
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of temperature and relative humidity on secondary organic aerosol formation from ozonolysis of Δ3-carene†
Ditte
Thomsen‡
 a,
Emil Mark
Iversen‡
a,
Jane Tygesen
Skønager
a,
Yuanyuan
Luo
b,
Linjie
Li
c,
Pontus
Roldin
df,
Michael
Priestley
cg,
Henrik B.
Pedersen
e,
Mattias
Hallquist
c,
Mikael
Ehn
b,
Merete
Bilde
*a and
Marianne
Glasius
*a
a,
Emil Mark
Iversen‡
a,
Jane Tygesen
Skønager
a,
Yuanyuan
Luo
b,
Linjie
Li
c,
Pontus
Roldin
df,
Michael
Priestley
cg,
Henrik B.
Pedersen
e,
Mattias
Hallquist
c,
Mikael
Ehn
b,
Merete
Bilde
*a and
Marianne
Glasius
*a
aDepartment of Chemistry, Aarhus University, 8000 Aarhus C, Denmark. E-mail: glasius@chem.au.dk; bilde@chem.au.dk
bInstitute for Atmospheric and Earth System Research (INAR), University of Helsinki, 00014 Helsinki, Finland
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, 41296 Gothenburg, Sweden
dDivision of Physics, Lund University, 22100 Lund, Sweden
eDepartment of Physics, Aarhus University, Aarhus C, 8000, Denmark
fIVL Swedish Environmental Research Institute, 21119 Malmö, Sweden
gIVL Swedish Environmental Research Institute, 41133 Gothenburg, Sweden
First published on 29th November 2023
Abstract
This study investigates the effects of temperature and relative humidity (RH) on the formation of secondary organic aerosol (SOA) from Δ3-carene, a prevalent monoterpene in boreal forests. Dark ozonolysis experiments of 10 ppb Δ3-carene were conducted in the Aarhus University Research on Aerosol (AURA) atmospheric simulation chamber at temperatures of 0, 10, and 20 °C. Under dry conditions (RH < 2%), the SOA formation in terms of both particle number and mass concentration shows minimal temperature dependence. This is in contrast to previous findings at higher initial concentrations and suggests an effect of VOC loading for Δ3-carene. Interestingly, the mass fraction of key oxidation products (cis-3-caric acid, cis-3-caronic acid) exhibit a temperature dependence suggesting continuous condensation at lower temperatures, while evaporation and further reactions over time become more favourable at higher temperatures. The oxygen-to-carbon ratios in the particle phase and the occurrence of highly oxygenated organic molecules (HOM) in the gas phase show modest increases with higher temperatures. Predictions from the Aerosol Dynamics and Gas- and Particle-Phase Chemistry Kinetic Multilayer Model (ADCHAM) agrees with the experimental results regarding both physical particle properties and aerosol composition considering the experimental uncertainties. At high RH (∼80%, 10 °C), a considerable increase in the particle nucleation rate and particle number concentration is observed compared to experiments under dry conditions. This is likely due to enhanced particle nucleation resulting from more stable cluster formation of water and inorganics at increased RH. However, RH does not affect the particle mass concentration.
Environmental significanceAtmospheric aerosols are known to affect both human health and climate, but the extent of their effects remains a large uncertainty in models of current and future climate. One such uncertainty lies in the limited understanding of the effect of temperature and relative humidity on the formation of secondary organic aerosol (SOA). In this work, we elucidate how temperature and relative humidity affect SOA formation and key particle properties for an important yet understudied monoterpene, Δ3-carene. |
1 Introduction
Secondary organic aerosol (SOA) constitutes a significant fraction of organic particulate matter in the atmosphere1 and affects climate2,3 and human health.4 An important source of SOA is biogenic volatile organic compounds (BVOCs),1,3,5e.g. monoterpenes including α-pinene and Δ3-carene. The total annual emission of monoterpenes is estimated to be ∼127 Tg C per year, where α-pinene is estimated to account for ∼50 Tg C per year.6 Globally, Δ3-carene is estimated to have lower emission than α-pinene,7 but in some environments like the boreal forest, the emission is similar to α-pinene.8,9 BVOCs are rapidly oxidised in the atmosphere via reactions initiated by O3, NO3, OH and may be further oxidised by autoxidation.3,10–12 Oxidation of BVOC leads to products with volatilities spanning several orders of magnitude,12,13 often described by the volatility framework and volatility classes developed by Donahue et al.13Extremely low volatile organic compounds (ELVOCs) condense onto essentially any existing aerosol particle surface.12,13 Highly oxygenated organic molecules (HOM), are compounds formed via autoxidation, which include at least 6 oxygen atoms.13 A substantial fraction of HOM formed from monoterpenes, such as the dimers C18–20H28–32O10–18, are likely ELVOCs. Since the discovery of HOM in 2014, they have been investigated due to their significant contribution to both formation and further growth of SOA.10,12,14–16 These compounds are detected with online atmospheric pressure inlet mass spectrometers.12 In the particle phase, covalently bonded dimers of carboxylic acid monomeric units are identified [e.g. ref. 17–26] and their molecular formulae indicate that they also classify as ELVOCs. These dimers have been hypothesised to form in the gas phase perhaps involving RO2 + RO2 chemistry or stabilised Criegee intermediates,24,27,28 through esterification19,29,30 or oligomerisation18,31 in the particle phase or most recently through acyl peroxy radicals for the C15–C19 particle phase dimers.32 These compounds are identified using offline mass spectrometric methods, such as liquid chromatography coupled to mass spectrometry [e.g. ref. 22, 25, 26 and 33–35].
Semi-volatile organic compounds (SVOCs) partition between the gas and the particle phase and therefore exist in considerable fractions in both phases. SVOCs include compounds such as organic acids [e.g. ref. 25, 26 and 36–38]. These compounds have been identified with offline mass spectrometric methods since the 1990s17,36,39 and more recently with online spectrometric methods like the filter inlet for gases and aerosols and chemical ionisation mass spectrometers (FIGAERO-CIMS).40
The most-emitted monoterpene α-pinene and its SOA formation has been studied for more than 25 years [e.g. ref. 17, 36 and 41–44], but new findings still occur [e.g. ref. 25, 26 and 45–49] in particular at low temperatures. Kristensen et al.25,26 observed that the SOA formation from α-pinene is strongly dependent on temperature with decreasing temperatures yielding a higher particle number concentration and a higher particle mass concentration in the temperature range from −15 to 20 °C. Later studies investigated the temperature effects on HOM formation and found that it decreases with decreasing temperature both in the range from −15 to 20 °C45 and from −50 to 25 °C.46 Surdu et al.49 investigated the effect of relative humidity on SOA yield and chemical composition from ozonolysis of α-pinene at −10 and −30 °C. They observed an increase in the SOA yield with increasing relative humidity and attributed this enhancement to increased partitioning of semi-volatile organics.
The emission of Δ3-carene can be almost as large as the emission of α-pinene in boreal forests.8 In contrast to α-pinene SOA, the SOA formation from Δ3-carene has only received little attention.34–36,42,50,51 Previously, the SOA formation from Δ3-carene has been assumed to be analogous to the SOA formation from α-pinene,52 because of the similarities in their chemical structures. In a series of experiments in the Aarhus University Research on Aerosol (AURA) atmospheric simulation chamber, Thomsen et al.,34 however, find differences in the formation of SOA from α-pinene and Δ3-carene. Ozonolysis of Δ3-carene results in increased particle mass concentrations and larger particles compared to ozonolysis of α-pinene under similar experimental conditions.34
The formation of SOA from monoterpenes is affected by environmental conditions like temperature25,26,45,46 and relative humidity.42,53,54 The effects of temperature34,42 and humidity42,55 on the SOA formation from oxidation of Δ3-carene were previously studied at different VOC loadings. The SOA yield and the particle number concentration from ozonolysis of Δ3-carene increased with decreasing temperature at 25 and −30 °C43 and from 20 to 0 °C.34 Jonsson et al.42 reported a small increase in the particle mass concentration and particle number concentration with increasing RH, whereas Li et al.55 found that the particle number concentration decreases with increasing RH, while the particle mass concentration is relatively constant. HOM formation from Δ3-carene oxidation is still understudied55 and more research is needed to understand the processes and effects of temperature and RH on HOM formation. Furthermore, the volatility distribution of oxidation products from Δ3-carene has not been studied.
Here, we present an overview of a measurement campaign carried out at the AURA atmospheric simulation chamber to further elucidate the oxidation and aerosol formation from the dark ozonolysis of Δ3-carene: the Aarhus Chamber Campaign on Evaporation, Partitioning, and Terpene Oxidation, ACCEPTO. We discuss particle phase properties and composition with a focus on the influence of temperature and relative humidity. For the first time, the chemistry and SOA formation during Δ3-carene ozonolysis is modelled with the Aerosol Dynamics and Gas- and Particle-Phase Chemistry Kinetic Multilayer Model (ADCHAM)56 and compared to the experimental results from the AURA atmospheric simulation chamber. The formation of HOM and the partitioning of volatile organic products are presented in two separate accompanying papers.
2 Methods
2.1 Chamber experiments
Dark ozonolysis experiments of Δ3-carene were performed in the AURA atmospheric simulation chamber. Kristensen et al.25 provide a detailed description of the specifications of AURA. In short, AURA consists of a ∼5 m3 cuboid Teflon bag fixed in a temperature-controlled room. The temperature inside the chamber may be regulated from −16 °C to 26 °C. All instruments used in the current study were placed outside the chamber in an air-conditioned laboratory at 22 °C. The general experimental procedure was (1) filling the chamber with clean air using a zero-air generator (model 737-14, Aadco Instruments, Inc.), (2) injecting ∼180 ppb O3 using an ozone generator (model 610, Jelight Company, Inc.), and (3) injecting 10 or 20 ppb Δ3-carene ((+)-3-carene, Sigma-Aldrich, 98.5%) to the AURA chamber through a glass manifold with a heated (>30 °C) flow of nitrogen gas (10 L min−1). Experiment time 0 minutes corresponds to injection of Δ3-carene marking the start of the experiment. The initial VOC concentration in ppb is calculated based on the injected volume of Δ3-carene. Table 1 presents an overview of the Δ3-carene ozonolysis experiments performed during the ACCEPTO campaign.| Date | ID | [VOC]0 (ppb) | [O3]0 (ppb) | T avg (°C) | RHavg (%) | Max. concentrations | SOA yield (%) | |
|---|---|---|---|---|---|---|---|---|
| Number (×104 cm−3) | Mass (μg m−3) | |||||||
| a This experiment was performed after the main campaign and a pump was added to simulate the airflows of the FIGAERO-CIMS and the NO3-CIMS. | ||||||||
| 220202 | 0A | 10 ± 5 | 159 ± 15 | 0.1 ± 0.1 | 1.6 ± 1.6 | 3.1 | 3.1 | 5 ± 3 |
| 220113 | 10A | 10 ± 5 | 174 ± 15 | 10.1 ± 0.1 | 0 ± 0 | 2.9 | 3.2 | 7 ± 4 |
| 220204 | 10B | 10 ± 5 | 171 ± 15 | 10.1 ± 0.1 | 0.4 ± 0.8 | 3.8 | 7.4 | 13 ± 7 |
| 220427 | 10Ca | 10 ± 5 | 169 ± 15 | 10.2 ± 0.1 | 0.6 ± 1.0 | 2.7 | 5.7 | 9 ± 5 |
| 220124 | 10D | 10 ± 5 | 174 ± 15 | 10.2 ± 0.1 | 78 ± 2 | 18.1 | 3.6 | 4 ± 2 |
| 220131 | 10E | 20 ± 5 | 169 ± 15 | 10.2 ± 0.1 | 76 ± 1 | 17.2 | 20.6 | 14 ± 7 |
| 220111 | 20A | 10 ± 5 | 181 ± 15 | 20.2 ± 0.1 | 0 ± 0 | 2.9 | 5.5 | 9 ± 5 |
| 220205 | 20B | 10 ± 5 | 181 ± 15 | 20.2 ± 0.1 | 0 ± 0 | 3.3 | 7.4 | 11 ± 6 |
The temperature and RH were measured in the centre of the chamber with a probe (HC2-C04, Rotronic AG). Five additional temperature sensors (ADT740, Analog Devices) measured the temperature at five different locations inside the temperature-controlled room around the chamber to check for a uniform temperature throughout the chamber. The ozone concentration and the concentrations of nitrogen oxides (NO and NO2) were monitored with a UV photometric (O342 Module, Environnement S. A.) and a chemiluminescent monitor (AC32M, Environnement S. A.), respectively.
