
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA UV-Vis method for investigation of gallium(III) complexation kinetics with NOTA and TRAP chelators: advantages, limitations and comparison with radiolabelling†
Viktor
Lebruška
 a,
Tereza
Dobrovolná
a,
Tereza
Gemperle
a,
Vojtěch
Kubíček
*a,
Susanne
Kossatz
b and
Petr
Hermann
a
a,
Tereza
Dobrovolná
a,
Tereza
Gemperle
a,
Vojtěch
Kubíček
*a,
Susanne
Kossatz
b and
Petr
Hermann
a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: kubicek@natur.cuni.cz; Tel: +420221951436
bDepartment of Nuclear Medicine, TUM University Hospital and Central Institute for Translational Cancer Research (TranslaTUM), School of Medicine, Technical University Munich (TUM), Einsteinstrasse 25, 81675, Munich, Germany
First published on 3rd October 2024
Abstract
An easy and cheap method for measurement of GaIII complexation kinetics was developed. The method is based on UV-Vis quantification of non-complexed chelators after the addition of CuII ions at individual time points. The method was evaluated using established ligands, H3nota and H6notPPr, and was utilized to study the kinetics of GaIII complexation with four new symmetric derivatives of 1,4,7-triazacyclononane bearing methylphosphonate/phosphinate pendant arms – TRAP ligands. Chelators bearing ethoxy groups (H3L1) or 2,2,2-trifluoroethyl groups (H3L2) on the phosphorus atoms showed fast formation (t99% = 21 and 10 min, respectively, at pH 2.0) and efficient radiolabelling which were comparable to the previously reported chelators bearing the 2-carboxyethyl group (H6notPPr). Chelators bearing (N,N-dibenzyl-amino)methyl (H3L3) and aminomethyl (H3L4) substituents showed a significantly slower complexation (t99% = 4.4 and 3.6 h, respectively, at pH 2.0) and inefficient radiolabelling, mainly at room temperature or low pH. This was caused by protonation of the amino groups of the pendant arms leading to coulombic repulsion between the GaIII ion and the positively charged protonated amines. The trends in complexation rates determined by the UV-Vis method correlated well with the results of the 68Ga radiolabelling study.
Introduction
Current medicine uses various imaging techniques based on ionizing radiation. One of them is positron emission tomography (PET), a non-invasive technique used for selective imaging of tissues, typically in combination with magnetic resonance imaging (MRI) or computed tomography (CT). PET utilizes positron-emitting radioisotopes. The emitted positron annihilates with an electron from the surrounding tissue and generates a photon pair. The simultaneous formation and detection of two photons offers high sensitivity and resolution. Detection of the photons allows for the reconstruction of the radioisotope distribution in tissues.Production of the most common β+ radioisotopes is based on cyclotrons. This represents a drawback, due to the high costs of purchase and maintenance of the cyclotrons. Thus, portable radioisotope generators attract increasing attention. Among them, commercial 68Ga/68Ge generators provide a source of the positron emitter 68Ga (89% β+; τ1/2 = 67.7 min; Emax(β+) = 1.90 MeV) for PET imaging.1,2 They are available from several vendors and versatile in their use, making 68Ga one of the most widely used imaging radioisotopes.
The free GaIII aqua-ion exhibits non-specific deposition in tissues and, thus, it must be bound in thermodynamically stable and kinetically inert complexes which are often conjugated to biologically active vectors to ensure their specific accumulation in tissues. There are several important parameters for the chelator to be used as a 68Ga carrier, such as charge, hydrophilicity/hydrophobicity, very fast, selective and quantitative GaIII complexation, and solubility and stability in body fluids. To meet these requirements, the chelator must be finely designed in terms of the number and kind of donor groups, overall complex charge, geometry of the coordination sphere, etc.
Commonly used chelators for GaIII complexation are analogues of H4dota (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and H3nota (1,4,7-triazacyclononane-1,4,7-triacetic acid). However, H4dota is not optimal for GaIII complexation as its macrocyclic cavity is too large and the number of donor atoms exceeds the number (six) of GaIII coordination sites.3 In addition, GaIII complexes with H4dota and its derivatives often show slow complexation/radiolabelling and limited in vivo stability. In contrast, the compact ligand cavity of H3nota is well pre-organized for the octahedral coordination of small ions and a lot of chelators derived from 1,4,7-triazacyclononane (tacn) have been used for GaIII complexation.4,5 Promising properties were reported for tacn-based chelators with three methylphosphinate pendant arms, called TRAP chelators.6 The phosphinic acid pendant arms become deprotonated at a pH lower than the carboxylic pendants and ring nitrogen atoms of phosphinate-bearing macrocycles are less basic than those in acetate-bearing H3nota. Therefore, phosphonate/phosphinate chelators mostly coordinate GaIII faster and at a lower pH than their acetate analogue.7–10 Various TRAP chelators bearing different groups attached to the phosphorus atom have been synthesized and studied for complexation of the GaIII ion. The chelators bearing 2-carboxyethyl or hydroxomethyl groups (H6notPPr, H3notPhm and H4nopo in Fig. 1) showed a fast complexation and a very good radiolabelling efficiency,6,9,11 whereas GaIII binding by TRAP chelators bearing hydrogen atoms or phenyl groups (H3notPH and H3notPPh) was much less effective.12 However, factors influencing the GaIII complexation are not fully understood.
 | ||
| Fig. 1 Discussed chelators (the colors and pictograms serve an easier orientation in the Results section). | ||
As the complexation rate is a key property of chelators used as carriers of radioisotopes, significant attention is usually focused on the measurement of complexation kinetics. The most common method used to follow complexation kinetics is UV-Vis spectroscopy. However, the GaIII ion shows no UV-Vis absorption. The complexation kinetics of such complexes must be studied by a different method. Therefore, previously reported data on the formation of GaIII complexes were mostly obtained by 31P and/or 71Ga NMR spectroscopy.3,12–14 However, the NMR experiments require high concentrations of the studied compounds and long data acquisitions. The acquisition times are typically at least minutes and, thus, only reaction kinetics running in timespans of tenths of minutes can be investigated by NMR. In addition, kinetic experiments are time-consuming which is often incompatible with the available NMR time. Thus, the published kinetic data are scarce and only a few complexation mechanistic studies have been reported.12,14,15
Another option for studying the complexation of ions having no UV-Vis absorption is by using indirect methods using a “visualization agent”.16–19 The experiments were mostly performed by the addition of complexing dyes (chelatometric indicators) into the reaction mixture. However, in such arrangement, the dye must be used in excess and, thus, it binds the metal ion and changes its rate of complexation by the studied ligand. This problem is typically solved by variation of the dye concentration and the linear extrapolation of reaction rates to zero dye concentration. However, the extrapolation introduces significant uncertainty as the linearity might not be maintained at low dye concentrations.
