
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multinuclear beryllium amide and imide complexes: structure, properties and bonding†
Deniz F.
Bekiş‡
 ,
Lewis R.
Thomas-Hargreaves‡
,
Sergei I.
Ivlev
and
Magnus R.
Buchner
*
,
Lewis R.
Thomas-Hargreaves‡
,
Sergei I.
Ivlev
and
Magnus R.
Buchner
*
Anorganische Chemie, Nachwuchsgruppe Hauptgruppenmetallchemie, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: magnus.buchner@chemie.uni-marburg.de; Fax: +49 (0)64212825669; Tel: +49 (0)6421 2825668
First published on 27th August 2024
Abstract
The beryllium amide and imide complexes [Be(HNMes)2]3, [(py)2Be(HNMes)2], [Be(HNDipp)2]2, [Be(NPh2)(μ2-HNDipp)]2 and [Be(NCPh2)2]3 have been prepared and characterised with NMR and IR spectroscopy as well as single crystal X-ray diffraction. Analysis of the localised molecular orbitals (LMOs) and intrinsic atomic orbital (IAO) atomic charges in the framework of the intrinsic bond orbital (IBO) localization method revealed a covalent bonding network consisting of 2-electron–2-centre and 2-electron–3-centre σ bonds, in which one electron pair of the anionic N-donor ligands is involved. The electron deficiency at the beryllium atoms is partially compensated through additional electron donation from the lone pair at the nitrogen atoms.
1 Introduction
The chemistry of low valent s-block and early p-block metals has received a lot of attention in the last two decades.1–4 Most of these low valent compounds were isolated utilising N-donor ligands.5–8 This is not the case, however, for the lightest group 2 metal, beryllium, of which low valent compounds have, with one exception,9 only been realised with C-donor ligands.10–16 There are only few described reductions of N-donor complexes of beryllium, which resulted in the formation of beryllates.17–19 Considering the huge number of beryllium complexes with N-donor ligands that have been recently published,20–35 it can be assumed that reduction attempts have been performed on some of these systems but did not yield defined compounds. Furthermore, no generally applicable reducing agent to obtain low valent beryllium has been found yet, and each ligand system requires special reduction conditions.10–16 Therefore, it is evident that too little is known about the properties of beryllium compounds to rationally select ligands and reducing agents. For this reason we have been studying simple beryllium halide complexes and organoberyllium compounds extensively.36–44Considering the abundance of N-donor complexes in main group metal chemistry, it is no surprise that there are many reports on heteroleptic organo beryllium amines, amides and some imides of the types (R3N)BeX2 (X = Cl, Br, I),54,55 [(R2N)BeR′]n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56–59 and [(R2C
56–59 and [(R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)BeR′]n, respectively.60 However, there are only few described homoleptic beryllium N-donor compounds. To the best of our knowledge [Be(NH3)4]2+ (I, Fig. 1)45,46 and [Be(py)4]2+61 are the only homoleptic beryllium compounds in which the metal is only coordinated by amines. In homoleptic beryllium amides, the coordination number of the central atom varies. In [Be(N(SiMe3)2)2] (II) the beryllium atom is dicoordinated,47–50 and the Be–N bond exhibits partial double bond character.50 Similar bonding has also been described in a beryllium imido complex.62 Reaction of BeCl2 with dialkyl amines and subsequent treatment with organolithium compounds gives homoleptic [Be(NR2)2]1–3, which are described as mono-, di- and trimeric in solution.63 [Be(NiPr2)2]1–2 is described to be isolatable as a mono- and a dimeric isomer, however their reactivity with CS2 is identical.64 With a sterically demanding imide dinuclear [Be(NCtBu2)2]2 (III) was obtained and its structure and spectroscopic properties investigated,51,52 while utilisation of small dimethylamide results in the formation of trinuclear [Be(NMe2)2]3 (IV).53
N)BeR′]n, respectively.60 However, there are only few described homoleptic beryllium N-donor compounds. To the best of our knowledge [Be(NH3)4]2+ (I, Fig. 1)45,46 and [Be(py)4]2+61 are the only homoleptic beryllium compounds in which the metal is only coordinated by amines. In homoleptic beryllium amides, the coordination number of the central atom varies. In [Be(N(SiMe3)2)2] (II) the beryllium atom is dicoordinated,47–50 and the Be–N bond exhibits partial double bond character.50 Similar bonding has also been described in a beryllium imido complex.62 Reaction of BeCl2 with dialkyl amines and subsequent treatment with organolithium compounds gives homoleptic [Be(NR2)2]1–3, which are described as mono-, di- and trimeric in solution.63 [Be(NiPr2)2]1–2 is described to be isolatable as a mono- and a dimeric isomer, however their reactivity with CS2 is identical.64 With a sterically demanding imide dinuclear [Be(NCtBu2)2]2 (III) was obtained and its structure and spectroscopic properties investigated,51,52 while utilisation of small dimethylamide results in the formation of trinuclear [Be(NMe2)2]3 (IV).53
 | ||
| Fig. 1 Structurally authenticated mono-, di- and trinuclear beryllium complexes with N-donor ligands.45–53 | ||
Due to the large variety of the ligands in these homoleptic amide and imide complexes, no clear trends concerning the influence of electronic and steric properties of the ligands onto the constitution of the respective beryllium complexes can be derived. However, this knowledge is the first step for a rational design of beryllium complexes with N-donor ligands. Therefore, we prepared beryllium compounds with selected amido and imido ligands, to investigate their electronic and steric influence on the structure and spectroscopic properties of the derived complexes.
2 Results and discussion
For the synthesis of the desired beryllium amides, direct deprotonation of amines with beryllium organic compounds was chosen. This approach had already been established by Coates et al.57 Addition of 2,4,6-trimethylaniline (H2NMes) to a benzene solution of BeEt2 resulted in immediate gas evolution. However, full conversion could only be achieved by heating the reaction solution to 70 °C for 20 h. Even though this reaction is quantitative according to NMR spectroscopy, homoleptic beryllium amide [Be(HNMes)2]3 (1) could only be isolated in a moderate yield of 64% (Fig. 2). 1 is well soluble in benzene and crystals suitable for single crystals X-ray diffraction analysis could be obtained by slow solvent evaporation.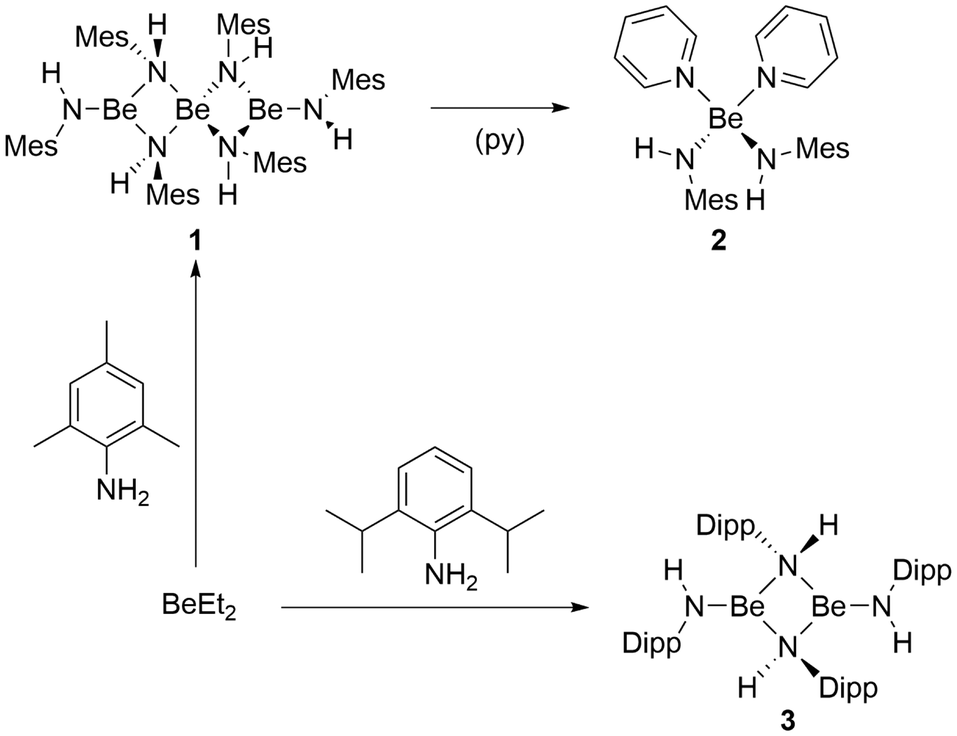 | ||
| Fig. 2 Reaction of diethyl beryllium with 2,4,6-trimethylaniline (H2NMes) or 2,6-diisopropylaniline (H2NDipp). | ||