Particle size distributions were measured with a scanning mobility particle sizer (SMPS) consisting of an electrostatic classifier (model 3082, Impactor 071CM, TSI Inc.) and a water-based condensation particle counter (CPC, model 3788, TSI Inc.). The electrostatic classifier was equipped with a long differential mobility column and operated with a Kr-85 neutraliser (TSI 3077A, TSI Inc.). The SMPS sampling time was 120 s (117 s upscan, 3 s downscan) with a purge time of 30 s to avoid carry-over of particles between scans. The sheath flow was 6.0 L min−1 and the aerosol flow was 0.6 L min−1. The lower cut-off diameter was 10 nm and the upper cut-off diameter was 421 nm. In experiments 10D and 10E, the aerosol stream was dried to an RH < 15% with a silica diffusion dryer (TSI Inc.) before entering the SMPS. The initial nucleation was monitored with a nano condensation nucleus counter (nCNC, Model A11, Airmodus Ltd.57) consisting of a particle size magnifier (Model A10, Airmodus Ltd.) and a butanol-operated CPC (Model A20, Airmodus Ltd.). The saturator flow was 0.245 L min−1 corresponding to a lower cut-off diameter of 1.7 nm. The nCNC measured for 30 minutes and was then disconnected from AURA. The chemical composition of the particles was monitored in several ways: (1) with a high-resolution time-of-flight aerosol mass spectrometer (AMS, Aerodyne Research, Inc.58), (2) with a Filter-Inlet for Gases and AEROsols Chemical Ionization Mass Spectrometer (FIGAERO-CIMS40), and (3) using a sequential spot sampler (Series 110 A, Aerosol Devices59,60) for detailed offline analysis using an ultra-high performance liquid chromatograph coupled to a mass spectrometer through an electrospray ionization inlet. The AMS was operated in V-mode with 1 minute averaging and a flow rate of 0.08 L min−1. In experiments 10D and 10E, the aerosol stream was dried before entering the AMS with an aerosol dryer (Aerodyne Research, Inc.). The gas phase was monitored with (1) a chemical ionization atmospheric pressure interface time-of-flight mass spectrometer (NO3-CIMS)61,62 and (2) the FIGAERO-CIMS. An overview of the instrument positions around the AURA chamber, the flows, and their sampling cycles are given in Section 1 in the ESI.† Experiment 10C was performed after the campaign without the FIGAERO-CIMS and the NO3-CIMS. To maintain similar flow conditions in the chamber without these instruments sampling as in the previous experiments, a pump with a similar flow to these two instruments removed air from the chamber to simulate sampling from the instruments.
All particle number concentrations are reported without wall-loss correction. All particle mass concentrations measured with the SMPS are wall-loss corrected according to Pathak et al.63 with an optimised fit of the decay of particle mass concentration. Section 2 in the ESI† describes the optimised fit and the following wall-loss correction. The uncertainty on the ozone concentration was estimated from the noise and the accuracy associated with the instrument. The uncertainty on the initial concentration of the VOC was estimated from previous experiments34,35 where the VOC concentration was measured with a proton transfer reaction mass spectrometer or a gas chromatograph with a flame ionisation detector. The absolute uncertainty for these experiments was 5 ppb, which is then also expected to be the same during the ACCEPTO campaign and it brings a noticeable uncertainty to the estimated SOA yields. The SOA yields are calculated according to Odum et al.64 and Pathak et al.63 The SOA yields are based on the aimed initial VOC concentrations of 10 ppb and the assumption that all Δ3-carene is consumed.34 The nucleation rate is calculated as described in Tomicic et al.65 and the growth rate for particles with diameter > 20 nm is calculated according to Kulmala et al.66 using the maximum-concentration method.67 An outlier test was performed for the mass fraction at 90 min acquired in the offline analysis in experiment 20A. The outlier test followed Fisher's least significance difference method described in Su et al.68
2.2 Particle sampling and offline analysis
Particle samples were collected continuously with a sequential spot sampler (Series 110 A, Aerosol Devices)59,60 in 30 minute intervals at a flow rate of 1.25 L min−1. The sequential spot sampler was operated with the following settings: conditioner temperature of 5 °C, initiator temperature of 35 °C, moderator temperature of 8 °C, nozzle temperature of 27 °C, and sample plate temperature of 35 °C. The particle samples were first extracted with 10% acetonitrile in MilliQ water (<0.05 μS cm−1), then with 50% acetonitrile in MilliQ water. The particle extracts were stored at 5 °C until analysis. The analysis was performed according to Thomsen et al.35 with an ultra-high performance liquid chromatograph (UHPLC) coupled to a quadrupole time-of-flight mass spectrometer (QTOF-MS, Compact, Bruker) through an electrospray inlet. The UHPLC method and QTOF-MS settings are described briefly in Section 6 in the ESI† and a detailed description is found elsewhere.34 Ozonolysis products of Δ3-carene are tentatively identified with authentic standards of α-pinene oxidation products by analyzing their compound-specific retention times34,36 and their mass spectral information e.g. fragmentation patterns and the molecular ions, [M−H]−.19,21,23,24,69,70 The α-pinene oxidation product standards were: cis-pinic acid (Aldrich), cis-pinonic acid (Aldrich, 98%), and diaterpenylic acid acetate (DTAA, kindly supplied by Y. Iinuma, TROPOS, Leipzig, Germany). Due to an extraction error, the data from particle samples from experiment 20B are not reported.2.3 FIGAERO-CIMS
A High-Resolution Time-of-Flight Chemical Ionisation Mass Spectrometer (HR-ToF-CIMS) combined with a Filter Inlet for Gases and AEROsols (FIGAERO, Aerodyne Research Inc.) was connected to the AURA chamber to measure both gas and particle phase oxidation products. A more detailed description of HR-ToF-CIMS with a FIGAERO inlet is found in Lopez-Hilfiker et al.40 During the ACCEPTO campaign, the flow rate of the gas phase into the ion molecule region (IMR) was 2 L min−1. Simultaneously, the particle phase sample was collected on PALL® PTFE membrane filter for 10 min with a flow of 3 L min−1. Fig. S.2† shows the sampling cycle of the FIGAERO-CIMS. The particle sample was subsequently desorbed by heated pure N2 (2 L min−1) into the IMR. The iodide (I−) ion produced from the ionisation of methyl iodide (CH3I) was delivered into the IMR in a flow of pure N2. The reagent ion (I−) and targeted compounds form molecular adduct ions in IMR and the adducts were detected by the HR-ToF-CIMS. The partitioning of detected compounds was calculated using the ion counts in the gas phase and the corresponding ion counts in the particle phase.2.4 Nitrate-CIMS
A NO3-CIMS (Tofwerk AG/Aerodyne Research, Inc.62) with nitrate (NO−3) as reagent ions were deployed for measuring highly oxygenated products of Δ3-carene oxidation in the gas phase. The sample molecules are detected either as deprotonated ions ([M−H]−) or more typically as clusters with reagent ions (M·NO−3). The instrument was configured to be capable of determining ions up to 960 Th with a mass resolving power of ∼8500 Th. In all experiments, except 10A and 20A, the instrument sampled 10 L min−1 from the chamber for the first 80 min after Δ3-carene injection, after which a cycle was started with 40 min without sampling alternating with 20 min of sampling. This was done to limit the amount of air drawn from the chamber. Fig. S.2† shows the sampling cycle of the CI-APi-TOF. In experiments 10A and 20A, air from the chamber was sampled with a flow rate of 5 L min−1 and directly mixed with an equally large clean airflow. In these cases, the sampling was continuous throughout the experiments. The NO3-CIMS data were preprocessed with tofTools MATLAB package (version 612).712.5 ADCHAM modelling
The Aerosol Dynamics and Gas- and Particle-Phase Chemistry Kinetic Multilayer Model ADCHAM72 was used to simulate the gas phase chemistry and secondary aerosol formation during selected ACCEPTO experiments. ADCHAM considers wall losses of particles and gases to the chamber walls and all aerosol dynamic processes. The particle number size distribution was represented by 200 size bins between 1.7 nm and 516 nm in diameter. Currently, no complete gas phase chemistry mechanism for ozonolysis and OH oxidation of Δ3-carene exists. Thus, in this work, we developed and implemented a Δ3-carene oxidation mechanism which is based on the Master Chemical Mechanism v3.3.1 (MCMv3.3.1) for α-pinene chemistry73,74 and the Peroxy Radical Autoxidation Mechanism (PRAM).75,76 The first reaction steps of the O3 and OH oxidation of Δ3-carene is represented by the mechanisms proposed by Wang et al.79 and Hantschke et al.,77 using the temperature-dependent Δ3-carene + OH reaction rate constant from Dillon et al.:78 2.48 × 10−11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(357/T) cm3 molecule−1 s−1. We could not find any empirically derived temperature-dependent reaction rate constant for ozonolysis of Δ3-carene in the literature. Hence, in this work we assume that the temperature-dependence for the Δ3-carene + O3 reaction rate constant is the same as for α-pinene in MCM: 3.7 × 10−16exp(−640/T) cm3 molecule−1 s−1. The implemented Δ3-carene + O3 reaction rate constant matches the reaction rate constant reported by Hantschke et al.77 at 300 K. The OH yield from the ozonolysis of Δ3-carene in the mechanism is 65%, which was based on the empirically estimated yield from Hantschke et al.77 of 65%. It can also be compared with the theoretical values of 56–59% from Wang et al.79 To be able to represent the observed HOM mass spectrum in ADCHAM we used a total ozonolysis molar yield of 2.5% for the first RO2 isomers (C10H15O4) that can undergo autoxidation and produce HOM in PRAM. In addition, we also included a minor (<0.4% molar yield) C9RO2 (C9H13O4) autoxidation pathway to HOM, in order to capture the observed mass peaks corresponding to the ions C9H12O9 and C9H14O9. A more detailed description of the new Δ3-carene gas phase chemistry mechanism is provided in Luo et al. (in prep.).
exp(357/T) cm3 molecule−1 s−1. We could not find any empirically derived temperature-dependent reaction rate constant for ozonolysis of Δ3-carene in the literature. Hence, in this work we assume that the temperature-dependence for the Δ3-carene + O3 reaction rate constant is the same as for α-pinene in MCM: 3.7 × 10−16exp(−640/T) cm3 molecule−1 s−1. The implemented Δ3-carene + O3 reaction rate constant matches the reaction rate constant reported by Hantschke et al.77 at 300 K. The OH yield from the ozonolysis of Δ3-carene in the mechanism is 65%, which was based on the empirically estimated yield from Hantschke et al.77 of 65%. It can also be compared with the theoretical values of 56–59% from Wang et al.79 To be able to represent the observed HOM mass spectrum in ADCHAM we used a total ozonolysis molar yield of 2.5% for the first RO2 isomers (C10H15O4) that can undergo autoxidation and produce HOM in PRAM. In addition, we also included a minor (<0.4% molar yield) C9RO2 (C9H13O4) autoxidation pathway to HOM, in order to capture the observed mass peaks corresponding to the ions C9H12O9 and C9H14O9. A more detailed description of the new Δ3-carene gas phase chemistry mechanism is provided in Luo et al. (in prep.).
