
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel(II) complexes with 14-membered bis-thiosemicarbazide and bis-isothiosemicarbazide ligands: synthesis, characterization and catalysis of oxygen evolution reaction†
Iuliana
Besleaga
 a,
Anastasia A.
Fesenko
b,
Anup
Paul
c,
Biljana
Šljukić
d,
Peter
Rapta
*e,
Armando J. L.
Pombeiro
*c,
Anatoly D.
Shutalev
*f and
Vladimir B.
Arion
*ag
a,
Anastasia A.
Fesenko
b,
Anup
Paul
c,
Biljana
Šljukić
d,
Peter
Rapta
*e,
Armando J. L.
Pombeiro
*c,
Anatoly D.
Shutalev
*f and
Vladimir B.
Arion
*ag
aUniversity of Vienna, Institute of Inorganic Chemistry, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: vladimir.arion@univie.ac.at
bA. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, 31 Leninsky Ave., 119071 Moscow, Russian Federation
cCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: pombeiro@tecnico.ulisboa.pt
dCenter of Physics and Engineering of Advanced Materials, Laboratory of Physics of Materials and Emerging Technologies, Chemical Engineering Department, Instituto Superior Técnico, Universidade de Lisboa, 1049-001, Lisbon, Portugal
eInstitute of Physical Chemistry and Chemical Physics, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Radlinského 9, SK-81237 Bratislava, Slovak Republic. E-mail: peter.rapta@stuba.sk
fN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky Ave., 119991 Moscow, Russian Federation. E-mail: anatshu@gmail.com
gInorganic Polymers Department, “Petru Poni” Institute of Macromolecular Chemistry, Aleea Gr. Ghica Voda 41 A, Iasi 700487, Romania
First published on 27th August 2024
Abstract
Design and development of novel, low-cost and efficient electrocatalysts for oxygen evolution reaction (OER) in alkaline media is crucial for lowering the reaction overpotential and thus decreasing the energy input during the water electrolysis process. Herein, we present the synthesis of new 14-membered bis-thiosemicarbazide and bis-isothiosemicarbazide macrocycles and their nickel(II) complexes characterized by spectroscopic techniques (1H and 13C NMR, IR, UV–vis), electrospray ionization mass spectrometry, single crystal X-ray diffraction, scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX) and cyclic voltammetry. Finally, the activity of nickel(II) complexes towards OER is reported. NiIILSEt delivered a current density of 10 mA cm−2 at the lowest overpotential of 350 mV with the lowest Tafel slope of 93 mV dec−1. The high performance of NiIILSEt might be attributed to its high surface area and thus abundant active sites with the observed low charge-transfer resistance enabling the effective current flow through the electrocatalyst. Square-planar coordination geometry and increment in Ni oxidation state are believed to favor its OER performance. Beside high activity towards OER, NiIILSEt demonstrated excellent long-term stability with continuous operation, advocating its possible application in commercial systems.
Introduction
Open-chain and macrocyclic potentially tetradentate bis-isothiosemicarbazide ligands were first reported more than 25 years ago.1,2 These organic compounds were assembled in the presence of metal ions favoring square-planar coordination geometry, as a rule Ni(II), but also Cu(II), Co(II), which in addition to square-planar coordination environment, could also accept 4 + 1 or 4 + 2 coordination geometry.3,4 Pentan-2,4-dione bis(S-alkylisothiosemicarbazones) were disclosed to act in some first row transition metal complexes as redox-active noninnocent ligands,5 adopt a series of protonation forms, as i.e., neutral species,6 monoanions,7 dianions8 or even trianions,9 stabilize particular metal ions in unusually high oxidation states, i.e., iron(IV),10 copper(III),11 or undergo 2e oxidation with formation of 14π electronic species with a definite oxidation state.12 Nickel(II) complexes with open-chain tetradentate ligands were also used as precursors for the assembly of macrocyclic complexes with both cis- and trans-arranged isothiosemicarbazide moieties when heated with acetylacetone (Hacac) and triethylorthoformate (CH(OEt)3) as both dehydrating and formylating agent.13 This kind of ligands is also quite reactive at central carbon atom of acac moiety and undergo dimerization through C–C bond formation14 or oxidation with formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group at the same carbon atom.15 Other related coordinated ligands exhibited similar reactivity.16–19 Redox noninnocent behavior was also disclosed for tetradentate ligands with N2O2 coordination environment20 and for some 14-membered hexaazamacrocyclic bis-semicarbazide ligands,21 shown in Chart 1.
O group at the same carbon atom.15 Other related coordinated ligands exhibited similar reactivity.16–19 Redox noninnocent behavior was also disclosed for tetradentate ligands with N2O2 coordination environment20 and for some 14-membered hexaazamacrocyclic bis-semicarbazide ligands,21 shown in Chart 1.
 | ||
| Chart 1 3d metal complexes with noninnocent ligands. | ||
The nickel(II) complexes with tetradentate open-chain and 14-membered macrocyclic ligands can be in particular instances demetallated with isolation of free ligands22,23 used for coordination with other metal ions which are not suited as templates. This was not the case for nickel(II) complexes with 14-membered hexaazamacrocycles based on 2,4-diketones and isothiosemicarbazides, which could not be demetalated due to their high thermodynamic stability and kinetic inertness.13,24
Quite recently, several new bis-semicarbazide25,26 and bis-thiosemicarbazide27 macrocycles were reported (Chart 2), which were self-assembled upon acid-promoted dimerization of particular hydrazones. The synthesis of these macrocycles offers opportunities for the discovery of unexplored so far aspects of their coordination chemistry and useful application of metal complexes.
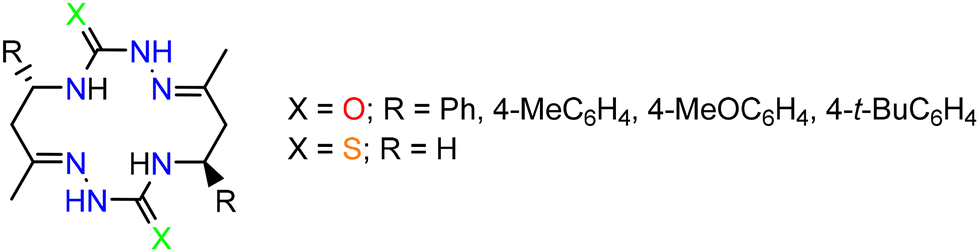 | ||
| Chart 2 Macrocyclic compounds reported recently. | ||
Herein we report on the synthesis and comprehensive characterization of new 14-membered bis-thiosemicarbazide and bis-isothiosemicarbazide macrocycles and of their nickel(II) complexes (Chart 3) by spectroscopic methods (1H and 13C NMR, IR, UV–vis), ESI mass spectrometry, single crystal X-ray diffraction (SC-XRD) and cyclic voltammetry. In contrast to metal complexes with open-chain chelating ligands, the complexes with polyazamacrocyclic ligands derived from isothiosemicarbazides are characterized by higher thermodynamic stability and kinetic inertness due to macrocyclic effect. Some of them remain intact in concentrated acids, e.g. HCl.
 | ||
| Chart 3 Compounds reported in this work. | ||
We also investigated the electrocatalytic activity of nickel(II) complexes towards oxygen evolution reaction (OER) that plays a vital role in operation of energy conversion devices such as water electrolyzers, unitized regenerative fuel cells and rechargeable metal–air batteries.28–30 At the moment, electrocatalysts based on noble metal oxides IrO2 and RuO2 exhibit the best overall performance towards OER, but high costs hamper their use in commercial devices. Consequently, various non-noble metal OER electrocatalysts have been designed and developed, delivering promising performance.29,31 However, it is still challenging to achieve excellent performance at low overpotentials using these non-noble metal electrocatalysts which limits the overall efficiency of the above-mentioned devices. The high overpotential and consequent high energy input necessary to drive OER comes from the complexity of OER as a 4-electron transfer process. Thus, quest for novel, advanced electrocatalysts for OER continues. Use of transition metal complexes has been recently suggested, though often combined with highly conductive, high-surface area carbon nanostructured materials.32–34
Results and discussion
Synthesis of H2LS and H2LSEt
Quite recently, we reported27 an unprecedented self-assembly of 14-membered bis-thiosemicarbazone and 28-membered tetrakis-thiosemicarbazone macrocycles via acid-promoted cyclooligomerization of 4-(3-oxobutyl)thiosemicarbazide hydrazone.35 We hypothesized that, under acidic conditions, hydrazones of other 4-(γ-oxoalkyl)thiosemicarbazides could be converted into new polyazamacrocyclic compounds as strong metal-binding chelators. However, the reaction of methyl-substituted β-isothiocyanatoketone 1 with hydrazine afforded thiosemicarbaside ketone 2 only as intermediate that spontaneously converted into its cyclic isomeric form, namely 1-amino-6-hydroxy-4-methyl-5,6-trimethylenehexahydropyrimidine-2-thione 3 (Scheme 1).36 At the same time, the 1H NMR spectrum of 3 revealed the presence of a small amount of the open-chain form 2. We envisioned that due to the ring-open chain isomerism this pyrimidine could react with an excess hydrazine to form the hydrazone of compound 2, which is expected to be a precursor to polyazamacrocycle.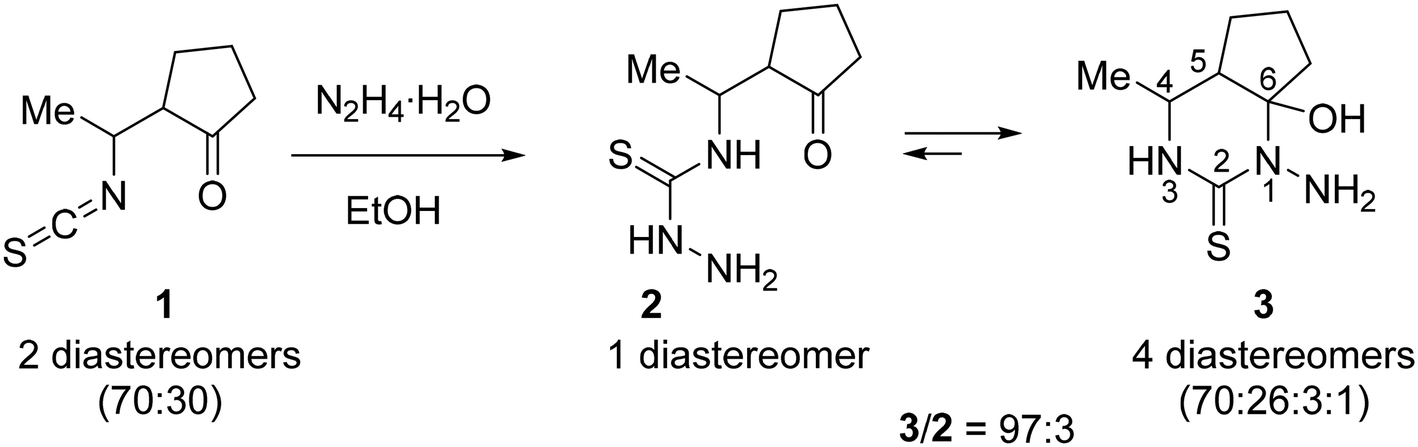 | ||
Scheme 1 Synthesis of a 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 equilibrium mixture of pyrimidine 3 and its acyclic isomer 2. :3 equilibrium mixture of pyrimidine 3 and its acyclic isomer 2. | ||
The starting material, a 97:3 mixture of 3 and its acyclic isomer 2, was prepared in 82% yield by the reaction of freshly distilled isothiocyanato ketone 1 (two diastereomers, 70:30) with hydrazine hydrate in MeCN according to the reported procedure (Scheme 1).36 It should be noted that both recrystallization of the mixture from MeCN and standing of solution of this mixture in DMSO-d6 at room temperature for 13 days practically did not change the 3:2 ratio (Fig. S1 in the ESI†), which indicates that these isomers reached an equilibrium.
