
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polarized metal–metal multiple bonding and reactivity of phosphinoamide-bridged heterobimetallic group IV/cobalt compounds
Nathanael H.
Hunter
 and
Christine M.
Thomas
*
and
Christine M.
Thomas
*
Department of Chemistry and Biochemistry, The Ohio State University, 100 W, 18th Ave, Columbus, OH 43210, USA. E-mail: thomasc@chemistry.ohio-state.edu
First published on 3rd September 2024
Abstract
Heterobimetallic complexes are studied for their ability to mimic biological systems as well as active sites in heterogeneous catalysts. While specific interest in early/late heterobimetallic systems has fluctuated, they serve as important models to fundamentally understand metal–metal bonding. Specifically, the polarized metal–metal multiple bonds formed in highly reduced early/late heterobimetallic complexes exemplify how each metal modulates the electronic environment and reactivity of the complex as a whole. In this Perspective, we chronicle the development of phosphinoamide-supported group IV/cobalt heterobimetallic complexes. This combination of metals allows access to a low valent Co−I center, which performs a rich variety of bond activation reactions when coupled with the pendent Lewis acidic metal center. Conversely, the low valent late transition metal is also observed to act as an electron reservoir, allowing for redox processes to occur at the d0 group IV metal site. Most of the bond activation reactions carried out by phosphinoamide-bridged M/Co−I (M = Ti, Zr, Hf) complexes are facilitated by cleavage of metal–metal multiple bonds, which serve as readily accessible electron reservoirs. Comparative studies in which both the number of buttressing ligands as well as the identity of the early metal were varied to give a library of heterobimetallic complexes are summarized, providing a thorough understanding of the reactivity of M/Co−I heterobimetallic systems.
Introduction
To quote the late Prof. Malcolm Chisholm, “Anything one can do, two can do too – and it's more interesting.”1 The expansion of coordination compounds to contain more than one transition metal center represents a rapidly intensifying area of interest in inorganic chemistry. Such bimetallic complexes have been used to study metal–metal cooperativity,2–10 and to give insight into the activity of both metalloenzymes11–15 and heterogeneous catalysts.16,17 The two metals in a bimetallic complex need not be directly linked via a metal–metal bond, but can be held in close proximity by a dinucleating or bridging ligand framework. For example, the dithiolate-bridged Ni–Fe and Ni–Pd complexes published by Darensbourg et al.18–21 are an excellent showcase of heterobimetallic biomimetic activity and open the door to the discussion of many novel and interesting site-differentiated multimetallic metal clusters and their widespread reactivity.22–25 However, this Perspective will instead more narrowly focus on early/late heterobimetallic complexes with direct polarized interactions between the two metals and the reactivity unique to such metal–metal bonds. In particular, the authors hope to provide a personal account of the development of phosphinoamide-linked early/late heterobimetallic compounds featuring metal–metal multiple bonds and their reactivity patterns, in hopes of providing fundamental insight into the structure/function relationships that correlate metal–metal bonding and reactivity.Overview of metal–metal bonding
First, an introduction to metal–metal bonds, particularly those between first-row metals, is warranted. It would be a travesty to start a discussion about metal–metal bonding without crediting the late F. A. Cotton.26 While the covalency of single bonds between metals had been studied,27,28 and metal–metal multiple bonding had been postulated,29 it was Cotton et al. that first discovered examples of metal–metal double,30 triple,31 and quadruple bonds,32 all with tetrachlororhenate(III)-based systems. These discoveries sparked thousands of reports of metal–metal multiple bonds,4,26,33–43 but the hypothetical metal–metal quintuple bond remained elusive until the first Cr–Cr quintuple bond was reported by Power et al. in 2005.44 To aid in quantifying the degree of metal–metal bonding using structural parameters alone, Cotton developed a metric known as the Formal Shortness Ratio (FSR).26 The FSR of a multimetallic molecule is defined as the distance between two metal centers divided by the sum of the single bond atomic radii (Pauling's R1)45 of each metal atom component. This definition is sufficiently general to be applied for comparative purposes across a broad range of systems with different metal–metal combinations and allows a general correlation between bond length and bond order, even among heterobimetallic complexes. However, distance alone is not sufficient to describe the covalent interactions between two metal centers as there is a broad variance in FSR between metal–metal bonds of different orders.26 As an example, Power's Cr–Cr quintuple-bonded complex has a metal–metal distance (1.8351(4) Å, FSR = 0.77) that is comparable to many of the previously reported Cr–Cr quadruple-bonded complexes.26,44 It is for this reason that analysis of the electronic structure is also required to provide a complete understanding of metal–metal bonding from an orbital overlap perspective.The degree of metal–metal bonding in bimetallic complexes is dependent upon the local symmetry of each metal and the d orbital population dictated by each metal's formal oxidation state. For example, Fig. 1 depicts the differences in orbital configuration and population as well as metal–metal bond order as a function of symmetry and orbital population. In a hypothetical ligandless low-spin bimetallic CrICrI dication (Fig. 1, left), a maximum bond order of five could be obtained, thus inspiring the low-coordination number in Power's Cr–Cr quintuple bond. More commonly, multiple dinucleating ligands are employed to support bimetallic systems. Bimetallic systems of D4h symmetry are the most ubiquitous, and carboxylate, triazenate, or amidinate bridging ligands have been used to form “paddlewheel” [M2L4] complexes with most of the first-row transition metals, including vanadium,46–52 chromium,50,53,54 iron,55,56 cobalt,57–60 nickel,61–65 and copper,61,66,67 with metal–metal bond orders ranging from 0 to 4. In D4h symmetry the degeneracy of the δ orbitals is broken, resulting in population of δ* antibonding orbitals in compounds with more than 8d electrons and a maximum bond order of 4 (Fig. 1, middle). Moreover, the high-spin configurations of many first row bimetallic complexes result in lower bond orders due to the population of multiple M–M antibonding orbitals (Fig. 1, right).33,56 In the diiron paddlewheel case, the high spin configuration also leads to a Jahn–Teller distortion from D4h to D2d symmetry. Heterobimetallic paddlewheels have also been reported, such as the example reported by Tonks et al. wherein (2-diphenylphosphido)pyrrolide linkers aid in the formation of a Ti–Fe triple bond.68
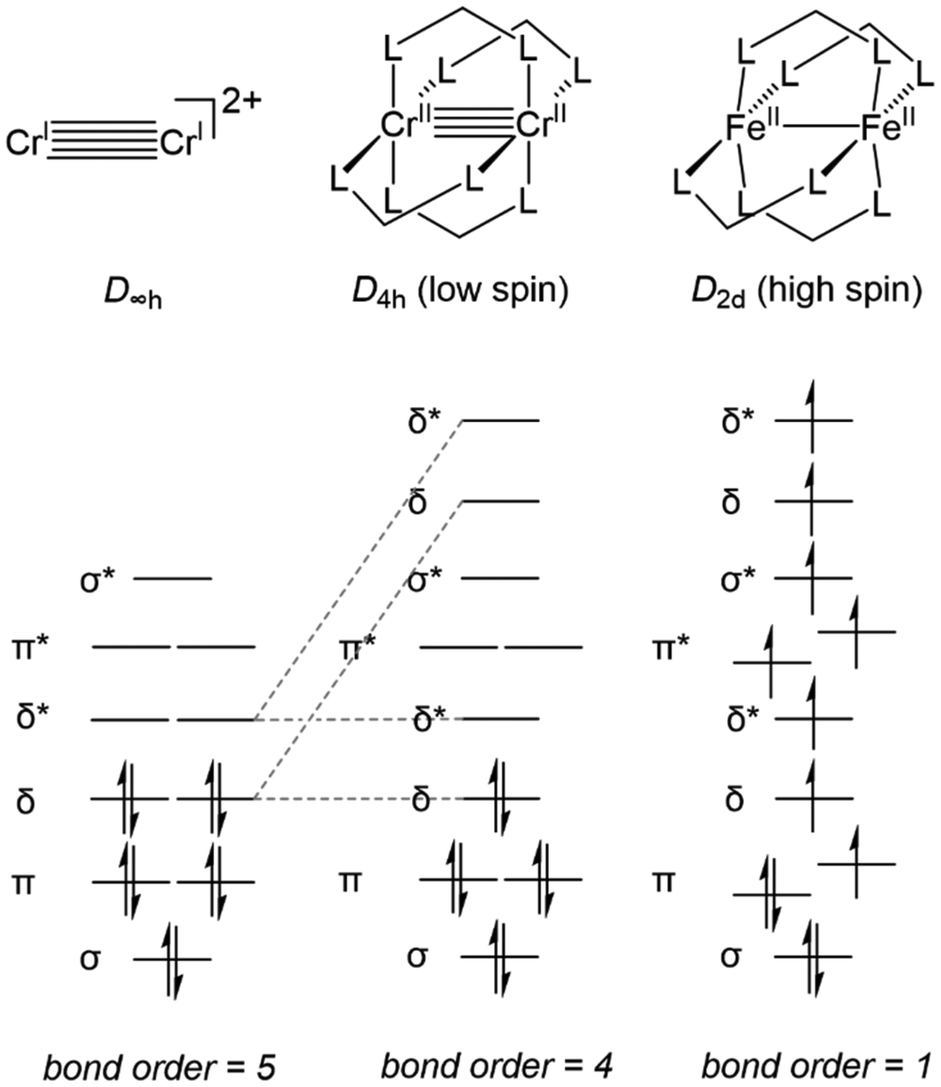 | ||
| Fig. 1 A comparison of the frontier molecular orbitals and metal–metal bond order of an idealized D∞h metal–metal quintuple-bonded system, a low spin D4h paddlewheel complex, and a high spin D2d paddlewheel complex. L^L represents a dinucleating monoanionic ligand. | ||
Lower symmetry trigonal D3h and C3v systems do not suffer from the “early” population of antibonding orbitals owing to the weaker C3-symmetric ligand field and maintained degeneracy of the orbitals of δ symmetry (Fig. 2A), and have been shown to support metal–metal multiple bonding among first-row transition metals and in heterobimetallic systems.4,33,60,69 Many different C3-symmetric ligand systems have been utilized to develop early/late heterobimetallic complexes.69,70,71,72,33 Multiple bonds are common with these systems not only due to their favorable symmetry, but also because of their electronic properties. The Lewis acidic nature of the early transition metal serves to withdraw electron density from the late transition metal via metal–metal dative donor–acceptor interactions, which results in more facile reduction.33 Upon reduction, the atomic orbitals of the late transition metal rise in energy, resulting in more orbital overlap and an increase in metal–metal bond order. Orbitals of the proper symmetry to form metal–metal δ bonds are often populated in reduced early/late heterobimetallic compounds. However, due to the differences in electronegativity and orbital energies of the two metals, these orbitals, which also have poor spatial overlap, typically remain non-bonding and localized on each metal (Fig. 2B).