In order to avoid the above-mentioned limitations, we developed a new method for UV-Vis quantification of the reaction progress. It is a batch method based on addition of CuII ions in excess to the reaction solution at individual time points. The complexation of CuII ions by the remaining free ligand quenches the GaIII complexation and the quantification of the formed CuII complex allows us to determine the GaIII complexation extent. The method was first evaluated using established ligands, H3nota and H6notPPr (Fig. 1) and, furthermore, it was utilized to study the kinetics of GaIII complexation with four new symmetric derivatives of tacn bearing methylphosphonate/phosphinate pendant arms – TRAP ligands – to evaluate the effect of the phosphorus atom substituents (Fig. 1). The ligands H3L1 bearing ethoxy groups (i.e. phosphonate monoester moiety), H3L2 2,2,2-trifluoro-ethyl groups and H3L3N,N-dibenzyl-aminomethyl groups are chelators with an increased lipophilicity. The effect of the hydrophilic and positively charged amino groups was evaluated comparing H3L3 and H3L4 bearing aminomethyl groups. To the best of our knowledge, this is the first work systematically studying the kinetics of the GaIII complexation.
Methodology
The UV-Vis method for investigation of GaIII complexation kinetics
In order to gather data necessary for the evaluation of GaIII complexation kinetics, we sought a suitable and cheap method, and UV-Vis spectroscopy is the method of the first choice. However, neither the GaIII ion nor the studied chelators showed any absorption bands in the UV-Vis region. Thus, an indirect method must be used. The previously reported indirect methods used dyes to visualize metal ions.16–19 The dyes were added directly to the reaction mixture so the metal ion was not present in the solution in a free (i.e. aqua-ion) form but as a complex with the dye. The metal ion–dye interaction significantly changed the availability of the metal ion for complexation by the investigated chelator. To overcome this issue, we propose a batch method in which the “visualization” agent was not added into the reaction mixture but it was added into the analyzed solution just before the UV-Vis measurement and it also quenches the studied complexation reaction. We first tested several dyes (e.g. Arsenazo III, xylenol orange) to visualize the “free” metal aqua-ion and none of them was suitable for the batch method due to a long time necessary to reach full equilibrium after addition of the dye. Finally, we developed a method, which did not visualize the free metal ion but the free ligand by the addition of a CuII ion. The added CuII was immediately bound by the free ligand resulting in the formation of a complex with significant UV-Vis absorption allowing precise quantification of the reaction extent.The method was first tested on the parent ligand, H3nota. The solution containing GaIII and H3nota was incubated at 30 °C under desired conditions, an aliquot of the solution was taken at each time point and quickly mixed with an excess of CuII ions. The CuII ion is known to show very fast complexation by H3nota.20,21 This reaction is several orders of magnitude faster than the reaction of H3nota with the GaIII ion12 and, thus, any free chelator immediately bound CuII ions. After the CuII addition, the complexation processes were immediately stopped and the extent of the reaction was evaluated by quantification of the CuII complex concentration which equaled the concentration of the free non-complexed chelator. Thus, the progress of the reaction with GaIII could be easily quantified from the intensity of the absorption band of the formed CuII complex. The ligand-to-metal charge transfer (LMCT) band at ∼270 nm was used for the quantification as it showed a significantly higher intensity than a broad d–d transition band in the visible region. An example of the original kinetic data and their treatment is shown in Fig. S1.†
After the addition of CuII salt, the metal ion was present in an excess over the chelator. Thus, possible transmetallations after the quenching of the reaction had to be excluded. To ensure this, each CuII and GaIII complex was treated with ten-times excess of the other metal ion and no transmetallation was observed for 30 min under the same conditions as those used for the quenching reactions; this time period provided enough time to perform the UV-Vis measurement of the solution. The CuII ion did not replace the GaIII ion in the complex due to thermodynamic reasons (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K[Ga(nota)] = 29.6,12 logK[Cu(nota)] = 23.320), whereas the GaIII ion did not replace the CuII ion in its complex due to kinetic reasons; GaIII complexation by the free ligand is relatively slow and it is even much slower if the ligand cavity is blocked by the CuII ion (the transmetallation reaction is extremely slow).
K[Ga(nota)] = 29.6,12 logK[Cu(nota)] = 23.320), whereas the GaIII ion did not replace the CuII ion in its complex due to kinetic reasons; GaIII complexation by the free ligand is relatively slow and it is even much slower if the ligand cavity is blocked by the CuII ion (the transmetallation reaction is extremely slow).
To demonstrate that the method can also be used for TRAP ligands and to independently validate the suggested UV-Vis method, GaIII complexation with H3L2 was followed at pH 1 by 1H NMR under identical conditions (Fig. S2†). Ligand H3L2 was chosen as the methylene signal of its trifluoroethyl group was not affected by excitation sculpting used to suppress the water signal. The rate constants were almost identical in both experiments (NMR: kobs = 2.8(2) × 10−4 s−1; UV-Vis: kobs = 2.7(2) × 10−4 s−1) and, thus, the direct NMR measurement fully confirmed the accuracy of the UV-Vis method used.
The developed method is not universal and has several limitations:
(i) The most important requirements are a suitable CuII complexation rate and transmetallation rates in both directions. The CuII complexation rate must be at least two orders of magnitude faster than that of the metal ion in question so that the progress of complexation with the studied metal ion is negligible during the formation of the CuII complex. Additionally, transmetallation must be slow enough to avoid any mutual transformation of the species during the sample manipulation and spectral measurements. The knowledge of CuII complexation and transmetallation kinetics does require additional preliminary experiments. However, detailed quantitative data are not necessary to obtain, and qualitative experiments can be easily and quickly obtained using UV-Vis measurements.
(ii) The ligand should form complexes of sufficient stability with both, CuII and the metal ion in question. However, the stability of the CuII complex might be lower than that of the studied metal ion as the decisive factors are the complexation and transmetallation rates.
(iii) Thermodynamic stability of the complexes is important for quantitative formation of complexes, mainly under strongly acidic conditions. This requirement must be met for the studied metal ion but it is not crucial for the CuII complex. Quenching of the reaction can be done with CuII excess and in solution buffered at a different pH from that of the kinetic experiments. This was also our approach. To ensure quantitative CuII complexation, CuII solution buffered at pH 3 was used for quenching the reaction and the consequent spectral measurements. Our experiments showed that there is no influence of pH of the CuII solution (tested in the range 2–4) on the data obtained.
(iv) Another factor is the ligand-to-metal stoichiometry. In the optimal case, the stoichiometry of the CuII complex and the complex of the studied metal ions should be the same. However, this is not an absolute requirement as the relative change in spectra is the measure of the reaction progress and it should not be dependent on the complex stoichiometry.
The method could be used not only for the investigation of the GaIII complexation but also for kinetic investigations of other metal ions having no UV-Vis absorption. The requirements described above indicate that the method shows high potential mainly for the complexation of trivalent metal ions with macrocyclic ligands. Trivalent metal ions form typically complexes much more slowly than CuII and their complexes with macrocyclic ligands are often kinetically inert and, thus, they undergo only slow dissociation and transmetallation reactions. The prospective members of this group are ScIII, YIII, InIII, LaIII or LuIII, all of them being of high radiochemical interest.
The high intensity of the CT band of the studied CuII complexes allows us to perform the experiments in millimolar or even sub-millimolar concentrations of the reactants. The method could be used for kinetic measurements performed at various ligand-to-metal ratios. There is no limit to the metal ion excess used. However, a high ligand excess would decrease the sensitivity of the method as the amount of GaIII-bound ligand is too small with respect to the overall amount of the ligand bound in the CuII complex formed after quenching. The limit of the method regarding rates of studied processes is given by the time required to transfer a precise volume of the studied solution to the solution containing CuII ions. This can be managed in a few seconds and allows the method to be used for investigation kinetics with half-times significantly lower than one minute. As mentioned earlier, the advantage of the described batch quenching method is that CuII ions are not present in the reaction mixture in the course of the investigated complexation process and, therefore, do not alter the complexation reaction and measured complexation rate. A disadvantage of the experiment is that it requires the continuous presence of the researcher through the measurements. However, this disadvantage could be overcome by automation using e.g. flow injection analysis.