According to 1H NMR spectroscopic data, thiosemicarbazide 2 is a single diastereomer, while pyrimidine 3 is a mixture of 4 diastereomers with a significant predominance of two of them (70:26:3:1). Since the ratio of the two main isomers in 3 is approximately equal to the isomer ratio in the starting isothiocyanate 1 (see Scheme 1), it is obvious that these isomers differ in the relative configuration at the C4 and C5 atoms. The 1H NMR spectrum of 3 in DMSO-d6 showed that the major diastereomer had configuration 4R*,5S*,6R* with equatorial orientation of the Me group and axial positions of the OH group and the C5 substituent, providing a cis-junction of hexahydropyrimidine and cyclopentane rings.37 The first minor diastereomer of 3 (26%) had configuration 4R*,5R*,6R* with axial orientation of the OH group and equatorial positions of the Me group and the C5 substituent, providing a trans-junction of hexahydropyrimidine and cyclopentane rings.38
The prepared equilibrium mixture of pyrimidine 3 and its acyclic isomer 2 was used for the synthesis of the two most suitable macrocycle precursors, namely, hydrazone and oxime of 4-[1-(2-oxocyclopentyl)ethyl]thiosemicarbazide (4 and 5, respectively, in Scheme 2).
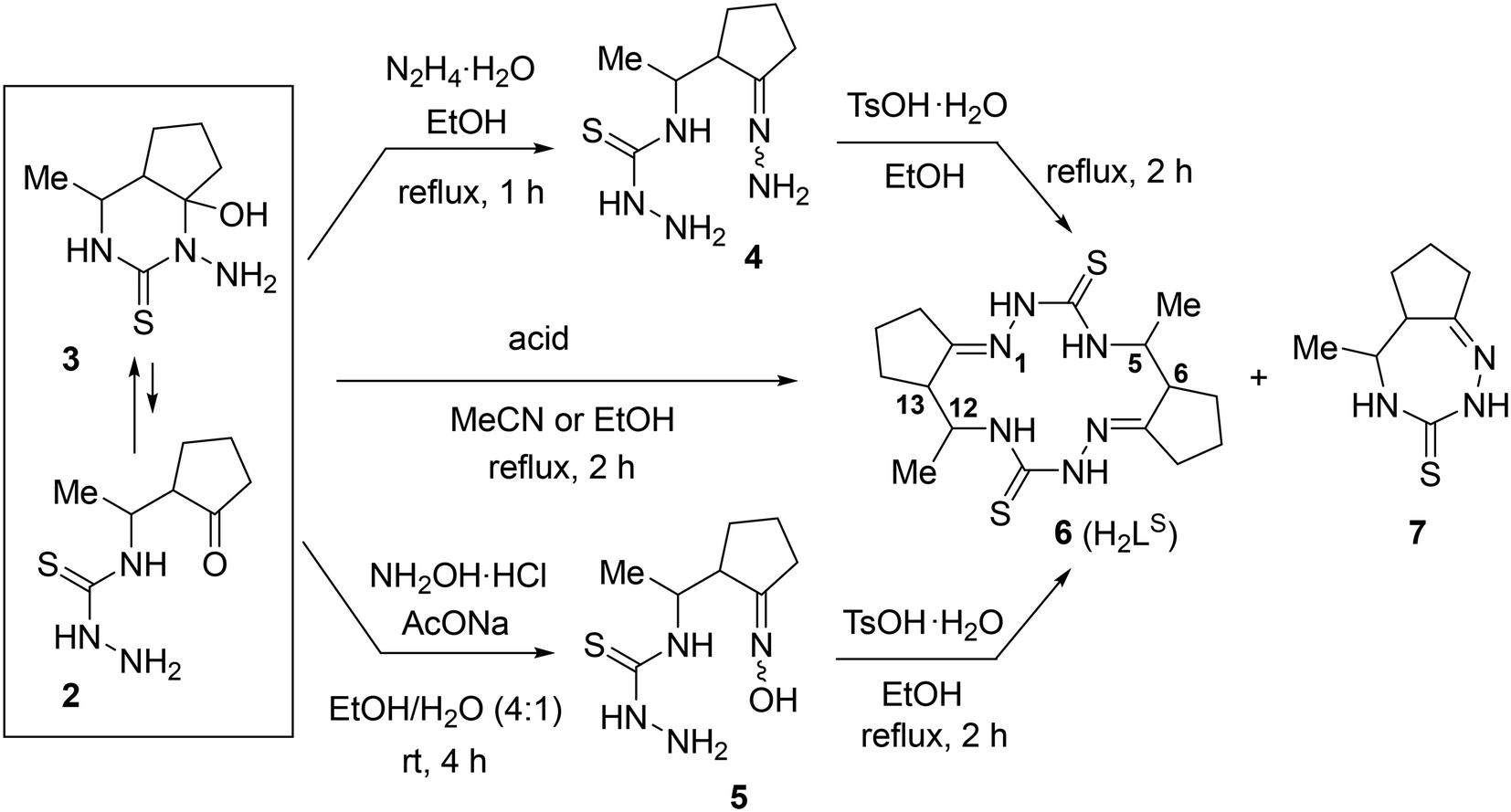 | ||
| Scheme 2 Synthesis of 14-membered cyclic bis-thiosemicarbazone 6 (H2LS). | ||
We found that the 3 + 2 mixture readily and completely reacts with excess hydrazine hydrate in refluxing EtOH to give the expected hydrazone 4 as an oil. After many failed attempts to solidify the hydrazine 4, we finally succeeded to isolate it as a hygroscopic solid foam by extracting the oily product with chloroform, washing the extract with brine and evaporating the solvent under reduced pressure.39
According to 1H NMR spectrum, hydrazone 4 was formed as a mixture of three stereoisomers in a ratio of 46:45:9. Since we were unable to obtain this compound in analytically pure form due to its high hygroscopicity and lability, we used it in the cyclization step without additional purification.
Oxime 5 was prepared as a hygroscopic solid foam by reaction of the 3 + 2 mixture with NH2OH·HCl in the presence of NaOAc in EtOH/H2O at room temperature followed by work up (see Experimental part). 1H NMR spectrum of 5 in DMSO-d6 showed that it was a mixture of four stereoisomers in a ratio of 55:26:11:7. This product was also used in the macrocyclization step without further purification.
The macrocyclization of hydrazone 4 and oxime 5 was performed in refluxing EtOH by using TsOH·H2O as an acidic promoter (Scheme 2). In contrast to the cyclization of hydrazone of 4-(3-oxobutyl)thiosemicarbazide which, under similar conditions, was converted into a 92:8 mixture of 14- and 28-membered polyazamacrocycles,27 hydrazone 4 was cyclized with exclusively high selectivity to give only the 14-membered cyclic bis-thiosemicarbazone 6 (H2LS). This product precipitated from the reaction mixture and was isolated in 83% yield and high purity (>98%) (Table 1, entry 1). Although the starting material was a mixture of stereoisomers (vide supra) and there were 4 stereocenters in the macrocycle 6, it was formed with excellent stereoselectivity. According to 1H NMR spectrum, the crude macrocycle 6 was a mixture of several diastereomers with a huge predominance of the isomer with the configuration 5R*,6R*,12R*,13R* (≥94 mol%). Under similar conditions, oxime 5 was converted into macrocycle 6 with very high stereoselectivity, but with a lower yield (64%, entry 2).
a
| Entry | Substrateb | Promoter (equiv) | Solvent | Conc. of substrate mmol mL−1 | Products distributionc (%) | Isolated yield (%) | |
|---|---|---|---|---|---|---|---|
|
6d |
7 (isomer ratio) | ||||||
|
a All reactions were carried out in EtOH or MeCN under reflux for 2 h.
b The starting material was either a 97:3 mixture of compounds 3 and 2, or hydrazone 4 (solid foam), or oxime 5 (solid foam) (see the discussion).
c According to 1H NMR spectroscopic data for the crude products.
d The major stereoisomer (90–94 mol%) with (7E,14E)-(5R*,6R*,12R*,13R*)-configuration plus a set of small amounts of other stereoisomers (total 6–10 mol% according to 1H NMR data).
e Not less than 35% unidentified by-products was also formed (1H NMR data).
|
|||||||
| 1 | 4 | TsOH·H2O (1.10) | EtOH | 0.35 | 100 | 0 | 83 |
| 2 | 5 | TsOH·H2O (1.12) | EtOH | 0.18 | 100 | 0 | 64 |
| 3 | 3 + 2 | NH2OH·HCl (1.25) | EtOH | 0.30 | 100 | 0 | 92 |
| 4 | 3 + 2 | NH2OH·HCl (1.26) | MeCN | 0.25 | 82 | 18 (72:28) |
88 |
| 5 | 3 + 2 | NH2OH·HCl (0.75) | EtOH | 0.23 | 95 | 5 (63:37) |
89 |
| 6 | 3 + 2 | NH2OH·HCl (0.26) | EtOH | 0.21 | 78 | 22 (71:29) |
87 |
| 7 | 3 + 2 | NH2C(O)NHNH2.HCl (1.27) | EtOH | 0.25 | 90 | 10 (63:37) |
87 |
| 8 | 3 + 2 | Pyridine (1.27), TsOH·H2O (1.25) | EtOH | 0.32 | 28 | 72 (85:15) |
86 |
| 9 | 3 + 2 | AcOH (1.25) | EtOH | 0.27 | Traces | 100 (37:63) |
—e |
Despite the successful synthesis of 6 from hydrazone 4 and oxime 5, the use of these starting materials was inconvenient due to their high hygroscopicity and the inability to reliably purify them (vide supra). We hypothesized that in situ generation of these compounds from the 3 + 2 mixture in the presence of acid could lead to the formation of the target macrocycle 6. Indeed, treatment of the 97:3 mixture of 3 and 2 with hydroxylamine hydrochloride in refluxing EtOH produced the bis-thiosemicarbazone 6 in 92% yield and with high diastereoselectivity (entry 3). Apparently, the first step of this transformation involves deprotonation of NH2OH·HCl in the presence of thiosemicarbazides 2 or/and 3 as bases followed by reaction of NH2OH formed with 2, which is in equilibrium with 3, to afford oxime 5. Then, the two molecules of oxime 5 react with each other and the obtained dimeric species undergoes cyclization to give 6.
The successful NH2OH·HCl promoted synthesis of 6 prompted us to study the macrocyclization of the 3 + 2 mixture using other promoters and other reaction conditions. However, all the results obtained were worse than those described above. For example, under the above conditions, but with MeCN as a solvent, a 82:18 mixture of macrocycle 6 and triazepinethione 7 (two diastereomers, 72:28) was prepared (entry 4). Decrease of the amount of NH2OH·HCl from 1.25 equiv (entry 4) to 0.75 equiv (entry 5), and then to 0.26 equiv (entry 6) also reduced the reaction selectivity (6/7 = 100:0, 95:5, and 78:22, respectively). Semicarbazide hydrochloride was shown to be a less selective promoter than hydroxylamine hydrochloride (entry 7 vs. entry 3). Pyridinium tosylate also catalyzed the macrocyclization of the 3 + 2 mixture, but the formed product contained 72% of triazepinethione 7 and only 28% of macrocycle 6 (entry 8). The use of AcOH as a promoter resulted in the predominant formation of triazepinethione 7 (entry 9).
Thus, very convenient synthesis of 14-membered macrocycle 6 (H2LS) involving treatment of readily available isomeric mixture of compounds 3 and 2 with NH2OH·HCl was developed. Macrocycle was formed in excellent yield and purity, including diastereomeric purity (vide supra). It should be noted that we scaled up this reaction to a 40.8 mmol loading (multi-gram scale) with no loss of product purity and even a slight increase in yield (up to 97%).
