
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adducts of Lewis acidic stibanes with phosphane chalcogenides†
Jonas
Krieft
 ,
Hannah
Koch
,
Beate
Neumann
,
Hans-Georg
Stammler
,
Jan-Hendrik
Lamm
,
Andreas
Mix
and
Norbert W.
Mitzel
*
,
Hannah
Koch
,
Beate
Neumann
,
Hans-Georg
Stammler
,
Jan-Hendrik
Lamm
,
Andreas
Mix
and
Norbert W.
Mitzel
*
Chair of Inorganic and Structural Chemistry, Center for Molecular Materials CM2, Faculty of Chemistry, Bielefeld University, Universitätsstraße 25, 33615 Bielefeld, Germany. E-mail: mitzel@uni-bielefeld.de
First published on 14th September 2024
Abstract
Starting from ClSbRF2 (RF = C2F5, 3,5-(CF3)2C6H3) and H(E)P(tBu)2 (E = O, S), we prepared the oxy- and sulphanediyl-bridged adducts RF2Sb(Cl)–E–(H)P(tBu)2, which are stable against the elimination of HCl. The different electron-withdrawing substituents and chalcogen bridging units influence the size of the Sb–E–P angle. ClSb(C2F5)2 and nBu3SnH react to give HSb(C2F5)2, which seems to interact weakly with H(O)P(tBu)2 in solution as observed via NMR. All products were characterised by NMR spectroscopy and all the stable ones were additionally characterised by X-ray diffraction and elemental analyses.
Introduction
The neutralisation reaction of Lewis acids and bases can be suppressed by steric shielding, as observed by Brown et al. 82 years ago by contrasting the reaction of 2,6-lutidine with BF3 and the absence of a reaction with the bulkier B(CH3)3.1 These inter- or even intramolecular Lewis acid/base combinations still have untapped reaction potential. The investigation of such “Frustrated Lewis Pairs” (FLP) was decisively initiated and promoted by Stephan and Erker, who presented the first FLP systems by combining boron-containing Lewis acids and phosphorus-based Lewis bases.2 Within the last two decades, this high level of interest has led to an ever-expanding repertoire of functional building blocks, e.g. those based on tetrels,3–6 rare-earth metals7 or d-block elements,8 in addition to the Lewis acid centres of boron and aluminium,9 which are usually considered to be the most common. A wide range of reactions such as C–H activation, hydrogen and small molecule activation and CO2 reduction are accessible with these systems.10,11 The use of strongly electron-withdrawing pentafluoroethyl groups allowed the monitoring of intramolecular FLP systems of type (F5C2)nECH2P(tBu)2 (E = Si, Ge, Sn with n = 3 and Sb with n = 2) and the observation of the activation of substrates such as CO2, H2 and unsaturated systems typical of some of these FLPs.4–6,12–14 The bis(pentafluoroethyl)stibanyl function, (F5C2)2Sb–, with its lone pair at the Sb(III) atom, represents a soft Lewis acid whose reactivity follows Pearson's HSAB concept.13–15 These results complement the work by Gabbaï et al. on antimony-based Lewis acid systems, such as stibonium cations and various stiboranes for the detection and transport of (fluoride) anions.16We have succeeded in chelating various halides and (nitrogen-based) Lewis bases in the form of pnictogen bonding with anthracene-based systems, which bear –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Sb(C2F5)2 functions.17,18
C–Sb(C2F5)2 functions.17,18
Stephan and Erker provided a proficient insight into the crucial importance of the bridging unit and the orientation of the (interacting) opposing functions in intramolecular FLPs with respect to dihydrogen cleavage or adduct formation.11 The study of chalcogen-based spacers has so far received less attention. Only a few examples from Stephan,19 Wang,20 and our own research group exist in the form of B/Al/Si–O–P FLPs.21,22
In this work we aim to combine our established bis(pentafluoroethyl)stibanyl function ((F5C2)2Sb–) with the chalcogen spacer motif and investigate the adducts formed.
Results and discussion
Starting from MesSbCl2 (1) and following synthesis of (F5C2)2SbMes (2, Mes = mesityl, Scheme 1), we have optimised the synthesis of the previously presented antimony reagent (F5C2)2SbCl (3),17 finally avoiding the occurrence of a mixture with the cleavage product from the deprotection of the protected precursors. Unlike toluene, which is formed by cleaving a toluyl protecting group, mesitylene, which is formed by reacting 2 with hydrogen chloride, can be separated by distillation due to its higher boiling point difference from 3. | ||
| Scheme 1 Synthesis of 3 and 5 from MesSbCl2 (1, Mes = mesityl) over the protected precursors 2 and 4. | ||
The reported synthesis23 of 1 yielded crystals, suitable for X-ray diffraction experiments. The solid-state structure of 1 (Fig. 1) with its three molecules in the asymmetric unit shows Sb–Cl bond lengths of 2.376(1)–2.395(1) Å and Sb–C bond lengths of 2.134(3)–2.150(2) Å. The Cl–Sb–Cl angles are smaller (92.0(1)–95.3(1)°) than the Cl–Sb–C angles (96.9(1)–101.6(1)°).