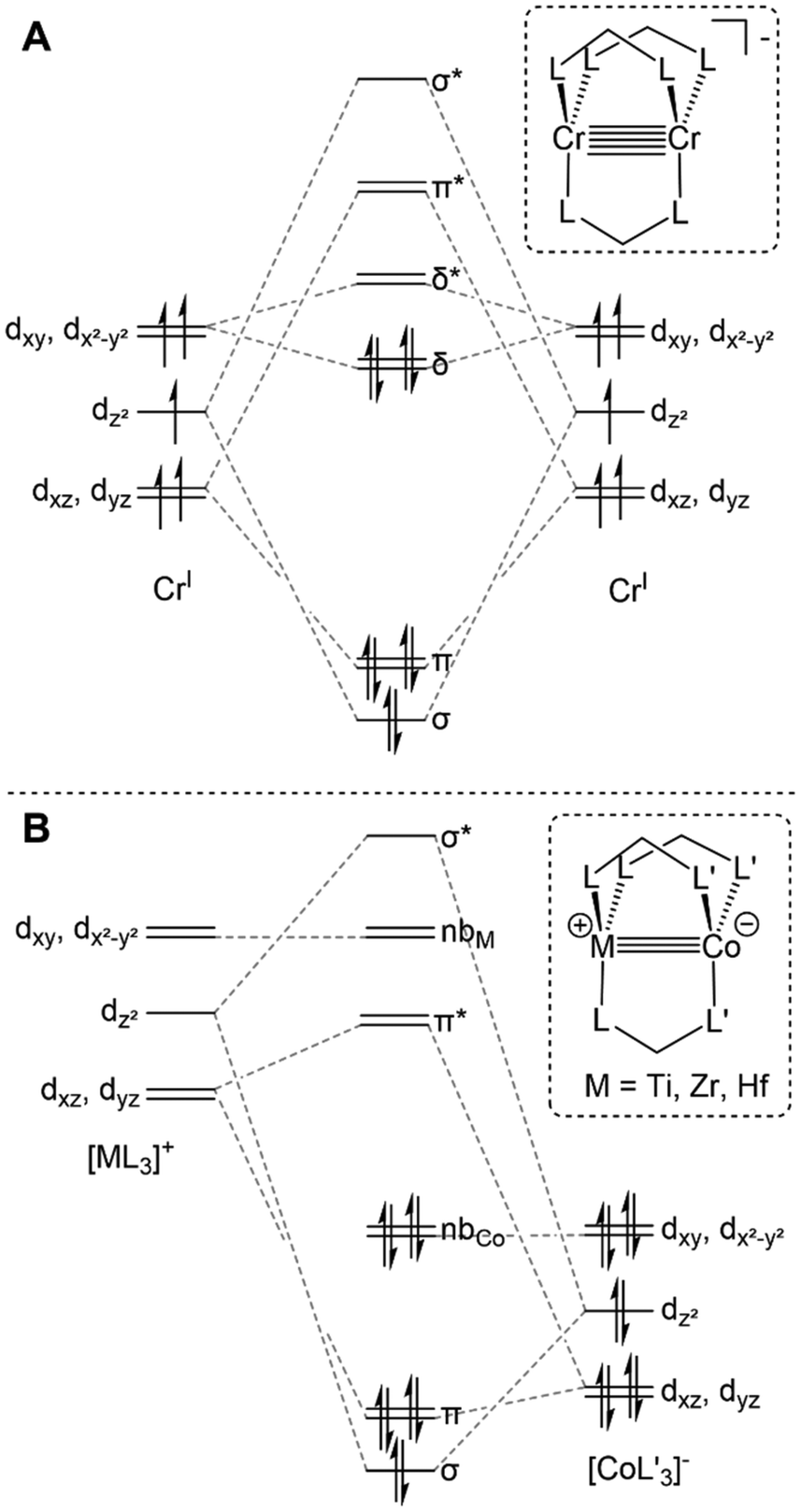 | ||
| Fig. 2 Qualitative molecular orbital diagrams depicting the metal–metal bonding in representative trigonal homobimetallic (A) and early/late heterobimetallic (B) complexes. L^L represents a bidentate monoanionic ligand that is either symmetric (A) or asymmetric (B) with different donor types. | ||
Metal–metal bonding in early/late heterobimetallic complexes
Interest in early/late heterobimetallic complexes has fluctuated since the 1980s,2,73–77 initially inspired by efforts to mimic the so-called “strong metal–support interactions” observed between late transition metal catalysts and metal oxide supports such as titania.17,78,79 Within this research area, there are several particularly noteworthy examples of early/late heterobimetallic compounds featuring polarized metal–metal bonds. While not formally assigned as a metal–metal multiple bond, the bonding between titanium and iron in Gade's HC(Me2SiNTol)3TiFe(CO)2Cp (A) compound was described as a combination of donor–acceptor interactions with both σ and π character (Fig. 3), demonstrating that there is a driving force for such interactions in the absence of bridging ligands.80 Wolczanski et al. leveraged the hard/soft acid/base preferences of alkoxyphosphine ligands to stabilize multiple bonds between group IV metals and late transition metals.70,81–84 For example, the C3 symmetric Ti/Rh heterobimetallic complex Ti(OC(CH3)2CH2PPh2)3Rh (B) was found to have a record-breaking short Ti–Rh distance of 2.2142(11) Å (FSR = 0.86) (Fig. 3).70 A computational investigation found that the metal–metal bonding consisted of three (one σ and two π) well-defined polarized dative bonds that Wolczanski chose to represent using a solid line and two arrows. We have adopted a similar depiction of bonding to represent both bond order and the donor/acceptor qualities of the metal–metal interactions throughout this Perspective and in most of our published work.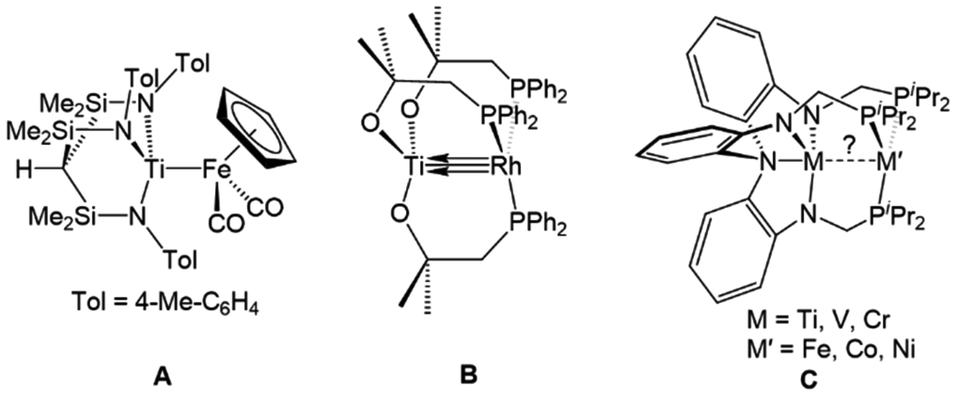 | ||
| Fig. 3 Selected examples of early/late heterobimetallic complexes featuring metal–metal (multiple) bonds reported by Gade (A),80 Wolczanski (B),70 and Lu (C).72 | ||
The examples most closely related to those discussed herein are a series of C3-symmetric trigonal lantern complexes studied by Lu and coworkers utilizing the heptadentate “double-decker” ligand [N(o-(NCH2PiPr2)C6H4)3]3−.72,85 A series of late transition metal Ti/M,86 V/M,87 and Cr/M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 88–90 complexes (M = Fe, Co, Ni) were investigated, with several metal–metal pairs giving rise to metal–metal multiple bonding in their fully reduced states (C, Fig. 3).72 Similar systems using the double-decker ligand motif to link late transition metals with main group metals and lanthanides have given rise to catalytic systems for carbon dioxide reduction to formate,91–93 the hydrogenation of alkenes and alkynes,94,95 and the hydrodefluorination of aryl fluorides.96
88–90 complexes (M = Fe, Co, Ni) were investigated, with several metal–metal pairs giving rise to metal–metal multiple bonding in their fully reduced states (C, Fig. 3).72 Similar systems using the double-decker ligand motif to link late transition metals with main group metals and lanthanides have given rise to catalytic systems for carbon dioxide reduction to formate,91–93 the hydrogenation of alkenes and alkynes,94,95 and the hydrodefluorination of aryl fluorides.96
These examples reported by Wolczanski and Lu illustrate the utility of heterobifunctional ligands with a hard anionic donor and a soft neutral donor, which preferentially bind to high-valent early metals and low-valent late metals, respectively. This binding site differentiation is key to stabilizing two disparate metals in a single framework, leading to the facile formation of metal–metal multiple bonds.
