DOI:
10.1039/D4DT02386B
(Paper)
Dalton Trans., 2024,
53, 17620-17628
Utilisation of in situ formed cyano-bridged coordination polymers as precursors of supported Ir–Ni alloy nanoparticles with precisely controlled compositions and sizes†
Received
22nd August 2024
, Accepted 30th September 2024
First published on 15th October 2024
Abstract
Ir–Ni alloys supported on SiO2 have been reported to show high catalytic activity for styrene hydrogenation; however, precise control of compositions and sizes of the Ir–Ni alloys is difficult when conventional metal salts are used as precursors. Furthermore, the concomitant formation of unalloyed Ni nanoparticles disturbs quantitative discussion about Ir–Ni alloy compositions. We report herein a preparation method of Ir–Ni alloys with precisely controlled compositions on SiO2 using Ni(NO3)2 and an Ir complex possessing CN− ligands, [Ir(CN)6]3− or [Ir(ppy)2(CN)2]− (ppy = 2-phenylpyridine), as precursors. The in situ formation of cyano-bridged coordination polymers involving Ir and Ni promotes the formation of Ir–Ni alloys, whose compositions are virtually the same as expected from the amounts of Ir and Ni used for the preparation, after heat treatment under H2. The use of [Ir(ppy)2(CN)2]− as the precursor resulted in the formation of smaller Ir–Ni alloy particles than those with [Ir(CN)6]3− related to the structures of the formed coordination polymers.
Introduction
Hydrogenation of olefins and aromatic compounds catalysed by supported metal catalysts is widely used in industrial processes for petro-, coal-, and fine-chemicals.1–9 For example, supported Pt- or Pd-based catalysts are most frequently used,10–12 and Rh- and Ir-based catalysts have also emerged as a new class of catalyst.13–17 The catalytic behaviour of metal catalysts can be modified by forming alloys with a foreign metal.18–23 Classically, the forms of alloys can be categorized into two groups: one is solid solutions in which two metals are completely disordered in a solid; and the second is intermetallic alloys in which two different metals are periodically aligned similarly to molecules. Recently, more sophisticated structures such as core–shell have been adopted for rational catalysis enhancement.24–27 The catalytic activity and selectivity of single-atom alloys attract much attention because of their high potential with precise control of electronic and geometric structures around the single atom.7,28–40
Single-atom alloys of Ni and Ir metals as guest and host metals, respectively (Ir–Ni alloy), loaded on SiO2 have been reported to exhibit high catalytic activity for styrene hydrogenation selective for ethylbenzene.41 The styrene hydrogenation is industrially important because styrene formed during gasoline pyrolysis provides troublesome gums, resulting in the choking of pipelines.42,43 The Ir–Ni alloy catalysts have been prepared by the sequential impregnation method using H2IrCl6 and Ni(NO3)2 as precursors in previous reports.41 The prepared Ir–Ni alloys supported on SiO2 exhibited catalytic activity for styrene hydrogenation that was significantly higher than that for unalloyed monometallic catalysts.41 A proposed reason for the high catalytic activity is the formation of half-hydrogenated styrene adsorbed on both a Ni single atom and an adjacent Ir atom surrounded by H species adsorbed on Ir metals (Scheme 1).44 As a result, the activation energy for the hydrogenation of styrene decreased by about 20 kJ mol−1.44 The composition of the Ir–Ni alloy has been reported to influence the catalytic activity, although the unalloyed Ni metal concomitantly generated on the SiO2 surfaces was catalytically inactive (Scheme S1†). The precise control of the Ir–Ni alloy composition is hardly possible when preparation methods with conventional precursors are used. In situ formation of multinuclear complexes containing both Ir and Ni by sequential impregnation could be a promising method to avoid the formation of unalloyed Ni metals.
 |
| | Scheme 1 The proposed catalytic mechanism for styrene hydrogenation on an Ir–Ni alloy.44 | |
Metal complexes with cyano ligands such as hexacyanometallates are known to coordinate with a foreign metal ion to form heterometallic multinuclear complexes.45–52 The cyano ligand can be removed by thermal treatments below ∼300 °C so that cyano-bridged complexes can be converted into metal oxides and/or alloys.53 For example, Fe–Co alloy/carbon composites have been reported to be prepared by in situ pyrolysis of cyano-bridged Fe and Co complexes with precisely controlled Fe/Co ratios.11 Therefore, cyano-bridged iridium and nickel complexes could be promising precursors for Ir–Ni alloys with precisely controlled compositions.
We report herein the preparation of Ir–Ni alloys supported on metal oxide supports (MAOB = SiO2, SiO2-Al2O3, TiO2) by using Ni(NO3)2 and an Ir complex, i.e., [Ir(CN)6]3− with six cyano ligands or [Ir(ppy)2(CN)2]− (ppy = 2-phenylpyridine) with two cyano ligands, as precursors (Scheme 2). First, a series of Ir–Ni alloys supported on SiO2 were prepared by using [Ir(CN)6]3−, which forms a three-dimensional reticular structure with Ni2+. Next, Ir–Ni alloys supported on MAOB were furnished by using [Ir(ppy)2(CN)2]−, which forms a one–dimensional linear structure with Ni2+. Moreover, the catalytic activity of Ir–Ni alloys supported on MAOB was evaluated for styrene hydrogenation. This is the first report using cyano-bridged Ir–Ni complexes as precursors of Ir–Ni alloys with precisely controlled compositions supported on metal oxides.