It should be also stressed that although the starting material, pyrimidine 3, was mainly a mixture of (4R*,5S*,6R*)- and (4R*,5R*,6R*)-diastereomers in a ratio of 70:26, macrocycle 6 was mainly obtained as the only diastereomer with the configuration 5R*,6R*,12R*,13R*. This nontrivial result can be explained by epimerization of (R*,S*)-isomers in isomeric mixtures of the acyclic intermediates (e.g., oxime 5) under reaction conditions to the corresponding (R*,R*)-isomers, followed by dimerization of the latter and cyclization. It is also of note that the dimerization proceeds highly selectively involving molecules with the same relative configuration of the stereocenters to give predominantly (5R*,6R*,12R*,13R*)-diastereomer of macrocycle 6, but not its (5R*,6R*,12S*,13S*)-diastereomer. Apparently, the rate of the dimerization of molecule pair with the same relative configurations (R,R + R,R or S,S + S,S) is essentially higher than that of molecule pair with the opposite relative configurations (R,R + S,S or S,S + R,R). It should be mentioned, however, that in addition to the (5R*,6R*,12R*,13R*)-isomer, small amounts of some other stereoisomers are also formed, but their total content did not exceed 10% (Fig. S2 in the ESI†). After recrystallization of the crude product 6 from nBuOH, the major diastereomer was isolated in pure form.
The structure of (5R*,6R*,12R*,13R*)-6 was unambiguously confirmed by 1D and 2D NMR spectra (Fig. S3A–S3H in the ESI†), ESI mass spectrometry (Fig. S5 and S6 in the ESI†), elemental analysis, infrared spectra (Fig. S8 in the ESI†), as well as by single crystal X-ray diffraction (vide infra). In ESI (+) mass spectrum a characteristic peak with m/z 367.25 was attributed to the ion [H2LS + H]+ (Fig. S5 in the ESI†), while in the ESI (−) spectrum the peak with m/z 365.17 was attributed to the ion [H2LS-H]− (Fig. S6 in the ESI†). The number of resonances in 1H and 13C{1H} NMR spectra of (5R*,6R*,12R*,13R*)-6 is in agreement with C2 molecular symmetry of the compound in solution (Fig. S3A in the ESI†). A high value (11.0 Hz) of vicinal coupling between the H-5 and H-6 protons and between the H-12 and H-13 protons provides evidence that they are pairwise antiperiplanar, and therefore, this compound adopts trans-arrangement of substituents at the C-5 and C-6 atoms, as well as at the C-12 and C-13 atoms. These data and the results of the NOESY experiment (Fig. S3H in the ESI†) indicated (5R*,6R*,12R*,13R*)-configuration of the isolated macrocycle.
The crude macrocycle 6 was further alkylated by reaction with excess iodoethane in the presence of NaH in dry MeCN at room temperature to give the desired 14-membered bis-isothiosemicarbazone 8 (H2LSEt) as a diastereomeric mixture with a strong predominance of (7E,14E)-(5R*,6R*,12R*,13R*)-diastereomer (≥90%) in 98% yield (Scheme 3). Recrystallization of the crude 8 from EtOH afforded this diastereomer in analytically pure form.
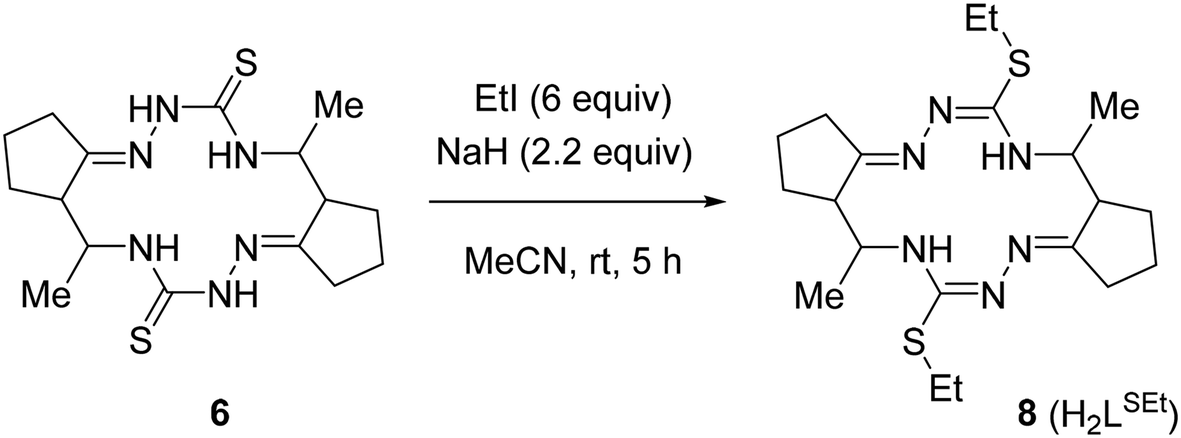 | ||
| Scheme 3 Synthesis of 14-membered cyclic bis-isothiosemicarbazone 8 (H2LSEt). | ||
The structure of (5R*,6R*,12R*,13R*)-8 was unambiguously established by 1D and 2D NMR spectroscopy (Fig. S4A–S4M in the ESI†), ESI mass spectrometry (Fig. S7 in the ESI†), infrared spectra (Fig. S9 in the ESI†), elemental analysis, as well as by single crystal X-ray diffraction (vide infra). The bis-isothiosemicarbazide macrocycle H2LSEt showed in the positive ion mass spectra a peak with m/z 423.25 corresponding to the ion [H2LSEt + H]+ (Fig. S7 in the ESI†). The 1H and 13C{1H} NMR spectra of this compound are in agreement with C2 molecular symmetry of 8 in solution. Like for 6, a high value (10.3 Hz) of vicinal coupling between the H-5 and H-6 protons and between the H-12 and H-13 protons indicates that these protons are pairwise antiperiplanar. The presence of vicinal coupling (3.4 Hz) of the H-5 and H-12 protons with the N(4)H and N(13)H protons, respectively, indicates that the tautomeric structures of the two isothiosemicarbazone fragments correspond to those presented in Scheme 3.
Synthesis of Ni(II) complexes
By heating the reaction mixture of H2LS and NiCl2·6H2O in DMF in the presence of Et3N in 1:1:2 molar ratio the complex NiLS was isolated as red microcrystalline product in 53% yield. The ESI mass spectrum measured in positive ion mode (Fig. S10 in the ESI†) showed a peak with m/z 423.16 and 445.11 attributed to [NiLS + H]+ and [NiLS + Na]+ respectively, while that recorded in the negative ion mode (Fig. S11 in the ESI†) revealed a peak with m/z 421.00 assigned to [NiLS-H]−. The complex is diamagnetic. The number of resonances in 1H and 13C NMR spectra (Fig. S12 and S13 in the ESI†) is in accord with its C2-molecular symmetry in solution. IR and UV–vis spectra of NiLS are presented in Fig. S14 and S15 in the ESI.†
The bis-isothiosemicarbazide macrocycle H2LSMe was generated by treatment of H2LS with NaH in dry DMF under argon and subsequent addition of iodomethane. The ligand generated in situ was further reacted with Ni(OAc)2·4H2O to give a red microcrystalline product of NiLSMe in 53% yield. The methylation of both thione sulfur atoms in H2LS was confirmed by ESI (+) mass spectrum (Fig. S16 in the ESI†), in which a peak with m/z 451.21 was observed, which could be easily attributed to the ion [NiLSMe + H]+. In ESI (−) mass spectrum (Fig. S17 in the ESI†), a peak with m/z 449.02 was assigned to the ion [NiLSMe-H]−. The SMe protons were seen at 2.49 ppm, while the 13C resonance of these two groups at 16.35 ppm. As for H2LS, the number of resonances in 1H and 13C NMR spectra (Fig. S18 and S19 in the ESI†) is in agreement with its C2-molecular symmetry. Fig. S20 and S21 in the ESI† show the IR and UV–vis spectra of NiLSMe.
In contrast to NiLSMe, the complex NiLSEt was prepared by direct reaction of H2LSEt with Ni(OAc)2·4H2O in the presence of 2 equiv of Et3N in dry and anoxic DMF in 63% yield. The complex formation was evidenced by mass spectra, in which the peak with m/z 479.20 was attributed to [NiLSEt + H]+ in positive mode, while that with m/z 477.06 in the negative ion mode to [NiLSEt-H]− (Fig. S22 and S23 in the ESI†). The 1H and 13C NMR spectra (Fig. S24 and 25 in the ESI†) provided further evidence for the formation of NiLSEt and its C2-molecular symmetry in solution. Other spectroscopic data for NiLSEt are displayed in Fig. S26 and S27 in the ESI.† The optical spectra of NiLS, NiLSMe and NiLSEt in the visible region of the spectrum are characterized by low intensity d–d band with maxima at 526, 530 and 529 nm, respectively (Fig. S15, S21 and S27 in the ESI†), in accord with their square-planar coordination geometry for low-spin d8 electronic configuration (S = 0).
In addition, single crystals of X-ray diffraction quality of NiLS and NiLSEt were obtained from mother liquor in a Schlenk tube upon standing at +4 °C, while those of NiLSMe by diffusion of diethyl ether into the mother liquor at room temperature.
