
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chemically induced, room temperature, single source precursor to CuS (covellite) nanomaterials: synthesis and reactivity of [Cu(S2CNHBz)]n†
Siqiao
Huang
a,
Xiang
Xu
ab,
Jagodish C.
Sarker
 a,
David
Pugh
a and
Graeme
Hogarth
*a
a,
David
Pugh
a and
Graeme
Hogarth
*a
aDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 IDB, UK. E-mail: graeme.hogarth@kcl.ac.uk
bInstitute of Pharmaceutical Sciences, King's College London, Franklin Wilkins Building, Stamford Street, London SE1 9NH, UK
First published on 8th October 2024
Abstract
Addition of two equivalents of NaS2CNHBz to CuSO4 affords the yellow diamagnetic coordination polymer [Cu(S2CNHBz)]n (1), resulting from intramolecular electron-transfer and concomitant formation of the thiourea, (BzNH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S. 1 reacts with PPh3 and 1,1′-bis(diphenylphosphino)ferrocene (dppf) in CH2Cl2 to give monomeric [Cu(κ2-S2CNHBz)(PPh3)2] (2) and [Cu(κ2-S2CNHBz)(κ2-dppf)] (3), respectively, both of which have been crystallographically characterised. While 1 is thermally stable in dimethylsulfoxide (DMSO) up to ca. 70 °C, addition of nBuNH2 to 1 leads to its rapid decomposition to afford CuS (covellite) nanomaterials; indeed in neat nBuNH2, covellite formation is rapid at room temperature. Thus, 1 serves as an effective low-temperature base-induced single source precursor to covellite nanomaterials.
S. 1 reacts with PPh3 and 1,1′-bis(diphenylphosphino)ferrocene (dppf) in CH2Cl2 to give monomeric [Cu(κ2-S2CNHBz)(PPh3)2] (2) and [Cu(κ2-S2CNHBz)(κ2-dppf)] (3), respectively, both of which have been crystallographically characterised. While 1 is thermally stable in dimethylsulfoxide (DMSO) up to ca. 70 °C, addition of nBuNH2 to 1 leads to its rapid decomposition to afford CuS (covellite) nanomaterials; indeed in neat nBuNH2, covellite formation is rapid at room temperature. Thus, 1 serves as an effective low-temperature base-induced single source precursor to covellite nanomaterials.
The single source precursor (SSP) approach to nanoscale materials has been extensively developed over the past two decades1–3 with dithiocarbamate complexes4 being widely utilised as SSPs for the synthesis of metal-sulfide nanomaterials.5 Nanoscale copper chalcogenides are of particular interest due to their novel physical properties.6 A variety of stoichiometric phases are accessible, ranging for the sulfides from copper-rich (Cu2S) to sulfur-rich (CuS). Importantly, these materials have phase-dependent direct/indirect bandgaps in the range of 1.1–2.0 eV and consequently are utilised for applications such as optical filters, solar cells, photovoltaics, optical imaging devices and super-ionic materials.6
Copper–dithiocarbamate chemistry has been extensively developed with Cu(I), Cu(II) and Cu(III) complexes being accessible.7 Most prevalent are Cu(II) dialkyl-dithiocarbamate complexes, [Cu(κ2-S2CNR2)2], which are air and moisture stable dark brown solids, easily prepared upon addition of Cu(II) salts to NaS2CNR2 in water or MeOH.7 Both Cu(I) and Cu(II) dialkyl-dithiocarbamate complexes have been effectively utilised as SSPs, both towards copper sulfides and also copper-containing ternary and quaternary sulfides.5,7 The former is exemplified by the work of Alivisatos and co-workers, who utilised the thermal decomposition of [Cu(κ2-S2CNEt2)2] to prepare copper-rich djurleite (Cu1.94S) quantum dots (QDs) with tuneable localized surface plasmon resonances (LSPRs).8,9 Recent studies have identified the so-called Cu(II) sulfide, covellite (CuS), as the material of choice for QD applications.10–12 It has a relatively small window of thermal stability, losing sulfur above ca. 130 °C (ref. 13) and consequently, dialkyl-dithiocarbamate SSPs with their relatively high decomposition temperatures are barely suitable for its formation.5 Consequently, the design of low-temperature SSPs to covellite, and indeed other ternary and quaternary metal sulfide nanomaterials, is of significant interest.