 |
| | Scheme 2 Partial structures of in situ formed coordination polymers containing Ni and (a) [Ir(CN)6]3− (Ir(CN)6) or (b) [Ir(ppy)2(CN)2]− (Ir(CN)2). | |
Experimental section
Materials
All chemicals were obtained from chemical companies and used without further purification unless otherwise noted. Potassium hexacyanoiridium(III) (K3[Ir(CN)6]) was obtained from Kishida Chemical Co., Ltd. Potassium cyanide (KCN), nickel(II) nitrate hexahydrate (Ni(NO3)2, 99.9%), acetonitrile (CH3CN), ethanol (C2H5OH, 99.5%), toluene, and n-dodecane were obtained from FUJIFILM Wako Pure Chemical Corporation. Iridium(III) chloride hydrate (IrCl3·xH2O), 2-phenyl pyridine (ppy) and tetra-n-butylammonium chloride (TBACl) were purchased from Tokyo Chemical Industry Co., Ltd. SiO2 obtained from Fuji Silysia Chemical Ltd was calcined before use in air at 973 K for 3 h. TiO2 and SiO2-Al2O3 were provided by the Catalysis Society of Japan (JRC-SAL-4 and JRC-TIO-4). Ion-exchange resin was purchased from the Organo Corporation. Ultrapure water was produced using a Thermo Fisher Scientific Barnstead™ Smart2Pure™, with an electrical conductance of 18.2 MΩ cm. (n-Bu4N)[Ir(ppy)2(CN)2] was synthesized using the reported procedure.54
Preparation of Ir–Ni/SiO2 using [Ir(CN)6]3− and Ni2+ as precursors (Ir(CN)6-Ni/SiO2-X)
A series of Ir–Ni/SiO2 catalysts using H3[Ir(CN)6] and Ni(NO3)2 as precursors were prepared by the sequential incipient wetness method. The calcined SiO2 (412 mg) impregnated with an aqueous solution containing H3[Ir(CN)6] (0.10 mmol, 330 μL), which was obtained by passing a K3[Ir(CN)6] solution through a cation exchange resin, was dried in vacuo at room temperature for 1 h. Then, the H3[Ir(CN)6]/SiO2 thus achieved was impregnated with an aqueous solution of Ni(NO3)2 (0.017–0.14 mmol, 330 μL), followed by drying in vacuo at room temperature for 1 h. The obtained powder was heated under H2 (30 mL min−1) at 773 K for 15 min just prior to catalytic tests. The series of catalysts was named Ir(CN)6-Ni/SiO2-X, where X indicates the amount of Ni relative to the total amounts of Ni and Ir used for the preparation (=Ni/(Ir + Ni)).
Preparation of Ir–Ni/SiO2 using [Ir(ppy)2(CN)2]− and Ni2+ as precursors (Ir(CN)2-Ni/SiO2-X)
Ir–Ni alloy catalysts supported on SiO2 were prepared by the incipient wetness method similar to the method used for Ir(CN)6-Ni/SiO2-X. [Ir(ppy)2(CN)2]−/SiO2 was prepared by impregnation of the calcined SiO2 (100 mg, calcined at 973 K, 3 h) with an acetonitrile solution of (n-Bu4N)[Ir(ppy)2(CN)2] (20.8 μmol, 80 μL). After drying in vacuo at room temperature for 1 h, [Ir(ppy)2(CN)2]−/SiO2 was then immersed in an acetonitrile solution of Ni(NO3)2 (3.6–45 μmol, 80 μL), followed by drying in vacuo at room temperature for 1 h. The obtained powder was reduced to form an Ir–Ni alloy under H2 (30 mL min−1) at 773 K for 15 min just prior to use. The series of catalysts was named Ir(CN)2-Ni/SiO2-X, where X indicates the amount of Ni relative to the total amounts of Ni and Ir used for the preparation (=Ni/(Ir + Ni)).
Preparation of Ir–Ni/SiO2-Al2O3 and Ir–Ni/TiO2 using [Ir(ppy)2(CN)2]− and Ni2+ as precursors (Ir(CN)2-Ni/MAOB-X)
Ir–Ni/SiO2-Al2O3 and Ir–Ni/TiO2 were prepared by the incipient wetness method used for Ir(CN)2-Ni/SiO2. A support of SiO2-Al2O3 or TiO2 (70 mg) calcined at 973 K for 3 h prior to use was impregnated with an acetonitrile solution of (n-Bu4N)[Ir(ppy)2(CN)2] (14.6 μmol, 60 μL), followed by drying in vacuo at room temperature for 1 h. Then, Ni was loaded on [Ir(ppy)2(CN)2]−/SiO2-Al2O3 or TiO2 by impregnation with an acetonitrile solution of Ni(NO3)2 (5.31–20.5 μmol, 60 μL), followed by drying in vacuo at room temperature for 1 h. The catalysts were reduced under H2 flow (30 ml min−1) at 773 K for 15 min just before use. The obtained catalysts were named Ir(CN)2-Ni/SiO2-Al2O3-X or Ir(CN)2-Ni/TiO2-X, where X indicates the relative amount of Ni to the total amounts of Ni and Ir used for the preparation (=Ni/(Ir + Ni)).
Catalytic activity for styrene hydrogenation
Catalytic activity was investigated in a stainless-steel autoclave with an inserted glass vessel. The typical reaction conditions of styrene hydrogenation were as follows: a reduced catalyst (20 or 50 mg) was added to a mixture of styrene (200 mmol) and methanol (30 g) in a glass vessel in air. Then, the glass vessel sealed in a stainless jacket was heated to 303 K under a pressurized H2 atmosphere (8 MPa) with magnetic stirring (500 rpm) for a certain time period. After the reaction, the resulting liquid phase was washed with ethanol (10 g). The products were analysed using an FID-GC equipped with a TC–WAX column. The products were quantified by using n-dodecane as an internal standard. The initial rates were calculated based on the formed amount of ethylbenzene divided by the reaction times, because no products other than ethylbenzene were detected.
Physical measurements
Powder X-ray diffraction (PXRD) patterns were recorded using a Rigaku MiniFlex 600. Incident X-ray radiation was produced by a Cu X-ray tube operating at 40 kV and 15 mA with Cu Kα radiation (λ = 1.54 Å). The scan rate was 1° min−1 from 2θ = 40–48°. Silicon was used as the internal standard, providing the (111) peak at 2θ = 47.46°. X-ray photoelectron spectroscopy (XPS) analyses were performed using a Shimadzu ESCA-3400HSE. The incident radiation was Mg-Kα X-rays (1253.6 eV) at 200 W. The samples were mounted on a stage with double-sided carbon tape. The binding energy of each element was corrected by the C 1s peak (284.6 eV) from the carbon tape. TEM images of nanoparticles mounted on a copper microgrid coated with elastic carbon were obtained using a JEOL JEM-2100 operated at 200 kV with an X-ray energy dispersive (EDX) spectrometer. Particle size was determined by averaging over 100 particles in TEM images. The atomic ratios of Ir and Ni were determined using a Malvern PANalytical Epsilon 1 X-ray fluorescence (XRF) spectrometer.