Phosphinoamide-linked early/late heterobimetallic complexes
The first examples of early/late heterobimetallic complexes linked by phosphinoamide ligands were reported by Nagashima and coworkers.97–100 In contrast to the ligands described above, phosphinoamide ligands feature two distinct coordination sites (amide and phosphine) directly joined through a single covalent bond. This ensures that the two metal centers are held in close proximity, promoting orbital overlap and metal–metal interactions. Combining early metal phosphinoamide compounds with late transition metal precursors, heterobimetallic bis(phosphinoamide) and tris(phosphinoamide) compounds such as Cl2Ti(tBuNPPh2)2PtX2 (D, X = Cl, Me), [Cl2Ti(tBuNPPh2)2M(η3-allyl)]+ (E, M = Ni, Pd, Pt), ClZr(iPrNPPh2)3Mo(CO)3 (F), and ClZr(iPrNPPh2)3CuCl (G) were synthesized (Fig. 4).97–100 The cationic Ti/M allyl complexes E were found to be active catalysts for allylic amination reactions, and the authors found that the appended Lewis acidic Ti center increased the electrophilicity of the M-allyl fragment and was crucial for achieving catalysis, as monometallic Ni, Pd, and Pt phosphine complexes with similar structures were unable to achieve catalytic turnover.98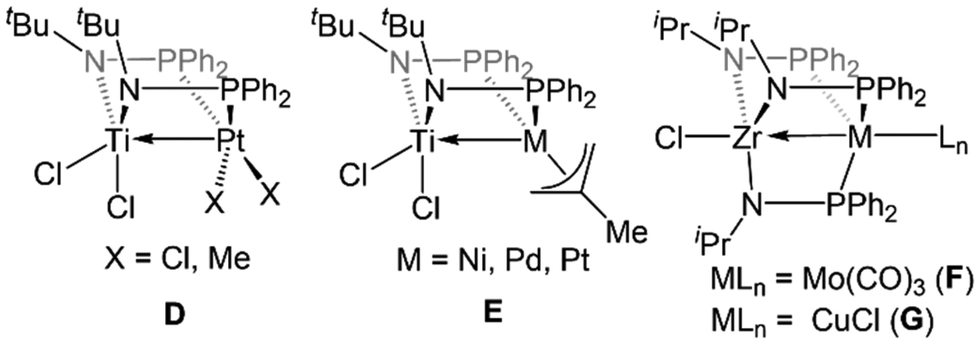 | ||
| Fig. 4 Phosphinoamide-linked heterobimetallic complexes reported by Nagashima and coworkers.97–100 | ||
Drawing inspiration from the precedents set by Nagashima et al., our group set out to synthesize a wider variety of phosphinoamide-supported early/late heterobimetallic complexes and explore their redox properties and reactivity. Using three phosphinoamide ligands to bridge an early and late metal gives the previously discussed C3 symmetry about the bimetallic core. The trigonal ligand field and the proximity of the two metal centers facilitated the construction of early/late heterobimetallic complexes bearing metal–metal multiple bonds.33,71 We have reported a wide variety of tris(phosphinoamide) and bis(phosphinoamide) early/late heterobimetallics using late first-row transition metals, including Cr/M,101 V/M,102–104 Nb/M,105–107 Ti/M,104 and U/Co108 combinations (M = Fe, Co, Ni, Cu). However, the most heavily studied metal–metal combinations to date are the group IV M/Co complexes (M = Ti, Zr, Hf) and that is where we will focus this Perspective. Trends in metal–metal bonding will be described in the context of the unique reactivity enabled by these compounds and their polarized metal–metal multiple bonds and enhanced redox behavior.
Zr/Co (phosphinoamide) complexes
Metal–metal bonding in tris(phosphinoamide) Zr/Co complexes
The Zr/Co pair initially sparked our interest owing to the availability of multiple common oxidation states ranging from CoIII to Co−I for cobalt, combined with the relative inertness of zirconium to redox processes. The tendency of Zr to remain in the d0 ZrIV state was initially posited (and later confirmed)109 to lend simplicity to the assignment of redox processes to one metal or the other while ensuring that the Lewis acidity of the early metal center remains unperturbed by redox events alone. Our foray into tris(phosphinoamide) complexes began in 2009, when our group reported a series of three ZrIV/CoI tris(phosphinoamide) compounds, each distinguished by the substituents on the amide and phosphine donors, ClZr(iPrNPPh2)3CoI (1), ClZr(MesNPiPr2)3CoI (2), and ClZr(iPrNPiPr2)3CoI (3) (Mes = 2,4,6-trimethylphenyl, Fig. 5).110 The metal–metal distances in 1–3 range from 2.73 Å (1) to 2.63 Å (2 and 3), corresponding to FSRs of 1.00–1.04, and the metal–metal bonding in these compounds is best described as a dative Co→Zr bond. Cyclic voltammetry experiments demonstrated that the potentials for one and two-electron reduction of the Co(I) center in bimetallic complex 1 decreased by 0.84 and 1.01 V, respectively, as compared to a Co monometallic species with an otherwise identical coordination environment about the Co center, (iPrNHPPh2)3CoI. Even the most electron-donating variant of the ligand set (3) could be reduced at potentials more than 0.5 V more positive than the aforementioned monometallic Co species. This seminal discovery demonstrated the remarkable impact of a Lewis acidic early metal ion in facilitating redox processes at an appended late metal center, demonstrating the utility of early/late heterobimetallic frameworks for accessing highly reduced and reactive species.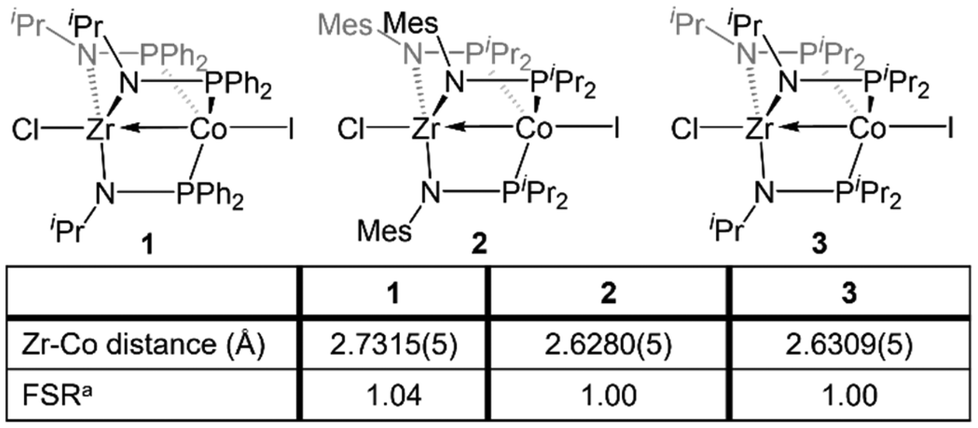 | ||