1 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) with two formula units per unit cell. The trinuclear compound comprises one central beryllium atom, which is connected to two terminal beryllium atoms via two μ2-bridging amido nitrogen atoms, each. The coordination sphere of the terminal beryllium atoms is completed by an additional amido ligand. This results in a linear trinuclear beryllium amide, in which the central beryllium atom is tetra-coordinated while the terminal beryllium atoms are only tri-coordinated, as depicted in Fig. 3. This structural motif is known from [Be(NMe2)2]3,53 [BePh2]340 and [Be(O2tBu)]3.65 The molecules of 1 are heavily disordered in the crystal, which leads to a huge variation in bond lengths and angles. The Be–Be–Be angle in 1 is with 170.8(4)–174.9(5)° close to the one in [Be(O2tBu)]3 (175.5(2)°),65 slightly smaller than in [Be(NMe2)2]3 (179.985°)53 and significantly larger than in [BePh2]3 (141.72(8)°).40 The Be⋯Be distance in 1 is with 2.247(11)–2.379(9) Å comparable to [Be(NMe2)2]3 (2.300(9) Å)53 and [Be(O2tBu)]3 (2.2836(17) Å),65 which is considerably above the sum of the single bond covalent radii of beryllium (1.02 Å).66 It should be noted that the Be⋯Be separations in [BePh2]3 (2.033(2) & 2.047(2) Å) are considerably shorter than in 1.40 The Be–N bonds to the central beryllium atom are with 1.756(3)–1.768(3) Å longer than those of the terminal beryllium atoms to the non-bridging amido ligands (1.540(11)–1.604(9) Å), while the atom separations of the terminal beryllium atoms to the bridging nitrogen atoms are due to their huge variation (1.482(13)–1.885(9) Å) identical to the other Be–N bond lengths in 1. These atomic separations compare well to those found in [Be(NMe2)2]3 (Be2–Nμ 1.653(7) Å, Be1/3–N1/6 1.573(11) Å, Be1/3–Nμ 1.785(4) Å; labelling scheme in analogy to Fig. 3)53 and are by tendency shorter than in related heteroleptic compounds.65,67 While the bridging nitrogen atoms are pseudo-tetrahedrally coordinated, the terminal nitrogen atoms exhibit a planar coordination sphere. To evaluate this unexpected structural feature and to understand the bonding situation in 1, localised molecular orbital (LMO) and intrinsic atomic orbital (IAO) atomic charge analysis was performed.68,69
(2) with two formula units per unit cell. The trinuclear compound comprises one central beryllium atom, which is connected to two terminal beryllium atoms via two μ2-bridging amido nitrogen atoms, each. The coordination sphere of the terminal beryllium atoms is completed by an additional amido ligand. This results in a linear trinuclear beryllium amide, in which the central beryllium atom is tetra-coordinated while the terminal beryllium atoms are only tri-coordinated, as depicted in Fig. 3. This structural motif is known from [Be(NMe2)2]3,53 [BePh2]340 and [Be(O2tBu)]3.65 The molecules of 1 are heavily disordered in the crystal, which leads to a huge variation in bond lengths and angles. The Be–Be–Be angle in 1 is with 170.8(4)–174.9(5)° close to the one in [Be(O2tBu)]3 (175.5(2)°),65 slightly smaller than in [Be(NMe2)2]3 (179.985°)53 and significantly larger than in [BePh2]3 (141.72(8)°).40 The Be⋯Be distance in 1 is with 2.247(11)–2.379(9) Å comparable to [Be(NMe2)2]3 (2.300(9) Å)53 and [Be(O2tBu)]3 (2.2836(17) Å),65 which is considerably above the sum of the single bond covalent radii of beryllium (1.02 Å).66 It should be noted that the Be⋯Be separations in [BePh2]3 (2.033(2) & 2.047(2) Å) are considerably shorter than in 1.40 The Be–N bonds to the central beryllium atom are with 1.756(3)–1.768(3) Å longer than those of the terminal beryllium atoms to the non-bridging amido ligands (1.540(11)–1.604(9) Å), while the atom separations of the terminal beryllium atoms to the bridging nitrogen atoms are due to their huge variation (1.482(13)–1.885(9) Å) identical to the other Be–N bond lengths in 1. These atomic separations compare well to those found in [Be(NMe2)2]3 (Be2–Nμ 1.653(7) Å, Be1/3–N1/6 1.573(11) Å, Be1/3–Nμ 1.785(4) Å; labelling scheme in analogy to Fig. 3)53 and are by tendency shorter than in related heteroleptic compounds.65,67 While the bridging nitrogen atoms are pseudo-tetrahedrally coordinated, the terminal nitrogen atoms exhibit a planar coordination sphere. To evaluate this unexpected structural feature and to understand the bonding situation in 1, localised molecular orbital (LMO) and intrinsic atomic orbital (IAO) atomic charge analysis was performed.68,69
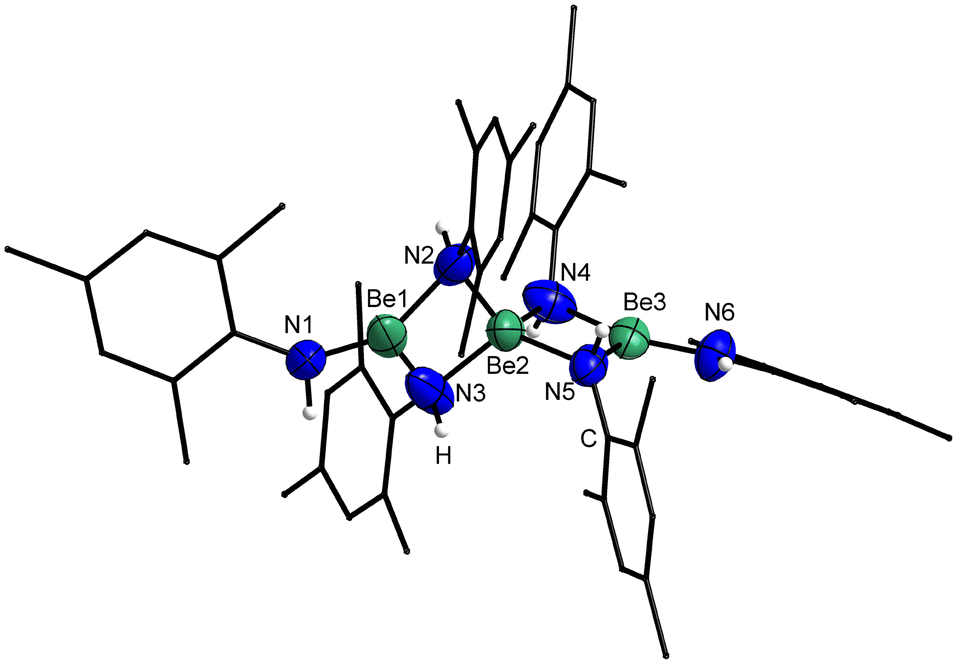 | ||
| Fig. 3 Solid state structure of 1. Ellipsoids are depicted at 70% probability at 100 K, while hydrogen atoms are shown isotropic with arbitrary radii. Carbon bound hydrogen atoms are omitted and carbon atoms shown as wire-frame for clarity. Selected bond lengths (Å) and angles (°): Be⋯Be 2.247(11)–2.379(9), Be1/3–Nμ 1.482(13)–1.885(9), Be2–Nμ 1.756(3)–1.768(3), Be1/3–N1/6 1.540(11)–1.604(9); Be1/3–Nμ–Be2 78.0(4)–90.7(4), Nμ–Be2–Nμ 92.7(1)–118.6(2), Nμ–Be1/3–Nμ 94.9(4)–101.5(6), N1/6–Be1/3–Nμ 111.1(8)–146.6(8), N1/6–Be1/3–Be2 162.8(9)–167.1(7), Be1–Be2–Be3 170.8(4)–174.9(5), N1/6–Be1/3–Nμ–Be2 173.8(19)–179.7(6); angle between Be1/3N3 planes: 89.2(2)–89.9(2)°. | ||
All structures of non-deprotonated ligands and complexes were optimized at the DFT-PBE0/def2-TZVP level of theory, and the resulting bond lengths and angles in the gas-phase are in good agreement with the experimental results. Therefore, the applied computational model can confidently be used for bonding analysis. On the basis of the obtained Kohn–Sham DFT wave functions, LMOs and IAO atomic charges were obtained by using the intrinsic bond orbital (IBO) localization method.68 The IAO atomic charges of the beryllium atoms in 1 are 1.48 to 1.51, while those at the nitrogen atoms range from −0.96 to −1.03 (ESI Fig. S27†). These charges are in line with the electronegativity difference of the two bonding partners. Comparison of the deprotonated ligand in 1 and free H2NMes (ESI Fig. S28†) shows increased negative charge at the nitrogen atoms in the deprotonated coordinated ligand, as would be expected. While the charge at the carbon atom bound to the nitrogen atom of the terminal amido ligands is not significantly different to the one calculated for free H2NMes, this carbon charge is significantly reduced in the bridging amido ligands. Additionally, the charge of the bridging nitrogen atoms is significantly more negative than in their terminal counterparts. LMO analysis of 1 reveals two sets of bonds between the terminal beryllium atoms and the non-bridging nitrogen atoms as depicted in Fig. 4(a) and (b). The prior is a σ Be–N bond, which is slightly polarised towards the nitrogen atom, which reflects the electronegativity difference between the bonding partners. The latter is a π interaction, which is mostly localised on the nitrogen atom and can be interpreted as donation of the lone pair at nitrogen into the empty p orbital of beryllium. This is supported by the fact that only p-orbitals of beryllium and nitrogen are involved, while in the σ Be–N bond also considerable s-orbital contribution is present (see ESI Table S3† for further details). This π interaction is also the cause for the planarisation at the terminal nitrogen atoms. The interaction of the bridging nitrogen atoms with the beryllium atoms consists of four very similar 2-electron–3-centre σ bonds, which contain significant s and p orbital contributions from all bonding partners (Fig. 4c and ESI Table S4†). These bonds are polarised towards the nitrogen atom, but less than in the terminal σ Be–N bonds. Also there is significantly stronger polarisation towards the central beryllium atom. Additionally there are two π type 2-electron–3-centre bonds, which are highly polarised towards the nitrogen atom as well as two further interactions of the terminal beryllium atoms with one of the bridging nitrogen atoms, each (Fig. 4d and ESI Table S5†). These Be–N interactions are also highly polarised towards nitrogen and are mainly composed of one p atomic orbital of nitrogen. All N–C bonds in 1 are σ bonds and no indication for π delocalisation over the N–C bond into the aryl systems was found (ESI Fig. S38 and Table S6†). While there is no N–C bond polarisation in the terminal amido ligands, in the bridging ligands these bonds are slightly polarised towards nitrogen. In non-coordinated H2NMes, the N–C bond is slightly more polarised towards the nitrogen atom, and also shows no sign of π delocalisation (ESI Fig. S39, S40 and Table S7†).