Results and discussion
In situ formation of Ir(CN)6-Ni and Ir(CN)2-Ni complexes on SiO2 confirmed by IR spectroscopy
The formation of cyano-bridged complexes was confirmed by mixing Ni2+ and [Ir(CN)6]3− or [Ir(ppy)2(CN)2]− in solution. The IR measurements of the resulting precipitates indicated the higher wavenumber shift of the CN stretching band compared with that of [Ir(CN)6]3− or [Ir(ppy)2(CN)2]− (Fig. S1†). The CN stretching band of K3[Ir(CN)6] that appeared at 2127 cm−1 shifted to 2175 cm−1 due to the formation of a CN-bridging structure.55 Similarly, the CN stretching band of (n-Bu4N)[Ir(ppy)2(CN)2] observed at 2092 and 2101 cm−1 shifted to 2122 cm−1 with a shoulder peak around 2100 cm−1 by the reaction with Ni2+. In addition, powder X-ray diffraction (PXRD) for the precipitates obtained by the reaction of [Ir(CN)6]3− with Ni2+ evidenced the formation of cubic structures, so called Prussian blue analogues, with a 3D reticular structure (Fig. S2†). The 1D structure of [Ir(ppy)2(CN)2]- with Ni2+ is appropriate under consideration of the molecular structure; however, further investigation is necessary.
A series of coordination polymers containing Ir and Ni with different Ni/Ir ratios was supported on SiO2 surfaces by the two-step impregnation method where [Ir(CN)6]3− was impregnated first and then Ni(NO3)2 in SiO2, because simple one-step impregnation may result in the formation of insoluble coordination polymers apart from the SiO2 surfaces. The in situ formation of a coordination polymer was confirmed by IR measurements (Fig. 1). The CN stretching band, ν(CN), of [Ir(CN)6]3− supported on SiO2 (Ir(CN)6/SiO2) appeared at the same position as K3[Ir(CN)6] at 2129 cm−1 (Fig. 1a).55 The impregnation of an aqueous Ni(NO3)2 solution with Ir(CN)6/SiO2, in which the ratio of Ni/(Ir + Ni) = 0.15, provided a new ν(CN) at 2179 cm−1 (Fig. 1b). Such a high wavenumber shift of ν(CN) is generally observed when the CN bridging structure forms;55 thus, the coordination polymer with an Ir–CN–Ni bridged structure formed on the SiO2 surfaces. Such a high wavenumber shift of ν(CN) was also observed when [Ir(ppy)2(CN)2]−/SiO2 (Ir(CN)2/SiO2) was impregnated with Ni(NO3)2, where the ν(CN) shifted to 2113 and 2117 cm−1 from the 2098 cm−1 observed for Ir(CN)2/SiO2 alone (Fig. 1c and d). Therefore, the two-step impregnation method allowed the formation of cyano-bridged coordination polymers containing Ir3+ and Ni2+ on SiO2. The cyano-bridged coordination polymers thus formed were reduced by H2 at higher temperatures to achieve Ir–Ni alloys.
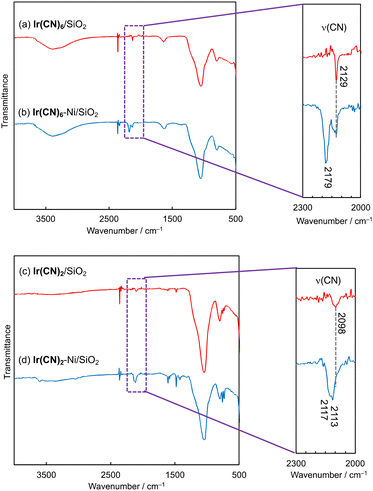 |
| | Fig. 1 IR spectra of (a) Ir(CN)6/SiO2, (b) Ir(CN)6-Ni/SiO2-0.15, (c) Ir(CN)2/SiO2 and (d) Ir(CN)2-Ni/SiO2. | |
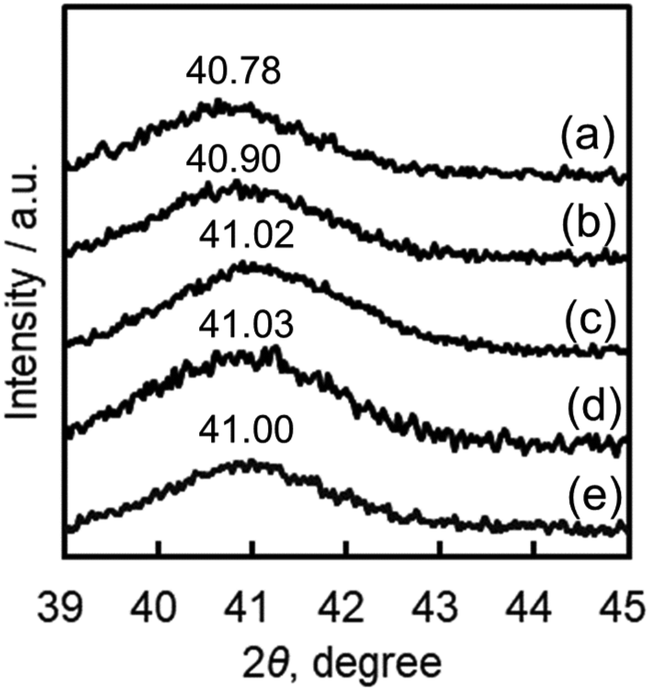
Compositions and sizes of Ir–Ni alloy particles in Ir(CN)6-Ni/SiO2-X
The loading amounts of Ir were fixed to 4.6% of SiO2 in a series of Ir(CN)6-Ni/SiO2-X where X represents the amounts of Ni relative to the total amounts of Ir and Ni [=Ni/(Ir + Ni)] in moles. The values of X determined by XRF were almost the same as expected from the used amounts of Ni and Ir precursors (Table 1). Then, the Ni contents in Ir–Ni alloys of a series of catalysts were estimated by powder X-ray diffraction (PXRD) (Fig. 2 and S3†). The (111) peak of Ir metal appearing at 40.70° was shifted to a higher angle (between 40.7° and 41.0°) by increasing X, indicating that the Ir–Ni alloys readily formed. The Ni contents in Ir–Ni alloys estimated from Vegard's law are tabulated in Table 1 together with the relative Ni amounts determined by XRF. The Ni contents in Ir–Ni alloys estimated from Vegard's law were almost the same as the X values for Ir(CN)6-Ni/SiO2-X with X = 0.05–0.15. However, no further increase of Ni contents in Ir–Ni alloys was observed with X values higher than 0.20. The maximum Ni content of 0.14 in Ir–Ni alloys implied that one Ni2+ ion was mainly surrounded by six [Ir(CN)6]3−, in which Ni/(Ir + Ni) was 0.14 (=1/(1 + 6)). No appearance of PXRD peaks assignable to Ni metal implied that Ni2+ ions not bound to [Ir(CN)6]3− were atomically dispersed on the SiO2 support or the formed amorphous phase.
