
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binuclear methylphosphinidine complexes of cyclopentadienylruthenium carbonyls: effects of the higher ligand field strength of ruthenium derivatives relative to iron derivatives†
Oleg
Rudenco
a,
Alexandru
Lupan
 *a,
Radu
Silaghi-Dumitrescu
a and
R. Bruce
King
*b
*a,
Radu
Silaghi-Dumitrescu
a and
R. Bruce
King
*b
aFaculty of Chemistry and Chemical Engineering, Babeș-Bolyai University, Cluj-Napoca, Romania. E-mail: alexandru.lupan@ubbcluj.ro
bDepartment of Chemistry, University of Georgia, Athens, Georgia 30602, USA. E-mail: rbking@chem.uga.edu
First published on 14th October 2024
Abstract
The structures and energetics of the binuclear methylphosphinidene complexes of cyclopentadienylruthenium carbonyls of the type [MePRu2(CO)nCp2] (n = 4, 3, 2, 1) have been investigated for comparison with their previously studied iron analogues. For the tetracarbonyls and tricarbonyls [MePM2(CO)nCp2] (n = 4, 3) substituting ruthenium for iron has relatively little effect on the energetically preferred structures. Thus such structures have two-electron donor bridging MeP groups with no metal–metal bond for the tetracarbonyls and a metal–metal single bond for the tricarbonyls. This leads to favored 18-electron configurations for both ruthenium atoms in all cases. However, the higher ligand field strengths of ruthenium complexes relative to analogous iron complexes have major effects on the energetically preferred structures for the dicarbonyls and monocarbonyls [MePM2(CO)nCp2] (M = Fe, Ru; n = 2, 1). Thus the 11 lowest energy structures for the dicarbonyl [MePFe2(CO)2Cp2] are triplet or quintet spin state structures whereas the 6 lowest energy structures for the ruthenium analogue [MePRu2(CO)2Cp2] are all singlet structures. These low-energy singlet [MePRu2(CO)2Cp2] structures include species in which both ruthenium atoms attain the favored 18-electron configurations in different ways: either by a Ru–Ru single bond and an agostic C–H–Ru interaction from the methyl group, a Ru–Ru single bond and a four-electron donor bridging MeP ligand with P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ru double bonds, or a formal RuRu double bond with a two-electron donor bridging MeP ligand. The 8 lowest energy structures for the diiron monocarbonyl [MePFe2(CO)Cp2] are all triplet or quintet spin structures whereas the lowest energy structure for the diruthenium monocarbonyl [MePRu2(CO)Cp2] by more than 20 kcal mol−1 is a singlet structure with a formal RuRu double bond and bridging CO and four-electron donor MeP groups. Thermochemical information predicts such monocarbonyl derivatives to be the dominant binuclear decarbonylation products of the tricarbonyls [RPRu2(CO)3Cp2] by thermal or photochemical methods.
Ru double bonds, or a formal RuRu double bond with a two-electron donor bridging MeP ligand. The 8 lowest energy structures for the diiron monocarbonyl [MePFe2(CO)Cp2] are all triplet or quintet spin structures whereas the lowest energy structure for the diruthenium monocarbonyl [MePRu2(CO)Cp2] by more than 20 kcal mol−1 is a singlet structure with a formal RuRu double bond and bridging CO and four-electron donor MeP groups. Thermochemical information predicts such monocarbonyl derivatives to be the dominant binuclear decarbonylation products of the tricarbonyls [RPRu2(CO)3Cp2] by thermal or photochemical methods.
1. Introduction
Alkyl- and arylphosphinidenes provide examples of species that are not isolable in the free state but that can occur as stable ligands in transition metal complexes, particularly as bridges between two metal atoms.1 In such environments they can function either as two-electron donors or four electron donors. A two-electron donor phosphinidene ligand bridging a pair of metal atoms is similar to a bridging carbonyl group by forming P–M single bonds with each of the metal atoms (Fig. 1). This leaves a stereochemically active phosphorus lone pair so that the phosphorus atom lies significantly outside of the [M2C(alkyl)] plane. Experimental examples of two-electron donor phosphinidene ligands in binuclear cyclopentadienylmetal carbonyl chemistry include iron and ruthenium derivatives of the type [(μ-RP)M2(μ-CO)(CO)2Cp2] (Cp = η5-C5H5; M = Fe,2 R = cyclohexyl, phenyl, mesityl, and 2,4,6-tBu3C6H2; M = Ru,3 R = phenyl). Such species are related to the well-known [Cp2M2(μ-CO)2(CO)2] (M = Fe,4–6 Ru7,8) with a phosphinidene ligand replacing one of the carbonyl groups. | ||
| Fig. 1 Two-electron donor phosphinidene ligands and their occurrence in cyclopentadienylmetal carbonyl chemistry of iron and ruthenium. | ||
Phosphinidene ligands can also serve as four-electron donor bridging ligands through involvement of the phosphorus lone pair. Such a bridging four-electron donor phosphinidene ligand has either PM double bonds or P → M dative bonds to each of the metal atoms (Fig. 2). Four-electron donor bridging phosphinidene ligands ideally have the phosphorus atom in the [M2C(alkyl)] plane and shorter PM or P → M distances than the P–M distances of two-electron donor bridging phosphinidene ligands. A four-electron donor bridging phosphinidene ligand is found in the experimentally known molybdenum complex [(μ-2,4,6-tBu3C6H2P)Mo2(CO)4Cp2].9
 | ||
| Fig. 2 Four-electron donor phosphinidene ligands and their occurrence in cyclopentadienylmetal carbonyl chemistry of molybdenum. | ||
Decarbonylation of the binuclear cyclopentadienyliron carbonyl [Cp2Fe2(μ-CO)2(CO)2] using different methods yields two stable products of interest (Fig. 3). Thus photolysis of [Cp2Fe2(μ-CO)2(CO)2] gives a tricarbonyl [Cp2Fe2(μ-CO)3], which is of interest in representing a stable triplet state organometallic molecule with a formal FeFe σ + 2/2π double bond similar to the OO double bond in dioxygen.10–12 However, pyrolysis of [Cp2Fe2(μ-CO)2(CO)2] gives a very stable tetranuclear derivative [Cp4Fe4(μ3-CO)4] consisting of a central Fe4 tetrahedron in which each face is bridged by a μ3-CO carbonyl group.13 A binuclear [Cp2Fe2(CO)2] derivative is a possible intermediate in the formation of the tetranuclear [Cp4Fe4(μ3-CO)4]. A Fe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe formal triple bond is required for each iron atom to have the favored 18-electron configuration in such a [Cp2Fe2(CO)2] derivative.14,15 Density functional theory studies on [Cp2Fe2(CO)n] (n = 4, 3, 2, 1) systems are consistent with these experimental results.16
Fe formal triple bond is required for each iron atom to have the favored 18-electron configuration in such a [Cp2Fe2(CO)2] derivative.14,15 Density functional theory studies on [Cp2Fe2(CO)n] (n = 4, 3, 2, 1) systems are consistent with these experimental results.16
 | ||
| Fig. 3 Decarbonylation products of [Cp2Fe2(μ-CO)2(CO)2]. | ||
Experimental studies show the decarbonylation of the binuclear phosphinidene cyclopentadienyliron carbonyl complexes [(μ-RP)Fe2(μ-CO)(CO)2Cp2] to be much more complicated than that of [Cp2Fe2(μ-CO)2(CO)2] discussed above.17 In particular, trinuclear and tetranuclear decarbonylation products were found in which the phosphinidene phosphorus lone pair becomes involved in the metal–ligand bonding. Furthermore, the structure of the decarbonylation product was found to be dependent on the alkyl or aryl group of the phosphinidene ligand.