In this work, we used the method to investigate the rates of complexation reactions of GaIII with H3nota and several phosphinic acid analogues. The H3nota derivatives and their complexes were expected to behave analogously to H3nota, except for the complexation rate. The phosphinic acid derivatives have a similar difference in GaIII and CuII stability constants as H3nota (e.g. logK[Ga(notPPr)] = 26.24 and logK[Cu(notPPr)] = 16.85)6 and the CuII–GaIII mutual transmetallation reactions are very slow. The method allowed the use of relatively low sub-millimolar concentrations of the reactants. As radiochemical labelling is typically performed under a large chelator excess, the measurements described in this work were performed under a ligand excess; however, only a two-fold ligand excess was used to maintain a high sensitivity of the measurements.
Results and discussion
Synthesis of chelators and GaIII complexes
Chelators were synthesized by Mannich-type reactions of the corresponding organophosphorus precursors with tacn (1,4,7-triazacyclononane) and paraformaldehyde (Fig. 2). All chelators were characterized by NMR, MS and EA (Fig. S3–S6†). The ligand H3L1 was prepared by a two-step procedure. In the first step, fully esterified intermediate 1 was synthesized by a Mannich-type reaction with an excess of triethyl phosphite (used as a solvent). Ion-exchange workup yielded a crude intermediate 1 (∼90% yield) which was subsequently treated with aq. NaOH to selectively hydrolyze one ester group. Purification on a strong cation exchanger followed by lyophilization yielded H3L1 as a hygroscopic powder in 47% yield (based on tacn). Ligand H3L1 was earlier synthesized by a different approach.22 In the first step, 1 was prepared by the reaction of tacn with diethyl phosphite and paraformaldehyde in benzene and the second step was similar to our procedure. Our method achieved a slightly higher yield than the reported one (41% based on tacn), while eliminating the use of carcinogenic benzene as a solvent.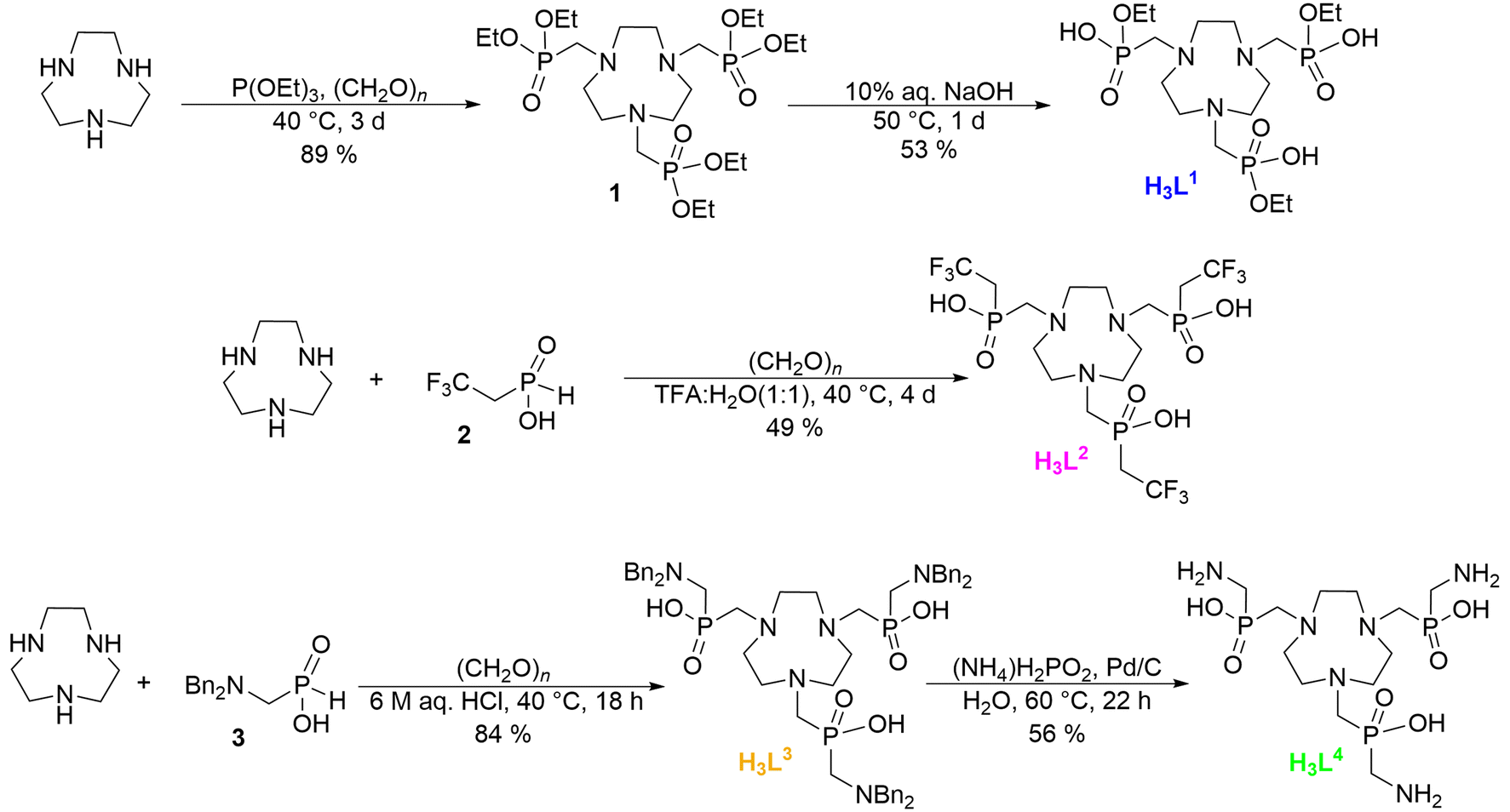 | ||
| Fig. 2 Synthesis of the studied chelators. | ||
The ligand H3L2 was synthesized in a single step according to a published procedure.23 Starting from tacn with a slight excess of (2,2,2-trifluoroethyl)-H-phosphinic acid (2) and paraformaldehyde, H3L2 was formed with 85% conversion according to 31P NMR. A side-product was the macrocycle bearing two pendant arms. After chromatography on silica and ion exchange workup, the product was obtained in 71% yield.
