
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2,3-Bis(2-pyridyl)thieno[3,4-b]pyrazine and its ruthenium(II) complexes: a new bidentate bridging ligand for enhanced metal–metal communication†
Kristine L.
Konkol
,
Wyatt D.
Wilcox
and
Seth C.
Rasmussen
 *
*
Department of Chemistry and Biochemistry, North Dakota State University, Department 2508, PO Box 6050, Fargo, North Dakota 58108-6050, USA. E-mail: seth.rasmussen@ndsu.edu
First published on 27th September 2024
Abstract
A new bidentate bridging ligand, bis(2-pyridyl)thieno[3,4-b]pyrazine is reported, along with its mono- and bi-metallic Ru(II) complexes as representative examples. Spectroscopic, electrochemical and X-ray crystallographic characterization of these species is reported, with the separation of the two Ru(III)/Ru(II) couples of the bimetallic complex suggesting better metal–metal communication than classical polypyridyl analogues.
Introduction
The assembly of multiple metal centers into supramolecular constructs allows for the production of multimetallic systems capable of a variety of useful light- and/or redox-induced functions, thus finding applications in light harvesting, conversion of light into chemical or electrical energy, sensing, and photo-catalysis.1–3 A primary advantage of this approach is that careful selection of the molecular components can allow for fine-tuning and production of specific desired properties for the given application. Furthermore, suitable choices of metal-based components and bridging ligands, coupled with design of the supramolecular structure, can provide the occurrence of interesting and useful multi-component processes such as energy transfer along predetermined pathways, photoinduced charge separation, or multielectron exchange at a predetermined potential.2The choice of bridging ligand employed to connect the individual metal-based components is a critical component of such multimetallic assemblies.3,4 Technically, any species capable of donating pairs of electrons to two separate metal centers can serve as a bridging ligand, but bridging ligands capable of multidentate coordination provide greater stability of the multimetallic species during excitation. In addition, ligands providing a conjugated path between metals can promote electronic coupling, which changes the electronic properties of the bridged systems compared to the individual single metal systems.4 Such π-conjugated ligands are also often redox and spectroscopically active, commonly possessing empty low-lying π*-orbitals and filled high-energy π-orbitals that serve to mediate electron and energy transfer between bridged metals.3,4 As a consequence, this makes the nature of the bridging ligand key to the redox, spectroscopic, and photochemical properties of these multi-metallic complexes.4
Polypyridyl ligands are a widely used class of polydentate ligands.4–6 As such, it is not surprising that various polypyridyl systems have served as very successful bridging ligands, providing the desirable properties described above.3,4 Among the most widely studied of these are 2,3-bis(2-pyridyl)pyrazine (dpp) and its extended analogues 2,3-bis(2-pyridyl)quinoxaline (dpq) and 2,3-bis(2-pyridyl)benzo[g]quinoxaline (dpb) (Fig. 1).3,4 Efforts to further tune these bridging ligands have included functionalized derivatives or covalently linking the pendant pyridines together to reduce conformational flexibility, but the study of other ring systems fused to the pyrazine core has been very limited and only includes pyridopyrazine analogues of dpq (dpPP and dpPP′, Fig. 1).7,8 The fused-ring thiophene analogue of quinoxaline, thieno[3,4-b]pyrazine (TP), has been known since 1957,9 although its primary application to date has been as a monomer for low band gap conjugated polymers.10 The basicity of the TP nitrogens was determined in 2002,11 and was shown to be essentially identical to both pyrazine and quinoxaline (pKa = 0.55 vs. 0.57 and 0.56, respectively). Still, the metal coordination of TP has remained largely unexplored.
 | ||
| Fig. 1 Common bidentate polypyridyl bridging ligands and heterocyclic analogues. | ||
