
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-ion magnetism in novel Btp-based cobalt complexes of different charge†
Gustavo
Rama-Martínez‡
 a,
Marcelo
Osorio-Celis‡
a,
Yolanda
Sabater-Algarra‡
a,
Diego
Sánchez-Brunete
a,
Antonio L.
Llamas-Saiz
c,
Eugenia P.
Quirós-Díez
a,
M. Eugenio
Vázquez
b,
Miguel
Vázquez López
a and
María del Carmen
Giménez López
*a
a,
Marcelo
Osorio-Celis‡
a,
Yolanda
Sabater-Algarra‡
a,
Diego
Sánchez-Brunete
a,
Antonio L.
Llamas-Saiz
c,
Eugenia P.
Quirós-Díez
a,
M. Eugenio
Vázquez
b,
Miguel
Vázquez López
a and
María del Carmen
Giménez López
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS) and Departamento de Química Inorgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: maria.gimenez.lopez@usc.es
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
cUnidade de Raios X. Área de Infraestruturas de Investigación, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
First published on 12th November 2024
Abstract
The magnetic behavior of single-ion metal complexes may be influenced by the nature and composition of the secondary coordination sphere that can be composed of solvent molecules and counterions bound through non-covalent interactions. However, achieving precise control over the outer-coordination sphere of these magnetic complexes to demonstrate its influence on their magnetic properties presents a challenge. A strategy for varying the number of counterions, while simultaneously preserving the arrangement of the ligand atoms around the metal center without altering its oxidation state, is to adjust the overall formal charge of the complex. This adjustment could lead to changes in the magnetic properties of single-ion metal complexes. In this study, we present two novel ligands featuring the coordinating unit Btp (2,6-bis(1,2,3-triazol-4-yl)pyridine). These ligands are equipped with functional groups that can potentially undergo deprotonation. By carefully selecting the solvents used during the crystallization process of the complexes, we can tune at will the charge of the complexes, thus modifying the composition of the CoII complexes’ outer-coordination sphere. We show that, by modifying these conditions, we can tailor the secondary coordination sphere of both charged (mono- and dicationic) and neutral anisotropic CoII metal complexes to show field-induced single-ion magnetism, influencing in turn the size of the barrier to reversal of the magnetization and their slow relaxation process.
Introduction
3d single-ion magnets (SIMs) are a sub-class of monometallic single-molecule magnets in which the magnetic properties of these compounds originate from a single ion within a ligand field.1 These systems have attracted increasing attention over the last twenty years for their potential applications in high-density data storage,2 quantum computing3 and spintronics.4 Although lanthanide ions have been the focus of most of the research in this area,5 3d transition metal complexes are another major paramagnetic source for the construction of SIMs.6Magnetic properties of these SIMs depend mainly on the d-orbital splitting, which in turn depends on various factors related to the metal ion (i.e., its oxidation state and the principal quantum number of its d-valence orbitals), the nature of the coordinated ligand(s), or the geometry around the metal ion, among others. So far most of the 3d metal complexes reported displaying SIM behavior exhibit several shared characteristics for their first coordination sphere, including low coordination numbers and reduced geometries that may be induced through bulky groups, as well as an efficient quantum tunneling of the magnetization (QTM) in the absence of an applied direct current (dc) field.7
The outer-sphere, or secondary coordination sphere, of a metal ion refers to any molecule—mainly from the solvent(s)—or counterions that interact with the metal complex through supramolecular forces.8 It is known that these outer-sphere interactions can affect the metal-binding properties of the ligand(s) and can even cause structural distortions in the metal complex, thus affecting its reactivity9 and some of its properties such as, for example, those related to magnetism.10
In this context, although the properties of SIMs are essentially based on discrete molecules, the metal complexes in single crystals are not isolated, but are part of a highly organized superstructure together with solvent molecules and counterions, where every interaction counts for the final magnetic behavior of the system as a whole. The importance of the secondary coordination sphere in SIMs has been described previously.11,12 However, the development of methods to control these external interactions in a magnetic context is still a relatively unexplored matter.13
In this work we report the synthesis of two new 2,6-bis(1,2,3-triazol-4-yl)pyridine (Btp) ligands. This ligand is broadly studied in other chemical disciplines, in fact this scaffold has been studied in spin crossover magnetic systems.14–16 However, no reported literature was found for systems based in other metals suitable for SIM behavior. As well as the synthesis of tBuOOCBtp and HOOCBtp (Scheme 1), here we report the X-ray crystal structures and the magnetic properties of a serie of anisotropic CoII complexes derived from these ligands exhibiting field-induced single-ion magnetism: [Co(tBuOOCBtp)2](ClO4)2 (1), [Co(HOOCBtp)(OOCBtp)]2(ClO4)2·6H2O (2), [Co(OOCBtp)2]·CH3CN·3H2O (3).
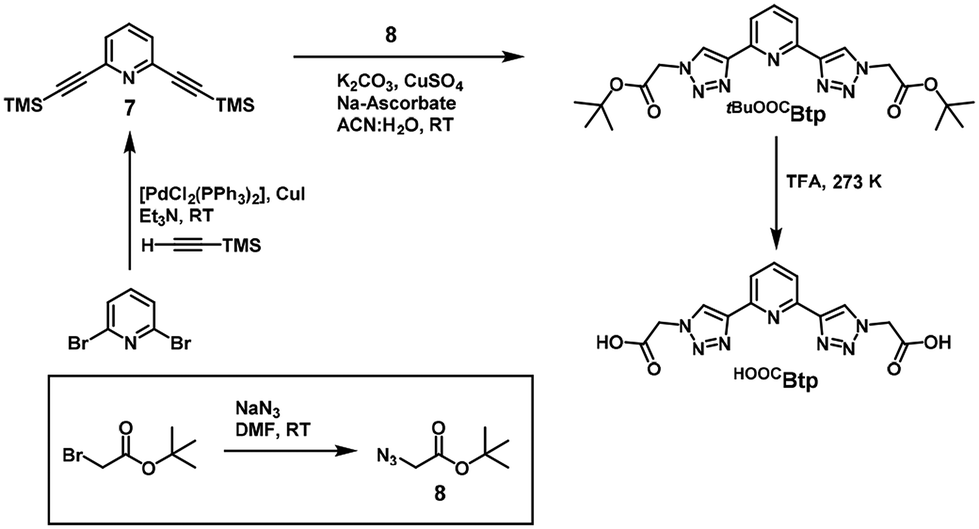 | ||
| Scheme 1 Synthetic scheme of the ligands tBuOOCBtp and HOOCBtp. | ||
We discuss here how the precise selection of terminal functionalizations of the tridentate metal-binding motif, combined with the appropriate choice of solvents during the crystallization process, allows for the tuning of the formal charge of the anisotropic complex, while preserving the arrangement of ligand atoms around the metal center without altering the metal oxidation state. Consequently, this approach adjusts the composition of the outer coordination sphere of CoII complexes in single crystals, which could influence the size of the barrier to reversal of magnetization and their slow relaxation process.
The study and understanding of these structural differences would aid in comprehending the variances observed in the magnetic behavior of the compounds. Thus, we will endeavor to establish correlations and/or trends between structural parameters, such as single ion anisotropy and orbital splitting, among other factors. Understanding these relationships may shed light on the modulation of energy barriers and the mechanisms underlying slow relaxation of the magnetization.17,18
Results and discussion
Synthesis of the Btp ligands
Btp derivatives are a class of terdentate ligands with high versatility in terms of functionalization and metal-binding properties,19 and that have been widely used in coordination20 and supramolecular chemistry,21 including applications in magnetochemistry.16 Since they contain triazole units in their structure, which are generated by click chemistry (CuAAC), they are considered members of the “click ligands” or “clickates”.15Two novel Btp ligands, named tBuOOCBtp and HOOCBtp, were synthesized here by CuAAC methods (Scheme 1). The former is equipped with two tert-butyl acetate terminal arms, whereas the latter is the corresponding acetic acid analog. Both ligands were obtained pure and characterized by reversed phase ultra-high performance liquid chromatography (RP-UHPLC), electrospray ionization mass spectrometry (ESI-MS), and 1H- and 13C-NMR spectroscopy (see ESI, Fig. S1–S13†).