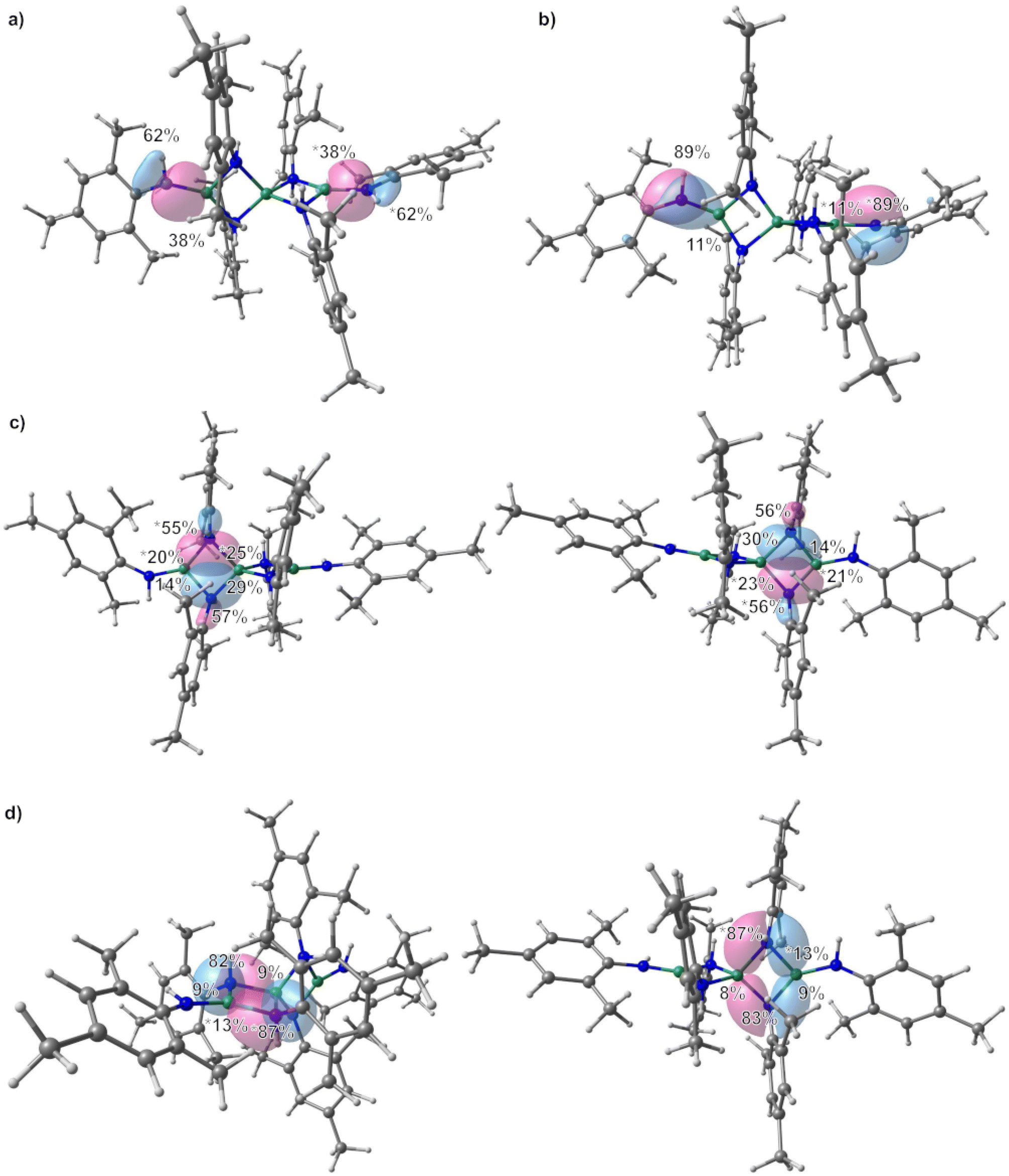 | ||
| Fig. 4 Localized molecular orbitals (LMOs) showing the Be–N bonds of 1, with a contour value for the LMO isosurface of 0.05 a.u. Percentages indicate the contribution of each atom to the LMO. The larger the percentage, the more polarized the bond. If the summation does not add up to 100%, then other atoms contribute less than 2% to the LMO. To distinguish between the two LMOs per picture one contribution is marked with an asterisks, each. Be: green, N: blue, C: grey, H: white; red: positive phase, blue: negative phase. | ||
One- and two-dimensional 1H and 13C NMR experiments show that the solid state structure of 1 is also retained in solution. However, two isomers with a ratio of roughly two-to-one are present in solution. According to phase selective NOESY experiments, 1 is dynamic in solution and not only the expected exchange between inequivalent methyl and aryl proton signals is observed, but also exchange between the mesityl and NH groups of the two isomers. Interestingly the latter exchange is more pronounced than exchange between terminal and bridging amide units. The exchange between terminal and bridging units is comparable to [BePh2]340 and is in contrast to [Be(OtBu)2]3, which exhibits no dynamic behaviour in solution.65 Due to the dynamic behaviour in solution, no 9Be NMR signals of the tri-coordinated beryllium nuclei could be observed. These signals are expected to be broad due to the low symmetry around the quadrupolar 9Be nuclei70,71 and the dynamic behaviour of 1 broadens these signals to an extent that they are not observable with the applied NMR experiment. The 9Be NMR signals of the central beryllium atoms of the major and minor isomer are observed at 6.6 ppm (ω1/2 = 7.0 Hz) and 5.3 ppm (ω1/2 = 5.6 Hz), respectively. These chemical shifts are in line with other beryllium amido complexes72 and the line widths are indicative for tetra-coordinated beryllium nuclei with high symmetry.71
To evaluate the stability of 1 towards coordinating ligands, it was dissolved in pyridine (py). This led to immediate formation of [(py)2Be(HNMes)2] (2, Fig. 2) as evident from the 1H and 13C NMR spectra. This reaction is in line with similar reactions performed with heteroleptic beryllium amides of the constitution [(R2N)BeR′]n73 as well as the formation of [(py)2BePh2] from [BePh2]3 in pyridine.74 The 9Be NMR signals of 2 are found at 8.1 (ω1/2 = 288.0 Hz) and 6.8 (ω1/2 = 131.7 Hz) in C6D6 and pyridine-d5, respectively. These chemical shifts are typical for tetra-coordinated pyridine adducts of beryllium.74 However, the broad linewidths might indicate partial dissociation of one pyridine ligand in solution. Slow solvent evaporation from a benzene solution of 2 resulted in the formation of crystals suitable for single crystal X-ray diffraction analysis. 2 crystallises in the monoclinic space group I2/a (15) with four formula units per unit cell. The complex comprises a beryllium atom with a pseudo-tetrahedral coordination sphere, coordinated by two amido and two pyridine ligands as depicted in Fig. 5.