We have recently reported that the carboxylic-acid derivative [Cu{κ2-S2CN(CH2CO2H)2}2] decomposes at ca. 90 °C (in water) to afford CuS (covellite) nanomaterials, the relatively low decomposition temperature being associated with a non-C–N cleavage pathway.14 A second strategy for the synthesis of low temperature SSPs towards metal-sulfides is to utilise primary amine-derived dithiocarbamates, [M(κ2-S2CNHR)2], and this approach has been successfully applied for the formation of nanoscale nickel,15 zinc16,17 and cadmium17–19 sulfides. Such species, which also form in situ via amine-exchange when [M(κ2-S2CNR2)2] are decomposed in long chain amines such as oleylamine,14,20–22 eliminate RNCS (under basic conditions), a process initiated by deprotonation with concomitant formation of the dithiocarbimate complexes [M(κ2-S2CNR)2]2−.23 For copper, however, despite contrary reports,24–32 complexes of the type [Cu(κ2-S2CNHR)2] or [Cu(κ2-S2CNHAr)2] have not been established. Indeed, a well-developed synthesis of organic isothiocyanates (RNCS) utilises the in situ formation of M(κ2-S2CNHR/Ar) (M = alkali metal), which eliminate organic isothiocyanate,33 a process that can be accelerated upon addition of copper salts.34 Interestingly, the unwanted by-product in this transformation is copper sulfide. We thus set out to investigate whether primary amine-derived dithiocarbamate salts of copper had any significant stability, and if so to harness them as low-temperature SSPs for the synthesis of covellite nanomaterials.
We initially screened a range of primary alkyl and aryl amine-derived dithiocarbamate salts with Cu(II) salts. There are some differences in the outcomes of these reactions depending upon the primary amine used, but there is a group of primary alkylamines that behave in a similar manner, and in this contribution, we focus on BzNH2. Dropwise addition of an aqueous solution of CuSO4·5H2O to a well-stirred aqueous solution of two equivalents of NaS2CNHBz in air results in the formation of a heavy bright yellow precipitate. After complete addition, filtration followed by sequential washing with water, MeOH and THF and drying at ca. 60 °C afforded [Cu(S2CNHBz)]n (1) as a bright yellow solid in ca. 70–80% yield. A pertinent experimental observation was the immediate formation of a dark brown color upon addition of each drop of CuSO4·5H2O to the pale-yellow NaS2CNHBz solution, which dissipated rapidly (over ca. 1–2 s). We interpret this observation as resulting from the initial formation of [Cu(κ2-S2CNHBz)2], which rapidly converts via an intramolecular electron-transfer to the isolated product. The fate of the oxidised dithiocarbamate was established as the thiourea (BzNH)2CS (see below and the ESI†), thus allowing a reaction pathway to be established (Scheme 1).