 | ||
| Fig. 1 Molecular structure of 1 in the solid state with three molecules in the asymmetric unit. For clarity, only one molecule is depicted and the hydrogen atoms are omitted. Ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Sb(1)–Cl(1) 2.395(1), Sb(1)–Cl(2) 2.381(1), Sb(1)–C(1) 2.150(2), Sb(2)–Cl(3) 2.387(1), Sb(2)–Cl(4) 2.376(1), Sb(2)–C(10) 2.134(3), Sb(3)–Cl(5) 2.381(1), Sb(3)–Cl(6) 2.377(1), Sb(3)–C(19) 2.143(2); Cl(1)–Sb(1)–Cl(2) 92.0(1), Cl(1)–Sb(1)–C(1) 97.5(1), Cl(2)–Sb(1)–C(1) 101.4(1), Cl(3)–Sb(2)–Cl(4) 94.5(1), Cl(3)–Sb(2)–C(10) 101.6(1), Cl(4)–Sb(2)–C(10) 97.0(1), Cl(5)–Sb(3)–Cl(6) 95.3(1), Cl(5)–Sb(3)–C(19) 96.9(1), Cl(6)–Sb(3)–C(19) 100.4(1). | ||
The solid-state structure of liquid 2 (Fig. 2) was determined via in situ crystallisation on the diffractometer. The Sb(1)–C(1) bond lengths in 1 and 2 are quite the same. The other two Sb–C bonds in 2 are about 0.1 Å longer than the Sb(1)–C(1) bond.
 | ||
| Fig. 2 Molecular structure of 2 in the solid state. Ellipsoids are set at 50% probability. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sb(1)–C(1) 2.152(1), Sb(1)–C(10) 2.240(1), Sb(1)–C(12) 2.246(1); C(1)–Sb(1)–C(10) 98.3(1), C(1)–Sb(1)–C(12) 103.1(1), C(10)–Sb(1)–C(12) 92.7(1). | ||
The NMR data for 2 and 3 in CD2Cl2 show the typical splitting pattern for pentafluoroethylstibanyl functions.17,18,24 The 13C{1H} NMR spectra show triplets of quartets (2: 121.8 ppm, 3: 126.4 ppm, CF2) and quartets of triplets (2: 120.4 ppm, 3: 120.3 ppm CF3) and the 19F NMR spectra contain multiplets of the CF3 unit (2: −83.2 ppm, 3: −81.3 ppm) and broad multiplets of the AB-spin systems of the CF2 units (2: −106.4/−107.2 ppm, 3: −111.8/−112.3 ppm), respectively.
We have now succeeded in determining the solid-state structure of 3, which is liquid at room temperature. A single crystal, suitable for X-ray diffraction, was grown on the diffractometer by in situ crystallisation. After establishing a solid–liquid equilibrium at 235 K and manually melting all but a tiny seed crystal (using a piece of wire for warming), the sample was cooled to 100 K to achieve its complete crystallization. The molecular structure of 3 (Fig. 3) shows an Sb–Cl bond length of 2.356(1) Å. As is common for trisubstituted pnictogen atoms in the oxidation state +III, the antimony atom is trigonal-pyramidal coordinated with angles involving the Sb atom ranging between 92.8(1)° and 88.5(1)°.
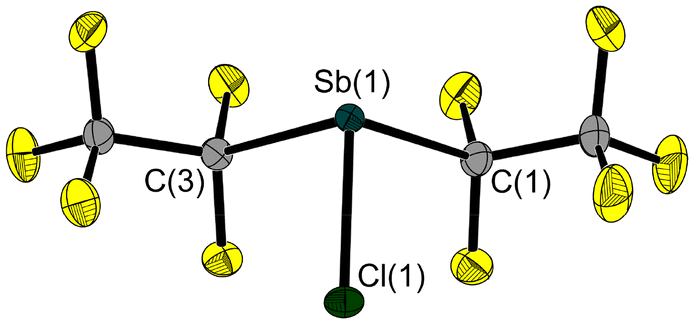 | ||
| Fig. 3 Molecular structure of 3 in the solid state. Ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Sb(1)–Cl(1) 2.356(1), Sb(1)–C(1) 2.252(1), Sb(1)–C(3) 2.243(1); C(1)–Sb(1)–Cl(1) 92.8(1), C(3)–Sb(1)–Cl(1) 91.2(1), C(1)–Sb(1)–C(3) 88.5(1). | ||
In order to investigate the influence of the electron-withdrawing substituents on the structure of Sb–E–P systems, we also synthesised the mesityl-protected Fxyl2SbMes (4, Fxyl = bis(trifluoromethyl)phenyl) starting from dichloromesitylstibane (1, Scheme 1). Subsequent deprotection of compound 4 with hydrogen chloride resulted in Fxyl2SbCl (5, Scheme 1). The aryl-type Fxyl substituent allows the use of less stringent conditions for the cleavage of the mesityl protecting group.