Synthesis of H3L3 and H3L4 started from [(N,N-dibenzyl)-aminomethyl]-H-phosphinic acid (3).24 Using a slight excess of 3 and paraformaldehyde, H3L3 was formed quantitatively according to 31P NMR and it was isolated in 84% yield after ion exchange workup and lyophilization. H3L4 was synthesized by reductive cleavage of the benzyl groups of H3L3 on the Pd/C catalyst. The reaction is very sensitive as the utilization of inappropriate conditions (temperature, H2 gas pressure, pH, solvent, hydrogen source) easily led to N–C–P fragment cleavage and pendant arm degradation. Quantitative debenzylation with minimal degradation was reached in diluted aqueous ammonia using ammonium hypophosphite as a hydrogen source and at room temperature. After catalyst filtration and ion exchange workup, H3L4 was isolated by lyophilization in 56% yield (based on H3L3). Solutions of Ga–L1 and Ga–L2 complexes were prepared by stirring a freshly prepared suspension of Ga(OH)3 in the chelator solution overnight. The Ga(OH)3 excess was filtered off through a microfilter, yielding stock solutions of the complexes. The Ga–L4 solution was prepared by mixing the chelator solution with an excess of Ga(NO3)3 at pH 3.5. Then, pH was raised to 5.6 leading to the precipitation of the excessive GaIII in the form of Ga(OH)3 which was filtered off. The solutions of Ga–L1, Ga–L2 and Ga–L4 complexes were characterized by NMR and HRMS (Fig. S7–S12†). Chelator H3L3 formed an insoluble complex with GaIII precipitating from the reaction mixture. The Ga–L3 complex was not characterized by spectroscopic techniques due to its very low solubility in common solvents.
1H NMR spectra of the studied GaIII complexes contained a number of split or broad signals (Fig. S7, S9 and S11†). This could be ascribed to the decreased ligand symmetry after the GaIII complexation and/or the presence and interconversion of isomers of the complexes. In complexes of NOTA-like ligands, the pendant arms are twisted clock-wise or anticlock-wise with the respect to the macrocrocycle chelate ring conformation and, thus, the complexes are chiral.25 As a result, the hydrogen atoms in all ligand methylene groups become nonequivalent. In addition, the coordination of phosphinates or phosphonate monoesters to a metal ion introduces other chirality centers on the phosphorus atoms.6,12,26–28 Thus, the complexes often form a mixture of diastereoisomers in solution that typically undergo mutual interconversion. The isomeric composition and the interconversion were difficult to study by 1H NMR due to the mentioned signal broadening and splitting. The 31P and 19F NMR (for Ga–L2) spectra provided better information on the speciation of the complexes in solution.
The Ga–L1 complex showed two major signals in the 31P NMR spectra at room temperature. These diastereomers could not be distinguished in 71Ga NMR due to the very similar coordination environment formed by the chelator in all species. The presence of two (or more) diastereomers also resulted in very complicated 1H NMR spectra. At an elevated temperature (90 °C, Fig. S7b†), the spectra became simpler confirming the mutual interconversion of the diastereomers. However, the broad signals showed that the interconversion proceeded on the NMR time scale even at an elevated temperature. In contrast, only one dominant signal was found in 31P NMR spectra of the Ga–L2 and Ga–L4 complexes. The different diastereomeric composition of complexes of each chelator could result from a mutual interaction of the pendant arms (e.g. with assistance of hydrogen bonds) or from the preferential formation of some diastereomers in the course of complexation which might be related to the geometry of complexation intermediates. The obtained results agreed with the previously reported formation of the diastereomeric mixture in the complexes of H3nota and H4dota analogues with the phosphonate monoester pendant arms, whereas complexes of ligands with the phosphinate pendants preferentially formed one diastereomer.12,27–29
Protonation constants of ligands
A key step of metal ion complexation by NOTA-like ligands is deprotonation of the macrocycle amino groups. Thus, the protonation constants of H3L1 and H3L4 were determined by potentiometry. The results are compiled and compared with data from the literature for other ligands in Tables 1 and S1.† The protonation constants of H3L3 were not determined due to the limited ligand solubility in the neutral pH region.Ka) protonation constants of the studied and similar ligands and stability constants of their complexes (I = 0.1 M (NMe4)Cl, 25 °C)
| Species | H3nota12,20 | H6notPPr6 |
H3L1a |
H3L223 |
H3L4 | H3notPhm12 |
H3notPH12 |
|---|---|---|---|---|---|---|---|
| a For a full set of stability constants see Table S2.† | |||||||
| HL | 13.17 | 11.48 | 11.54 | 10.23 | 11.21 | 11.47 | 10.48 |
| H2L | 5.74 | 5.44 | 3.43 | 2.86 | 8.84 | 3.85 | 3.28 |
| H3L | 3.22 | 4.84 | 1.40 | — | 8.16 | 1.30 | ∼1.1 |
| H4L | 1.96 | 4.23 | — | — | 7.30 | ||
| H5L | 0.70 | 3.45 | — | — | 2.09 | ||
| H6L | — | 1.66 | — | — | — | ||
| ∑logKN |
18.91 | 14.93 | 14.97 | 13.09 | 13.30 | 15.32 | 13.76 |
| logK[Ga(L)] |
29.63 | 26.24 | 20.5 | — | — | 23.3 | 21.91 |
| logK[Cu(L)] |
23.33 | 16.85 | 14.7 | — | — | 15.53 | 13.43 |
In all ligands, the first protonation was localized on the macrocycle amine groups and occurred in the alkaline region (10–13). The first protonation constant of H3nota was significantly higher than that of all phosphinate ligands. It agreed with the common trend in ligand basicity where amino groups in amino-phosphinates are less basic than those in amino-carboxylates.30 H3L2 showed the lowest basicity due to the electron-withdrawing effect of the fluorine atoms extending to the ring nitrogen atoms. The further protonation(s) of the ligands occurred in the acidic region. In H3L1, H3L2 and the simple analogues H3notPhm and H3notPH, it corresponded to the second macrocycle amine protonation. In H3nota and H6notPPr showing several protonation constants in the acidic region, it corresponded to the protonation of the carboxylate groups and to the second protonation of the macrocycle amines. The phosphinate-bound methylamino groups of H3L4 were protonated in the weakly alkaline region (pH 7–9) and the close values of all three aminomethyl protonation constants indicated that the protonations are more or less independent. The phosphinic acid groups were highly acidic and phosphinates were protonated only in strongly acidic solutions. Thus, not more than one protonation constant corresponding to the protonation of the phosphinate groups of each ligand could be determined by potentiometry.
GaIII complexation kinetic data
The UV-Vis method described above was used to investigate GaIII complexation kinetics with H3nota, H6notPPr and the title chelators H3L1–H3L4. The plots showing absorbance at the maximum of the absorption band as a function of time are shown in Fig. S13 and S14.† The measurements were carried out at a molar ratio of Ga:L of 1:2 and at 30 °C. The times required for quantitative complexation and the rate constants are shown in Fig. 3 and Table S3.† The complexation kinetics of H6notPPr was studied between pH 0 and 2 as the complexation was too fast at pH > 2. At pH 2, H6notPPr showed the fastest complexation among the studied chelators. H3nota showed a significantly slower complexation compared to H6notPPr and its complexation rate was studied between pH 1 and 2.5. The experiments were in a good agreement with the previously reported superiority of H6notPPr over H3nota in GaIII complexation rates.12 The complexation kinetics of H3L1 and H3L2 was studied at pH 0.5–2.5. Both chelators showed fast complexation at pH < 1.6, although the half-times are higher than those of H6notPPr. At pH > 2, the complexation of H3L1 and H3L2 was significantly slower than that of H6notPPr and the half-times of the reaction were more comparable to that of H3nota. The complexation rates of H3L3 and H3L4 were studied at pH 1.0–3.0 and 1.6–3.0, respectively. Their complexation rates were comparable. However, these reactions were significantly slower than those of the other phosphinate chelators. This might be ascribed to the presence of a positive charge on the protonated aminomethyl groups of the phosphinate pendant arms. The inhibitory effect of the protonated pendants was so significant that both chelators showed even a slower complexation than H3nota at almost all pH points.