3 Results and discussion
3.1 Particle formation and growth
Fig. 1 shows the formation and growth of particles over time obtained from the SMPS data. The upper panels (I–III) relate to investigating the temperature dependence of the SOA formation from ozonolysis of Δ3-carene. The lower panels (IV–VI) relate to the investigation of the effect of relative humidity on SOA formation from ozonolysis of Δ3-carene. Consistent with previous observations,34,35 the particle number concentration generally increases steeply a few minutes after the injection of Δ3-carene. After the maximum particle number concentration is reached, a decay ascribed to wall loss and coagulation is observed. The particle mass concentration and particle mode diameter also grow and reach relatively constant values after about 150 minutes. The full experimental details are presented in Section 3 in ESI.†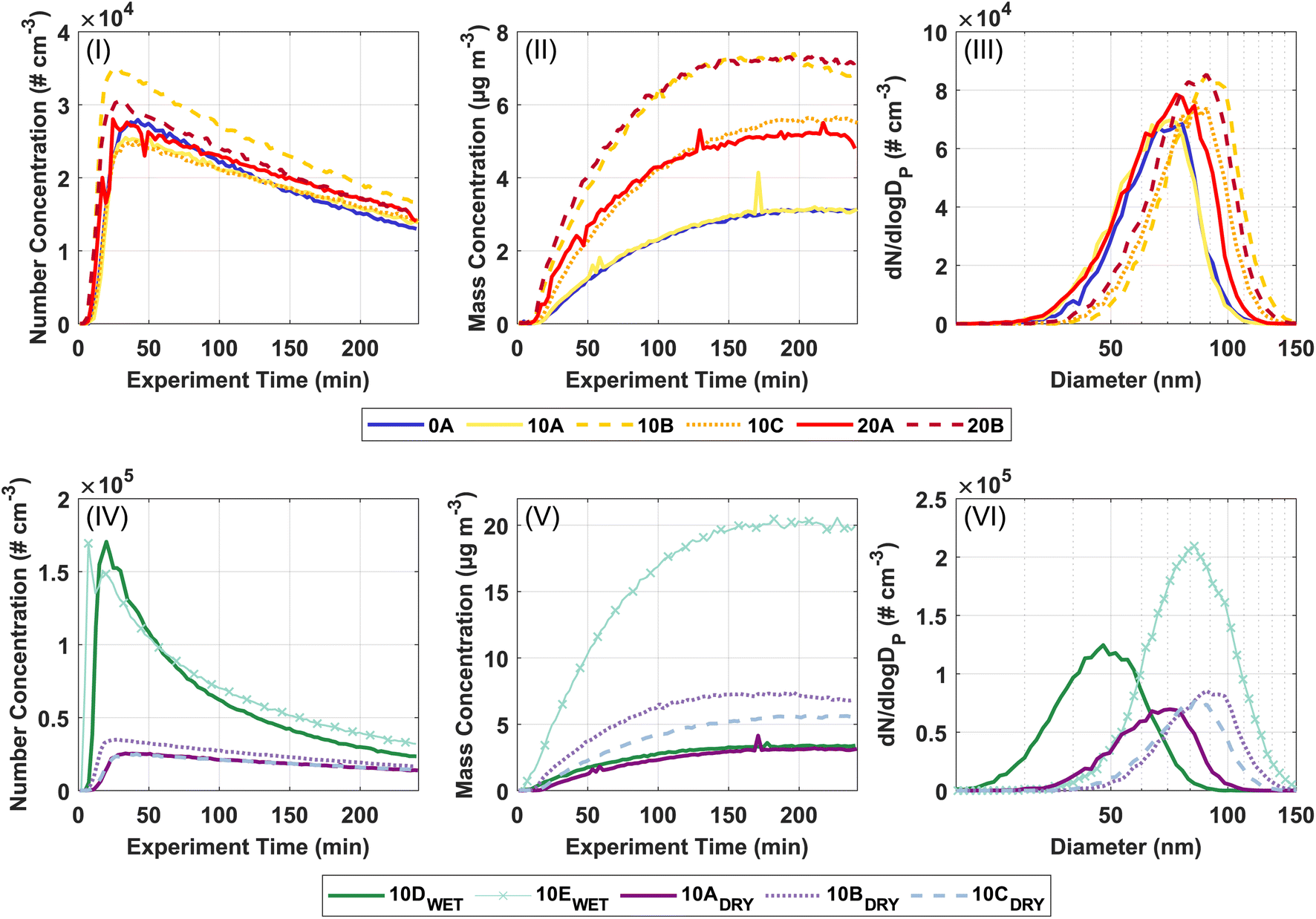 | ||
| Fig. 1 Panels (I–III) refer to experiments conducted in order to investigate temperature dependence. Panels (IV–VI) refer to experiments conducted in order to investigate the effect of humidity. Experiment 10EWET is marked with × as it was performed with a starting concentration of Δ3-carene of 20 ppb instead of 10 ppb. (I and IV) Particle number concentrations in # cm−3. (II and V) SMPS derived particle mass concentrations in μg m−3. (III and VI) Particle size distributions at maximum particle mass concentration. Note that experiment 10A–C is plotted in both series. | ||
The particle number concentrations at 0, 10, and 20 °C at 0% RH are in the range (2.7–3.8) × 104 # cm−3. Across all temperatures, the measured particle number concentrations are almost equal. However, the particle mass concentrations in experiments 10A–C and 20A–B fluctuate. This variation in particle mass concentrations – especially pronounced at 10 °C – makes it difficult to draw any firm conclusions in relation to temperature trends, however, this points towards that the temperature effect on ozonolysis of Δ3-carene is not very strong at the current experimental conditions. The variation in particle mass concentration may in part arise from small differences in the initial concentration of VOC as suggested by the result of the ADCHAM model (Section 3.3) or in part, the low VOC loadings where the concentration of trace compounds, e.g. ammonia or sulfuric acid,80,81 might play a significant role in the formation of particles.
The average particle mode diameter for SOA for all experiments at the maximum particle mass is 80 ± 9 nm, which again indicates that the temperature does not strongly affect the SOA formation from ozonolysis of Δ3-carene. Table 2 shows the nucleation rates and growth rates for all experiments. Neither the nucleation rates nor growth rates show a strong temperature dependence under dry conditions and are across all temperatures between 33.8–59.4 # cm−3 s−1 and 35.1–51.7 nm h−1. In contrast, Simon et al.46 observed increasing nucleation rates with decreasing temperatures for α-pinene oxidation. This illustrates that ozonolysis of Δ3-carene exhibits a less pronounced temperature dependence compared to ozonolysis of α-pinene under dry conditions.
It is interesting to compare our data with previous observations of ozonolysis of monoterpenes in the AURA simulation chamber.25,26,34 Ozonolysis of α-pinene exhibits a clear temperature dependence showing increasing particle number concentrations and particle mass concentrations with decreasing temperature at both 10 and 50 ppb.25,26 However, the temperature effect is less pronounced for the ozonolysis of 10 ppb of α-pinene than at 50 ppb. Ozonolysis of 50 ppb Δ3-carene show some temperature dependency, but not to the same degree as for ozonolysis of α-pinene.34 It thus seems reasonable that the lower VOC concentration in this study leads to a weaker temperature effect for ozonolysis of Δ3-carene. However, drawing any definitive conclusions about the effect of temperature on the particle number concentrations and particle mass concentrations is hindered by the uncertainty in the initial VOC concentration and that only one experiment was performed at 0 °C.
Regarding the influence of relative humidity, ozonolysis of 10 ppb Δ3-carene at 0% RH (low RH, experiments 10A–C) and at 80% RH (high RH, experiment 10D) result in comparable particle mass concentrations. Meanwhile, the particle number concentrations from ozonolysis of Δ3-carene at low RH are in the range (2.7–3.8) × 104 # cm−3, while the resulting particle number concentration at the high RH is 18.1 × 104 # cm−3. The drastic increase in the particle number concentration at high RH is also observed in experiment 10E, where ozonolysis of 20 ppb Δ3-carene at 80% RH results in a similar particle number concentration of 17.2 × 104 # cm−3 and a higher particle mass concentration consistent with the increased VOC loading. The high particle number concentrations observed in both experiments at high RH indicate that RH influences nucleation. This is also clear from the nucleation rates (Table 2) for experiments 10D and 10E, which are considerably higher than in the dry experiments.
While we cannot separate individual contributions to nucleation, growth, and resulting particle mass concentration based on these measurements, it is known that humid conditions enhance the nucleation driven by inorganics.80,82 No inorganics are added to the AURA chamber during these experiments, but direct background measurements show that sulfuric acid is present in very low concentrations (Fig. S.8†). The level of H2SO4 is similar in all experiments. However, in experiments with increased RH, water should stabilise the H2SO4 clusters leading to higher cluster formation rates compared to dry experiments.56,83,84
The particle growth rate is 43 ± 6 nm h−1 at the low RH and 20.6 nm h−1 at the high RH. The smaller particle diameter (48 nm) at high RH compared to low RH (80 ± 9 nm) naturally follows from similar particle mass concentrations but a much higher particle number concentration at high RH.
Previous observations of the effect of RH on SOA formation show conflicting results. Jonsson et al.42 report an increasing particle number concentration with increasing RH in agreement with this work and suggest that it is a mechanistic effect. In contrast, Bonn et al.53 find a decreasing particle number concentration with increasing RH, but do not pursue an explanation of the observation. The discrepancy in the observations may be due to different experimental conditions, as Jonsson et al.42 use ∼29 ppb Δ3-carene and ∼2000 ppb O3 in a flow tube setup, while Bonn et al.53 use 500 ppb of both α-pinene and O3 in a spherical glass reactor.85,86
The effect of humidity is often observed as a considerable increase in the particle mass concentration due to water uptake. During the ACCEPTO experiments carried out at high RH, the particles were dried prior to measurement with the SMPS and the AMS and hence this water uptake is not visible in our observations. Jonsson et al.42 measure the particle size distributions with no drying of the particles and observe an increase in the particle mass concentration. They explain ∼30% of their growth with water uptake and state that the additional growth observed is due to a mechanistic effect of water affecting the product distribution.
3.2 Aerosol composition
Three approaches to characterise the aerosol phase have been applied in this study: (1) AMS, (2) FIGAERO-CIMS, and (3) offline analysis using UHPLC-QTOF-MS. AMS yields information on the general oxidation state of the particle phase in the form of hydrogen-to-carbon (H/C) ratios and oxygen-to-carbon (O/C) ratios. The FIGAERO-CIMS provides information on partitioning. The analysis of particles collected yields both qualitative and quantitative information on aerosol composition.The O/C ratio is often used to characterize the degree of oxidation of compounds in the particle phase. Fig. 2 shows a van Krevelen plot with the H/C ratio as a function of the O/C ratio from AMS data from each experiment. A slight temperature trend is observed with an increasing O/C ratio with increasing temperature for dry experiments. Previous studies with ozonolysis of Δ3-carene at 50 ppb34 and 100 ppb35 and ozone concentration of about 200 ppb, respectively, both show O/C ratios of about 0.4. In this work, the O/C ratio for experiments 10A–C lies at about 0.45–0.5, which indicates a more oxidised particle phase. This likely stems from the lower VOC loading in this work, while the ozone concentration is similar to the previous studies.34,35 It seems that there is an effect of relative humidity on the H/C ratio for the ozonolysis of 10 ppb Δ3-carene. For the low RH experiments at 10 °C, the H/C ratio is between 1.55 and 1.6; for experiment 10D with high RH, the H/C ratio range between 1.6 and 1.7 over the duration of the experiment. An increasing H/C in ambient AMS measurements has previously been suggested to arise from hydrolysis.87 However, an increase in the H/C ratio for ozonolysis of 20 ppb Δ3-carene at 80% RH (10E) is not observed, which might suggest that the increase in H/C ratio for the ozonolysis of 10 ppb Δ3-carene does not arise from hydrolysis. The O/C ratio is either similar (10D) or lower (10E) at high relative humidity compared to dry conditions (10A-C). Surdu et al.49 observe decreasing O/C ratios with increasing RH. This decrease in O/C is suggested to arise from an increase in moderately oxygenated compounds (C10H16O2–5), while more oxygenated compounds (C10H16O6–8) are unaffected. No such effect is observed in offline chemical analysis (Fig. 4) nor in the online measurements from the FIGAERO (Fig. S.9†).