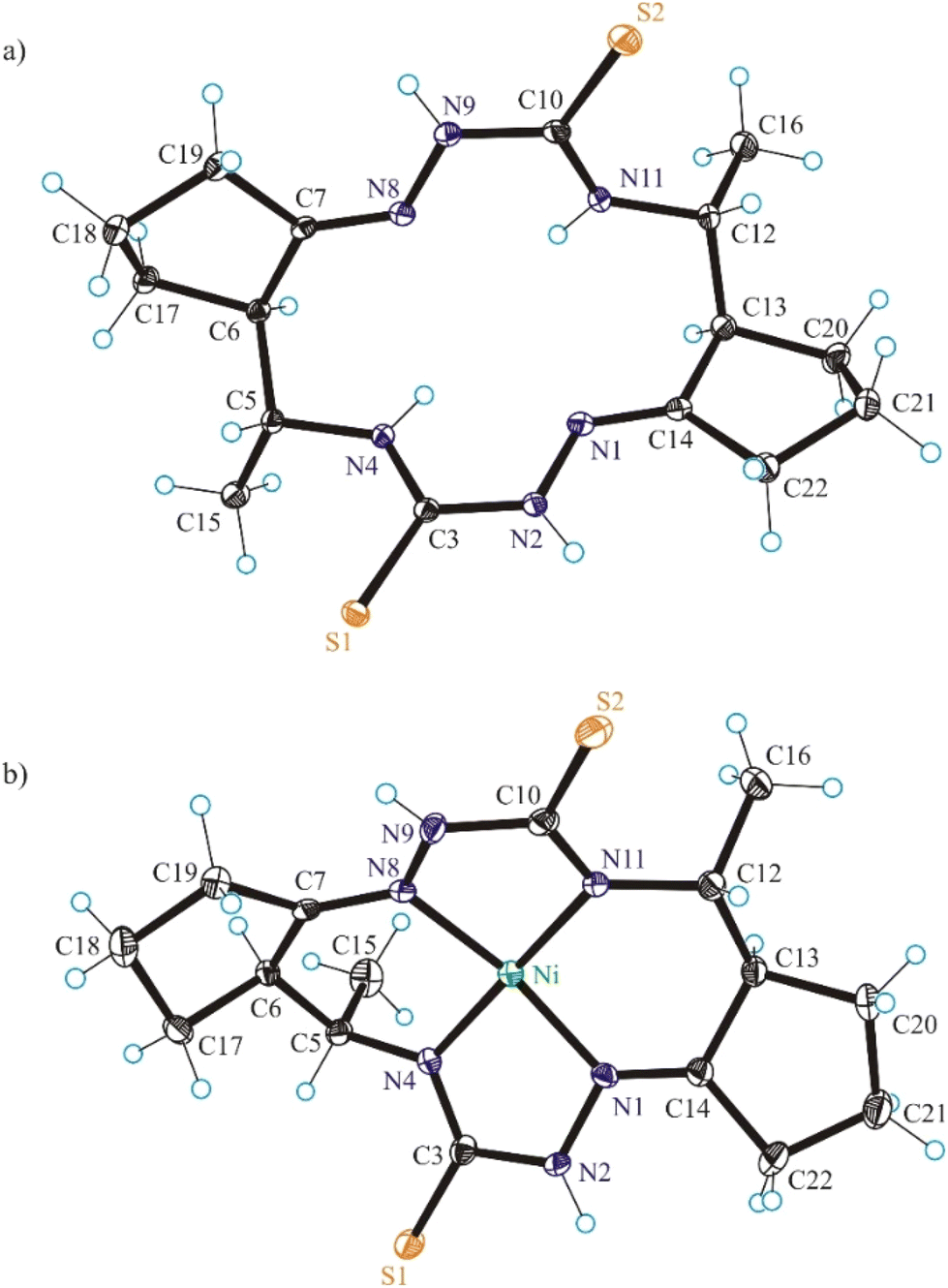 | ||
| Fig. 1 ORTEP views of (a) H2LS and (b) NiIILS as diastereomers with 5R,6R,12R,13R-configuration. The thermal ellipsoids are drawn at 50% probability level. Selected bond distances (Å) and bond angles (°) in H2LS: N1–N2 1.3853(11), N2–C3 1.3662(11), C3–S1 1.6943(9), C3–N4 1.3316(12), N4–C5 1.4673(11), C5–C6 1.5491(13), C6–C7 1.5183(13), C7–N8 1.2806(11), N8–N9 1.3818(12), N9–C10 1.3723(12), C10–S2 1.6853(10), C10–N11 1.3294(13), N11–C12 1.4599(11), C12–C13 1.5439(15), C13–C14 1.5199(13), C14–N1 1.2780(12); ΘN1–N2–C3–N4 = –0.62(13), ΘN8–N9–C10–N11 = 8.24(12); in NiIILS: Ni–N1 1.854(3), Ni–N4 1.863(3), Ni–N8 1.871(2), Ni–N11 1.873(3), N1–N2 1.394(4), N2–C3 1.393(4), C3–S1 1.695(3), C3–N4 1.316(4), N4–C5 1.469(4), C5–C6 1.527(4), C6–C7 1.508(4), C7–N8 1.282(4), N8–N9 1.397(3), N9–C10 1.381(4), C10–S2 1.707(3), C10–N11 1.312(4), N11–C12 1.477(4), C12–C13 1.529(4), C13–C14 1.509(4), C14–N1 1.279(4); ΘN1–N2–C3–N4 = 12.5(4), ΘN8–N9–C10–N11 = 6.4(4). | ||
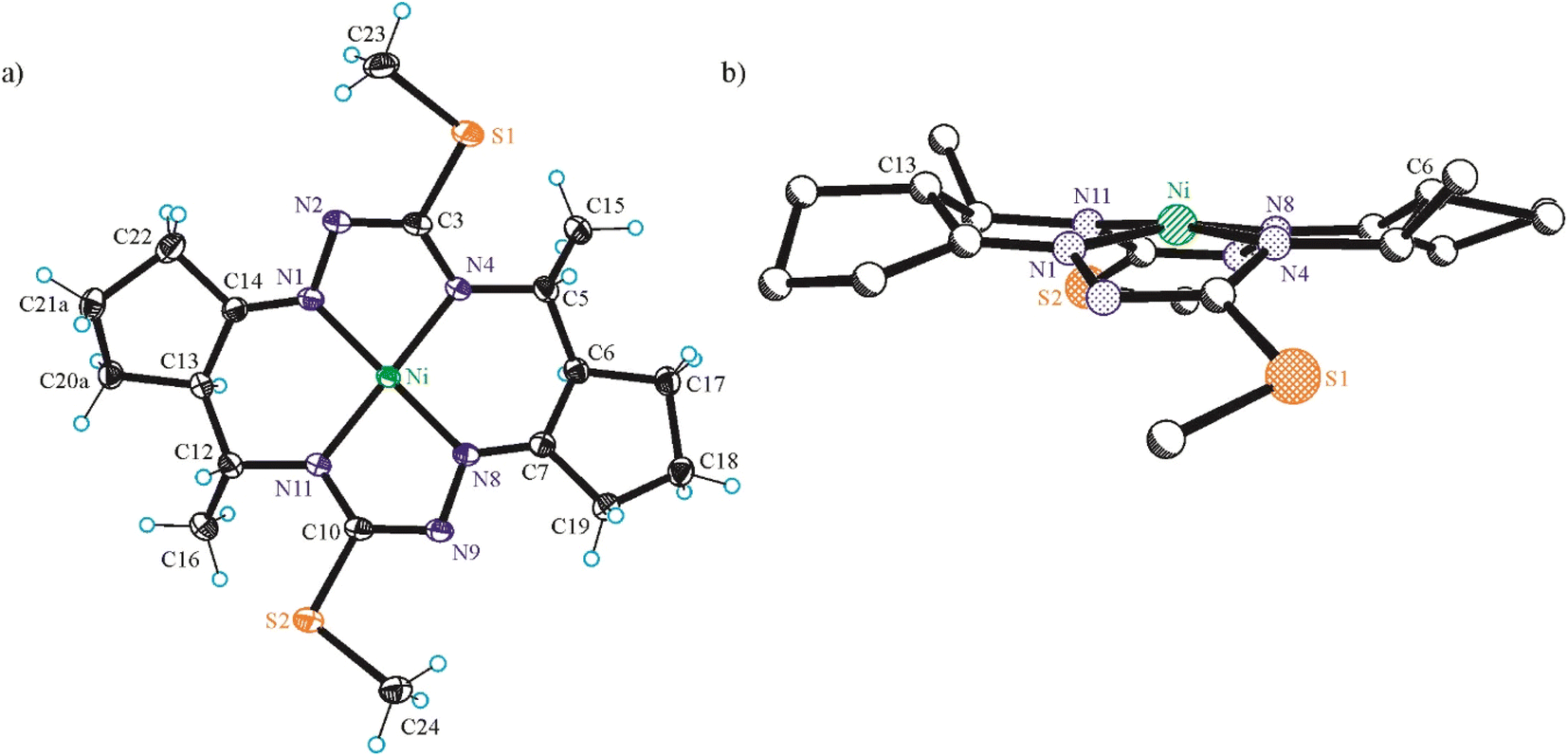 | ||
| Fig. 2 ORTEP view of (a) NiIILSMe as diastereomer with 5R,6R,12R,13R-configuration and the thermal ellipsoids drawn at 50% probability level, and (b) ball-and-stick projection of the same molecule perpendicular to the NiN4 mean plane. Selected bond distances (Å) and torsion angles (°): Ni–N1 1.8471(11), Ni–N4 1.8845(10), Ni–N8 1.8486(11), Ni–N11 1.8868(10), N1–N2 1.4008(14), N2–C3 1.3239(17), C3–S1 1.7717(12), C3–N4 1.3447(16), N4–C5 1.4703(16), C5–C6 1.5370(17), C6–C7 1.5030(17), C7–N8 1.2854(16), N8–N9 1.3987(14), N9–C10 1.3242(17), C10–S2 1.7754(13), C10–N11 1.3370(16), N11–C12 1.4702(16), C12–C13 1.5295(19), C13–C14 1.5037(18), C14–N1 1.2839(16); ΘN1–N2–C3–N4 = 5.75(16), ΘN8–N9–C10–N11 = 2.55(16). | ||
 | ||
| Fig. 3 ORTEP view of (a) H2LSEt and (b) NiIILSEt as diastereomers with 5R,6R,12R,13R-configuration. The thermal ellipsoids are drawn at 50% probability level. Selected bond distances (Å) and bond angles (°) in H2LSEt: N1–N2 1.413(2), N2–C3 1.293(3), C3–S1 1.777(2), C3–N4 1.343(3), N4–C5 1.463(3), C5–C6 1.539(3), C6–C7 1.516(3), C7–N8 1.276(3), N8–N9 1.411(2), N9–C10 1.297(3), C10–S2 1.764(2), C10–N11 1.349(3), N11–C12 1.455(3), C12–C13 1.544(3), C13–C14 1.520(3), C14–N1 1.274(3); ΘN1–N2–C3–N4 = −1.1(3), ΘN8–N9–C10–N11 = −0.8(3); in NiIILSEt: Ni–N1 1.8476(15), Ni–N4 1.8729(14), Ni–N8 1.8535(15), Ni–N11 1.8767(14), N1–N2 1.396(2), N2–C3 1.322(2), C3–S1 1.7717(18), C3–N4 1.340(2), N4–C5 1.468(2), C5–C6 1.534(2), C6–C7 1.503(2), C7–N8 1.282(2), N8–N9 1.4028(19), N9–C10 1.322(2), C10–S2 1.7709(18), C10–N11 1.339(2), N11–C12 1.470(2), C12–C13 1.536(3), C13–C14 1.503(3), C14–N1 1.282(2); ΘN1–N2–C3–N4 = 1.6(2), ΘN8–N9–C10–N11 = 4.1(2). | ||
The conformation of the macrocycle in NiIILS is better described in terms of the component chelate rings formed upon binding to central atom. While the two 5-membered rings are quite similar and adopt envelope conformations (see Table S1†), the two six-membered rings shown in Chart 4 adopt two different conformations A and B according to classification proposed by Curtis40 for macrocyclic compounds (see Table S2 in the ESI†), which have two secondary amine and two imine donor atoms. The two conformations, A and B, can be distinguished in the complex by atom displacements from the MN2 plane (Tables S1and S2 in the ESI,†Chart 4), atom C(4) being displaced further on the same side of this plane than C(3) for A, and less than for B. So, the 6-membered chelate ring NiN4C5C6C7N8 adopted conformation A, while NiN11C12C13C14N1 the conformation B.
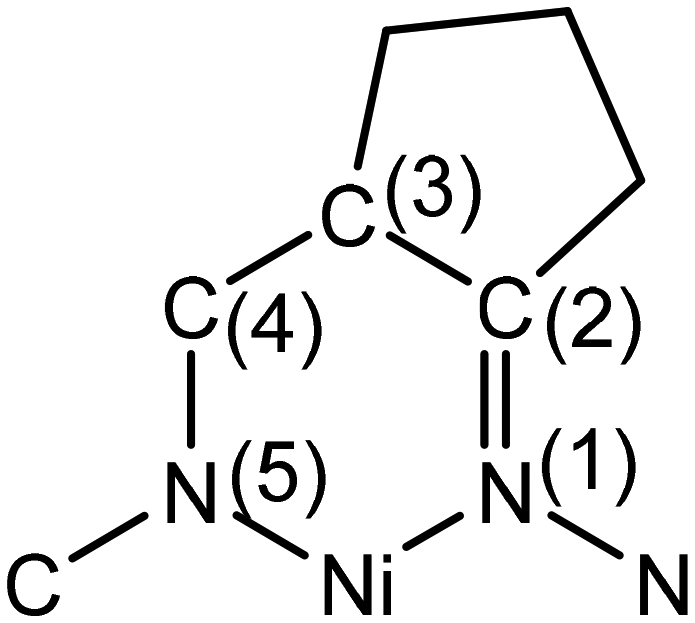 | ||
| Chart 4 Chelate rings present in NiIILS. | ||
The free macrocycle H2LSEt and coordinated macrocycic ligands in NiIILSMe and NiIILSEt (Fig. 2 and 3) have a saddle-shaped distortion from planarity, i.e., with five-membered chelate rings NiNCNN tilted to one side of the molecular plane, and the six-membered rings tilted to the other side. A consequence of the saddle shape of the metal-free macrocycles is that lone pairs of the four cavity N atoms are directed out of the N4 plane, while in the nickel(II) complexes the lone pairs lie in the NiN4 plane. Generally, the macrocycles in complexes NiIILSMe and NiIILSMe are more “symmetric” than in NiIILS.