| Fig. 5 Tris(phosphinoamide) linked ZrIV/CoI compounds featuring dative Co→Zr bonds. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. | ||
Complexes 1–3 readily undergo two-electron reduction, affording dinitrogen-bound ZrIV/Co−I species when reduced under a dinitrogen atmosphere.110,111 The PPh2-substituted complex 1 is reduced to afford an anionic complex [{ClZr(iPrNPPh2)3CoN2}2Na(THF)4][Na(THF)6] (4) that retains the Zr-bound halide (Fig. 6). Although high quality single crystal X-ray diffraction data could not be obtained for 4, its formulation and connectivity were inferred by comparison of its spectroscopic features to a Hf/Co analogue (vide infra).112 In contrast, the two PiPr2-substituted compounds 2 and 3 generate zwitterionic species (THF)Zr(MesNPiPr2)3CoN2 (5) and Zr(iPrNPiPr2)3CoN2 (6) via loss of two equivalents of sodium halide salt (Fig. 6).111 Reduction products 5 and 6 feature Zr–Co distances that are dramatically shortened upon reduction, with FSRs of 0.90 and 0.89, respectively. Complex 2 can be reduced in the absence of dinitrogen to afford the N2-free complex (THF)Zr(MesNPiPr2)3Co (7), which has an even shorter Zr–Co distance (2.1432(13) Å, FSR = 0.82).111
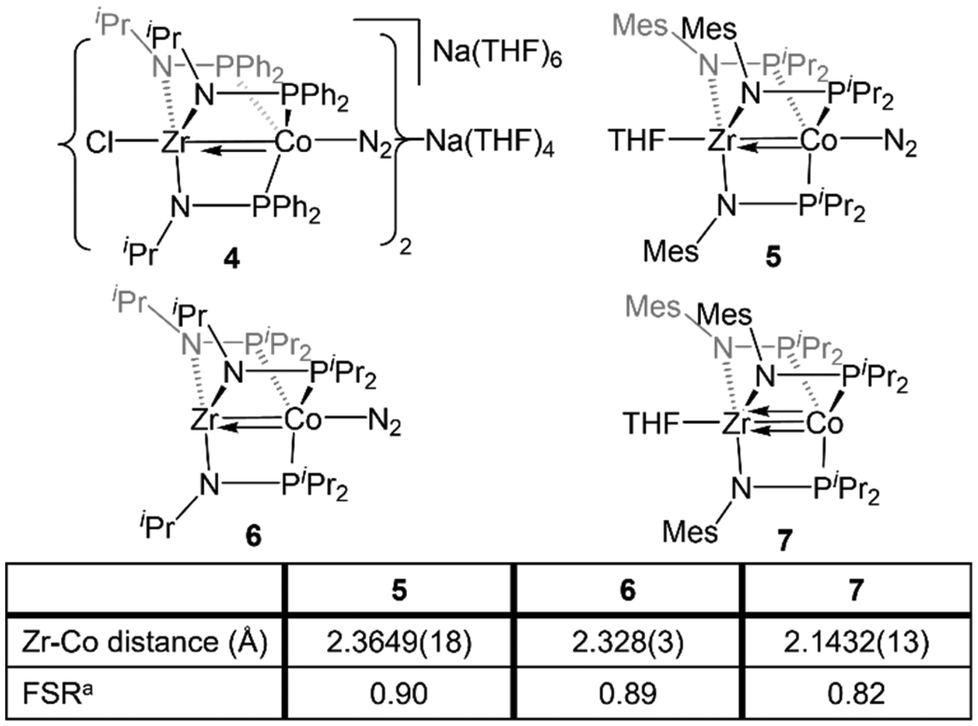 | ||
| Fig. 6 Tris(phosphinoamide) linked ZrIV/Co−I compounds featuring metal–metal multiple bonds. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. | ||
A computational analysis of the frontier molecular orbitals of 5–7 revealed that the decrease in metal–metal distance results from an increase in Zr–Co bond order upon reduction. A representative frontier molecular orbital diagram of 7 is shown in Fig. 7A, revealing a metal–metal triple bond comprised of one σ bonding orbital and two π bonding orbitals. All three metal–metal bonding orbitals are polarized, with significantly more orbital contribution from the Co center. The two highest occupied molecular orbitals are Co-centered dxy and dx2−y2 orbitals of δ-symmetry. Although the formation of metal–metal δ bonds is symmetry-allowed, δ-bonding is inherently weaker as a result of poorer orbital overlap and, due to the energy differences between the Zr and Co atomic orbitals, δ bonding is negligible in these heterobimetallic systems. The net result is a (σ)2(π)4(Conb)4 electronic configuration and a metal–metal triple bond in 7 (Fig. 7A).33,109,111,113 Although the dxz and dyz orbitals in ZrIV/CoI complex 2 were also of the correct symmetry to form metal–metal π bonds, such bonding interactions only form upon reduction of the cobalt center to Co−I, as this raises the energy of the cobalt-centered orbitals such that overlap with the higher energy dxz and dyz orbitals on Zr becomes possible. Due to the highly reduced nature of the cobalt center in 7 and its C3 symmetry, the dxz and dyz orbitals on cobalt are also of the proper symmetry and energy to bind dinitrogen, generating 5 upon exposure to N2. Examination of the frontier molecular orbitals shows that although the (σ)2(π)4(Conb)4 electronic configuration is retained in 5, significant mixing of the M–M π bonding orbitals with the dinitrogen π* orbitals decreases the metal–metal orbital overlap and results in a bond order closer to two (Fig. 7B).111 As a result of competition between Zr and N2 for the electron density in Co's dxz and dyz orbitals, the apical dinitrogen ligand in 5 is rather labile and samples of 5 revert to 7in vacuo.111 The oxidation states of compounds 5–7 are best described as ZrIV/Co−I and therefore these compounds undergo a myriad of oxidative chemistry,109 facilitated by the open coordination sites or labile ligands on both the Zr and Co apical sites.
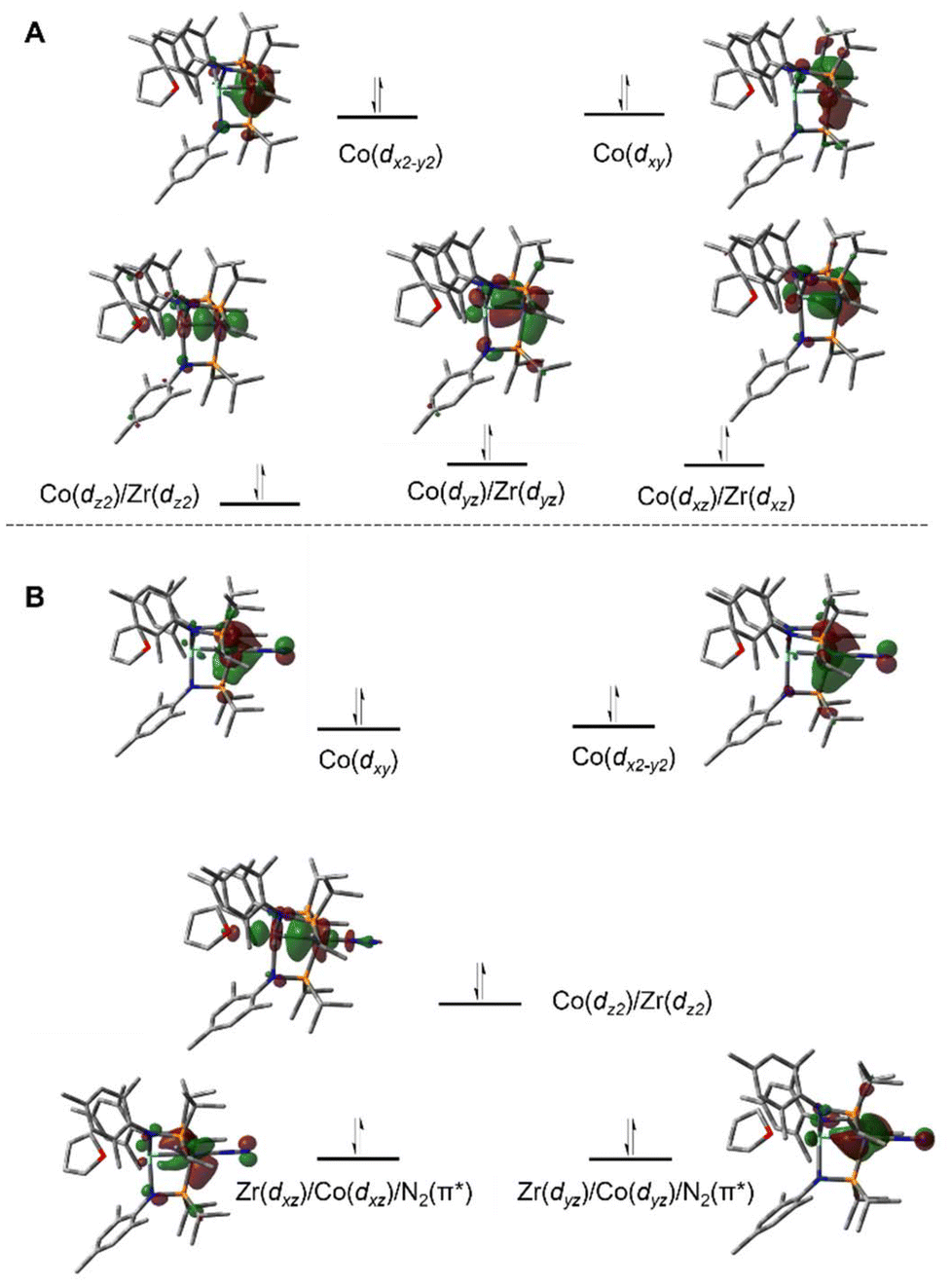 | ||
| Fig. 7 Pictorial representations of the frontier MO diagrams of 7 (A) and 5 (B) derived from DFT calculations (ωB97XD/def2SVP). | ||
Reactivity of tris(phosphinoamide) Zr/Co complexes
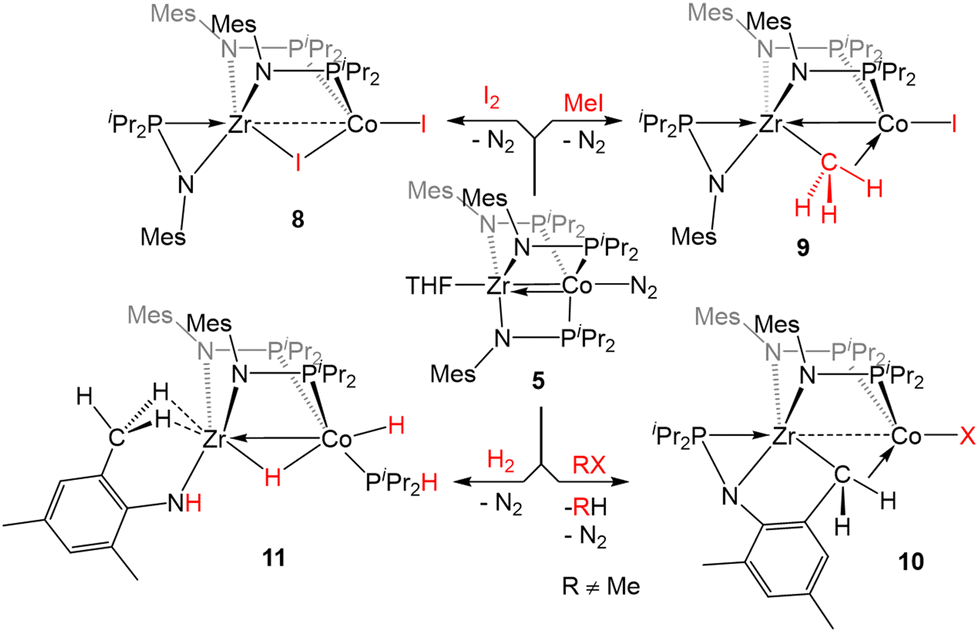 | ||
| Scheme 1 Examples of two-electron oxidative addition reactions of 5. | ||