 | ||
| Fig. 3 The times necessary for 99% complexation, t99%, with the studied chelators as a function of pH (cchel = 0.4 mM, cGa = 0.2 mM, 30 °C, pH 0.25–3.0). The lines serve only as guides to the eye. | ||
The complexation of metal ions by macrocyclic chelators with coordinating pendant arms proceeds in two steps.31 The first step is the formation of an intermediate out-of-cage complex in which the metal ion is bound only to the donor atoms in the pendant arms and the macrocycle amine group(s) are protonated. The second step is the deprotonation of the macrocycle amines with simultaneous transfer of the metal ion into the macrocyclic cavity, forming the final in-cage complex. This step is typically the rate-limiting step and is base-catalyzed. Thus, the complexation rate increases with the increasing pH which agrees with the results observed here. The ligand deprotonation in the rate-limiting step indicates that the macrocycle basicity is an important factor. The high basicity of the ring amino groups in H3nota might explain its slow complexation compared to the H6notPPr, H3L1 and H3L2 complexation. However, the ligand basicity did not correlate with the differences in complexation rates among the phosphinate ligands. Thus, an important role had to be ascribed to the thermodynamic stability of the out-of-cage intermediate as the overall reaction rate was also proportional to its concentration. The high stability and, consequently, the high out-of-cage complex abundance might be the reason for the fastest H6notPPr complexation among the studied ligands as the phosphinato-propionate pendant arms allow for chelating coordination in the out-of-cage complex.6 A structure showing such coordination was previously reported for the out-of-cage GdIII complex of the H4dota analogue bearing four phosphinato-propionate pendant arms.32 On the other hand, the pendant arm amino groups in H3L3 and H3L4 were protonated in acidic solutions. The low affinity of the GaIII ion to the nitrogen donor groups and its repulsion with the positively charged protonated amines probably resulted in low stability and, consequently, in low abundance of the out-of-cage GaIII–H3L3 and GaIII–H3L4 complexes. It explains the slow in-cage complex formation for these ligands.
In the very low concentrations of all reactants during radiolabelling, the higher abundance (i.e. higher thermodynamic stability) of the out-of-cage complexes was a very important parameter as it allows a close approach of the diluted metal radioisotope to the ligand cavity. The difference in accessibility of the out-of-cage complexes was highlighted in the radiolabelling experiments discussed below.
Radiolabelling with 68Ga
The 68Ga radiolabelling properties of the studied chelators were compared to those of H3nota and H6notPPr both being the current standards for 68Ga radiolabelling. The radiolabelling was performed at various pH values, temperatures, and chelator concentrations according to the established protocols.6,33,34 The radiolabelling yields (RCYs) as a function of pH (at 95 °C) are shown in Fig. 4A. Chelators H3L1 and H3L2 behaved very similar to H6notPPr – their RCYs were almost quantitative at pH > 1 and they decreased to ∼20% at pH 0.5. Ligands H3L3 and H3L4 behaved similar to H3nota and they show lower RCYs than H3L1 and H3L2. They required pH > 2.0 to achieve good radiolabelling; however, even under the most optimal conditions at pH 3, their RCYs did not exceed ∼95% after five minutes. The differences between the chelators were most pronounced at pH 1 where H3L1 and H3L2 achieved quantitative RCYs whereas the radiolabelling of H3L3 and H3L4 does not exceed 30%. | ||
| Fig. 4 Radiolabelling of the studied chelators with 68Ga (20–30 MBq ∼ 0.2–0.3 pmol 68Ga, 95 °C) as a function of pH (A: cchel = 10 μM ∼ nchel = 1 nmol, Ga:L ∼ 1: 5000, t = 5 min), chelator concentration (B: 95 °C, pH 3.0, t = 5 min), temperature (C: cchel = 10 μM ∼ nchel = 1 nmol, Ga:L ∼ 1:5000, pH 3, t = 5 min) and time (D: 95 °C, cchel = 10 μM ∼ nchel = 1 nmol, Ga:L ∼ 1:5000, pH 1.0). | ||
The RCYs as a function of the chelator concentration (or more correctly, a function of chelator molar excess over a molar amount of the metal radionuclide) at 95 °C are shown in Fig. 4B. H3L1 and H3L2 were labelled with high RCYs at concentrations >0.1 μM which was comparable to that of H6notPPr. Their RCYs at a concentration of 0.1 μM were over 90% and the RCY decreased rapidly at concentrations lower than 0.01 μM. H3L3 and H3L4 exhibited significantly lower radiochemical yields at low concentrations. H3L4 showed a slightly higher RCY than H3L3 but RCYs of both ligands steeply decreased at concentrations lower than 1 μM and the efficiency of both chelators was comparable to that of H3nota.
The RCYs as a function of temperature at pH 3 are shown in Fig. 4C. The H3L1 and H3L2 as well as H3nota and H6notPPr exhibited high radiochemical yields even at low temperatures (>80% at 35 °C). In contrast, RCYs of H3L3 and H3L4 were good only at high temperatures (<70 °C) and significantly decreased with decreasing temperature. The RCY changes with temperature were similar for both chelators, H3L3 and H3L4.
The role of time in the radiolabelling process was followed at decreased pH (pH = 1) to emphasize differences between the chelators. The experiments were conducted between 2.5 min and 20 min as the times are relevant for practical radiolabelling with 68Ga (Fig. 4D). Ligands H3L1, H3L2 and H6notPPr showed an almost quantitative radiolabelling even after 2.5 minutes. In contrast, H3L3 and H3L4 were radiolabelled significantly slower and had radiochemical yields of ∼80% even after twenty minutes and the benzylated derivative H3L3 reacted slightly slower. However, all chelators achieved higher radiochemical yields than H3nota showing by far the slowest radiolabelling at such a low pH.