 |
| | Fig. 2 Powder X-ray diffraction peaks of Ir(CN)6-Ni/SiO2-X [X = Ni/(Ni + Ir)] in the Ir(111) region. X = (a) 0.05, (b) 0.10, (c) 0.15, (d) 0.20 and (e) 0.25. | |
Table 1 The relative amounts of Ni to the total amounts of (Ir + Ni) in Ir(CN)6-Ni/SiO2-X determined by XRF, Ni contents in Ir–Ni alloys in Ir(CN)6-Ni/SiO2-X determined by PXRD, and the positions of PXRD peaks appearing around 40.70° assignable to the Ir(111) peak
| Catalyst |
Ni/(Ir + Ni) |
Peak position (2θ / °) |
| XRF |
PXRD |
|
Ir(CN)
6
-Ni/SiO2-0.05 |
0.05 |
0.05 |
40.78 |
|
Ir(CN)
6
-Ni/SiO2-0.10 |
0.11 |
0.10 |
40.90 |
|
Ir(CN)
6
-Ni/SiO2-0.15 |
0.15 |
0.14 |
41.02 |
|
Ir(CN)
6
-Ni/SiO2-0.20 |
0.19 |
0.14 |
41.03 |
|
Ir(CN)
6
-Ni/SiO2-0.25 |
0.25 |
0.14 |
41.00 |
The Ir–Ni alloys formed on SiO2 were observed by TEM. The sizes estimated by Scherrer's equation were as large as ∼50 nm based on the peak appearing around 40.70° in PXRD. TEM observations of Ir(CN)6-Ni/SiO2-0.20 indicated that the sizes of the Ir–Ni alloys were widely distributed from 5 to 70 nm (Fig. 3 and S4a†). Thus, the size of the Ir–Ni alloy nanoparticles was hardly controlled by using [Ir(CN)6]3− as a precursor although the compositions could be well controlled.
 |
| | Fig. 3 Transmission electron microscope (TEM) images of Ir(CN)6-Ni/SiO2-0.20 in (a) wide and (b) magnified views. | |
The composition uniformity of Ir–Ni alloy particles was further confirmed by the EDX line spectra for multiple single particles in TEM observations. The Ni contents in the three single particles of Ir(CN)6-Ni/SiO2-0.4 were 0.17, 0.19 and 0.13, which were close to the maximum Ni content of 0.14 determined by PXRD measurements (Fig. S5†). The frequency of smaller particles in Ir(CN)6-Ni/SiO2-0.4 increased compared with that in Ir(CN)6-Ni/SiO2-0.2, suggesting that excess Ni formed unalloyed small Ni particles (Fig. S4†).
Catalytic activity of Ir(CN)6-Ni/SiO2-X for styrene hydrogenation
The catalytic activity of Ir(CN)6-Ni/SiO2-X was evaluated for styrene hydrogenation performed in an autoclave at 303 K for 15–60 min under a pressurised hydrogen atmosphere (8 MPa) (Fig. 4a). No products other than ethylbenzene were detected in solution. The concentrations of the formed ethylbenzene were linearly increased up to 60 min for a series of Ir(CN)6-Ni/SiO2-X (Fig. 4b).
 |
| | Fig. 4 (a) Reaction conditions of catalytic styrene hydrogenation. Catalytic activity of Ir(CN)6-Ni/SiO2-X for styrene hydrogenation. (b) Time courses of Ir(CN)6-Ni/SiO2-0.05 (×), Ir(CN)6-Ni/SiO2-0.10 (▲), Ir(CN)6-Ni/SiO2-0.15 (■), Ir(CN)6-Ni/SiO2-0.20 (◆) and Ir(CN)6-Ni/SiO2-0.25 (●). (c) Initial reaction rates (<60 min) plotted against the relative amounts of Ni used for the preparation (X). | |
The initial rates determined from the slopes of the plots (=formed ethylbenzene concentrations divided by reaction times) increased with increasing the X of Ir(CN)6-Ni/SiO2-X up to 0.15, while no further increase was observed when X was higher than 0.15 (Fig. 4c). Therefore, an increase of Ni contents in the Ir–Ni alloy is critical to achieve catalytic activity, and unalloyed Ni species deposited on SiO2 are inactive for the reaction.26 Further increase of Ni contents to higher than 0.15 in the Ir–Ni alloys is promising to enhance the catalytic activity.
Furthermore, the ethylbenzene formation rates lower than 0.09 mmol L−1 s−1 with Ir(CN)6-Ni/SiO2-X catalysts were inferior to that observed with Ir–Ni/SiO2 prepared by a conventional impregnation method, 0.52 mmol L−1 s−1,41 because of the larger sizes of the Ir–Ni particles. The formation of larger particles was partly due to the easy formation of coordination polymers with a 3D reticular structure at higher Ni contents. Therefore, an Ir precursor to form a non-reticular structure with Ni2+ seems to be promising to reduce the size of Ir–Ni nanoparticles. [Ir(ppy)2(CN)2] (ppy = 2-phenylpyridine) would be an appropriate candidate because only two CN− ligands are available for polymerization with Ni2+ (Scheme 2b). The ppy chelate ligand strongly directs the formation of 1D polymer chains by disturbing the formation of a 3D reticular structure.
