS (HR-ToF-AMS) independently confirms these same trends in the SO4 fragmentation pattern, however, which eliminates the concern that this result could be an artifact of the unit-mass fragmentation table. These SV HR-ToF-AMS data will be presented in a future publication.
4 Conclusions
We present high time resolution observations of PM mass and composition from downtown Fairbanks made over the course of three years and across all four seasons. These measurements are important for better understanding air quality issues in a city that has dealt with problematic levels of air pollution over the course of decades.We show how different aerosol species vary over the course of the day, between different types of day (weekday vs. weekend), and between and within seasons. Identifying the diurnal pattern of different aerosol components sheds light on their sources, and is a major novel aspect of this study. We also deconvolve the dominant non-refractory aerosol species-OA-into different factors, which reflect source contributions. We estimate that, at a minimum, half of the OA comes from primary emissions sources during winter, and thus may be addressable through various mechanisms (emissions controls, behavior changes, etc.). Some fraction of the second-most abundant aerosol species by mass-SO4-may also be addressable through similar mechanisms.
Because Fairbanks has such perennial air quality problems during winter, focusing on modifiable emissions sources makes sense. However, we show, as others have before, that pollutant concentrations are also strongly controlled by meteorological conditions (e.g., temperature, wind speed) as well. In Fairbanks, it truly takes both strong local emissions and low dispersion levels to create the air quality issues typical of wintertime. This interplay between dispersion and emissions creates a potential strategy for mitigating the most-polluted periods by reducing emissions when dispersion is lowest, which has led FNSB to institute a tiered “burn ban” system limiting wood stove use during forecasted temperature inversions.
Lastly, this study helps put the ALPACA campaign15 into context, which will provide many more-detailed insights on air quality issues in Fairbanks from winter 2022. We show here that wintertime concentrations in 2022 are similar to the two prior years, and that chemical composition and diurnal patterns are broadly similar as well. Thus, insights from ALPACA should be broadly applicable to wintertime in Fairbanks, as well as cold, dark urban places more generally.
Author contributions
R. J. W. and J. M. planned experimental design. M. B., J. R. C., J. M., and R. J. W. set up ACSM for data collection in Fairbanks. M. B., J. R. C., J. M., and E. S. R. monitored the ACSM during the course of data collection. E. S. R. and P. F. D. analyzed data and wrote the manuscript, with significant input from all other co-authors. J. M. and W. S. organized ALPACA 2022.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank University of Alaska Fairbanks and the Geophysical Institute for logistical support, and we thank Fairbanks for welcoming and engaging with this research. We owe a great debt to the on-going work done by ADEC for maintaining and operating the regulatory monitoring network in Fairbanks, which was very useful both for this manuscript and ALPACA as a whole. Thanks as well to ADEC for the use of the instrument trailer at the NCORE monitoring site, without which this study would not have been possible. Funding support for E. S. R. and P. F. D. comes from the U.S. National Science Foundation's (NSF) Navigating the New Arctic (NNA) Program (grant nos. NNA-90086753 and NNA-1927750). J. R. C. and J. M. were supported by the NSF Atmospheric & Geospace Sciences (AGS) Program (grant no. AGS-2029747). M. B. and R. J. W. were supported by NSF AGS (grant no. AGS-2029730) and the NSF NNA (grant no. NNA-1927778). M. C.-M. and W. S. were supported by NSF NNA (grant no. 1927750).References
- Alaska Department of Environmental Convservation, State Implementation Plan, 2016, https://dec.alaska.gov/air/anpms/communities/fbks-pm2-5-moderate-sip/ Search PubMed.
- American Lung Association, State of the Air Report, 2021 Search PubMed.
- J. Schmale, S. R. Arnold, K. S. Law, T. Thorp, S. Anenberg, W. R. Simpson, J. Mao and K. A. Pratt, Earth’s Future, 2018, 6, 1385–1412 CrossRef.
- W. Simpson, et al., ALPACA White Paper, 2019, https://alpaca.community.uaf.edu/wp-content/uploads/sites/758/2019/05/ALPACA-whitepaper.pdf Search PubMed.
- U.S. Department of Agriculture, Wood Energy for Residential Heating in Alaska: Current Conditions, Attitudes, and Expected Use, 2010 Search PubMed.
- R. A. Kotchenruther, Atmos. Environ., 2016, 142, 210–219 CrossRef CAS.
- T. Ward, B. Trost, J. Conner, J. Flanagan and R. K. M. Jayanty, Aerosol Air Qual. Res., 2012, 12, 536–543 CrossRef CAS.
- Y. Wang and P. K. Hopke, Aerosol Air Qual. Res., 2014, 14, 1875–1882 CrossRef CAS.
