
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simply accessible organometallic system to gauge electronic properties of N-heterocyclic carbenes†
Francis
Bru
 a,
Rex S. C.
Charman
b,
Laurens
Bourda
a,
Kristof
Van Hecke
a,
Laurence
Grimaud
c,
David J.
Liptrot
b and
Catherine S. J.
Cazin
*a
a,
Rex S. C.
Charman
b,
Laurens
Bourda
a,
Kristof
Van Hecke
a,
Laurence
Grimaud
c,
David J.
Liptrot
b and
Catherine S. J.
Cazin
*a
aDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281, 9000 Ghent, Belgium. E-mail: Catherine.Cazin@ugent.be
bDepartment of Chemistry, Faculty of Science, University of Bath, Claverton Down, Bath BA2 7AY, UK
cLaboratoire des Biomolécules, LBM, Département de Chimie, Ecole Normale Supérieure, PSL University, Sorbonne Université, CNRS, 75005 Paris, France
First published on 18th September 2024
Abstract
The intricate σ and π-bonding of N-heterocyclic carbenes (NHCs) to metals and the need to quantify their electronic properties to rationalize reactivity of complexes have resulted in the creation of numerous methodologies to understand the NHC–metal interaction which are, as we now show, flawed. Our search for a unified, easily accessible system to gauge these fundamental properties has resulted in the discovery of two systems that highlight the flaws present in existing systems and provide a more accurate measure of the NHC ligand electronic properties.
Introduction
N-heterocyclic carbenes (NHCs) have been a most prevalent ligand class in organometallic chemistry over the last 25 years due to their tuneable steric and electronic properties.1 To understand the inner workings of transition metal (TM) catalysis and to subsequently explain and predict the reactivity of TM complexes, the quantification of ligand stereo-electronic properties is key. This has been attempted over the years through the development of several models designed to assist in delineating the fine balance of such steric and electronic contributions to the nature of the bonding involved between a NHC and a metal (Fig. 1). Quantifying ligand electronic parameters in TM complexes has been mainly approached through the use of the Tolman Electronic Parameter (TEP) where the donating capability of a given ligand L is measured using the A1 infrared frequency of the CO-bond in the corresponding [Ni(CO)3(L)] complex.2 This strategy is simple as the synthesis of the Ni-complexes is straightforward, the use of an IR-spectrometer is commonplace, and the wide applicability of this method to a large number of NHC and phosphorus ligands has been demonstrated.2,3 Using this experimental technique, the carbonyl stretching frequencies of a vast number of the most commonly used NHC ligands have been measured.3,4 However, this method also possesses disadvantages such as the high toxicity of the Ni-precursor, the dominance of the π-interaction in this model whereas the σ-donation plays the largest role in NHC donor properties, and the relatively small numerical range where the IR-data lie.3 Additionally, the TEP model has difficulties with the bulkiest NHCs, which can distort the geometry of the otherwise tetrahedral complexes or even lead to the elimination of an additional CO ligand leading to [Ni(L)(CO)2] complexes.4,5 | ||
| Fig. 1 Approaches to determine NHC electronic properties. | ||
To circumvent the use of the toxic [Ni(CO)4], alternative carbonyl bearing systems have been designed. [IrCl(CO)2(NHC)] complexes,6 and their Rh congeners,7,8 have shown good correlation with data from the nickel system, thus allowing extraction of TEP data from these alternative systems. However, this approach is still plagued by the limited range of the IR-measurements and, in some cases by decomposition through the dissociation of one of the carbonyl ligands.9,10 Furthermore, sterically demanding ligands have been reported to induce modulations in the CO stretching frequency through intramolecular interactions11 and the generation of a charged field across the CO ligand, which generates an “internal Stark effect”.12 This effect can cause incorrect ligand electronic parameter interpretations.