Compositions and particles sizes of Ir–Ni alloy particles in Ir(CN)2-Ni/SiO2-X
A series of Ir(CN)2-Ni/SiO2-X catalysts was prepared by using [Ir(ppy)2(CN)2]− as a precursor for reducing the particle sizes of Ir–Ni alloys. The values of X determined by XRF agreed well with the used amounts of Ir and Ni precursors within experimental error for a series of Ir(CN)2-Ni/SiO2-X (Table 2). PXRD measurements of the series of catalysts indicated that the peaks appearing around 41° became broader compared with those of Ir(CN)6-Ni/SiO2-X (Fig. 5 and S6†). Two possible reasons for the peak broadening are the formation of small-sized crystallites and alloy formation with gradually changed compositions. The particle sizes of the Ir–Ni alloys in a series of Ir(CN)2-Ni/SiO2-X estimated by Scherrer's equation were smaller than ∼10 nm, although the particle size depends on the X value. For example, the estimated sizes of the Ir–Ni alloys of Ir(CN)2-Ni/SiO2-0.10 and -0.15 were as small as 6.8 and 1.9 nm, respectively. The average particle sizes of Ir–Ni alloys confirmed by the TEM observation were ∼8.5(±4.4) and ∼4.4(±2.4) nm, respectively (Fig. 6 and S7†). The small difference in the sizes determined by PXRD and TEM for each sample demonstrates the homogeneity of Ir–Ni alloy compositions. The Ni contents in Ir–Ni alloys were estimated by PXRD (Table 2 and Fig. 5). The maximum Ni concentration in the Ir–Ni alloys estimated from Vegard's law reached 0.29, which is double that obtained for the catalyst prepared by using [Ir(CN)6]3− and Ni2+ as precursors. The maximum Ni contents suggested the formation of linear Ni[Ir(ppy)2(CN)2]2, in which the maximum theoretical Ni content was 0.33. The in situ formation of the one-dimensional coordination polymer is beneficial not only for reducing the size of Ir–Ni alloy nanoparticles but also for increasing the maximum Ni contents in the Ir–Ni alloy.
 |
| | Fig. 5 Powder X-ray diffraction peaks of (a) Ir(CN)2-Ni/SiO2-0.05, (b) Ir(CN)2-Ni/SiO2-0.10, (c) Ir(CN)2-Ni/SiO2-0.15, (d) Ir(CN)2-Ni/SiO2-0.20, (e) Ir(CN)2-Ni/SiO2-0.3, (f) Ir(CN)2-Ni/SiO2-0.4 and (g) Ni/SiO2 as a reference, in the Ir(111) region. | |
 |
| | Fig. 6 Transmission electron microscope (TEM) images of (a and b) Ir(CN)2-Ni/SiO2-0.1 and (c and d) Ir(CN)2-Ni/SiO2-0.15. The white arrows indicate the Ir–Ni particles. The white arrows indicate the Ir–Ni alloy particles. | |
Table 2 The amounts of Ni relative to the total amounts of (Ir + Ni) in Ir(CN)2-Ni/SiO2-X determined by XRF, Ni contents in Ir–Ni alloys in Ir(CN)2-Ni/SiO2-X determined by PXRD, the positions of PXRD peaks appearing around 40.70° assignable to the Ir(111) peak, and particles sizes estimated from the PXRD line width
| Catalysts |
Ni/(Ir + Ni) |
Peak position (2θ /°) |
Particle size/nm |
| XRF |
PXRD |
|
Ir(CN)
2
-Ni/SiO2-0.05 |
0.05 |
0.07 |
41.00 |
9.0 |
|
Ir(CN)
2
-Ni/SiO2-0.1 |
0.10 |
0.11 |
41.11 |
6.8 |
|
Ir(CN)
2
-Ni/SiO2-0.15 |
0.14 |
0.15 |
41.32 |
1.9 |
|
Ir(CN)
2
-Ni/SiO2-0.2 |
0.18 |
0.20 |
41.53 |
2.8 |
|
Ir(CN)
2
-Ni/SiO2-0.3 |
0.29 |
0.29 |
41.89 |
10.1 |
|
Ir(CN)
2
-Ni/SiO2-0.4 |
0.38 |
0.25 |
41.72 |
8.1 |
The Ni contents of three single particles of Ir(CN)2-Ni/SiO2-0.4 were 0.23, 0.22 and 0.24, which were close to the 0.25 determined by PXRD measurements (Fig. S8†). The formation of particles larger than 12 nm was rarely observed in Ir(CN)2-Ni/SiO2-0.4, also supporting the formation of smaller Ni particles (Fig. 7). These results suggest that the Ir–Ni particles formed on SiO2 surfaces have uniform Ni contents, and the excess Ni formed tiny Ni particles.