 | ||
| Scheme 1 Proposed formation of [Cu(S2CNHBz)]n (1) and (BzNH)2CS. | ||
The bright yellow colour of 1 immediately suggested a Cu(I) complex, and its diamagnetic nature was confirmed by a magnetic susceptibility measurement, while elemental analysis is consistent with a copper–dithiocarbamate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The IR spectrum shows a strong sharp absorption at 3221 cm−1 consistent with the presence of a non-hydrogen bonded N–H moiety. Unfortunately, it is insoluble in non-coordinating solvents but slowly dissolves with stirring in DMSO, the lemon-yellow solid dissolving to give an orange-brown solution. Both 1H and 13C{1H} NMR spectra in d6-DMSO are consistent with the Cu(I) assignment, with sharp resonances being seen. Notable features are the presence of a resonance at δ 10.90 assigned to the NH group in the 1H NMR spectrum, and a singlet at 204.3 ppm in the 13C{1H} spectrum associated with the quaternary backbone carbon. The insolubility of 1 in non-coordinating solvents suggests that it is a coordination polymer, dissolving to form a DMSO solvate. Copper(I) dithiocarbamates [Cu(μ3-S2CNR2)]4 are normally tetrahedral clusters7,35–37 with good solubility in organic solvents. Liu and co-workers have recently reported the molecular structure of a xanthate analogue, namely [Cu(S2COPri)]∞, which is a 1D coordination polymer with the xanthate spanning four different copper centres in a κ4–μ1,μ3-fashion,38 while [Cu(S2COMe)]∞ is a 2D coordination polymer with the xanthate bound in a κ4–μ2,μ2-fashion.39 While preparing this manuscript, Jung and co-workers reported the molecular structure of [Cu{μ3-S2CNH(CH2CH2OH)}]4, which adopts a tetrahedral geometry.40 No yields were given, nor characterising data or solubility information, the authors simply stating that the complex was separated from the THF washing of the insoluble yellow precipitate initially formed from the reaction. Thus, the precise structure of 1 remains undetermined and we are continuing our efforts to obtain crystals of 1 suitable for X-ray crystallography.
:1. The IR spectrum shows a strong sharp absorption at 3221 cm−1 consistent with the presence of a non-hydrogen bonded N–H moiety. Unfortunately, it is insoluble in non-coordinating solvents but slowly dissolves with stirring in DMSO, the lemon-yellow solid dissolving to give an orange-brown solution. Both 1H and 13C{1H} NMR spectra in d6-DMSO are consistent with the Cu(I) assignment, with sharp resonances being seen. Notable features are the presence of a resonance at δ 10.90 assigned to the NH group in the 1H NMR spectrum, and a singlet at 204.3 ppm in the 13C{1H} spectrum associated with the quaternary backbone carbon. The insolubility of 1 in non-coordinating solvents suggests that it is a coordination polymer, dissolving to form a DMSO solvate. Copper(I) dithiocarbamates [Cu(μ3-S2CNR2)]4 are normally tetrahedral clusters7,35–37 with good solubility in organic solvents. Liu and co-workers have recently reported the molecular structure of a xanthate analogue, namely [Cu(S2COPri)]∞, which is a 1D coordination polymer with the xanthate spanning four different copper centres in a κ4–μ1,μ3-fashion,38 while [Cu(S2COMe)]∞ is a 2D coordination polymer with the xanthate bound in a κ4–μ2,μ2-fashion.39 While preparing this manuscript, Jung and co-workers reported the molecular structure of [Cu{μ3-S2CNH(CH2CH2OH)}]4, which adopts a tetrahedral geometry.40 No yields were given, nor characterising data or solubility information, the authors simply stating that the complex was separated from the THF washing of the insoluble yellow precipitate initially formed from the reaction. Thus, the precise structure of 1 remains undetermined and we are continuing our efforts to obtain crystals of 1 suitable for X-ray crystallography.
Formation of 1 occurs via an intramolecular electron transfer from the dithiocarbamate to the Cu(II) centre (Scheme 1), likely following initial coordination, hence the observation of an initially formed brown bis(dithiocarbamate) intermediate. While dithiocarbamate radicals generated from secondary amines dimerise to afford thiuram disulfides,41 oxidation of primary amine dithiocarbamate salts is known to result in mixtures of the corresponding isothiocyanates, RNCS, and thioureas (RNH)2CS, with the latter being favoured at high pH.42 We thus worked-up the MeOH wash and NMR spectroscopy and X-ray diffraction showed that (BzNH)2CS was generated (see ESI†) in full accordance with the proposed intramolecular electron-transfer and subsequent reactivity of the BzNHCS2 radical (Scheme 1).