- R. Davey, Characterizing Wintertime Aerosol Composition and Sulfate Formation in Fairbanks, Alaska, 2020, http://hdl.handle.net/11122/11261 Search PubMed.
- A. Moon, U. Jongebloed, K. K. Dingilian, A. J. Schauer, Y.-C. Chan, M. Cesler-Maloney, W. R. Simpson, R. J. Weber, L. Tsiang, F. Yazbeck, S. Zhai, A. Wedum, A. J. Turner, S. Albertin, S. Bekki, J. Savarino, K. Gribanov, K. A. Pratt, E. J. Costa, C. Anastasio, M. O. Sunday, L. M. D. Heinlein, J. Mao and B. Alexander, ACS ES&T Air, 2024, 1, 139–149 Search PubMed.
- L. Ye and Y. Wang, Atmosphere, 2020, 11, 1203 CrossRef CAS.
- K. M. Shakya and R. E. Peltier, Environ. Sci. Technol., 2013, 47, 9332–9338 CrossRef CAS PubMed.
- P. L. Joyce, R. v. Glasow and W. R. Simpson, Atmos. Chem. Phys., 2014, 14, 7601–7616 CrossRef.
- Alaska Department of Environmental Convservation, Design Values FNSB, 2021, https://dec.alaska.gov/air/air-monitoring/community-data/fnsb-summary-pm25/ Search PubMed.
- W. R. Simpson, J. Mao, G. J. Fochesatto, K. S. Law, P. F. DeCarlo, J. Schmale, K. A. Pratt, S. R. Arnold, J. Stutz, J. E. Dibb, J. M. Creamean, R. J. Weber, B. J. Williams, B. Alexander, L. Hu, R. J. Yokelson, M. Shiraiwa, S. Decesari, C. Anastasio, B. D'Anna, R. C. Gilliam, A. Nenes, J. M. S. Clair, B. Trost, J. H. Flynn, J. Savarino, L. D. Conner, N. Kettle, K. M. Heeringa, S. Albertin, A. Baccarini, B. Barret, M. A. Battaglia, S. Bekki, T. Brado, N. Brett, D. Brus, J. R. Campbell, M. Cesler-Maloney, S. Cooperdock, K. C. d. Carvalho, H. Delbarre, P. J. DeMott, C. J. Dennehy, E. Dieudonne, K. K. Dingilian, A. Donateo, K. M. Doulgeris, K. C. Edwards, K. Fahey, T. Fang, F. Guo, L. M. D. Heinlein, A. L. Holen, D. Huff, A. Ijaz, S. Johnson, S. Kapur, D. T. Ketcherside, E. Levin, E. Lill, A. R. Moon, T. Onishi, G. Pappaccogli, R. Perkins, R. Pohorsky, J.-C. Raut, F. Ravetta, T. Roberts, E. S. Robinson, F. Scoto, V. Selimovic, M. O. Sunday, B. Temime-Roussel, X. Tian, J. Wu and Y. Yang, ACS ES&T Air, 2024, 1, 200–222 Search PubMed.
- H. N. Tran and N. Mölders, Atmos. Res., 2011, 99, 39–49 CrossRef CAS.
- M. Cesler Maloney, W. R. Simpson, T. Miles, J. Mao, K. S. Law and T. J. Roberts, J. Geophys. Res.: Atmos., 2022, 127, e2021JD036215 CrossRef CAS.
- N. R. Council, The Ongoing Challenge of Managing Carbon Monoxide Pollution in Fairbanks, Alaska, The national academies press technical report, 2002 Search PubMed.
- E. S. Robinson, M. Cesler-Maloney, X. Tan, J. Mao, W. Simpson and P. F. DeCarlo, Environ. Sci.: Atmos., 2023, 3, 568–580 CAS.
- N. L. Ng, S. C. Herndon, A. Trimborn, M. R. Canagaratna, P. L. Croteau, T. B. Onasch, D. Sueper, D. R. Worsnop, Q. Zhang, Y. L. Sun and J. T. Jayne, Aerosol Sci. Technol., 2011, 45, 780–794 CrossRef CAS.
- V. Crenn, J. Sciare, P. L. Croteau, S. Verlhac, R. Fröhlich, C. A. Belis, W. Aas, M. Äijälä, A. Alastuey, B. Artiñano, D. Baisnée, N. Bonnaire, M. Bressi, M. Canagaratna, F. Canonaco, C. Carbone, F. Cavalli, E. Coz, M. J. Cubison, J. K. Esser-Gietl, D. C. Green, V. Gros, L. Heikkinen, H. Herrmann, C. Lunder, M. C. Minguillón, G. Monik, C. D. O'Dowd, J. Ovadnevaite, J.-E. Petit, E. Petralia, L. Poulain, M. Priestman, V. Riffault, A. Ripoll, R. Sarda-Estève, J. G. Slowik, A. Setyan, A. Wiedensohler, U. Baltensperger, A. S. H. Prévôt, J. T. Jayne and O. Favez, Atmos. Meas. Tech., 2015, 8, 5063–5087 CrossRef CAS.