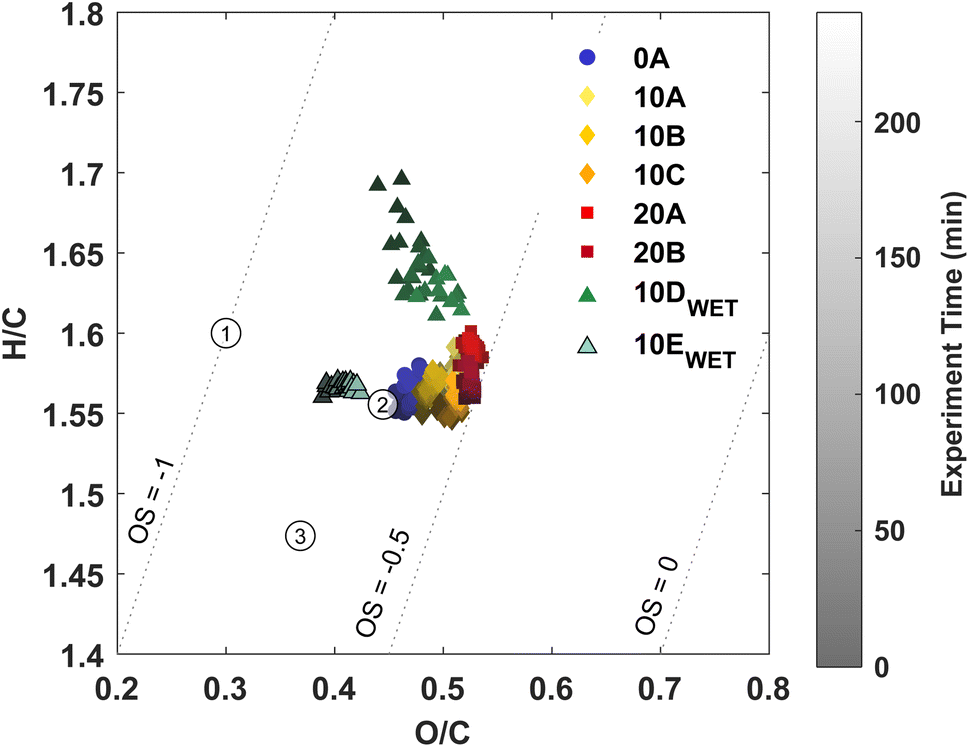 | ||
| Fig. 2 A van Krevelen plot with the general oxidation state of the particle phase. Three oxidation states of known compounds from ozonolysis of Δ3-carene are shown. (1) cis-3-Caric acid, (2) cis-3-caronic acid, and (3) dimer MW368. | ||
In the offline analysis, four carboxylic acids, cis-3-caric acid, cis-3-caronic acid, OH–3-caronic acid (10-hydroxy-3-caronic acid), and a carboxylic acid with molecular weight (MW) 170 g mol−1, are tentatively identified. MW170 is tentatively identified as 3-caralic acid or nor-3-caronic acid.34 In addition to the four carboxylic acids, one dimer with MW 368 g mol−1 was tentatively identified. The presence of these five ozonolysis products is in line with previous work,34 where Thomsen et al. identified ∼50% of the total particle mass and find that ∼2/3 of the identified particle mass arise from cis-3-caric acid in SOA formed from ozonolysis of 50 ppb Δ3-carene. Fig. S.10† shows the development of the estimated concentrations of ozonolysis products cis-3-caric acid and cis-3-caronic acid over time in experiments 0A, 10A, 10B, 10C, and 20A as mass fractions compared to the SMPS particle mass concentration without wall-loss correction. Mass fractions are presented in order to avoid overinterpretation of mass differences between experiments and to indirectly correct for wall loss. A substantial mass fraction of cis-3-caric acid (∼30%) is found in the particle phase. The condensation of cis-3-caric acid has previously been suggested to be more efficient than cis-pinic acid,34 which might explain this large fraction of cis-3-caric acid in the particle phase. Fig. S.12† shows the development in the mass fractions of OH–3-caronic acid and the estimated mass fraction of MW170. OH–3-caronic acid constitutes ∼10% of the particle mass. MW170 is only detected in experiments 10B and 10C after 90 minutes. A gradual build-up from 0.1% of the total particle mass to 1.4% of the total particle mass is observed.
Fig. 3 shows the mass fractions of the ozonolysis products cis-3-caric acid, cis-3-caronic acid, and OH–caronic acid for experiments 0A, 10A, 10B, 10C, and 20A. The correlation coefficients, R2, from linear least squares regression were between 0.68 and 0.92 at 0 and 20 °C. Overall, an increase with time in the mass fraction of cis-3-caric acid, cis-3-caronic acid, and OH–3-caronic acid is observed at 0 °C. In contrast, at 20 °C, a decrease of cis-3-caric acid, cis-3-caronic acid, and OH–3-caronic acid is observed. At 10 °C, no apparent trend is observed. This is consistent with R2 generally being lower for 10 °C experiments. It is interesting that the fractions of the three carboxylic acids increase with time at 0 °C, while the fractions decrease with time at 20 °C. This suggests that condensation of the three carboxylic acids is continuous at lower temperatures, while at higher temperatures, evaporation back into the gas phase or further reactions removing these species are favourable, and other reaction products not detected by the present analysis become more prominent in the particle phase.
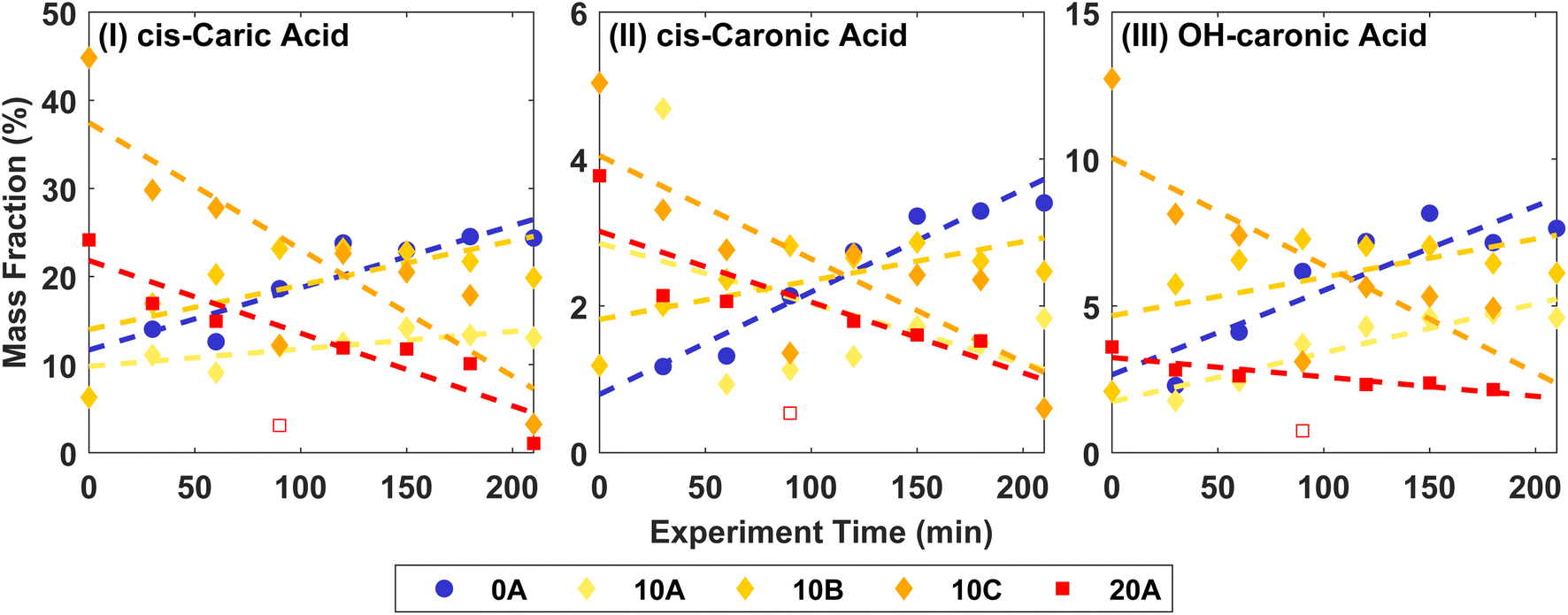 | ||
| Fig. 3 Mass fractions of (I) cis-3-caric acid, (II) cis-3-caronic acid, and (III) OH–3-caronic acid at 0, 10, and 20 °C. The dashed lines show linear least squares regression to the experimental data, where the open point for 20A is omitted in the fit as it is an outlier (Fisher's least significance difference method,68). | ||
Fig. 4 shows the development in the mass fractions of cis-3-caric acid and cis-3-caronic acid over time in experiments 10A–E to investigate the effect of RH. Ozonolysis of 10 ppb Δ3-carene at 0% RH results in a mass fraction of cis-3-caric acid between 10 and 35%, while ozonolysis of 10 ppb Δ3-carene at 80% RH results in a mass fraction of cis-3-caric acid between 10 and 70%. After 90 minutes, the mass fraction of cis-3-caric acid formed at 80% RH exceeds the mass fraction of cis-3-caric acid formed at 0% RH. The increase in cis-3-caric acid could potentially be linked to enhanced partitioning of cis-3-caric acid as the bulk diffusivity increases and the viscosity decreases with increasing RH.49
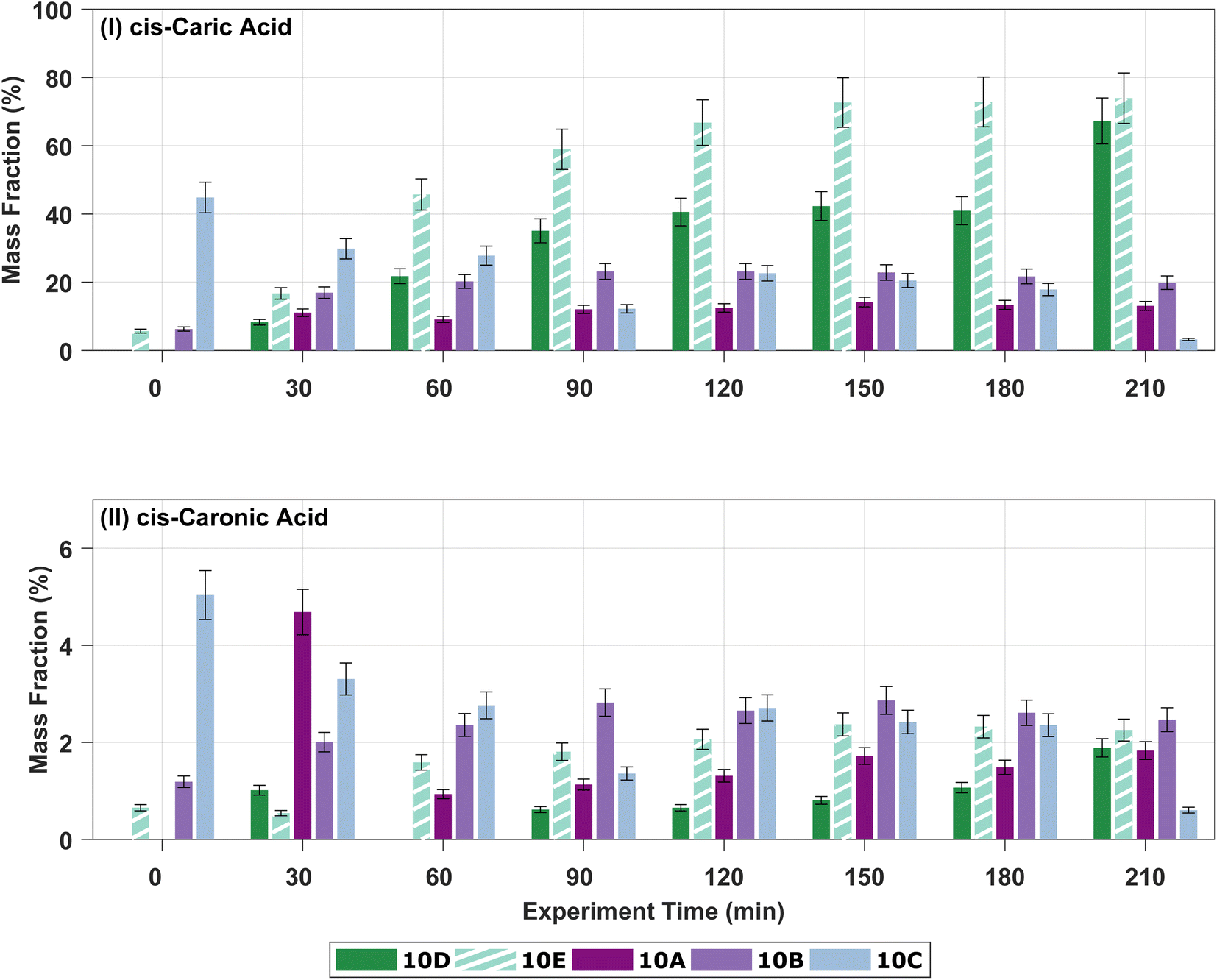 | ||
| Fig. 4 Mass fractions of ozonolysis products of Δ3-carene at 0% RH (10A–C) and 80% RH (10D–E). (I) The evolution over time of cis-3-caric acid with an estimated uncertainty of ±10%. (II) The evolution over time of cis-3-caronic acid with an estimated uncertainty of ±10%. | ||
Surprisingly, we observe that the mass fraction of cis-3-caronic acid found in the particle phase at 80% RH is lower than the mass fraction of cis-3-caronic acid at 0% RH. Surdu et al.49 discussed the effect of water on the gas-particle partitioning of semi-volatiles in detail. Water vapour tends to have little effect on the gas phase chemistry, but if water were to react with a Criegee intermediate a higher concentration of cis-3-caronic acid would be expected. They deem this a negligible contribution in good agreement with our observations of a lower mass fraction of cis-3-caronic acid with increasing RH. Ma et al.88 found that the formation of cis-pinonic acid from ozonolysis of α-pinene is indeed water dependent and Tillmann et al.54 stress that the effect of water on the oxidation products from α-pinene is not understood to fully describe the effects on the SOA formation and its dependency on water. Tillmann et al.54 also observe a humidity-dependent gain in SOA yields at −20 and −30 °C and explain the growth as arising from water-dependent chemistry in the formation of oxidation products and rule out physical water uptake. Fig. S.12† shows the development over time of OH–3-caronic acid for experiments 10A–E. No effect of humidity is observed for the presence of OH–3-caronic acid in the particle phase. Meanwhile, MW170 is only observed in the low RH experiments 10B and 10C and also in the high RH experiment with the higher VOC loading, 10E. These somewhat contradictory observations for four semi-volatile oxidation products stress that it is important to study the effect of humidity on oxidation pathways of BVOCs and the subsequent SOA formation and growth.