Complex 1 is converted into soluble mononuclear species upon addition of phosphines. For example, addition of ca. 2 equivalents of PPh3 to a CH2Cl2 suspension in air results in rapid dissolution and discoloration to afford clear solutions from which [Cu(κ2-S2CNHBz)(PPh3)2] (2) is isolated in high yield. Similar reactivity was observed with diphosphines, and the addition of CH2Cl2 to a mixture of 1 and 1,1′-bis(diphenylphosphino)ferrocene (dppf) resulted in the rapid formation of [Cu(κ2-S2CNHBz)(κ2-dppf)] (3) (Scheme 2). Both 2 and 3 were characterised by X-ray crystallography (Fig. 1) proving the stoichiometry of 1. The structure of 2 is similar to that of [Cu(κ2-S2CNHPh)(PPh3)2], formed upon the reaction of [Cu(κ2-BH4)(PPh3)2] with PhNCS,43,44 and the structure of 3 is similar to that of [Cu{κ2-S2CNBz(CH2py)}(κ2-dppf)] formed upon addition of the secondary amine-dithiocarbamate salt to [Cu(κ2-dppf)(μ-Br)]2.45 Spectroscopic data are in full accordance with the solid-state structures. In the 1H NMR spectra, the NH proton is observed at ca. δ 7.50 and in the 31P{1H} NMR spectra each shows a singlet associated with phosphine coordination.
 | ||
| Scheme 2 Reactivity of 1 towards PPh3 and dppf. | ||
 | ||
| Fig. 1 Molecular structures of (a) [Cu(κ2-S2CNHBz)(PPh3)2] (2) and (b) [Cu(κ2-S2CNHBz)(κ2-dppf)] (3). | ||
As articulated in the introduction, a key aim of our research is to design and prepare low temperature single source precursors (SSPs) towards nanoscale metal sulfides.5 The decomposition of SSPs is normally thermally induced and thus we initially assessed the thermal stability of 1 in the solid-state via TGA-DSC measurements (Fig. 2). The most pertinent features are its stability until ca. 135 °C and then decomposition to CuS at ca. 300 °C in a two-stage process, the first of which involves a ca. 18% mass loss (ca. 44 amu) that is possibly due to the elimination of carbon monosulfide (CS).
 | ||
| Fig. 2 TGA and DSC traces of the thermal decomposition of 1. | ||
In previous work, we have shown that secondary-amine derived dithiocarbamate complexes decompose at significantly lower temperatures in primary amine solutions than they do in the solid state, being associated with amine-exchange and the in situ generation of primary-amine derivatives.14,20–22 Complex 1 is stable in DMSO, up to ca. 70 °C, but heating at 90 °C for 3 h resulted in slow decomposition with concomitant formation of CuS (covellite). Decomposition was greatly accelerated upon addition of a small amount of nBuNH2 to a DMSO solution. Thus, at room temperature the solution initially darkens, possibly due to the formation of an amine adduct [Cu(S2CNHBz)(NH2nBu)n], and then slowly (ca. 12 h) becomes lighter with the concomitant deposition of a fine precipitate, indicative of the formation of CuS. The rate of decomposition is linked to the pH of the solution, indicating that the decomposition is base induced. Complex 1 dissolves in neat nBuNH2 (pH ca. 14) to initially (ca. 5 min) afford a brown solution, akin to that described above in DMSO, but over ca. 1 h this discolours with the deposition of a dark green precipitate, shown by PXRD to be nanoscale covellite (Fig. 3). As might be expected, diffraction peaks are very broad, indicative of the small particle size estimated at between 10 and 18 nm (av. 14.5 nm) by the Scherrer method. Thus, the decomposition of 1 is base-induced, likely being activated by the removal of the acidic N–H proton, with the formation of a dithiocarbimate complex46 which decomposes via elimination of BzNCS.