Preparation of CoII complexes derived from the Btp ligands
By using UV-vis and fluorescence spectroscopy, the formation constants of the CoII complexes with tBuOOCBtp and HOOCBtp were studied in solution. As shown in the ESI (Fig. S14–S18†), all data sets were processed to extract the corresponding titration profiles and fitted using the DynaFit software,22 which has been extensively used to calculate the formation constants of classic coordination compounds and other complex systems, such as metallopeptides.23,24 The titration profiles, obtained through both spectroscopic techniques, were successfully fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (M:L) binding model using DynaFit.18 The values obtained for the global association constants calculated from both spectroscopic techniques for the homoleptic compounds with tBuOOCBtp and HOOCBtp are very similar, and only slightly smaller in comparison with other cobalt complexes with terdentate ligands reported in the literature demonstrating a strong metal–ligand interaction (Table 1).25,26
:2 (M:L). The global formation constants β2) were calculated by the equation β2 = K1·K2; where K1 and K2 are the stepwise formation constants
:2 (M:L) binding model using DynaFit.18 The values obtained for the global association constants calculated from both spectroscopic techniques for the homoleptic compounds with tBuOOCBtp and HOOCBtp are very similar, and only slightly smaller in comparison with other cobalt complexes with terdentate ligands reported in the literature demonstrating a strong metal–ligand interaction (Table 1).25,26
:2 (M:L). The global formation constants β2) were calculated by the equation β2 = K1·K2; where K1 and K2 are the stepwise formation constants
| UV-vis | Fluorescence | |||||
|---|---|---|---|---|---|---|
| logK1 |
logK2 |
logβ2 |
logK1 |
logK2 |
logβ2 | |
| CoIIvs.tBuOOCBtp | 3.96 | 3.91 | 7.87 ± 0.04 | 4.00 | 3.99 | 7.99 ± 0.04 |
| CoIIvs.HOOCBtp | 3.96 | 3.69 | 7.65 ± 0.07 | 3.99 | 3.79 | 7.78 ± 0.07 |
Considering the data obtained from UV-vis and fluorescence, the formation of the cobalt complexes with varying charges (ranging from dicationic to neutral) were prepared in solution by mixing a CoII perchlorate salt with the corresponding ligand (tBuOOCBtp and HOOCBtp) in a 1:2 ratio. After allowing the resulting solution to slowly evaporate at room temperature under a constant flow of nitrogen, single crystals suitable for X-ray diffraction studies were collected. When a 1:1 mixture of CH3OH/CH3CN was used as the solvent medium, clear orange single crystals of the homoleptic CoII complex derived from tBuOOCBtp of formula [Co(tBuOOCBtp)2](ClO4)2 (1) were collected. It is worth noting that both the formula and the structure depicted for 1 in Fig. 1 do not include solvent information. The crystallographic data of 1 indicate the presence of molecules of solvent in the crystal structure (one cavity filled with solvent with a volume of 354 Å3 per unit cell), which is further supported by the gradual weight loss below 100 °C observed from the thermogravimetric measurements (Fig. S19a†). However, attempts to model the disordered solvent were unsuccessful. The SQUEEZE procedure from the PLATON program was used to account for the contribution of the disordered molecules’ electron density to the measured experimental crystal diffraction data.27 Within the cavity, we identified a total of 73 electrons, which corresponds to an approximate maximum occupancy of 4 methanol molecules, which agrees with elemental analysis performed on the crystals of 1. Therefore, as a reasonable approximation, we assumed that the crystallographic asymmetric unit of the void (half of the total cavity volume) might contain 2 methanol molecules an the crystal stoichiometry is C42H54CoN14O8, 2(ClO4), 2(CH4O) (Table S1†).
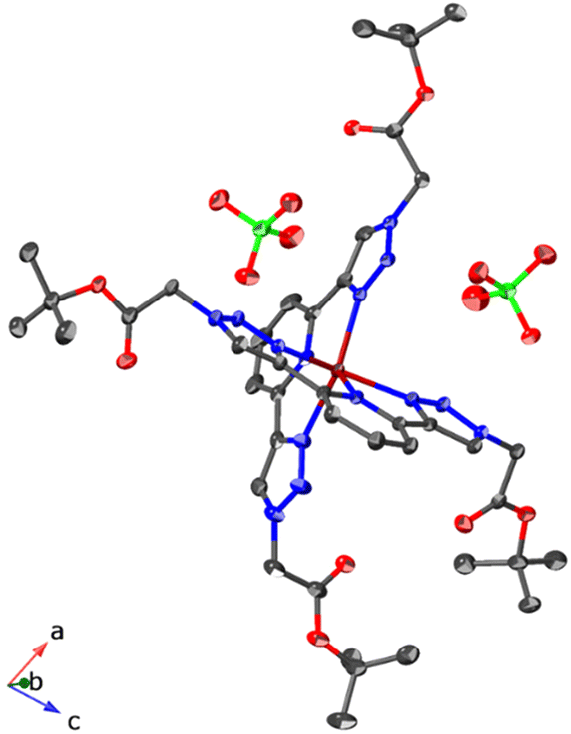 | ||
| Fig. 1 Thermal ellipsoids (30% probability ellipsoids) representation for the crystal structure of [Co(tBuOOCBtp)2](ClO4)2 (1) without the contribution of the disordered solvent. X-Ray crystal diffraction data can be found in the ESI.† Hydrogens and disordered atoms have been omitted for clarity. Carbon is represented in grey, nitrogen in blue, oxygen in red, chlorine in light green, and cobalt in brown. | ||
We have observed that by choosing the crystallization solvent and in presence of the divalent metal ion, we can deprotonate the carboxylic groups of HOOCBtp coordinating the metal, tuning the formal charge of the resulting metal complexes. Thus, using a low-deprotonating solvent mixture (CH3OH/CH3CN in a 1:1 ratio), clear light orange single crystals of a monocationic CoII complex of formula [Co(HOOCBtp)(OOCBtp)]2(ClO4)2·6H2O (2) were obtained, in which only one of the HOOCBtp ligands in the complex is monodeprotonated (referred to as OOCBtp). By using a more strongly-deprotonating solvent mixture (CH3CN:H2O in a 1:1 ratio), translucent orange single crystals of the neutral homoleptic CoII complex of formula [Co(OOCBtp)2]·CH3CN·3H2O (3) were isolated with both HOOCBtp ligands monodeprotonated.
Thermogravimetric analysis (TGA) was performed on the collected single crystals of the obtained CoII complexes under dry air in the range of 40–1000 °C with a scan rate of 2 °C min−1 (Fig. S19†). The gradual weight loss observed below 110 °C for all the compounds agrees with the presence of solvated molecules ascertained by X-ray single-crystal diffraction. After desolvation, the weight loss with increasing temperature for compounds 2 and 3 occurs in three consecutive steps, while compound 1 shows an additional weight loss step at 180 °C, involving the loss of the tert-butyl groups. After 250 °C all the compounds exhibit a weight loss corresponding to the thermal decomposition of ClO4− anions and the carboxylic groups. The complete decomposition of the Btp ligands to yield Co2O3 takes place in two consecutive steps between 300–550 °C for 1, 300–500 °C for 2, and 300–450 °C for 3. The solid residue isolated at 1000 °C corresponds to Co3O4.
Crystal structures of Btp derived CoII complexes
Single-crystal X-ray diffraction was used to determine the structures of the compound 1–3 at 100 K, all of which crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). The asymmetric unit cell for compounds 1 and 3 contains only one CoII metal center, while two crystallographically independent CoII centers are found for compound 2. In all cases, the CoII metal centers are coordinated by two Btp ligand units, resulting in a pseudo-octahedral CoN6 coordination environment around the metal ion (Fig. 1–3). The Co–N bond distances found for all the complexes (2.0933(14)–2.1811(16) Å for 1, 2.072(5)–2.184(16) Å for 2 and 2.0913(14)–2.1637(16) Å for 3) are above 2 Å and typical of CoII centres in high spin (Scheme S1 and Table S2†).28 The deviation of the CoN6 coordination environment from a regular octahedral geometry is expected to influence the magnetic properties of the metal complexes. This deviation becomes evident when analyzing the distortion indices Σ and Θ for the CoII complexes (1–3). Σ measures the deviation of a metal ion from an ideal octahedral geometry considering the sum of the deviation of the 12 cis N–Co–N angles from 90°, while Θ indicates its distortion from an octahedral towards a trigonal prismatic structure as the sum of the 24 N–Co–N angles measured on the projection of two triangular faces of the octahedron along their common pseudo-threefold axis (Scheme S2†). Both indices were obtained using the OctaDist program29 and their values deviate significantly from Σ = Θ = 0, which would indicate a perfect octahedral geometry (Table S3†).30,31 The deviation of CoN6 from a regular octahedral coordination geometry can also be observed by examining the Ntriazole–Co–Ntriazole angles within each of the coordinated ligands, as well as the Npyridine–Co–Npyridine angle involving the two coordinated ligands. These angles deviate significantly from the ideal 180° symmetry (parameter Ψ (ranging from 152.18(4)–150.15(6)°) and φ (ranging from 166.35(7)–173.22(1)°)), respectively (Scheme S3 and Table S3†). We also observed for 1–3 a deviation from 90°, expected for a perfect octahedral environment, for the dihedral angle between the planes containing the atoms of the tBuOOCBtp and HOOCBtp ligands coordinating the metal (parameter θ in Table S3†). By the careful analysis of all these parameters for the CoII complexes (1–3), a strongly distorted octahedral geometry can be inferred.