The solid-state structures of 4 (Fig. 4) and 5 (Fig. 5) also show the striking trigonal-pyramidal structural motif around the antimony atom. The Sb–Cl bond in compound 4 is significantly longer (2.504(1) Å) compared to that in 3 (0.15 Å), which is due to the pentafluoroethyl groups in 3. In general, all Sb–E bonds in 4 and 5 are longer than the corresponding bonds in the pentafluoroethyl analogues 2 and 3.
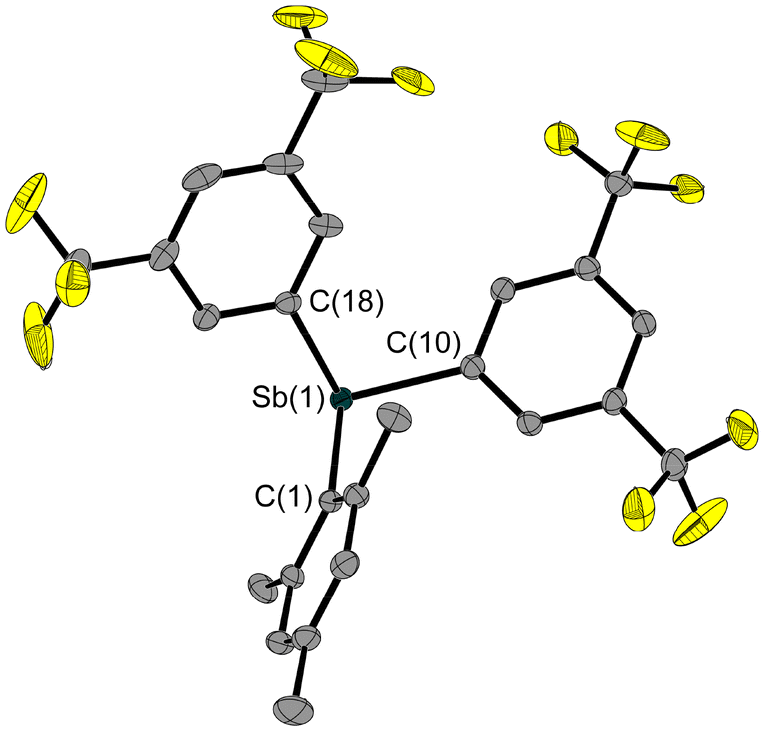 | ||
| Fig. 4 Molecular structure of 4. Ellipsoids are set at 50% probability; hydrogen atoms and the disorder of one trifluoromethyl group are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sb(1)–C(1) 2.160(2), Sb(1)–C(10) 2.162(2), Sb(1)–C(18) 2.167(2); C(1)–Sb(1)–C(10) 99.4(1), C(1)–Sb(1)–Cl(18) 96.1(1), C(10)–Sb(1)–C(18) 97.7(1). | ||
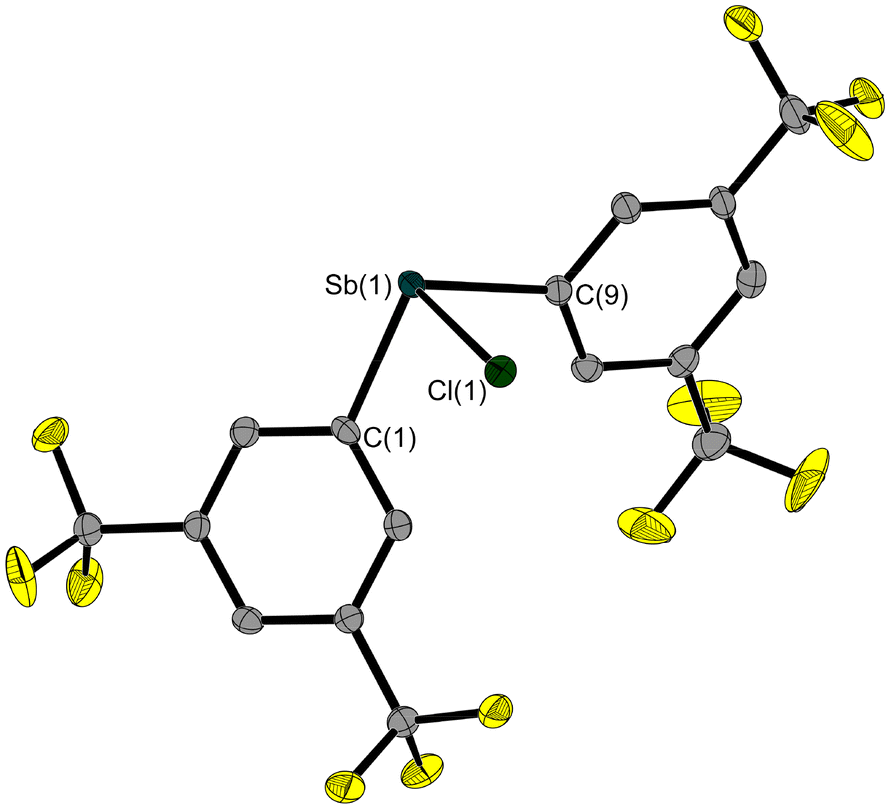 | ||
| Fig. 5 Molecular structure of 5 in the solid state. Ellipsoids are set at 50% probability; hydrogen atoms and the disorder of one trifluoromethyl group are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sb(1)–Cl(1) 2.504(1), Sb(1)–C(1) 2.151(3), Sb(1)–C(9) 2.152(3); C(1)–Sb(1)–Cl(1) 91.0(1), C(9)–Sb(1)–Cl(1) 87.8(1), C(1)–Sb(1)–C(9) 99.3(1). | ||
The reaction of 3 with di-tert-butylphosphane oxide afforded the oxy-bridged compound 6 in almost quantitative yield. In contrast to Wang and Stephan, who observed the formation of the aforementioned oxy bridge and also the elimination of hydrogen chloride in the reaction of chloroboranes with di-mesitylphosphane oxide19,20 and di-tert-butylphosphane oxide,19 respectively, we observed the formation of the Sb–O bond but did not observe the release of hydrogen chloride (Scheme 2).
 | ||
| Scheme 2 Synthesis of the pentafluoroethyl-substituted adducts 6 and 7 from 3, and the Fxyl-substituted adducts 8 and 9 from 5, with H(O)PtBu2 and H(S)PtBu2, respectively. | ||
Because of the non-spontaneous elimination of HCl, we attempted to abstract the chlorine atom and the hydrogen atom of 6 under more severe conditions by conversion with lithium and potassium hexamethyldisilazide, trityl lithium and n-butyl lithium and various amines, respectively; in all cases the corresponding NMR spectra showed only unselective decomposition or, in the case of the amines, no reaction.