Herein the synthesis and characterization of 2,3-bis(2-pyridyl)thieno[3,4-b]pyrazine (dpTP, Fig. 1) is reported. In order to illustrate its ability to act as an effective bridging ligand, its mono- and bi-metallic Ru(II) complexes [(bpy)2Ru(dpTP)](PF6)2 and [{(bpy)2Ru}2(dpTP)](PF6)4 (with bpy = 2,2′-bipyridine) are also reported, which represent the first known metal complexes of dpTP. For the purpose of evaluating the potential of this new polypyridyl bridging ligand, dpTP and its Ru(II) complexes will be fully compared to the classical bridging ligands dpp, dpq, and dpb, as well as their analogous Ru(II) complexes.12–21
Results and discussion
Ligand synthesis and characterization
The ligand dpTP was synthesized as outlined in Scheme 1. These methods are based on the well-developed production of 3,4-diaminothiophene (2)22via the reduction of 2,5-dibromo-3,4-dinitrothiopene (1),23 which then allows the condensation of 2 with 2,2′-pyridil to give dpTP as a yellow solid in yields of 86–88%. It must be pointed out that the synthesis of dpTP was previously included in a 2004 study on the use of microwave heating for the pyrazine condensation step.24 In this way, dpTP was produced as a brown solid in lower yields (77%). Furthermore, comparison of the reported data with the material reported here confirms reduced purity by the previous methods.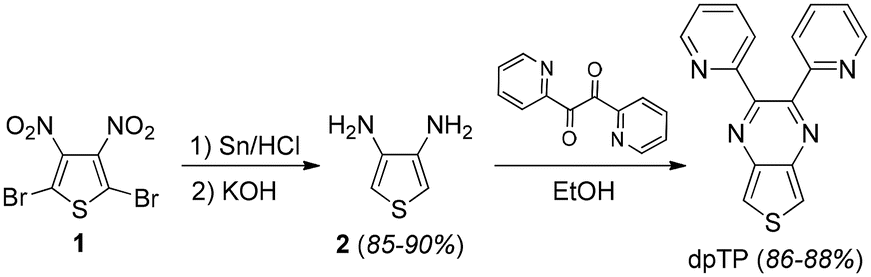 | ||
| Scheme 1 Synthesis of dpTP. | ||
Single crystals of dpTP were grown from ethanol in order to determine its crystal structure, with the ellipsoid plot given in Fig. 2. The fused-ring dpTP core is consistent with that previously reported for Me2TP,11 with minor bond length deviations of ca. 0.007–0.008 Å found in the pyrazine ring that can be attributed to conjugation with the 2-pyridyl moieties (see ESI†). In comparison to dpq, the TP core of dpTP is much more planar than the quinoxaline of dpq, the latter exhibiting a distortion from planarity of 8.4°.25 In comparison, the TP unit of dpTP shows only a slight twist of ca. 4° within the pyrazine ring (Fig. 2B). In addition, the deviation of the two pyridyl substituents from the plane of the TP core is significantly reduced, with the torsional angle of the C–C bond connecting the pyridyl rings to the pyrazine (i.e., C(7)–C(5)–C(6)–C(12)) found to be 16.9° in comparison to 30.3° for dpq.25
 | ||
| Fig. 2 Face (A) and edge (B) ellipsoid plots of dpTP at the 50% probability level. | ||
Characterization by cyclic voltammetry (CV) shows that the LUMO of dpTP (E½ of its reduction) falls below that of dpb. Thus, the following trend in the first reduction (vs. Ag/Ag+) is found: −2.12 V (dpp)26> −2.05 V (dpq)27> −1.77 (dpb)27> −1.70 V (dpTP). This also agrees well with the known acceptor abilities of TP vs. quinoxaline.28 A further oxidation at 2.15 V is seen for dpTP, with HOMO levels of the other ligands too deep to measure.
Synthesis of Ru(II) complexes
In order to evaluate its effectiveness as a bridging ligand for multimetallic complexes, dpTP was then reacted with cis-Ru(bpy)2Cl2 (bpy = 2,2′-bipyridine) under fairly standard conditions. As outlined in Scheme 2, the reaction with 1.5 equivalents of the Ru(II) precursor successfully generated the monometallic complex [(bpy)2Ru(dpTP)](PF6)2 as a crystalline purple solid in 68% yield. The induced asymmetry of the coordinated dpTP can be clearly seen in the NMR signals of the thiophene α-protons (see ESI†). The singlet of the free ligand at 8.29 ppm splits into two doublets upon coordination, one shifting downfield to 8.50 ppm, with the other undergoing a more significant shift upfield to 7.20 ppm. The coupling constant (J = 3.4 Hz) between these doublets is in close agreement with that of inequivalent thiophene α-protons.29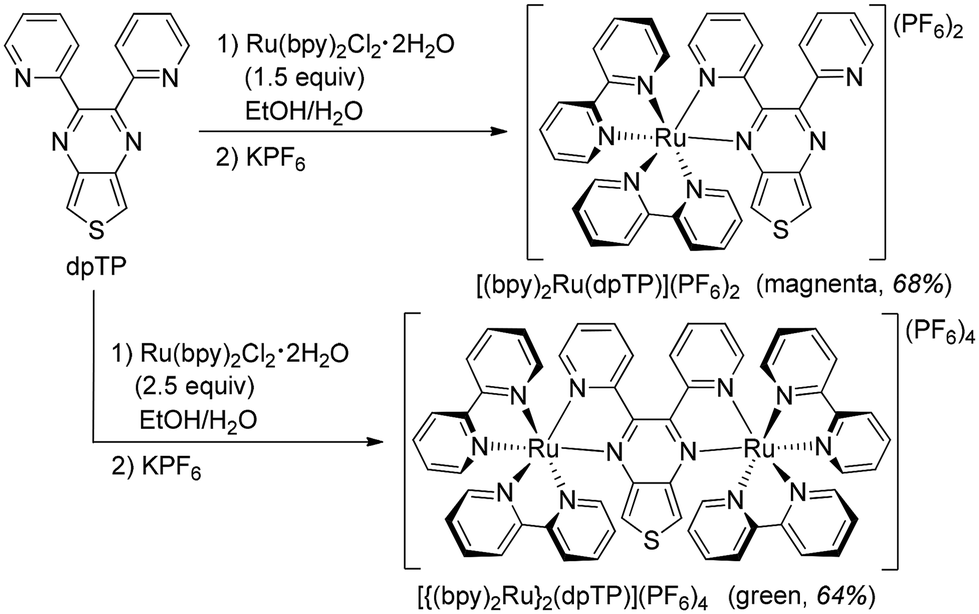 | ||
| Scheme 2 Synthesis of Ru(II) complexes of dpTP. | ||
In the same way, the application of 2.5 equivalents of the Ru(II) precursor under similar conditions then led to the isolation of the bimetallic complex [{(bpy)2Ru}2(dpTP)](PF6)4 as a crystalline green solid in 64% yield. Here, the coordinated dpTP becomes symmetric again, as the two doublets observed for the thiophene protons in the 1H NMR are replaced by a singlet appearing at 7.38 ppm (see ESI†). Both Ru(II) complexes are stable species, thus allowing detailed characterization of their structural, optical, and electronic properties.