In order to gain some insight into possible intermediates for the decarbonylation of [(μ-RP)Fe2(μ-CO)(CO)2Cp2] derivatives we undertook a density functional theory study of the simplest such derivatives, namely the methylphosphinidene derivatives [(μ-MeP)Fe2(CO)nCp2] (n = 4, 3, 2, 1).18 The low-energy structures of the unsaturated dicarbonyl and monocarbonyl [(μ-MeP)Fe2(CO)nCp2] (n = 2, 1) systems were all found to be high-spin triplet and quintet structures rather than the singlet structures with iron–iron double and triple bonds found in their [Cp2Fe2(CO)n+1] (n = 2, 1) analogues. The observation of low-energy high-spin [(μ-MeP)Fe2(CO)nCp2] (n = 2, 1) structures with significant spin density on the iron atoms suggests mechanistic possibilities for forming new iron–iron bonds leading to the experimentally observed trinuclear and tetranuclear clusters.
Substituting ruthenium for iron in the [(μ-MeP)M2(CO)nCp2] derivatives is expected to lead to stronger ligand fields thereby disfavoring the high spin structures observed as low-energy structures for the iron derivatives. Accordingly, we have now used similar density functional methods to investigate the ruthenium derivatives [(μ-MeP)Ru2(CO)nCp2] (n = 4, 3, 2, 1) related to the experimentally known3 [PhPRu2(μ-CO)(CO)2Cp2]. In contrast to the iron systems, we find that the low-energy structures for the unsaturated ruthenium derivatives [(μ-MeP)Ru2(CO)nCp2] (n = 2, 1) are singlet spin state structures with examples of structures with RuRu formal double bonds and/or four-electron donor methylphosphinidine ligands. These theoretical studies predict that decarbonylation by photolysis or pyrolysis of ruthenium derivatives [(μ-RP)Ru2(μ-CO)(CO)2] is likely to lead to very different products than that of the corresponding iron derivatives.
2. Theoretical methods
The initial [MePRu2(CO)nCp2] chemical structures studied in this work have been designed by considering a [MePRu2Cp2] unit followed by systematic placement of carbonyl groups as terminal and/or bridging ligands (coordinating to the metal ions through the carbon as well as both the carbon and oxygen atoms). Various Cp–Ru–CO orientations were also considered. This led to 95 different [MePRu2(CO)nCp2] (n = 1 to 4) starting structures computed as singlets, triplets, and quintets.Full geometry optimizations were performed by using the PBE1PBE DFT functional19 and the def2-TZVP basis set as implemented in the Gaussian 09 software package20 applying an ultrafine integration grid and tight convergence criteria. The energies include the zero-point and thermal corrections at 273 K. The nature of the stationary points was characterized by their harmonic vibrational frequencies. Saddle point structures with imaginary vibrational frequencies were reoptimized by following the normal modes to ensure that genuine minima were obtained. The ν(CO) frequencies reported in the paper are scaled with a factor of 0.96. All of the lowest-energy structures have substantial HOMO–LUMO gaps ranging from 3.44 to 4.23 eV (Table S8 in the ESI†).
Only the lowest energy and thus potentially chemically significant structures are presented in detail in this paper. A larger number of structures of higher energy is presented in the ESI.† The optimized structures are designated as Ru2PCOn-mX, where n designates the number of carbonyl groups, m designates the energy ordering in terms of relative energy as compared to the global minimum of each family, and X designates the spin state as S (singlet), T (triplet), or Q (quintet).