3.3 Modelling
According to the ADCHAM simulations, approximately 1/3 of all Δ3-carene was oxidised by OH and 2/3 by O3 in the dry experiments 20A–B, 10A–B and 0A. At the start of the experiments, the modelled maximum OH concentration was in the range (1.0–1.4) × 106 molecules cm−3. The highest OH concentrations were reached in the 20A–B experiments and the lowest in the 0A experiment. In all simulated experiments, more than 90% of all Δ3-carene was consumed within 3 hours.Fig. 5(I) and (II) show both the modelled and measured ozone concentrations for experiments 10A and 10B. From the measured ozone concentration, an initial Δ3-carene concentration is estimated as the ozone decay at the beginning of an experiment depends on the Δ3-carene concentration. The estimated initial Δ3-carene concentrations for experiments 10A and 10B are 6.5 and 14 ppb, respectively. The estimated absolute uncertainty of the Δ3-carene concentration of 5 ppb gives a range of Δ3-carene concentrations spanning from 5 to 15 ppb. For this range of initial Δ3-carene concentrations, the model predicts ozone concentrations as outlined by the shaded area in Fig. 5. The calculated initial Δ3-carene concentrations are both within the estimated uncertainty on the injected amount of Δ3-carene. By using the estimated initial Δ3-carene concentrations, the model is able to reproduce the measured ozone concentration apart from spikes and scattering of the data. In both experiments, the ozone concentration decreases faster after about two hours. This is expected to arise from leaks of surrounding air into the chamber and therefore included in the ADCHAM model as a dilution factor. Fig. 5(III) shows the modelled SOA mass concentration and the AMS mass concentration. The modelled results are based on the estimated initial Δ3-carene concentrations of 6.5 and 14 ppb for experiments 10A and 10B, respectively. The shaded area represents model predictions of the full range of possible initial Δ3-carene concentrations according to the absolute uncertainty related to the concentration. With the estimated Δ3-carene concentration, the ADCHAM model captures the evolution in the SOA mass concentration within the experimental uncertainties. The model results from experiments 0A, 20A and 20B are presented in Section 7 in the ESI.† At both 0 and 20 °C, both the ozone concentrations and the SOA mass concentrations are captured by the model in agreement with experiments 10A and 10B.
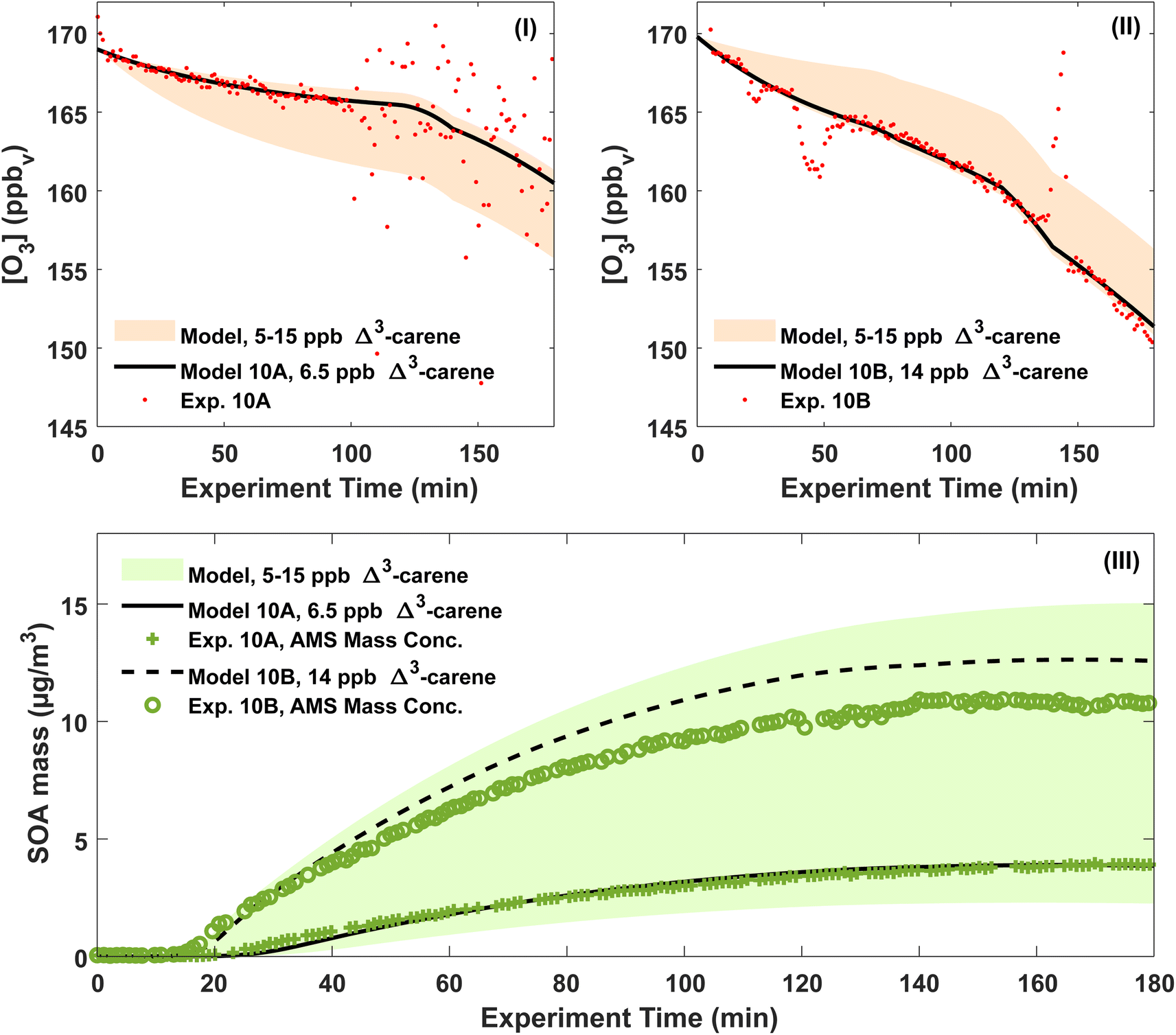 | ||
| Fig. 5 (I and II) Measured and modelled ozone concentrations in experiments 10A and 10B. The ozone concentrations were used to calculate initial Δ3-carene concentrations of 6.5 ppb and 14 ppb for experiments 10A and 10B respectively. (III) The measured and modelled SOA mass concentrations for experiments 10A and 10B. The model assumes initial concentrations as calculated from the ozone concentrations. | ||
The modelled SOA mass yields are generally higher than the experimentally derived values shown in Table S.1.† This is expected because the modelled SOA yields use the actual consumed amount of Δ3-carene estimated by the model, while the experimentally derived SOA yields assume that the consumed concentration of Δ3-carene is 10 ppb.
The modelled, particle wall-loss corrected SOA yields range from 11 (20A) to 20% (10B) and are influenced both by the temperature and the amount of reacted Δ3-carene. SOA yields generally decrease with increasing temperature and increase with the amount of reacted VOC [e.g. ref. 25, 26, 34, 43, 63 and 89]. According to the modelled ozone trends, it is likely that experiments 0A and 10A had lower initial Δ3-carene concentrations, ∼5 and ∼6.5 ppb, respectively, while experiments 10B and 20B had higher initial Δ3-carene concentrations, ∼14 ppb (Fig. 5, S.14 and S.15†). We also used the model to calculate SOA mass yields for an idealised atmospheric simulation chamber without dilution and wall losses of particles and gases. These modelled, atmospherically relevant SOA mass yields range between 16% at 20 °C and 32% at 0 °C for experiments where 10 ppb Δ3-carene is oxidised by ozone and OH without the presence of UV light.
Consistent with the observations, the modelled relative SOA mass fractions of cis-3-caric acid and OH–3-caronic acid increased with decreasing temperatures. The cis-3-caric acid mass fractions ranged between 17% and 26% and the OH–3-caronic acid mass fractions between 2% and 5% at the end of the experiments. Meanwhile, cis-3-caronic acid has a negligible contribution to the modelled SOA mass, except for experiment 0A where cis-3-caronic acid contributes to 2% of the SOA mass. The modelled SOA O/C ratios were 0.58–0.60 in experiments 20A–B, 0.52–0.55 in experiments 10A–B and 0.50 in experiment 0A. Thus, it is mainly in the 20A–B experiments that the model O/C ratios are substantially higher than the observed experimental values (Fig. 2).
3.4 Highly oxygenated organic molecules
HOMs are produced rapidly via autoxidation after injecting Δ3-carene into the AURA chamber. Most HOMs are low or extremely low-volatility organic compounds (ELVOCs), and they can both form new particles and condense onto chamber walls or particles contributing to further particle growth.10,12,90 Therefore, the observed HOM concentrations in the chamber are the results of the balance between formation and loss. A variety of products in the gas phase were detected by the NO3-CIMS, including both HOM monomers mostly with 8–10 carbon atoms and HOM dimers with 18–20 carbon atoms.Fig. 6 shows the time series of two selected HOM monomers C10H14O9 and C10H16O9 in experiments 20B, 10B, and 0A. At the beginning of each experiment, HOM are rapidly produced while the condensation sink from new particles is still low, thus we find C10H14O9 and C10H16O9 grow by more than an order of magnitude within 10 min. After around 20 min, C10H14O9 and C10H16O9 start to decrease because of the increasing condensation sink and loss rate of HOM.