Crystal structure of the bimetallic complex
Single crystals of [{(bpy)2Ru}2(dpTP)](PF6)4·4CH3CN were grown via the slow evaporation of CH3CN solutions, allowing the determination of its crystal structure. The bimetallic complex crystallizes as a racemic mixture of the chiral Δ,Δ- and Λ,Λ-isomers, with views of the packing of these racemic pairs given in Fig. 3. This is fairly unusual in that nearly all known crystal structures of [{(bpy)2Ru}2BL]n+ (BL = bridging ligand) complexes are of the meso diastereoisomer.13b The structure of rac-[{(bpy)2Ru}2dpp](PF6)4·4H2O has been reported,20a but this was accomplished by first separating the diastereoisomer via cation-exchange chromatography prior to crystallization. For the dpTP complexes here, 1H NMR of the bimetallic complex does show a minor species that could indicate the presence of a small amount of the meso diasteroisomer. This possibility is further supported by the observed spectral broadening seen for this minor species, as meso diastereoisomers of dpb has previously been found to exhibit greater broadening in comparison with the rac form.13b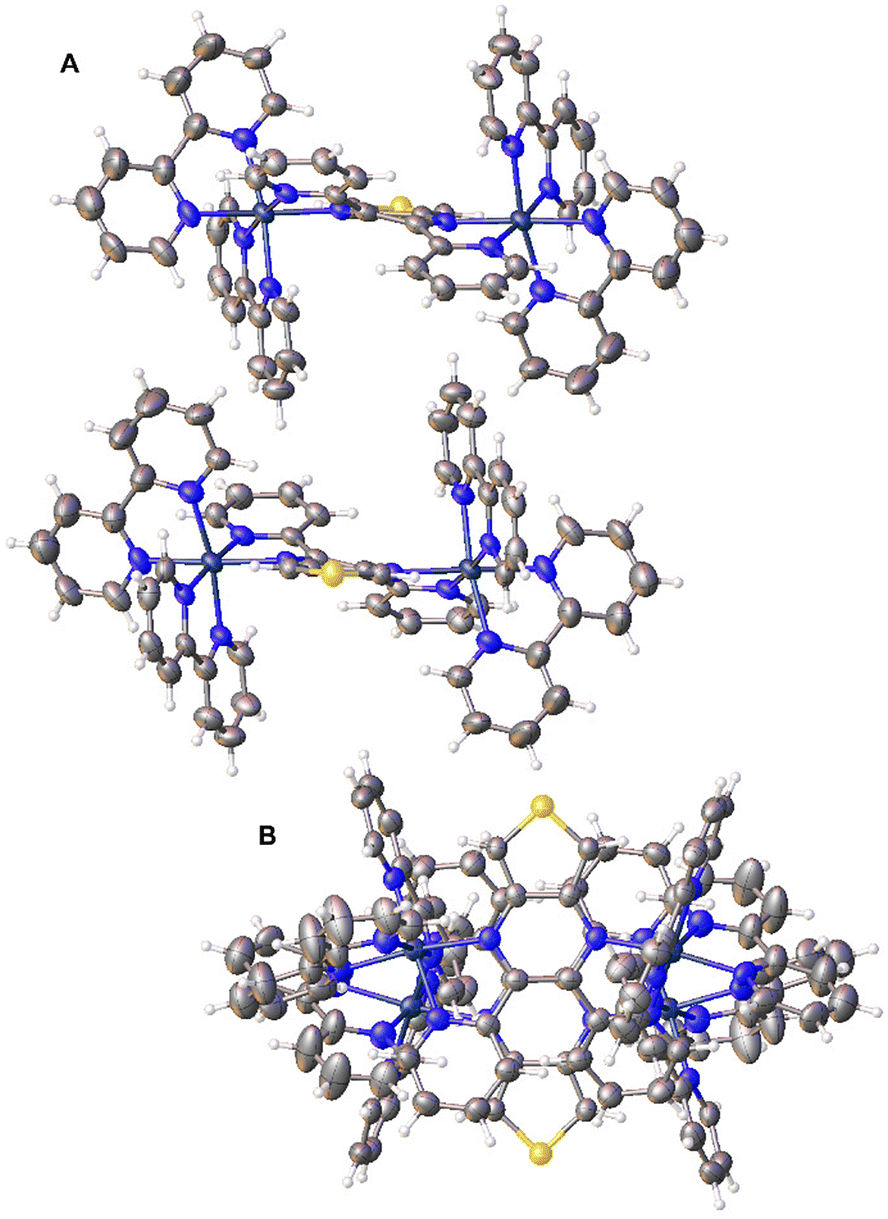 | ||
| Fig. 3 Side (A) and top (B) views of a racemic pair of [{(bpy)2Ru}2(dpTP)]4+ complexes (thermal ellipsoids shown at the 50% probability level). | ||
An isolated ellipsoid plot of just the Λ,Λ-[{(bpy)2Ru}2(dpTP)]4+ isomer is shown in Fig. 4 and select bond distances for both dpTP and [{(bpy)2Ru}2(dpTP)]4+ are given in Table 1. The complex exhibits Ru–N bond lengths of 2.079 and 2.042 Å for the coordination of the dpTP pyrazine and pyridines, respectively, in agreement with related bimetallic complexes.13b The metals reside in distorted octahedral environments and the average bite angle to dpTP is 77.52°, a bit smaller than the 79–80° bite angle for the external bpy ligands.
 | ||
| Fig. 4 Face (A) and edge (B) ellipsoid plots of Λ,Λ-[{(bpy)2Ru}2(dpTP)]4+ at the 50% probability level. | ||
| Parameter | dpTP | [{(bpy)2Ru}2(dpTP)]4+ |
|---|---|---|
| Ru1–N1 | 2.079(5) | |
| Ru1–N3 | 2.042(5) | |
| S1–C1 | 1.688(6) | 1.694(6) |
| C1–C2 | 1.365(7) | 1.368(9) |
| C2–C3 | 1.432(7) | 1.451(11) |
| C2–N1 | 1.374(6) | 1.380(8) |
| N1–C5 | 1.300(6) | 1.335(8) |
| C5–C6 | 1.468(7) | 1.445(13) |
| C5–C7 | 1.488(6) | 1.472(9) |
| C7–C8 | 1.401(6) | 1.394(9) |
| C8–C9 | 1.376(6) | 1.373(10) |
| C9–C10 | 1.378(6) | 1.394(11) |
| C10–C11 | 1.384(6) | 1.368(10) |
| C11–N3 | 1.324(6) | 1.354(9) |
| N3–C7 | 1.335(6) | 1.356(8) |
As with other analogous Ru bimetallic complexes, coordination of the bridging ligand results in some distortion.13b,20a As shown in Table 1, most of the bonds within the TP pyrazine undergo elongation (by up to 0.035 Å), although the bond between the carbons containing the 2-pyridyl moieties shortens by ca. 0.02 Å. At the same time, the distortion from planarity within the central TP core is slightly reduced to ca. 3.7°, while the torsional angle of the C–C bond connecting the pyridyl rings to the pyrazine nearly doubles in comparison to the free ligand (31.2° vs. 16.9°). This increase is largely due to enhanced sterics resulting from the necessary conformation of the pyridyl rings to permit chelation to the metal center, which in turn introduces hindrance between the protons at the 3-position of the pyridyl rings.