3. Results and discussion
3.1. Structures of the tetracarbonyl [MePRu2(CO)4Cp2]
The metal atoms in the tetracarbonyls [MePM2(CO)4Cp2] (M = Fe, Ru) with a two-electron donor bridging MeP ligand have the favored 18-electron configuration without the need for a metal–metal bond. Thus the low-energy structures for the ruthenium tetracarbonyl derivative [MePRu2(CO)4Cp2] are singlet structures that are very similar to those of its iron analogue18 (Fig. 4 and Table 1). The lowest energy such structure Ru2PCO4-1S has a long non-bonding Ru⋯Ru distance of ∼4.20 Å and all terminal CO groups. The next higher energy [MePRu2(CO)4Cp2] structure, namely Ru2PCO4-2S at 11.4 kcal mol−1 in energy above Ru2PCO4-1S, also has a long non-bonding Ru⋯Ru distance of ∼4.28 Å. However, one of the CO groups in Ru2PCO4-2S bridges an Ru–P bond whereas the other three CO groups remain terminal groups. The CO group in Ru2PCO4-2S bridging a Ru–P bond exhibits a low ν(CO) frequency, namely 1734 cm−1. This ν(CO) frequency is not only more than 200 cm1 below the terminal ν(CO) frequencies at 1958, 2008, and 2056 cm−1 in Ru2PCO4-2S, similar to its iron analogue, but also ∼100 cm−1 below the ν(CO) frequencies of μ-CO groups bridging Ru–Ru bonds in the [MePRu2(CO)3Cp2] structures discussed below. The WBI values of 0.74 to 0.78 for the Ru–P bonds in Ru2PCO4-1S and Ru2PCO4-2S bonds are consistent with formal single bonds. | ||
| Fig. 4 The two lowest energy [MePRu2(CO)4Cp2] structures lying within 20 kcal mol−1 of the lowest energy structure. | ||
| Structure (symmetry) | ΔE | Ru–Ru interaction | Ru–P interactions | P distance (Å) | ΔE Fe | ||
|---|---|---|---|---|---|---|---|
| Distance | WBI | Distances | WBI | Above Ru2C plane | Analog | ||
| Ru2PCO4-1S (C1) | 0.0 | 4.202 | 0.02 | 2.45, 2.45 | 0.78, 0.76 | 0.79 | 1.1 |
| Ru2PCO4-2S (C1) | 11.4 | 4.277 | 0.04 | 2.33, 2.37 | 0.74, 0.75 | 0.14 | 8.8 |
The lowest four unoccupied molecular orbitals of Ru2PCO4-1S (Fig. 5) have dominant components on the metal centers, suggesting a formal description of di-Ru(II) bound to a di-anionic phosphinidene.
 | ||
| Fig. 5 Lowest unoccupied orbitals of Ru2PCO4-1S. | ||
3.2. Structures of the tricarbonyl [MePRu2(CO)3Cp2]
The metal atoms in the tricarbonyls [MePM2(CO)3Cp2] (M = Fe, Ru) with a two-electron donor bridging MeP ligand have the favored 18-electron configuration if there is a metal–metal formal single bond. The three lowest-energy [MePRu2(CO)3Cp2] structures, namely the three stereoisomers Ru2PCO3-1S, Ru2PCO3-2S, and Ru2PCO3-3S, all lying within 5 kcal mol−1 of energy, are of this type with Ru–Ru single bond distances of ∼2.67 Å having WBI values of 0.33 and corresponding very closely to the three lowest-energy structures of their iron analogues [MePFe2(CO)3Cp2] (Fig. 6 and Tables 2 and 3). The two-electron donor bridging MeP ligands in these three structures have Ru–P single bond distances of ∼2.36 Å with the phosphorus atom ∼1 Å outside of the Ru2C(methyl) plane indicating a stereochemically active lone pair. One of the three CO groups in each of the three structures bridges the Ru–Ru bond. The remaining two CO groups are terminal CO groups with one on each ruthenium atom. The bridging CO groups in each of the three structures Ru2PCO3-1S, Ru2PCO3-2S, and Ru2PCO3-3S exhibit a ν(CO) frequency around 1820 cm−1 which is significantly lower than the terminal ν(CO) frequencies at 1980 cm−1 and higher (Table 3) Both Ru2PCO3-1S and Ru2PCO3-3S have the pair of Cp rings in cis orientations relative to the Ru–Ru bond. However, in Ru2PCO3-1S the terminal CO groups are cis relative to the MeP bridge whereas in Ru2PCO3-3S the terminal CO groups are trans to the MeP bridge and the bridging MeP group. Structure Ru2PCO3-2S has the two Cp rings and the two terminal CO groups in trans orientations relative to the Ru–Ru bond. The WBI values for the Ru–P bonds in Ru2PCO3-1S, Ru2PCO3-2S, and Ru2PCO3-3S all fall in the range 0.77 to 0.80 consistent with formal single bonds. | ||
| Fig. 6 The four lowest energy [MePRu2(CO)3Cp2] structures lying within 31 kcal mol−1 of the lowest energy structure. | ||
| Structure (symmetry) | ΔE | Ru–Ru interaction | Ru–P interactions | P distance (Å) | ΔE Fe | ||
|---|---|---|---|---|---|---|---|
| Distance | WBI | Distances | WBI | Above Ru2C plane | Analog | ||
| Ru2PCO3-1S (Cs) | 0.0 | 2.761 | 0.33 | 2.35, 2.35 | 0.80, 0.80 | 0.95 | 0.0 |
| Ru2PCO3-2S (C1) | 2.1 | 2.774 | 0.33 | 2.35, 2.37 | 0.77, 0.79 | 1.01 | 3.3 |
| Ru2PCO3-3S (Cs) | 4.2 | 2.767 | 0.33 | 2.37, 2.37 | 0.77, 0.77 | 1.07 | 4.8 |
| Ru2PCO3-4S (C1) | 15.2 | 4.098 | 0.06 | 2.15, 2.38 | 0.73, 1.46 | 0.06 | — |
| Structure | ν(CO) frequencies, cm−1 |
|---|---|
| Ru2PCO3-1S | 1822(557), 1981(262), 2014(1286) |
| Ru2PCO3-2S | 1818(556), 1978(1257), 1990(191) |
| Ru2PCO3-3S | 1818(561), 1993(254), 2026(1411) |
| Ru2PCO3-4S | 1956(1138), 2000(743), 2045(676) |
The lowest unoccupied molecular orbitals of Ru2PCO3-1S (Fig. 7) include an empty σ* Ru–Ru antibonding component (LUMO) alongside an occupied σ orbital (HOMO−4), confirming a net order of 1 with respect to the Ru–Ru interaction. Also, the fact that four LUMO's are located on the metals confirms a formal description of di-Ru(II) and implicitly a di-anionic phosphinidene.