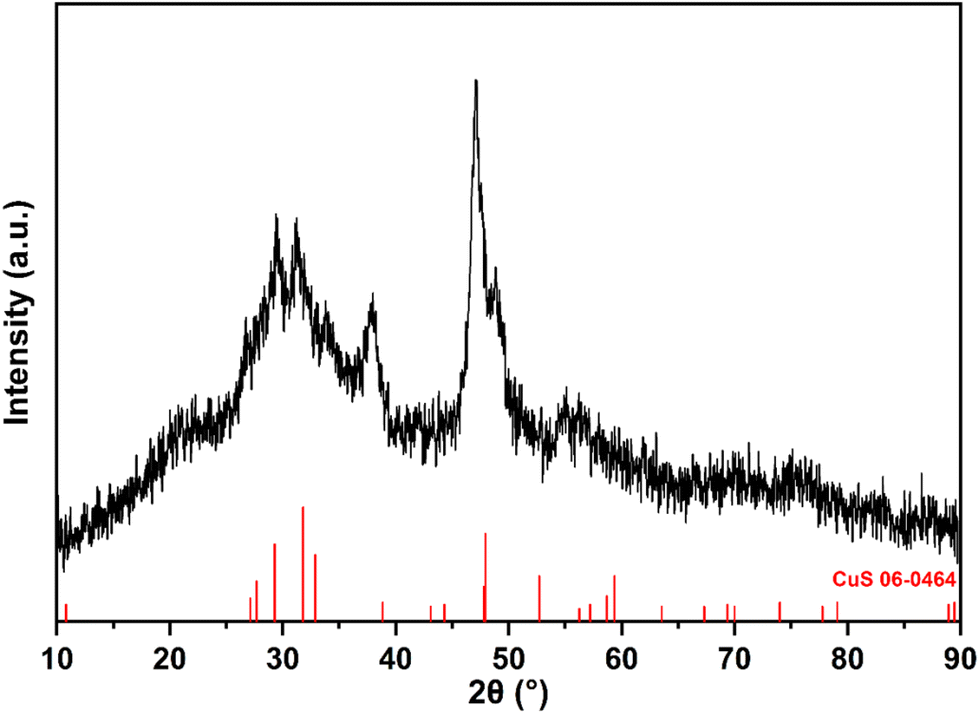 | ||
| Fig. 3 Powder X-ray diffractogram of covellite (CuS) nanoparticles formed from the room temperature decomposition of 1 in nBuNH2. | ||
In this contribution we have unambiguously shown that the addition of the primary amine-dithiocarbamate NaS2CNHBz to Cu(II) salts cleanly affords the Cu(I) dithiocarbamate [Cu(S2CNHBz)]n (1) as a result of a rapid intramolecular electron-transfer from the unstable Cu(II) complex [Cu(κ2-S2CNHBz)2]. Several older papers47–49 report a similar formation of Cu(I) complexes from dithiocarbamates derived from amino acids, but none was fully characterized. It has also been reported that ligand to metal charge transfer in [Cu(κ2-S2CNEt2)2] is photochemically activated50,51 and a very recent article reports that, upon prolonged sitting in DMSO, [Cu{κ2-S2CN(CH2CO2H)2}2] is reduced to afford the related Cu(I) complex.52 Indeed, this latter observation is fully consistent with our earlier findings on the low-temperature decomposition of this Cu(II) complex, as amine solutions turn yellow upon warming.14 Thus, ligand to metal electron-transfer in Cu(II)-dithiocarbamate complexes may be widespread. We are in the process of screening the reactivity of a range of other primary amine-derived dithiocarbamates with Cu(II) salts. Preliminary results suggest that while the outcome of the reaction is dependent upon the nature of the substituents, there are a large group of primary amines that behave in the same way as that described here for benzylamine.
Author contributions
G. H. devised and supervised the project, the main conceptual ideas and proof outline. S. H., X. X. and G. H. performed synthesis, characterisation, and decomposition work. D. P. carried out single crystal X-ray diffraction studies. J. C. S. and X. X. supervised S. H. G. H. wrote the initial manuscript and all authors contributed to editing to generate the final version.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for 2, 3 and (BzNH)2CS have been deposited at the CCDC under 2378475–2378477.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Commonwealth Scholarship Commission (JCS) and the Chinese Scholarship Council (XX) for PhD studentships and King's College London for UG project funding (SH). We also thank the KCL Chemistry Department facilities for technical support, and the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for 2.References
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS.