The results of 68Ga radiolabelling showed the same trends as those obtained from the UV-Vis complexation studies. In both experiments, TRAP-ligands dominated over H3nota at pH < 1.5. However, H3nota showed a higher relative acceleration of 68Ga labelling with increasing pH than the TRAP-ligands, which might be related to the deprotonation of the carboxylate groups in H3nota in the studied pH range. Similarly, protonation of P-methylamino groups in the pendant arms of H3L3 and H3L4 resulted in a much less efficient radiolabelling compared to the other TRAP-ligands. The H3nota, H3L3 and H3L4 showed also much slower radiolabelling at low ligand concentrations whereas H6notPPr, H3L1 and H3L2 dominated at low pH as well as at low ligand concentrations. These results indicate that protonation of the pendant arms which prevents the formation of the out-of-cage intermediates is a key negative factor for the complexation rates and, mainly, for the radiolabelling yields. Thus, chelators able to form relatively stable/abundant out-of-cage complexes, H6notPPr and H3nopo, exhibited the best radiolabelling efficiency.6,10,11
Conclusion
A new method for the determination of GaIII complexation rates with NOTA-like chelators at μM–mM concentrations (“chemical conditions”) was developed and validated. The method is based on UV-Vis quantification of the CuII complex formed with a non-complexed chelator at individual time points. The method is an easy and cheap tool to follow the kinetics of complexation reactions of metal ions having no UV-Vis absorption under equimolar conditions as well as under a ligand or a metal ion excess. The method was used to study the kinetics of complexation reactions of the GaIII ion with four phosphi(o)nate analogues of H3nota and the complexation rates were compared with those for GaIII–H3nota and GaIII–H6notPPr systems. Radiolabelling with 68Ga was studied for the same chelators. The data from the chemical and radiochemical experiments followed the same trends and, thus, the “chemical” data could reasonably predict the chelator behavior in radiolabelling. The results further pointed to the importance of the groups attached to the phosphorus atoms in the complexation kinetics/radiolabelling. The H6notPPr bearing coordinating carboxylate groups showed the fastest complexation and the highest radiochemical yields. In contrast, complexes of the aminomethylphosphinate derivatives were formed slowly due to electrostatic repulsion between the protonated amino groups and the GaIII cation resulting in a low abundance of out-of-cage intermediates. The complexation rates and the radiolabelling efficiencies of the ligands bearing the non-coordinating and non-charged substituents (ethoxy or 2,2,2-trifluoroethyl) were between those of H6notPPr and the aminomethylphosphinate derivatives. It indicates that the right choice of substituents on the phosphorus atoms plays a crucial role in the complexation kinetics.Experimental
Materials and methods
Commercially available chemicals with synthetic purity were used as received. The [(N,N-dibenzylamino)methyl]-H-phosphinic acid (3),24 (2,2,2-trifluoroethyl)-H-phosphinic (2) and H3L2 were synthesized according to the literature.23 The characterization NMR measurements (1H, 13C{1H}, 19F, 31P, 31P{1H}, 71Ga) were performed on a Bruker Avance III HD (9.4 T, 400 MHz). 1H and 13C{1H} characterization of H3L3 was performed on a Bruker Avance III (14.3 T, 600 MHz) and 31P and 31P{1H} characterization was performed on a Varian VNMRS300 (7.0 T, 300 MHz). The 1H and 13C{1H} spectra were referenced to an internal standard tBuOH in D2O (δH 1.24 ppm; δC 30.3 ppm); 19F spectra were referenced to an external standard containing 0.1 M TFA (δF −75.51 ppm); 31P/31P{1H} spectra were referenced to external 85% aq. H3PO4 (δP 0.0 ppm); 71Ga spectra were referenced to Ga(NO3)3 (δGa 0.0 ppm) in 8 M HNO3/D2O or to [Ga(OH)4]− (δGa 222.4 ppm) in 1 M NaOD/D2O. The mass spectra were recorded on a Waters ACQUITY QDa spectrometer equipped with an electrospray ionization source and a quadrupole detector. The HR-MS spectra (negative mode, electrospray ionization) were recorded on a Bruker APEX-Q FT-MS.Synthesis
:1). The impurities were eluted with EtOH, and the product was eluted with EtOH/ammonia solution. The product-containing fraction was evaporated in a vacuum to yield a yellowish oil (8.02 g, 89%).
1H NMR (CDCl3): δ 1.29 (CH3–CH2, t, 3JHH 7.1 Hz, 18H), 2.93 (N–CH2–CH2–N, s, 12H), 2.96 (N–CH2–P, d, 2JHP 9.2 Hz, 6H), 4.08 (m, 12H, CH3–CH2). 31P{1H} NMR: δ 26.4 (s). MS+: 580.2 [M + H]+ (calc. 579.3).
1H NMR (D2O, pD 1.0): δ 1.29 (CH3–CH2, t, 3JHH 7.1 Hz, 9H), 3.37 (N–CH2–P, d, 2JHP 11.0 Hz, 6H), 3.59 (N–CH2–CH2–N, s, 12H), 4.02 (CH3–CH2, m, 6H). 13C{1H} (D2O, pD 1.0): δ 16.7 (CH3–CH2, d, 3JCP 5.7 Hz), 51.7 (N–CH2–CH2–N, d, 3JCP 6.0 Hz), 52.4 (N–CH2–P, d, 1JCP 138.5 Hz), 62.5 (CH3–CH2, d, 2JCP 6.1 Hz). 31P (D2O, pD 1.0) δ 14.4 (bs). 31P{1H} (D2O, pD 1.0) δ 14.35 (s). Elemental analysis (C15H35N3NaO9P3·1.5H2O, Mr 527.37): C 33.09% (33.25%), H 7.04% (7.07%), N 7.72% (7.45%). MS+: 496.3 [M + H]+, 991.7 [2M + H]+. MS−: 494.2 [M − H]− (calc. 495.2).
1H NMR (D2O, pD 1.9): δ 2.98 (N–CH2–P, d, 6H, 2JHP 7 Hz,), 3.18 (P–CH2–NBn2, d, 6H, 2JHP 9 Hz), 3.30 (N–CH2–CH2–N, s, 12H), 4.41 (CH2–Ph, s, 12H), 7.45–7.54 (H-arom., m, 30H). 13C{1H} NMR (D2O, pD 1.9): δ 50.8 (P–CH2–NBn2, d, 1JCP 87 Hz), 52.9 (N–CH2–CH2–N, s), 57.0 (N–CH2–P, d, 1JCP 101 Hz), 59.9 (CH2–Ph, s), 129.3 (C-arom., s), 130.2 (C-arom., s), 131.1 (C-arom., s), 132.2 (C-arom., s). 31P NMR (D2O, pD 1.9): δ 20.7 (m, 2JHP 7 Hz, 2JHP 9 Hz). 31P{1H} (D2O, pD 1.9): δ 20.7 (s). Elemental analysis (C54H69N6O6P3·3H2O, Mr 1045.1): C 62.1 (61.8), H 7.2 (7.0), N 8.0 (8.0), MS+: 991.1 [M + H]+, 1013.0 [M + Na]+, 1035.1 [M + 2Na − H]+, MS−: 989.63 [M − H]− (calc. 990.4).
1H NMR (D2O, pD 8): δ 3.05 (P–CH2–NH2, d, 6H, 2JHP 9.0 Hz), 3.14 (N–CH2–CH2–N, s, 12H), 3.22 (N–CH2–P, d, 6H, 2JHP 6.4 Hz). 13C{1H} (D2O, pD 8): δ 38.9 (P–CH2–NH2, d, 1JCP 91 Hz), 51.8 (N–CH2–CH2–N, d, 3JCP 4.3 Hz), 54.7 (N–CH2–P, d, 1JCP 99.3 Hz). 31P (D2O, pD 8): δ 27.6 (m, 2JHP 9.0 Hz, 2JHP 6.4 Hz). 31P{1H} (D2O, pD 8): δ 27.6 (s). Elemental analysis (C12H33N6O6P3·2.5H2O, MR 495.4): C 29.1 (29.2), H 7.7 (7.3), N 17.0 (16.8). MS+: 451.36 [M + H]+, 473.28 [M + Na]+, 489.24 [M + K]+, 901.50 [M + H]+. MS−: 249.00 [M − 2H]2−, 449.27 [M − H]− (calc. 450.2).