 | ||
| Fig. 7 Frontier molecular orbitals of Ru2PCO3-1S. | ||
Our previous study of the [MePFe2(CO)3Cp2] system18 revealed three triplet structures within 9 kcal mol−1 of the lowest-energy isomer analogous to Ru2PCO3-1S. For the analogous ruthenium [MePRu2(CO)3Cp2] system no triplet structures were found within 31 kcal mol−1 of the lowest energy isomer Ru2PCO3-1S relating to the higher ligand field strength in ruthenium complexes relative to their iron analogues. However, in the ruthenium system [MePRu2(CO)3Cp2] a fourth singlet structure Ru2PCO3-4S was found with an energy of 15.2 kcal mol−1 relative to that of Ru2PCO3-1S (Fig. 6 and Tables 2 and 3). An analogous iron structure was not found in the [MePFe2(CO)3Cp2] system. Structure Ru2PCO3-4S differs from its three lower energy isomers in having a long non-bonding Ru⋯Ru distance of ∼4.10 Å. Furthermore, the bridging MeP ligand in Ru2PCO3-4S is a four-electron donor with a formal PRu double bond of length 2.15 Å and a WBI value of 1.46 to the ruthenium atom bearing a single terminal CO group and a dative P → Ru single bond of length 2.38 Å and WBI value of 0.73 to the ruthenium atom bearing two terminal CO groups. Furthermore, the phosphorus atom in the bridging MeP ligand of Ru2PCO3-4S lies only 0.06 Å out of the Ru2C(methyl) plane confirming the participation of its lone pair in the ligand–metal bonding.
3.3. Structures of the dicarbonyl [MePRu2(CO)2Cp2]: major differences between the ruthenium and iron systems
The metal atoms in the dicarbonyls [MePM2(CO)2Cp2] (M = Fe, Ru) with a two-electron donor bridging MeP ligand require a formal MM double bond in order to attain the favored 18-electron configuration in a singlet spin state structure. However, for the iron system [MePFe2(CO)2Cp2] no fewer than 11 triplet and quintet spin state isomers are found of lower energy than the lowest energy singlet structure lying 9.7 kcal mol−1 above the lowest energy higher spin state structure. The corresponding ruthenium system [MePRu2(CO)2Cp2] is very different since six singlet structures are found with lower energies than the lowest energy triplet structure Ru2PCO2-7T lying 15.3 kcal mol−1 above the global minimum Ru2PCO2-1S (Fig. 8 and Tables 4 and 5). Also for the iron system [MePFe2(CO)2Cp2] six quintet spin state structures were found within 8 kcal mol−1 of the lowest energy structure whereas for the analogous ruthenium system no quintet structures were found within 30 kcal mol−1 of the lowest energy structures. These differences are a clear reflection of the higher ligand field strength in complexes of the second row metal ruthenium relative to its first row analogue iron. This also suggests that the nature of the products formed by photochemical or thermal decarbonylation of [RPRu2(CO)3Cp2] derivatives is likely to be very different than those formed by decarbonylation of their iron analogues.
 | ||
| Fig. 8 The 12 lowest energy [MePRu2(CO)2Cp2] structures lying within 30 kcal mol−1 of the lowest energy structure. The C–H–Ru agostic interactions in Ru2PCO2-1S and Ru2PCO2-3S are marked with dash lines. | ||
| Structure (symmetry) | ΔE | Ru–Ru interaction | Ru–P interactions | P distance (Å) | ΔE Fe | ||
|---|---|---|---|---|---|---|---|
| Distance | WBI | Distances | WBI | Above Ru2C plane | Analog | ||
| Ru2PCO2-1S (C1) | 0.0 | 2.752 | 0.37 | 2.29, 2.34 | 0.79, 0.82 | 1.29 | |
| Ru2PCO2-2S (C1) | 2.7 | 2.731 | 0.36 | 2.26, 2.34 | 1.03, 0.84 | 0.88 | 9.7 |
| Ru2PCO2-3S (C1) | 4.1 | 2.772 | 0.33 | 2.29, 2.36 | 0.80, 0.81 | 1.37 | 14.5 |
| Ru2PCO2-4S (C1) | 6.6 | 3.022 | 0.38 | 2.18, 2.19 | 1.15, 1.13 | 0.49 | |
| Ru2PCO2-5S (Cs) | 11.3 | 3.088 | 0.38 | 2.17, 2.18 | 1.08, 1.20 | 0.44 | |
| Ru2PCO2-6S (Cs) | 13.7 | 2.506 | 0.57 | 2.38, 2.38 | 0.78, 0.78 | 1.10 | |
| Ru2PCO2-7T (Cs) | 15.3 | 2.496 | 0.44 | 2.37, 2.37 | 0.76, 0.76 | 1.11 | 7.0 |
| Ru2PCO2-8T (Cs) | 17.7 | 2.751 | 0.46 | 2.23, 2.30 | 0.77, 0.97 | 0.78 | |
| Ru2PCO2-9T (Cs) | 18.1 | 2.748 | 0.33 | 2.24, 2.28 | 0.94, 0.89 | 0.78 | 0.0 |
| Ru2PCO2-10T (C2v) | 19.4 | 2.624 | 0.34 | 2.28, 2.28 | 0.88, 0.88 | 0.78 | 7.0 |
| Ru2PCO2-11T (Cs) | 23.4 | 4.053 | 0.09 | 2.21, 2.22 | 1.07, 1.13 | 0.08 | 8.6 |
| Ru2PCO2-12T (C2) | 25.6 | 4.031 | 0.10 | 2.22, 2.22 | 1.11, 1.09 | 0.01 | 6.9 |
| Structure | ν(CO) frequencies, cm−1 |
|---|---|
| Ru2PCO2-1S | 1810(563), 1977(960) |
| Ru2PCO2-2S | 1801(584), 1987(771) |
| Ru2PCO2-3S | 1807(588), 1972(730) |
| Ru2PCO2-4S | 1952(1692), 1964(63) |
| Ru2PCO2-5S | 1955(715), 1981(1373) |
| Ru2PCO2-6S | 1804(789), 1880(541) |
| Ru2PCO2-7T | 1827(1027), 1857(256) |
| Ru2PCO2-8T | 1944(1485), 1958(128) |
| Ru2PCO2-9T | 1811(607), 1987(846) |
| Ru2PCO2-10T | 1812(919), 1847(259) |
| Ru2PCO2-11T | 1955(1268), 1968(670) |
| Ru2PCO2-12T | 1959(1302), 1976(1280) |