The intermetallic Ru–Ru distance of 6.972 Å is longer than that found in analogous bimetallic complexes (6.813–6.887 Å).13b,20a This difference is the result of slightly longer Ru–N(pyrazine) bond distances, coupled with a more linear arrangement of the metals with the plane of the bridging TP core (see Fig. 4B). For both metals, the deviation from the plane of the TP unit is only ca. 6–9°. In comparison, other complexes of classical bridging ligands adopt a more puckered geometry in which the metal centers reside slightly below the central plane of the bridging ligand,13b,20a thus allowing the two metals to shorten the intermetallic distance. Overall, the enhanced planarity of both the central TP unit and the Ru–TP–Ru path should have a noticeable effect on the electronic communication provided by dpTP.
Absorption spectroscopy
The ligand dpTP and its Ru(II) complexes were then characterized by UV-visible spectroscopy, with the resulting spectra given in Fig. 5. Comparison to the analogous complexes of the classical bridging ligands is also given in Table 2, in which care was taken to use collected literature values using identical conditions to the dpTP results reported herein. In addition to the typical π–π* transitions observed in conventional ligands, dpTP also exhibits a charge-transfer (CT) band at 332 nm which provides its observed yellow color. This CT band is typical of TPs and corresponds to a transition from a thiophene-localized HOMO to a LUMO of greater pyrazine contribution.9 As with Ru(II) complexes of the classical bridging ligands, both Ru(II) complexes of dpTP exhibit a low energy transition which is assigned as a metal-to-ligand charge transfer (MLCT) from the Ru(II) center to the π* of the dpTP ligand. More unusual, however, is that while complexes of the classical ligands typically exhibit a featureless MLCT band, both dpTP complexes also exhibit a lower energy shoulder. At this point, it is unclear if these low-energy features correspond to separate transitions or are just vibrational components of the same electronic transition (the energetic separation in both cases is 2500–2600 cm−1).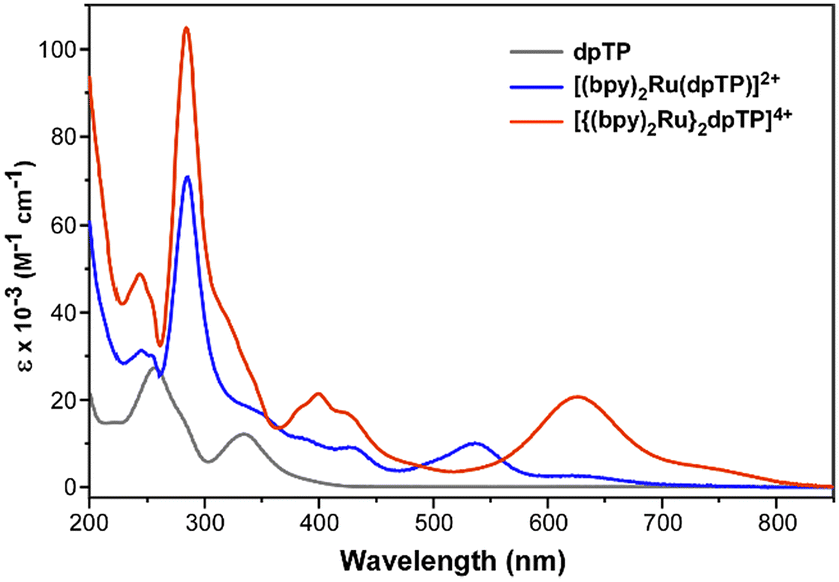 | ||
| Fig. 5 Absorption spectra of dpTP and its Ru(II) complexes. | ||
| BL | [(bpy)2Ru(BL)]2+ | [{(bpy)2Ru}2BL]4+ |
|---|---|---|
| a In CH3CN, (sh) = shoulder. b Ref. 17. c Ref. 14. d Ref. 21. e Ref. 13b, at −35 °C. | ||
| dppb | 284, 434, 468 (sh) | 284, 425, 526 |
| dpqc | 284, 348 (sh), 427 (sh), 515 | 283, 382 (sh), 399, 423 (sh), 605 |
| dpb | 284, 320, 410, 551d | 340, 372, 409, 425, 644, 765 (sh)e |
| dpTP | 284, 348 (sh), 430, 535, 625 (sh) | 284, 325 (sh), 400, 628, 745 (sh) |
As can be seen in Table 2, however, the low energy transitions of the dpTP complexes are considerably red-shifted in comparison to most of the analogous complexes of the classical bridging ligands, with the absorbance of the bimetallic dpTP complex extending out to nearly 850 nm. The exception is the bimetallic complex of dpb, which exhibits its MLCT at slightly lower energy than the dpTP analogue. Still, this further supports the low energy nature of the dpTP LUMO, with the trend in lowest energy transitions given in Table 2 closely following the trend in bridging ligand LUMO energies discussed above.