The propensity of reduced tris(phosphinoamide) ZrIV/Co−I species for C–X bond activation was harnessed for catalytic Kumada coupling reactions of alkyl halides with alkyl Grignard reagents.116 Through computational mechanistic studies, it was found that the extremely reducing nature of the [ZrCo]3+ bimetallic core enables the catalysis, as the lowest energy pathway begins with a net oxidative addition achieved by a one-electron reduction of the alkyl halide followed by a radical recombination event with the resulting open-shell ZrIV/Co0 intermediate.117 The ZrIV/CoI oxidative addition product then undergoes transmetallation with the alkyl Grignard reagent, a phosphine dissociation event, and subsequent reductive elimination to form a C–C bond and regenerate the ZrIV/Co−I catalyst.117 The bimetallic nature of the Zr/Co system was also found to favor reductive elimination via the withdrawal of electron density from Co by the neighboring Lewis acidic ZrIV center. Neither the oxidative addition, nor the reductive elimination were found to be energetically accessible with a cobalt monometallic analogue.117
Complex 5 was also found to readily oxidatively add the C–H bonds of terminal alkynes. When exposed to two equivalents of TMSC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH or PhCCH at low temperatures, an adduct of the E-isomer of the dimerized enyne was formed (12, Scheme 2).118 Although TMSCCH addition to 5 led to exclusive formation of the E-isomer, even at room temperature, all three potential enyne isomers (E, Z, and gem) were generated when the addition of PhCCH to 5 was carried out at room temperature. It was hypothesized, and later determined computationally,119 that this reaction takes place via an initial oxidative addition of the terminal C–H bond of the alkyne, followed by insertion of a second equivalent of alkyne into the resulting cobalt hydride bond, and then reductive elimination to arrive at the enyne adduct. This mechanism was supported by trapping a C–H activated bridging acetylide intermediate, (η2-iPr2PNMes)Zr(μ-MesNPiPr2)2(μ-CCTMS)Co(H)(CNtBu) (13), via the addition of tBuNC to the mixture of TMSCCH to 5.118
CH or PhCCH at low temperatures, an adduct of the E-isomer of the dimerized enyne was formed (12, Scheme 2).118 Although TMSCCH addition to 5 led to exclusive formation of the E-isomer, even at room temperature, all three potential enyne isomers (E, Z, and gem) were generated when the addition of PhCCH to 5 was carried out at room temperature. It was hypothesized, and later determined computationally,119 that this reaction takes place via an initial oxidative addition of the terminal C–H bond of the alkyne, followed by insertion of a second equivalent of alkyne into the resulting cobalt hydride bond, and then reductive elimination to arrive at the enyne adduct. This mechanism was supported by trapping a C–H activated bridging acetylide intermediate, (η2-iPr2PNMes)Zr(μ-MesNPiPr2)2(μ-CCTMS)Co(H)(CNtBu) (13), via the addition of tBuNC to the mixture of TMSCCH to 5.118
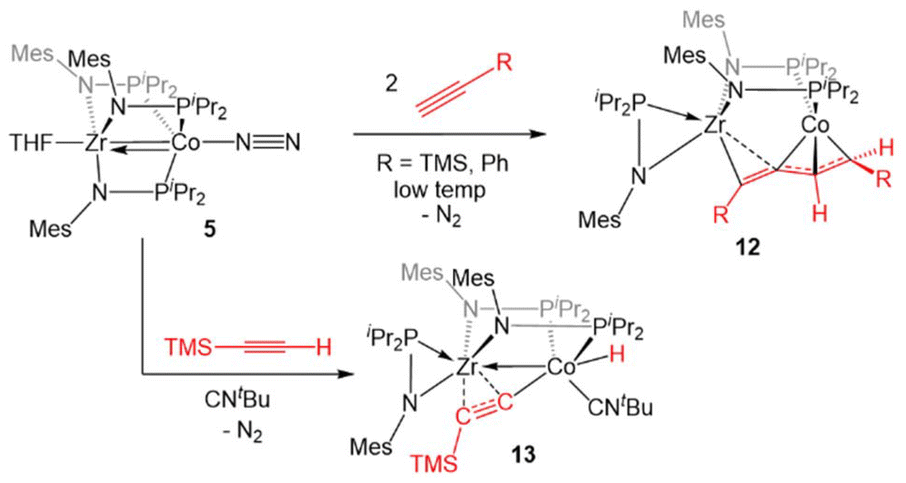 | ||
| Scheme 2 Reactions of terminal alkynes with 5. | ||
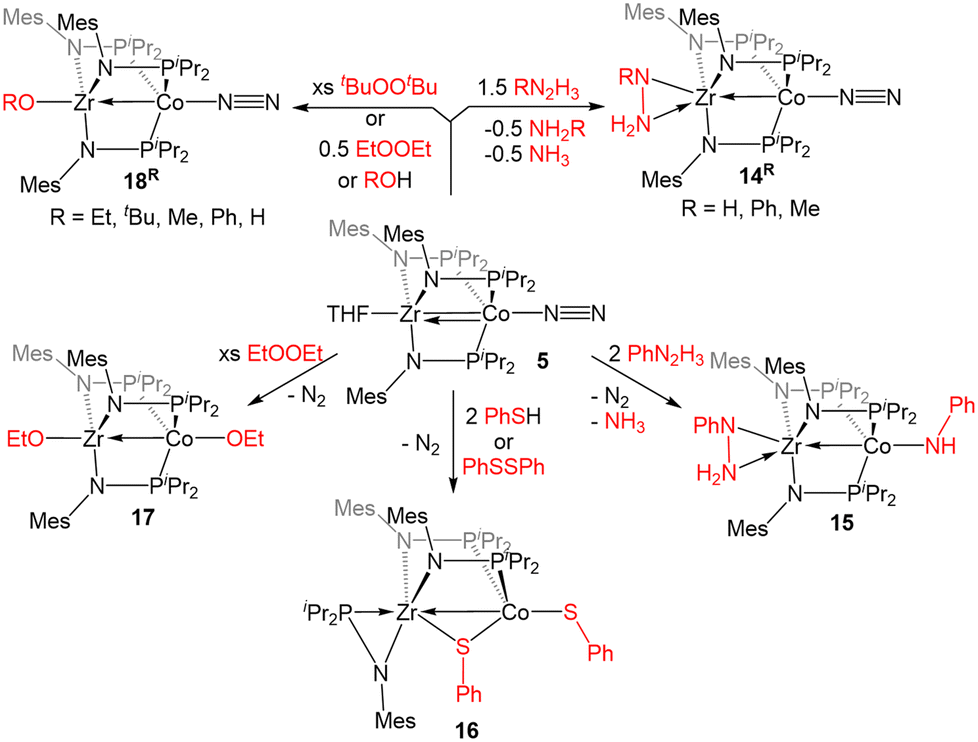 | ||
| Scheme 3 Examples of E–E and E–H bond activation promoted by 5 (E = N, O, S). | ||
The reactivity of 5 with sulfur-containing substrates was also investigated (Scheme 3). The reaction between 5 and two equivalents of thiophenol or one equivalent of diphenyl disulfide yielded ZrIV/CoI product (η2-MesNPiPr2)Zr(μ-MesNPiPr2)2(μ-SPh)Co(SPh) (16).121 A similar oxidative process occurred with excess diethyl peroxide but with the conservation of the C3 symmetry of the complex, generating (EtO)Zr(MesNPiPr2)3Co(OEt) (17). By using 0.5 equivalents of the peroxide, or by using the bulkier di-tert-butyl peroxide, a single alkoxy group was added at Zr, resulting in a ZrIV/Co0 complex, (RO)Zr(MesNPiPr2)3Co(N2) (R = Et (18Et), tBu (18tBu)). Therefore, peroxide O–O bond cleavage proceeds through a one-electron process, whereas S–S and S–H bond activation processes appear to proceed through concerted two-electron redox processes. One-electron oxidation products analogous to 18R were also obtained through the addition of alcohols or H2O to 5.121
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E double bonds (E = N, O, S).