The three lowest-energy [MePRu2(CO)2Cp2] structures, namely Ru2PCO2-1S, Ru2PCO2-2S, and Ru2PCO2-3S, lying within 5 kcal mol−1 of energy, are a related series of singlet stereoisomers with single bridging CO and MeP groups (Fig. 8 and Table 4). The bridging MeP group is a two-electron donor as indicated by Ru–P single bond distances of ∼2.27 and ∼2.35 Å with WBI values in the range 0.79 to 1.03 and phosphorus distances 0.88 to 1.37 Å above the Ru2C(methyl) plane. The Ru–Ru bonds in these three structures are clearly formal single bonds with Ru–Ru distances ranging from 2.73 to 2.77 Å and WBIs ranging from 0.33 to 0.37 Å. In both Ru2PCO2-1S and Ru2PCO2-3S there are C–H–Ru agostic interactions with Ru–H distances of 1.826 and 1.836 Å, respectively, of one of the methyl hydrogens to the ruthenium atom not bearing a terminal CO group. Such an agostic interaction effectively results in the donation of the electron pair forming a methyl C–H bond to the nearby ruthenium atom. This combined with the two-electron donor MeP ligands, the usual two-electron donor CO groups, and a single Ru–Ru bond gives each ruthenium atom in Ru2PCO2-1S and Ru2PCO2-3S the favored 18-electron configuration. Structure Ru2PCO2-1S has a cis orientation of the Cp rings and a trans orientation of the CO groups whereas Ru2PCO2-3S has a trans orientation of the Cp rings and a cis orientation of the CO groups. Structure Ru2PCO2-2S does not have a similar C–H–Ru agostic interaction and both of its CO groups are the usual two-electron donor CO groups. Therefore the ruthenium atom in Ru2PCO2-2S not bearing the terminal CO group has only a 16-electron configuration.
The next two singlet [MePRu2(CO)2Cp2] structures, namely Ru2PCO2-4S and Ru2PCO2-5S, lying 6.6 and 11.3 kcal mol−1 can be interpreted as having formal Ru–Ru single bonds and four-electron donor bridging MeP groups thereby giving each ruthenium atom the favored 18-electron configuration (Fig. 8 and Table 4). The lengths of the Ru–Ru single bonds in Ru2PCO2-4S and Ru2PCO2-5S are 3.022 and 3.088 Å, respectively, corresponding to WBI values of 0.38. These Ru–Ru single bond distances in Ru2PCO2-4S and Ru2PCO2-5S without a bridging CO group are ∼0.3 Å longer than the Ru–Ru distances in Ru2PCO2-1S, Ru2PCO2-2S, and Ru2PCO2-3S with a bridging CO group. The PRu double bonds from the four-electron donor bridging MeP group to the ruthenium atom of lengths 2.18 Å are clearly shorter than the P–Ru single bonds of lengths ∼2.3 Å in several of the structures discussed above. The WBIs of these PRu double bonds ranging from 1.08 to 1.20 are somewhat higher than the WBIs of the P–Ru single bonds in the carbonyl-richer [MePRu2(CO)nCp2] (n = 4, 3) structures discussed above.
Similarly to what is seen for Ru2PCO3-1S, the lowest unoccupied molecular orbitals of Ru2PCO2-1S (Fig. 9) include an empty σ* Ru–Ru antibonding component (LUMO) consistent with a net order of 1 with respect to the Ru–Ru interaction, and four LUMO's located on the metals consistent with a formal description of di-Ru(II) and implicitly a di-anionic phosphinidene.
 | ||
| Fig. 9 Frontier molecular orbitals of Ru2PCO2-1S. | ||
The [MePRu2(CO)2Cp2] structure Ru2PCO2-6S, lying 13.7 kcal mol−1 in energy above Ru2PCO2-1S, is the lowest energy structure clearly having the formal RuRu double bond required to give each ruthenium atom the favored 18-electron configuration in a structure with a two-electron donor bridging MeP group (Fig. 8 and Table 4). In Ru2PCO2-6S both CO groups bridge the relatively short RuRu double bond of length 2.596 Å with a WBI of 0.57 significantly higher than the WBI of the Ru–Ru single bonds found in other structures. The Ru–P single bonds in Ru2PCO2-6S have typical lengths of 2.38 Å and WBI values of 0.78 with the phosphorus atom 1.10 Å outside the Ru2C(methyl) plane.
The next six [MePRu2(CO)2Cp2] structures from Ru2PCO2-7T to Ru2CO2-12T in energy above the six singlet structures Ru2PCO2-1S to Ru2CO2-6S are triplet structures ranging in energy from 15.3 to 25.6 kcal mol−1 above Ru2PCO-1S (Fig. 8 and Table 3). Five of these six triplet [MePRu2(CO)2Cp2] structures have counterparts in low-energy structures in the analogous iron system [MePFe2(CO)2Cp2].18 Structure Ru2PCO2-7T is the triplet counterpart of the singlet structure Ru2PCO2-6S with a RuRu double bond of length 2.496 Å with a WBI of 0.44. This gives each ruthenium atom in Ru2PCO2-7T the favored 18-electron configuration leaving the two unpaired electrons in a RuRu double bond of the σ + 2/2π type analogous to the OO double bond in dioxygen. Structure Ru2PCO2-7T can be related to the experimentally known10–12 triplet binuclear cyclopentadienyliron carbonyl derivative [Cp2Fe2(μ-CO)3] by replacement of iron with ruthenium and replacement of one of the bridging CO groups with a two-electron donor bridging MeP group. In addition Ru2PCO2-7T can be contrasted with Ru2PCO2-10T, also with a bridging two-electron donor bridging MeP group and two bridging μ-CO groups, but with a formal Ru–Ru single bond of length 2.624 Å having a WBI of 0.34. Thus in Ru2PCO2-10T the triplet spin state arises from a 17-electron configuration of each ruthenium atom rather than from the σ + 2/2π RuRu double bond in Ru2CO2-7T.