Electrochemistry
Electrochemical characterization of both dpTP complexes resulted in the CVs shown in Fig. 6. The monometallic complex exhibits a single Ru(II) oxidation at 1.07 V, with three ligand-based reductions at −1.01, −1.72, and −1.96 V. In comparison to the free ligand, donation of electron density to the Ru(II) center upon coordination results in a significantly shift of the dpTP reduction from −1.70 to −1.01 V. Donation of electron density to both Ru(II) centers in the bimetallic complex further shifts the reduction to even lower negative potential (−0.62 V), followed by a second dpTP reduction at −1.32 V. Two sequential reductions of the bridging ligand before reduction of the bpy ligands in [{(bpy)2Ru}2dpTP]4+ agrees well with previous assignments for bimetallic complexes of dpp and dpq.16b This is also consistent with the fact that the free dpTP undergoes reduction at much less negative potential than free bpy (−1.70 vs. −2.38 V vs. Ag/Ag+),30 with the difference between the 1st and 2nd reduction of the monometallic complex in close agreement with the difference between the reduction of the free ligands. In the bimetallic complex, the coordination of both metals to dpTP should result in a further shift in the reduction potential of dpTP, but should not cause further shifts to the bpy reduction. This is exactly what is observed, with the 3rd reduction of the bimetallic complex closely matching the 2nd reduction of monometallic complex. | ||
| Fig. 6 Cyclic voltammograms of [(bpy)2Ru(dpTP)]2+ and [{(bpy)2Ru}2dpTP]4+. | ||
Although the reduction of the bpy ligands in the monometallic complex occurs sequentially, the bimetallic complex exhibits two overlapping bpy reductions, logically corresponding to the reduction of one bpy from each Ru(II) center. As these correspond to the 3rd and 4th 1e− reductions, the neutral species is generated which exhibits reduced solubility, causing adsorption on the electrode as evidenced by the desorption spike observed in Fig. 6 upon reoxidation. It was found that this could be avoided by running the CV in DMF, thus allowing characterization of all four bpy reductions (see ESI†).
Table 3 compares the electrochemical data of [{(bpy)2Ru}2dpTP]4+ with the analogous complexes of the classical bridging ligands. As with the previous spectroscopic data, care was taken to use literature values using identical conditions to the dpTP results reported herein, with the only differences entailing the reference electrode used. This is particularly important as it has been found that changes in both solvent and electrolyte can impact the measured differences in the first and second oxidation of bridged bimetallic complexes.31 In contrast, while differences in reference electrode can result in a constant shift in measured potentials, it would result in no change when comparing the separation of two measured potentials.
| BL | Oxidation | Reduction | |||
|---|---|---|---|---|---|
| E ½ (6+/5+) (V) | E ½ (5+/4+) (V) | ΔEox (mV) | E ½ (4+/3+) (V) | E ½ (3+/2+) (V) | |
| a In CH3CN/Bu4NPF6, with potentials vs. Ag/Ag+. Literature values converted to Ag/Ag+. b Ref. 16b. c Ref. 13b. | |||||
| dppb | 1.36 | 1.16 | 200 | −0.94 | −1.44 |
| dpqb | 1.43 | 1.25 | 180 | −0.62 | −1.40 |
| dpbc | 1.33 | 1.13 | 200 | −0.57 | −1.22 |
| dpTP | 1.35 | 1.12 | 230 | −0.62 | −1.32 |
Table 3 shows that the first metal oxidation occurs at nearly the same potential for all species, suggesting little difference in electron donation to the metals from the bridging ligand. Of greater interest, however, are the potentials of the second oxidation. While all complexes exhibit well separated oxidations of the bridged Ru(II) centers, the complexes vary in terms of the extent of that separation. The observed separation between the two metal oxidations (ΔEox in Table 3) is viewed to be indicative of the stability of the mixed-valence intermediate,31 and has been ascribed to the electrostatic and electronic effects brought about by the proximity of the two metals and the shared π-system between them.14 As shown in Table 3, Ru(II) centers bridged by dpTP exhibit the greatest separation, indicative of stronger coupling between the two metals and improved metal–metal communication. This extent of electronic coupling has been related to the structural and electronic properties of the bridging ligand,26 with emphasis on the π* (LUMO) of the ligand that dictates ligand-mediated interactions. In addition, the nature of the bridging ligand plays an important role in electronic coupling by controlling the orientation and distance between the bridged metal centers.31
Experimental
General
Unless otherwise specified, all reactions were carried out under nitrogen atmosphere with reagent grade materials. All glassware was oven-dried, assembled hot, and cooled under a dry nitrogen stream before use. 3,4-Diaminothiophene22 and cis-Ru(bpy)2Cl2·2H2O32 were synthesized according to literature procedures. Acetonitrile was distilled over CaH2 under N2. Chromatographic separations were performed using standard column methods with silica gel (230–400 mesh) or alumina (neutral). HRMS (ESI-TOF) and elemental analysis was performed in-house. 1H and 13C NMR spectra were completed on 400 MHz spectrometer. All NMR data was referenced to residual solvent peaks and peak multiplicity reported as follows: s = singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, ddd = doublet of doublet of doublets, m = multiplet.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 hexanes:EtOAc → 100% EtOAc) to yield a bright yellow solid (86–88% yield). Mp. 165 °C (dec.). 1H NMR (CDCl3): δ 8.32 (ddd, J = 4.9, 1.9, 1.2 Hz, 2H), 8.14 (s, 2H), 7.92 (dt, J = 7.8, 1.2 Hz, 2H), 7.80 (td, J = 7.8, 1.9 Hz, 2H), 7.22 (ddd, J = 7.8, 4.9, 1.2 Hz, 2H), this NMR data agrees well with literature values.31H NMR (d6-acetone): δ 8.29 (s, 2H), 8.19 (ddd, J = 4.9, 1.7, 1.2 Hz, 2H), 8.00 (dt, J = 7.8, 1.2 Hz, 2H), 7.91 (td, J = 7.8, 1.7 Hz, 2H), 7.28 (ddd, J = 7.8, 4.9, 1.2 Hz, 2H). 13C NMR (d6-acetone): δ 159.3, 153.8, 148.9, 142.3, 137.5, 124.6, 123.9, 119.7. HRMS: m/z 291.0694 [M + H]+ (calcd for C16H11N2S 291.0704); 313.0540 [M + Na]+ (calcd for C16H10N2NaS 313.0524).