(Table S1†). The asymmetric unit cell for compounds 1 and 3 contains only one CoII metal center, while two crystallographically independent CoII centers are found for compound 2. In all cases, the CoII metal centers are coordinated by two Btp ligand units, resulting in a pseudo-octahedral CoN6 coordination environment around the metal ion (Fig. 1–3). The Co–N bond distances found for all the complexes (2.0933(14)–2.1811(16) Å for 1, 2.072(5)–2.184(16) Å for 2 and 2.0913(14)–2.1637(16) Å for 3) are above 2 Å and typical of CoII centres in high spin (Scheme S1 and Table S2†).28 The deviation of the CoN6 coordination environment from a regular octahedral geometry is expected to influence the magnetic properties of the metal complexes. This deviation becomes evident when analyzing the distortion indices Σ and Θ for the CoII complexes (1–3). Σ measures the deviation of a metal ion from an ideal octahedral geometry considering the sum of the deviation of the 12 cis N–Co–N angles from 90°, while Θ indicates its distortion from an octahedral towards a trigonal prismatic structure as the sum of the 24 N–Co–N angles measured on the projection of two triangular faces of the octahedron along their common pseudo-threefold axis (Scheme S2†). Both indices were obtained using the OctaDist program29 and their values deviate significantly from Σ = Θ = 0, which would indicate a perfect octahedral geometry (Table S3†).30,31 The deviation of CoN6 from a regular octahedral coordination geometry can also be observed by examining the Ntriazole–Co–Ntriazole angles within each of the coordinated ligands, as well as the Npyridine–Co–Npyridine angle involving the two coordinated ligands. These angles deviate significantly from the ideal 180° symmetry (parameter Ψ (ranging from 152.18(4)–150.15(6)°) and φ (ranging from 166.35(7)–173.22(1)°)), respectively (Scheme S3 and Table S3†). We also observed for 1–3 a deviation from 90°, expected for a perfect octahedral environment, for the dihedral angle between the planes containing the atoms of the tBuOOCBtp and HOOCBtp ligands coordinating the metal (parameter θ in Table S3†). By the careful analysis of all these parameters for the CoII complexes (1–3), a strongly distorted octahedral geometry can be inferred.
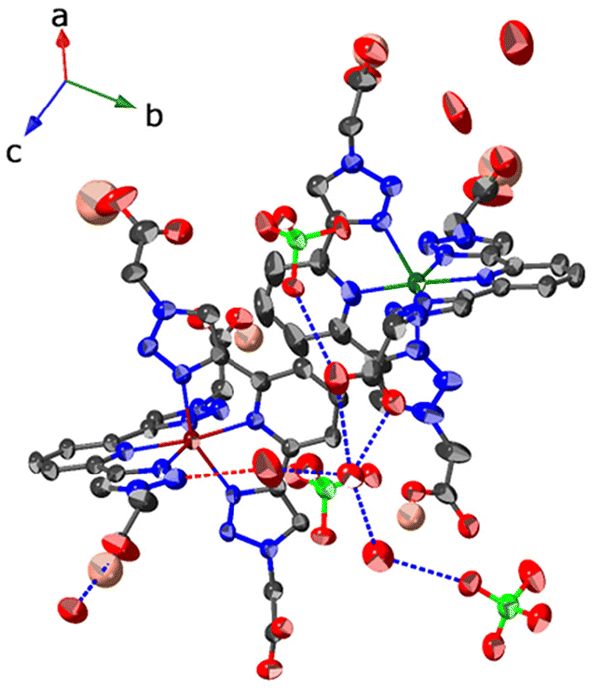 | ||
| Fig. 2 Thermal ellipsoids (30% probability ellipsoids) representation for the crystal structure of [Co(HOOCBtp)(OOCBtp)]2(ClO4)2·6H2O (2). The asymmetric unit contains two metal complexes that interact through π–π stacking interactions between the pyridine rings. X-Ray crystal diffraction data can be found in the ESI.† Hydrogen bonds between oxygen atoms are depicted as blue dashed lines and between oxygen and nitrogen atoms in red dashed lines. Hydrogens are omitted for clarity, except for the carboxylic groups, and the disordered atoms are also omitted, except for one perchlorate, which has an occupancy factor of 0.5. Carbon is represented in grey, nitrogen in blue, oxygen in red, chorine in light green, cobalt in brown (Co1) and green (Co2) and hydrogen in pink. | ||
 | ||
| Fig. 3 Thermal ellipsoids (30% probability ellipsoids) representation for the crystal structure of [Co(OOCBtp)2]·CH3CN·3H2O (3). X-Ray crystal diffraction data can be found in the ESI.† Hydrogens are omitted for clarity, except for the carboxylic groups and the disordered atoms are also omitted. Hydrogen bonds between oxygen atoms are depicted as blue dashed lines. Carbon is represented in grey, nitrogen in blue, oxygen in red and cobalt in brown. | ||
Although all the compounds 1–3 contain solvated molecules, the designed CoII complexes do not interact with the solvent in the same way. It is evident that compound 1 is more hydrophobic compared to compounds 2 and 3. This difference is attributed to the inability of the tBuOOCBtp ligand to form strong, directional hydrogen bonds with the solvent molecules (Fig. 1). Consequently, it can be inferred that the influence of the solvent on complex 1 is very limited, and its magnetic properties are likely to remain unaffected by the presence of the solvent.
In contrast, HOOCBtp is equipped with two carboxylic acids that can provide hydrogen-bonding sites for solvent molecules in the molecular packing. When the mixture used in the crystallization is CH3CN/CH3OH 1:1, only one of the carboxylic acids in one of the two HOOCBtp units of the metal complex is deprotonated, giving rise to the monocationic metal complex ion 2, which contains one perchlorate counterion in their second coordination sphere (Fig. 2). However, when CH3CN/H2O in 1:1 ratio is used in the crystallization process, two carboxyl groups, one from each of the HOOCBtp units are deprotonated, resulting in the formation of the neutral complex 3 (Fig. 3). In the HOOCBtp-derived complexes, hydrogen bonds are formed between water molecules in the second coordination sphere and the carboxylic groups (protonated or not) of the ligands (see Fig. 1–3). Interestingly, the presence of water molecules is a constant element in both HOOCBtp-derived complexes (2 and 3), regardless of whether water was used in the medium mixture for crystallization. Thus, it is hypothesized that the presence of water in the crystals can be attributed to the use of both undried organic solvents (CH3OH and CH3CN) and hydrated metal salts. Nevertheless, the presence of stronger secondary interactions (H-bonds) in 2 and 3 may have a stronger influence on the relaxation processes.
Regarding the role of the ClO4− counterions in the second coordination sphere of the metal complexes (Fig. 1 and 2), the obvious difference is the number of perchlorates between the dicationic (1) and the monocationic (2) complex. For the former, the interaction is solely through non-directional electrostatic interactions, while for the latest, the interaction with the protonated acid group of the metal complexes can be established through a direct (O–H⋯OCl) or a water mediated (O–HCOOH⋯–OCl–⋯H–OCOOH) hydrogen bonding (Fig. 2 and Table S4†).