- T. Joo, Y. Chen, W. Xu, P. Croteau, M. R. Canagaratna, D. Gao, H. Guo, G. Saavedra, S. S. Kim, Y. Sun, R. Weber, J. Jayne and N. L. Ng, ACS Earth Space Chem., 2021, 5, 2565–2576 CrossRef CAS.
- J. Peck, L. A. Gonzalez, L. R. Williams, W. Xu, P. L. Croteau, M. T. Timko, J. T. Jayne, D. R. Worsnop, R. C. Miake-Lye and K. A. Smith, Aerosol Sci. Technol., 2016, 50(8), 781–789 CrossRef CAS.
- A. M. Middlebrook, R. Bahreini, J. L. Jimenez and M. R. Canagaratna, Aerosol Sci. Technol., 2011, 46, 258–271 CrossRef.
- W. Hu, P. Campuzano-Jost, D. A. Day, P. Croteau, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop and J. L. Jimenez, Aerosol Sci. Technol., 2017, 51(6), 735–754 CrossRef CAS.
- N. L. Ng, S. C. Herndon, A. Trimborn, M. R. Canagaratna, P. L. Croteau, T. B. Onasch, D. Sueper, D. R. Worsnop, Q. Zhang, Y. L. Sun and J. T. Jayne, Aerosol Sci. Technol., 2011, 45, 780–794 CrossRef CAS.
- J. R. Campbell, M. Battaglia, K. Dingilian, M. Cesler-Maloney, J. M. S. Clair, T. F. Hanisco, E. Robinson, P. DeCarlo, W. Simpson, A. Nenes, R. J. Weber and J. Mao, Environ. Sci. Technol., 2022, 56, 7657–7667 CrossRef CAS PubMed.
- E. S. Robinson, T. B. Onasch, D. Worsnop and N. M. Donahue, Atmos. Meas. Tech., 2017, 10, 1139–1154 CrossRef CAS.
- P. Paatero and U. Tapper, Environmetrics, 1994, 5, 111–126 CrossRef.
- I. M. Ulbrich, M. R. Canagaratna, Q. Zhang, D. R. Worsnop and J. L. Jimenez, Atmos. Chem. Phys., 2009, 9, 2891–2918 CrossRef CAS.
- F. Canonaco, M. Crippa, J. G. Slowik, U. Baltensperger and A. S. H. Prévôt, Atmos. Meas. Tech., 2013, 6, 3649–3661 CrossRef.
- Q. Zhang, M. R. Alfarra, D. R. Worsnop, J. D. Allan, H. Coe, M. R. Canagaratna and J. L. Jimenez, Environ. Sci. Technol., 2005, 39, 4938–4952 CrossRef CAS PubMed.
- N. L. Ng, M. R. Canagaratna, J. L. Jimenez, Q. Zhang, I. M. Ulbrich and D. R. Worsnop, Environ. Sci. Technol., 2011, 45, 910–916 CrossRef CAS PubMed.
- M. Crippa, P. F. DeCarlo, J. G. Slowik, C. Mohr, M. F. Heringa, R. Chirico, L. Poulain, F. Freutel, J. Sciare, J. Cozic, C. F. D. Marco, M. Elsasser, J. B. Nicolas, N. Marchand, E. Abidi, A. Wiedensohler, F. Drewnick, J. Schneider, S. Borrmann, E. Nemitz, R. Zimmermann, J.-L. Jaffrezo, A. S. H. Prévôt and U. Baltensperger, Atmos. Chem. Phys., 2013, 13, 961–981 CrossRef.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Atmos. Chem. Phys., 2011, 11, 12049–12064 CrossRef.
- C. J. Hennigan, A. P. Sullivan, J. L. Collett and A. L. Robinson, Geophys. Res. Lett., 2010, 37, L09806 CrossRef.
- V. A. Lanz, M. R. Alfarra, U. Baltensperger, B. Buchmann, C. Hueglin and A. S. H. Prévôt, Atmos. Chem. Phys., 2007, 7, 1503–1522 CrossRef CAS.
- W. Hu, D. A. Day, P. Campuzano-Jost, B. A. Nault, T. Park, T. Lee, P. Croteau, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop and J. L. Jimenez, ACS Earth Space Chem., 2018, 2, 410–421 CrossRef CAS.