E double bonds (E = N, O, S).
Our first foray into CE bond activation was the discovery that the ZrIV/Co−I heterobimetallic complex 7 readily cleaved the CO bond of carbon dioxide (Scheme 4). Exposure of 7 to one equivalent of CO2 at low temperature resulted in oxidative addition of a CO double bond across the Zr–Co bond with concomittant dissociation of one of the phosphinoamide ligands from Co, generating (η2-iPr2PNMes)Zr(μ-MesNPiPr2)2(μ-O)Co(CO) (19).122 Cyclic voltammetry of 19 revealed a reversible redox event at −1.85 V (vs. Fc/Fc+) and subsequent reduction of 19 with 1 equiv. Na/Hg restored the tris(phosphinoamide) core and afforded the Co0/ZrIV-oxo anion [(THF)3Na][(O)Zr(MesNPiPr2)3Co(CO)] (20).122 Using excess Na/Hg to reduce 19 in the presence of an additional equivalent of CO2 gave a mixture of products, including the carbonate complex [(THF)2Na]2[(CO3)Zr(MesNPiPr2)3Co(CO)] (21).122 Lastly, reaction of 19 with PhSiH3 resulted in the formation of (PhH2SiO)Zr(MesNPiPr2)3Co(H)(CO) (22) via cleavage of an Si–H bond across the Co–O bond and reassociation of the third phosphinoamide ligand with Co (Scheme 4).122 Subsequent studies further explored the fundamental reaction steps involved in CO2 reduction and carbonate formation using heterobimetallic Zr/Co systems, but realization of a catalytic cycle for CO2 reduction/functionalization was hindered by the harsh conditions required to cleave Zr–O bonds once formed.123
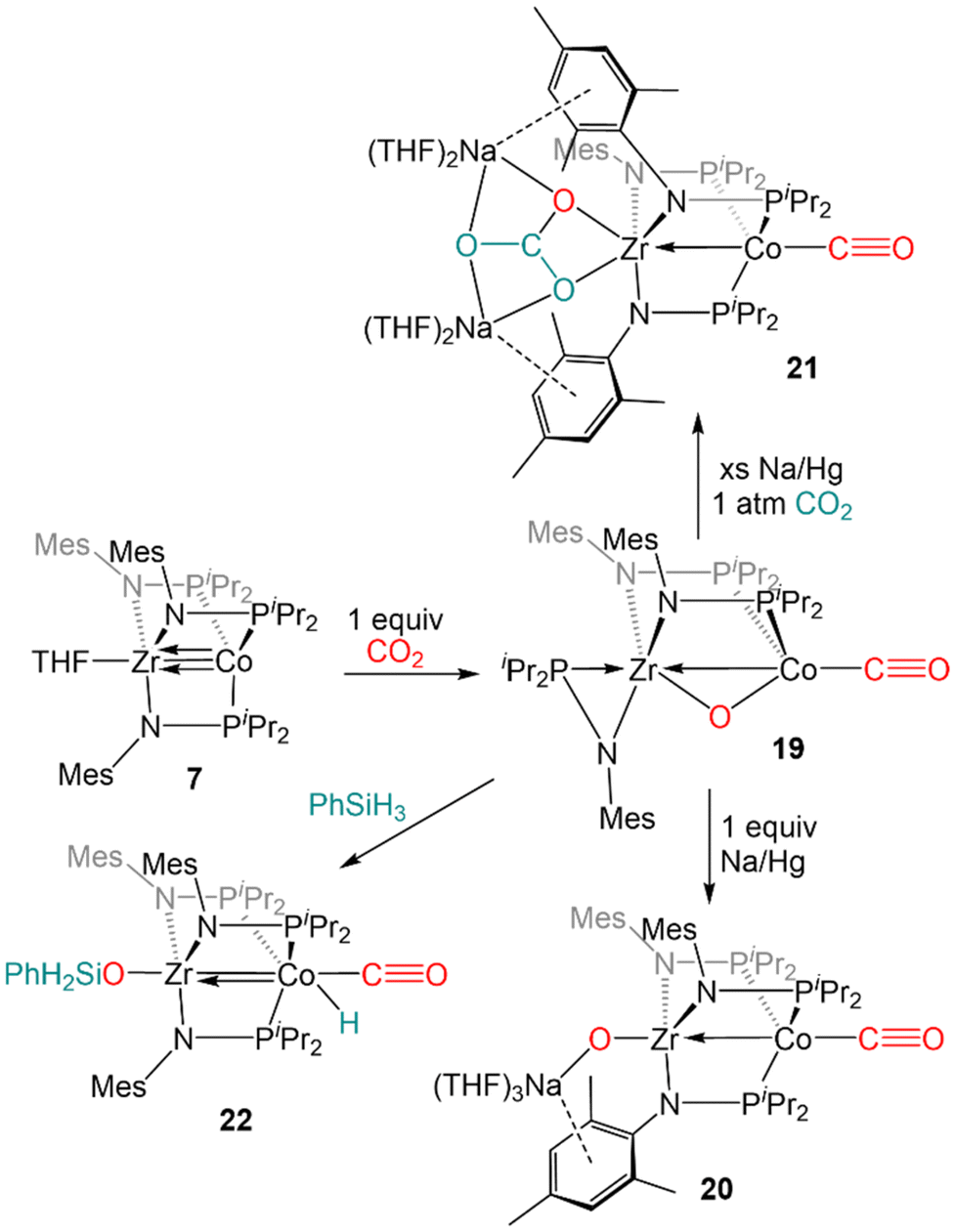 | ||
| Scheme 4 CO2 reduction reactions promoted by 7. | ||
Inspired by the cleavage of CO bonds in CO2, the reactivity of 5 with diaryl ketones was investigated (Scheme 5). Benzophenone and its derivatives react with 5 at room temperature to give ZrIV/Co0 complexes with a ketyl radical bound at Zr via single-electron transfer from Co to the Zr-bound ketone.124–126 In solution, the benzophenone product (Ph2C˙O)Zr(MesNPiPr2)3CoN2 (23) was found to be in equilibrium with an isobenzopinacol-coupled tetrametallic product 24, in which dimerization has occurred through the para position of the aryl ring of one Zr/Co complex and the ketyl carbon of a second equivalent of 23.124,127 In the case of para-methyl benzophenone or fluorenone derivatives, dimerization of the Zr-bound ketyl radical does not occur and the monomeric ketyl radical species can be isolated and structurally characterized. However, placing an electron-donating dimethylamino group at the para position results in no electron-transfer or activation of the CO double bond, with formation of a ZrIV/Co−I ketone adduct instead.125,126 Ketyl radical species such as 23 were proposed as key intermediates in the catalytic hydrosilylation of ketones performed by 5.124 Upon heating the equilibrium mixture of 23 and 24, the ketone CO double bond was cleaved in a similar fashion to CO2 to afford the μ-oxo/carbene complex (η2-MesNPiPr2)Zr(μ-MesNPiPr2)2(μ-O)CoCPh2 (25) in 80% yield, with concomitant formation of a byproduct resulting from cleavage of the phosphinoamide P–N bond.128
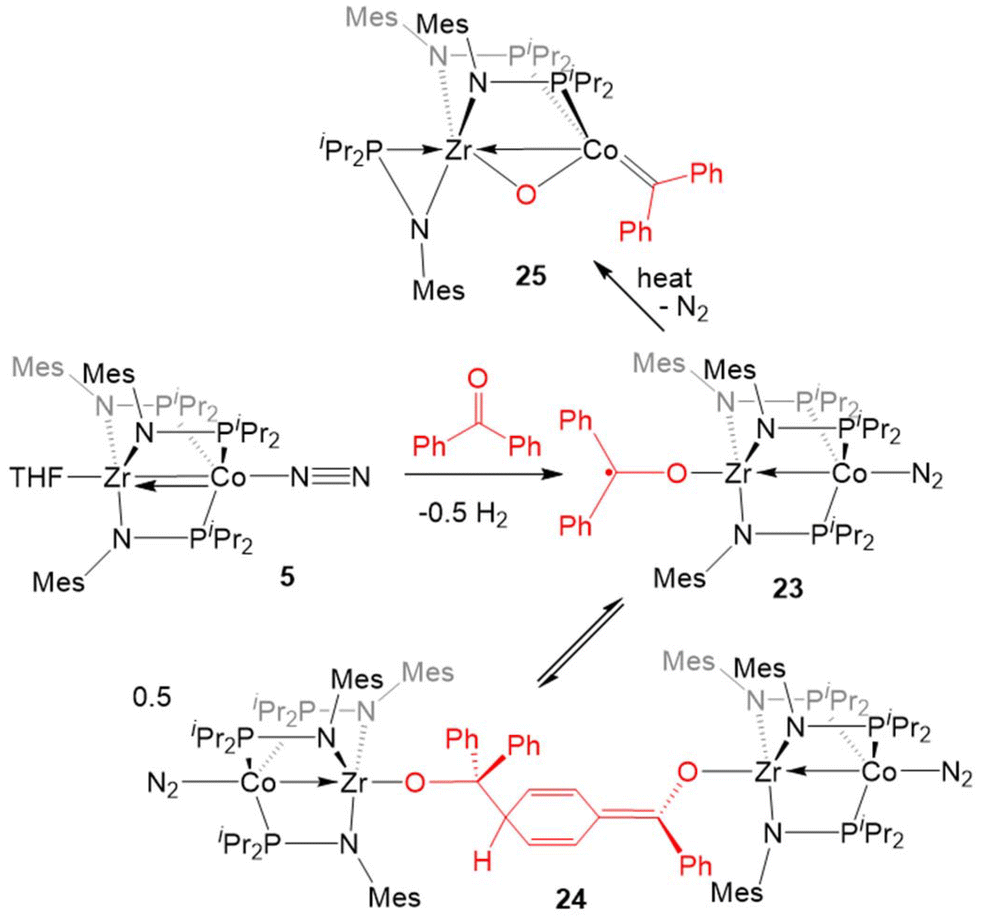 | ||
| Scheme 5 Reactions of 5 with ketones. | ||
An analogous reaction with thiobenzophenone, however, led to the partial activation of the CS bond to generate a product with a μ-κ1η2 binding mode of the thione (26, Scheme 6).125 The elongation of the C–S bond and the pyramidalization of the thioketone carbon in 26 provide evidence for the activation of the substrate upon binding across the two metal centers, but a short Zr–Co bond distance (2.3857(2) Å; FSR = 0.91) and the diamagnetic nature of the compound suggest that 26 is best described as a ZrIV/Co−I adduct of thiobenzophenone.