In the crystal packing, the CoII complexes are arranged in either cationic or neutral layers for 1–2 and 3, respectively, where the shortest Co–Co distances are found (7.63(6) Å for 1, 7.79(1) Å for 2 and 7.31(1) Å for 3) with interlayer Co–Co separations ranging from 11.04(1) Å for 2 to 13.97(8) Å for 1 (Table S6†). For 1, the [Co(tBuOOCBtp)2]2+cationic layers are stacked along the a-axis via van der Waals forces between the tert-butyl arms of the Btp ligands (Fig. 4). Within the layer, the [Co(tBuOOCBtp)2]2+ complexes are interacting by pyridine–pyridine and pyridine–triazole π–π interactions along the b-axis (Table S7†). The analysis of π–π interactions for all three compounds has been conducted taking into account the distance between centroids, the dihedral angle between the planes containing the centroids, and the slippage. Here, we have taken into consideration distances up to 4.0 Å, angles below 20°, and minimized slippages.32 The growth of the cationic layer along the c direction is facilitated via van der Waals forces between the tert-butyl arms. The ClO4− anions fill the interstitial spaces in the structure (Fig. S20†). For compound 2, the cationic layers are stacked along the a-axis through hydrogen bonding involving the water molecules, the ClO4− anions and the carboxylic groups (Fig. 5 and Fig. S21, Table S4†). Within the layer, the two crystallographic independent [Co(HOOCBtp)(OOCBtp)]+ complexes interact by π–π interactions along c-axis and hydrogen bonding involving ClO4− anions and the carboxylic groups from the Btp units along b-axis (Fig. S21 and Table S8†). For compound 3, the [Co(OOCBtp)2] neutral layers are stacked along the b-axis, connected through hydrogen bond via the water molecules present in the second coordination sphere (OCOOH⋯H–Ow–H⋯OCOOH) (Fig. 6 and Table S5†). Within the layers, the [Co(OOCBtp)2] complexes are connected by π–π interactions between pyridine–pyridine and pyridine–triazole groups of the Btp ligands along both the a and c-axis (Table S9†).
 | ||
| Fig. 4 Ball and stick representation of the cationic layers of the CoII complexes stacked along the a-axis in the crystal packing of [Co(tBuOOCBtp)2](ClO4)2 (1). Hydrogens and anions are omitted for clarity. π–π interactions are highlighted using black dashed lines. Carbons are in grey, nitrogen in blue, oxygen in red and cobalt in dark brown. | ||
 | ||
| Fig. 5 Ball and stick representation of the cationic layers of the CoII complexes growing along the c-axis in the crystal packing of [Co(HOOCBtp)(OOCBtp)]2(ClO4)2·6H2O (2). Hydrogens and anions are omitted for clarity, except for the hydrogens of the carboxylic acids. π–π interactions are highlighted using black dashed lines. Carbons are in grey, nitrogen in blue, oxygen in red, hydrogen in pink and cobalt in green and dark brown. Water molecules are represented using red spheres. | ||
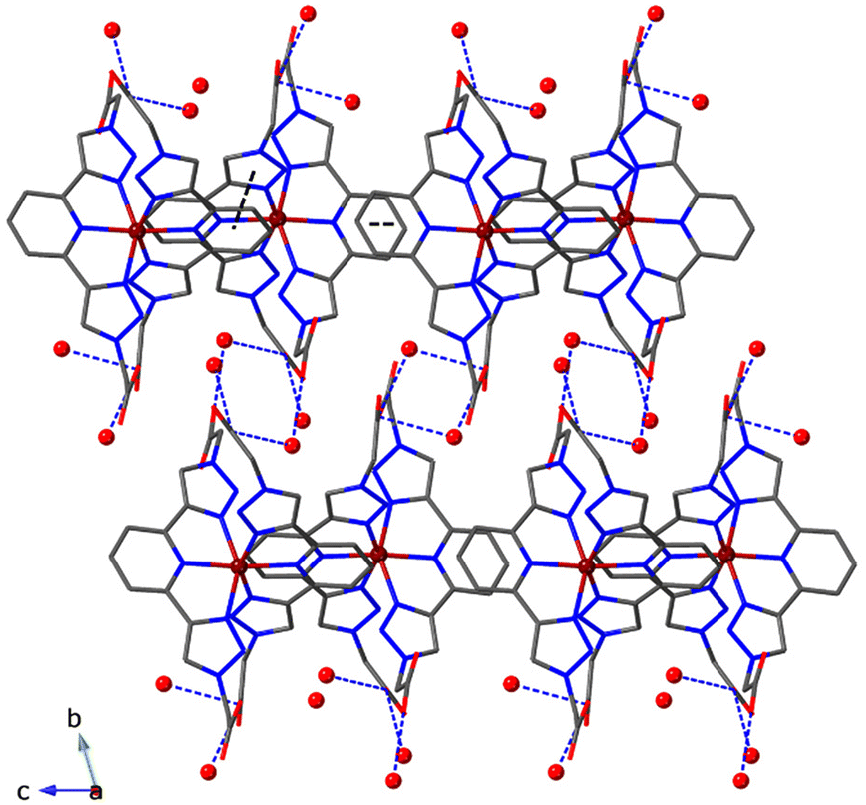 | ||
| Fig. 6 Ball and stick representation of the cationic layers of the CoII complexes stacked along the b-axis in the crystal packing of [Co(OOCBtp)2]·CH3CN·3H2O (3). Hydrogens are omitted for clarity. π–π interactions are highlighted using black dashed lines and H-bonds with blue dashed lines. Carbons are in grey, nitrogen in blue, oxygen in red and cobalt in dark brown. Oxygen atoms of the water molecules are depicted as red spheres. | ||
Magnetic studies
Field-dependence of the magnetization up to 5 T for 1–3 at 2, 3, 5 and 7 K was also done (Fig. S23†). Magnetization values continuously increase with the applied magnetic field for all the CoII complexes. No complete saturation of the magnetization is observed at 2 K under an applied field of 5 T (2.50 μB for 1, 5.38 μB for 2 and 2.83 μB for 3). The observation of non-saturation of magnetization with a high-field indicates the presence of magnetic anisotropy for all the complexes, which is further confirmed by the lack of superposition of the magnetization values in the reduced magnetization plots (M vs. H/T) between 2–7 K (Fig. S24†).36 This observation suggests that ZFS is achieved through a slight structural distortion around the CoII ion centers, resulting in a reduction of the three-fold symmetry. By using the PHI software package,37 an estimation of the anisotropy parameters of 1 and 3 can be obtained from the temperature and field-dependent magnetization data. Because of the presence of two crystallographically independent Co(II) centers, the estimation of the anisotropy parameters for 2 was not considered. By fixing the ge-factor values for the CoII ion centers obtained from the χMT measurements at 300 K (2.71 (1) and 2.81 (3) emu K mol−1) in the PHI software, the resulting fit shows positive axial ZFS parameters for all of them (D = 29.05 cm−1 and E = 0.82 cm−1 for 1; D = 13.29 cm−1 and E = 0.12 cm−1 for 3) (note: fitted curves extracted from PHI can be found in Fig. S25–S28†). This correlates well with the trend observed for the dihedral angles (θ) (91.35(4)° (1) < 99.52(5)/80.19(8)° (2) < 105.67(7)° (3)).
 | ||
| Fig. 7 Temperature-dependence of the out-of-phase ac susceptibility (χ′′) under a dc applied field at different frequencies between 110 to 10 kHz for 1 (a), 2 (b) and 3 (c). Frequency-dependence of the out-of-phase ac susceptibility (χ′′) under a dc applied field in the range of 3–5 K for 1 (d), 2–4.75 K for 2 (e), and in the range of 2–5.75 K for 3 (f). Dots corresponds to the experimental data and line correspond to the fitting obtained by CC-FIT software. Note that for 1 and 3 only the relaxation process at higher temperatures was fitted. Arrhenius plots constructed from data for 1 (g), 2 (h) and 3 (i). The solid blue line represents a fit to the two processes simultaneously (Raman and direct), while the green line only to the Orbach process. Error bars (red) are extracted from CC-FIT analysis. | ||
At a minimum, a maximum for the temperature and frequency dependence χM′′ is observed for all the complexes at these fields, suggesting at least one slow relaxation of the magnetization and a field-induced SIM behavior at low temperatures. It is worth noting that two relaxation processes can be clearly observed for both 1 and 3. As seen in Fig. 7, the frequency dependence χM′′ maxima were observed at all temperatures below 5 and 5.75 K for 1 and 3 (for the relaxation processes at higher temperatures in both) and 4.75 K for 2. Additionally, the observed peaks for the temperature dependence χM′′ (5.25 K (1), 4.75 K (2) and 5.75 K (3) at 10 kHz) increases in intensity and shift to low temperatures when frequency decreases from 10 kHz to 100 Hz for 1 and 2, while for 3 decreases in intensity and shift to low temperatures when frequency decreases from 10 kHz to 100 Hz.
Cole–Cole plots were constructed from the χM′ and χM′′ frequency-dependent ac data and fitted using the conventional generalized Debye model with the CC-FIT software package.38 For 1 and 3 only the relaxation process at higher temperatures was considered for the fitting. The fitted values of χT (isothermal susceptibility), χs (adiabatic susceptibility), τ and α are summarized in Table S10.† For 1–3, the curves show typical semicircles at low temperatures (Fig. S36†). A wider range for the distribution of the relaxation time (α) values is observed in the low-temperature regime for 1 (0.08–0.24) compared to the distribution of α values found for 2 (0.04–0.15) and 3 (0.05–0.12).