In compound 6, both the chlorine substituent on the pentafluoroethylstibanyl function and the hydrogen atom of the phosphane precursor are still present. The latter can be detected particularly well in the proton NMR spectrum of compound 6, which contains a doublet with a characteristically large 1JP,H coupling constant of 447 Hz at a chemical shift of 6.03 ppm.
The excellent crystallisation behaviour enabled us to investigate the structure of 6 using X-ray diffraction. Its solid-state structure (Fig. 6) shows the adduct formation via the Sb–O bond and a bent Sb–O–P backbone. This has a rather obtuse angle at 147.5(1)°, while the O–Sb–Cl angle is even more obtuse at 167.9(1)°, and therefore close to linearity. The antimony atom is bisphenoidally surrounded by its substituents due to its lone pair.
 | ||
| Fig. 6 Molecular structures of 6, 7, 8 and 9. Ellipsoids are set at 50% probability; hydrogen atoms except those of P–H and the disordered parts of 6, 8 and 9 are omitted for clarity. Selected bond lengths [Å] and angles [°]: 6: Sb(1)–Cl(1) 2.470(1), Sb(1)–O(1) 2.273(2), Sb(1)–C(1) 2.267(3), Sb(1)–C(3) 2.285(4), P(1)–O(1) 1.503(2); O(1)–Sb(1)–Cl(1) 167.9(1), O(1)–Sb(1)–C(1) 81.8(1), O(1)–Sb(1)–C(3) 82.2(1), C(1)–Sb(1)–Cl(1) 89.0(1), C(1)–Sb(1)–C(3) 88.6(1), C(3)–Sb(1)–Cl(1) 89.8(1), Sb(1)–O(1)–P(1) 147.5(1); 7: Sb(1)–Cl(1) 2.486(1), Sb(1)–S(1) 2.751(1), Sb(1)–C(1) 2.260(1), Sb(1)–C(3) 2.267(1), P(1)–S(1) 2.010(1); Cl(1)–Sb(1)–S(1) 169.1(1), C(1)–Sb(1)–Cl(1) 89.6(1), C(1)–Sb(1)–S(1) 87.9(1), C(1)–Sb(1)–C(3) 98.5(1), C(3)–Sb(1)–Cl(1) 86.8(1), C(3)–Sb(1)–S(1) 83.2(1), Sb(1)–S(1)–P(1) 98.5(1); 8: Sb(1)–Cl(1) 2.505(2), Sb(1)–O(1) 2.348(5), Sb(1)–C(1) 2.168(9), Sb(1)–C(9) 2.183(9), P(1)–O(1) 1.525(6); Cl(1)–Sb(1)–O(1) 170.4(2), C(1)–Sb(1)–Cl(1) 90.8(2), C(1)–Sb(1)–O(1) 84.0(3), C(1)–Sb(1)–C(9) 95.0(3), C(9)–Sb(1)–Cl(1) 90.1(2), C(9)–Sb(1)–O(1) 82.3(3), Sb(1)–O(1)–P(1) 131.0(4); 9: Sb(1)–Cl(1) 2.520(1), Sb(1)–S(1) 2.829(1), Sb(1)–C(1) 2.157(2), Sb(1)–C(9) 2.147(2), P(1)–S(1) 1.995(1); Cl(1)–Sb(1)–S(1) 171.4(1), C(1)–Sb(1)–Cl(1) 88.2(1), C(1)–Sb(1)–S(1) 87.0(1), C(1)–Sb(1)–C(9) 97.4(1), C(9)–Sb(1)–Cl(1) 90.4(1), C(9)–Sb(1)–S(1) 83.3(1), Sb(1)–S(1)–P(1) 95.2(1). | ||
The distance between the chlorine substituent and the hydrogen atom, whose position has been refined, may be an indication of why intramolecular elimination of hydrogen chloride does not occur spontaneously.