Other methods have been developed that avoid the use of IR-spectroscopy for the determination of the NHC electronic properties. Computational methods have successfully predicted TEP values in the nickel-carbonyl and rhodium-carbonyl systems.13–16 However, these calculations require a careful selection of the method and need a significant amount of computing power.17,18 These approaches rely on basic architectures that may contain additional steric interactions distorting the true nature of the M–NHC bonding. An additional approach has made use of the pKa values of NHC-salts, and these can be linked to the electronic properties of the ligand.19–22 These methods, however, do not involve the complex bonding picture created when a metal is involved. Additional methods have made use of NMR spectroscopy such as the chemical shift in the 13C NMR spectrum of the carbene carbon of the iPr-bimy (iPr-bimy = N,N′-diisopropylbenzimidazolin-2-ylidene) ligand in trans-[PdBr2(iPr-bimy)(L)] complexes.23,24 This method requires the synthesis of specific Pd complexes through a multistep sequence, a major drawback. Other NMR-based strategies have used the 31P NMR resonance of carbene-phosphinidene adducts,25 the 77Se NMR of carbene-selenium adducts,26 and most recently the use of 1H NMR data of azolium salts.27,28 Lastly, a method showing promise in determining electronic properties is one that uses electrochemistry. The Lever electronic parameter (LEP)29 is an electrochemical parameter (EL) measuring the ability of a ligand to stabilize certain metal oxidation states. This parameter can be correlated to ligand electronic properties, with smaller EL values being a result of more donating ligands.30 Data on the electrochemical evaluation of ligand donor properties have shown that this approach is often superior in terms of accuracy and precision compared to the carbonyl-based systems. However, only complexes that show a (quasi-)reversible redox couple in the available electrochemical window of the solvent can be used,29,31 which makes the classical Ni, Rh, and Ir-carbonyl systems unsuitable as these exhibit irreversible redox events. Additionally, redox-active ligands or substituents are considered non-innocent and cannot be evaluated using this electrochemical approach.32 Albrecht and co-workers have proposed Fe(II) piano-stool complexes, that also bear a CO ligand, that could be used to correlate electrochemical and vibrational data of a very limited number of monodentate and specially designed bidentate NHCs.33 Plenio and co-workers have reported two studies in which the [MCl(NHC)(cod)] (M = Rh or Ir; cod = cyclooctadiene) precursors of the [MCl(CO)2(NHC)] complexes were used as electrochemical probes, wherein they show that the redox behaviour of the precursors is closely related to the vibrational data of their carbonyl counterparts and that both Ir and Rh systems show a linear correlation in terms of redox behavior.7,34 Interestingly, the trends observed contradict the electronic parameter trend observed in the Ni system. Also noteworthy is that this correlation makes use of two systems, the cod and carbonyl systems. The dataset for these Ir and Rh systems has been expanded but, to our knowledge, no study has gathered data on the more classical NHCs10,35 and no study has attempted to explain why one or the other of the metals would best be suited for pure ligand electronic parameter determination.
Results and discussion
There exists, at present, no single system where at least two distinct spectroscopic handles can be used to gauge the properties of a broad range of ligands. The existing systems suffer from (1) toxicity (e.g. Ni(CO)4), (2) a complex synthetic sequence and most importantly, (3) the existence of additional steric repulsive interactions that lead to distortion in complex geometries that subsequently can lead to erroneous interpretation of pure electronic properties. To circumvent these issues, we selected an initial system that would offer a dual probe for ligand electronic properties. The approach uses [Rh(NHC)(acac)(CO)] complexes containing a carbonyl ligand for infrared measurements which also possess a reversible electrochemical redox event (Fig. 2). | ||
| Fig. 2 Cyclic voltammogram of [Rh(IPr)(acac)(CO)]. Conditions: 25 °C, 2 mM of analyte, 0.1 M NBu4PF6 in CH2Cl2, Pt work and auxiliary electrode, scan rate 100 mV s−1. Referenced to the decamethylferrocene couple determined at 0.15 V vs. Ag/Ag+ under the same conditions. | ||
The presence of these two measurables within the same system allows the verification of the existence of relationships between the two observables. The rhodium complexes are easily synthesized using a previously reported procedure.36 Using literature infrared data for the three carbonyl systems (i.e. [Ni(NHC)(CO)3], [IrCl(CO)2(NHC)] and [RhCl(CO)2(NHC)], Table 1) permits assessment of the usefulness of the new electrochemical probe. The selection of the [Rh(acac)(NHC)(CO)] system appears judicious, as even the use of very large NHCs results in isolable complexes and the redox potentials of these metal complexes prove reversible. As an example, the cyclic voltammogram of [Rh(IPr)(acac)(CO)] is presented in Fig. S1.† Voltammograms of other complexes in this system have similar appearances. The voltammogram of [Rh(IPr)(acac)(CO)] exhibits a single redox event, which is attributed to the quasi-reversible RhI/RhII couple. The quasi-reversibility of this redox event is confirmed by performing the CV at varying scan rates, observing a fixed E1/2 but a small shift of the peak potentials (see ESI, Fig. S3†).37 The peak-to-peak separation is quite large, ranging from 84 mV to 194 mV. This is in contrast with to the results for the [Rh(NHC)(cod)Cl] complexes that range from 74 to 92 mV.7 The presence of these quasi-reversible redox events permits the determination of E1/2 values which in turn are linked to the electron donating properties of the NHC ligands.