The extracted relaxation times were subsequently used to represent the Arrhenius plot (lnτ vs. T−1) (Fig. 7, green line). To examine the Orbach relaxation process, eqn (1) was employed.
| τ−1 = τ0−1·e−Ueff/KBT | (1) |
A fit to the linear portion of the data (at higher temperature values) affords the effective relaxation barriers (Ueff/KB) and pre-exponential factors (τ0−1) shown in Table S11.† From the trend observed for the experimental data in the lnτ vs. T−1 plot, it can be suggested that more than one relaxation process is involved.
To examine whether other relaxation processes existed, direct (AT) and Raman (CTn), were considered in addition to Orbach (or Arrhenius relaxation) using the following expression and considering that QTM relaxation is suppressed by an applied magnetic field:
| τ−1 = A·T + C·Tn + τ0−1·e−Ueff/KBT(2) |
The best fit parameters obtained for 1–3 are shown in Table 2. The observed values suggest that the primary relaxation processes for all compounds are Raman and Direct processes, as indicated by the minimized residuals. However, Orbach relaxation was also considered as a high-temperature process. Despite investigating both Orbach and Raman relaxation mechanisms, the low τ0 values and unconventional exponential ‘n’ parameters in Raman led us to regard these findings as less significant. Therefore, direct process seems to be predominant at low temperatures for 1–3 and Raman for 2, which is attributed to low-energy vibrations in the molecular lattice.
| H applied (T) | C (s−1 K−n) | n | A (s−1 K−1) | |
|---|---|---|---|---|
| 1 | 0.3 | 18.69 | 4.57 | 1.64 × 10−4 |
| 2 | 0.3 | 1.60 × 103 | 2.33 | 2.38 × 10−4 |
| 3 | 0.7 | 0.479 | 6.19 | 3.09 × 10−5 |
Direct mechanism has not been extensively found for CoII octahedral complexes, and although some cases are reported.39–41 It is important to highlight the complexity of the relaxation process in cobalt systems by covering Orbach process with Raman and Direct processes, this last dominant usually below 3 K. This fact is influenced by the spin entanglement found in Co(II) SIMs.42 It is also worth mentioning that typical values for n can range from 1–6 (if relaxation mechanism occurs through acoustic or optical phonon interactions), 7 (usually for non-Kramer ions) or 9 (for Kramer ions).43,44
Magneto-structural correlations
Considering the single-crystal X-ray diffraction analysis of 1–3, together with their magnetic data, several magneto-structural correlations can be inferred. The χMT values obtained for 1–3 confirm that both HOOCBtp and tBuOOCBtp exhibit a high-spin configuration for the same coordination number with a structural distortion around the metal as shown in Scheme S1.†45 In this context, the deviation from an ideal octahedral geometry result in unequally occupied orbitals giving rise to an appreciable magnetic anisotropy. Axial distortions associated to ZFS are obvious when looking at Co–Npyridine distances.Compounds 1 and 3 clearly have shorter distances for Co–Npyridine bonds (2.1025(14) and 2.0933(14) Å for 1 and 2.0913(14) and 2.0919(14) Å for 3), indicating a negative axial distortion. Interestingly, higher energy barriers (Ueff/KB) are found (16.11 and 9.64 cm−1 for 1 and 3, respectively; Table S11†) for those complexes with positive D values estimated from the static measurements, using the software package PHI, (29.05 cm−1 (1) and 13.29 cm−1 (3)). The dominant process for the spin reversal magnetization in 1 and 3 at low temperatures is a direct relaxation process, as shown by the fit of the lnτ vs. T−1 plot. This process may be induced by a small rhombic contribution (0.82 cm−1 (1) and 0.12 cm−1 (3)), which is in agreement with the largest distortion of the φ angle (N1–Co1–N51) from the ideal value of 180° (166 and 169°, respectively). In addition, a large distortion of the octahedral geometry towards a trigonal prismatic environment is observed for both compounds. It is hypothesized this distortion may be triggered by the extent of the π–π interactions between the Btp ligands. Nonetheless, other factors seem to have a large influence in the SIM behavior of these CoII complexes, such as their formal charge caused by the deprotonation of HOOCBtp, since 3 is the only neutral complex in the series and, therefore, without counterions in their unit cell.
For the case of 2, a much larger difference between the denoted Co–Npyridine distances for the two crystallographic independent metal complexes in the asymmetric unit (Co1–N1_2 and Co1–N1_1: 2.098(4) and 2.079(4) Å; Co2–N1_3 and Co2–N1_4: 2.099(5) and 2.072(5) Å) is observed, which could be attributed to an unequal negative axial distortion. The energy barrier found in this case is slightly smaller (8.82 cm−1, Table S11†). Additionally, the smaller distortion of the φ angle (N1_1–Co1–N1_2; N1_3–Co2–N1_4) from the ideal value of 180° (173 and 172°, respectively), gives an idea of the very small rhombic contribution if any.
Conclusions
Single crystals of three CoII metal complexes comprising two new Btp ligands (tBuOOCBtp and HOOCBtp) (1–3), were obtained and their structures solved through X-ray diffraction studies. We demonstrate the possibility of tuning the charge of the resulting metal complexes, achieving neutral, monocationic, and dicationic forms, all while preserving the arrangement of the ligand atoms around the metal center that does not change its oxidation state. This control is attained through precise selection of the terminal functionalization for the Btp ligands, and the solvent mixture employed during crystallization.The crystal structures of all complexes depicted a pseudo-octahedral CoN6 coordination environment around the metal ion, with deviations from ideal octahedral geometry. Static magnetic measurements unveiled significant χMT values, indicating the presence of unquenched orbital angular momentum. Field-dependent magnetization studies confirmed the presence of magnetic anisotropy, while dynamic magnetic studies revealed slow relaxation of magnetization and field-induced single-ion magnet behavior at low temperatures.
From magneto-structural correlations, it is evident that reducing the molecular symmetry increases the anisotropy of the CoII center and, consequently, an increase in the energy barrier would be expected. For instance, the dihedral angle between the ligand planes (θ), which is expected to be 90° in a regular octahedral complex, increases from 91.35° in 1 to 105.67° in 3. However, the observed energy barriers (Table S11†) follow the order 16.11 cm−1 (for 1) > 9.64 cm−1 (for 3) > 8.82 cm−1 (for 2). From frequency-dependence of the χM′′ under a dc applied magnetic field (Fig. 5d–f) can be clearly observed that the relaxation of 3 is much faster than that observed for 2 and 3, and dominated by a temperature-independent process. To explain this, we need to carefully consider both intramolecular factors (such as those promoted by the larger dihedral angle in 3) and intermolecular interactions, which result from the smaller Co–Co separation in 3 compared to 1 and 2, leading to increased dipole–dipole interactions. It is important to note that all three compounds display distinct and intricate relaxation processes, which are influenced by both Raman and direct mechanisms, similar to what has been observed in other Co(II) complexes.
In conclusion, understanding the complex interplay between structural distortions and alterations in the secondary coordination sphere is essential for elucidating the factors influencing the single-ion magnetism observed in transition metal coordination compounds. This study highlights the challenges in attributing the observed single-ion magnet behavior to a single factor and emphasizes the need for further research in this field to fully comprehend these intricate relationships.
Experimental
General
All chemicals and solvents from commercial sources were used as received. HOOCBtp and tBuOOCBtp ligands were synthesized as described below and characterized using HPLC-MS, NMR and MS-ESI-TOF. CAUTION: sodium azide and alkyl azides are potentially explosive. The synthesis should be carried out in small quantities and the precursors should be handled with care. Metal complexes 1–3 were obtained using a Schlenk line by the methods described below and subsequently crystallized under a very small constant flow of nitrogen at room temperature. Single crystals of 1–3 were characterized by SCXRD, MS-ESI-TOF, thermogravimetric analysis and CHN elemental analysis. High Resolution Mass spectrometry data were acquired on a Bruker MicrOTOF (ESI-TOF).Synthesis of ligands
:ether, 7:3) to afford 7 as a white solid (2.51 g, 96% yield). 1H-NMR (500 MHz, CDCl3, δ): 7.55 (t, 3J = 7.8 Hz, 1H), 7.34 (d, 3J = 7.8 Hz, 2H), 0.22 (s, 18H); 13C-NMR (126 MHz, CDCl3, δ): 143.40 (C), 136.32 (CH), 126.73 (CH), 103.19 (C), 95.46 (C), 0.26 (CH3); DEPT-135 (126 MHz, CDCl3, δ): 136.32 (CH), 126.73 (CH), 0.26 (CH3); MS-ESI: calculated for [C15H22NSi2]+ 272.13, found 272.17.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
:H2O (1:1). The mixture was stirred under nitrogen atmosphere for 16 h at room temperature, then a saturated solution of NH4Cl was added and the mixture was stirred for 30 min. The white solid was isolated by filtration, washed with isopropyl alcohol and deionized water and freeze-dried to give tBuOOCBtp as a white solid (893.6 mg, 88% yield). 1H-NMR (500 MHz, DMSO-d6, δ): 8.67 (s, 2H), 8.00 (s, 3H), 5.39 (s, 4H), 1.46 (s, 18H); 13C-NMR (126 MHz, DMSO-d6, δ): 116.19 (C), 149.81 (C), 147.06 (C), 138.38 (CH), 124.96 (CH), 118.51 (CH), 82.52 (C), 51.14 (CH2), 27.64 (CH3); DEPT-135 (126 MHz, DMSO-d6, δ): 138.38 (CH), 124.96 (CH), 118.51 (CH), 51.14(CH2), 27.64 (CH3); MS-ESI: m/z calculated for [C13H12N7O4]+ 442.22, found 442.24.