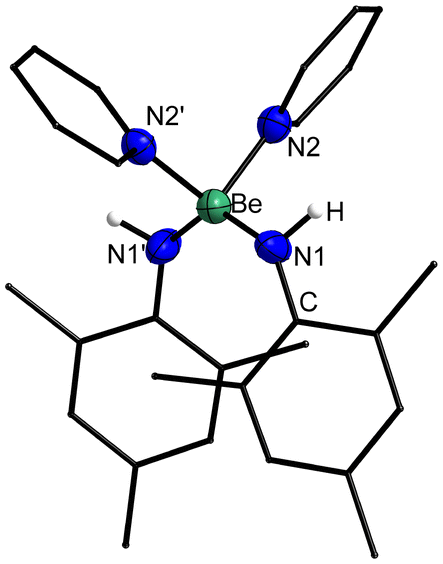 | ||
| Fig. 5 Solid state structure of 2. Ellipsoids are depicted at 70% probability at 100 K, while hydrogen atoms are shown isotropic with arbitrary radii. Carbon bound hydrogen atoms are omitted and carbon atoms shown as wire-frame for clarity. Selected bond lengths (Å) and angles (°): Be–N1 1.6557(14), Be–N2 1.8312(19); N1–Be–N1′ 131.5(1), N2–Be–N2′ 98.4(1), N1–Be–N2 103.41(6)–107.75(6). | ||
The Be–N bonds to the amido ligands in 2 are, with 1.6557(14) Å slightly longer than those to the non-bridging amides in 1. This would be expected due to the higher coordination number at the beryllium atom in 2. The Be–N bonds to the pyridine ligands are significantly longer at 1.8312(19) Å, which is in accordance with the dative nature of this bond. The respective Be–N bonds in [(py)2BePh2] (1.800(3) Å)74 are slightly shorter, which might indicate that amides are better donor ligands for beryllium than aryls. The N–Be–N angles in 2 deviate strongly from the ideal tetrahedron angle. This is caused by the significantly higher steric demand of the amido in comparison to the pyridine ligands.
In 2, the IAO atomic charge at beryllium is with 1.49 comparable to those found in 1, while the charge at the amido nitrogen is slightly less negative (−0.93, ESI Fig. S29†). The IAO atomic charge at the pyridine nitrogen atom is, with −0.35 significantly lower than in free pyridine (−0.28, ESI Fig. S30†). LMO analysis of 2 reveals two interactions between the amido ligand and the beryllium atom. One is a σ Be–N bond, which is highly polarised towards the nitrogen atom (Fig. 6a and ESI Table S8†) and is composed of s and p atomic orbital contributions of both atoms. The second interaction (Fig. 6b) is even more polarised towards nitrogen, and is almost exclusively composed of p atomic orbitals, meaning it can be interpreted as a slight donation of the lone pair at nitrogen towards beryllium. Again, no π delocalisation into the aryl system of the amido ligand was observed, and the N–C σ bond polarisation is comparable to the bridging amido ligands in 1. The Be–N bond to the pyridine ligands is of σ symmetry (Fig. 6c) and the bond polarisation is in between those found for the two Be–N interactions with the amido ligand in 2.
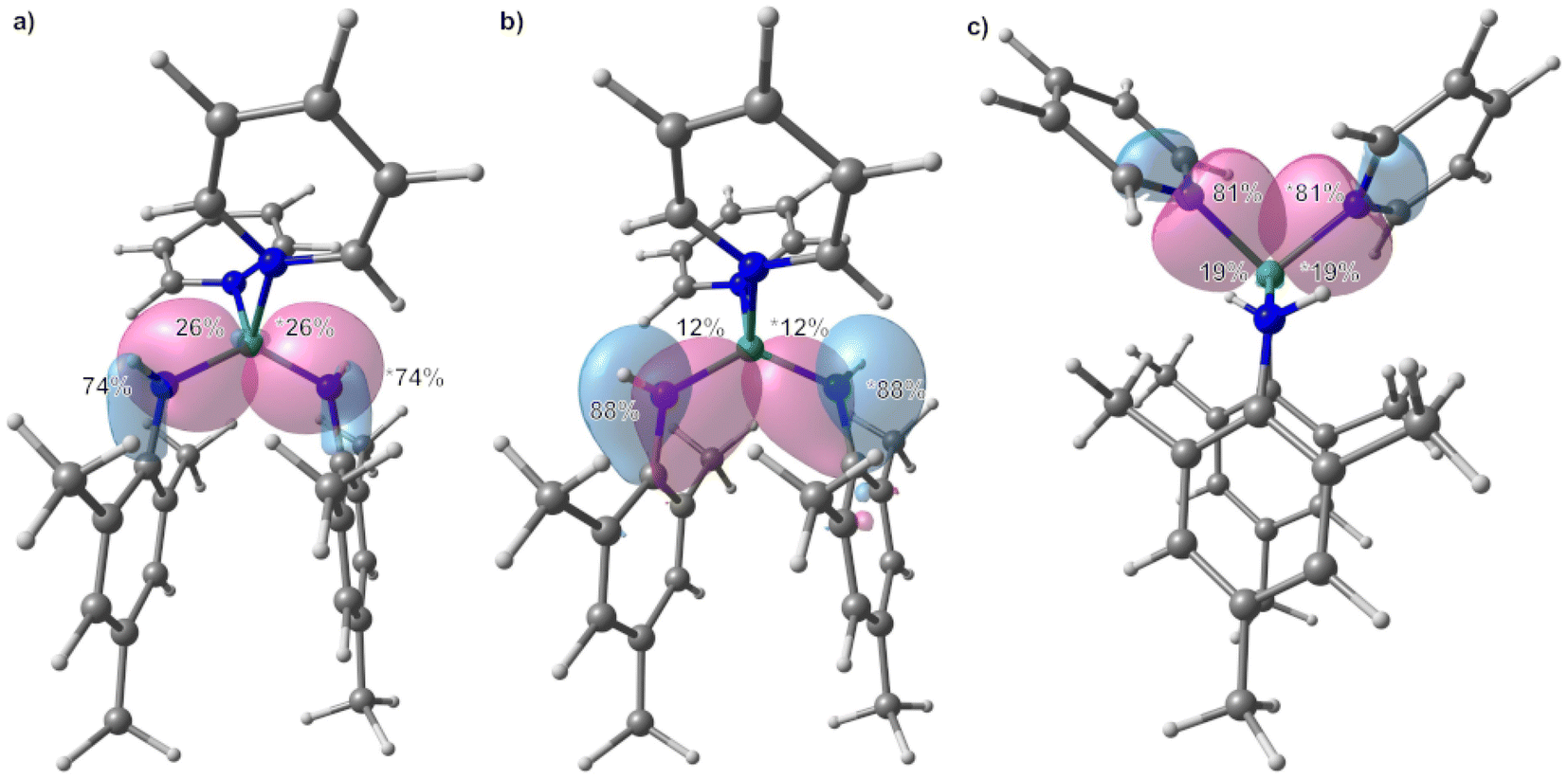 | ||
| Fig. 6 Localized molecular orbitals (LMOs) showing the Be–N bonds of 2, with a contour value for the LMO isosurface of 0.05 a.u. Percentages indicate the contribution of each atom to the LMO. The larger the percentage, the more polarized the bond. If the summation does not add up to 100%, then other atoms contribute less than 2% to the LMO. To distinguish between the two LMOs per picture one contribution is marked with an asterisks, each. Be: green, N: blue, C: grey, H: white; red: positive phase, blue: negative phase. | ||
In analogy to the reactivity of H2NMes, treatment of a benzene solution of BeEt2 with 2,6-diisopropylaniline (H2NDipp) led to immediate gas evolution. But again, full conversion was only achieved through heating to 70 °C for 20 h. Even though this reaction is quantitative according to NMR spectroscopy, homoleptic beryllium amide [Be(HNDipp)2]2 (3) could only be isolated in a moderate yield of 55% (Fig. 2). 3 is well soluble in benzene and crystals suitable for single crystal X-ray diffraction analysis could be obtained by slow solvent evaporation. The complex crystallises in the monoclinic space group P21/c (14) with two formula units per unit cell. Dinuclear 3 comprises two beryllium atoms which are μ2-bridged by two amido nitrogen atoms (Fig. 7). Each beryllium atom is additionally coordinated by a terminal amido ligand. The Be⋯Be, Be–N2 and Be–N1 distances are, with 2.269(2), 1.6879(14)–1.7350(14) and 1.5567(14) Å, respectively, comparable to 1. The two terminal amido ligands are tilted by approximately 20° out of the plane of the four-membered Be–N–heterocycle, which is presumably due to steric repulsion. While the bridging nitrogen atoms of 3 are pseudo-tetrahedrally coordinated, the terminal nitrogen atoms exhibit trigonal planar coordination spheres. These coordination geometries are in analogy to 1.