:1 mixture of EtOH and H2O (50 mL) was then added, and the solution was heated to reflux with stirring for 3 h. The reaction was then cooled to room temperature and poured into a stirred concentrated (ca. 1 M) aqueous solution of KPF6, whereby a precipitate instantaneously formed. Stirring was continued for 10 min, after which the precipitate was collected via vacuum filtration, washed with H2O until the filtrate ran clear (ca. 500 mL), and then washed with Et2O. The crude solid was purified on alumina column chromatography (50:50 toluene:acetone → acetone) to yield a magenta solid (68%). Mp. 195 °C (dec.). HRMS: m/z 352.0523 [M2+] (calcd for C36H26N8RuS 352.0527). Elem. Anal. Calc. for C36H26N8F12P2RuS·C3H6O: C, 44.54; H, 3.07; N, 10.65. Found: C, 44.35; H, 3.32; N, 11.14. See ESI† for 1H NMR.
:1 mixture of EtOH and H2O (100 mL) was then added, and the solution was heated to reflux with stirring for 3 h. The reaction was then cooled to room temperature and poured into a stirred concentrated (ca. 1 M) aqueous solution of KPF6, whereby a precipitate instantaneously formed. Stirring was continued for 10 min, after which the precipitate was collected via vacuum filtration, washed with H2O until the filtrate ran clear (ca. 500 mL), and then washed with Et2O. The crude solid was purified on alumina column chromatography (50:50 toluene:acetone → acetone) to yield a green solid (64%). Mp. 210 °C (dec.). HRMS: m/z 279.5371 [M4+] (calcd for C56H42N12Ru2S 279.5372). Elem. Anal. Calc. for C56H42N12F24P4Ru2S: C, 39.63; H, 2.49; N, 9.90. Found: C, 39.96; H, 2.20; N, 9.89. See ESI† for 1H NMR.
:50 hexanes:EtOAc → 100% EtOAc) to yield a bright yellow solid (86–88% yield). Mp. 165 °C (dec.). 1H NMR (CDCl3): δ 8.32 (ddd, J = 4.9, 1.9, 1.2 Hz, 2H), 8.14 (s, 2H), 7.92 (dt, J = 7.8, 1.2 Hz, 2H), 7.80 (td, J = 7.8, 1.9 Hz, 2H), 7.22 (ddd, J = 7.8, 4.9, 1.2 Hz, 2H), this NMR data agrees well with literature values.31H NMR (d6-acetone): δ 8.29 (s, 2H), 8.19 (ddd, J = 4.9, 1.7, 1.2 Hz, 2H), 8.00 (dt, J = 7.8, 1.2 Hz, 2H), 7.91 (td, J = 7.8, 1.7 Hz, 2H), 7.28 (ddd, J = 7.8, 4.9, 1.2 Hz, 2H). 13C NMR (d6-acetone): δ 159.3, 153.8, 148.9, 142.3, 137.5, 124.6, 123.9, 119.7. HRMS: m/z 291.0694 [M + H]+ (calcd for C16H11N2S 291.0704); 313.0540 [M + Na]+ (calcd for C16H10N2NaS 313.0524).
:1 mixture of EtOH and H2O (50 mL) was then added, and the solution was heated to reflux with stirring for 3 h. The reaction was then cooled to room temperature and poured into a stirred concentrated (ca. 1 M) aqueous solution of KPF6, whereby a precipitate instantaneously formed. Stirring was continued for 10 min, after which the precipitate was collected via vacuum filtration, washed with H2O until the filtrate ran clear (ca. 500 mL), and then washed with Et2O. The crude solid was purified on alumina column chromatography (50:50 toluene:acetone → acetone) to yield a magenta solid (68%). Mp. 195 °C (dec.). HRMS: m/z 352.0523 [M2+] (calcd for C36H26N8RuS 352.0527). Elem. Anal. Calc. for C36H26N8F12P2RuS·C3H6O: C, 44.54; H, 3.07; N, 10.65. Found: C, 44.35; H, 3.32; N, 11.14. See ESI† for 1H NMR.
:1 mixture of EtOH and H2O (100 mL) was then added, and the solution was heated to reflux with stirring for 3 h. The reaction was then cooled to room temperature and poured into a stirred concentrated (ca. 1 M) aqueous solution of KPF6, whereby a precipitate instantaneously formed. Stirring was continued for 10 min, after which the precipitate was collected via vacuum filtration, washed with H2O until the filtrate ran clear (ca. 500 mL), and then washed with Et2O. The crude solid was purified on alumina column chromatography (50:50 toluene:acetone → acetone) to yield a green solid (64%). Mp. 210 °C (dec.). HRMS: m/z 279.5371 [M4+] (calcd for C56H42N12Ru2S 279.5372). Elem. Anal. Calc. for C56H42N12F24P4Ru2S: C, 39.63; H, 2.49; N, 9.90. Found: C, 39.96; H, 2.20; N, 9.89. See ESI† for 1H NMR.