O).
:H2O (1:1). The mixture was stirred under nitrogen atmosphere for 16 h at room temperature, then a saturated solution of NH4Cl was added and the mixture was stirred for 30 min. The white solid was isolated by filtration, washed with isopropyl alcohol and deionized water and freeze-dried to give tBuOOCBtp as a white solid (893.6 mg, 88% yield). 1H-NMR (500 MHz, DMSO-d6, δ): 8.67 (s, 2H), 8.00 (s, 3H), 5.39 (s, 4H), 1.46 (s, 18H); 13C-NMR (126 MHz, DMSO-d6, δ): 116.19 (C), 149.81 (C), 147.06 (C), 138.38 (CH), 124.96 (CH), 118.51 (CH), 82.52 (C), 51.14 (CH2), 27.64 (CH3); DEPT-135 (126 MHz, DMSO-d6, δ): 138.38 (CH), 124.96 (CH), 118.51 (CH), 51.14(CH2), 27.64 (CH3); MS-ESI: m/z calculated for [C13H12N7O4]+ 442.22, found 442.24.
Synthesis of HOOCBtp
In a 100 mL round bottom flask, containing 513.0 mg of tBuOOCBtp (1.16 mmol), 5.0 mL of cold TFA were added. The solution was stirred in an ice bath for 1 h, then 70 mL of Et2O were added dropwise under vigorous stirring and a powdered white solid appeared. The solid was isolated by filtration, washed with Et2O and deionized water and freeze-dried at −45 °C to afford HOOCBtp as a white powder. (376.9 mg, 98.5% yield). 1H-NMR (500 MHz, DMSO-d6, δ): 13.49 (br, 2H), 8.66 (s, 2H), 7.99 (m, 3H), 5.39 (s, 4H); 13C-NMR (126 MHz, DMSO-d6, δ): 168.54, 149.85, 147.06, 138.39, 124.96, 118.48, 50.74; DEPT-135 (126 MHz, DMSO-d6, δ): 138.39 (CH), 124.96 (CH), 118.48 (CH), 50,74 (CH2); MS-ESI: m/z calculated for [C13H12N7O4]+ 330.09, found 330.11.Synthesis of metal complexes
:CH3CN (1:1). The resultant solution was degassed using Ar before adding the corresponding amount of metallic salt (41.4 mg of Co(ClO4)2·6H2O, 0.11 mmol, 0.50 eq.) dissolved in 2 mL of the same solvent mixture. The reaction mixture was stirred under Ar atmosphere for 30 minutes and then the solution was transferred to an Erlenmeyer flask and the solvent was left to slowly evaporate under atmospheric pressure at room temperature using a small flow of N2. Clear orange crystals were obtained for 1 (87.8 mg, 68% yield). (HR)-MS-ESI-TOF: calculated for [C42H54ClCoN14O12] [M + ClO4]+ 1041.3066, found 1041.3130; [C42H54CoN14O8] [M]2+ 470.6785, found 470.1592. Elemental analysis found (calculated for [Co(tBuOOCBtp)2](ClO4)2·2CH3OH): 43.24(43.54) % for C, 5.41(5.56) % for H and 16.26 (16.45) % for N.
:CH3CN (1:1). The resultant suspension was degassed using Ar before adding the corresponding amount of metallic salt (66.7 mg of Co(ClO4)2·6H2O, 0.18 mmol, 0.50 eq.) dissolved in 2 mL of the same solvent mixture. The reaction mixture was stirred under Ar atmosphere for 30 minutes and then the solution was transferred to an Erlenmeyer flask and the solvent was left to slowly evaporate under atmospheric pressure at room temperature using a small flow of N2. Clear light orange crystals were obtained for [Co(HOOCBtp)(OOCBtp)]ClO4·3H2O (2) (77.2 mg, 49% yield). (HR)-MS-ESI-TOF: calculated for [C26H22ClCoN14O12] [M + H + ClO4]+ 816.0562, found 816.0513; [C26H21CoN14O8] [M]+ 716.0999, found 716.1102, [C26H22CoN14O8] [M + H]2+ 358.5533, found 358.5598. Elemental analysis found (calculated for [Co(HOOCBtp)(OOCBtp)]ClO4·3H2O): 35.82 (35.90) % for C, 3.25 (3.13) % for H and 22.82 (22.54) % for N.
:CH3CN (1:1) mixture. The resultant suspension was degassed using Ar before adding the corresponding amount of metallic salt (66.7 mg of Co(ClO4)2·6H2O, 0.18 mmol, 0.50 eq.) dissolved in 2 mL of the of the same solvent mixture. The reaction mixture was stirred under Ar atmosphere for 30 minutes and then the solution was transferred to an Erlenmeyer flask and the solvent was left to slowly evaporate under atmospheric pressure at room temperature using a small flow of N2. Traslucent orange crystals were obtained for [Co(OOCBtp)2]·CH3CN·3H2O (3) (77.3 mg, 53% yield). (HR)-MS-ESI-TOF: calculated for [C26H21CoN14O8] [M + H]+ 716.0999, found 716.1154, [C26H22CoN14O8] [M + 2H]2+ 358.5533, found 358.5468. Elemental analysis found (calculated for [Co(OOCBtp)2]·CH3CN·3H2O): 41.02 (41.49) % for C, 3.50 (3.61) % for H and 26.03 (25.92) % for N.
SCXRD measurements
Single-crystal X-ray diffraction was measured on a Bruker D8 Venture Photon III C14 κ – geometry diffractometer system equipped with an Incoatec high brillance IμS 3.0 microsource (MoKα, λ = 0.71073 Å). Crystallographic data for all the complexes were deposited in the Cambridge Structural Database (CSD) with the following deposit numbers: CCDC 2281065 for 1; CCDC 2281066 for 2; CCDC 2281061 for 3.†Magnetic measurements
All magnetic measurements were performed solely on polycrystalline samples composed of ground single crystals packed on a plastic capsule using a Quantum Design MPMS-XL-5 SQUID magnetometer and a Quantum Design PPMS-5 both equipped with a 5 T magnet. After magnetic measurements powder X-ray diffraction measurements performed at the ground single crystals (compound 1 and 2) show no evidence of grinding causing structural changes on the compounds (Fig. S37†). Comparison with the simulated data from single-crystal X-ray diffraction measurements taken at 100 K suggests that the additional small peaks observed may be attributed to slight desolvation or the presence of a very minor impurity. Diamagnetic corrections were applied using tabulated Pascal constants. Small diamagnetic contribution from the plastic capsule was also considered. Variable-temperature susceptibility experiments were carried out in the temperature range 2–400 K under a static magnetic field of 0.1 and 1 T. Field-dependence magnetization measurements up to 5 T were performed at different temperatures between 2–7 K. At low temperatures ac susceptibility data were recorded with an oscillating field of 3.95 G amplitude at frequencies between 0.1 and 10 kHz. The optimum static fields for ac measurements were determined by measuring the frequency dependence of the in-phase and out-of-phase component of the susceptibility at 10 kHz under the application of dc fields ranging from 0 to 1.1 T.Author contributions
GRM designed the synthetic route and synthetized the ligand with the assistance of MOC and DSB; GRM and DSB performed the spectroscopic titrations; GRM and DSB calculate the association constants of the metal complexes; MOC and DSB prepared the metal complexes and isolate the single crystals; ALLS performed the single crystal X-Ray diffraction experiments and solved and refined the structures; EPQD, MOC and MCGL analyzed the crystal structures; MCGL, YSA and MOC analyzed the magnetic data; MCGL carried out the conceptualization and magneto-structural correlations. MEV designed the ligand, supervised the spectroscopic data fit and reviewed the manuscript draft together with MVL; MCGL, MEV and MVL funding acquisition; GRM, MOC and YSA writing-original draft preparation. MCGL writing-reviewing and editing the final manuscript. All authors commented on draft versions and approved the final version.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compound 1–3 have been deposited at the Cambridge Structural Database (CSD) with the following deposit numbers: CCDC 2281065 for 1; CCDC 2281066 for 2; CCDC 2281061 for 3 and can be obtained from https://www.ccdc.cam.ac.uk/structures/?Conflicts of interest
There are no conflicts to declare.Acknowledgements
GRM, MOC and EPQD thanks Xunta de Galicia for postdoctoral (grant 2018-PG020) and predoctoral (ED481A-2019/210 and ED481-2020/155) fellowships. YSA, GRM, MOC, EPQD and MCGL acknowledge financial support from European Research Council through the ERC-STG (NANOCOMP-679124). MCGL, MVL and MEV thank the Xunta de Galicia (grant ED431C 2021/29; ED431C 2024/05, the Oportunius Program (GAIN)) for financial support. MVL thanks Ideas Semilla 2021 – IDEAS211154VÁZQ for financial support. MVL, MEV and MCGL thank grants CNS2023-145421, PID2021-127341OB-I00, PID2021-127857NB-I00, PID2021-127702NB-I00 and TED2021-131451BC21 funded by MCIN/AEI/10.13039/501100011033 and by ERDF A way of making Europe. MCGL acknowledges financial support from the Ministry of Science of Spain (RYC-2016-20258). Authors thank J. M. Martínez-Agudo for technical assistance in the magnetic data acquisition.References
- M. Feng and M.-L. Tong, Single Ion Magnets from 3d to 5f: Developments and Strategies, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M. A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Magnetic memory of a single-molecule quantum magnet wired to a gold surface, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, Toward Molecular 4f Single-Ion Magnet Qubits, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- A. R. Rocha, V. M. García-Suárez, S. W. Bailey, C. J. Lambert, J. Ferrer and S. Sanvito, Towards Molecular Spintronics, Nat. Mater., 2005, 4, 335–339 CrossRef CAS PubMed.