| L= | [NiL(CO)3] | [IrL(CO)2Cl] | [RhL(CO)2Cl] |
|---|---|---|---|
| IPr | 2051.54 | 2023.938 | 2037.548 |
| SIPr | 2052.24 | 2024.938 | |
| IMes | 2050.74 | 2023.138 | 2037.649 |
| SIMes | 2051.54 | 2024.638 | 2040.57 |
| IPrCl | 2055.139 | 2028.338 | |
| IPrOMe | 2049.940 | 2023.440 | |
| IPent | 2049.341 | ||
| IHept | 2048.642 | ||
| 6Mes | 202950 | ||
| 7Mes | 202850 | ||
| 6Dipp | 2018.246 | ||
| ItBu | 2022.338 | 203651 | |
| IAd | 2021.638 | ||
| aNHC | 2009.947 | ||
| CAACCy | 204643 | 2035.552 | |
| IPr* | 2052.744 | 2025.642 | |
| IPr*OMe | 2051.145 |
As the [Rh(NHC)(acac)(CO)] complexes exhibit a quasi-reversible redox event, comparing two very different probes, based on different handles of the complex, for electronic ligand property determination from the same complex is now possible. These data are presented in Table 2. To extend our study, five new compounds in this family have been synthesized and characterized, to test our hypothesis with bulkier NHCs. Comparison of the carbonyl-stretching frequency and the E1/2 value of the RhI/II-redox couple shows a strikingly linear correlation (Fig. 3), with the abnormal (aNHC) cousin as the sole outlier. However, when comparing this system with the TEP data gathered on the Ir-dicarbonyl system, a clear lack of correlation exists for the abnormal NHC (aNHC) and the ring expanded NHC (re-NHC) 6Dipp (see ESI, Fig. S5†).
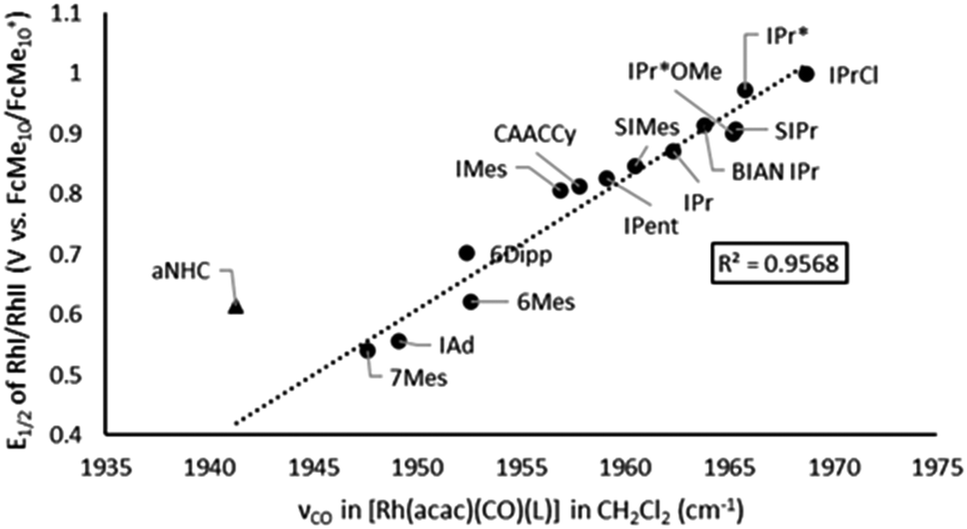 | ||
| Fig. 3 Comparison of the υCO and the E1/2 values measured of [Rh(NHC)(acac)(CO)] complexes. | ||
| RhI/RhII redox couplea | IR νCO valueb | ||
|---|---|---|---|
| NHC | E 1/2 (V) | ΔE (mV) | ν CO (cm−1) |
| a Conditions: 25 °C, 2 mM of analyte, 0.1 M NBu4PF6 in dichloromethane, Pt work and auxiliary electrode, scan rate 100 mV s−1. Potentials are reported referenced to an internal standard of the FcMe10/FcMe10+-couple determined at 0.1 V vs. the Ag/Ag+ reference electrode under the same conditions. Results calculated as the average of 3 separate experiments with estimated standard deviation in parentheses. b Recorded in dichloromethane. | |||
| IPr | 0.870 (0.002) | 159 (8) | 1962.3 |
| SIPr | 0.907 (0.003) | 140 (9) | 1965.3 |
| IMes | 0.805 (0.003) | 147 (7) | 1956.9 |
| SIMes | 0.845 (0.003) | 108 (6) | 1960.5 |
| IPrCl | 0.998 (0.008) | 100 (11) | 1968.7 |
| IPent | 0.825 (0.005) | 111 (10) | 1959.1 |
| BIAN IPr | 0.914 (0.004) | 120 (16) | 1963.8 |
| 6Mes | 0.622 (0.003) | 101 (5) | 1952.6 |
| 7Mes | 0.540 (0.002) | 99 (8) | 1947.6 |
| 6Dipp | 0.702 (0.004) | 103 (9) | 1952.4 |
| IAd | 0.555 (0.003) | 84 (8) | 1949.1 |
| aNHC | 0.615 (0.005) | 156 (15) | 1941.3 |
| CAACCy | 0.811 (0.002) | 108 (10) | 1957.8 |
| IPr* | 0.972 (0.004) | 194 (7) | 1965.8 |
| IPr*OMe | 0.900 (0.003) | 169 (10) | 1965.2 |
We performed further experiments to explain the observed discrepancies. The reported infrared data47 have led to classifying the re-NHCs as remarkable electron donors, even stronger than cyclic(alkyl)(amino)carbenes (CAACs). However, in our correlation with the electrochemical data, these are outliers. It is remarkable that the infrared data of the [Rh(NHC)(CO)2Cl] system change dramatically when expanding the ring size from 5 (SIMes, Rh-νavCO = 2040.5 cm−1) to 6 (6Mes, Rh-νavCO = 2029 cm−1), but no real change is seen for the next expansion to 7 (7Mes, Rh-νavCO = 2028 cm−1). The same trend is observed in the Ir-carbonyl system when comparing SIPr and 6Dipp. This can be explained by the increased steric strain of the NHC when expanding the ring.50 The expansion forces the N-substituents closer to the metal center, leading to an increased length of the carbene-metal bond. Additionally, Nachaev described that re-NHCs bearing bulky N-substituents can crowd the carbene carbon, which destabilizes the metal complex.51