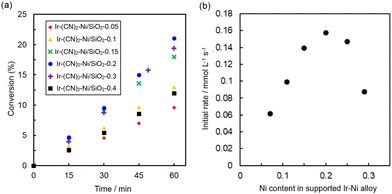 |
| | Fig. 7 Catalytic activity of Ir(CN)2-Ni/SiO2-X for styrene hydrogenation. (a) Time course of styrene conversion catalysed by Ir(CN)2-Ni/SiO2-0.05 (◆), Ir(CN)2-Ni/SiO2-0.1 (▲), Ir(CN)2-Ni/SiO2-0.2 (×), Ir(CN)2-Ni/SiO2-0.2 (●), Ir(CN)2-Ni/SiO2-0.3 (+) and Ir(CN)2-Ni/SiO2-0.4 (■). (b) Initial rates as a function of Ni contents (=Ni/(Ir + Ni)) in the Ir–Ni alloys determined by PXRD. | |
Catalysis evaluation of Ir(CN)2-Ni/SiO2-X for styrene hydrogenation
The catalytic activity of a series of Ir(CN)2-Ni/SiO2-X was evaluated for styrene hydrogenation. The initial rates were determined from the ethylbenzene formation rates for 60 min (Fig. 7). The maximum initial reaction rates increased to almost double those of Ir(CN)6-Ni/SiO2-X. The higher catalytic activity of Ir(CN)2-Ni/SiO2-X compared with that of Ir(CN)6-Ni/SiO2-X indicates that the smaller size of Ir–Ni alloy particles is beneficial for achieving high catalytic activity. The initial rate became higher with increasing the Ni contents in the Ir–Ni alloy up to 0.20, reaching the maximum initial rate of 0.16 mmol L−1 s−1 (Fig. 7b). However, the initial rates decreased at higher Ni contents in the Ir–Ni alloys. The higher Ni contents could reduce the number of isolated Ni atoms on Ir metal surfaces.44
Support effect of Ir(CN)2-Ni/MAOB-X
The particle sizes were rather small for Ir(CN)2-Ni/SiO2 compared with Ir(CN)6-Ni/SiO2; however, the enhancement of the initial rate was less than double, indicating that a strong support effect can be expected for smaller Ir–Ni nanoparticles. Thus, Ir–Ni alloys loaded on other supports, MAOB (SiO2-Al2O3 or TiO2), were examined for a series of catalysts prepared with [Ir(ppy)2(CN)2]− as a precursor. The PXRD measurements for a series of Ir(CN)2-Ni/MAOB-X indicated that the Ni concentrations in Ir–Ni alloys estimated by Vegard's law agreed with those expected from the amounts used for the catalyst preparation (Fig. 8). The Ni contents in Ir–Ni alloys are summarised in Table 3, although the diffraction peaks were broad for Ir(CN)2-Ni/SiO2-Al2O3-X. The nanoparticles of Ir–Ni alloys formed on Ir(CN)2-Ni/SiO2-Al2O3-0.2 and Ir(CN)2-Ni/TiO2-0.2 were observed by TEM as shown in Fig. 9. The Ir–Ni alloys formed on each support are indicated by the white arrows. The Ir–Ni alloys are tiny nanoparticles smaller than 2 nm in Ir(CN)2-Ni/SiO2-Al2O3-0.2. The average particle size is 5.1(±2.6) nm as estimated from the TEM image of Ir–Ni/SiO2-Al2O3-0.2 (Fig. 9a and S9†). The size is almost the same as that calculated using the Scherrer equation with the PXRD peak width. In addition, islands smaller than 20 nm were observed for Ir(CN)2-Ni/TiO2-0.2 (Fig. 9b). Thus, Ir–Ni alloys with precisely controlled compositions and small sizes were obtained by using [Ir(ppy)2(CN)2]− as a precursor regardless of supports.
 |
| | Fig. 8 Powder X-ray diffraction peaks of Ir(CN)2-Ni/MAOB-X [MAOB-X = (a) SiO2-Al2O3-0.1, (b) SiO2-Al2O3-0.2, (c) SiO2-Al2O3-0.3, (d) TiO2-0.1, (e) TiO2-0.2, (f) TiO2-0.21, and (g) TiO2-0.3] in the Ir(111) region. | |
 |
| | Fig. 9 Transmission electron microscopy (TEM) images of (a) Ir(CN)2-Ni/SiO2-Al2O3-0.2 and (b) Ir(CN)2-Ni/TiO2-0.20. The white arrows indicate the Ir–Ni alloy particles. | |
Table 3 The amounts of Ni relative to the total amounts of (Ir + Ni) in Ir(CN)2-Ni/MAOB-X determined by XRF, Ni contents in Ir–Ni alloys in Ir(CN)2-Ni/MAOB-X determined by PXRD, and the positions of PXRD peaks appearing around 40.70° assignable to the Ir(111) peak
| Catalyst |
Ni/(Ir + Ni) |
Peak position (2θ /°) |
| XRF |
PXRD |
|
Ir(CN)
2
-Ni/SiO2-Al2O3-0.1 |
0.10 |
0.10 |
41.01 |
|
Ir(CN)
2
-Ni/SiO2-Al2O3-0.2 |
0.20 |
0.18 |
41.42 |
|
Ir(CN)
2
-Ni/SiO2-Al2O3-0.3 |
0.30 |
0.28 |
41.48 |
|
Ir(CN)
2
-Ni/TiO2-0.1 |
0.10 |
0.11 |
41.13 |
|
Ir(CN)
2
-Ni/TiO2-0.18 |
0.20 |
0.18 |
41.43 |
|
Ir(CN)
2
-Ni/TiO2-0.21 |
0.21 |
0.21 |
41.32 |
|
Ir(CN)
2
-Ni/TiO2-0.3 |
0.30 |
0.29 |
41.53 |
Catalysis of Ir(CN)2-Ni/MAOB-X for styrene hydrogenation
The catalytic activity of Ir(CN)2-Ni/MAOB-X was evaluated for styrene hydrogenation. The initial rate calculated from the conversions for 60 min became the maximum at a Ni concentration of around 0.2 (Fig. 10). Such a volcano-type relationship between the initial rate and the Ni contents in Ir–Ni alloys is similar to that observed for Ir(CN)2-Ni/SiO2-X, although Ir(CN)2-Ni/SiO2-X catalysts were more active than Ir(CN)2-Ni/SiO2-Al2O3-X and Ir(CN)2-Ni/TiO2-X. In particular, the catalytic activity of Ir(CN)2-Ni/TiO2-X was much lower than those of Ir(CN)2-Ni/SiO2-X and Ir(CN)2-Ni/SiO2-Al2O3-X. The lower catalytic activity seemed to be due to larger island sizes; however, Ir–Ni alloy particles in Ir(CN)6-Ni/SiO2-X larger than 50 nm showed a certain activity. Therefore, the electronic states of Ir and Ni on TiO2 and SiO2 seemed to be more important than particle sizes.