X-ray diffraction
X-ray quality crystals of dpTP and [{(bpy)2Ru}2(dpTP)](PF6)4·4CH3CN were grown by the slow evaporation of ethanol and acetonitrile solutions, respectively. The X-ray intensity data of the crystals were measured at either 110 or 296 K on a CCD-based X-ray diffractometer system equipped with a Cu X-ray tube (λ = 1.54178 Å) operated at 2000 W of power. The detector was placed at a distance of 5.047 cm from the crystal. Frames were collected with a scan width of 0.3° in ω and exposure time of 10 s per frame and then integrated with the Bruker SAINT software package using an arrow-frame integration algorithm. The unit cell was determined and refined by least-squares upon the refinement of XYZ-centeroids of reflections above 20σ(I). The structure was refined using the Bruker SHELXTL (Version 5.1) Software Package. The crystallographic data for dpTP and [{(bpy)2Ru}2(dpTP)](PF6)4·4CH3CN is given in Table 4.| dpTP | [{(bpy)2Ru}2(dpTP)](PF6)4·4CH3CN | |
|---|---|---|
| a R 1 = ∑(||Fo| − |Fc||)/∑|Fo|, wR2 = [∑(w(Fo2 − Fc2)2)/∑(Fo2)2]1/2. | ||
| CCDC | 2363377 | 2363376 |
| Chemical formula | C16H10N4S | C56H42N12Ru2S,4(C2H3N),4(F6P) |
| Formula weight | 290.34 | 1861.31 |
| Temperature (K) | 296 | 110 |
| Space group | P21/c | C2/c |
| a (Å) | 15.1841(10) | 31.2206(11) |
| b (Å) | 7.5290(4) | 13.5395(5) |
| c (Å) | 11.9534(7) | 18.7558(6) |
| α (°) | 90.00 | 90.00 |
| β (°) | 97.518(4) | 102.904(2) |
| γ (°) | 90.00 | 90.00 |
| V (Å3) | 1354.78(14) | 7728.1(5) |
| Z | 4 | 4 |
| D calc (g cm−3) | 1.423 | 1.600 |
| μ (mm−1) | 2.098 | 5.176 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0833 | R 1 = 0.0742 |
| wR2 = 0.2228 | wR2 = 0.2115 | |
| R indices (all data)a | R 1 = 0.0985 | R 1 = 0.0786 |
| wR2 = 0.2355 | wR2 = 0.2152 | |
Optical spectroscopy
UV-vis spectra were measured on a dual beam scanning UV-visible-NIR spectrophotometer in 1 cm quartz cuvettes. Samples were prepared as dilute CH3CN solutions. Extinction coefficients were determined via standard Beer–Lambert relationships using at least five standard solutions of different concentrations.Electrochemistry
Electrochemical measurements were carried out in a three-electrode cell using a platinum disc working electrode, platinum wire auxiliary electrode, and Ag/Ag+ reference electrode All potentials are referenced to a Ag/Ag+ reference electrode (0.01 M AgNO3/0.1 M TBAPF6 in CH3CN; 0.251 V vs. SCE),33 calibrated to the ferrocene/ferrocenium redox couple (100 mV vs. Ag/Ag+). Supporting electrolyte consisted of 0.10 M tetrabutylammonium hexafluorophosphate (TBAPF6) in dry CH3CN. Solutions were deoxygenated by sparging with argon prior to each scan and blanketed with argon during the measurements. All measurements were collected at a scan rate of 100 mV s−1.Conclusions
A new bidentate polypyridyl bridging ligand (dpTP) has been reported that utilizes a fused-ring thieno[3,4-b]pyrazine core. The ligand dpTP provides a LUMO energy on par with the classical ligand dpb, combined with a structural size between the ligands dpp and dpq. In its application to bimetallic complexes, this combination allows effective control of the orientation between the bridged metals and effective metal–ligand interactions, resulting in improved metal–metal communication. As such, this new addition to the widely applied family of polypyridyl bridging ligands holds significant promise for future multimetallic applications.Author contributions
K. L. K.—methodology, investigation, formal analysis, writing—review and editing; W. D. W.—investigation, writing—review and editing; S. C. R.—conceptualization, methodology, validation, formal analysis, resources, data curation, writing—original draft preparation, writing—review and editing, supervision, project administration, funding acquisition.Data availability
Data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank North Dakota State University (NDSU) and ND EPSCoR for support of this research, as well as Dr Angel Ugrinov (NDSU) for collection of the X-ray crystal data, and NSF-CRIF (CHE-0946990) for the purchase of the departmental XRD instrument.References
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765–838 RSC.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed.
- K. J. Brewer, S. Swavey, R. L. Williams, Z. Fang and E. R. Bullock, Proc. SPIE – Int. Soc. Opt. Eng., 2001, 4512, 53–64 CrossRef CAS.
- H. M. Rogers, S. M. Arachchige, K. J. Brewer and S. Swavey, in Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, ed. J. Reedijk, Elsevier, Waltham, MA, 2014, pp. 1–21 Search PubMed.
- F. Dumur, E. Dumas and C. R. Mayer, in Targets in Heterocyclic Systems: Chemistry and Properties, ed. O. A. Attanasi and D. Spinelli, Italian Society of Chemistry, Rome, 2007, vol. 11, pp. 70–103 Search PubMed.
- R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC.
- T. J. Simpson and K. C. Gordon, Inorg. Chem., 1995, 34, 6323–6329 CrossRef CAS.
- A. T. Vallina, H. Stoeckli-Evans, A. Neels, J. Ensling and S. Decurtins, Inorg. Chem., 2003, 42, 3374–3382 CrossRef CAS PubMed.
- S. C. Rasmussen, M. E. Mulholland, R. L. Schwiderski and C. A. Larsen, J. Heterocycl. Chem., 2012, 49, 479–493 CrossRef CAS.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394–11410 RSC.
- D. D. Kenning, K. A. Mitchell, T. A. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS PubMed.