This is further emphasized by the inability to synthesize the [Rh(7Dipp)(cod)Cl] complex.50 The increased steric strain can therefore lead to an over-estimation of the donating ability of this class of NHCs when using traditional carbonyl-based systems, such as the issues previously described for bulky NHCs in the nickel-carbonyl system. Moreover, intramolecular interactions and the activation of methyl C–H bonds located on the wingtips, due to their proximity to the metal has been described for these re-NHCs in complexes with Ni,53 Ru,54 Rh55 and Ir.56 And finally, the internal Stark effect can also be increased by the higher proximity of the wingtip substituent. These three factors lead us to believe that previous estimations of electronic properties of the re-NHCs, along with this [Rh(NHC)(acac)Cl] system, have been tainted by the high steric demands of ligand in these complexes and that these properties have therefore been overestimated.
To further support this argument, variable temperature NMR experiments were performed on the [Rh(6Dipp)(acac)(CO)] complex, clearly showing that the expansion of the heterocycle results in hindered rotation of the NHC as both CH and CH3 peaks of the iPr fragments show peak splitting upon cooling (ESI, Fig. S11 and S12†). Additionally, crystals suitable for X-ray diffraction studies were obtained by slow diffusion of pentane in a saturated chloroform solution.57 The crystal structure of this complex (Fig. 4) shows an out of plane distortion (a ∼15° tilt) of the rhodium–carbene bond relative to the plane formed by the N–C–N atoms of the NHC. The buckling of the carbene-metal bond affects the donating properties of the carbene and distorts both the electrochemical and the infrared data measured on the Rh-complexes. Such issues are liable to be present in measurements using the Rh system with ligands of equivalent or larger steric demand than 6Dipp. We thus decided to explore an alternative, less sterically encumbered system from which to extract pure electronic parameters using electrochemistry. The easily made, linear and sterically uncluttered [Cu(NHC)Cl] system was next examined to provide an answer to the question of whether the ancillary ligand cluttering distorts the analyses. Most copper complexes shown here were easily synthesized following our simple published procedure.58 The [Cu(CAACCy)Cl] complex was made using the ‘built-in-base’ procedure, using Cu2O as a copper source under anaerobic conditions.59 Lastly, the sensitive ring-expanded NHCs were prepared under anaerobic conditions using the recently published microwave assisted procedure.60‡
 | ||
| Fig. 4 Crystal structure of [Rh(6Dipp)(acac)(CO)]. Deviation from planarity: torsion mean N2C1N1 with Rh1: 165°; distance Rh1 to mean N2C1N1 plane: 0.428 Å (H atoms omitted for clarity). | ||
To emphasize the reduced steric effects of the copper complexes, suitable crystals for XRD analysis of the [Cu(7Dipp)Cl] complex were grown.57 In contrast with the [Rh(6Dipp)(acac)(CO)] complex, the [Cu(7Dipp)Cl] complex (bearing the further expanded ring) shows a linear structure with the copper atom still within the N–C–N plane (ESI, Fig. S14†), emphasizing that very bulky ligands do not appear to present distortions in these linear Cu-complexes. We thus expect these systems to be free of the influence of abnormal carbene-metal bonding to which we attribute the conclusions of the Rh system with respect to large, re-NHC ligands.