[ GaL1 ]: Ga(NO3)3·xH2O (40 mg, ∼100 μmol) was dissolved in water (5 ml) and concentrated aq. ammonia (1 ml) was added. The precipitate was separated by centrifugation, repeatedly washed with water and centrifuged. The resulting material was suspended in a solution of H3L1·1.5H2O (20.1 mg, 38 μmol) in water (2 ml). The mixture was stirred at room temperature overnight. The excess of the solid Ga(OH)3 was filtered off with a microfilter and the filtrate was evaporated in a vacuum yielding [GaL1] as a hygroscopic oil (32 mg). The complex was characterized by 1H, 13C{1H}, 31P, 31P{1H}, 71Ga NMR and MS (Fig. S7 and S8†).
1H NMR (D2O, pD 7.6): δ 1.31 (CH3–CH2, m, 9H), 3.0–3.7 (N–CH2–P + N–CH2–CH2–N, m, 18 H), 4.15 (CH3–CH2, m, 6H). 13C{1H} (D2O, pD 7.6): δ 16.4 (CH3–CH2, m), 53.1 (m), 55.3 (m), 56.6 (m), 63.7 (m), 64.6 (m). 31P (D2O, pD 7.6): δ 20.7 (bs), 21.2 (bs). 31P{1H} (D2O, pD 7.6): δ 20.7 (bs), 21.2 (bs). 71Ga (D2O, pD 7.6): 120.3 (s, ν1/2 = 218 Hz). HR-MS−: 560.06087 [M − H]− (calc. 560.06127).
[ GaL2 ]: Ga(NO3)3·xH2O (40 mg, ∼100 μmol) was dissolved in water (5 ml) and concentrated aq. ammonia (1 ml) was added. The precipitate was separated by centrifugation and repeatedly washed with water and centrifuged. The resulting material was suspended in a solution of H3L2 (20.3 mg, 33 μmol) in water (2 ml) and the mixture was stirred at room temperature overnight. The solid Ga(OH)3 was filtered off with a microfilter, and water was evaporated in a vacuum yielding 25.6 mg of pure [GaL2] as a hygroscopic oil. The complex was characterized by 1H, 13C{1H}, 19F, 31P, 31P{1H} and 71Ga NMR and MS (Fig. S9 and S10†).
1H NMR (D2O, pD 5.4): δ 2.98 (P–CH2–CF3, dq, 2JHP 16.0, 3JHF 11.0 Hz, 6H), 3.08–3.60 (N–CH2–P, N–CH2–CH2–N, m, 18H). 13C{1H} (D2O, pD 5.4): δ 37.5 (P–CH2–CF3, dq, 1JCP 96.5 Hz, 2JCF 29.5 Hz), 54.2 (N–CH2–CH2–N, s), 57.6 (N–CH2–CH2–N, d, 3JCP 13 Hz) 60.8 (N–CH2–P, d, 1JCP 92.0 Hz), 126.8 (P–CH2–CF3, qd, 1JCF 276.0 Hz, 2JCP 3 Hz). 31P (D2O, pD 5.4): δ 30.67 (bs). 31P{1H} (D2O, pD 5.4): δ 30.67 (q, 3JFP 11 Hz). 19F (D2O, pD 5.4): −57.29 (m, 3JFP 11.1 Hz, 3JFH 11.0 Hz). 71Ga (122 MHz): 136.2 (s, ν1/2 = 236 Hz). HR-MS−: 673.99115 [M − H]− (calc. 673.99173).
[ GaL3 ]: H3L3·3H2O (25.0 mg 24 μmol) was dissolved in water (1 ml) and Ga(NO3)3·xH2O (8.2 mg, ∼27 μmol) was added. The formed precipitate was filtered, washed with water and dried on air. A low complex solubility disabled common spectral characterization. MS+: 1057.8 [M + H]+ (calc. 1056.4).
[ GaL4 ]: H3L4·2.5H2O (12.5 mg, 25 μmol) was dissolved in water (1 ml) and Ga(NO3)3·xH2O (11.2 mg, ∼37 μmol) was added. The pH was adjusted to pH 3.5 with aq. LiOH. The mixture was stirred at 50 °C overnight and pH was adjusted to 5.6 with aq. LiOH. Precipitated Ga(OH)3 was filtered off with a microfilter and water was evaporated in a vacuum yielding 16.5 mg of pure [GaL4] as a hygroscopic oil. The complex was characterized by 1H, 13C{1H},31P, 31P{1H} and 71Ga NMR and MS (Fig. S11 and S12†).
1H NMR (D2O, pD 7.6): δ 2.9–3.6 (m). 13C{1H} (D2O, pD 7.6): δ 39.7 (P–CH2–NH2, d, 1JCP 104.6 Hz), 53.1 (N–CH2–CH2–N, s), 56.5 (N–CH2–CH2–N, d, 3JCP 13 Hz), 56.9 (N–CH2–P, d, 1JCP 82.4 Hz). 31P (D2O, pD 7.6): δ 38.68 (m). 31P{1H} (D2O, pD 7.6): δ 38.68 (s). 71Ga (D2O, pD 7.6): 137.6 (s, ν1/2 = 320 Hz). HR-MS−: 515.06157 [M − H]− (teor. 515.06227).
Potentiometry
Potentiometry was carried out according to the previously published procedures; for the preparation of the stock solutions, equipment, electrode system calibration, titration procedures and data treatment, see ref. 3 and 35. Throughout the paper, pH means −log[H+]. The concentration protonation constants were determined in 0.1 M (NMe4)Cl at 25.0 °C with pKw = 13.81 by in-cell titrations from data obtained in the pH range 1.6–12 with ∼40 points per titration and four parallel titrations (cL = 0.004 M). The constants were calculated with the OPIUM program.36Formation kinetics
The formation kinetics of GaIII complexes was studied in aqueous solution at 30 °C, cchel = 0.4 mM and cGa = 0.2 mM. The experiments at pH 0.25–1.50 were performed in aq. HClO4 (pH was calculated using the HClO4 concentration), at pH 2.00 in aq. 0.1 M betaine/HCl buffer, and at pH values of 2.50 and 3.00 in aq. 0.1 M HEPES/HCl buffer. At each time point, the solution aliquot (400 μl) was transferred into a vial with CuSO4 solution (400 μl, 1.25 mM) in aq. 1 M HEPES/HCl buffer (pH 3). The mixture was incubated for 5 min at room temperature and UV-Vis spectra were recorded (190–400 nm) on a Specord 50 Plus system (Analytic Jena). The rate constants and the reaction times were obtained from the absorbance at 265 nm (280 nm in the case of H3L3) according to eqn (1).| A = A∞ + (A0 − A∞) × e−kt | (1) |
1H NMR experiments were performed on the GaIII–H3L2 system at 30 °C (cchel = 0.4 mM, cGa = 0.2 mM and pH 1.00 given by aq. 0.1 M HClO4) on a Bruker Avance III (14.3 T, 600 MHz). The water signal was suppressed by excitation sculpting. The rate constant and the reaction times were obtained from the integral intensity of the P–CH2–CF3 signal of the free chelator according to eqn (2).
| I = I∞ + (I0 − I∞) × e−kt | (2) |
Radiolabelling
Radiolabelling was studied in triplicate by a manual synthesis under different conditions (pH, chelator concentration, temperature and time). A 68Ge/68Ga-generator (Eckert & Ziegler) was eluted with 1.0 M aq. HCl. A fraction of 1.25 ml with the highest activity of 300–450 MBq was added to 2 M aq. HEPES/HCl (60 μl). Then, pH was adjusted with 1 M aq. HCl and 1 M aq. NaOH (the final pH 0.5–4). A solution aliquot (90 μl, 20–30 MBq) was transferred to a preheated Eppendorf tube containing an aqueous solution of the chelator (10 μl). The resulting chelator concentrations were 10−5–10−8 M. Incorporation of 68Ga was determined by radio-TLC on TLC-paper impregnated with silicagel (Agilent Technologies, mobile phase 1:1 1 M aq. (NH4)OAc:MeOH (v/v)). The radioactivity incorporation was evaluated using a BIOSCAN TLC scanner with Bio-chrom Lite software.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support from the Technology Agency of the Czech Republic (TAČR TH78020003) and the Grant Agency of the Charles University (GAUK 1501/2024) is acknowledged. The stay of V. L. at Technical University Munich was supported by the Foundation of the Faculty of Science of Charles University (Prague). We thank Dr J. Havlíčková for performing the potentiometric titrations.References
- G. I. Gleason, Int. J. Appl. Radiat. Isot, 1960, 8, 90 CrossRef PubMed.