Square-planar coordination geometry is observed in all three complexes studied by SC-XRD. Comparison of the bond lengths around low-spin Ni(II) in NiIILS, NiIILSMe and NiIILSEt shows that Ni–Nhydrazine bonds are commonly shorter than Ni–Nthioamide bonds, as also observed in the most part of square-planar nickel(II) complexes based on isothiosemicarbazide.13
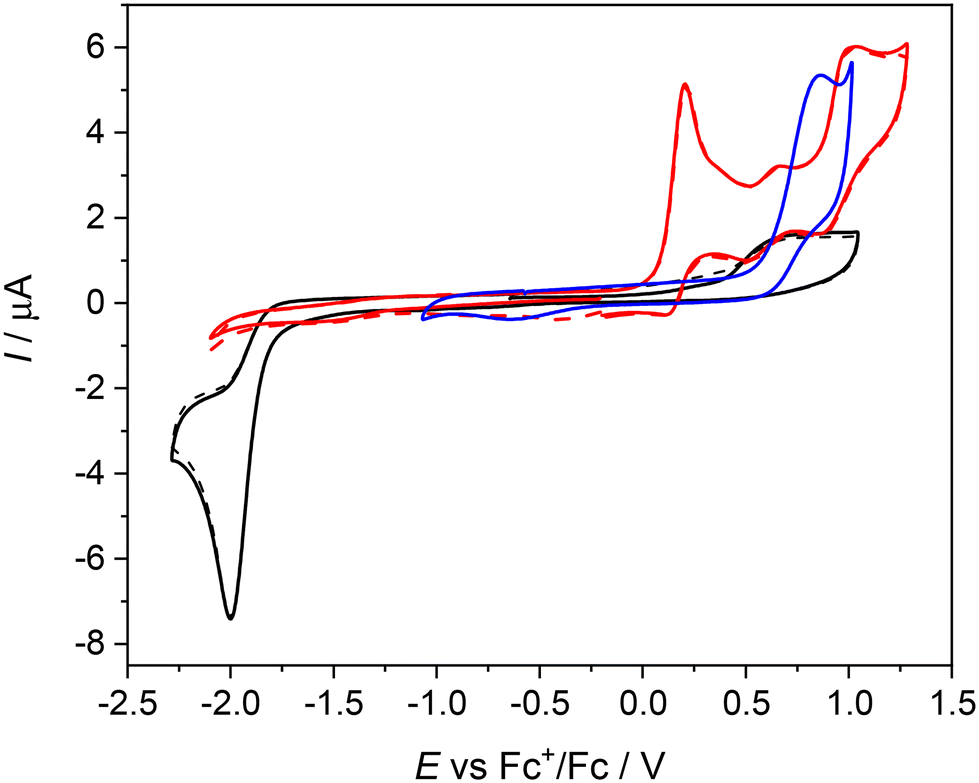 | ||
| Fig. 4 Cyclic voltammograms of NiIILS (black traces, solid trace – the first scan, dashed trace – the second scan), NiIILSMe (red traces), and of H2LS (blue trace) in 0.2 M nBu4NPF6/CH2Cl2 both in anodic and cathodic parts (Pt-disc working electrode, scan rate 100 mV s−1). | ||
 | ||
| Fig. 5 Cyclic voltammograms of NiIILSEt (red traces, solid trace – the first scan, dashed trace – the second scan), and of H2LSEt (blue trace) in 0.2 M nBu4NPF6/CH2Cl2 both in anodic and cathodic parts (Pt-disc working electrode, scan rate 100 mV s−1). | ||
The first oxidation peak is nearly chemically irreversible (but with a hint of small counter peak observed in a back scan) and following this peak a new minor reversible redox couple appears upon oxidation at E′pa = +0.66 V vs. Fc+/Fc. As the sample was rigorously purified and analyzed before voltammetric measurements we attribute this new minor peak to the redox active follow up product formed in the region of the first nearly irreversible oxidation. The second main oxidation peak at +1.01 V shows much higher degree of chemical reversibility. Very similar redox behavior was found for NiIILSEt (see red traces in Fig. 5) with E1pa = +0.24 V and E′pa = +0.70 V vs. Fc+/Fc. Again, a minor anodic peak at around 0.7 V indicates the follow up product after the first oxidation step in solution leading to the formation of new redox active species. The first oxidation potential of NiIILSEt is substantially negatively shifted in comparison to the corresponding metal-free proligand H2LSEt with E1pa = +0.75 V vs. Fc+/Fc indicating a strong influence of the central atom. Consequently, we can assume that the one-electron oxidized complex represents a system with a substantially noninnocent character of the ligand.
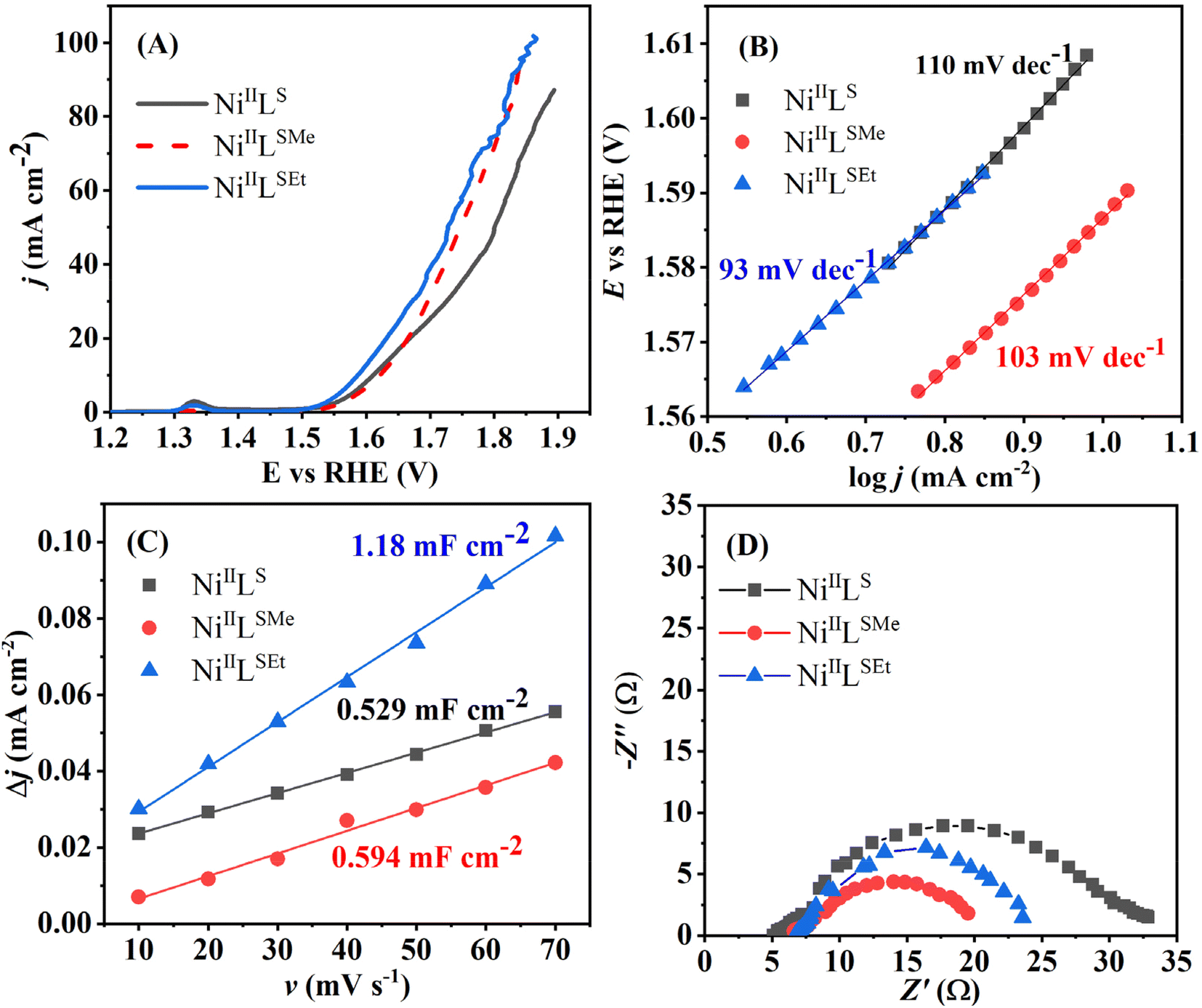 | ||
| Fig. 6 LSVs of NiIILSEt, NiIILSMe and NiIILS studied in a form of a thin film on a glassy carbon support at 10 mV s−1 (A) with the corresponding Tafel plots (B), double-layer capacitance determination plots (Δj vs. v) (C) and Nyquits plots at 1.67 V vs. RHE (D). All measurements were done in 1 M KOH. | ||
The current density at the overpotential of 400 mV was the highest one in the case of NiIILSEt (22.4 mA cm−2), followed by NiIILS (15.2 mA cm−2) and then NiIILSMe (13.9 mA cm−2). IrO2 delivered ca. 35 times lower current density than NiIILSEt at the same overpotential value.33
Tafel analysis of the intrinsic OER activity of the complexes was further performed. Tafel slope, reflecting the rate of increase of current density with potential, was found to be the lowest in the case of OER at NiIILSEt (93 mV dec−1), followed by NiIILSMe (103 mV dec−1) and then NiIILS (110 mV dec−1) (Fig. 6B). The value determined for NiIILSEt was comparable to that of IrO2 (96 mV dec−1).32
Scanning electron microscopy (SEM) studies of the as-prepared complexes illustrate different morphologies (Fig. 7). NiIILSEt is composed of cuboids and rod-like particles, while NiIILS displays a rod-like morphology as well. Particles of NiIILSMe were observed to be notably larger than in the case of the two other complexes. Energy-dispersive X-ray spectroscopy (EDX) analysis of the three Ni(II)-complexes confirmed the presence of Ni, C, N, S and O elements.
 | ||
| Fig. 7 SEM images of studies Ni(II)-complexes at magnifications of 5000× (upper row) and 10000× (lower row) with EDX spectra of NiIILSEt. | ||
Double-layer capacitance (Cdl) of NiIILSEt (1.18 mF cm−2) was determined to be ca. double that of NiIILSMe (0.594 mF cm−2) and NiIILS (0.529 mF cm−2) (Fig. 6C). This suggests that NiIILSEt possess large surface area, offering abundant active sites for surface catalytic reactions and thus contributing to its high activity towards OER. Electrochemical active surface area (ECSA) is then calculated by dividing the Cdl value with a capacitance of 40 μF previously reported for Ni-based OER catalysts.44 The highest ECSA of 29.5 cm2 was evaluated for NiIILSEt, well above those displayed by NiIILSMe (14.85 cm2) and NiIILS (13.22 cm2). Corresponding roughness factors were determined to be 118 for NiIILSEt, 59.4 for NiIILSMe and 52.8 for NiIILS.
Nyquist plots (Fig. 6D) reveal similar values of solution resistance (Rs) of 5–7 Ω suggesting the comparable surface porosity of the three materials. On the other hand, the charge-transfer resistance (Rct) of NiIILSEt (16.8 Ω) was comparable to that of NiIILSMe (15.3 Ω), but almost twice lower than that of NiIILS (29.9 Ω). This further suggests the fastest electron-transfer capability of NiIILSEt and NiIILSMe and accounts for their higher current densities reached.