To our delight, [Cu(NHC)Cl] complexes also exhibit a quasi-reversible redox couple that can be used as a probe for the electron-donating properties of the NHC ligand. As an example, the cyclic voltammogram of [Cu(IPr)Cl] is presented in Fig. S2.† For the Cu-complex, the voltammogram exhibits three electrochemical events in the potential window. The redox couple of peaks 1 and 1′ is attributed to the quasi-reversible CuI/CuII couple. The peak-to-peak separation (ΔE) of this couple is large, ranging from 100 to 216 mV, depending on the coordinated NHC, and is like that of other copper–NHC complexes reported in the literature.61–64 The quasi-reversibility of this redox event is confirmed by performing voltammograms at varying scan rates (see ESI, Fig. S4†). Moving to more positive potentials, a second irreversible oxidation wave (peak 2) is observed. Lastly, a redox couple of peaks 3 and 3′ is seen when scanning negative potentials. This couple is the result of the CuI-complex being reduced to metallic copper at peak 3, followed by stripping the Cu0 back off the electrode in the returning scan, which is also seen for dinuclear copper(I)–NHC complexes65 and exploited in the electrodeposition of copper described by Grujicic and Pesic.66 Reversing the scan before reaching peak 3 results in the disappearance of peak 3′, confirming their correlation.
The linear Cu-complexes permit the analysis of a broad range of NHCs, and interestingly also of very bulky congeners. Twenty NHCs were investigated in this study, including the most employed NHCs. The results of these measurements are presented in Table 3. Originally, acetonitrile was used as solvent as it is the most broadly employed solvent for cyclic voltammetry due to its large potential window.67 However, the complexes containing very bulky NHC ligand (i.e. IAd, IPr* and IPr**) proved to be insoluble in this solvent. Therefore, all complexes, including the very bulky ones, were measured in dichloromethane (DCM). However, this resulted in the observation that two of the investigated ligands (i.e. IPent and IHept) no longer exhibit a reversible couple in this solvent (ESI Fig. S9 and S10†). The linear relation between these two solvent systems is presented in Fig. S6† and a ranking from most to least donating NHC using the [Cu(NHC)Cl] system is shown in Fig. 5.
 | ||
| Fig. 5 Ranking from most to least donating NHCs according to the electrochemical analysis of the CuI/II redox-couple of [Cu(NHC)Cl] complexes in MeCN. aPredicted values due to insolubility issues. | ||
| CuI/CuII redox couple in MeCNa | CuI/CuII redox couple in DCMa | |||
|---|---|---|---|---|
| NHC | E 1/2 (V) | ΔE (mV) | E 1/2 (V) | ΔE (mV) |
| a Conditions: 25 °C, 1 mM of analyte, 0.1 M NBu4PF6 in acetonitrile or dichloromethane, Pt work and auxiliary electrode, scan rate 100 mV s−1. Potentials are reported referenced to an internal standard of the Fc/Fc+-couple determined at 0.13 V in MeCN and 0.46 V in DCM vs. the Ag/Ag+ reference electrode. Results calculated as the average of 3 separate experiments with estimated standard deviation in parentheses. b Complexes did not dissolve in the electrolyte solution. c Irreversible in DCM. | ||||
| IPr | 0.950 (0.004) | 100 (12) | 0.401 (0.006) | 156 (17) |
| SIPr | 0.981 (0.002) | 139 (18) | 0.414 (0.004) | 146 (18) |
| IMes | 0.914 (0.008) | 112 (18) | 0.390 (0.008) | 114 (10) |
| SIMes | 0.967 (0.006) | 149 (20) | 0.408 (0.005) | 132 (9) |
| IPrCl | 1.058(0.010) | 119 (19) | 0.447 (0.008) | 108 (15) |
| IPrMe | 0.837 (0.012) | 175 (28) | 0.353 (0.009) | 154 (10) |
| IPrOMe | 0.938 (0.004) | 154 (12) | 0.398 (0.012) | 153 (13) |
| IPent | 0.938 (0.010) | 190 (14) | —c | —c |
| IHept | 0.934 (0.010) | 252 (21) | —c | —c |
| BIAN IPr | 0.932 (0.008) | 196 (12) | 0.390 (0.007) | 190 (18) |
| 6Mes | 0.899 (0.003) | 160 (8) | 0.381 (0.003) | 125 (14) |
| 7Mes | 0.888 (0.006) | 172 (6) | 0.370 (0.006) | 169 (10) |
| 6Dipp | 0.942 (0.012) | 186 (14) | 0.396 (0.008) | 166 (8) |
| 7Dipp | 0.928 (0.010) | 83 (13) | 0.389 (0.012) | 178 (12) |