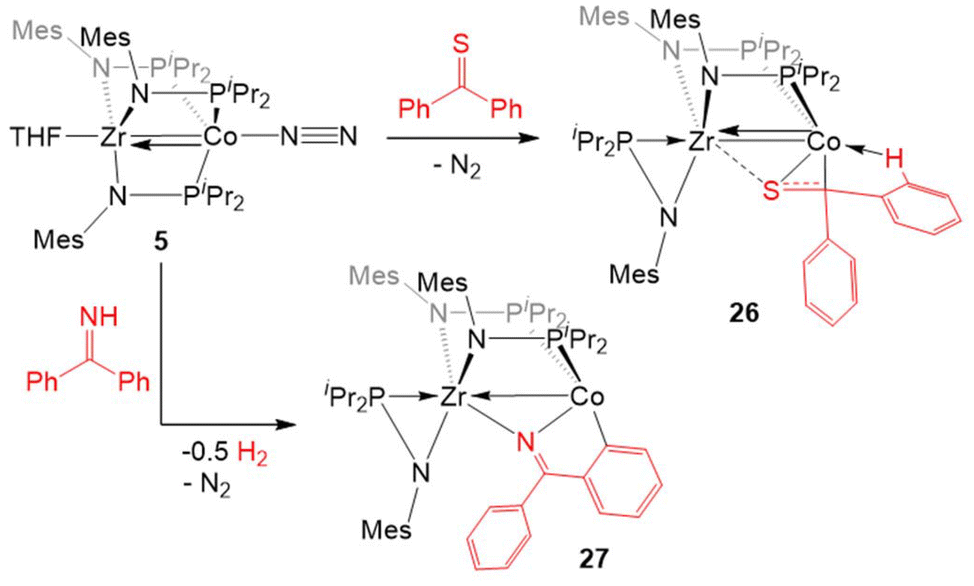 | ||
| Scheme 6 Representative reactions of 5 with diaryl thioketones and imines. | ||
The reaction of 5 with benzophenone imine gave an entirely different result. Rather than cleaving the CN bond, both N–H and C–H activation occurred to generate a product in which the deprotonated imine is bridging Zr and Co and a 5-membered ring is formed through ortho-C–H activation of one of the imine-derived phenyl rings (27, Scheme 6).125 The proposed reaction pathway includes the oxidative addition of the imine N–H bond to form a putative cobalt hydride, which is then lost as dihydrogen in the subsequent cyclometallation of the imine phenyl substituent.125 In support of such a reaction sequence, addition of fluorenoneimine to 5 generates an isolable hydride species from which cyclometallation is prevented due to the rigidity of the fused arene rings.
Zr(iPrNPiPr2)3CoNTol (28, Tol = p-tolyl) via oxidative nitrene transfer.129 This zwitterionic bimetallic compound features a triply bound imido ligand at each C3-symmetric metal center and no metal–metal interaction (Zr–Co distance = 3.1734(3) Å, FSR = 1.21). Alternatively, addition of the bulkier mesityl azide to 6 produced a diimido species (η2-iPrNPiPr2)Zr(μ-iPrNPiPr2)2(μ-NMes)CoNMes with one of the imido ligands bridging the Zr and Co centers and one of the phosphinoamide ligands dissociated from Co and bound η2 to Zr (29).129 Alkyl azides are notably less reactive than their aryl counterparts,130 and that trend was also observed in their reactivity with the bimetallic system. While dinitrogen was easily extruded from aryl azides, tert-butyl azide reacted with 6 to form (η2-iPrNPiPr2)Zr(μ-iPrNPiPr2)2(μ2-κ1,η3-NtBu)Co (30).129
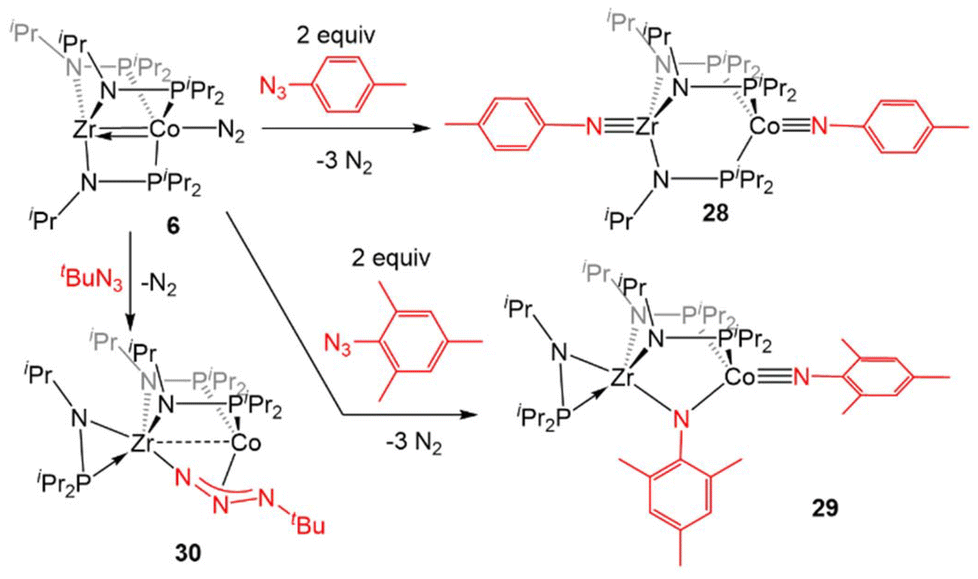 | ||
| Scheme 7 Reactions between 6 and organic azides. | ||
Complex 5 is also reactive towards oxidative O atom transfer from pyridine N-oxide (Scheme 8). Treatment of 5 with pyridine N-oxide affords the bridging oxo complex, (η2-MesNPiPr2)Zr(μ-MesNPiPr2)2(μ-O)Co(py) (31, py = pyridine), with pyridine displacing dinitrogen at the apical cobalt site and one phosphinoamide ligand dissociated from Co. In a reaction similar to that of the CO2 activation product 19 described above, the bridging oxo ligand in 31 was converted to a Zr-bound triphenylsiloxide ligand upon addition of Ph3SiH, affording (Ph3SiO)Zr(MesNPiPr2)3Co(H)(N2) (32).131 In contrast to 22, the lability of the N2 ligand on 32 permitted insertion of unsaturated substrates such as PhCCH, PhCN, and CO2 into the Co–H bond (e.g.33, Scheme 8).131
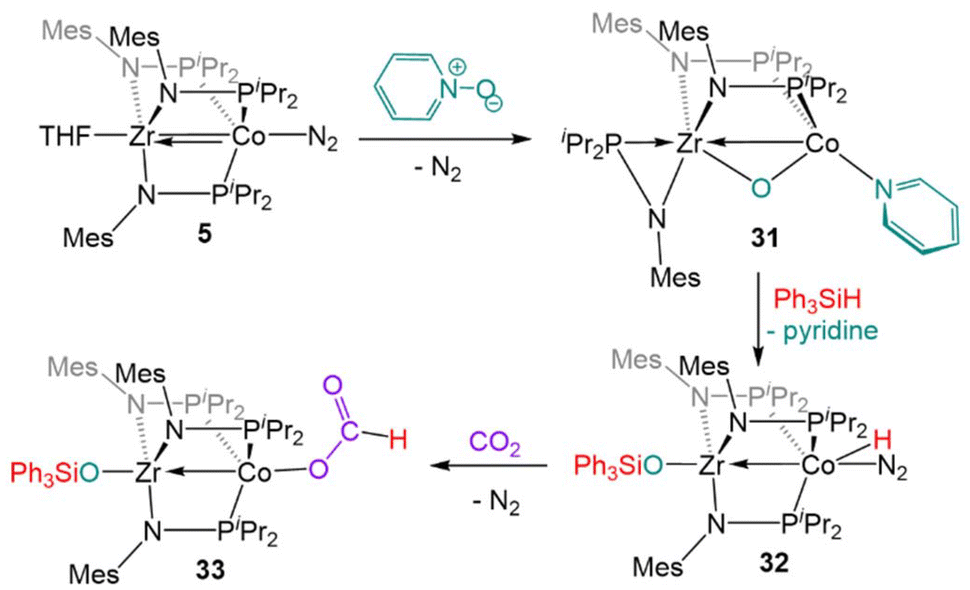 | ||
| Scheme 8 The two-electron oxidation of 5 with pyridine-N-oxide, subsequent reaction with silane to generate a ZrIV/CoI hydride complex, and a representative insertion reaction with CO2. | ||
Most of the examples of the reactivity of 5–7 provided so far involve the dissociation of the dinitrogen ligand to allow for reactivity at the Co center. Replacement of the labile dinitrogen ligand with tBuNC, which is sterically bulky and binds more tightly than N2, gives the ZrIV/Co−I complex (THF)Zr(MesNPiPr2)3Co(CNtBu) (34), where the coordination sphere about Co remains saturated, forcing substrate coordination to occur at Zr (Scheme 9). Although ZrIV is d0 and redox inert, the appended Co−I center allows redox processes to occur. In this case, it is useful to consider complexes such as 34 as ZrIV complexes with a redox-active Co−I containing metalloligand.132