As stability of the electrocatalysts is crucial for practical applications, continuous cycling was conducted. Current density was observed to continuously increase within the first 40 cycles (Fig. 8A) slightly decreasing with further cycling. The observed increase of current density suggests increment of Ni oxidation state during the stability test45 with Ni oxidation peak shifting somewhat positively from 1.47 V to 1.50 V. This further confirms that the Ni represents the active sites for OER. Namely, different reconstruction processes might occur during cycling under OER conditions involving formation of Ni oxidized species.41,46 Reversible reconstruction processes involve potential-dependent phase changes, the dissolution/redeposition of surface Ni atoms, and the adsorption/desorption of OER intermediates.47 Redox transformations of catalytic centers and the generation/refilling of vacancies proceed in parallel. Irreversible transformations may include phase changes and morphological or structural alterations.
 | ||
| Fig. 8 CVs of NiIILSEt during continuous cycling (A, 1st–100th cycle) along with the Raman spectra (B) and SEM images with EDX spectra (C) of NiIILSEt working electrode before and after chronoamperometric test at 1.67 V. | ||
Namely, nickel oxide (NiO) or α-phase of nickel hydroxide (α-Ni(OH)2) are formed in alkaline media.47,48 Generated α-Ni(OH)2 comprises layers with H2O molecules or OH− ions in the interlayer space. It typically converts to β-Ni(OH)2 phase as a result of its dissolution and recrystallization in aqueous media. Generated β-Ni(OH)2 is the closest-packed, hexagonal structure of Ni2+ and OH− ions, with no ions in the interlayer space. Under OER conditions, i.e., at more positive potentials, this phase usually reversibly transforms to nickel oxyhydroxide (β-NiOOH) with Ni in the oxidation state of +3. Moreover, a few γ-Ni(OH)2 phases comprising much larger interlayer distances, can also be present. Under OER conditions, α-Ni(OH)2 can reversibly transforms to γ-NiOOH. In addition, β-NiOOH can be irreversibly overcharged in highly alkaline media forming the γ-NiOOH. The presence of Ni2+ (hydroxide species) along with Ni3+ species has been reported to be fundamental for active-site formation in OER catalysis by Ni-based materials.49,50
NiIILSEt working electrode was additionally analyzed before and after the electrochemical tests by using SEM-EDX and Raman spectroscopy, Fig. 8B and C. Minor changes in the electrode surface and elements composition could be observed. As mentioned, in case of heterogeneous complexes as catalysts such as those studied herein, leaching of metal ions and their subsequent redeposition might occur and cause a reactivation effect.51 On the other hand, in case of homogeneous complexes as catalysts for water oxidation (i.e., catalysts present in the aqueous phase), their degradation and formation of transition metal oxides/oxyhydroxides on the electrode surface have been reported.52,53
The herein synthesized material is assumed to undergo dynamic reconstruction under oxidative OER conditions, forming an active oxyhydroxide layer. These phase transformations, which are dependent on the applied potential, further lead to structural changes with the formation of a new surface layer, changes in the oxidation state of interfacial Ni cations, and number of lattice oxygen vacancies. These open the possibility of OER proceeding by both conventional adsorbate evolution mechanism (involving oxygen-containing intermediates from the electrolyte) and the lattice-oxygen mediated mechanism (involving lattice oxygen vacancies) with the latter being associated with increased OER activity of Ni-based materials.47
Conclusion
Successful synthesis of new 14-membered bis-thiosemicarbazide and bis-isothiosemicarbazide macrocycles and their nickel(II) complexes is reported and the results are supported by spectroscopic data (1H and 13C NMR, IR, UV–vis), ESI-MS and SC-XRD analysis. Moreover, potential application of the prepared complexes for electrocatalysis of OER in alkaline media was scrutinized for the first time. NiIILSEt delivered a current density of 10 mA cm−2 and 50 mA cm−2 at low overpotential of 350 mV and 480 mV in KOH electrolyte of moderate concentration (1 M), with Tafel slope of 93 mV dec−1. In addition, NiIILSEt exhibited high stability during long-term operation. Considering the exhibited high performance for OER along with the synthesis directly scalable to industrial level and the use of only non-precious metals, the herein prepared Ni complexes are suggested as promising candidates for alkaline water electrolysis.Experimental
Chemicals
Triethylamine, NiCl2·6H2O, NaH, NH2OH·HCl, TsOH·H2O, iodomethane, iodoethane, Ni(OAc)2·4H2O were purchased from commercial suppliers and used without further purification. All solvents and liquid reagents purchased from commercial sources and used for the synthesis of metal-free macrocycles were distilled prior use. Petroleum ether had a distillation window 40–70 °C. Anhydrous MeCN was obtained according to the standard procedure. Pyrimidine 3 was prepared according to the reported procedure.36:3 mixture of compounds 3 and 2 (0.180 g, 0.90 mmol),36 NH2OH·HCl (0.078 g, 1.12 mmol), and EtOH (3 mL) at room temperature. The resulting mixture was refluxed under stirring on a hot plate magnetic stirrer for 2 h. At the beginning of heating, a solution quickly forms, from which after 3 min a white solid begins to precipitate. After completion of the reaction, the solvent was removed under reduced pressure, the solid residue was triturated with H2O until suspension formed, and cooled to 0 °C. The precipitate was filtered, thoroughly washed with ice-cold water, petroleum ether, and dried to give compound 6 (0.150 g, 92%; white solid) as a mixture of (7E,14E)-(5R*,6R*,12R*,13R*)-diastereomer (≥90%) with small amounts of some other diastereomers. Compound 6 with similar purity was also prepared by method A using multi-gram loadings. For example, the treatment of a 97:3 mixture of compounds 3 and 2 (8.213 g, 40.80 mmol) with NH2OH·HCl (3.555 g, 51.16 mmol) in refluxing EtOH (120 mL) for 2 h gave 6 (7.226 g, 97%) as a white solid. An analytically pure sample of (7E,14E)-(5R*,6R*,12R*,13R*)-6 (2.09 g; white solid) was obtained after crystallization of crude 6 (2.95 g) from butan-1-ol (140 mL). Anal. Calcd for C16H26N6S2, %: C, 52.43; H, 7.15; N, 22.93. Found, %: C, 52.34; H, 7.38; N, 23.13. Mp 261 °C (decomp., black foam; butan-1-ol) (heating rate close to the melting point is about 1 °C per 15–20 s; at a lower heating rate, only decomposition occurs without melting). IR (KBr) ν, cm−1: 3287 (br vs), 3208 (br s) (ν NH), 1653 (m) (ν CN), 1542 (br vs), 1519 (s), 1491 (s), 1478 (s) (thioamide-II), 1215 (s), 1183 (s), 1138 (s); 1H NMR spectrum (600.13 MHz, DMSO-d6) δ: 10.08 (2 × 1H, s, 2 NH-N), 8.72 (2 × 1H, d, 3J = 5.8 Hz, 2 NH-CH), 3.87 (2 × 1H, ddq, 3J = 11.0, 3J = 6.1, 3J = 5.8 Hz, H-5 and H-12), 2.64 (2 × 1H, dddd, 3J = 11.0, 3J = 8.0, 3J = 4.4, 4J = 1.6 Hz, H-6 and H-13), 2.40–2.46 (2 × 1H, m, HA in 2 CH2CN), 2.31–2.37 (2 × 1H, m, HB in 2 CH2CN), 1.93–1.99 (2 × 1H, m, HA in 2 CH2CH2CH2CN), 1.69–1.81 (2 × 2H, m, 2 CH2CH2CH2CN), 1.63–1.68 (2 × 1H, m, HB in 2 CH2CH2CH2CN), 1.33 (2 × 3H, d, 3J = 6.1 Hz, 2 CH3); 13C NMR spectrum (150.90 MHz, DMSO-d6) δ: 176.57 (C-3, C-10), 160.07 (C-7, C-14), 52.12 (C-5, C-12), 47.73 (C-6, C-13), 28.94 (2 CH2CH2CH2CN), 28.76 (2 CH2CH2CH2CN), 22.77 (2 CH2CH2CH2CN), 20.31 (2 CH3); EIMS spectrum: m/z 368 [3, (M + 2)+], 367 [6, (M + 1)+], 366 (33, M+), 291 (4), 250 (3), 210 (14), 184 (37), 183 (100), 182 (19), 169 (13), 168 (38), 167 (42), 154 (8), 150 (24), 142 (10), 141 (11), 140 (14), 128 (10), 125 (19), 124 (26), 123 (50), 115 (40), 110 (42), 109 (37), 108 (41), 106 (24), 97 (55), 86 (52), 82 (66), 81 (57), 80 (64), 69 (58), 67 (83), 60 (83), 55 (62), 53 (54), 44 (80), 41 (90). HRMS (ESI-TOF) m/z calcd for C16H27N6S2 [M + H]+ 367.1733, found 367.1729.
Method B (Table 1, entry 1): A solution of a 97:3 mixture of compounds 3 and 2 (3.192 g, 15.86 mmol) and N2H4·H2O (7.944 g, 158.69 mmol) in EtOH (50 mL) was stirred under reflux for 1 h, then the solvent was removed under reduced pressure at 60 °C. The oily residue was co-evaporated with toluene (3 × 25 mL), dissolved in CHCl3 (60 mL), and washed with brine (3 × 5 mL). The solvent was removed under reduced pressure, the residual foam was dried in a vacuum desiccator over P2O5 overnight. The obtained crude hydrazone 4 (3.592 g, 16.68 mmol) was dissolved in EtOH (35 mL) upon warming, then solid TsOH·H2O (3.499 g, 18.39 mmol) and EtOH (15 mL) were added, and the mixture was stirred under reflux for 2 h. The resulting suspension was evaporated to dryness under reduced pressure, the solid residue was triturated with saturated aqueous solution of NaHCO3 until suspension formed, and cooled. The precipitate was filtered, thoroughly washed with ice-cold H2O, petroleum ether, and dried to give compound 6 (2.530 g, 83%) as a mixture of (5R*,6R*,12R*,13R*)-diastereomer (≥94%) with small amounts of some other diastereomers.
Method C (Table 1, entry 2): To a stirred solution of NH2OH·HCl (0.115 g, 1.65 mmol) and NaOAc·3H2O (0.228 g, 1.68 mmol) in H2O (1 mL) was added a 97:3 mixture of compounds 3 and 2 (0.268 g, 1.33 mmol), followed by EtOH (4 mL), and the reaction mixture was stirred at room temperature. Approximately 3 h after the beginning of the reaction, the solid was completely dissolved. After 4.5 h, the solution was evaporated to dryness under reduced pressure, the residue was dissolved in CHCl3 (15 mL), and washed with Н2О (3 × 5 mL), brine (2 × 5 mL). The solvent was removed under reduced pressure, the residual foam was dried in a vacuum desiccator over P2O5 overnight. To the obtained crude oxime 5 (0.160 g, 0.74 mmol) were added TsOH·H2O (0.157 g, 0.83 mmol) and EtOH (4 mL), and the mixture was stirred under reflux for 2 h. At the beginning of heating, a solution quickly forms, from which after 11 min a white solid begins to precipitate. After completion of the reaction, the solvent was removed under reduced pressure, the solid residue was triturated with saturated aqueous solution of NaHCO3 until suspension formed, and cooled to 0 °C. The precipitate was filtered, thoroughly washed with ice-cold water, petroleum ether, and dried to give compound 6 (0.087 g, 64%) as a mixture of (7E,14E)-(5R*,6R*,12R*,13R*)-diastereomer (≥90%) with small amounts of some other diastereomers.