| ItBu | 0.875 (0.012) | 121 (8) | 0.378 (0.012) | 147 (8) |
| IAd | —b | —b | 0.353 (0.005) | 154 (10) |
| aNHC | 0.916 (0.012) | 136 (28) | 0.383 (0.014) | 216 (42) |
| CAACCy | 0.810 (0.010) | 125 (12) | 0.340 (0.001) | 115 (19) |
| IPr* | —b | —b | 0.421 (0.008) | 176 (24) |
| IPr** | —b | —b | 0.415 (0.006) | 178 (18) |
However, the electrochemical data for this Cu-system show a clear lack of correlation with the electrochemical data collected for the Rh-system (Fig. 6A). The correlation between both electrochemical probes is mostly distorted by bulky NHCs such as IAd, the re-NHCs, and the backbone modified BIAN IPr. As previously described, bulky substituents can also have a steric effect on the carbonyl bond, making it weaker as the steric strain increases. This leads to mistakenly considering these bulky NHCs as better donating ligands, which is showcased in the correlation as most of these ligands are considered more donating in the more sterically encumbered [Rh(NHC)(acac)(CO)] system compared to the linear [Cu(NHC)Cl] system. Additionally, the backbone functionalized BIAN IPr ligand causes the largest deviation of reversibility in the electrochemical investigation of its Rh-acac-complex, showing a very broad re-reduction peak (ESI, Fig. S8†), suggesting that this large π-system modification on the backbone is non-innocent and not suitable for this evaluation method. This highlights the copper electrochemical system as a better suited system to assess electrochemical properties of sterically more demanding ligands. Interestingly, the Rh-electrochemical data are also distorted by this steric influence, as the linear correlation of υCO and E1/2 on the same probe extends to these bulky NHCs as well. This lends credence to the proposal that in these systems, buckling of the metal out of the N–C–N plane significantly distorts the metal–ligand bonding.
 | ||
| Fig. 6 Comparison of the E1/2 values measured for [Cu(NHC)Cl] complexes with: (A) E1/2 values measured for [Rh(NHC)(acac)(Co)] complexes; (B) νCO reported values for [Ni(NHC)(CO)3]; (C) νCO reported values for [IrCl(CO)2(NHC)]; and (D) νCO reported values for [RhCl(CO)2(NHC)]. | ||
Comparison of our copper electrochemical data with that of the Ni-system results in a linear correlation (Fig. 6B). Additional NHC ligands can be included in the comparison by comparing with the iridium-carbonyl system (Fig. 6C), resulting again in a linear correlation with two major outliers, namely the abnormal NHC and the ring-expanded 6Dipp. As discussed above, this iridium system suffers from the same steric hindrance issues, explaining this ring-expanded NHC being observed as an outlier.
The cyclic voltammogram of the [Cu(aNHC)Cl] complex shows a very different course to the other systems, having a less defined CuI/CuII-redox couple, exhibiting additional peaks, and changing dramatically upon consecutive scans (see ESI, Fig. S7†). This, together with the lack of correlation in the [Rh(NHC)(acac)Cl] system, leads us to believe that this abnormal class of NHCs may not be suited to be evaluated using our electrochemical approach.
Lastly, the copper electrochemical data were compared to that of the rhodium-carbonyl system (Fig. 6D). The correlation of our electrochemical data deviates further from linearity in this comparison. This Rh system exchanges the two most common NHCs, IPr and IMes, in their donating properties. The Ir- and Ni-carbonyl system both assess IMes as the higher donor, while the Rh-carbonyl system ranks IPr over IMes. However, the incorporation of the Rh-carbonyl system enables the inclusion of two other re-NHC to further investigate their behaviour. In this comparison, the lack of correlation of the re-NHCs is even clearer, placing them as the highest donating ligands, even beyond the CAAC family. This leads to the conclusion that the copper electrochemical system is more informative, considering the re-NHC less donating than CAACs, by removing any influences of steric bulk in the evaluation method.