- T. Trindade and P. O'Brien, Chem. Mater., 1997, 9, 523–530 CrossRef CAS.
- W. E. Buhro, Adv. Mater. Opt. Electron., 1996, 6, 175–184 CrossRef CAS.
- G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71–561 CrossRef CAS.
- J. C. Sarker and G. Hogarth, Chem. Rev., 2021, 121, 6057–6123 CrossRef CAS.
- C. Coughlan, M. Ibanez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS.
- G. Hogarth and D. C. Onwudiwe, Inorganics, 2021, 9, 70 CrossRef CAS.
- Y. Wu, C. Wadia, W. Ma, B. Sadtler and A. P. Alivisatos, Nano Lett., 2008, 8, 2551–2555 CrossRef CAS.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS.
- W. Liang and M. Whangbo, Solid State Commun., 1993, 85, 405–408 CrossRef CAS.
- H. T. Evans and J. A. Konnert, Am. Mineral., 1976, 61, 996–1000 CAS.
- M. Liu, Y. Liu, B. Gu, X. Wei, G. Xu, X. Wang, M. T. Swihart and K. Yong, Chem. Soc. Rev., 2019, 48, 495–4965 Search PubMed.
- P. Roy and S. K. Srivastava, CrystEngComm, 2015, 17, 7801–7815 RSC.
- P. B. Mann, I. J. McGregor, S. Bourke, M. Burkitt-Gray, S. Fairclough, M. T. Ma, G. Hogarth, M. Thanou, N. Long and M. Green, Nanoscale Adv., 2019, 1, 522–526 RSC.
- N. Hollingsworth, A. Roffey, H. U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. De Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- D. C. Onwudiwe, T. Arfin, C. A. Strydom and R. J. Kriek, Electrochim. Acta, 2013, 109, 809–817 CrossRef CAS.
- A. A. Memon, M. Afzaal, M. A. Malik, C. Q. Nguyen, P. O'Brien and J. Raftery, J. Chem. Soc., Dalton Trans., 2006, 4499–4505 RSC.
- C. E. Morrison, F. Wang, N. P. Rath, B. M. Wieliczka, R. A. Loomis and W. E. Buhro, Inorg. Chem., 2017, 56, 12920–12929 CrossRef CAS.
- L. H. Van Poppel, T. L. Groy and M. T. Caudle, Inorg. Chem., 2004, 43, 3180–3188 CrossRef CAS.
- A. Roffey, N. Hollingsworth, H. U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. De Leeuw, Nanoscale, 2016, 8, 11067–11075 RSC.
- A. Roffey, N. Hollingsworth, H. U. Islam, W. Bras, G. Sankar, N. H. De Leeuw and G. Hogarth, Nanoscale Adv., 2019, 1, 2965–2978 RSC.
- H. U. Islam, A. Roffey, N. Hollingsworth, W. Bras, G. Sankar, N. H. De Leeuw and G. Hogarth, Nanoscale Adv., 2020, 2, 798–807 RSC.
- C. C. Hadjikostas, G. A. Katsoulos, M. P. Sigalas, C. A. Tsipis and N. V. Duffy, Polyhedron, 1989, 8, 2637–2639 CrossRef CAS.
- W. O. Foye, I. B. van de Workeen and J. D. Matthes, J. Am. Pharm. Assoc., 1958, 47, 4556–4558 Search PubMed.
- M. Tariquea and M. Aslam, Asian J. Chem., 2010, 22, 2031–2034 Search PubMed.
- N. Torres-Gómez, A. R. Vilchis-Nestor, R. M. Gómez-Espinosa and I. García-Orozco, Adv. Mater. Res., 2014, 976, 164–168 Search PubMed.
- A. P. Klein and E. S. Sattely, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1910–1915 CrossRef CAS.
- E. I. Duran-Garcia, J. Martinez-Santana, N. Torres-Gomez, A. R. Vilchis-Nestor and I. Garcia-Orozco, Mater. Chem. Phys., 2021, 269, 124743 CrossRef CAS.