N), 1562 (vs) (NC-NH), 1306 (s), 1140 (m); 1H NMR spectrum (600.13 MHz, DMSO-d6) δ: 8.14 (2 × 1H, d, 3J = 3.4 Hz, 2 NH-CH), 3.22 (2 × 1H, ddq, 3J = 10.3, 3J = 6.0, 3J = 3.4 Hz, H-5 and H-12), 2.99 (2 × 1H, dq, 2J = 13.0, 3J = 7.3 Hz, HA in 2 SCH2), 2.93 (2 × 1H, dq, 2J = 13.0, 3J = 7.3 Hz, HB in 2 SCH2), 2.61–2.67 (2 × 1H, m, HA in 2 CH2CN), 2.45 (2 × 1H, dddd, 3J = 10.3, 3J = 8.1, 3J = 5.3, 4J = 1.7 Hz, H-6 and H-13), 2.31–2.37 (2 × 1H, m, HB in 2 CH2CN), 1.94–2.00 (2 × 1H, m, HA in 2 CH2CH2CH2CN), 1.69–1.76 (2 × 1H, m, HA in 2 CH2CH2CH2CN), 1.55–1.66 (2 × 2H, m, HB in 2 CH2CH2CH2CN and HB in 2 CH2CH2CH2CN), 1.29 (2 × 3H, d, 3J = 6.0 Hz, 2 CH3), 1.26 (2 × 3H, t, 3J = 7.3 Hz, CH3 in 2 SEt); 13C NMR spectrum (150.90 MHz, DMSO-d6) δ: 170.18 (C-7, C-14), 159.94 (C-3, C-10), 50.86 (C-5, C-12), 47.85 (C-6, C-13), 29.19 (2 CH2CH2CH2CN), 28.90 (2 CH2CH2CH2CN), 23.70 (2 SCH2), 22.71 (5-CH3, 12-CH3), 22.33 (2 CH2CH2CH2CN), 14.42 (CH3 in 2 SEt).
Synthesis of complexes
NiIILS. To a suspension of H2LS (110 mg, 0.3 mmol) in dry DMF (3 mL) under argon was added triethylamine (83.7 μL, 0.6 mmol). The solution was stirred at room temperature for 15 min, then nickel(II) chloride hexahydrate (74.7 mg, 0.3 mmol) was added and the reaction mixture was heated at 90 °C for 1 h. Afterwards, the obtained solution was cooled down to room temperature. The red precipitate was filtered off, washed with cold methanol (5 × 1 mL) and dried in vacuo. Yield: 79 mg, 53%. Anal. Calcd for C16H24N6S2NiCl2 (Mr = 423.22), %: C, 45.41; H, 5.72; N, 19.86; S, 15.15%. Found, %: C, 45.32; H, 5.72; N, 19.63; S, 15.13. ESI-MS in MeCN/MeOH +1% H2O (positive): m/z 423.16 [NiLS + H]+, 445.11 [NiLS + Na]+. ESI-MS in ACN/MeOH +1% H2O (negative): m/z 421.00 [NiLS-H]−. 1H NMR (700 MHz, DMSO-d6) δ, ppm: 10.50 (s, 2H, 2 and 9), 3.10–3.06 (m, 2H, 5 and 12), 2.73–2.68 (m, 4H, 6 and 13, (6- and 13-CH2-CH2-CH2)b), 2.45–2.39 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 2.08–2.05 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 1.89–1.85 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 1.65–1.58 (m, 2H, (6- and 13-CH2-CH2-CH2)b), 1.50–1.44 (m, 2H, (6- and 13-CH2-CH2-CH2)b), 1.33 (d, 6H, 5- and 12-CH3). Due to the splitting of the signals of the methyl groups, those co-planar with CH 6 and 13 are indicated with an italic a as subindex. 13C NMR (151 MHz, DMSO-d6) δ, ppm: 181.62 (3 and 10), 165.82 (7 and 14), 55.44 (6 and 13), 51.63 (5 and 12), 31.18 (6- and 13-CH2-CH2-CH2), 30.53 (6- and 13-CH2-CH2-CH2), 23.23 (6- and 13-CH2-CH2-CH2), 21.16 (5- and 12-CH3). Single crystals suitable for X-ray diffraction study were obtained from mother liquor in a Schlenk tube upon standing at 4 °C overnight. UV–visible (MeOH), λmax, nm (ε, M−1 cm−1): 256sh; 271(16764), 300sh, 356(38256), 526(226). IR (ATR, selected bands,  ): 3319, 3070, 2876, 2646, 2322, 1660, 1546, 1446, 1362, 1277, 1199, 1084, 890, 647 cm−1.
): 3319, 3070, 2876, 2646, 2322, 1660, 1546, 1446, 1362, 1277, 1199, 1084, 890, 647 cm−1.
NiIILSMe. To a suspension of sodium hydride (14.4 mg, 0.6 mmol) in dry DMF (1.5 mL) under argon was added a suspension of H2LS (110 mg, 0.3 mmol) in dry DMF (1.5 mL). The obtained solution was stirred at room temperature for 1 h, then iodomethane (37.35 μL, 0.6 mmol) was added and the solution was further stirred for 3 h. Then, nickel(II) acetate tetrahydrate (24.88 mg, 0.1 mmol) was added and the solution was heated at 90 °C for 2 h. The reaction mixture was cooled down to room temperature. The red precipitate was filtered off, washed with methanol (5 × 1 mL) and dried in vacuo. Yield: 72 mg, 53%. Anal. Calcd for C18H28N6S2Ni (Mr = 451.28), %: C, 47.91; H, 6.25; N, 18.62; S, 14.21%. Found, %: C, 47.72; H, 6.22; N, 18.47; S, 14.09. ESI-MS in MeCN/MeOH +1% H2O (positive): m/z 451.21 [NiLSMe + H]+. ESI-MS in ACN/MeOH +1% H2O (negative): m/z 449.11 [NiLSMe-H]−. 1H NMR (600 MHz, CDCl3) δ 2.99–2.82 (m, 4H, 5 and 12, (6- and 13-CH2-CH2-CH2)b), 2.58–2.51 (m, 2H, 6 and 13), 2.49 (s, 6H, 3- and 10-S-CH3), 2.44 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 2.08 (dt, 2H, (6- and 13-CH2-CH2-CH2)a), 1.97–1.86 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 1.67–1.56 (m, 2H, (6- and 13-CH2-CH2-CH2)b), 1.51–1.39 (m, 2H, (6- and 13-CH2-CH2-CH2)b), 1.32 (d, J = 5.8 Hz, 6H, 5- and 12-CH3). Due to the splitting of the signals of the methyl groups, those co-planar with CH 6 and 13 are indicated with an italic a as subindex. 13C NMR (151 MHz, CDCl3) δ 169.41 (3 and 10), 166.26 (7 and 14), 56.69 (6 and 13), 50.70 (5 and 12), 31.38 (6- and 13-CH2-CH2-CH2), 30.66 (6- and 13-CH2-CH2-CH2), 23.68 (6- and 13-CH2-CH2-CH2), 23.53 (5- and 12-CH3), 16.35 (3- and 10-S-CH3). Single crystals suitable for X-ray diffraction study were obtained by slow diffusion of diethyl ether into the mother liquor. UV–visible (MeOH), λmax, nm (ε, M−1 cm−1): 216(204849), 236sh, 286(16152), 318(6163), 400(4564), 530(222). IR (ATR, selected bands,  ): 2955, 2925, 2867, 2831, 1463, 1286, 1142, 1089, 1012, 888, 828, 782, 653 cm−1.
): 2955, 2925, 2867, 2831, 1463, 1286, 1142, 1089, 1012, 888, 828, 782, 653 cm−1.
NiIILSEt. To a solution of H2LSEt (63 mg, 0.15 mmol) in dry DMF (3 mL) under argon was added triethylamine (36.9 μL, 0.3 mmol). The solution was stirred at room temperature for 15 min, then nickel(II) acetate tetrahydrate (37.3 mg, 0.15 mmol) was added and the reaction mixture was stirred at room temperature for 1 h. Afterwards, the volume of solution was reduced to 1/3 by concentration in vacuo and allowed to stand at 4 °C overnight. The red precipitate was filtered off, washed with cold methanol (3 × 2 mL) and dried in vacuo. Yield: 45 mg, 63%. Anal. Calcd for C20H32N6S2Ni (Mr = 479.33), %: C, 50.11; H, 6.73; N, 17.53; S, 13.38%. Found, %: C, 49.79; H, 6.68; N, 17.39; S, 13.33. ESI-MS in MeCN/MeOH +1% H2O (positive): m/z 479.20 [NiLSEt + H]+. 1H NMR (600 MHz, CDCl3) δ, ppm: 3.14–3.01 (m, 4H, 3- and 10-S-CH2-CH3), 2.97–2.85 (m, 4H, 5 and 12, (6- and 13-CH2-CH2-CH2)b), 2.56–2.49 (m, 2H, 6 and 13), 2.43 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 2.08 (dt, 2H, (6- and 13-CH2-CH2-CH2)a), 1.94–1.86 (m, 2H, (6- and 13-CH2-CH2-CH2)a), 1.64–1.57 (m, 2H, (6- and 13-CH2-CH2-CH2)b), 1.45 (ddd, 2H, (6- and 13-CH2-CH2-CH2)b), 1.32 (dd, 5- and 12-CH3, 3- and 10-S-CH2-CH3). Due to the splitting of the signals of the methyl groups, those co-planar with CH 6 and 13 are indicated with an italic a as subindex. 13C NMR (151 MHz, CHCl3) δ, ppm: 168.83(3 and 10), 165.93(7 and 14), 56.68(6 and 13), 50.71(5 and 12), 31.41(6- and 13-CH2-CH2-CH2), 30.63(6- and 13-CH2-CH2-CH2), 27.83(3- and 10-S-CH2-CH3), 23.76(5- and 12-CH3), 23.63(6- and 13-CH2-CH2-CH2), 15.00(3- and 10-S-CH2-CH3). Single crystals suitable for X-ray diffraction study were obtained from mother liquor in a Schlenk tube upon standing at 4 °C overnight. UV–visible (MeOH), λmax, nm (ε, M−1 cm−1): 216(23232), 238sh, 289(18880), 319(7249), 402(53858), 529(301). IR (ATR, selected bands,  ): 2957, 1450, 1370, 1333, 1297, 1245, 1142, 1093, 1006, 777 cm−1.
): 2957, 1450, 1370, 1333, 1297, 1245, 1142, 1093, 1006, 777 cm−1.