 | ||
| Fig. 7 Solid state structure of 3. Ellipsoids are depicted at 70% probability at 100 K, while hydrogen atoms are shown isotropic with arbitrary radii. Carbon bound hydrogen atoms are omitted and carbon atoms shown as wire-frame for clarity. Selected bond lengths (Å) and angles (°): Be⋯Be 2.269(2), Be–N2/2′ 1.6879(14)–1.7350(14), Be–N1 1.5567(14); Be–N2–Be 83.05(7), N2–Be–N2′ 96.95(7), N2–Be–N1/1′ 117.26(8) & 143.43(9), N1–Be–Be′ 161.36(11), N1–Be–N2–Be 159.9(2) & 166.66(9), N1–Be–Be′–N1′ 180.0(2). | ||
The solid state structure of 3 is retained in solution, as evident from two signal sets of 2,6-diisopropylphenylamido ligands in a one-to-one ratio in the 1H and 13C NMR spectra. No indications of dynamic behaviour were observed in these NMR experiments at ambient temperature. The 9Be NMR chemical shift and linewidths of 3 are, with δ = 12.2 ppm and ω1/2 = 281.6 Hz, in the typical range of tri-coordinated beryllium amide complexes.71,72
In 3 the IAO atomic charges at the beryllium atoms are with 1.51 comparable to 1, which also applies for the charge at the terminal nitrogen atoms of −0.95 (ESI Fig. S31†). The charge at the bridging nitrogen atoms of 3 is with −1.01 comparable to those found in 1. Just as in 1, the deprotonated ligands in 3 show increased negative charge at the nitrogen atoms in comparison to free H2NDipp (ESI Fig. S32†). The IAO atomic charge at the nitrogen bound carbon atom of the terminal amido ligand is not significantly different to free H2NDipp, while this charge is slightly reduced in the bridging amido ligand. Analogous to 1, LMO analysis of 3 gives a σ Be–N bond to the terminal amido ligands (Fig. 8a), which is composed of s and p orbitals of both atoms (ESI Table S11†) and is even less polarized than in 1. While a π interaction between nitrogen and beryllium was found in 1, a 2-electron–3-centre π bond is found in 3 (Fig. 8b), which is mostly localised on the nitrogen atom and can be interpreted as donation of the lone pair at nitrogen into the empty p orbital of beryllium as well as into the aromatic π system. The bridging nitrogen atoms of 3 form two 2-electron–3-centre σ bonds with the beryllium atoms, which are polarised towards nitrogen, as well as to one of the beryllium atoms each (Fig. 8c). Two further interactions of one of the beryllium atoms with one of the bridging nitrogen atoms, each are found (Fig. 8d and ESI Table S12†), which are highly polarised towards nitrogen and are mainly composed of one p atomic orbital of nitrogen. These bridging interactions are comparable to the analogous bonds in 1. Interestingly, two agostic interactions between beryllium and the CH groups of two iso-propyl moieties were calculated (Fig. 8e and ESI Table S13†). The N–C bonds in 3 are σ bonds and, besides the above mentioned 2-electron–3-centre π bond, no indication for further π delocalisation over the N–C bonds into the aryl systems was found (ESI Fig. S42 and Table S14†). The N–C bond polarisation in 3 and comparison of 3 to non coordinated H2NDipp (ESI Fig. S43, S44 and Table S15†) gives analogous results to 1.
 | ||
| Fig. 8 Localized molecular orbitals (LMOs) showing the Be–N bonds of 3, with a contour value for the LMO isosurface of 0.05 a.u. Percentages indicate the contribution of each atom to the LMO. The larger the percentage, the more polarized the bond. If the summation does not add up to 100%, then other atoms contribute less than 2% to the LMO. To distinguish between the two LMOs per picture one contribution is marked with an asterisks, each. Be: green, N: blue, C: grey, H: white; red: positive phase, blue: negative phase. | ||
To evaluate the influence of the nature of the amide ligands more finely, a comparison between two different amido ligands bound in the same complex was necessary. Therefore, a benzene solution of [(NPh2)BePh]259,65 was reacted with H2NMes. After one hour of sonication, this gave heteroleptic [Be(NPh2)(μ2-HNDipp)]2 (4, Fig. 9) in quantitative yield, according to NMR spectroscopy. The constitution of 4 in solution could be derived from 1H and 13C NMR spectra, which show one signal set for a diphenylamido and a 2,6-diisopropylphenylamido ligand, each, and no sign for dynamic behaviour at ambient temperature. The 9Be NMR shift of 12.4 ppm and line width of ω1/2 = 275.5 Hz are indicative for tri-coordinated beryllium nuclei.71,72
 | ||
| Fig. 9 Reaction of [(NPh2)BePh]259,65 with 2,6-diisopropylaniline (H2NDipp). | ||
4 dissolves readily in benzene, and crystals suitable for single crystal X-ray diffraction could be grown by slow evaporation of this solvent. The compound crystallises in the monoclinic space group P21/c (14) with two formula units per unit cell. Heteroleptic dinuclear 4 is composed of two beryllium atoms, which are μ2-bridged by the nitrogen atoms of two 2,6-diisopropylphenylamido ligands as depicted in Fig. 10. Each beryllium atom is additionally coordinated by one diphenylamido ligand. The Be⋯Be distance is with 2.263(4) Å identical to the one in 3 but longer than in [(NPh2)BePh]2.65 The Be–N2 and Be–N1 distances are with 1.705(2) & 1.706(2) and 1.574(2) Å, respectively, comparable to 1, 3 and [(NPh2)BePh]2.65 Unlike in 3, the two terminal amido ligands in 4 are only slightly tilted (aprox. 7°) out of the plane of the four-membered Be–N–heterocycle. The terminal nitrogen atoms of 4 exhibit trigonal planar coordination spheres, while the bridging nitrogen atoms are pseudo-tetrahedrally coordinated, like in 1 and 3.
 | ||
| Fig. 10 Solid state structure of 4. Ellipsoids are depicted at 70% probability at 100 K, while hydrogen atoms are shown isotropic with arbitrary radii. Carbon bound hydrogen atoms are omitted and carbon atoms shown as wire-frame for clarity. Selected bond lengths (Å) and angles (°): Be⋯Be 2.263(4), Be–N2/2′ 1.705(2) & 1.706(2), Be–N1 1.574(2); Be–N2–Be′ 83.1(1), N2–Be–N2′ 96.9(1), N2–Be–N1 125.2(1) & 137.9(1), N1–Be–Be′ 173.37(17), N1–Be–N2–Be′ 177.4(2) & 177.8(2), N1–Be–Be′–N1′ 180.0(10). | ||
The IAO atomic charges at the beryllium atoms of 4 are with 1.50–1.51 identical to those found in 3 (ESI Fig. S33†). This also applies for the charges at the bridging nitrogen atoms of the 2,6-diisopropylphenylamido ligands, with −1.02–1.03. The charges at the nitrogen atoms of the diphenylamido ligands are with −0.75–0.79 the least negative charges found. Again, these charges are significantly more negative than in free H2NDipp and HNPh2 (ESI Fig. S34†) and the charges at the nitrogen bound carbon atoms do not change considerably. The charge at the nitrogen atoms of the diphenylamido ligands in [(NPh2)BePh]2 is −0.86 (ESI Fig. S35†) and, therefore, slightly lower than in 4. This is presumably caused by the bridging coordination mode in [(NPh2)BePh]2. The charge at the beryllium atom in [(NPh2)BePh]2 is with 1.45 slightly lower than in the other complexes, which might indicate better donor properties of the phenylido ligand compared to the amido ligands. The LMO analysis of 4 reveals essentially the same bonding modes as in 3. However, the 2-electron–3-centre σ bonds from the bridging nitrogen to the two beryllium atoms are less polarised between the two beryllium atoms and are more comparable to the bridging interactions in 1. This is also the case for the other two interactions which are mainly composed of p orbitals at the bridging nitrogen atoms (ESI Fig. S45 and Table S16†). While the Be–N σ bond to the terminal diphenylamido ligands is essentially identical to the respective one in 3, there is only one 2-electron–3-centre π interaction on one side and some donation of the lone pair at the nitrogen atoms into the p orbitals of the beryllium atoms on the other side of the molecule (ESI Fig. S46 and Table S17†). The C–N bonds are exclusively of σ symmetry in 4 and free HNPh2 with no indication for π delocalisation into the aromatic ring (ESI Fig. S47–S49 and Tables S18–S19†). In [(NPh2)BePh]2 the Be–C bonds are exclusively of σ symmetry, with almost no bond polarisation (ESI Fig. S50 and Table S20†). The bridging interaction between the nitrogen atoms and the beryllium atoms consists of two 2-electron–3-centre σ bonds, which are basically identical to those in 4 (ESI Fig. S51 and Table S21†). However, there are two additional 2-electron–3-centre interactions, which are composed mainly of one of the p orbitals at the nitrogen atoms each (ESI Fig. S51 and Table S21†). This might indicate that the poorer donating properties of the phenylido ligands are compensated through multi-centre bonding between beryllium and nitrogen. The N–C bonds in 4 and [(NPh2)BePh]2 are very similar (ESI Fig. S52 and Table S22†).