- (a) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Lanthanide Single-Molecule Magnets, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (b) S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Molecular single-ion magnets based on lanthanides and actinides: Design considerations and new advances in the context of quantum technologies, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- (a) Y. Rechkemmer, F. D. Breitgoff, M. Van der Meer, M. Atanasov, M. Hakl, M. Orlita, P. Neugebauer, F. Neese, B. Sarkar and J. Van Slageren, A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier, Nat. Commun., 2016, 7, 10467 CrossRef CAS PubMed; (b) P. C. Bunting, M. Atanasov, E. Damgaard-Møller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. Van Slageren, F. Neese and J. R. Long, Science, 2018, 362, 7319 CrossRef PubMed.
- X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, Two-Coordinate Co(II) Imido Complexes as Outstanding Single-Molecule Magnets, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed.
- J. Martí-Rujas and F. Guo, Dehydrohalogenation reactions in second-sphere coordination complexes, Dalton Trans., 2021, 50, 11665–11680 RSC.
- L. R. Widger, C. G. Davies, T. Yang, M. A. Siegler, O. Troeppner, G. N. L. Jameson, I. Ivanović-Burmazović and D. P. Goldberg, Dramatically Accelerated Selective Oxygen-Atom Transfer by a Nonheme Iron(IV)-Oxo Complex: Tuning of the First and Second Coordination Spheres, J. Am. Chem. Soc., 2014, 136, 2699–2702 CrossRef CAS PubMed.
- (a) V. Jornet-Mollá, Y. Duan, C. Giménez-Saiz, Y. Tang, P. Li, F. M. Romero and R. Xiong, A Ferroelectric Iron(II) Spin Crossover Material, Angew. Chem., 2017, 129, 14240–14244 CrossRef; (b) M. C. Giménez-López, M. Clemente-León and C. Giménez-Saiz, Unravelling the spin-state of solvated [Fe(bpp)2]2+ spin-crossover complexes: structure–function relationship, Dalton Trans., 2018, 47, 10453–10462 RSC.
- (a) W. Phonsri, P. Harding, L. Liu, S. G. Telfer, K. S. Murray, B. Moubaraki, T. M. Ross, G. N. L. Jameson and D. J. Harding, Solvent modified spin crossover in an iron(III) complex: phase changes and an exceptionally wide hysteresis, Chem. Sci., 2017, 8, 3949–3959 RSC; (b) A. Galet, A. B. Gaspar, M. C. Muñoz and J. A. Real, Influence of the Counterion and the Solvent Molecules in the Spin Crossover System [Co(4-terpyridone)2]Xp·nH2O, Inorg. Chem., 2006, 45, 4413–4422 CrossRef CAS PubMed.
- (a) R. Herchel, P. Zoufalý and I. Nemec, The effect of the second coordination sphere on the magnetism of [Ln(NO3)3(H2O)3]· (18-crown-6) (Ln = Dy and Er), RSC Adv., 2019, 9, 569–575 RSC; (b) J. Jung, O. Cador, K. Bernot, F. Pointillart, J. Luzon and B. L. Guennic, Influence of the supramolecular architecture on the magnetic properties of a DyIII single-molecule magnet: an ab initio investigation, Beilstein J. Nanotechnol., 2014, 5, 2267–2274 CrossRef PubMed; (c) Z.-B. Hu, Z.-Y. Jing, M.-M. Li, L. Yin, Y.-D. Gao, F. Yu, T.-P. Hu, Z. Wang and Y. Song, Important Role of Intermolecular Interaction in Cobalt(II) Single-Ion Magnet from Single Slow Relaxation to Double Slow Relaxation, Inorg. Chem., 2018, 57, 10761–10767 CrossRef CAS PubMed.
- D. A. Reed, T. J. Hochuli, N. A. Gadjieva, S. He, R. A. Wiscons, A. K. Bartholomew, A. M. Champsaur, M. L. Steigerwald, X. Roy and C. Nuckolls, Controlling Ligand Coordination Spheres and Cluster Fusion in Superatoms, J. Am. Chem. Soc., 2022, 144, 306–313 CrossRef CAS PubMed.
- M. Ostermeier, M.-A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Complexes of Click-Derived Bistriazolylpyridines: Remarkable Electronic Influence of Remote Substituents on Thermodynamic Stability as well as Electronic and Magnetic Properties, Chem. – Eur. J., 2010, 16, 10202–10213 CrossRef CAS PubMed.
- J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, The btp [2,6-bis(1,2,3-triazol-4-yl)pyridine] binding motif: a new versatile terdentate ligand for supramolecular and coordination chemistry, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC.
- I. Capel Berdiell, D. J. Davies, J. Woodworth, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Structures and Spin States of Iron(II) Complexes of Isomeric 2,6-Di(1,2,3-triazolyl)pyridine Ligands, Inorg. Chem., 2021, 60, 14988–15000 CrossRef CAS PubMed.
- M. A. Palacios, I. F. Díaz-Ortega, H. Nojiri, E. A. Suturina, M. Ozerov, J. Krzystek and E. Colacio, Tuning magnetic anisotropy by the π-bonding features of the axial ligands and the electronic effects of gold(I) atoms in 2D {Co(L)2[Au(CN)2]2}n metal–organic frameworks with field-induced single-ion magnet behaviour, Inorg. Chem. Front., 2020, 7, 4611–4630 RSC.
- Z.-Y. Ding, Y.-S. Meng, Y. Xiao, Y.-Q. Zhang, Y.-Y. Zhu and S. Gao, Probing the influence of molecular symmetry on the magnetic anisotropy of octahedral cobalt(II) complexes, Inorg. Chem. Front., 2017, 4, 1909–1916 RSC.
- (a) Q. V. C. van Hilst, N. R. Lagesse, D. Preston and J. D. Crowley, Functional metal complexes from CuAAC “click” bidentate and tridentate pyridyl-1,2,3-triazole ligands, Dalton Trans., 2017, 47, 997–1002 RSC; (b) E. P. McCarney, C. S. Hawes, J. A. Kitchen, K. Byrne, W. Schmitt and T. Gunnlaugsson, A Lanthanide Luminescent Cation Exchange Material Derived from a Flexible Tricarboxylic Acid 2,6-Bis(1,2,3-triazol-4-yl)pyridine (btp) Tecton, Inorg. Chem., 2018, 57, 3920–3930 CrossRef CAS PubMed; (c) R. A. S. Vasdev, D. Preston and J. D. Crowley, Functional metallosupramolecular architectures using 1,2,3-triazole ligands: it's as easy as 1,2,3 “click”, Dalton Trans., 2017, 46, 2402–2414 RSC.