110, 1650, 2420 and 722 frames were measured, each for 30, 2, 3, 30 and 35 s over 0.12, 0.5, 0.360, 0.360 and 2° scan width, respectively. Crystal data, data collection parameters, and structure refinement details are given in Table S1 in the ESI.† The structures were solved by direct methods and refined by full-matrix least-squares techniques. Non-H atoms were refined with anisotropic displacement parameters. H atoms were inserted in calculated positions and refined with a riding model. The following computer programs and hardware were used: structure solution, SHELXS-2014 and refinement, SHELXL-2014;54 molecular diagrams, ORTEP;55 computer, Intel CoreDuo. CCDC no.: 2324332 (H2LS), 2324333 (H2LSEt), 2324334 (NiLS), 2324335 (NiLSMe) and 2324336 (NiLSEt).†
Author contributions
Iuliana Besleaga – data curation; formal analysis; investigation; methodology; writing – original draft; Anastasia A. Fesenko – data curation; investigation; methodology; Anup Paul – data curation; investigation; methodology, sofware, writing-original draft; Biljana Šljukić – investigation; formal analysis; writing – original draft; visualisation; Peter Rapta – investigation; methodology; writing – original draft; Armando J. L. Pombeiro – writing – review and editing; supervision; funding acquisition; Anatoly Shutalev – investigation; writing – review and editing; supervision, and Vladimir B. Arion – conceptualization; funding acquisition; investigation; project administration; writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds H2LS, H2LSEt, NiLS, NiLSMe and NiLSEt have been deposited at the CCDC under accession numbers 2324332, 2324333, 2324334, 2324335 and 2324336† and can be obtained from CCDC e-mail: deposit@ccdc.cam.ac.uk.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Austrian Science Fund via grant no. I4729. A. A. F. acknowledges the Russian Science Foundation (grant no. 23-23-00324) for financial support of the research on the synthesis of macrocyclic compounds. A. P. and B. Š. acknowledge Instituto Superior Técnico and Fundação para a Ciência e a Tecnologia (FCT, Portugal) for the Scientific Employment contract (Contrato No: IST-ID/197/2019, DOI: 10.54499/DL57/2016/CP1384/CT0081 and IST-ID/156/2018) under Decree-Law no. 57/2016, of August 29. This work has also been partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal, through projects UIDB/00100/2020 (https://doi.org/10.54499/UIDB/00100/2020), UIDP/00100/2020 (https://doi.org/10.54499/UIDP/00100/2020), and LA/P/0056/2020 (https://doi.org/10.54499/LA/P/0056/2020) of Centro de Química Estrutural. The authors would also like to thank associate professor Nemanja Gavrilov, Faculty of Physical Chemistry, University of Belgrade, for SEM/EDX analysis. PR thanks the Science and Technology Assistance Agency (grant APVV-19-0024) and grant agency VEGA (contract 1/0392/24) for the financial support. V. B. A. thanks the Ministry of Research, Innovation and Digitalization for financial support via project no. PNRR-III-C9-2023-I8-99/31.07.2023 within the National Recovery and Resilience Plan (Romania).References
- Yu. A. Simonov, V. K. Beĺskii, N. V. Gerbeleu, S. G. Shova and V. B. Arion, Dokl. Akad. Nauk SSSR, 1985, 285, 620–624 Search PubMed.
- N. V. Gerbeleu, Yu. A. Simonov, V. B. Arion, S. G. Shova and T. I. Malinovskii, Dokl. Akad. Nauk SSSR, 1985, 283, 633–637 CAS.
- N. V. Gerbeleu, V. A. Kogan, V. B. Arion, V. V. Lukov and K. M. Indrichan, Zh. Neorg. Khim., 1989, 34, 107–111 CAS.
- V. B. Arion, Yu. A. Simonov, N. V. Gerbeleu, A. A. Dvorkin, D. I. Gradinaru and T. I. Malinovskii, Dokl. Akad. Nauk SSSR, 1992, 325, 502–507 CAS.
- V. Arion, K. Wieghardt, T. Weyhermueller, V. M. Leovac and A. Rufinska, Inorg. Chem., 1997, 36, 661–669 CrossRef CAS.
- U. Knof, Die Koordinationschemie von “Non-Innocent” Bis(S-Alkylisothiosemicarbazonat)-Liganden Mit Übergangsmetallionen, PhD thesis, Ruhr-Universität Bochum, 1995 Search PubMed.
- V. B. Arion, P. D. Beer, M. G. B. Drew and P. Hopkins, Polyhedron, 1998, 18, 451–458 CrossRef.
- N. V. Gerbeleu, V. B. Arion, Yu. A. Simonov, V. E. Zavodnik, K. I. Turta, S. S. Stavrov, A. A. Pasinsky, O. G. Ellert, D. I. Gradinaru and M. S. Birca, Inorg. Chim. Acta, 1992, 202, 173–181 CrossRef CAS.
- V. M. Leovac, R. Herak, B. Prelesnik and S. R. Niketic, Dalton Trans., 1991, 2295–2299 RSC.
- N. V. Gerbeleu, Yu. A. Simonov, V. B. Arion, V. M. Leovac, K. I. Turta, K. M. Indrichan, D. I. Gradinaru, V. E. Zavodnik and T. I. Malinovsky, Inorg. Chem., 1992, 31, 3264–3268 CrossRef CAS.
- U. Knof, T. Weyhermüller, T. Wolter and K. Wieghardt, J. Chem. Soc., Chem. Commun., 1993, 726–728 RSC.
- U. Knof, T. Weyhermüller, T. Wolter, K. Wieghardt, E. Bill, C. Butzlaff and A. X. Trautwein, Angew. Chem., Int. Ed. Engl., 1993, 32, 1635–1638 CrossRef.
- V. B. Arion, Coord. Chem. Rev., 2019, 387, 348–397 CrossRef CAS.
- V. B. Arion, N. V. Gerbeleu, V. G. Levitsky, Yu. A. Simonov, A. A. Dvorkin and P. N. Bourosh, J. Chem. Soc., Dalton Trans., 1994, 1913–1916 RSC.
- N. V. Gerbeleu, Yu. A. Simonov, V. B. Arion, V. E. Zavodnik, D. E. Gradinaru and K. M. Indrichan, Zh. Neorg. Khim., 1991, 36, 906–913 CAS.
- J. K. Bilyj, M. J. Riley and P. V. Bernhardt, Dalton Trans., 2018, 47, 2018–2030 RSC.
- J. K. Bilyj, N. V. Silajew and P. V. Bernhardt, Dalton Trans., 2021, 50, 612–623 RSC.
- J. K. Bilyj, N. V. Silajew, G. R. Hanson, J. R. Harmer and P. V. Bernhardt, Dalton Trans., 2019, 48, 15501–15514 RSC.
- J. K. Bilyj, J. R. Harmer and P. V. Bernhardt, Eur. J. Inorg. Chem., 2018, 4731–4741 CrossRef CAS.
- V. B. Arion, S. Platzer, P. Rapta, P. Machata, M. Breza, D. Vegh, J. Telser, S. Shova, T. C. O. Mac Leod and A. J. L. Pombeiro, Inorg. Chem., 2013, 52, 7524–7540 CrossRef CAS PubMed.
- A. Dobrov, A. Fesenko, A. Yankov, I. Stepanenko, D. Darvasiova, M. Breza, P. Rapta, L. M. D. R. S. Martins, A. J. L. Pombeiro, A. D. Shutalev and V. B. Arion, Inorg. Chem., 2020, 59, 10650–10664 CrossRef CAS PubMed.
- V. B. Arion, P. Rapta, J. Telser, S. S. Shova, M. Breza, C. Luspai and J. Kozisek, Inorg. Chem., 2011, 50, 2918–2931 CrossRef CAS PubMed.
- V. L. Goedken and M. C. Weiss, Inorg. Synth., 1980, 20, 115–119 CrossRef.
- N. V. Gerbeleu, V. B. Arion and J. Burgess, Template Synthesis of Macrocyclic Compounds, Wiley-VCH, Weinheim, 1999 Search PubMed.
- A. D. Shutalev, A. A. Fesenko, O. M. Kuzmina, A. N. Volov, D. V. Albov, V. V. Chernyshev and I. A. Zamilatskov, Tetrahedron Lett., 2014, 55, 5481–5485 CrossRef CAS.
- A. D. Shutalev, A. A. Fesenko, A. N. Yankov, V. A. Tafeenko and V. V. Chernyshev, J. Mol. Struct., 2017, 1150, 349–357 CrossRef CAS.
- A. A. Fesenko, M. S. Grigoriev, V. B. Arion and A. D. Shutalev, J. Org. Chem., 2022, 87, 15722–15731 CrossRef CAS PubMed.
- J.-T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian and Z.-Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- D. Mladenović, A. Mladenović, D. M. F. Santos, A. B. Yurtcan, Š. Miljanić, S. Mentus and B. Šljukić, J. Electroanal. Chem., 2023, 946, 117709 CrossRef.
- J. Milikić, A. Nastasić, M. Martins, C. A. C. Sequeira and B. Šljukić, Batteries, 2023, 9, 394 CrossRef.
- S. Li, Y. Gao, N. Li, L. Ge, X. Bu and P. Feng, Energy Environ. Sci., 2021, 14, 1897–1927 RSC.
- G. Gupta, F. Gusmão, A. Paul, B. Šljukić, D. M. F. Santos, J. Lee, M. F. C. Guedes Da Silva, A. J. L. Pombeiro and C. Y. Lee, Dalton Trans., 2024, 53, 5001–5009 RSC.
- A. Paul, K. Radinović, S. Hazra, D. Mladenović, B. Šljukić, R. A. Khan, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Molecules, 2022, 27, 7323 CrossRef CAS PubMed.
- W. Lv, Y. Li, F. Li, X. Lan, Y. Zhang, L. Du, Q. Zhao, D. L. Phillips and W. Wang, J. Am. Chem. Soc., 2019, 141, 17482–17486 CrossRef CAS PubMed.
- 4-(3-Oxobutyl)thiosemicarbazide hydrazone was prepared by the reaction of 4-isothiocyanatobutan-2-one with an excess hydrazine.
- A. A. Fesenko and A. D. Shutalev, Tetrahedron, 2016, 72, 2560–2573 CrossRef CAS.
- This conclusion is based on the vicinal couplings of the 4-H proton with the N(3)H and 5-H protons (3JN(3)H,4-H = 0, 3J4-H,5-H = 3.6 Hz), as well as the presence of the long-range coupling between the N(3)H and 5-H protons (4JN(3)H,5-H = 1.8 Hz). In addition, the characteristic nOe correlations of the OH proton with the 4-H and 5-H protons were observed in the 1H–1H NOESY spectrum of this isomer.
- This was proved by the values of the following coupling constants in its 1H NMR spectrum in DMSO-d6: 3JN(3)H,4-H = 0, 3J4-H,5-H = 10.9, 4J5-H,OH = 0.7 Hz.
- This solidification step was necessary to completely remove trace amounts of hydrazine hydrate, the presence of which significantly affects the subsequent macrocyclization reaction.
- Coordination Chemistry of Macrocyclic Compounds, ed. G. A. Melson, Springer US, Boston, MA, 1979, pp. 219–344 Search PubMed.
- M. Retuerto, F. Calle-Vallejo, L. Pascual, P. Ferrer, Á. García, J. Torrero, D. Gianolio, J. L. G. Fierro, M. A. Peña, J. A. Alonso and S. Rojas, J. Power Sources, 2018, 404, 56–63 CrossRef CAS.
- C. P. K. Prabhu, K. R. Naveen and J. Hur, RSC Appl. Interfaces, 2024, 1, 301–312 RSC.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- W.-D. Zhang, Q.-T. Hu, L.-L. Wang, J. Gao, H.-Y. Zhu, X. Yan and Z.-G. Gu, Appl. Catal., B, 2021, 286, 119906 CrossRef CAS.
- S. Kalantarifard, S. I. Allakhverdiev and M. M. Najafpour, Int. J. Hydrogen Energy, 2020, 45, 33563 CrossRef CAS.
- N. Hales, T. J. Schmidt and E. Fabbri, Curr. Opin. Electrochem., 2023, 38, 101231 CrossRef CAS.
- D. M. F. Santos, B. Sljukic, L. Amaral, D. Maccio, A. Saccone and C. A. C. Sequeira, J. Electrochem. Soc., 2014, 161, F594 CrossRef CAS.
- M. M. Najafpour and H. Feizia, Dalton Trans., 2018, 47, 6519–6527 RSC.
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287–1298 RSC.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- P. Garrido-Barros, S. Grau, S. Drouet, J. Benet-Buchholz, C. Gimbert-Suriñach and A. Llobet, ACS Catal., 2019, 9, 3936–3945 CrossRef CAS.
- J.-W. Wang, P. Sahoo and T.-B. Lu, ACS Catal., 2016, 6, 5062–5068 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, ORNL–6895, 369685, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra, ESI mass spectra of the metal-free macrocycles, spectroscopic characterization of Ni(II) complexes, crystallographic data for both metal-free macrocycles and Ni(II) complexes. CCDC 2324332–2324336. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02182g |
| This journal is © The Royal Society of Chemistry 2024 |