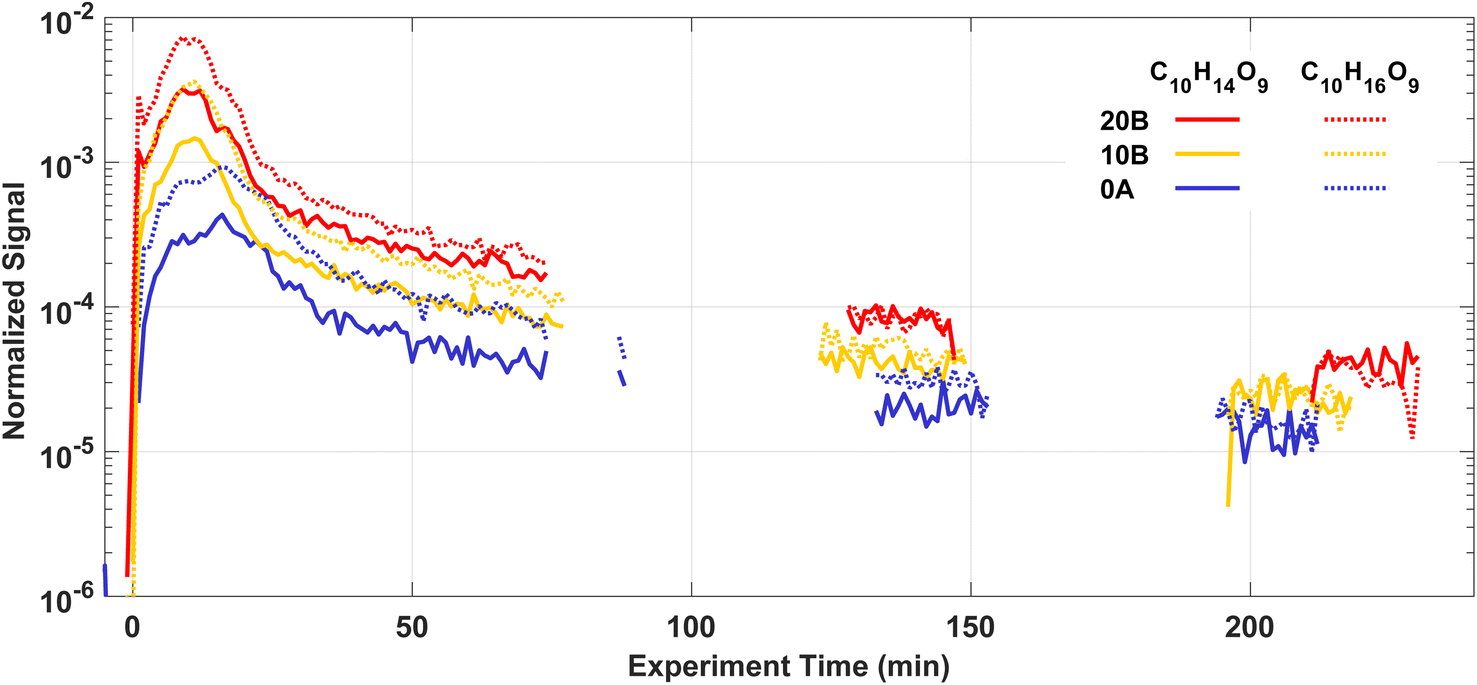 | ||
| Fig. 6 Temporal behaviours of two main HOM monomers: C10H14O9 and C10H16O9 from Δ3-carene ozonolysis at different temperatures. Signal intensities were normalised to the signal of reagent ions. Data were averaged over 1 min. Periods without signal correspond to the sampling cycle of the NO3-CIMS. | ||
The autoxidation of RO2 to form HOM is temperature dependent, where low temperatures decrease the autoxidation process, and hence are expected to result in less HOM formation.91,92 From Fig. 6, it is clear that the temperature at which the Δ3-carene ozonolysis took place had a considerable impact on the HOM concentrations. The signal intensities of C10H14O9 and C10H16O9 were up to one order of magnitude higher at 20 °C compared to 0 °C 10 min after the injection of Δ3-carene. A similar result with an even stronger enhancement of HOM formation at 20 °C was found for ozonolysis of α-pinene by Quéléver et al.45 The relative abundance of C10H14O9 and C10H16O9 signals increase slightly with decreasing temperatures indicating the potentially different distributions of HOM at different temperatures. More details of HOM formation and the influence of temperature and RH will be reported in a following paper.
3.5 Partitioning of 3-caronic acid
From the high-resolution peak fitting of the collected FIGAERO-CIMS dataset, one of the dominant detected compounds is C10H16O3. The time trends of C10H16O3 in the gas and particle phases of experiments 0A and 20B are shown in Fig. 7. The molecular formula (C10H16O3) may have different structures, e.g. isomers which FIGAERO-CIMS cannot distinguish, but this molecular formula corresponds to caronic acid, one of the dominant products from Δ3-carene ozonolysis as also identified in the offline analysis (Fig. 4 and S.10†). Thus, here C10H16O3 is discussed as caronic acid. The total signals of caronic acid increase with experiment time and peak at around 2 h and then decrease, primarily due to wall loss of both particles and gases in the chamber. Comparison of experiments 0A and 20B shows that an increase in temperature leads to an increase in the caronic acid formation between experiments at 0 and 20 °C.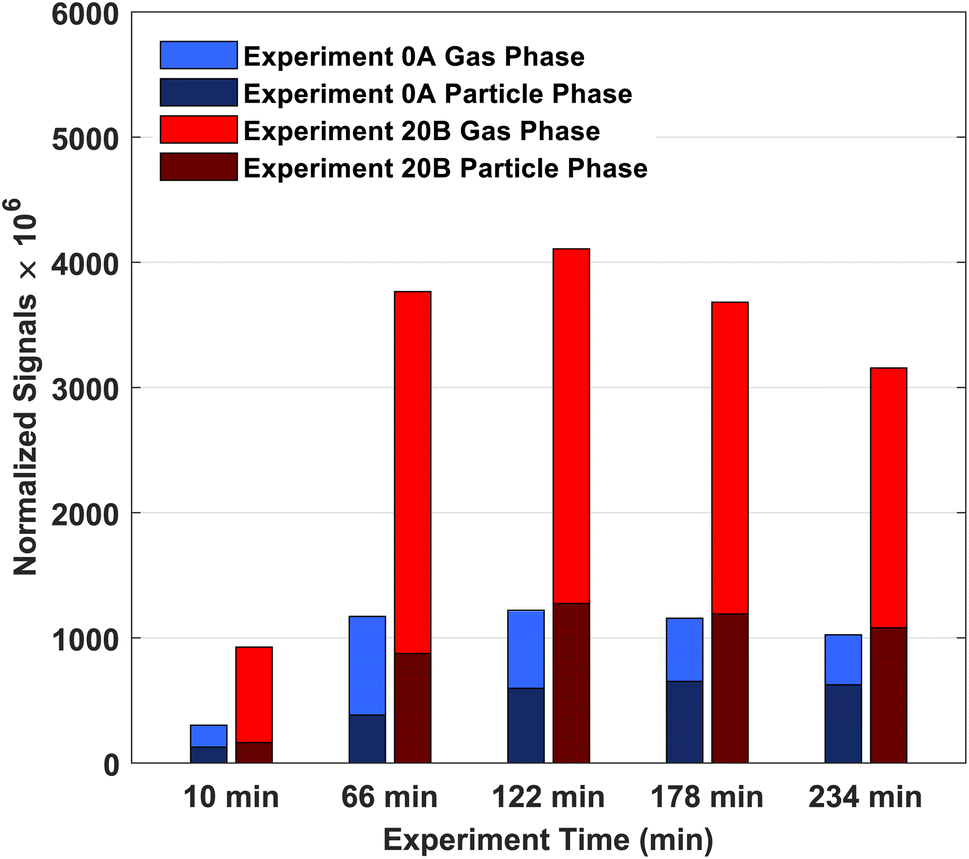 | ||
| Fig. 7 Normalised signals of ozonolysis product (C10H16O3) of Δ3-carene in experiments 0A and 20B at experiment times 0–10 min, 56–66 min, 112–122 min, 168–178 min, and 224–234 min. Signals are normalised by reagent I− signal and multiplied with 106. | ||
The particle fraction of each species can be expressed as particle-phase fraction (Fp), which is a proxy of volatility.93 The Fp of caronic acid is calculated from the normalised signals in the gas and the particle phases according to eqn (1) at each time point.
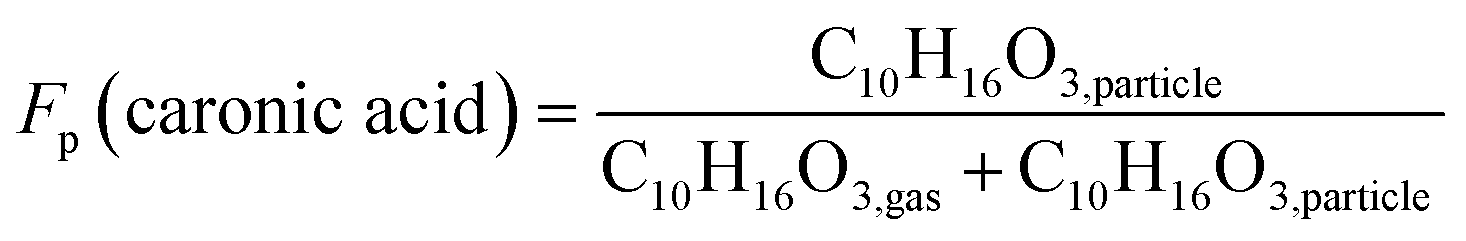 | (1) |
The average Fp values of caronic acid from experiments 0A and 20B are 0.48 and 0.26, respectively, showing a decrease as the temperature increase. The Fp values of caronic acid from the chamber study of Δ3-carene ozonolysis are close to other reports of observed Fp values of pinonic acid (0.1–0.3).94,95 The increase in Fp with decreasing temperature is expected as condensation of organics is enhanced with decreasing temperature.63,89,96,97 It is also in line with previous research for semi-volatile oxidation products from both ozonolysis of α-pinene25,26 and Δ3-carene.34 Fig. S.11† shows the concentration of cis-3-caronic acid quantified with the offline analysis of experiment 20A. According to the ADCHAM model experiment 20B was performed with an increased VOC loading (14 ppb, Table 1 and Section 3.3), which will increase the resulting fraction of products compared to experiment 20A (10 ppb, Table 1 and Section 3.3). In agreement with the FIGAERO data, the concentration of cis-3-caronic acid measured by offline analysis in the particle phase at 20 °C exceeds that of cis-3-caronic acid in the particle phase at 0 °C until ∼120 min. At 150 minutes, the concentration of cis-3-caronic acid is higher at 0 °C than at 20 °C. A more extensive investigation of the partitioning of caronic acid and other ozonolysis products produced in the two experiments will be discussed in a separate paper.
4 Conclusion
In the ACCEPTO campaign, multiple state-of-the-art analysis methods for both gas phase and particle phase species were applied to investigate the formation and composition of SOA from Δ3-carene.The SOA formation from ozonolysis of Δ3-carene does not show a strong effect of temperature in particle number concentration or SOA yields, which is in agreement with previous research on the SOA formation from Δ3-carene. This is a surprising finding as the formation of SOA from the structurally similar monoterpene α-pinene exhibits increasing particle number concentrations and particle mass concentrations with decreasing temperature.
A slight temperature dependence is, however, observed in the composition of the particle phase, where the mass fractions of the tentatively identified carboxylic acids cis-3-caric acid, cis-3-caronic acid, and OH–caronic acid show an increase over time at 0 °C and a decrease over time at 20 °C. In the gas phase, an increase in the signal intensity from caronic acid is observed with increasing temperature. Likewise, the signal intensity from caronic acid in the particle phase increases slightly with increasing temperature. The formation of the HOM monomers, C10H14O9 and C10H16O9, are enhanced with increasing temperature in agreement with previous observations for ozonolysis of α-pinene.
A very large increase in the particle number concentration and the nucleation rate is observed with increasing RH (80% vs. 0%), while the particle mass concentration is not affected suggesting that the nucleation is driven by inorganics in these experiments.
The modelled SOA mass concentrations are within the experimental uncertainties of all experiments and in general, agree well with the experimental observations.
In conclusion, the experimental results indicate that the SOA formation from Δ3-carene is not as dependent on temperature as the SOA formation from α-pinene, but that there is an effect of the presence of water vapour. However, the ADCHAM model simulations of idealised dry atmospheric simulation chamber conditions, i.e. without wall losses or chamber dilution effects, show a factor of 2 increase in the SOA mass yields when the temperature decreases from 20 to 0 °C. Thus, further studies are needed to explain the mechanisms of the temperature and water-dependent chemistry for Δ3-carene ozonolysis and to quantify the corresponding SOA mass yield dependencies.
Data availability
The data is available upon request. All data is available from the ATMO-ACCESS Database of Atmospheric Simulation Chamber Studies at the following DOIs: https://doi.org/10.25326/TBWC-R133, https://doi.org/10.25326/62Y1-3K47, https://doi.org/10.25326/DT8P-XF52, https://doi.org/10.25326/D1A9-G032, https://doi.org/10.25326/V4X9-EX29, https://doi.org/10.25326/6XQN-5852, https://doi.org/10.25326/MWVJ-TT47, and https://doi.org/10.25326/BF7F-F767.Author contributions
Conceptualisation: DT, EMI, HBP, PR, ME, MH, MG, and MB. Investigation: DT, EMI, JTS, YL, LL, and MP. Formal analysis: DT, EMI, JTS, YL, LL, and MP. ADCHAM model calculations: PR. Visualisation: DT, EMI, JTS, YL, LL, and PR. DT, EMI, JTS, YL, LL, MP, PR, HBP, MH, ME, MB, and MG took part in the scientific discussion. Writing – original draft: DT, EMI, JTS with large contributions from YL, LL, and PR. Writing – review & editing: all authors. Data curation: DT, EMI, JTS, MB, MG.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The ACCEPTO campaign is part of a project that is supported by the European Commission under the Horizon 2020 – Research and Innovation Framework Programme, through the ATMO-ACCESS Integrating Activity under grant agreement no. 101008004. We thank the Independent Research Fund Denmark (0136-00345B, 8021-00355B), the Carlsberg Foundation, Danish Agency for Higher Education and Science (ACTRIS-DK, 5072-00032B), Aarhus University, the Danish National Research Foundation through the Center of Excellence for Chemistry of Clouds (Grant Agreement No. DNRF172), the Research Council of Finland, the Swedish Research Council (Formas project no. 2018-01745-COBACCA), the Swedish Research Council VR (project no. 2019-05006) and the Crafoord Foundation (project no. 20210969) for their financial support. We thank laboratory manager Mads Mørk Jensen for his assistance in the laboratory. We thank Eva R. Kjærgaard and Þuríður Nótt Björgvinsdottír for their help during the campaign. We thank the workshops at the Department of Chemistry, Aarhus University for technical support.References
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll and et al. , Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. Connors, C. Péan and S. Berger, et al., in Climate Change 2021: the Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2021 Search PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys and et al. , The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi and et al. , Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini and et al. , Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel and et al. , A global model of natural volatile organic compound emissions, J. Geophys. Res.: Atmos., 1995, 100, 8873–8892 CrossRef CAS.