The Sb–O bond length of 2.273(2) Å is within the expected range for Sb–O bonds.14 The formation of this Sb–O bond slightly lengthens the P–O bond (1.503(2) Å) compared to that of the reactant H(O)P(tBu)2 (1.482(2) Å),25 while the Sb–Cl bond in 6 (2.470(1) Å) is significantly longer than that in 3 (2.356(1) Å).
We synthesised H(S)P(tBu)2 from hydrogen sulphide and di-tert-butylchlorophosphane in the same way as H(O)P(tBu)2.21 As in 6, a doublet with a large coupling constant (420 Hz), at 5.77 ppm in the 1H NMR spectrum, is detected for the sulphanediyl-bridged compound 7.
In the solid-state structure of 7 (Fig. 6), the large difference in conformation compared to compound 6 is immediately apparent; the Sb–S–P backbone has an angle of 98.5(1)°, which is almost 50° smaller. However, the E–Sb–Cl angles in 6 and 7 are quite similar (Table 1). As expected, the bonds involving the larger sulphur atom rather than the oxygen atom are about 0.5 Å longer. The Sb–Cl bond is also more extended compared to that in 3 and 6. The structure of the central bridging unit thus follows a known behaviour: the central chalcogen-including angle is narrower in sulphanediyl- than in oxy-bridged molecules, as can be seen in the reported gas-phase structures of distibanyl oxide (Me2Sb)2O (131.1(2)°)27/distibanyl sulfide (Me2Sb)2O (104.5(5)°)28 and disiloxane (H3Si)2O (144.1(9)°)29/disilthiane (H3Si)2S (108.7(3)°).30 Substitution with electronegative groups leads to an increase in these angles (H3SiOSiH3: 144.1(9)°,29 Cl3SiOSiCl3: 146(4)°,31 F3SiOSiF3: 155.7(2)°).32
| Sb–E–P [°] | Cl–Sb–E [°] | Sb–E [Å] | Sb–Cl [Å] | P–E [Å] | |
|---|---|---|---|---|---|
| H(O)P(tBu)2 | — | — | — | — | 1.482(2)23 |
| H(S)P(tBu)2 | — | — | — | — | 1.967(1)24 |
| 3 | — | — | — | 2.356(1) | — |
| 6 (E = O) | 147.5(1) | 167.9(1) | 2.273(2) | 2.470(1) | 1.503(2) |
| 7 (E = S) | 98.5(1) | 169.1(1) | 2.751(1) | 2.486(1) | 2.010(1) |
| 5 | — | — | — | 2.504(1) | — |
| 8 (E = O) | 131.0(4) | 170.4(2) | 2.348(5) | 2.505(2) | 1.525(6) |
| 9 (E = S) | 95.2(1) | 171.4(1) | 2.829(1) | 2.520(1) | 1.995(1) |
The oxy- and sulphanediyl-bridged adducts 8 and 9 were synthesised from 7 in the same way as compounds 6 and 7 (Scheme 2). As for 6 and 7, the proton and 31P NMR spectra confirm the presence of the hydrogen atom of the phosphonium function in 8 and 9 (Table 2).
| 1H P–H [ppm] | 1 J P,H [Hz] | 31P{1H} P–H [ppm] | |
|---|---|---|---|
| H(O)P(tBu)2 | 6.01 | 424 | 64.7 |
| H(S)P(tBu)2 | 5.82 | 418 | 75.0 |
| 6 (E = O) | 6.03 | 447 | 71.6 |
| 7 (E = S) | 5.77 | 420 | 72.3 |
| 8 (E = O) | 5.85 | 436 | 70.2 |
| 9 (E = S) | 5.69 | 420 | 74.2 |
As expected, the molecular structure of 8 (Fig. 6) contains the previously observed bent structural motif with an obtuse Sb–O–P angle, which is 16° narrower than in compound 6. At the same time, the Cl–Sb–E angle is slightly more compressed at 170.4(2)°. Compared to 6, all bonds containing oxygen and chlorine are longer.
The molecular structure of 9 (Fig. 6) matches the expectations, based on the structural parameters of 6, 7 and 8. The sulphanediyl-bridging unit results in an Sb–S–P angle close to 90°, which is the smallest of all the structures discussed here (Table 1). This is in line with a narrowing of this angle due to the substitution with the Fxyl substituents compared to the pentafluoroethyl group.
However, the Cl–Sb–S angle is the most obtuse with 171.4(1)°. The Sb–S/Cl bonds are even longer than in 6, 7 and 8. Only the P–S bond is shorter than in the pentafluoroethyl-substituted analogue (Table 1).
The NMR data show a high field shift for the P–H unit in proton NMR for O → S and (F5C2) → (Fxyl), respectively. For the 31P resonances a low-field shift in the same order is observed.
Starting from 3 and nBu3SnH, we have succeeded in synthesising and isolating hydridostibane HSb(C2F5)2 (10, Scheme 3). In addition to the analogous pentafluoroethyl-substituted nitrogen,33 phosphorus34 and bismuth35 analogues, 10 is a highly reactive volatile compound that fumes strongly on contact with traces of moisture or oxygen and decomposes slowly at temperatures above −30 °C. However, an in situ-grown single crystal of 10 allowed for the determination of its solid-state structure (Fig. 7). In comparison with 3, the Sb–C bond lengths in 10 are slightly shorter. Due to the significantly lower steric demand of the hydrogen atom instead of the chlorine atom, the C–Sb–C angle in 10 is about 9° larger.
 | ||
| Scheme 3 Synthesis of 10 from 3 and nBu3SnH. | ||
 | ||
| Fig. 7 Molecular structure of the hydridostibane HSb(C2F5)2 (10) in the solid state. Ellipsoids are set at 50% probability. The disorder of one pentafluoroethyl group is not shown. Selected bond lengths [Å] and angles [°]: Sb(1)–C(1) 2.219(1), Sb(1)–C(3) 2.214(1); C(1)–Sb(1)–C(3) 94.4(1). | ||
As with the other homologues with this substitution pattern, the proton resonance at 7.28 ppm in C6D6 (8.11 ppm in CD2Cl2) shows a complex splitting pattern. This is caused by coupling to the magnetically non-equivalent fluorine atoms of the CF2 unit and to the trifluoromethyl groups. Fig. 8 shows that the experimental (top) and the simulated (bottom) multiplet patterns are in good agreement.36 The underlying fitted coupling constants for the AA′BB′H spin system in 10 and their assignment are also given.
 | ||
| Fig. 8 Optimised coupling constants of 10 (left) and highlighted area of the experimental and simulated Sb–H region of the 1H NMR spectrum of 10 in C6D6 (right).34 | ||
In the IR spectrum of gaseous 10, we detected a band at 1919 cm−1 for the Sb–H vibration, which is within the typical range for this type of stretching frequency.37
The Gutmann–Beckett test allows an assessment to be made about the Lewis acidity of the compounds. The so-called acceptor number (AN) is calculated based on the 31P NMR shift of the adduct of the Lewis acid with OP(Et)3.38 With AN values of 52.3 (3), 52.5 (5) and 29.8 (10), the stibanes 3 and 5 are in the upper mid-range of Gutmann's original scale from 0 (for hexane) to 100 (for SbCl5); according to the test, 10 is significantly less Lewis acidic than 3 and 5.
We tried to react hydridostibane 10 with the phosphane oxide H(O)P(tBu)2 and the phosphane sulphide H(S)P(tBu)2, expecting the formation of the adducts (C2F5)2Sb(H)–O–P(H)(tBu)2 and (C2F5)2Sb(H)–S–P(H)(tBu)2, which would be the formally hydrogen-loaded FLPs. In the samples of these reactions we detected all the NMR signals of the reactants, but only the reaction of the sample containing the phosphane oxide showed a slight deviation in the NMR shifts relative to the reactants. For the mixture of 10 with phosphane sulphide there is no evidence of an interaction or reaction. However, attempts to isolate the product of the conversion of 10 with H(O)P(tBu)2 failed and undefinable decomposition products were observed.
The mixture of 10 with H(O)P(tBu)2 was also investigated by diffusion NMR experiments. The hydrodynamic volumes of 10 and the phosphane oxide in the mixture were found to be only slightly larger compared to those of the pure reactants. This does not allow for any clear conclusions to be drawn about the reaction of the two reactants.
Fxyl2SbH could not be synthesised from 7 and nBu3SnH by a procedure analogous to that for 10, so it was not possible to investigate any adducts that might be formed.
Conclusions
The presented RF2Sb(Cl)–E–(H)P(tBu)2 (RF = C2F5, 3,5-(CF3)2C6H3; E = O, S) systems, based on the corresponding chlorostibanes and phosphane chalcogenides, were found to be stable against HCl elimination. Even attempts to abstract hydrogen chloride under more severe reaction conditions failed. The angle of the bridging Sb–E–P unit in the adducts is significantly narrower for compounds with the higher homologue sulphur than for oxygen. The pentafluoroethyl groups cause a widening of this angle, as can be seen from the comparison with the Fxyl groups on the antimony atom in the corresponding solid-state structures. The hydridostibane HSb(C2F5)2, obtained from the reaction of ClSb(C2F5)2 with nBu3SnH, seems to interact weakly with di-tert-butylphosphane oxide as revealed by slight shifts of the resonances in the NMR spectra. For a mixture of HSb(C2F5)2 with di-tert-butylphosphane sulphide, comparable behaviour was not observed.Author contributions
J. Krieft: investigation, methodology, validation, visualization, and writing (original draft); H. Koch: investigation (supporting synthesis); J.-H. Lamm: investigation (SCXRD); B. Neumann: investigation (SCXRD); H.-G. Stammler: investigation (SCXRD); A. Mix: investigation (diffusion NMR); and N. W. Mitzel: funding acquisition, project administration, supervision, reviewing and editing.Data availability
The data published in this contribution are available as the ESI,† submitted with the manuscript. Crystallographic data have been deposited with the Cambridge Crystal Structure Database (CCDC).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Marco Wißbrock for recording NMR spectra and Barbara Teichner for performing elemental analyses. This work was supported by Deutsche Forschungsgemeinschaft (grant MI477/44-1, project number 461833739).Notes and references
- H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325–329 CrossRef CAS.
- (a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS; (c) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (d) D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC; (e) S. Döring, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Organometallics, 1998, 17, 2183–2187 CrossRef.
- T. Thorwart, D. Hartmann and L. Greb, Chem. – Eur. J., 2022, 28, e202202273 CrossRef CAS.
- B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416–13419 CrossRef CAS.
- T. A. Kinder, R. Pior, S. Blomeyer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2019, 25, 5899–5903 CrossRef CAS PubMed.
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114–5118 CrossRef CAS PubMed.
- (a) M. Pieper, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2017, 46, 5326–5336 RSC; (b) D. Salusso, G. Grillo, M. Manzoli, M. Signorile, S. Zafeiratos, M. Barreau, A. Damin, V. Crocellà, G. Cravotto and S. Bordiga, ACS Appl. Mater. Interfaces, 2023, 15, 15396–15408 CrossRef CAS.
- (a) R. C. Neu, E. Otten, A. Lough and D. W. Stephan, Chem. Sci., 2011, 2, 170–176 RSC; (b) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826–8829 CrossRef CAS; (c) Z. Chen, Y. Ye, X. Feng, Y. Wang, X. Han, Y. Zhu, S. Wu, S. Wang, W. Yang, L. Wang and J. Zhang, Nat. Commun., 2023, 14, 2000 CrossRef CAS PubMed; (d) K. Chang, X. Wang, Z. Fan and X. Xu, Inorg. Chem., 2018, 57, 8568–8580 CrossRef CAS; (e) P. Jochmann and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 9831–9835 CrossRef CAS.
- (a) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 CrossRef CAS; (b) L. Keweloh, H. Klöcker, E.-U. Würthwein and W. Uhl, Angew. Chem., Int. Ed., 2016, 55, 3212–3215 CrossRef CAS; (c) P. Federmann, R. Müller, F. Beckmann, C. Lau, B. Cula, M. Kaupp and C. Limberg, Chem. – Eur. J., 2022, 28, e202200404 CrossRef CAS; (d) A. F. G. Maier, S. Tussing, T. Schneider, U. Flörke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef CAS; (e) K. Samigullin, I. Georg, M. Bolte, H.-W. Lerner and M. Wagner, Chem. – Eur. J., 2016, 22, 3478–3484 CrossRef CAS PubMed; (f) H. Li, A. J. A. Aquino, D. B. Cordes, F. Hung-Low, W. L. Hase and C. Krempner, J. Am. Chem. Soc., 2013, 135, 16066–16069 CrossRef CAS PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS; (b) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef; (c) D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC; (d) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS; (e) C. Manankandayalage, D. K. Unruh and C. Krempner, Chem. – Eur. J., 2021, 27, 6263–6273 CrossRef CAS PubMed; (f) O. J. Metters, S. J. K. Forrest, H. A. Sparkes, I. Manners and D. F. Wass, J. Am. Chem. Soc., 2016, 138, 1994–2003 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- P. Holtkamp, D. Poier, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2021, 27, 3793–3798 CrossRef CAS PubMed.