The next two triplet [MePRu2(CO)2Cp2] structures Ru2PCO2-11T and Ru2PCO2-12T, lying 23.4 and 25.6 kcal mol−1, respectively, in energy above Ru2PCO2-1S, are a pair of stereoisomers with trans and cis orientations, respectively, of the single terminal CO group on each ruthenium atom (Fig. 8 and Table 4). The long Ru⋯Ru distances of ∼4.04 Å in Ru2PCO2-11T and Ru2PCO2-12T suggest the lack of a formal ruthenium–ruthenium bond. The presence of the phosphorus atom less than 0.1 Å out of the Ru2C(methyl) plane suggests a four-electron donor bridging MeP group. The PRu distances of 2.22 Å with WBI values ranging from 1.07 to 1.13 can be interpreted as double bonds from the phosphorus atom to each ruthenium atom. The combination of these double bonds, a terminal CO group on each ruthenium atom, and the Cp ligand gives each ruthenium atom in Ru2PCO2-11T and Ru2PCO2-12T a 17-electron configuration consistent with a binuclear triplet.
3.4. Structures of the monocarbonyl [MePRu2(CO)Cp2]
The potential energy surface of the monocarbonyl [MePRu2(CO)Cp2] is remarkably simple since a single structure Ru2PCO-1S is found to lie a full 24 kcal mol−1 below the next lowest energy structure (Fig. 10). The short PRu distances of 2.17 Å with WBI values of 1.2 suggest the possibility of formal double bonds corresponding to a four-electron donor bridging MeP group. Interpreting the RuRu distance of 2.73 Å with a WBI of 0.49 somewhat higher than the WBI values below 0.4 for Ru–Ru single bonds in singlet structures as a formal double bond gives each ruthenium atom in Ru2PCO-1S the favored 18-electron configuration. The single CO group in Ru2PCO-1S exhibits a ν(CO) frequency of 1783 cm−1 consistent with its bridging position. The iron analogue of Ru2PCO-1S in the [MePFe2(CO)Cp] system is the lowest energy singlet structure but lies 16.7 kcal mol−1 above the quintet spin state global minimum. This is another example of the effect of the larger ligand field strengths in ruthenium complexes relative to analogous iron complexes.
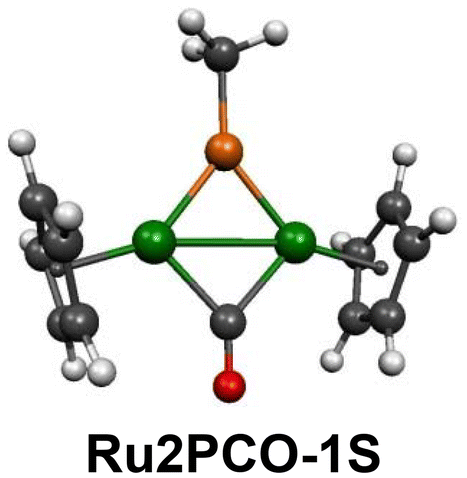 | ||
| Fig. 10 The [MePRu2(CO)Cp2] structure lying ∼24 kcal mol−1 below the next lowest energy structure. | ||
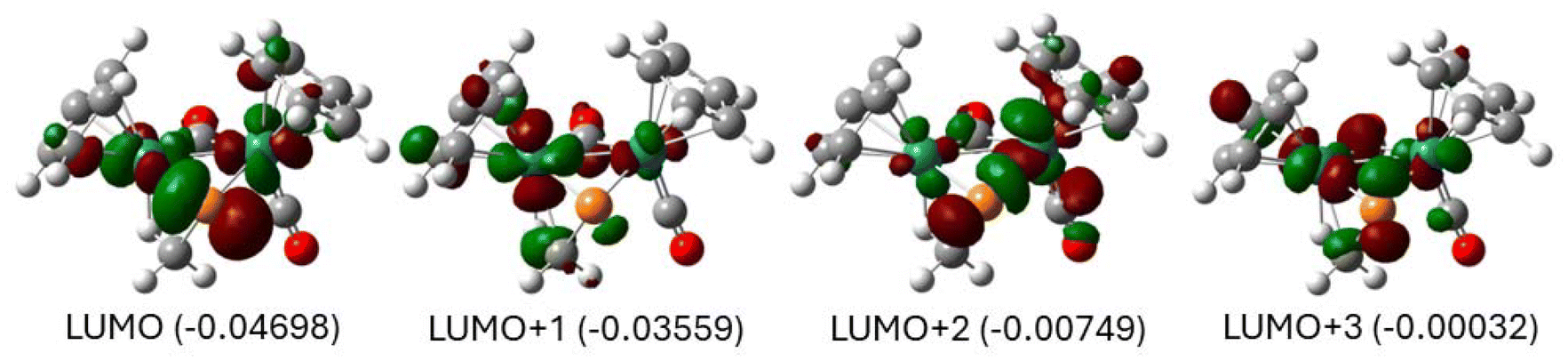
Of the frontier molecular orbitals of Ru2PCO-1S (Fig. 11), the four lowest-energy LUMOs are located on the ruthenium atoms consistent with two d6 centers. This formal di-Ru(II) description implies the phosphinidene ligand is acting as a dianion. The orbitals in Fig. 11 also indicate that the phosphorus atom is essentially non-hybridized, as expected.21 The spatial distribution of LUMO+1 suggests a two-electron three-center interaction of the two metals with a phosphorus p lone pair. In this interaction, the LUMO+1 is the π* component, while the other two orbitals, HOMO−2 and HOMO−6, display π Ru–Ru bonding character. On the other hand, orbitals HOMO−1 and HOMO−5 indicate that the bond order for the σ interaction between the two metals is zero, as the antibonding HOMO−1 is doubly occupied. Thus, the net bond order for the Ru–Ru interaction is 1, arising from a π bond and with no σ bonding.