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J.-F. Müller, U. Kuhn, P. Stefani and W. Knorr, Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 2014, 14, 9317–9341 CrossRef.
- J. Bäck, J. Aalto, M. Henriksson, H. Hakola, Q. He and M. Boy, Chemodiversity of a Scots pine stand and implications for terpene air concentrations, Biogeosciences, 2012, 9, 689–702 CrossRef.
- J. L. Fry, D. C. Draper, K. J. Zarzana, P. Campuzano-Jost, D. A. Day, J. L. Jimenez and et al. , Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 2013, 13, 8585–8605 CrossRef.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen and et al. , A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin and et al. , Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- N. M. Donahue, J. H. Kroll, S. N. Pandis and A. L. Robinson, A two-dimensional volatility basis set – part 2: diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 2012, 12, 615–634 CrossRef CAS.
- F. Bianchi, J. Tröstl, H. Junninen, C. Frege, S. Henne, C. R. Hoyle and et al. , New particle formation in the free troposphere: a question of chemistry and timing, Science, 2016, 352, 1109–1112 CrossRef CAS PubMed.
- U. Molteni, M. Simon, M. Heinritzi, C. R. Hoyle, A.-K. Bernhammer, F. Bianchi and et al. , Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 2019, 3, 873–883 CrossRef CAS.
- O. Peräkylä, T. Berndt, L. Franzon, G. Hasan, M. Meder, R. R. Valiev and et al. , Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 2023, 145, 7780–7790 CrossRef PubMed , PMID: 36995167..
- T. Hoffmann, R. Bandur, U. Marggraf and M. Linscheid, Molecular composition of organic aerosols formed in the α-pinene/O3 reaction: implications for new particle formation processes, J. Geophys. Res.: Atmos., 1998, 103, 25569–25578 CrossRef CAS.
- M. P. Tolocka, M. Jang, J. M. Ginter, F. J. Cox, R. M. Kamens and M. V. Johnston, Formation of Oligomers in Secondary Organic Aerosol, Environ. Sci. Technol., 2004, 38, 1428–1434 CrossRef CAS PubMed.
- F. Yasmeen, R. Szmigielski, R. Vermeylen, Y. Gómez-González, J. D. Surratt, A. W. H. Chan, J. H. Seinfeld, W. Maenhaut and M. Claeys, Mass spectrometric characterization of isomeric terpenoic acids from the oxidation of α-pinene, β-pinene, d-limonene, and Δ3-carene in fine forest aerosol, J. Mass Spectrom., 2011, 46, 425–442 CrossRef CAS PubMed.
- K. Kristensen, K. L. Enggrob, S. M. King, D. R. Worton, S. M. Platt, R. Mortensen and et al. , Formation and occurrence of dimer esters of pinene oxidation products in atmospheric aerosols, Atmos. Chem. Phys., 2013, 13, 3763–3776 CrossRef.
- K. Kristensen, T. Cui, H. Zhang, A. Gold, M. Glasius and J. D. Surratt, Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity, Atmos. Chem. Phys., 2014, 14, 4201–4218 CrossRef.
- K. Kristensen, M. Bilde, P. P. Aalto, T. Petäjä and M. Glasius, Denuder/filter sampling of organic acids and organosulfates at urban and boreal forest sites: gas/particle distribution and possible sampling artifacts, Atmos. Environ., 2016, 130, 36–53 CrossRef CAS.
- K. Kristensen, Å. K. Watne, J. Hammes, A. Lutz, T. Petäjä, M. Hallquist, M. Bilde and M. Glasius, High-Molecular Weight Dimer Esters Are Major Products in Aerosols from α-Pinene Ozonolysis and the Boreal Forest, Environ. Sci. Technol. Lett., 2016, 3, 280–285 CrossRef CAS.
- C. Mohr, F. D. Lopez-Hilfiker, T. Yli-Juuti, A. Heitto, A. Lutz, M. Hallquist and et al. , Ambient observations of dimers from terpene oxidation in the gas phase: implications for new particle formation and growth, Geophys. Res. Lett., 2017, 44, 2958–2966 CrossRef CAS.
- K. Kristensen, L. N. Jensen, M. Glasius and M. Bilde, The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alpha-pinene, Environ. Sci.: Processes Impacts, 2017, 19, 1220–1234 RSC.
- K. Kristensen, L. N. Jensen, L. L. J. Quéléver, S. Christiansen, B. Rosati, J. Elm and et al. , The Aarhus Chamber Campaign on Highly Oxygenated Organic Molecules and Aerosols (ACCHA): Particle Formation, Organic Acids, and Dimer Esters from α-Pinene Ozonolysis at Different Temperatures, Atmos. Chem. Phys., 2020, 20, 12549–12567 CrossRef CAS.
- G. Hasan, V.-T. Salo, R. R. Valiev, J. Kubečka and T. Kurtén, Comparing Reaction Routes for 3(RO⋯OR’) Intermediates Formed in Peroxy Radical Self- and Cross-Reactions, J. Phys. Chem. A, 2020, 124, 8305–8320 CrossRef CAS PubMed.
- V.-T. Salo, R. Valiev, S. Lehtola and T. Kurtén, Gas-Phase Peroxyl Radical Recombination Reactions: A Computational Study of Formation and Decomposition of Tetroxides, J. Phys. Chem. A, 2022, 126, 4046–4056 CrossRef CAS PubMed.
- J. Hamilton, A. Lewis, J. Reynolds, L. Carpenter and A. Lubben, Investigating the composition of organic aerosol resulting from cyclohexene ozonolysis: low molecular weight and heterogeneous reaction products, Atmos. Chem. Phys., 2006, 6, 4973–4984 CrossRef CAS.
- L. Müller, M.-C. Reinnig, J. Warnke and T. Hoffmann, Unambiguous identification of esters as oligomers in secondary organic aerosol formed from cyclohexene and cyclohexene/α- pinene ozonolysis, Atmos. Chem. Phys., 2008, 8, 1423–1433 CrossRef.
- S. Gao, N. L. Ng, M. Keywood, V. Varutbangkul, R. Bahreini, A. Nenes and et al. , Particle Phase Acidity and Oligomer Formation in Secondary Organic Aerosol, Environ. Sci. Technol., 2004, 38, 6582–6589 CrossRef CAS PubMed.
- Y. Zhao, M. Yao, Y. Wang, Z. Li, S. Wang, C. Li and H. Xiao, Acylperoxy Radicals as Key Intermediates in the Formation of Dimeric Compounds in α-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 2022, 56, 14249–14261 CrossRef CAS PubMed , PMID: 36178682..
- Y. Zhang, Y. Chen, Z. Lei, N. E. Olson, M. Riva, A. R. Koss and et al. , Joint Impacts of Acidity and Viscosity on the Formation of Secondary Organic Aerosol from Isoprene Epoxydiols (IEPOX) in Phase Separated Particles, ACS Earth Space Chem., 2019, 3, 2646–2658 CrossRef CAS.
- D. Thomsen, J. Elm, B. Rosati, J. T. Skønager, M. Bilde and M. Glasius, Large Discrepancy in the Formation of Secondary Organic Aerosols from Structurally Similar Monoterpenes, ACS Earth Space Chem., 2021, 5, 632–644 CrossRef CAS.
- D. Thomsen, L. D. Thomsen, E. M. Iversen, T. N. Björgvinsdóttir, S. F. Vinther, J. T. Skønager, T. Hoffmann, J. Elm, M. Bilde and M. Glasius, Ozonolysis of α-Pinene and Δ3-Carene Mixtures: Formation of Dimers with Two Precursors, Environ. Sci. Technol., 2022, 56, 16643–16651 CrossRef CAS PubMed.
- M. Glasius, M. Lahaniati, A. Calogirou, D. Di Bella, N. R. Jensen, J. Hjorth, D. Kotzias and B. R. Larsen, Carboxylic Acids in Secondary Aerosols from Oxidation of Cyclic Monoterpenes by Ozone, Environ. Sci. Technol., 2000, 34, 1001–1010 CrossRef CAS.
- Y. Y. Zhang, L. Müller, R. Winterhalter, G. K. Moortgat, T. Hoffmann and U. Pöschl, Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter, Atmos. Chem. Phys., 2010, 10, 7859–7873 CrossRef CAS.
- M. Glasius and A. H. Goldstein, Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry, Environ. Sci. Technol., 2016, 50, 2754–2764 CrossRef CAS PubMed.
- J. Yu, D. R. Cocker III, R. J. Griffin, R. C. Flagan and J. H. Seinfeld, J. Atmos. Chem., 1999, 34, 207–258 CrossRef CAS.
- F. D. Lopez-Hilfiker, C. Mohr, M. Ehn, F. Rubach, E. Kleist, J. Wildt and et al. , A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 2014, 7, 983–1001 CrossRef.
- T. Hoffmann, J. R. Odum, F. Bowman, D. Collins, D. Klockow, R. C. Flagan and J. H. Seinfeld, Formation of Organic Aerosols from the Oxidation of Biogenic Hydrocarbons, J. Atmos. Chem., 1997, 26, 189–222 CrossRef CAS.
- Å. M. Jonsson, M. Hallquist and E. Ljungström, Impact of Humidity on the Ozone Initiated Oxidation of Limonene, Δ3-Carene, and α-Pinene, Environ. Sci. Technol., 2006, 40, 188–194 CrossRef PubMed , PMID: 16433350..
- Å. M. Jonsson, M. Hallquist and E. Ljungström, The effect of temperature and water on secondary organic aerosol formation from ozonolysis of limonene, and Δ3-carene and α- pinene, Atmos. Chem. Phys., 2008, 8, 6541–6549 CrossRef.
- B. Witkowski and T. Gierczak, Early stage composition of SOA produced by α-pinene/ozone reaction: α-acyloxyhydroperoxy aldehydes and acidic dimers, Atmos. Environ., 2014, 95, 59–70 CrossRef CAS.
- L. L. J. Quéléver, K. Kristensen, L. Normann Jensen, B. Rosati, R. Teiwes, K. R. Daellenbach and et al. , Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis, Atmos. Chem. Phys., 2019, 19, 7609–7625 CrossRef.
- M. Simon, L. Dada, M. Heinritzi, W. Scholz, D. Stolzenburg, L. Fischer and et al. , Molecular understanding of new-particle formation from α-pinene between −50 and +25 °C, Atmos. Chem. Phys., 2020, 20, 9183–9207 CrossRef CAS.
- Q. Ye, M. Wang, V. Hofbauer, D. Stolzenburg, D. Chen, M. Schervish, A. Vogel, R. L. Mauldin, R. Baalbaki, S. Brilke and et al. , Molecular composition and volatility of nucleated particles from α-pinene oxidation between −50 °C and +25 °C, Environ. Sci. Technol., 2019, 53, 12357–12365 CrossRef CAS PubMed.
- L. Caudillo, B. Rörup, M. Heinritzi, G. Marie, M. Simon, A. C. Wagner and et al. , Chemical composition of nanoparticles from α-pinene nucleation and the influence of isoprene and relative humidity at low temperature, Atmos. Chem. Phys., 2021, 21, 17099–17114 CrossRef CAS.
- M. Surdu, H. Lamkaddam, D. S. Wang, D. M. Bell, M. Xiao, C. P. Lee and et al. , Molecular Understanding of the Enhancement in Organic Aerosol Mass at High Relative Humidity, Environ. Sci. Technol., 2023, 57, 2297–2309 CrossRef CAS PubMed , PMID: 36716278..