 |
| | Fig. 10 (a) Time profiles of styrene conversion in styrene hydrogenation catalysed by Ir(CN)2-Ni/SiO2-Al2O3-X (X = 0.1 (■), 0.2 (●) and 0.3 (▲)) and Ir(CN)2-Ni/TiO2-X (X = 0.1 (+), 0.18 (×), 0.21 (−) and 0.3 (◆)) at 30 °C under H2 (8 MPa). (b) Initial rates for ethylbenzene formation as a function of Ni concentrations (=Ni/(Ir + Ni)) in Ir–Ni alloys of Ir(CN)2-Ir/MOx (MOx = SiO2 (●), SiO2-Al2O3 (▲) and TiO2 (■)). | |
The electronic states of Ir and Ni were investigated by X-ray photoelectron spectroscopy (XPS). The XPS measurements of Ir(CN)2-Ni/SiO2-0.20 revealed that Ir 4f7/2 and Ir 4f5/2 peaks appeared at 60.8 and 63.7 eV (Fig. 11a). The binding energy for Ir 4f7/2 lower than 61 eV evidenced that the Ir species are in the metallic state.56 Also, Ni 2p3/2 and Ni 2p1/2 peaks of Ir(CN)2-Ni/SiO2-0.20 appeared at 855.1 and 872.3 eV (Fig. 11b). The peak separation of 17.2 eV is close to that reported for Ni metal, 17.4 eV, rather than the 18.4 eV reported for NiO.56 However, the binding energy for 2p3/2 is more positive than that reported for Ni metal as 852.3 eV by 2.8 eV. Usually, such a high binding energy shift indicates the species are in higher oxidation states; however, intense satellite peaks characteristic for NiO species are missing. Therefore, the Ni species of Ir(CN)2-Ni/SiO2-0.20 are metallic but positively charged due to the higher electronegativity of Ir metal (2.22) than that of Ni metal (1.91).57
 |
| | Fig. 11 XPS spectra of Ir 4f and Ni 2p regions of (a and b) Ir(CN)2-Ni/SiO2-0.20 and (c and d) Ir(CN)2-Ni/TiO2-0.20. | |
Deconvolution of the overlapped Ir 4f and Ti 3s peaks of Ir(CN)2-Ni/TiO2-0.20 indicated that Ir 4f7/2 and Ir 4f5/2 peaks appeared at 61.4 and 64.6 eV (Fig. 11c). The Ir 4f peaks were shifted to a higher binding energy region by 0.6 and 0.9 eV compared with those of Ir 4f7/2 and 4f5/2 of Ir(CN)2-Ni/SiO2-0.20, indicating that the Ir metal on TiO2 is in a higher oxidation state than that on SiO2. The catalytic role of Ir metal has been reported as providing hydrogen atoms to an adsorbed styrene on a Ni atom.44 The electron deficient state of Ir results in deceleration of the dissociation of hydrogen molecules to hydrogen atoms. The Ni 2p3/2 peak of Ir(CN)2-Ni/TiO2-0.20 appears at 854.7 eV, which is lower than that of Ir(CN)2-Ni/SiO2-0.20 by 0.4 eV. The peak position of Ni 2p1/2 was uncertain due to overlapping with the Ti L3M23M23 Auger peak; however, the Ni in Ir(CN)2-Ni/TiO2-0.20 is more negatively charged than that on SiO2. The observed strong support effects could result from the electronic state change occurring in Ir–Ni alloys.
Conclusions
Ir–Ni alloy catalysts with precisely controlled compositions were prepared on SiO2 by using [Ir(CN)6]3− or [Ir(ppy)2(CN)2]− as an Ir precursor. Ni2+ was alloyed after reduction of in situ formed coordination polymers on the support. However, the maximum Ni contents in Ir–Ni alloys were limited to 0.14 and 0.29 with [Ir(CN)6]3− and [Ir(ppy)2(CN)2]−, respectively, because of compositions of coordination polymers thus formed. The particles sizes of Ir–Ni alloys using [Ir(ppy)2(CN)2]− as the precursor were smaller than those using [Ir(CN)6]3−, indicating that a one-dimensional structure is more suitable to prepare smaller Ir–Ni particles than a three dimensional structure. The reduction of Ir–Ni particles sizes enhanced the catalytic activity for styrene hydrogenation. The precise control of compositions and sizes of Ir–Ni particles was also possible for TiO2 and SiO2-Al2O3 supports. Thus, an appropriate choice of coordination polymers as precursors allows control of the compositions and sizes of alloy nanoparticles on metal oxide supports.
Author contributions
Y.Y.: conceptualization, methodology, writing – original draft, and writing – review and editing. M.N.: investigation, methodology, and writing – review and editing. T. Nakabayashi: investigation, methodology, and writing – review and editing. T. Nakazono: investigation, methodology, and writing – review and editing. H. L.: investigation, methodology, and writing – review and editing. P. C.: investigation, methodology, and writing – review and editing. M. T.: conceptualization, methodology, and writing – review and editing.
Data availability
Supporting data of this study are available within the paper. Further details regarding the data are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Grant-in-Aid for Scientific Research (B) (grant 22H01871) and (C) (grant 23K04770) and the Grant-in-Aid for Specially Promoted Research (23H05404) from the JSPS, University fellowship creation project for creating scientific and technological innovation (JPMJFS2138) from JST, and Strategic Reserach Promotion Project (Priority Research) 2024 from Osaka Metropolitan University.
References
- T. Li, D. Jia, S. Zhou, Z. Liu, J. Chen, T. Ban, A. Li, H. Li and H. Gao, Fuel, 2024, 373, 132329 CrossRef CAS.
- D. Zhao, X. Li, K. Zhang, J. Guo, X. Huang and G. Wang, Coord. Chem. Rev., 2023, 487, 215159 CrossRef CAS.
- J.-Y. Yan and G.-P. Cao, Macromolecules, 2023, 56, 3774–3808 CrossRef CAS.
- A. N. Marianov, Y. Jiang, A. Baiker and J. Huang, Chem. Catal., 2023, 3, 100631 CrossRef.
- B. Lakshminarayana, M. Selvaraj, G. Satyanarayana and C. Subrahmanyam, Catal. Rev., 2022, 66, 259–342 CrossRef.
- K. Liu, R. Qin and N. Zheng, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- M. A. Stoffels, F. J. R. Klauck, T. Hamadi, F. Glorius and J. Leker, Adv. Synth. Catal., 2020, 362, 1258–1274 CrossRef CAS PubMed.
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- S. Zhang, J. Li, Z. Xia, C. Wu, Z. Zhang, Y. Ma and Y. Qu, Nanoscale, 2017, 9, 3140–3149 RSC.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed.
- Y. Wu, S. Cai, D. Wang, W. He and Y. Li, J. Am. Chem. Soc., 2012, 134, 8975–8981 CrossRef CAS PubMed.
- H. Wang, A. M. Fiore, C. Fliedel, E. Manoury, K. Philippot, M. M. Dell'Anna, P. Mastrorilli and R. Poli, Nanoscale Adv., 2021, 3, 2554–2566 RSC.