- M. A. Agoro, J. Z. Mbese, E. L. Meyer and K. Onyenankeya, Mater. Lett., 2021, 285, 129191 CrossRef CAS.
- C. Chen, K.-W. Yang, L. Zhai, H.-H. Ding and J.-Z. Chigan, Bioorg. Chem., 2022, 118, 105474 CrossRef CAS.
- W. H. Mahdi, A. S. Faihan, H. M. Jirjes, A. M. Al-Mutairi, A. M. A. Alsheri, S. A. Alotaibi, A. M. Saleh, M. R. Hatshan, A. S. M. Al-Janabi, M. Kuniyil, B. Shaik and S. F. Adil, Int. J. Quantum Chem., 2032, 123, e27191 CrossRef.
- J. Nath, H. Ghosh, R. Yella and B. K. Patel, Eur. J. Org. Chem., 2009, 1849–1851 CrossRef CAS.
- H. M. Mesheram, S. Dale and J. S. Yadav, Tetrahedron Lett., 1997, 38, 8743–8744 CrossRef CAS.
- U. Mandapati, S. Pinapati and R. Rudraraju, Tetrahedron Lett., 2017, 58, 125–128 CrossRef CAS.
- D. Cardell, G. Hogarth and S. A. Faulkner, Inorg. Chim. Acta, 2006, 359, 1321–1324 CrossRef CAS.
- L. I. Victoriano and H. B. Cortés, J. Coord. Chem., 1996, 39, 231–239 CrossRef CAS.
- A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler Jr, X. Yang, G. A. Baker and J. R. Walensky, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS.
- P. V. V. N. Kishore, D.-R. Shi, J.-H. Liao, A. K. Gupta and C. W. Liu, Inorg. Chim. Acta, 2019, 496, 119068 CrossRef CAS.
- K. Tang, X. Jin, Y. Long, P. Cui and Y. Tang, J. Chem. Res. (S), 2000, 452–453 CrossRef CAS.
- H.-J. Lee, J.-H. Cha and D.-Y. Jung, J. Mol. Struct., 2023, 1275, 134666 CrossRef CAS.
- G. Hogarth, Mini-Rev. Med. Chem., 2012, 12, 1202–1215 CrossRef CAS.
- Z. Fu, W. Yuan, N. Chen, Z. Yang and J. Xu, Green Chem., 2019, 20, 4484–4491 RSC.
- C. Bianchini, C. A. Ghilardi, A. Meli, S. Midollini and A. Orlandini, J. Organomet. Chem., 1983, 255, C27–C30 CrossRef CAS.
- C. Bianchini, C. A. Ghilardi, A. Meli, S. Midollini and A. Orlandini, Inorg. Chem., 1985, 24, 932–939 CrossRef CAS.
- V. Kumar and S. Singh, J. Struct. Chem., 2021, 62, 1723–1731 CrossRef CAS.
- P. J. Heard, Y.-S. Tan, I. C. Yeo and E. R. T. Tiekink, Inorganics, 2021, 9, 71 CrossRef CAS.
- B. Marcías, M. V. Villa, A. Mateos and M. Páramo, J. Coord. Chem., 1998, 46, 71–77 CrossRef.
- B. Marcías, M. V. Villa and M. R. Rodriguez-Gallego, Transition Met. Chem., 1995, 20, 347–350 CrossRef.
- B. Marcías, M. V. Villa, E. Chicote, S. Martín-Velasco, A. Castineiras and J. Borrás, Polyhedron, 2002, 21, 1899–1904 CrossRef.
- B. G. Jeliazkova and N. D. Yordanov, Inorg. Chim. Acta, 1993, 203, 201–204 CrossRef CAS.
- B. G. Jeliazkova and M. A. Doicheva, Polyhedron, 1996, 15, 1277–1283 CrossRef CAS.
- V. Behling, B. Kintzel, J. Heinrich, M. Hingel, J. Meyer, S. Greiner and P. Liebing, Cryst. Growth Des., 2023, 23, 7777–7788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2378475–2378477. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02366h |
| This journal is © The Royal Society of Chemistry 2024 |