 | ||
| Fig. 11 Frontier molecular orbitals of Ru2PCO-1S. | ||
3.5. Thermochemistry
Table 6 lists the carbonyl dissociation energies (ΔH and ΔG) for carbonyl dissociation processes of the type [MePRu2(CO)nCp2] → [MePRu2(CO)n−1Cp2] + CO considering the lowest-energy structures, all of which are singlets. Such carbonyl dissociation processes are seen to be endothermic in all cases. However, carbonyl dissociation from the tetracarbonyl [MePRu2(CO)4Cp2] to give the tricarbonyl [MePRu2(CO)3Cp2] thereby forming a Ru–Ru bond is only weakly endothermic suggesting that this process can occur relatively easily. More significantly, combining the carbonyl dissociation energies for the tricarbonyl [MePRu2(CO)3Cp2] and the dicarbonyl [MePRu2(CO)2Cp2] shows that the disproportionation of the dicarbonyl into the tricarbonyl and monocarbonyl is significantly exothermic. This suggests that the decarbonylation of the stable tricarbonyl, such as the experimentally known3 [PhPRu2(μ-CO)(CO)2Cp], by photochemical or thermal methods is likely to lead directly to the monocarbonyl [MePRu2(CO)Cp2] for which structure Ru2PCO-1S is clearly a thermodynamic sink lying more than 20 kcal mol−1 below any of its isomers.| Reaction | ΔH | ΔG |
|---|---|---|
| [MePRu2(CO)4Cp2] (Ru2PCO4-1S) → [MePRu2(CO)3Cp2] (Ru2PCO3-1S) + CO | 13.1 | 3.8 |
| [MePRu2(CO)3Cp2] (Ru2PCO3-1S) → [MePRu2(CO)2Cp2] (Ru2PCO2-1S) + CO | 42.9 | 32.7 |
| [MePRu2(CO)2Cp2] (Ru2PCO2-1S) → [MePRu2(CO)Cp2] (Ru2PCO-1S) + CO | 27.5 | 14.1 |
| 2 [MePRu2(CO)2Cp2] → [MePRu2(CO)3Cp2] + [MePRu2(CO)Cp2] | −15.4 | −18.6 |
4. Conclusion
The potential energy surfaces for the tetracarbonyls [MePM2(CO)4Cp2] and the tricarbonyls [MePM2(CO)3Cp2] (M = Fe, Ru) are very similar for the iron and ruthenium systems. The energetically favored structures for both metals have a two-electron donor bridging MeP group without a direct metal–metal bond for the tetracarbonyls and a metal–metal single bond for the tricarbonyls thereby leading to the favored 18-electron metal configurations in all cases. The tetracarbonyls and tricarbonyls are thus saturated systems where the favored structures are not likely to be affected by the difference in ligand field strengths between analogous iron and ruthenium derivatives.The situation is very different for the dicarbonyls and monocarbonyls [MePM2(CO)nCp2] (n = 2, 1; M = Fe, Ru). For the iron dicarbonyl system [MePFe2(CO)2Cp2] the 11 lowest-energy structures are all triplet or quintet structures with the lowest-energy singlet isomer lying ∼10 kcal mol−1 above the overall lowest-energy structure. However, for the ruthenium dicarbonyl system [MePRu2(CO)2Cp2] the six lowest energy structures are all singlets with the lowest energy triplet isomer lying ∼15 kcal mol−1 above the lowest energy structure. This major difference between the preferred structure types for the iron and ruthenium derivatives of the type [MePM2(CO)nCp2] is clearly a manifestation of the effect of the higher ligand field strengths in ruthenium complexes relative to iron complexes.
A ruthenium derivative of the type [MePRu2(CO)2Cp] with a two-electron donor bridging MeP group requires a formal RuRu double bond for each ruthenium atom to have the favored 18-electron configuration. An example of such a structure is Ru2PCO2-6S (Fig. 8 and Table 4). However, this structure lies 13.7 kcal mol−1 above its lowest energy isomer Ru2PCO2-1S. Both Ru2PCO2-1S and the slightly higher energy Ru2PCO2-3S have only a Ru–Ru single bond. However, in both of these structures there is an agostic C–H–Ru interaction between one of the methyl hydrogens and the ruthenium atom. The resulting donation of the two electrons from a methyl C–H bond to a ruthenium atom combined with a Ru–Ru single bond can give both ruthenium atoms the favored 18-electron configuration. The ruthenium atoms in the [MePRu2(CO)2Cp] isomers Ru2PCO2-4S and Ru2CO2-5S acquire the favored 18-electron configuration in a still different manner, namely by having four-electron donor bridging MeP groups rather that two-electron donor MeP groups combined with an Ru–Ru single bond. The four-electron donor bridging MeP groups in Ru2PCO2-4S and Ru2CO2-5S exhibit relatively short PRu distances of ∼2.18 Å suggesting formal double bonds.
The effect of the higher ligand field strength of ruthenium complexes relative to analogous iron complexes is also exhibited in the monocarbonyl systems [MePM2(CO)Cp2] (M = Fe, Ru). For the iron system [MePFe2(CO)Cp2] the eight lowest energy structures are triplet and quintet spin state structures with the lowest energy singlet structure lying 16.7 kcal mol−1 above the lowest energy isomer. The same type of singlet [MePRu2(CO)Cp2] structure in the analogous ruthenium system, namely Ru2PCO-1S with bridging CO and MeP groups, is not only the lowest energy such structure but also lies more than 20 kcal mol−1 below the next lowest energy isomer. The ruthenium atoms in Ru2PCO-1S attain the favored 18-electron configuration through a formal RuRu double bond and its MeP bridge being a four-electron donor with PRu double bond distances of ∼2.15 Å. Thermochemical information on CO dissociation energies predict the [RPRu2(CO)Cp2] structures of the type Ru2PCO-1S to be the preferred product from the decarbonylation of [RPRu2(CO)3Cp2] derivatives by thermal or photochemical methods.
Data availability
The optimized coordinates of the structures presented in this manuscript are provided as a separate standard xyz-file which can be visualized with any visualiser program – we recommend using Mercury (from CCDC) which is a free program. The ESI† also contains the distance matrix between all the atoms, as provided by the Gaussian program. All of the Gaussian log files of all the structures presented in this manuscript are available upon request.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The computational facilities were provided by the Babeş-Bolyai University under project POC/398/1/1/124155 co-financed by the European Regional Development Fund (ERDF) through the Competitiveness Operational Program for Romania 2014–2020.References
- M. E. García, D. García-Vivó, A. Ramos and M. A. Ruiz, Phosphinidene-bridged bincuclear complexes, Coord. Chem. Rev., 2017, 330, 1–36 CrossRef.