- B. J. B. Nelson, J. D. Andersson, F. Wuest and S. Spreckelmeyer, EJNMMI Radiopharm. Chem., 2022, 7, 27 CrossRef PubMed.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, E. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960 CrossRef PubMed.
- P. R. W. J. Davey and B. M. Paterson, Molecules, 2023, 28, 203 CrossRef PubMed.
- M. Fellner, P. Riss, N. Loktinova, K. Zhernosekov, O. Thews, C. F. G. C. Geraldes, Z. Kovacs, I. Lukeš and F. Rösch, Radiochim. Acta, 2011, 99, 43 CrossRef CAS.
- J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem. – Eur. J., 2010, 16, 7174 CrossRef CAS PubMed.
- M. I. M. Prata, J. P. André, Z. Kovács, A. I. Takács, G. Tircsó, I. Tóth and C. F. G. C. Geraldes, J. Inorg. Biochem., 2017, 177, 8 CrossRef CAS PubMed.
- G. Máté, J. Šimeček, M. Pniok, I. Kertész, J. Notni, H.-J. Wester, L. Galuska and P. Hermann, Molecules, 2015, 20, 13112 CrossRef.
- J. Notni, J. Šimeček and H.-J. Wester, ChemMedChem, 2014, 9, 1107 CrossRef CAS PubMed.
- J. Šimeček, P. Hermann, H.-J. Wester and J. Notni, ChemMedChem, 2013, 8, 95 CrossRef.
- J. Šimeček, O. Zemek, P. Hermann, J. Notni and H.-J. Wester, Mol. Pharmaceutics, 2014, 11, 3893 CrossRef.
- J. Šimeček, M. Schulz, J. Notni, J. Plutnar, V. Kubíček, J. Havlíčková and P. Hermann, Inorg. Chem., 2012, 51, 577 CrossRef.
- J. Holub, M. Meckel, V. Kubíček, F. Rösch and P. Hermann, Contrast Media Mol. Imaging, 2015, 10, 122 CrossRef CAS.
- J.-F. Morfin and É. Tóth, Inorg. Chem., 2011, 50, 10371 CrossRef CAS PubMed.
- A. Vágner, A. Forgács, E. Brücher, I. Tóth, A. Maiocchi, A. Wurzer, H.-J. Wester, J. Notni and Z. Baranyai, Front. Chem., 2018, 6, 170 CrossRef.
- K. Kumar, C. A. Chang and M. F. Tweedle, Inorg. Chem., 1993, 32, 587 CrossRef.
- K. Kumar and M. F. Tweedle, Inorg. Chem., 1993, 32, 4193 CrossRef.
- H.-Z. Cai and T. A. Kaden, Helv. Chim. Acta, 1994, 77, 383 CrossRef.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535 RSC.
- V. Kubíček, Z. Böhmová, R. Ševčíková, J. Vaněk, P. Lubal, Z. Poláková, R. Michalicová, J. Kotek and P. Hermann, Inorg. Chem., 2018, 57, 3061 CrossRef.
- J. Kubinec, V. Širůčková, J. Havlíčková, J. Kotek, V. Kubíček, P. Lubal and P. Hermann, Eur. J. Inorg. Chem., 2022, e202200173 CrossRef.
- I. Lázár and A. D. Sherry, Synthesis, 1995, 453 CrossRef.
- F. Koucký, T. Dobrovolná, J. Kotek, I. Císařová, J. Havlíčková, A. Liška, V. Kubíček and P. Hermann, Dalton Trans., 2024, 53, 9267 RSC.
- J. Kotek, P. Lebdušková, P. Hermann, L. Vander Elst, R. N. Muller, C. F. G. C. Geraldes, T. Maschmeyer, I. Lukeš and J. A. Peters, Chem. – Eur. J., 2003, 9, 5899 CrossRef PubMed.
- D. A. Moore, P. E. Fanwick and M. J. Welch, Inorg. Chem., 1990, 29, 612 CrossRef.
- C. Broan, J. P. Cox, A. S. Craig, R. Kataky, D. Parker, A. Harrison, A. M. Randall and G. Ferguson, J. Chem. Soc., Perkin Trans. 2, 1991, 87 RSC.
- Z. Kotková, G. A. Pereira, K. Djanashvili, J. Kotek, J. Rudovský, P. Hermann, L. Vander Elst, R. N. Muller, C. F. G. C. Geraldes, I. Lukeš and J. A. Peters, Eur. J. Inorg. Chem., 2009, 119 CrossRef.
- G. A. Pereira, L. Ball, A. D. Sherry, J. A. Peters and C. F. G. C. Geraldes, Helv. Chim. Acta, 2009, 92, 2532 CrossRef.
- P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027 RSC.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287 CrossRef.
- E. Brücher, G. Tircsó, Z. Baranyai, Z. Kovács and A. D. Sherry, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Chichester, U.K., 2nd edn, 2013, pp. 157–208 Search PubMed.
- J. Šimeček, P. Hermann, J. Havlíčková, E. Herdtweck, T. Kapp, N. Engelbogen, H. Kessler, H.-J. Wester and J. Notni, Chem. – Eur. J., 2013, 19, 7748 CrossRef PubMed.
- J. Notni, J. Šimeček, P. Hermann and H.-J. Wester, Chem. – Eur. J., 2011, 17, 14718 CrossRef PubMed.
- M. Asti, G. De Pietri, A. Fraternali, E. Grassi, R. Sghedoni, F. Fioroni, F. Roesch, A. Versari and D. Salvo, Nucl. Med. Biol., 2008, 35, 721 CrossRef PubMed.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535 RSC.
- (a) M. Kývala and I. Lukeš, in International Conference Chemometrics ‘95, Pardubice, Czech Republic, 1995, p. 63; (b) M. Kývala, P. Lubal and I. Lukeš, in IX. Spanish-Italian and Mediterranean Congress on Thermodynamics of Metal Complexes (SIMEC 98), Girona, Spain, 1998. The full version of the OPIUM program is available (free of charge) on https://www.natur.cuni.cz/_kyvala/opium.html.
Footnote |
| † Electronic supplementary information (ESI) available: Examples of UV-Vis and NMR kinetic data, NMR and MS spectra of ligands and complexes, overall protonation constants, complexation rate constants and times. See DOI: https://doi.org/10.1039/d4dt02383h |
| This journal is © The Royal Society of Chemistry 2024 |