- S. M. Molnar, K. R. Neville, G. E. Jensen and K. J. Brewer, Inorg. Chim. Acta, 1993, 206, 69–76 CrossRef CAS.
- (a) D. M. D'Alessandro, L. S. Kelso and F. R. Keene, Inorg. Chem., 2001, 40, 6841–6844 CrossRef PubMed; (b) D. M. D'Alessandro, P. C. Junk and F. R. Keene, Supramol. Chem., 2005, 17, 529–546 CrossRef.
- D. P. Rillema and K. B. Mack, Inorg. Chem., 1982, 21, 3849–3854 CrossRef CAS.
- (a) C. H. Braunstein, A. D. Baker, T. C. Strekas and H. D. Gafney, Inorg. Chem., 1984, 23, 857–864 CrossRef CAS; (b) Y. Fuchs, S. Lofters, T. Dieter, W. Shi, R. Morgan, T. C. Strekas, H. D. Gafney and A. D. Baker, J. Am. Chem. Soc., 1987, 109, 2691–2697 CrossRef CAS; (c) O. Morgan, S. Wang, S. Bae, R. J. Morgan, A. D. Baker, T. C. Strekas and R. Engel, J. Chem. Soc., Dalton Trans., 1997, 3773–3776 RSC.
- (a) A. W. Wallace, W. R. Murphy Jr. and J. D. Petersen, Inorg. Chim. Acta, 1989, 166, 47–54 CrossRef CAS; (b) J. B. Cooper, D. B. MacQueen, J. D. Petersen and D. W. Wertz, Inorg. Chem., 1990, 29, 3701–3705 CrossRef CAS.
- K. Kalyanasundaram and Md. K. Nazeeruddin, Inorg. Chem., 1990, 29, 1888–1897 CrossRef CAS.
- (a) P. Ceroni, F. Paolucci, C. Paradisi, A. Juris, S. Roffia, S. Serroni, S. Campagna and A. J. Bard, J. Am. Chem. Soc., 1998, 120, 5480–5487 CrossRef CAS; (b) M. Marcaccio, F. Paolucci, C. Paradisi, S. Roffia, C. Fontanesi, L. J. Yellowlees, S. Serroni, S. Campagna, G. Denti and V. Balzani, J. Am. Chem. Soc., 1999, 121, 10081–10091 CrossRef CAS; (c) M. Carano, P. Ceroni, C. Fontanesi, M. Marcaccio, F. Paolucci, C. Paradisi and S. Roffia, Electrochim. Acta, 2001, 46, 3199–3206 CrossRef CAS; (d) H. B. Baudin, J. Davidsson, S. Serroni, A. Juris, V. Balzani, S. Campagna and L. J. Hammarström, Phys. Chem. A, 2002, 106, 4312–4319 CrossRef CAS.
- D. S. Seneviratne, Md. J. Uddin, V. Swayambunathan, H. B. Schlegel and J. F. Endicott, Inorg. Chem., 2002, 41, 1502–1517 CrossRef CAS PubMed.
- (a) D. M. D'Alessandro, A. C. Topley, M. S. Davies and F. R. Keene, Chem. – Eur. J., 2006, 12, 4873–4884 CrossRef PubMed; (b) D. M. D'Alessandro, P. H. Dinolfo, M. S. Davies, J. T. Hupp and F. R. Keene, Inorg. Chem., 2006, 45, 3261–3274 CrossRef PubMed.
- Q.-X. Zhou, W.-H. Lei, Y. Sun, J.-R. Chen, C. Li, Y.-J. Hou, X.-S. Wang and B.-W. Zhang, Inorg. Chem., 2010, 49, 4729–4731 CrossRef CAS PubMed.
- L. Wen, J. P. Nietfeld, C. M. Amb and S. C. Rasmussen, J. Org. Chem., 2008, 73, 8529–8536 CrossRef CAS PubMed.
- L. Wen and S. C. Rasmussen, J. Chem. Crystallogr., 2007, 37, 387–398 CrossRef CAS.
- Z. Zhao, D. D. Wisnoski, S. E. Wolkenberg, W. H. Leister, Y. Wang and C. W. Lindsley, Tetrahedron Lett., 2004, 45, 4873–4876 CrossRef CAS.
- S. C. Rasmussen, M. M. Richter, E. Yi, H. Place and K. J. Brewer, Inorg. Chem., 1990, 29, 3926–3932 CrossRef CAS.
- X. Cai, M. P. Donzello, E. Viola, C. Rizzoli, C. Ercolani and K. M. Kadish, Inorg. Chem., 2009, 48, 7086–7098 CrossRef CAS PubMed.
- M. I. J. Polson, S. L. Howell, A. H. Flood, A. K. Burrell, A. G. Blackman and K. C. Gordon, Polyhedron, 2004, 23, 1427–1439 CrossRef CAS.
- T. E. Anderson, E. W. Culver, F. Almyahi, P. C. Dastoor and S. C. Rasmussen, Synlett, 2018, 2542–2546 CAS.
- K. L. Konkol and S. C. Rasmussen, Organometallics, 2016, 35, 3234–3239 CrossRef CAS.
- N. E. Tokel-Takvoryan, R. E. Hemingway and A. J. Bard, J. Am. Chem. Soc., 1973, 95, 6582–6589 CrossRef CAS.
- D. M. D'Alessandro and F. R. Keene, Dalton Trans., 2004, 3950–3954 RSC.
- M. E. Marmion and K. J. Takeuchi, J. Am. Chem. Soc., 1988, 110, 1472–1480 CrossRef CAS.
- R. C. Larson, R. T. Iwamoto and R. N. Adams, Anal. Chim. Acta, 1961, 25, 371–374 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, X-ray crystallographic and electrochemical data. CCDC 2363376 and 2363377. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02495h |
| This journal is © The Royal Society of Chemistry 2024 |