- C. M. Alvarez, M. G. Alvarez, M. E. García, R. González, M. A. Ruiz, H. Hamidov and J. C. Jeffery, High-yield synthesis and reactivity of stable diiron complexes with bent-phosphinidene bridges, Organometallics, 2005, 24, 5503–5505 CrossRef CAS.
- S. Takemoto, Y. Kimura, K. Kammikawa and H. Matsuzaka, P–H bond addition to a dinuclear ruthenium imido complex: Synthesis and reactivitiy of an amido phosphide complex, Organometallics, 2008, 27, 1780–1785 CrossRef CAS.
- O. S. Mills, The crystal structure of dicyclopentadienyldiiron tetracarbonyl, Acta Crystallogr., 1958, 11, 620–623 CrossRef CAS.
- R. F. Bryan and P. T. Greene, Crystal structure of trans-di-μ-carbonyldicarbonyldi-π-cyclopentadienyldi-iron (Fe-Fe), a redetermination, J. Chem. Soc. A, 1970, 3064–3068 RSC.
- A. Mitschler, B. Rees and M. S. Lehmann, Electron density in bis(dicarbonyl-π-cyclopentadienyliron) at liquid-nitrogen temperature by X-ray and ue, J. Am. Chem. Soc., 1978, 100, 3390–3397 CrossRef CAS.
- E. O. Fischer and A. Vogler, Dimeric cyclopentadienylrutheniumdicarbonyl, Z. Naturforsch., 1962, 17b, 421–422 CrossRef CAS.
- O. S. Mills and J. P. Nice, Structure of bis(cyclopentadienyldicarbonylruthenium), J. Organomet. Chem., 1967, 9, 339–344 CrossRef CAS.
- A. M. Arif, A. H. Cowley, N. C. Norman, A. G. Orpen and M. Pakulski, Transition-metal phosphinidene complexes: Syntheses, structures, and bonding in dinuclear phosphinidene complexes containing 14- and 15-electron metal fragments, Organometallics, 1988, 7, 309–318 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, Mechanistic aspects of the photochemistry of metal-metal bonds—evidence for the intervention of two different primary photoproducts in the photochemistry of (η5-C5H5)2Fe2(CO)4, J. Am. Chem. Soc., 1980, 102, 7794–7795 CrossRef CAS.
- R. H. Hooker, K. A. Mahmoud and A. J. Rest, Spectroscopic evidence for the formation of tri-μ-carbonyl-bis{(η5-cyclopentadienyl)iron} on photolysis of bis[(dicarbonyl)η5-cyclopentadienyl)iron} in low-temperature matrices, J. Chem. Soc., Chem. Commun., 1983, 1022–1024 RSC.
- A. F. Hepp, J. P. Blaha, C. Lewis and M. S. Wrighton, Photochemistry of (η5-C5H5)2Fe2(CO)4 and related complexes in rigid matrices at low temperature—loss of carbon monoxide from the trans isomer to yield triply CO –bridged species, Organometallics, 1984, 3, 174–177 CrossRef CAS.
- R. B. King, Polynuclear cyclopentadienyl metal carbonyls of iron and cobalt, Inorg. Chem., 1966, 5, 2227 CrossRef CAS.
- F. A. Kvietok and B. E. Bursten, Stepwise photochemical CO loss from Cp2Fe2(CO)2(μ-CO)2 in low-temperature matrices—evidence for an unsupported FeFe triple bond, J. Am. Chem. Soc., 1994, 116, 9807–9808 CrossRef CAS.
- M. Vitale, M. E. Archer and B. E. Bursten, Theoretical and experimental studies of the unprecedented spin-dependent structures of Cp2Fe2(CO)2, the double-CO-loss product of Cp2Fe2(CO)4, Chem. Commun., 1998, 179–180 RSC.
- H. Y. Wang, Y. Xie, R. B. King and H. F. Schaefer, Unsaturation in binuclear cyclopentadienyliron carbonyls, Inorg. Chem., 2006, 45, 3384–3392 CrossRef CAS.
- M. A. Alvarez, M. E. García, R. González, A. Ramos and M. Ruiz, Synthesis and decarbonylation reactions of diiron cyclopentadienyl complexes with bent-phosphinidene bridges, Organometallics, 2011, 30, 1102–1115 CrossRef CAS.
- O. Rudenco, A. Lupan, R. Silagni-Dumitrescu and R. B. King, Decarbonylation products of binuclear methylphosphinidene complexes of cyclopentadienyliron carbonyls: triplet and quintet structures are favored energetically over singlet structures with iron-iron multiple bonding, ACS Omega, 2024, 9, 12125–12134 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- Gaussian09 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2009. The complete reference is given in the ESI.†.
- R. Silaghi-Dumitrescu and F. J. Carrascoza-Mayen, A twist in the anomeric effect, Stud. Univ. Babes-Bolyai, Chem., 2014, LIX(3), 95–101 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1. Initial [MePRu2(CO)nCp2] structures. Table S2. Distance table for the lowest-lying [Cp2Ru2PMe] structures. Table S3. Distance table for the lowest-lying [MePFe2(CO)Cp2] structures. Table S4. Distance table for the lowest-lying [MePRu2(CO)2Cp2] structures. Table S5. Distance table for the lowest-lying [MePRu2(CO)3Cp2] structures. Table S6. Distance table for the lowest-lying [MePRu2(CO)4Cp2] structures. Table S7. Orbital energies and HOMO/LUMO gaps. Complete Gaussian reference (ref. 20). Separated concatenated xyz-file containing the Cartesian coordinates of the optimized structures which can be visualized with a variety of software such as Mercury CCDC which is a free program. See DOI: https://doi.org/10.1039/d4dt02279c |
| This journal is © The Royal Society of Chemistry 2024 |