- E. L. D'Ambro, S. Schobesberger, C. J. Gaston, F. D. Lopez-Hilfiker, B. H. Lee, J. Liu and et al. , Chamber-based insights into the factors controlling epoxydiol (IEPOX) secondary organic aerosol (SOA) yield, composition, and volatility, Atmos. Chem. Phys., 2019, 19, 11253–11265 CrossRef.
- D. A. Day, J. L. Fry, H. G. Kang, J. E. Krechmer, B. R. Ayres, N. I. Keehan and et al. , Secondary Organic Aerosol Mass Yields from NO3 Oxidation of α-Pinene and Δ-Carene: Effect of RO2 Radical Fate, J. Phys. Chem. A, 2022, 126, 7309–7330 CrossRef CAS PubMed.
- M. Boy, D. Mogensen, S. Smolander, L. Zhou, T. Nieminen, P. Paasonen and et al. , Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 2013, 13, 3865–3879 CrossRef.
- B. Bonn and G. K. Moorgat, New particle formation during a- and b-pinene oxidation by O3, OH and NO3, and the influence of water vapour: particle size distribution studies, Atmos. Chem. Phys., 2002, 2, 183–196 CrossRef CAS.
- R. Tillmann, M. Hallquist, Å. M. Jonsson, A. Kiendler-Scharr, H. Saathoff, Y. Iinuma and T. F. Mentel, Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of α-pinene, Atmos. Chem. Phys., 2010, 10, 7057–7072 CrossRef CAS.
- X. Li, S. Chee, J. Hao, J. P. D. Abbatt, J. Jiang and J. N. Smith, Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 2019, 19, 1555–1570 CrossRef CAS.
- M. Sipilä, T. Berndt, T. Petäjä, D. Brus, J. Vanhanen, F. Stratmann and et al. , The Role of Sulfuric Acid in Atmospheric Nucleation, Science, 2010, 327, 1243–1246 CrossRef PubMed.
- J. Vanhanen, J. Mikkilä, K. Lehtipalo, M. Sipilä, H. E. Manninen, E. Siivola, T. Petäjä and M. Kulmala, Particle Size Magnifier for Nano-CN Detection, Aerosol Sci. Technol., 2011, 45, 533–542 CrossRef CAS.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken and et al. , Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 2006, 78, 8281–8289 CrossRef CAS PubMed.
- A. E. Fernandez, G. S. Lewis and S. V. Hering, Design and Laboratory Evaluation of a Sequential Spot Sampler for Time-Resolved Measurement of Airborne Particle Composition, Aerosol Sci. Technol., 2014, 48, 655–663 CrossRef PubMed.
- A. Eiguren Fernandez, G. S. Lewis, S. R. Spielman and S. V. Hering, Time-resolved Characterization of Particle Associated Polycyclic Aromatic Hydrocarbons using a Newly-developed Sequential Spot Sampler with Automated Extraction and Analysis, Atmos. Environ., 2014, 96, 125–134 CrossRef CAS PubMed.
- H. Junninen, M. Ehn, T. Petäjä, L. Luosujärvi, T. Kotiaho, R. Kostiainen and et al. , A highresolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 2010, 3, 1039–1053 CrossRef CAS.
- T. Jokinen, M. Sipilä, H. Junninen, M. Ehn, G. Lönn, J. Hakala, T. Petäjä, R. L. Mauldin III, M. Kulmala and D. R. Worsnop, Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 2012, 12, 4117–4125 CrossRef CAS.
- R. K. Pathak, C. O. Stanier, N. M. Donahue and S. N. Pandis, Ozonolysis of α-pinene at atmospherically relevant concentrations: temperature dependence of aerosol mass fractions (yields), J. Geophys. Res.: Atmos., 2007, 112, D03201 CrossRef.
- J. R. Odum, T. Hoffmann, F. Bowman, D. Collins, R. C. Flagan and J. H. Seinfeld, Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 1996, 30, 2580–2585 CrossRef CAS.
- M. Tomicic, M. Bødker Enghoff and H. Svensmark, Experimental study of H2SO4 aerosol nucleation at high ionization levels, Atmos. Chem. Phys., 2018, 18, 5921–5930 CrossRef CAS.
- M. Kulmala, T. Petäjä, T. Nieminen, M. Sipilä, H. E. Manninen, K. Lehtipalo and et al. , Measurement of the nucleation of atmospheric aerosol particles, Nat. Protoc., 2012, 7, 1651–1667 CrossRef CAS PubMed.
- K. E. J. Lehtinen and M. Kulmala, A model for particle formation and growth in the atmosphere with molecular resolution in size, Atmos. Chem. Phys., 2003, 3, 251–257 CrossRef CAS.
- X. Su, X. Yan and C.-L. Tsai, Linear regression, Wiley Interdiscip. Rev. Comput. Stat., 2012, 4, 275–294 CrossRef.
- M. Claeys, Y. Iinuma, R. Szmigielski, J. D. Surratt, F. Blockhuys, C. Van Alsenoy and et al. , Terpenylic Acid and Related Compounds from the Oxidation of α-Pinene: Implications for New Particle Formation and Growth above Forests, Environ. Sci. Technol., 2009, 43, 6976–6982 CrossRef CAS PubMed.
- A. Kahnt, R. Vermeylen, Y. Iinuma, M. Safi Shalamzari, W. Maenhaut and M. Claeys, Highmolecular-weight esters in α-pinene ozonolysis secondary organic aerosol: structural characterization and mechanistic proposal for their formation from highly oxygenated molecules, Atmos. Chem. Phys., 2018, 18, 8453–8467 CrossRef CAS.
- H. Junninen, PhD thesis, Faculty of Science, University of Helsinki, 2014.
- P. Roldin, A. C. Eriksson, E. Z. Nordin, E. Hermansson, D. Mogensen, A. Rusanen and et al. , Modelling non-equilibrium secondary organic aerosol formation and evaporation with the aerosol dynamics, gas- and particle-phase chemistry kinetic multilayer model ADCHAM, Atmos. Chem. Phys., 2014, 14, 7953–7993 CrossRef.
- S. M. Saunders, M. E. Jenkin, R. Derwent and M. Pilling, Protocol for the development of the master chemical mechanism, MCM v3 (part A): tropospheric degradation of nonaromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- M. Jenkin, J. Young and A. Rickard, The MCM v3. 3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- P. Roldin, M. Ehn, T. Kurtén, T. Olenius, M. P. Rissanen, N. Sarnela and et al. , The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 2019, 10, 1–15 CrossRef PubMed.
- W. Nie, C. Yan, L. Yang, P. Roldin, Y. Liu and A. L. Vogel, et al., NO at low concentration can enhance the formation of highly oxygenated biogenic molecules in the atmosphere, Nat. Commun., 2023, 14, 3347 CrossRef CAS PubMed.
- L. Hantschke, A. Novelli, B. Bohn, C. Cho, D. Reimer, F. Rohrer, R. Tillmann, M. Glowania, A. Hofzumahaus, A. Kiendler-Scharr and et al. , Atmospheric photooxidation and ozonolysis of Δ 3-carene and 3-caronaldehyde: rate constants and product yields, Atmos. Chem. Phys., 2021, 21, 12665–12685 CrossRef CAS.
- T. J. Dillon, K. Dulitz, C. Groß and J. N. Crowley, Temperature-dependent rate coefficients for the reactions of the hydroxyl radical with the atmospheric biogenics isoprene, alphapinene and delta-3-carene, Atmos. Chem. Phys., 2017, 17, 15137–15150 CrossRef CAS.
- L. Wang, Y. Liu and L. Wang, Ozonolysis of 3-carene in the atmosphere. Formation mechanism of hydroxyl radical and secondary ozonides, Phys. Chem. Chem. Phys., 2019, 21, 8081–8091 RSC.
- J. Kirkby, J. Duplissy, K. Sengupta, C. Frege, H. Gordon, C. Williamson and et al. , Ion-induced nucleation of pure biogenic particles, Nature, 2016, 533, 521–526 CrossRef CAS PubMed.
- E. M. Dunne, H. Gordon, A. Kürten, J. Almeida, J. Duplissy, C. Williamson and et al. , Global atmospheric particle formation from CERN CLOUD measurements, Science, 2016, 354, 1119–1124 CrossRef CAS PubMed.
- J. Almeida, S. Schobesberger, A. Kürten, I. K. Ortega, O. Kupiainen-Määttä, A. P. Praplan and et al. , Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 2013, 502, 359–363 CrossRef CAS PubMed.
- H. Henschel, J. C. A. Navarro, T. Yli-Juuti, O. Kupiainen-Määttä, T. Olenius, I. K. Ortega, S. L. Clegg, T. Kurtén, I. Riipinen and H. Vehkamäki, Hydration of Atmospherically Relevant Molecular Clusters: Computational Chemistry and Classical Thermodynamics, J. Phys. Chem. A, 2014, 118, 2599–2611 CrossRef CAS PubMed , PMID: 24678924..
- F. R. Rasmussen, J. Kubečka, V. Besel, H. Vehkamäki, K. V. Mikkelsen, M. Bilde and J. Elm, Hydration of Atmospheric Molecular Clusters III: Procedure for Efficient Free Energy Surface Exploration of Large Hydrated Clusters, J. Phys. Chem. A, 2020, 124, 5253–5261 CrossRef CAS PubMed , PMID: 32463668..
- O. Horie, P. Neeb and G. K. Moortgat, Ozonolysis of trans- and cis-2-butenes in low parts-per-million concentration ranges, Int. J. Chem. Kinet., 1994, 26, 1075–1094 CrossRef CAS.
- O. Horie, P. Neeb, S. Limbach and G. K. Moortgat, Formation of formic acid and organic peroxides in the ozonolysis of ethene with added water vapour, Geophys. Res. Lett., 1994, 21, 1523–1526 CrossRef CAS.
- C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. DeCarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer and P. Artaxo, A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 2010, 37, L08803 Search PubMed.
- Y. Ma, T. R. Willcox, A. T. Russell and G. Marston, Pinic and pinonic acid formation in the reaction of ozone with α-pinene, Chem. Commun., 2007, 1328–1330 RSC.
- H. Saathoff, K.-H. Naumann, O. Möhler, Å. M. Jonsson, M. Hallquist, A. Kiendler-Scharr, T. F. Mentel, R. Tillmann and U. Schurath, Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 2009, 9, 1551–1577 CrossRef CAS.
- T. Jokinen, T. Berndt, R. Makkonen, V.-M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop and et al. , Production of extremely low volatile organic compounds from biogenic emissions: measured yields and atmospheric implications, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS PubMed.
- M. Ehn, T. Berndt, J. Wildt and T. Mentel, Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 2017, 49, 821–831 CrossRef CAS.
- C. Frege, I. K. Ortega, M. P. Rissanen, A. P. Praplan, G. Steiner, M. Heinritzi, L. Ahonen, A. Amorim, A.-K. Bernhammer, F. Bianchi and et al. , Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 2018, 18, 65–79 CrossRef CAS.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 2006, 40, 2635–2643 CrossRef CAS PubMed , PMID: 16683603..
- A. Lutz, C. Mohr, M. Le Breton, F. D. Lopez-Hilfiker, M. Priestley, J. A. Thornton and M. Hallquist, Gas to particle partitioning of organic acids in the boreal atmosphere, ACS Earth Space Chem., 2019, 3, 1279–1287 CrossRef CAS.
- R. Yatavelli, H. Stark, S. Thompson, J. Kimmel, M. Cubison, D. Day, P. Campuzano-Jost, B. Palm, A. Hodzic, J. Thornton and et al. , Semicontinuous measurements of gas–particle partitioning of organic acids in a ponderosa pine forest using a MOVI-HRToF-CIMS, Atmos. Chem. Phys., 2014, 14, 1527–1546 CrossRef.
- T. M. Svendby, M. Lazaridis and K. Tørseth, Temperature dependent secondary organic aerosol formation from terpenes and aromatics, J. Atmos. Chem., 2008, 59, 25–46 CrossRef CAS.
- B. Warren, R. L. Austin and D. R. Cocker, Temperature dependence of secondary organic aerosol, Atmos. Environ., 2009, 43, 3548–3555 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Placement and sampling cycles of instruments, wall-loss correction, experimental details, background concentration of sulfuric acid, evolution in moderately oxygenated species, details of the offline analysis, time-evolution plots of ozonolysis products (PDF). See DOI: https://doi.org/10.1039/d3ea00128h |
| ‡ These authors contributed to the manuscript equally. |
| This journal is © The Royal Society of Chemistry 2024 |