- M. Tamura, N. Hayashigami, A. Nakayama, Y. Nakagawa and K. Tomishige, ACS Catal., 2021, 12, 868–876 CrossRef.
-
L. M. Martínez-Prieto, I. Cano and P. W. N. M. van Leeuwen, in Iridium Catalysts for Organic Reactions, ed. L. A. Oro and C.Claver, Springer International Publishing, 2021, pp. 397–454 Search PubMed.
- S. Hanf, L. A. Rupflin, R. Gläser and S. Schunk, Catalysts, 2020, 10, 510 CrossRef CAS.
- F. Corvaisier, Y. Schuurman, A. Fecant, C. Thomazeau, P. Raybaud, H. Toulhoat and D. Farrusseng, J. Catal., 2013, 307, 352–361 CrossRef CAS.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- D. C. Zhong, Y. N. Gong, C. Zhang and T. B. Lu, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC.
- X. Cai, H. Wang, Y. Tian, W. Ding and Y. Zhu, ACS Catal., 2024, 14, 11918–11930 CrossRef CAS.
- L. B. Belykh, N. I. Skripov, T. P. Sterenchuk, T. A. Kornaukhova, E. A. Milenkaya and F. K. Schmidt, Mol. Catal., 2022, 528, 112509 CrossRef CAS.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- Z. P. Wu, S. Shan, S. Q. Zang and C. J. Zhong, Acc. Chem. Res., 2020, 53, 2913–2924 CrossRef CAS PubMed.
- K. Murugesan, V. G. Chandrashekhar, C. Kreyenschulte, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2020, 59, 17408–17412 CrossRef CAS PubMed.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed.
- J. Kim, H. E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS PubMed.
- L. Liu and A. Corma, Trends Chem., 2020, 2, 383–400 CrossRef CAS.
- J. Han, J. Lu, M. Wang, Y. Wang and F. Wang, Chin. J. Chem., 2019, 37, 977–988 CrossRef CAS.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS PubMed.
- M. T. Darby, R. Réocreux, E. C. H. Sykes, A. Michaelides and M. Stamatakis, ACS Catal., 2018, 8, 5038–5050 CrossRef CAS.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2011, 11, 49–52 CrossRef PubMed.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- H. Feng, W. Liu, L. Wang, E. Xu, D. Pang, Z. Ren, S. Wang, S. Zhao, Y. Deng, T. Liu, Y. Yang, X. Zhang, F. Li and M. Wei, Adv. Sci., 2024, 11, e2304908 CrossRef PubMed.
- Y. Chen, R. Zhang, Z. Chen, J. Liao, X. Song, X. Liang, Y. Wang, J. Dong, C. V. Singh, D. Wang, Y. Li, F. D. Toste and J. Zhao, J. Am. Chem. Soc., 2024, 146, 10847–10856 CrossRef CAS PubMed.
- Z. Wang, X. Zhou, G. Wang, Q. Tong, H. Wan and L. Dong, Inorg. Chem., 2024, 63, 7241–7254 CrossRef CAS PubMed.
- H. Yu, W. Tang, K. Li, S. Zhao, H. Yin and S. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 6958–6969 CrossRef CAS PubMed.
- J.-q. Bai, M. Tamura, Y. Nakagawa and K. Tomishige, Chem. Commun., 2019, 55, 10519–10522 RSC.
- Z. Zhou, T. Zeng, Z. Cheng and W. Yuan, Ind. Eng. Chem. Res., 2010, 49, 11112–11118 CrossRef CAS.
- T. A. Nijhuis, F. M. Dautzenberg and J. A. Moulijn, Chem. Eng. Sci., 2003, 58, 1113–1124 CrossRef CAS.
- J.-q. Bai, M. Tamura, A. Nakayama, Y. Nakagawa and K. Tomishige, ACS Catal., 2021, 11, 3293–3309 CrossRef CAS.
- H. Yang, J. Liu, Z. Chen, R. Wang, B. Fei, H. Liu, Y. Guo and R. Wu, Chem. Eng. J., 2021, 420, 127671 CrossRef CAS.
- T. G. U. Ghobadi, E. Ozbay and F. Karadas, Chem. – Eur. J., 2021, 27, 3638–3649 CrossRef PubMed.
- H. Matsuoka, O. Tomita, H. Tabe, H. Suzuki, Y. Yamada and R. Abe, J. Photochem. Photobiol., A, 2022, 426, 113753 CrossRef CAS.
- Y. Yamada, K. Oyama, R. Gates and S. Fukuzumi, Angew. Chem., Int. Ed., 2015, 54, 5613–5617 CrossRef CAS PubMed.
- Y. Yamada, K. Oyama, T. Suenobu and S. Fukuzumi, Chem. Commun., 2017, 53, 3418–3421 RSC.
- M. Yamane, H. Tabe, M. Kawakami, H. Tanaka, T. Kawamoto and Y. Yamada, Inorg. Chem., 2020, 59, 16000–16009 CrossRef CAS PubMed.
- Y. Seki, H. Tabe and Y. Yamada, J. Phys. Chem. C, 2022, 126, 5564–5574 CrossRef CAS.
- H. Tabe, S. Yorozu and Y. Yamada, J. Phys. Chem. C, 2022, 126, 4365–4373 CrossRef CAS.
- Y. Shan, G. Zhang, W. Yin, H. Pang and Q. Xu, Bull. Chem. Soc. Jpn., 2022, 95, 230–260 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, D. Berner, S. Rivier, L. Zuppiroli and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 8790–8797 CrossRef CAS PubMed.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th edn, Wiley Interscience, 1986 Search PubMed.
-
J. F. Moulder, J. K. Chastain and J. C. Roger, Handbook of X-ray Photoelectron Spectroscopy, 1995 Search PubMed.
- B. Bal, S. Ganguli and M. Bhattacharya, J. Phys. Chem., 1984, 88, 4575–4577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic drawing, IR, PXRD, EDX line scanning spectra, and the size distribution of Ir–Ni alloy particles observed by TEM. See DOI: https://doi.org/10.1039/d4dt02386b |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Miho
Nishida
a,
Tatsuya
Nakabayashi
a,
Takashi
Nakazono
*ab,
Miho
Nishida
a,
Tatsuya
Nakabayashi
a,
Takashi
Nakazono