Conclusions
We have presented two new electrochemical probes for the determination of the electronic properties of NHCs. The first, a square planar, four-coordinate Rh-based system, bears a carbonyl ligand as an infrared handle, allowing to readily correlate carbonyl stretching frequencies and E1/2 values, validating our electrochemical approach except for very large NHCs. This method is the first of its kind, showing a wide applicability to assess the most common types of NHCs through cyclic voltammetry, whilst allowing a direct comparison to well-established IR-based methods. The good correlation between most of these data establishes the viability of electrochemical methods for assessing donor properties. The second system is based on linear Cu-complexes that removes any influence of the steric pressure brought about by ancillary ligand on orientation/bonding of NHC ligands to the metal, as well as being synthetically trivial and inexpensive. Both electrochemical probes exhibit a close relationship with the original TEP concept based on the nickel-carbonyl system, however we show that when increasing the ligand sterics, the Rh-probe displays significant distortions, leading to misinterpretation of electronic properties, especially when investigating ring-expanded NHCs. In contrast, the Cu system suffers from no such issues. We have presented a detailed comparison of literature infrared data and have described the drawbacks and the presence of outliers in both models. This has allowed a re-evaluation of the ring expanded NHC class. We argue that extending the now validated electrochemical approach to the sterically accessible Cu-system represents the first proper assessment of re-NHCs donor properties. The present study permits to take a step forward in understanding the complex nature of M-NHC bonding and will aid in the design of new and efficient ligands for catalytic applications.Author contributions
The manuscript was written through contributions of all authors.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data has been deposited at the CCDC under 2277155 and 2277789.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the FWO and the University of Ghent for funding. D. J. L. thanks the Royal Society for the support of a University Research Fellowship (URF\R1\191066). We wish to thank the University of Bath for funding and MC2 for use of their analytical facilities. Umicore AG and Johnson Matthey are gratefully thanked for gifts of materials.References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- C. A. Tolman, Catal. Chem. Rev., 1977, 77, 313–348 CAS.
- M. N. Hopkinson and F. Glorius, An Overview of NHCs, in N-Heterocyclic Carbenes in Organocatalysis, Wiley-VCH, Weinheim, 2019, pp. 1–35 Search PubMed.
- R. Dorta, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
- R. Dorta, E. D. Stevens, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 10490–10491 CrossRef CAS PubMed.
- A. R. Chianese, X. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef CAS.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- G. D. Frey, C. F. Rentzsch, D. von Preysing, T. Scherg, M. Mühlhofer, E. Herdtweck and W. A. Herrmann, J. Organomet. Chem., 2006, 691, 5725–5738 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- D. A. Valyaev, R. Brousses, N. Lougan, I. Fernández and M. A. Sierra, Chem. – Eur. J., 2011, 17, 6602–6605 CrossRef CAS PubMed.
- G. L. Parker, P. Van Lommel, N. Roig, M. Alonso and A. B. Chaplin, Chem. – Eur. J., 2022, e202202283 CrossRef CAS.
- L. Perrin, E. Clot, O. Eisenstein, J. Loch and R. H. Crabtree, Inorg. Chem., 2001, 40, 5806–5811 CrossRef CAS PubMed.
- R. Tonner and G. Frenking, Organometallics, 2009, 28, 3901–3905 CrossRef CAS.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- J. Mathew and C. H. Suresh, Inorg. Chem., 2010, 49, 4665–4669 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- N. Fey, B. M. Ridgway, J. Jover, C. L. McMullin and J. N. Harvey, Dalton Trans., 2011, 40, 11184–11191 RSC.
- E. M. Higgins, J. Armstrong, R. S. Massey, R. W. Alder and A. M. C. O'Donoghue, Chem. Commun., 2011, 47, 1559–1561 RSC.
- R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421–20432 CrossRef CAS PubMed.
- Y. J. Kim and A. Streitwieser, J. Am. Chem. Soc., 2002, 124, 5757–5761 CrossRef CAS PubMed.
- T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS PubMed.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- Q. Teng and H. V. Huynh, Dalton Trans., 2017, 46, 614–627 RSC.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- G. Meng, L. Kakalis, S. P. Nolan and M. Szostak, Tetrahedron Lett., 2019, 60, 378–381 CrossRef CAS.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416–2425 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- S. S. Fielder, M. C. Osborne, A. B. P. Lever and W. J. Pietro, J. Am. Chem. Soc., 1995, 117, 6990–6993 CrossRef CAS.
- E. L. Rosen, C. D. Varnado, A. G. Tennyson, D. M. Khramov, J. W. Kamplain, D. H. Sung, P. T. Cresswell, V. M. Lynch and C. W. Bielawski, Organometallics, 2009, 28, 6695–6706 CrossRef CAS.
- U. Siemeling, C. Färber, M. Leibold, C. Bruhn, P. Mücke, R. F. Winter, B. Sarkar, M. Von Hopffgarten and G. Frenking, Eur. J. Inorg. Chem., 2009, 31, 4607–4612 CrossRef.
- L. Mercs, G. Labat, A. Neels, A. Ehlers and M. Albrecht, Organometallics, 2006, 25, 5648–5656 CrossRef CAS.
- S. Leuthäußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- S. G. Guillet, G. Pisanò, S. Chakrabortty, B. H. Müller, J. G. de Vries, P. C. J. Kamer, C. S. J. Cazin and S. P. Nolan, Eur. J. Inorg. Chem., 2021, 34, 3506–3511 CrossRef.