- (a) Q. V. C. van Hilst, N. R. Lagesse, D. Preston and J. D. Crowley, Functional metal complexes from CuAAC “click” bidentate and tridentate pyridyl-1,2,3-triazole ligands, Dalton Trans., 2017, 47, 997–1002 RSC; (b) Y. Li, J. C. Huffman and A. H. Flood, Can terdentate 2,6-bis(1,2,3-triazol-4-yl)pyridines form stable coordination compounds?, Chem. Commun., 2007, 2692–2694 RSC.
- (a) E. P. McCarney, J. I. Lovitt and T. Gunnlaugsson, Mechanically Interlocked Chiral Self-Templated [2]Catenanes from 2,6-Bis(1,2,3-triazol-4-yl)pyridine (btp) Ligands, Chem. – Eur. J., 2021, 27, 12052–12057 CrossRef CAS PubMed; (b) L. Xu, Y. Li and Y. Li, Application of “Click” Chemistry to the Construction of Supramolecular Functional Systems, Asian J. Org. Chem., 2014, 3, 582–602 CrossRef CAS; (c) D. A. W. Ross, J. A. Findlay, R. A. S. Vasdev and J. D. Crowley, Can 2-Pyridyl-1,2,3-triazole “Click” Ligands be Used to Develop Cu(I)/Cu(II) Molecular Switches?, ACS Omega, 2021, 6, 30115–30129 CrossRef PubMed.
- (a) P. Kuzmič, Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase, Anal. Biochem., 1996, 237, 260–273 CrossRef PubMed; (b) P. Kuzmič, DynaFit–a software package for enzymology, Methods Enzymol., 2009, 467, 247–280 Search PubMed.
- J. Gómez-González, Y. Pérez, G. Sciortino, L. Roldan-Martín, J. Martínez-Costas, J. Maréchal, I. Alfonso, M. Vázquez López and M. E. Vázquez, Dynamic Stereoselection of Peptide Helicates and Their Selective Labeling of DNA Replication Foci in Cells, Angew. Chem., Int. Ed., 2021, 60, 8859–8866 CrossRef PubMed.
- J. Gómez-González, D. Bouzada, L. A. Pérez-Márquez, G. Sciortino, J.-D. Maréchal, M. Vázquez López and M. E. Vázquez, Stereoselective Self-Assembly of DNA Binding Helicates Directed by the Viral β-Annulus Trimeric Peptide Motif, Bioconjugate Chem., 2021, 32, 1564–1569 CrossRef PubMed.
- R. Shunmugam, G. J. Gabriel, K. A. Aamer and G. N. Tew, Metal-Ligand-Containing Polymers: Terpyridine as the Supramolecular Unit, Macromol. Rapid Commun., 2010, 31, 784–793 CrossRef CAS PubMed.
- A. Winter, M. Gottschaldt, G. R. Newkome and U. S. Schubert, Terpyridines and their Complexes with First Row Transition Metal Ions:Cytotoxicity, Nuclease Activity and Self-Assembly of Biomacromolecules, Curr. Top. Med. Chem., 2012, 12, 158–175 CrossRef CAS PubMed.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 9–18 CrossRef PubMed.
- (a) J. Vallejo, F. R. Fortea-Perez, E. Pardo, S. Benmansour, I. Castro, J. Krzystek, D. Armentano and J. Cano, Guest-dependent single-ion magnet behaviour in a cobalt(II) metal-organic framework., Chem. Sci., 2016, 7, 2286–2293 RSC; (b) V. Garcia-Lopez, F. J. Orts-Mula, M. Palacios-Corella, J. M. Clemente-Juan, M. Clemente-Leon and E. Coronado, Field-induced slow relaxation of magnetization in a mononuclear Co(II) complex of 2,6-bis(pyrazol-1-yl)pyridine functionalized with a carboxylic acid, Polyhedron, 2018, 150, 54–60 CrossRef CAS.
- (a) M. B.-L. Cointe, J. Hébert, C. Baldé, N. Moisan, L. Toupet, P. Guionneau, J. F. Létard, E. Freysz, H. Cailleau and E. Collet, Intermolecular control of thermoswitching and photoswitching phenomena in two spin-crossover polymorphs, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 064114 CrossRef; (b) J. A. Alonso, M. J. Martínez-Lopez, M. T. Casais and M. T. Fernández-Díaz, Evolution of the Jahn−Teller Distortion of MnO6 Octahedra in RMnO3 Perovskites (R = Pr, Nd, Dy, Tb, Ho, Er, Y): A Neutron Diffraction Study, Inorg. Chem., 2000, 39, 917–923 CrossRef CAS PubMed; (c) M. Marchivie, P. Guionneau, J.-F. Létard and D. Chasseau, Photo-induced spin-transition: the role of the iron(II) environment distortion, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25–28 CrossRef PubMed.
- M. A. Halcrow, Structure:function relationships in molecular spin-crossover complexes, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- J. K. McCusker, A. L. Rheingold and D. N. Hendrickson, Variable-Temperature Studies of Laser-Initiated 5T2 → 1A1 Intersystem Crossing in Spin-Crossover Complexes: Empirical Correlations between Activation Parameters and Ligand Structure in a Series of Polypyridyl Ferrous Complexes, Inorg. Chem., 1996, 35, 2100–2112 CrossRef CAS.
- C. Janiak, A critical account on Π–Π stacking in metal complexes with aromatic nitrogen-containing ligands, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- V. García-López, F. J. Orts-Mula, M. Palacios-Corella, J. M. Clemente-Juan, M. Clemente-León and E. Coronado, Field-induced slow relaxation of magnetization in a mononuclear Co(II) complex of 2,6-bis(pyrazol-1-yl)pyridine functionalized with a carboxylic acid, Polyhedron, 2018, 150, 54–60 CrossRef.
- F. E. Mabbs, D. J. David and J. Machin, Magnetism and transition metal complexes, Dover Publications, 2008 Search PubMed.
- R. Boča, Zero-field splitting in metal complexes, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- Y. Cui, Y. Xu, X. Liu, Y. Li, B. L. Wang, Y. Dong, W. Li and S. Lei, Field-Induced Single-Ion Magnetic Behavior in Two Mononuclear Cobalt(II) Complexes, Chem. – Asian J., 2019, 14, 2620–2628 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- D. Reta and N. F. Chilton, Uncertainty estimates for magnetic relaxation times and magnetic relaxation parameters, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- H. Zenno, F. Kobayashi, M. Nakamura, Y. Sekine, L. F. Lindoy and S. Hayami, Hydrogen bond-induced abrupt spin crossover behaviour in 1-D cobalt(II) complexes – the key role of solvate water molecules, Dalton Trans., 2021, 50, 7843–7853 RSC.
- N. Portolés-Gil, S. Gómez-Coca, O. Vallcorba, G. Marbán, N. Aliaga-Alcalde, A. López-Periago, J. A. Ayllón and C. Domingo, Single molecule magnets of cobalt and zinc homo- and heterometallic coordination polymers prepared by a one-step synthetic procedure, RSC Adv., 2020, 10, 45090–45104 RSC.
- Q. Yang, G.-L. Wang, Y.-Q. Zhang and J. Tang, Self-assembly of fish-bone and grid-like CoII-based single-molecule magnets using dihydrazone ligands with NNN and NNO pockets, Dalton Trans., 2022, 51, 13928–13937 RSC.
- S. Gómez-Coca, A. Urtizberea, E. Cremades, P. J. Alonso, A. Camón, E. Ruiz and F. Luis, Origin of slow magnetic relaxation in Kramers ions with non-uniaxial anisotropy, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- K. N. Shrivastava, Theory of Spin–Lattice Relaxation, Phys. Status Solidi., 1983, 117, 437–458 CrossRef CAS.
- A. Singh and K. N. Shrivastava, Optical-acoustic two-phonon relaxation in spin systems, Phys. Status Solidi, 1979, 95, 273–277 CrossRef CAS.
- (a) P. Gütlich, H. A. Goodwin and H. A. Goodwin, Recent Advances of Spin Crossover Research, Top. Curr. Chem., 2004, 23–47 Search PubMed; (b) R. Hogg and R. G. Wilkins, Exchange studies of certain chelate compounds of the transitional metals. Part VIII. 2,2′,2′′-terpyridine complexes, J. Chem. Soc., 1962, 341–350 RSC; (c) S. Klokishner and S. M. Ostrovsky, Exchange studies of certain chelate compounds of the transitional metals. Part VIII. 2,2′,2′′-terpyridine complexes Interplay of Jahn-Teller Ordering and Spin Crossover in Co(II) Compounds, Magnetochemistry, 2020, 6, 62–73 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary tables, and figures. CCDC 2281061, 2281065 and 2281066. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02338b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |