
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the remote catalytic functionalisation of ferrocenes
William
Erb

Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: william.erb@univ-rennes.fr
First published on 3rd September 2024
Abstract
While the catalytic activation of C–H bonds adjacent to a directing group is well developed in the ferrocene series, the functionalisation of a remote position has scarcely been explored. Here, we summarise the latest developments in this field in the broader context of 1,3-disubstituted ferrocene derivatives.
 William Erb | William Erb obtained his Ph.D. in organic chemistry in 2010 under the supervision of Prof. Jieping Zhu on the total synthesis of natural products and palladium-catalyzed reactions. During the next 4 years of post-doctoral studies he worked in various laboratories on different research projects ranging from organocatalysis to supramolecular chemistry with an emphasis on the development of new methodologies (University of Bristol, Prof. Varinder Aggarwal, UK – ESPCI, Prof. Janine Cossy, Paris – LCMT, Prof. Jacques Rouden, Caen – COBRA, Prof. Géraldine Gouhier, Rouen). He was appointed assistant professor at the University of Rennes (France) in 2015 where he is mainly working on the development of ferrocene functionalization toward original derivatives. |
Introduction
The serendipitous discovery of ferrocene in 19511,2 rejuvenated the area of organometallic chemistry and, 70 years later, derivatives of this stable organometallic still occupy a pivotal place in chemistry. However, the development of synthetic routes3–6 and applications7–10 is mainly limited to monosubstituted, 1,1′- and 1,2-disubstituted derivatives, while unusual substitution patterns remain barely investigated. The case of 1,3-disubstituted ferrocenes is especially representative of this trend, although this original scaffold is a key element in compounds used as liquid crystals,11 sensors,12,13 molecular machines14 and biologically active products.15 Pincer ligands,16,17 chalcogen bond donors18 and phosphine-gold catalysts19 are other representative examples of this family of compounds. Although these studies highlight the value of this scaffold, the development of 1,3-disubstituted ferrocenes remains well behind that of 1,1′- and 1,2-disubstituted analogues.This is due, at least in part, to the lack of straightforward synthetic approaches to these compounds. Apart from the construction of the ferrocene core itself, as initially reported by Hafner,20 common strategies to produce 1,3-disubstituted ferrocenes rely on the functionalisation of a ferrocene derivative (Scheme 1). This can be achieved using aromatic electrophilic substitution as described by Nesmeyanov, although hardly separable isomeric mixtures are obtained.21 Directed deprotolithiation using removable directing groups (DG), initially documented by Slocum22 and later revisited by Weissensteiner,23,24 Jaouen25 and Butler,26 has remained the preferred route to 1,3-disubstituted ferrocenes, especially in their stereopure form. Another approach, mainly developed by our group,27–34 relies on the “halogen dance” reaction,35–37 an isomerisation process promoted by a lithium amide and driven by the stability of lithiated intermediates during which a heavy halogen adjacent to a directing group moves to a remote position. A final approach, based on the remote deprotolithiation of ferrocene sulfides has also been reported by Brown.38 Although various 1,3-disubstituted ferrocenes have been made over the last few decades using these approaches, they suffer from either the number of steps involved or their lack of generality. It is therefore clear that the development of straightforward synthetic routes is needed to explore the chemical space around 1,3-disubstituted ferrocenes and unlock their full potential.
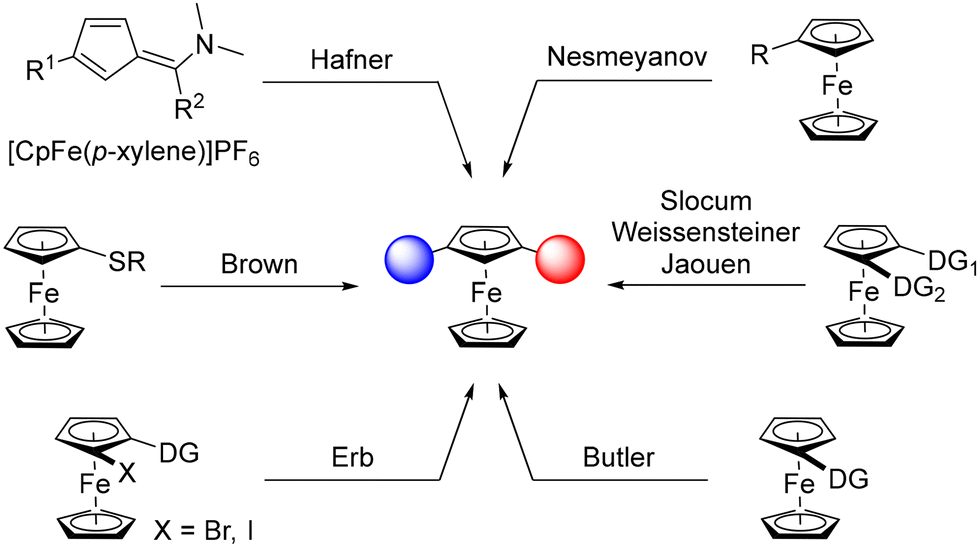 | ||
| Scheme 1 Main approaches toward 1,3-disubstituted ferrocenes. Cp: cyclopentadiene. | ||
Remote catalytic functionalisation of ferrocenes
Over the last few decades, remote catalytic C–H functionalisation has emerged as a powerful tool to introduce substituents in a site-selective, step- and atom-economical manner in the benzene series.39–42 Although this approach holds great potential for efficiently delivering 1,3-disubstituted ferrocenes, it has scarcely been studied until recently.In 2004, Plenio pioneered this strategy in the ferrocene series, with the reaction between substituted ferrocenes and bis(pinacolato)diboron (B2pin2) in the presence of [Ir(cod)(OMe)]2 (cod: 1,5-cyclooctadiene) and 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) under reflux in octane (Scheme 2).43 From bromoferrocene and carbomethoxyferrocene, the 3-borylated products 1a–b were isolated in moderate yields while substantial amounts of their 1′ isomers were also formed (28 and 31% yields, respectively). This side reaction on the unsubstituted cyclopentadienyl ring was suppressed when 1,1′-disubstituted ferrocenes were evaluated, and the corresponding products 2a–b were obtained in good yields. However, a ketone was barely compatible as 1,1′-diacetylferrocene gave only 15% of the corresponding borylated product 2c. The presence of electron-donating substituents led to a marked drop in reactivity, with 1,1′-dimethylferrocene 2d being isolated in a low 12% yield. The origin of the regioselectivity was not discussed at this stage.
 | ||
| Scheme 2 Iridium-catalysed remote borylation of ferrocene derivatives. | ||
It was only 20 years after Plenio's seminal work that progress was made in the field of remote catalytic functionalisation of ferrocene, with four studies published in 2023. The first was reported by Hu and Chen who revisited the iridium-catalysed borylation of 1,1′-disubstitued ferrocenes using a catalytic system consisting of [Ir(cod)(OMe)]2 and 2,2′-dipyridyl-(3-fluorophenyl)methane (L1) at 100 °C in methylcyclohexane (Scheme 3).44 Pleasingly, they were able to overcome the limitations encountered by Plenio43 as electron-rich derivatives bearing alkyls, aryls, silanes and sulfides now reacted smoothly to afford the desired products 2d–g in good yields. Electron-withdrawing substituents such as ester and bromine, provided that 3,4,7,8-tetramethyl-1,10-phenanthroline was used for the latter, were also tolerated and afforded the compounds 2a and 2b in 83 and 54% yields, respectively. However, the acetate and sulfone derivatives required the use of tetrahydrofuran (THF) to deliver the products 2h–i. To rationalise the excellent regioselectivities recorded, the authors postulated that the unfavourable steric interactions between the substituents and the bulky catalyst45 would favour activation of the remote C–H bond rather than the one adjacent to the substituent.
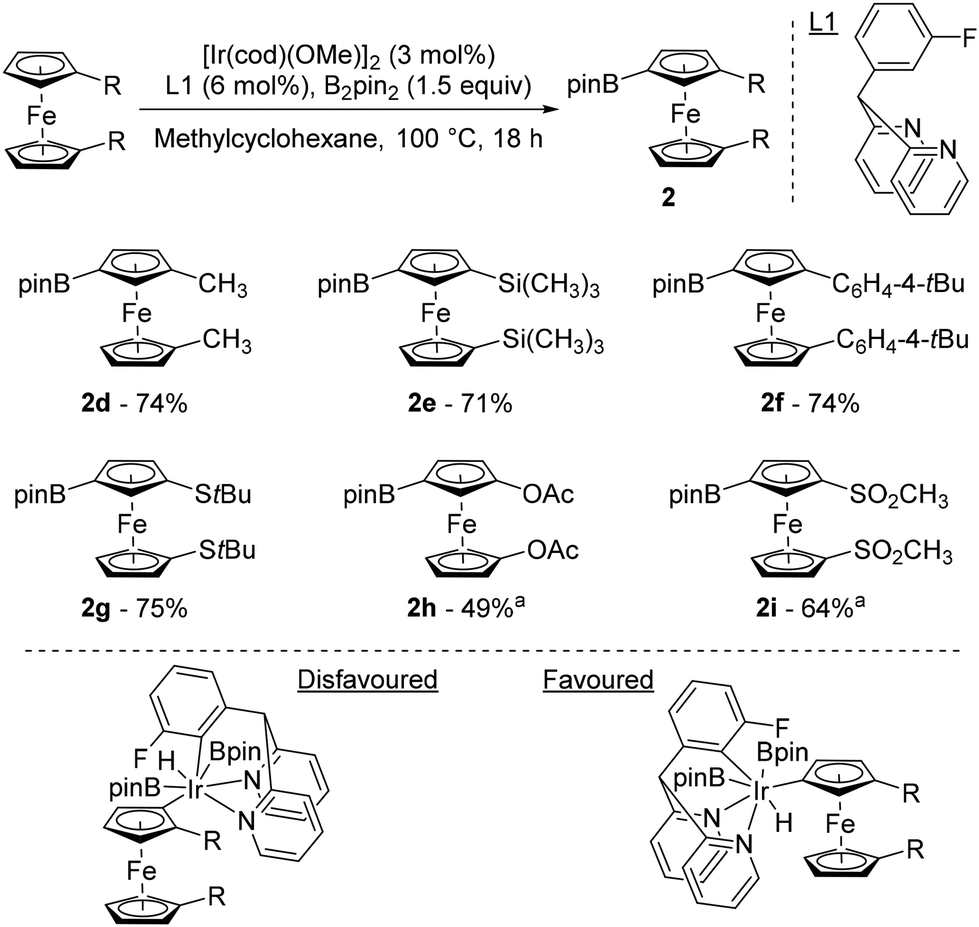 | ||
Scheme 3 Top: iridium-catalysed remote borylation of ferrocene derivatives. Bottom: putative origin of the observed regioselectivity. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction performed in THF. Reaction performed in THF. | ||
That same year, Madhavan and Kapur reported another approach to 1,3-disubstituted ferrocenes46 using the template strategy originally developed by Yu.47 Many challenges are associated with this approach: (1) controlling proximal vs. remote functionalisation; (2) forming a large thermodynamically unfavourable cyclophane-like pre-transition state ring; (3) performing functionalisation of the substituted ring; and (4) avoiding polyfunctionalisation. To address these issues, the authors attached a fluoropyridine-based template (R1) to aminomethylferrocene (compound 3) and optimised a catalytic system consisting of Pd(OAc)2, N-acetylglycine (N-Ac-Gly-OH) and AgOAc in 2,2,2-trifluoroethanol (TFE) at 60 °C (Scheme 4). Under these conditions, they were able to carry out remote alkenylation of their substrates with moderate to good yields and selectivities. The reaction tolerated various alkyl- (4a), aryl- (4b) and benzylacrylates (4c) as coupling partners, as well as acrylamides (4d), acrylonitrile (4e) and vinylsulfone (4f). More complex acrylates derived from glucofuranose, cholesterol, D-α-tocopherol and fenchol were also involved in the reaction, again with excellent regioselectivities but slightly lower yields. Cleavage of the template could be achieved using HCl under reflux with ethanol prior to the protection of the resulting primary amine.
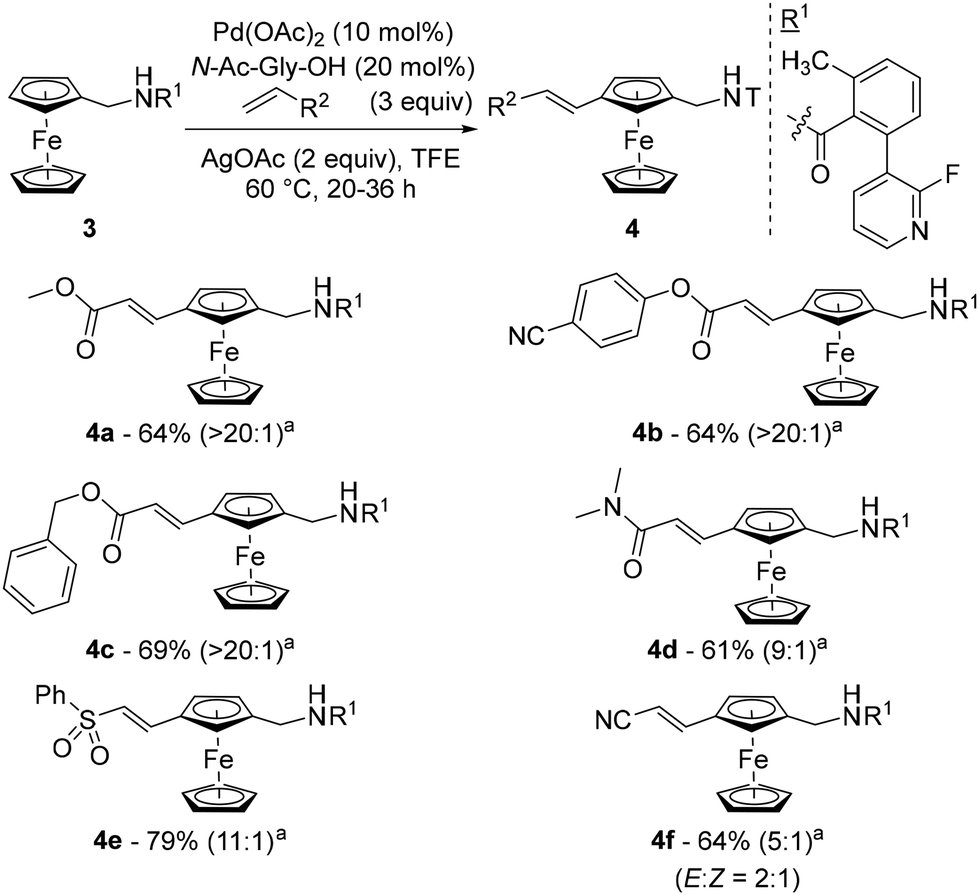 | ||
| Scheme 4 Representative examples of the remote functionalisation of ferrocene using the template-assisted approach. aRatio between the product functionalised at the 3-position and the other isomers, determined by HPLC of the crude reaction mixture. | ||
The structure of the template was of utmost importance as no product was formed in the absence of either the methyl group or the fluorine atom. While the former probably favours an optimal conformation of the template, the effect of the latter could be steric (also favouring an active conformation) or electronic (modulating the coordination capacity of the pyridine). Deuteration studies ruled out the C–H activation and β-elimination steps as rate-determining, while the stoichiometric reaction between substrate 3, Pd(OAc)2 and N-Ac-Gly-OH allowed identification of the palladacycle III by ESI-HRMS (Scheme 5). On the basis of these findings, the authors proposed the catalytic cycle depicted in Scheme 5.
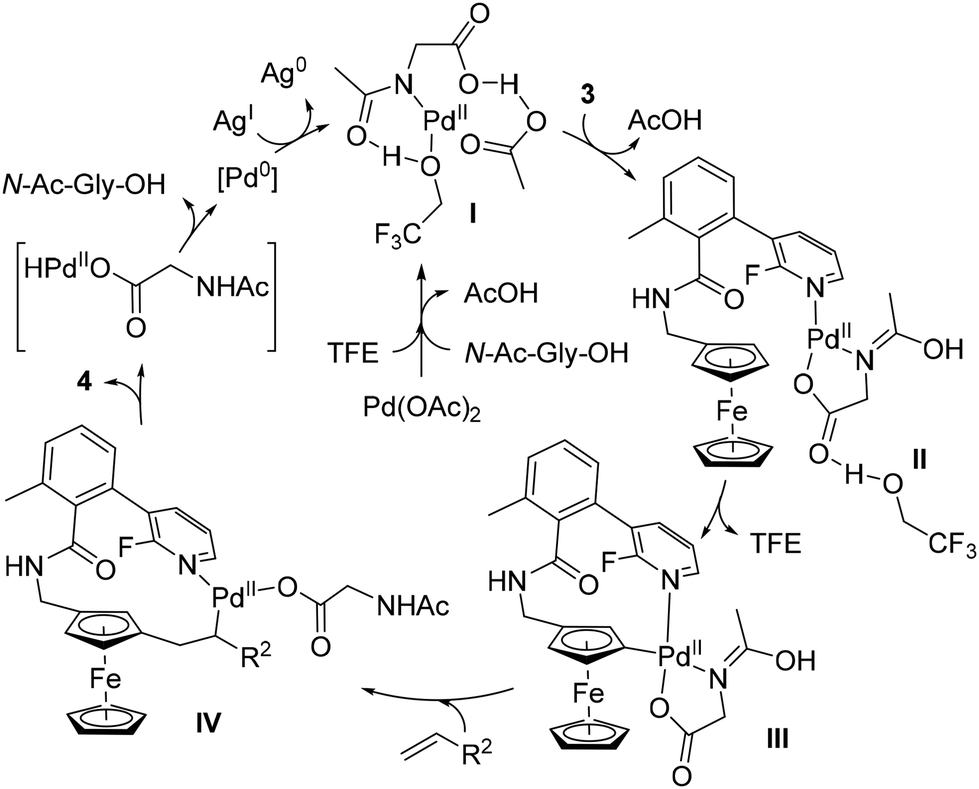 | ||
| Scheme 5 Proposed reaction mechanism for the remote C–H functionalisation of ferrocene by the template-assisted approach. | ||
An enantioselective version of this remote C–H activation was attempted using various chiral amino acids, (R)-BINOL and various (R)-BINOL-based phosphoric acids. However, these efforts were unsuccessful, with the best value of 9% ee having been reached with N-acetyl-alanine as the ligand.
In a research paper published online in December 2022 and not yet peer-reviewed,48 the same research group also explored a relay strategy inspired by the Catellani reaction49 and originally developed by Yu in the benzene series.50 In this process, a norbornene derivative was used as a relay to move a palladium intermediate adjacent to a directing group to a remote position. Starting from (dimethylaminomethyl)ferrocene (5), the authors identified a combination of Pd(OAc)2, norbornene (NBE1), ligand L2, tetra-n-butylammonium bromide (TBAB), silver trifluoroacetate (AgTFA) and potassium carbonate in dimethylformamide (DMF) as able to deliver the arylated ferrocenes 6 in moderate yields (Scheme 6, top). While the use of protected amino acids with norbornene was inefficient (10% yield at best), arylation occurred in 44% yield when N-Ac-Gly-OH was used in combination with 2-carbomethoxynorbornene, suggesting a subtle interaction between the norbornene relay and the ligand. Various substituted iodobenzenes were evaluated as coupling partners in yields ranging from 26 to 68% (compounds 6a–c) and the reaction was extended to iodinated pyridines (6d), indole, benzopyrazole (6e) and carbazole as well as some natural product derivatives.
 | ||
| Scheme 6 Representative examples of the remote functionalisation of ferrocene by the relay strategy. | ||
An asymmetric version of the reaction was developed using N-acetyl-L-phenylalanine (N-Ac-L-Phe-OH) in combination with 2-carbomethoxynorbornene (NBE2), affording the product 6a in 46% yield and 92% ee (Scheme 6, bottom). While all L-amino acids gave the same enantiomer, although not determined, of 6a, the use of N-Ac-D-Phe-OH surprisingly gave similar results, albeit with a reduced 80% ee.
The latest study concerning remote functionalisation in the ferrocene series was published by Cheng, Yu and Zhou in April 2023, although it was submitted a year earlier.51 In this work, the authors also explored the relay strategy initially developed in the benzene series.50 To address the differences between benzene and ferrocene, which can affect both palladation at position 3 and final relay elimination, they evaluated a library of racemic norbornene derivatives in combination with protected L-valine derivatives. Like Madhavan and Kapur, the use of 2-carbomethoxynorbornene and N-Boc-L-valine did not yield the expected product. However, minor modifications to norbornene had a major influence on the reaction outcome, with 1-carbomethoxynorbornene improving the yield by 19%. Optimisation of the reaction parameters led to the identification of the best catalytic system consisting of Pd(OAc)2, N-Boc-L-Val-OH, norbornene NBE3 and potassium carbonate in a DMF–DMSO mixture at 80 °C for 18 h (Scheme 7). The high enantioselectivity for the Sp enantiomers, revealed by X-ray diffraction analysis, arises solely from the chiral amino acid as the use of enantioenriched NBE3 did not influence the reaction outcome whereas N-Boc-D-valine gave the Rp enantiomer with 99% ee. Various substituted iodobenzenes (6a–b), benzothiophene, indole (6f), pyridine (6g) and quinoline were evaluated as coupling partners with yields ranging from 39 to 95%, and ee from 96 to 99%. Aryl bromides can also be used with unchanged enantioselectivities, albeit in reduced yields. The reaction has been extended to complex aryl iodides derived from drugs (6h) or natural products (6i) as well as ruthenocene derivatives (6j), again with good results. Deuteration studies revealed that activation of the adjacent C–H bond was irreversible and could therefore be the enantiodetermining step. The authors also found that the final protodepalladation step was favoured by a protic species such as the remaining water, and the catalytic cycle depicted in Scheme 8 was proposed.
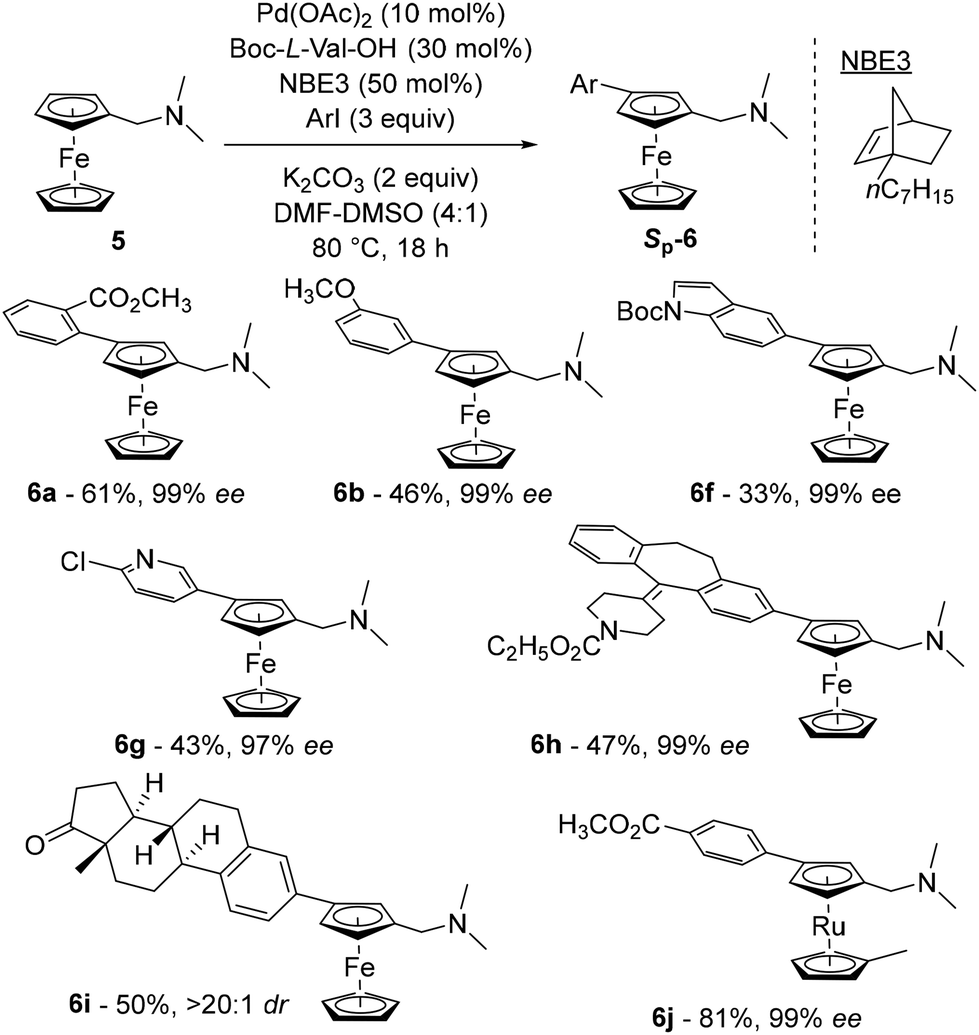 | ||
| Scheme 7 Representative examples of the asymmetric remote functionalisation of ferrocene by the relay strategy. | ||
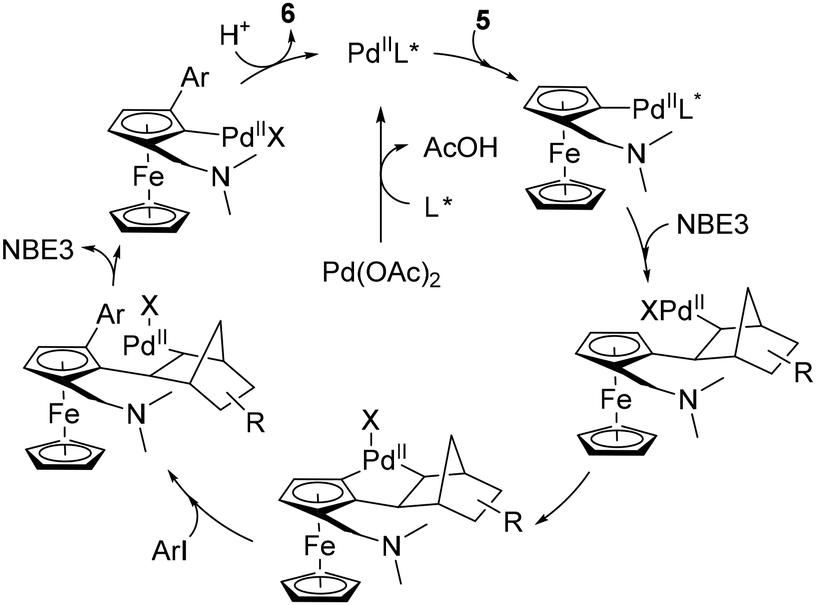 | ||
| Scheme 8 Proposed reaction mechanism for the remote asymmetric functionalisation of ferrocene using a transient mediator. | ||
Conclusions and perspectives
While Plenio's seminal paper on the remote catalytic functionalisation of ferrocene was published 20 years ago, it is only in recent years that we have witnessed elegant developments of this approach. However, there are still challenges ahead. One of these is to broaden the scope of coupling partners in these transformations. Indeed, while the pinacol ester can be converted into other substituents, alkenes and aryls are less amenable to other transformations. It would also be desirable to extend these reactions to other substituted ferrocenes. Although catalytic borylation currently appears to be unlimited in terms of ferrocene substituents, the template and relay strategies require ferrocenes bearing specific linkers or directing groups. Finally, the development of remote asymmetric functionalisation is still in its infancy. The relay strategy is currently the only approach capable of providing enantioenriched products, with the origin of the enantioselectivity residing in the asymmetric activation of a C–H bond adjacent to the directing group. Controlling the direct asymmetric activation of a C–H bond remote from a substituent is likely to be the next objective to be reached. To address these challenges, inspiration could be drawn from work on non-covalently bound templates40,41 based on metal coordination, hydrogen bonding or ionic interactions. These might be capable of enantioselectively directing a catalytic metal to the position 3 of ferrocene, as recently reported by Phipps in the benzene series.52 The development of these methodologies is expected to broaden the 1,3-disubstituted ferrocenes family, for applications in medicine,15,53 molecular machines14 and catalysis.19Author contributions
Writing – review and editing, W. E.Data availability
No primary research results were generated as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche (ANR; Ferrodance project ANR-19-CE07-0015-01). A CC-BY public copyright licence has been applied by the author and will be applied to all subsequent versions up to the Author's Accepted Manuscript, in accordance with the grant's open access conditions. W. E. thanks the Université de Rennes, Rennes Métropole and Prof. F. Mongin for their support, critical review of this document and valuable suggestions.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- I. R. Butler, Eur. J. Inorg. Chem., 2012, 2012, 4387–4406 CrossRef.
- D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668–5704 CrossRef CAS.
- S. Arae and M. Ogasawara, Tetrahedron Lett., 2015, 56, 1751–1761 CrossRef CAS.
- M. Korb and H. Lang, Eur. J. Inorg. Chem., 2022, e202100946 CrossRef CAS.
- R. Sun, L. Wang, H. Yu, Z.-U. Abdin, Y. Chen, J. Huang and R. Tong, Organometallics, 2014, 33, 4560–4573 CrossRef CAS.
- L. Cunningham, A. Benson and P. J. Guiry, Org. Biomol. Chem., 2020, 18, 9329–9370 RSC.
- B. Sharma and V. Kumar, J. Med. Chem., 2021, 64, 16865–16921 CrossRef CAS PubMed.
- G. Roy, R. Gupta, S. Ranjan Sahoo, S. Saha, D. Asthana and P. Chandra Mondal, Coord. Chem. Rev., 2022, 473, 214816 CrossRef CAS.
- R. Deschenaux and J.-L. Marendaz, J. Chem. Soc., Chem. Commun., 1991, 909–910 RSC.
- S. R. Collinson, T. Gelbrich, M. B. Hursthouse and J. H. R. Tucker, Chem. Commun., 2001, 555–556 RSC.
- J. Y. C. Lim and P. D. Beer, Eur. J. Inorg. Chem., 2017, 220–224 CrossRef.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612–5613 CrossRef.
- B. Ferber, S. Top, A. Vessières, R. Welter and G. Jaouen, Organometallics, 2006, 25, 5730–5739 CrossRef.
- E. J. Farrington, E. Martinez Viviente, B. S. Williams, G. van Koten and J. M. Brown, Chem. Commun., 2002, 308–309 RSC.
- A. A. Koridze, A. M. Sheloumov, S. A. Kuklin, V. Y. Lagunova, I. I. Petukhova, F. M. Dolgushin, M. G. Ezernitskaya, P. V. Petrovskii, A. A. Macharashvili and R. V. Chedia, Russ. Chem. Bull., 2002, 51, 1077–1078 CrossRef.
- V. Mamane, P. Peluso, E. Aubert, R. Weiss, E. Wenger, S. Cossu and P. Pale, Organometallics, 2020, 39, 3936–3950 CrossRef.
- U. Caniparoli, I. Escofet and A. M. Echavarren, ACS Catal., 2022, 12, 3317–3322 CrossRef PubMed.
- P. Bickert, B. Hildebrandt and K. Hafner, Organometallics, 1984, 3, 653–657 CrossRef.
- A. N. Nesmeyanov, E. V. Leonova, N. S. Kochetkova, A. I. Malkova and A. G. Makarovskaya, J. Organomet. Chem., 1975, 96, 275–278 CrossRef.
- D. W. Slocum, R. L. Marchal and W. E. Jones, J. Chem. Soc., Chem. Commun., 1974, 967–968 RSC.
- M. Steurer, K. Tiedl, Y. Wang and W. Weissensteiner, Chem. Commun., 2005, 4929–4931 RSC.
- M. Steurer, Y. Wang, K. Mereiter and W. Weissensteiner, Organometallics, 2007, 26, 3850–3859 CrossRef CAS.
- B. Ferber, S. Top, R. Welter and G. Jaouen, Chem. – Eur. J., 2006, 12, 2081–2086 CrossRef CAS PubMed.
- I. R. Butler, M. G. B. Drew, C. H. Greenwell, E. Lewis, M. Plath, S. Mussig and J. Szewczyk, Inorg. Chem. Commun., 1999, 2, 576–580 CrossRef CAS.
- A. Zirakzadeh, A. Herlein, M. A. Groß, K. Mereiter, Y. Wang and W. Weissensteiner, Organometallics, 2015, 34, 3820–3832 CrossRef.
- M. Tazi, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet and F. Mongin, Organometallics, 2017, 36, 4770–4778 CrossRef.
- W. Erb and T. Roisnel, Chem. Commun., 2019, 55, 9132–9135 RSC.
- M. Tazi, W. Erb, T. Roisnel, V. Dorcet, F. Mongin and P. J. Low, Org. Biomol. Chem., 2019, 27, 9352–9359 RSC.
- W. Erb, M. Wen, J.-P. Hurvois, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel, Eur. J. Inorg. Chem., 2021, 3165–3176 CrossRef.
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel and V. Dorcet, Organometallics, 2021, 40, 1129–1147 CrossRef CAS.
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel, Molecules, 2022, 27, 1798 CrossRef CAS PubMed.
- W. Erb, J.-P. Hurvois, Y. S. Halauko, V. E. Matulis and T. Roisnel, Inorg. Chem. Front., 2022, 9, 5862–5883 RSC.
- M. Schnürch, in Halogenated Heterocycles: Synthesis, Application and Environment, ed. J. Iskra, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 185–218 Search PubMed.
- W. Erb and F. Mongin, Tetrahedron, 2016, 72, 4973–4988 CrossRef CAS.
- K. Inoue and K. Okano, ChemCatChem, 2024, 16, e202400408 CrossRef CAS.
- C. Pichon, B. Odell and J. M. Brown, Chem. Commun., 2004, 598–599 RSC.
- A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS.
- U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef PubMed.
- A. Datta, A. Kollhofer and H. Plenio, Chem. Commun., 2004, 1508–1509 RSC.
- H. Zheng, C.-H. Liu, X.-Y. Wang, Y. Liu, B.-Z. Chen, Y.-C. Hu and Q.-A. Chen, Adv. Sci., 2023, 10, 2304672 CrossRef.
- M. R. Jones, C. D. Fast and N. D. Schley, J. Am. Chem. Soc., 2020, 142, 6488–6492 CrossRef PubMed.
- P. Gupta, S. Madhavan and M. Kapur, Angew. Chem., Int. Ed., 2023, 62, e202305278 CrossRef PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef.
- P. Gupta, P. C. Tiwari, S. Madhavan and M. Kapur, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-zc2vv.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581–585 CrossRef PubMed.
- L. Zhou, H.-G. Cheng, L. Li, K. Wu, J. Hou, C. Jiao, S. Deng, Z. Liu, J.-Q. Yu and Q. Zhou, Nat. Chem., 2023, 15, 815–823 CrossRef PubMed.
- G. R. Genov, J. L. Douthwaite, A. S. K. Lahdenperä, D. C. Gibson and R. J. Phipps, Science, 2020, 367, 1246–1251 CrossRef PubMed.
- N. J. Forrow, G. S. Sanghera and S. J. Walters, J. Chem. Soc., Dalton Trans., 2002, 3187–3194 RSC.
| This journal is © The Royal Society of Chemistry 2024 |