- Y. Zheng, X. Cheng, K. Liao, Y. Li, Y. J. Li, R.-J. Huang, W. Hu, Y. Liu, T. Zhu, S. Chen, L. Zeng, D. R. Worsnop and Q. Chen, Atmos. Meas. Tech., 2020, 13, 2457–2472 CrossRef CAS.
- D. Bhattu, P. Zotter, J. Zhou, G. Stefenelli, F. Klein, A. Bertrand, B. Temime-Roussel, N. Marchand, J. G. Slowik, U. Baltensperger, A. S. H. Prevot, T. Nussbaumer, I. E. Haddad and J. Dommen, Environ. Sci. Technol., 2019, 53, 2209–2219 CrossRef CAS PubMed.
- A. Trubetskaya, C. Lin, J. Ovadnevaite, D. Ceburnis, C. O'Dowd, J. J. Leahy, R. F. D. Monaghan, R. Johnson, P. Layden and W. Smith, Energy Fuels, 2021, 35, 4966–4978 CrossRef CAS PubMed.
- F. Fachinger, F. Drewnick, R. Giere and S. Borrmann, Atmos. Environ., 2017, 158, 216–226 CrossRef CAS.
- W. Aas, A. Mortier, V. Bowersox, R. Cherian, G. Faluvegi, H. Fagerli, J. Hand, Z. Klimont, C. Galy-Lacaux, C. M. B. Lehmann, C. L. Myhre, G. Myhre, D. Olivié, K. Sato, J. Quaas, P. S. P. Rao, M. Schulz, D. Shindell, R. B. Skeie, A. Stein, T. Takemura, S. Tsyro, R. Vet and X. Xu, Sci. Rep., 2019, 9, 953 CrossRef PubMed.
- Alaska Interagency Coordination Center, 2023, https://fire.ak.blm.gov/predsvcs/intel.php Search PubMed.
- U.S. Environmental Protection Agency, Wood Stove Emissions: Particle Size and Chemical Composition., U.S. environmental protection agency technical report, 2000 Search PubMed.
- E. Dovrou, C. Y. Lim, M. R. Canagaratna, J. H. Kroll, D. R. Worsnop and F. N. Keutsch, Atmos. Meas. Tech., 2019, 12, 5303–5315 CrossRef CAS.
- Y. Chen, L. Xu, T. Humphry, A. P. S. Hettiyadura, J. Ovadnevaite, S. Huang, L. Poulain, J. C. Schroder, P. Campuzano-Jost, J. L. Jimenez, H. Herrmann, C. O'Dowd, E. A. Stone and N. L. Ng, Environ. Sci. Technol., 2019, 53, 5176–5186 CrossRef CAS PubMed.
- W. Hu, P. Campuzano-Jost, D. A. Day, P. Croteau, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop and J. L. Jimenez, Aerosol Sci. Technol., 2017, 51, 735–754 CrossRef CAS.
- J. M. Moch, E. Dovrou, L. J. Mickley, F. N. Keutsch, Z. Liu, Y. Wang, T. L. Dombek, M. Kuwata, S. H. Budisulistiorini, L. Yang, S. Decesari, M. Paglione, B. Alexander, J. Shao, J. W. Munger and D. J. Jacob, J. Geophys. Res.: Atmos., 2020, 125, e2020JD032706 CrossRef CAS PubMed.
- G. M. Wolfe, J. Kaiser, T. F. Hanisco, F. N. Keutsch, J. A. d. Gouw, J. B. Gilman, M. Graus, C. D. Hatch, J. Holloway, L. W. Horowitz, B. H. Lee, B. M. Lerner, F. Lopez-Hilifiker, J. Mao, M. R. Marvin, J. Peischl, I. B. Pollack, J. M. Roberts, T. B. Ryerson, J. A. Thornton, P. R. Veres and C. Warneke, Atmos. Chem. Phys., 2016, 16, 2597–2610 CrossRef CAS PubMed.
- A. Fortems-Cheiney, F. Chevallier, I. Pison, P. Bousquet, M. Saunois, S. Szopa, C. Cressot, T. P. Kurosu, K. Chance and A. Fried, Atmos. Chem. Phys., 2012, 12, 6699–6721 CrossRef CAS.
- Z. Rao, Z. Chen, H. Liang, L. Huang and D. Huang, Atmos. Environ., 2016, 124, 207–216 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00008k |
| ‡ Now at: U.S. Army DEVCOM CBC, Aberdeen Proving Ground, Maryland, USA. |
| This journal is © The Royal Society of Chemistry 2024 |