Since some electron density is delocalised over the C–N–Be bonds in 3 and 4, we wanted to study the influence of more efficient delocalisation onto the structure of the corresponding beryllium complex. We therefore, reacted an etheral solution of Be(iBu)2 with diphenylmethaneimine (HNCPh2). This resulted in immediate gas evolution. However, complete conversion, according to NMR spectroscopy, was only achieved through heating to 65 °C over 20 h. This gave brightly yellow [Be(NCPh2)2]3 (5) in an isolated yield of 53% (Fig. 11). 5 is reasonably soluble in aromatic solvents and exhibits two singlets with a one-to-two ratio at 6.0 ppm (ω1/2 = 21.5 Hz) and 6.7 ppm (ω1/2 = 17.5 Hz) in the 9Be NMR spectrum. This is indicative for a trinuclear linear compound, which is also supported by the presence of two signal sets of diphenylmethaneimido ligands with one-to-two ratio in the 1H and 13C NMR spectra.
 | ||
| Fig. 11 Reaction of diisobutyl beryllium with diphenylmethanimine (HNCPh2). | ||
Crystals of 5 suitable for single crystal X-ray diffraction analysis could be grown by slow solvent evaporation from a saturated Et2O solution. Trinuclear 5 crystallises in the triclinic space group P (2) with two formula units per unit cell and comprises a central tetra-coordinated beryllium atom, which is μ2-bridged by four imide nitrogen atoms to two terminal beryllium atoms. These terminal beryllium atoms are further coordinated to one nitrogen atom of a non-bridging imido ligand each and exhibit a trigonal planar coordination sphere (Fig. 12). This structure is closely related to 1, however, 5 is significantly more linear with a Be–Be–Be angle of 179.1(1)°. While in 1 the angle between the trigonal planes of the terminal beryllium atoms is almost 90°, in 5 this angle is with 74.6(1)° significantly smaller. The Be⋯Be separations in 5 are with 2.330(3) and 2.339(3) Å comparable to those in 1 and by tendency longer than in the dinuclear compounds. The Be–N bonds to the terminal imido nitrogen atoms are with 1.502(3) and 1.507(3) Å shorter than those to the bridging nitrogen atoms. The Be–N distances from the central beryllium atom to the bridging nitrogen atoms are with 1.718(3)–1.728(3) Å considerably longer than those to the terminal beryllium atoms (1.668(3)–1.674(3) Å). All Be–N bonds in 5 are by tendency shorter than in complexes 1, 2, 3, 4 and [(NPh2)BePh]2. In the terminal imido ligands the nitrogen atoms are almost linearly coordinated, with Be–N–C angles of 175.8(2)° and 176.7(2)° and the CPh2 plane is standing almost perpendicular to the BeN3 plane. In contrast to this, the CPh2 plane of the bridging imido ligands is parallel to the NBe2 plane.
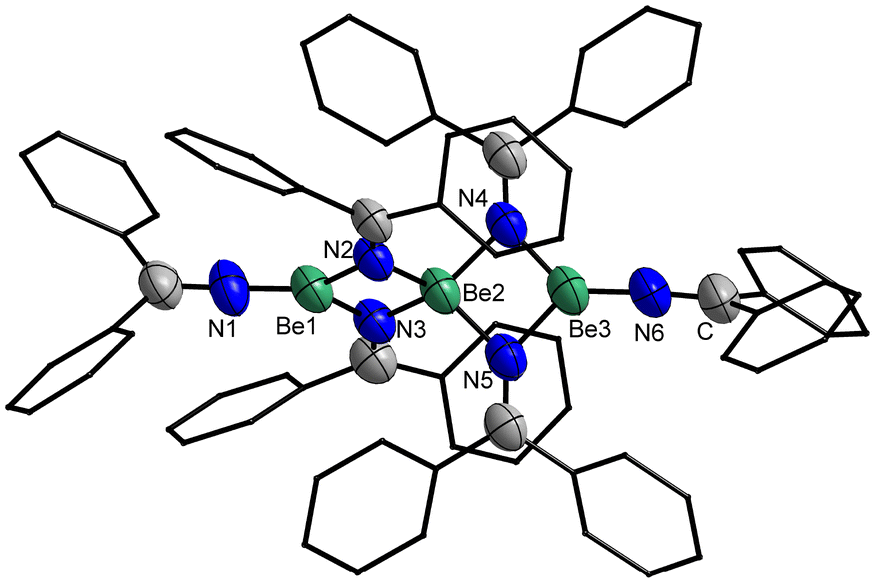 | ||
| Fig. 12 Solid state structure of 5. Ellipsoids are depicted at 70% probability at 100 K. Hydrogen atoms are omitted and phenyl carbon atoms shown as wire-frame for clarity. Selected bond lengths (Å) and angles (°): Be⋯Be 2.330(3) & 2.339(3), Be1/3–Nμ 1.668(3)–1.674(3), Be2–Nμ 1.718(3)–1.728(3), Be1/3–N1/6 1.502(3) & 1.507(3), Nμ–C 1.270(2)–1.280(2), N1/6–C 1.253(2), 1.264(2); Be1/3–Nμ–Be2 86.7(1)–87.3(1), Nμ–Be2–Nμ 91.2(1)–110.7(1), Nμ–Be1/3–Nμ 84.7(1)–95.2(1), N1/6–Be1/3–Nμ 130.5(2)–134.3(2), N1/6–Be1/3–Be2 176.7(2) & 179.6(1), Be1–Be2–Be3 179.1(1), N1/6–Be1/3–Nμ–Be2 176.2(3)–179.9(3); angle between Be1/3N3 planes: 74.6(1)°. | ||
The IAO atomic charges at the beryllium atoms of 5 are, with 1.49–1.51, comparable to those in all other investigated compounds (ESI Fig. S36†). The charges at the bridging nitrogen atoms are with −0.96 significantly more negative than at the terminal nitrogen atoms (−0.90), which is in line with the complexes described above. Also, the deprotonated ligands show significantly more negative partial charges at the nitrogen atoms in comparison to free HNCPh2 (ESI Fig. S37†). The IAO atomic charges at the nitrogen bound carbon atom of the terminal imido ligands and free HNCPh2 are almost identical, while the respective charges in the bridging imido ligands are slightly higher. For the interaction of the bridging nitrogen atoms with the beryllium atoms, the LMO analysis of 5 reveals four identical 2-electron–3-centre σ bonds, which compare well to those found in 1 (ESI Fig. S53 and Table S23†). Additionally, π type 2-electron–4-centre bonds were found, which are polarised towards the nitrogen atom, but do have comparable contributions of the nitrogen bound carbon atom as well as of the beryllium atoms (ESI Fig. S54 and Table S23†). The terminal Be–N bonds consist of one σ bond each, which exhibits a 60% polarisation towards nitrogen and is, therefore, comparable to those in the other compounds (ESI Fig. S55 and Table S24†). Additionally, however, there is a 2-electron–3-centre π bond, which is in comparison to the other complexes significantly more delocalised (N 76%, Be 10%, C 14%). All NC bonds consist of very slightly polarised σ bonds and π bonds that are polarised slightly more towards nitrogen (ESI Fig. S56–S58 and Table S25†). These two bonds are only slightly altered in 5 in comparison to free HNCPh2 (ESI Fig. S59, S60 and Table S26†). Interestingly, the lone pair at nitrogen in free HNCPh2 is slightly donating into the p orbital of the adjacent carbon atom. Such an interaction was not observed for the other ligands.
3 Conclusions
In summary we have prepared a set of closely related beryllium amide and imide complexes: [Be(HNMes)2]3 (1), [(py)2Be(HNMes)2] (2), [Be(HNDipp)2]2 (3), [Be(NPh2)(μ2-HNDipp)]2 (4) and [Be(NCPh2)2]3 (5). These compounds are all well soluble in aromatic solvents, which is in contrast to many related organo beryllium complexes. With exception of 1 and 2 no evidence for dynamic behaviour in solution was observed. IAO atomic charge analysis revealed no significant differences in the charge distributions of the multinuclear compounds or the mononuclear pyridine adduct 2. LMO analysis showed that the Be–N interactions to the terminal amido ligands mainly consist of covalent σ Be–N bonds, while the μ2-N bridging interactions are 2-electron–3-centre σ bonds. The electron deficiency at the beryllium atoms is partially compensated through additional donation from the lone pairs at the nitrogen atoms to the beryllium atoms. This results in a partial double bond character of the terminal Be–N bonds and is the reason for the planarisation of the terminal nitrogen atoms. In case of the bridging nitrogen atoms, the lone pair at nitrogen stays mainly located at the nitrogen atom and only slightly donates to one or two beryllium atoms. This multi-centre bonding is favoured if the 2-electron–3-centre σ bond is equally distributed over the participating beryllium atoms. π delocalisation over the terminal Be–N–C and bridging Be2–N–C bonds is possible, if the molecular geometry allows for it and can be enforced through introduction of imido ligands, which contain NC double bonds. However, this π delocalisation has no significant effect on the partial charge distribution at the beryllium and nitrogen atoms. In conclusion, alteration of the electronic properties of the N-donor ligands has little influence on the bonding situation. It is more likely that the steric demand of the ligands dictates the structure of the beryllium compounds, which is illustrated in the space filling models depicted in Fig. 13.