- J. Krieft, P. C. Trapp, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler, J.-H. Lamm and N. W. Mitzel, Chem. Sci., 2024, 15, 12118–12125 RSC.
- J. Krieft, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2024, 53, 11762–11768 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef CAS; (b) D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC; (c) C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 CrossRef CAS; (d) C.-H. Chen and F. P. Gabbaï, Dalton Trans., 2018, 47, 12075–12078 RSC; (e) G. Park, D. J. Brock, J.-P. Pellois and F. P. Gabbaï, Chem, 2019, 5, 2215–2227 CrossRef CAS; (f) M. Yang, M. Hirai and F. P. Gabbaï, Dalton Trans., 2019, 48, 6685–6689 RSC; (g) G. Park and F. P. Gabbaï, Chem. Sci., 2020, 11, 10107–10112 RSC; (h) D. You, B. Zhou, M. Hirai and F. P. Gabbaï, Org. Biomol. Chem., 2021, 19, 4949–4957 RSC; (i) V. M. Gonzalez, G. Park, M. Yang and F. P. Gabbaï, Dalton Trans., 2021, 50, 17897–17900 RSC; (j) B. L. Murphy and F. P. Gabbaï, J. Am. Chem. Soc., 2023, 145, 19458–19477 CrossRef CAS.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2023, 14, 13551–13559 RSC.
- D. Zhu, Z.-W. Qu and D. W. Stephan, Dalton Trans., 2020, 49, 901–910 RSC.
- Y. Wang, Z. H. Li and H. Wang, RSC Adv., 2018, 8, 26271–26276 RSC.
- L. Wickemeyer, N. Aders, A. Mix, B. Neumann, H.-G. Stammler, J. J. Cabrera-Trujillo, I. Fernández and N. W. Mitzel, Chem. Sci., 2022, 13, 8088–8094 RSC.
- L. Wickemeyer, L. Hartmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2022, e202202842 Search PubMed.
- M. Ates, H. J. Breunig, A. Soltani-Neshan and M. Tegeler, Z. Naturforsch., 1986, 41B, 321–326 CrossRef CAS.
- D. Naumann, G. Nowicki and K.-J. Sassen, Z. Anorg. Allg. Chem., 1997, 623, 1183–1189 CrossRef CAS.
- F. Dornhaus, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o657–o658 CrossRef CAS.
- G. Y. Li and W. J. Marshall, Organometallics, 2002, 21, 590–591 CrossRef CAS.
- A. Haaland, V. I. Sokolov, H. V. Volden, H. J. Breunig, M. Denker and R. Rösler, Z. Naturforsch., 1997, 52B, 296–300 CrossRef.
- A. Haaland, D. J. Shorokhov, V. I. Sokolov, H. V. Volden, H. J. Breunig, M. Denker and R. Rösler, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 136, 463–466 CrossRef.
- A. Almenningen, O. Bastiansen, V. Ewing, K. Hedberg and M. Traetteberg, Acta Chem. Scand., 1963, 17, 2455–2460 CrossRef CAS.
- A. Almenningen, K. Hedberg and R. Seip, Acta Chem. Scand., 1963, 17, 2264–2270 CrossRef CAS.
- W. Airey, C. Glidewell, A. G. Robiette and G. M. Sheldrick, J. Mol. Struct., 1971, 8, 413–422 CrossRef CAS.
- W. Airey, C. Glidewell, D. W. H. Rankin, A. G. Robiette, G. M. Sheldrick and D. W. J. Cruickshank, Trans. Faraday Soc., 1970, 66, 551 RSC.
- K. E. Peterman and J. M. Shreeve, Inorg. Chem., 1975, 14, 1223–1228 CrossRef CAS.
- A. V. Zakharov, Y. V. Vishnevskiy, N. Allefeld, J. Bader, B. Kurscheid, S. Steinhauer, B. Hoge, B. Neumann, H.-G. Stammler, R. J. F. Berger and N. W. Mitzel, Eur. J. Inorg. Chem., 2013, 3392–3404 CrossRef CAS.
- S. Solyntjes, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat'ev and B. Hoge, Chem. – Eur. J., 2017, 23, 1557–1567 CrossRef CAS PubMed.
- P. H. Budzelaar, gNMR Version 5.0, IvorySoft, 2006 Search PubMed.
- D. W. H. Rankin, C. A. Morrison and N. W. Mitzel, Structural Methods in Molecular Inorganic Chemistry, John Wiley & Sons, Inc., Chichester, West Sussex, U.K., 1st edn, 2013 Search PubMed.
- (a) M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS; (b) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367656–2367665. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02268h |
| This journal is © The Royal Society of Chemistry 2024 |