- N. Aristov and A. Habekost, World J. Chem. Educ., 2015, 3, 115–119 CAS.
- R. A. Kelly, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef CAS.
- F. Izquierdo, C. Zinser, Y. Minenkov, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, F. Nahra, C. S. J. Cazin and S. P. Nolan, ChemCatChem, 2018, 10, 601–611 CrossRef CAS.
- D. J. Nelson, A. Collado, S. Manzini, S. Meiries, A. M. Z. Slawin, D. B. Cordes and S. P. Nolan, Organometallics, 2014, 33, 2048–2058 CrossRef CAS.
- A. Collado, J. Balogh, S. Meiries, A. M. Z. Slawin, L. Falivene, L. Cavallo and S. P. Nolan, Organometallics, 2013, 32, 3249–3252 CrossRef CAS.
- S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 17358–17368 CrossRef CAS PubMed.
- U. S. D. Paul and U. Radius, Organometallics, 2017, 36, 1398–1407 CrossRef CAS.
- J. Balogh, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3259–3263 CrossRef CAS.
- S. Meiries, K. Speck, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 330–339 CrossRef CAS.
- D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172–6175 CrossRef CAS PubMed.
- G. Ung and G. Bertrand, Chem. – Eur. J., 2011, 17, 8269–8272 CrossRef CAS PubMed.
- T. Sato, Y. Hirose, D. Yoshioka and S. Oi, Organometallics, 2012, 31, 6995–7003 CrossRef CAS.
- C. A. Urbina-Blanco, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2010, 6, 1120–1126 CrossRef CAS PubMed.
- M. Iglesias, D. J. Beetstra, B. Kariuki, K. J. Cavell, A. Dervisi and I. A. Fallis, Eur. J. Inorg. Chem., 2009, 13, 1913–1919 CrossRef.
- D. Tapu, O. J. Buckner, C. M. Boudreaux, B. Norvell, M. Vasiliu, D. A. Dixon and C. D. McMillen, J. Organomet. Chem., 2016, 823, 40–49 CrossRef CAS.
- V. Lavallo, Y. Canac, A. DeHope, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 7236–7239 CrossRef CAS PubMed.
- C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahon and M. K. Whittlesey, Chem. Commun., 2010, 46, 5151–5153 RSC.
- R. Armstrong, C. Ecott, E. Mas-Marzá, M. J. Page, M. F. Mahon and M. K. Whittlesey, Organometallics, 2010, 29, 991–997 CrossRef CAS.
- A. Binobaid, K. J. Cavell, M. S. Nechaev and B. M. Kariuki, Aust. J. Chem., 2011, 64, 1141–1147 CrossRef CAS.
- N. Phillips, J. Rowles, M. J. Kelly, I. Riddlestone, N. H. Rees, A. Dervisi, I. A. Fallis and S. Aldridge, Organometallics, 2012, 31, 8075–8078 CrossRef CAS.
- CCDC 2277155 and 2277789† contain the supplementary crystallographic data for [Cu(7Dipp)Cl] and [Rh(6Dipp)(acac)(CO)].
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC.
- C. A. Citadelle, E. Le Nouy, F. Bisaro, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2010, 39, 4489–4491 RSC.
- J. W. Hall, D. Bouchet, M. F. Mahon, M. K. Whittlesey and C. S. J. Cazin, Organometallics, 2021, 40, 1252–1261 CrossRef CAS.
- D. Domyati, S. L. Hope, R. Latifi, M. D. Hearns and L. Tahsini, Inorg. Chem., 2016, 55, 11685–11693 CrossRef CAS PubMed.
- K. Glinton, R. Latifi, D. S. Cockrell, M. Bardeaux, B. Nguyen and L. Tahsini, RSC Adv., 2019, 9, 22417–22427 RSC.
- J. N. Melville and P. V. Bernhardt, Inorg. Chem., 2021, 60, 9709–9719 CrossRef CAS PubMed.
- X. Hu, I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2003, 125, 12237–12245 CrossRef CAS PubMed.
- M. M. Kimani, D. Watts, L. A. Graham, D. Rabinovich, G. P. A. Yap and J. L. Brumaghim, Dalton Trans., 2015, 44, 16313–16324 RSC.
- D. Grujicic and B. Pesic, Electrochim. Acta, 2005, 50, 4426–4443 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- D. J. Babula, R. S. C. Charman, J. A. Hobson, M. F. Mahon and D. J. Liptrot, Dalton Trans., 2024, 53, 3990–3993 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, NMR spectra, additional experiments. CCDC 2277155 and 2277789. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02584a |
| ‡ More recently, such complexes have been shown to be accessible through a convenient mechanochemical approach.68 |
| This journal is © The Royal Society of Chemistry 2024 |