 | ||
| Fig. 13 Space filling models of (a) [Be(HNMes)2]3 (1), (b) [Be(HNDipp)2]2 (3), (c) [Be(NPh2)(μ2-HNDipp)]2 (4), (d) [(NPh2)BePh]265 and (e) [Be(NCPh2)2]3 (5). Be: green, N: blue, C: grey, H: white. | ||
4 Experimental procedures
Caution! Beryllium and its compounds are categorized as human carcinogens and work with these substances is associated with severe health hazards.75 As the biochemical mechanisms that cause beryllium associated diseases are still unknown,76–78 special (safety) precautions are strongly advised.754.1 General experimental techniques
All manipulations were performed either under solvent vapor pressure or dry argon using a glovebox and Schlenk techniques. C6D6 was dried over NaK alloy, while for C6H6, n-pentane and Et2O sodium was used. Pyridine (py) and pyridine-d5 (py-d5) were dried over CaH2. All solvents were subsequently vacuum distilled before storage in an argon filled glovebox. BeEt2,26,27,79 Be(iBu)226,27,79 and [(NPh2)BePh]265 were prepared according to the literature. 2,4,6-Trimethylaniline (H2NMes), 2,6-diisopropylaniline (H2NDipp) and diphenylmethanimine (HNCPh2) were purchased from commercial vendors and vacuum distilled prior to use. All reactions were carried out in J. Young NMR tubes or PTFE sealed Schlenk tubes. All glassware was silylated according to the procedure in the ref. 80. Due to the expected extreme toxicity of the obtained compounds, no elemental analysis or mass spectrometry could be performed.
4.2 Synthesis and characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CPh2)2]3 (5).
Be(iBu)2 (123.2 mg, 1.00 mmol, 1 eq.) was dissolved in 5 ml Et2O and HNCPh2 (362.5 mg, 2.00 mmol, 2 eq.) was added. Immediately gas evolution was observed and the red reaction mixture was subsequently heated to 65 °C for 20 h. The solvent was removed in vacuo. The obtained yellow solid was washed with 5 ml n-pentane to give the title compound as a bright yellow solid in 53% (195.9 mg) isolated yield. Single crystals were obtained from an oversaturated Et2O solution. 1H NMR (500 MHz, C6D6) δ = 6.55–7.28 (m, 52H, CHAr), 7.56–7.65 (m, 8H, CHAr). 9Be NMR (42 MHz, C6D6) δ = 6.0 (s, ω1/2 = 21.5 Hz, 1Be), 6.7 (s, ω1/2 = 17.5 Hz, 2Be). 13C NMR (126 MHz, C6D6) δ = 127.0 (s, CHAr), 127.4 (s, CHAr), 128.6 (s, CHAr), 128.8 (s, CHAr), 129.2 (s, CHAr), 129.3 (s, CHAr), 129.4 (s, CHAr), 129.4 (s, CHAr), 129.7 (s, CHAr), 142.2 (s, CAr), 143.3 (s, CAr), 143.8 (s, CAr), 159.1 (s, CN), 178.7 (s, CN). IR (cm−1): 3351 (vw), 3302 (w), 3053 (w), 3025 (w), 2959 (m), 2921 (w), 2864 (w), 1732 (w), 1617 (m), 1595 (w), 1575 (w), 1483 (m), 1460 (n), 1434 (s), 1385 (w), 1356 (m), 1334 (w), 1316 (m), 1297 (w), 1273 (m), 1260 (s), 1230 (m), 1218 (w), 1189 (w), 1154 (w), 1116 (w), 1099 (w), 1052 (s), 1028 (s), 950 (m), 926 (m), 887 (w), 851 (s), 830 (w), 816 (w), 802 (s), 787 (w), 769 (s), 740 (vs), 693 (vs), 679 (m), 645 (w), 628 (s), 598 (w), 587 (w), 557 (w), 543 (s), 510 (m), 485 (w), 455 (w), 434 (w), 422 (m).
CPh2)2]3 (5).
Be(iBu)2 (123.2 mg, 1.00 mmol, 1 eq.) was dissolved in 5 ml Et2O and HNCPh2 (362.5 mg, 2.00 mmol, 2 eq.) was added. Immediately gas evolution was observed and the red reaction mixture was subsequently heated to 65 °C for 20 h. The solvent was removed in vacuo. The obtained yellow solid was washed with 5 ml n-pentane to give the title compound as a bright yellow solid in 53% (195.9 mg) isolated yield. Single crystals were obtained from an oversaturated Et2O solution. 1H NMR (500 MHz, C6D6) δ = 6.55–7.28 (m, 52H, CHAr), 7.56–7.65 (m, 8H, CHAr). 9Be NMR (42 MHz, C6D6) δ = 6.0 (s, ω1/2 = 21.5 Hz, 1Be), 6.7 (s, ω1/2 = 17.5 Hz, 2Be). 13C NMR (126 MHz, C6D6) δ = 127.0 (s, CHAr), 127.4 (s, CHAr), 128.6 (s, CHAr), 128.8 (s, CHAr), 129.2 (s, CHAr), 129.3 (s, CHAr), 129.4 (s, CHAr), 129.4 (s, CHAr), 129.7 (s, CHAr), 142.2 (s, CAr), 143.3 (s, CAr), 143.8 (s, CAr), 159.1 (s, CN), 178.7 (s, CN). IR (cm−1): 3351 (vw), 3302 (w), 3053 (w), 3025 (w), 2959 (m), 2921 (w), 2864 (w), 1732 (w), 1617 (m), 1595 (w), 1575 (w), 1483 (m), 1460 (n), 1434 (s), 1385 (w), 1356 (m), 1334 (w), 1316 (m), 1297 (w), 1273 (m), 1260 (s), 1230 (m), 1218 (w), 1189 (w), 1154 (w), 1116 (w), 1099 (w), 1052 (s), 1028 (s), 950 (m), 926 (m), 887 (w), 851 (s), 830 (w), 816 (w), 802 (s), 787 (w), 769 (s), 740 (vs), 693 (vs), 679 (m), 645 (w), 628 (s), 598 (w), 587 (w), 557 (w), 543 (s), 510 (m), 485 (w), 455 (w), 434 (w), 422 (m).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. R. B. thanks Prof. F. Kraus for moral and financial support as well as the provision of laboratory space. The DFG is gratefully acknowledged for financial support (BU2725/8-1).References
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- C. Czernetzki, T. Kunz, S. Huynh, A. Lamprecht, J. Sprenger, M. Finze, M. Arrowsmith and H. Braunschweig, Angew. Chem., Int. Ed., 2024, 63, e202401279 CrossRef CAS PubMed.
- I.-A. Bischoff, S. Danés, P. Thoni, B. Morgenstern, D. M. Andrada, C. Müller, J. Lambert, E. C. J. Gießelmann, M. Zimmer and A. Schäfer, Nat. Chem., 2024, 16, 1093–1100 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef.
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- G. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS PubMed.
- C. Czernetzki, M. Arrowsmith, F. Fantuzzi, A. Gärtner, T. Tröster, I. Krummenacher, F. Schorr and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 20776–20780 CrossRef CAS PubMed.
- J. T. Boronski, L. R. Thomas-Hargreaves, M. A. Ellwanger, A. E. Crumpton, J. Hicks, D. F. Bekiş, S. Aldridge and M. R. Buchner, J. Am. Chem. Soc., 2023, 145, 4408–4413 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, A. F. Roper and S. Aldridge, Nat. Chem., 2024, 16, 1295–1300 CrossRef CAS PubMed.
- C. Berthold, J. Maurer, L. Klerner, S. Harder and M. R. Buchner, Angew. Chem., Int. Ed., 2024, 63, e202408422 CrossRef CAS PubMed.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Chem. Commun., 2023, 59, 1453–1456 RSC.
- K. G. Pearce, H.-Y. Liu, S. E. Neale, H. M. Goff, M. F. Mahon, C. L. McMullin and M. S. Hill, Nat. Commun., 2023, 14, 8147 CrossRef CAS PubMed.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Organometallics, 2024, 43, 432–437 CrossRef CAS PubMed.
- R. Fleischer and D. Stalke, Inorg. Chem., 1997, 36, 2413–2419 CrossRef CAS PubMed.
- M. Niemeyer and P. P. Power, Inorg. Chem., 1997, 36, 4688–4696 CrossRef CAS.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and I. Mallov, Inorg. Chem., 2012, 51, 13408–13418 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2013, 42, 9720–9726 RSC.
- D. Naglav, D. Bläser, C. Wölper and S. Schulz, Inorg. Chem., 2014, 53, 1241–1249 CrossRef CAS PubMed.
- D. Naglav, B. Tobey, C. Wölper, D. Bläser, G. Jansen and S. Schulz, Eur. J. Inorg. Chem., 2016, 2424–2431 CrossRef CAS.
- M. Bayram, D. Naglav, C. Wölper and S. Schulz, Organometallics, 2016, 35, 2378–2383 CrossRef CAS.
- M. Bayram, D. Naglav, C. Wölper and S. Schulz, Organometallics, 2017, 36, 467–473 CrossRef CAS.
- D. Naglav, B. Tobey, K. Dzialkowski, D. Bäser, C. Wölper, G. Jansen and S. Schulz, Dalton Trans., 2018, 47, 12511–12515 RSC.
- A. Paparo and C. Jones, Chem. – Asian J., 2019, 14, 486–490 CrossRef CAS PubMed.
- A. Paparo, C. N. de Bruin-Dickason and C. Jones, Aust. J. Chem., 2020, 73, 1144–1148 CrossRef CAS.
- A. Paparo, S. P. Best, K. Yuvaraj and C. Jones, Organometallics, 2020, 4208–4213 CrossRef CAS.
- M. Müller, A. J. Karttunen and M. R. Buchner, Chem. Sci., 2020, 11, 5414–5422 RSC.
- A. Paparo, A. J. R. Matthews, C. D. Smith, A. J. Edwards, K. Yuvaraj and C. Jones, Dalton Trans., 2021, 50, 7604–7609 RSC.
- M. R. Buchner and M. Müller, Dalton Trans., 2021, 50, 7246–7255 RSC.
- C. Berthold, M. Müller, S. I. Ivlev, D. M. Andrada and M. R. Buchner, Dalton Trans., 2023, 52, 13547–13554 RSC.
- M. R. Buchner, M. Müller and S. S. Rudel, Angew. Chem., Int. Ed., 2017, 56, 1130–1134 CrossRef CAS PubMed.
- B. Scheibe and M. R. Buchner, Eur. J. Inorg. Chem., 2018, 2300–2308 CrossRef CAS.
- M. Müller and M. R. Buchner, Angew. Chem., Int. Ed., 2018, 57, 9180–9184 CrossRef PubMed.
- M. Müller and M. R. Buchner, Inorg. Chem., 2019, 58, 13276–13284 CrossRef PubMed.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2020, 26, 9915–9922 CrossRef PubMed.
- M. R. Buchner, L. R. Thomas-Hargreaves, L. K. Kreuzer, N. Spang and S. I. Ivlev, Eur. J. Inorg. Chem., 2021, 4990–4997 CrossRef CAS.
- M. R. Buchner, D. Cocic, S. I. Ivlev, N. Spang, M. Müller and R. Puchta, Dalton Trans., 2023, 52, 5287–5296 RSC.
- D. F. Bekiş, L. R. Thomas-Hargreaves, C. Berthold, S. I. Ivlev and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2023, 78, 165–173 CrossRef.
- M. R. Buchner, L. R. Thomas-Hargreaves, C. Berthold, D. F. Bekiş and S. I. Ivlev, Chem. – Eur. J., 2023, 29, e202302495 CrossRef CAS PubMed.
- F. Kraus, S. A. Baer, M. R. Buchner and A. J. Karttunen, Chem. – Eur. J., 2012, 18, 2131–2142 CrossRef CAS PubMed.
- M. Müller and M. R. Buchner, Chem. Commun., 2019, 55, 13649–13652 RSC.
- H. Bürger, C. Forker and J. Goubeau, Monatsh. Chem., 1965, 96, 597–601 CrossRef.
- A. H. Clark and A. Haaland, Acta Chem. Scand., 1970, 24, 3024–3030 CrossRef CAS.
- A. H. Clark and A. Haaland, J. Chem. Soc. D, 1969, 912–913 RSC.
- D. Naglav, A. Neumann, D. Blaser, C. Wolper, R. Haack, G. Jansen and S. Schulz, Chem. Commun., 2015, 51, 3889–3891 RSC.
- J. B. Farmer, H. M. M. Shearer, J. D. Sowerby and K. Wade, J. Chem. Soc., Chem. Commun., 1976, 160–161 RSC.
- B. Hall, J. B. Farmer, H. M. M. Shearer, J. D. Sowerby and K. Wade, J. Chem. Soc., Dalton Trans., 1979, 102–109 RSC.
- J. L. Atwood and G. D. Stucky, J. Am. Chem. Soc., 1969, 91, 4426–4430 CrossRef CAS.
- M. P. Dressel, S. Nogai, R. J. F. Berger and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 173–182 CrossRef CAS.
- M. R. Buchner, M. Müller and N. Spang, Dalton Trans., 2020, 49, 7708–7712 RSC.
- G. E. Coates, F. Glockling and N. D. Huck, J. Chem. Soc., 1952, 4512–4515 RSC.
- G. E. Coates and F. Glockling, J. Chem. Soc., 1954, 22–27 RSC.
- N. A. Bell, G. E. Coates and J. W. Emsley, J. Chem. Soc. A, 1966, 49–52 RSC.
- G. E. Coates and M. Tranah, J. Chem. Soc. A, 1967, 236–239 RSC.
- N. A. Bell, G. E. Coates and A. H. Fishwick, J. Organomet. Chem., 1980, 198, 113–120 CrossRef CAS.
- T. Hohl, T. Sinn and C. Hoch, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 509–516 CrossRef CAS.
- G. Wang, J. E. Walley, D. A. Dickie, A. Molino, D. J. Wilson and R. J. Gilliard, Angew. Chem., Int. Ed., 2021, 60, 9407–9411 CrossRef CAS PubMed.
- H. Nöth and D. Schlosser, Inorg. Chem., 1983, 22, 2700–2703 CrossRef.
- H. Nöth and D. Schlosser, Chem. Ber., 1988, 121, 1711–1713 CrossRef.
- L. R. Thomas-Hargreaves, C. Berthold, W. Augustinov, M. Müller, S. I. Ivlev and M. R. Buchner, Chem. – Eur. J., 2022, 28, e202200851 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- L. R. Thomas-Hargreaves, Y.-Q. Liu, Z.-H. Cui, S. Pan and M. R. Buchner, J. Comput. Chem., 2023, 44, 397–405 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- Partial charges were calculated with two different programs, IBOViewer and Turbomole. The latter results yielded a more ionic bonding situations in comparison to the results from IBOViewer and the performed Natural Populations Analysis (NPA) as reference. Due to technical problems in IBOViewer with 6, IAO analysis had to be performed with Turbomole. After adjusting the IBO reference basis set for Be in Turbomole, all results obtained in IBOViewer were reproduced and the IAO analysis for all compounds was performed. The changed parameters and all partial charges are given in the ESI.†.
- P. G. Plieger, K. D. John, T. S. Keizer, T. M. McCleskey, A. K. Burrell and R. L. Martin, J. Am. Chem. Soc., 2004, 126, 14651–14658 CrossRef CAS PubMed.
- J. K. Buchanan and P. G. Plieger, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 459–472 CrossRef CAS.
- H. Nöth and D. Schlosser, Eur. J. Inorg. Chem., 2003, 2003, 2245–2254 CrossRef.
- G. E. Coates and A. H. Fishwick, J. Chem. Soc. A, 1967, 1199–1204 RSC.
- L. R. Thomas-Hargreaves, M. Müller, N. Spang, S. I. Ivlev and M. R. Buchner, Organometallics, 2021, 40, 3797–3807 CrossRef CAS.
- M. R. Buchner and M. Müller, ACS Chem. Health Saf., 2023, 30, 36–43 CrossRef CAS.
- J. Elguero and I. Alkorta, Struct. Chem., 2023, 34, 391–398 CrossRef CAS.
- M. R. Buchner, Chem. Commun., 2020, 56, 8895–8907 RSC.
- M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 405–412 CrossRef CAS.
- H. Gilman and F. Schulze, J. Chem. Soc., 1927, 2663–2669 RSC.
- D. Naglav, M. R. Buchner, G. Bendt, F. Kraus and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 10562–10576 CrossRef CAS PubMed.
- MestReNova, Mestrelab Research S.L, Santiago de Compostela, Spain, 2011 Search PubMed.
- OPUS, Bruker Optik GmbH, Ettlingen, Germany, 2009 Search PubMed.
- X-Area, Stoe & Cie GmbH, Darmstadt, Germany, 2017 Search PubMed.
- APEX5 Crystallography Software Suite, Bruker AXS Inc., Madison, WI, USA, 2023 Search PubMed.
- SADABS/SAINT within APEX5, Bruker AXS Inc., Madison, WI, USA, 2023 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-2018/3, Göttingen, Germany, 2018 Search PubMed.
- G. M. Sheldrick, SHELXT-2018/2, Göttingen, Germany, 2018 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- TURBOMOLE V7.7, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, 2022 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- Chemcraft – Graphical Software for Visualization of Quantum Chemistry Computations. Version 1.8. https://www.chemcraftprog.com.
Footnotes |
| † Electronic supplementary information (ESI) available: Extended crystallographic data, IR and NMR spectra. CCDC 2373010–2373014. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02269f |
| ‡ Equal author contribution. |
| This journal is © The Royal Society of Chemistry 2024 |