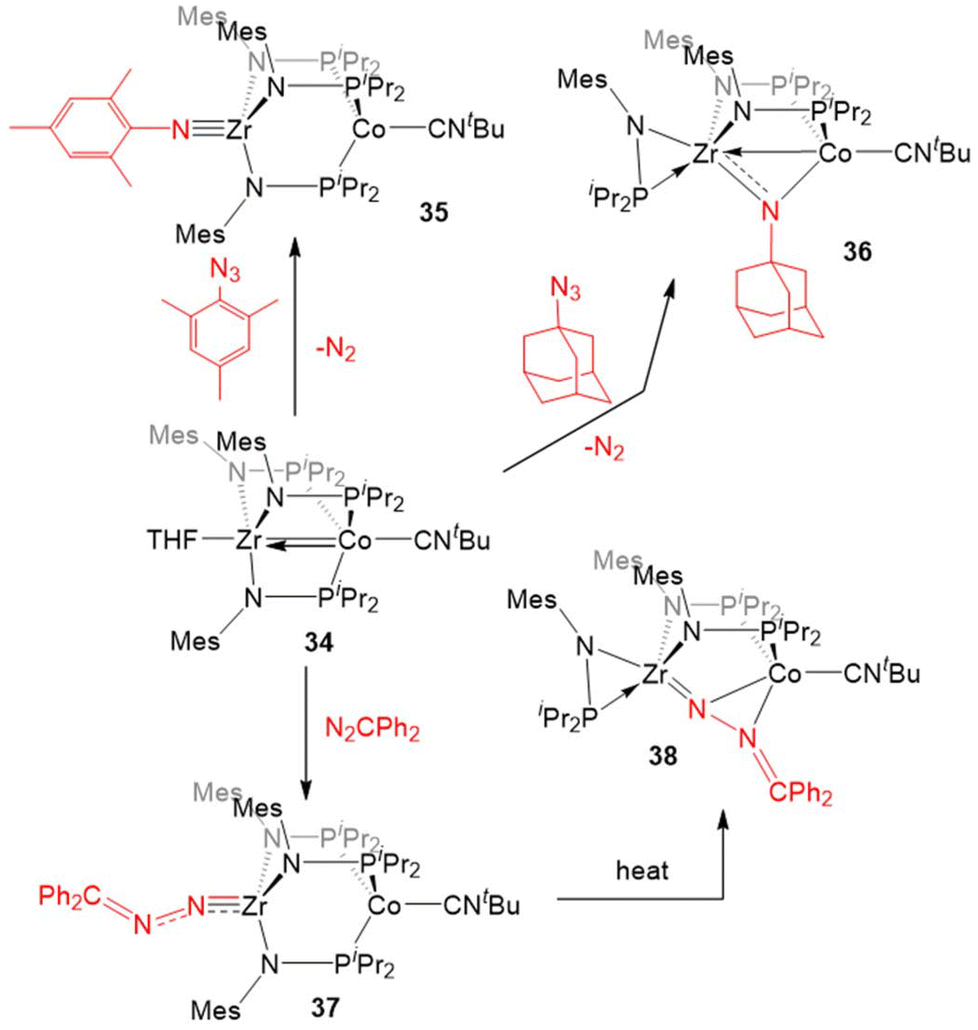 | ||
| Scheme 9 Examples of two-electron reactions promoted by 34. | ||
The “redox-active metalloligand” approach is best exemplified in reactions between 34 and oxidative group transfer reagents. As shown in Scheme 9, the addition of mesityl azide to 34, resulted in the formation of the terminal Zr-imido complex MesNZr(MesNPiPr2)3Co(CNtBu) (35). The bulkier adamantyl azide instead formed the bridging imido complex (η2-iPr2PNMes)Zr(μ-MesNPiPr2)2(μ-NMes)Co(CNtBu) (36). In both of these cases, two-electron processes were observed, which differs from the previously discussed reactions of organic azides with 6 (Scheme 7, vide supra), and shows the ability of tert-butyl isocyanide to partially “block” the late metal site.129,132 Diphenyl diazomethane reacts readily with 34 to form a ZrIV/CoI alkylidene hydrazido complex (Ph2CN2)Zr(MesNPiPr2)3Co(CNtBu) (37), wherein oxidation of the diazo reagent and formation of a Zr–N multiple bond results in cleavage of the metal–metal bond. Heating of 37 in an effort to promote N2 release resulted in isomerization to (η2-iPr2PNMes)Zr(μ-MesNPiPr2)2(μ-N2CPh2)Co(CNtBu) (38).132
Due to the oxophilic nature of the ZrIV center, the reducing nature of the Co−I center, and the stability of the complex imparted by both the bridging phosphinoamide ligands and the appended isocyanide moiety, 34 was uniquely suited to participate in reactions with O2 and O-atom donors (Scheme 10). Exposure of 34 to excess dry O2 resulted in a cobalt-centered two-electron oxidation and binding of the resulting η2-O22− ligand at Zr to generate the ZrIV/CoI compound (η2-O2)Zr(MesNPiPr2)3Co(CNtBu) (39).133
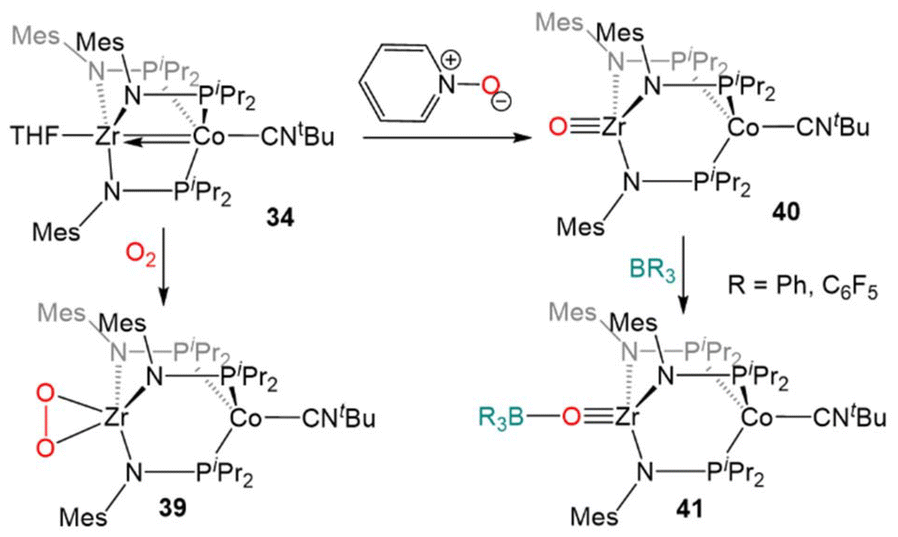 | ||
| Scheme 10 Two-electron oxidation of 34 using O2 or pyridine-N-oxide and subsequent nucleophilic reactivity of terminal Zr oxo complex 40. | ||
In contrast to the reaction of 5 with pyridine-N-oxide, the addition of pyridine-N-oxide to 34 afforded the terminal Zr oxo complex OZr(MesNPiPr2)3Co(CNtBu) (40).133 The isolation of 40 is notable as only one other neutral terminal Zr oxo compound had been previously isolated.134 The nucleophilicity of the oxo moiety in 40 was confirmed by its ability to readily bind Lewis acidic boranes to generate (R3BO)Zr(MesNPiPr2)3Co(CNtBu) (41, Scheme 10).133
The combination of the nucleophilicity of the Zr-oxo fragment and the redox activity of the appended CoI center opens new avenues for bimetallic cooperativity and the redox properties and further reactivity of 40 were, therefore, investigated (Scheme 11). The ZrIV/CoI oxo complex displays a reversible one-electron reduction at −1.71 V in its cyclic voltammogram, and could be reduced by the addition of Na/Hg amalgam to give the ZrIV/Co0 tetrametallic dimer Na2[OZr(MesNPiPr2)3Co(CNtBu)]2 (42).135 Another ZrIV/Co0 complex (Ph3CO)Zr(MesNPiPr2)3Co(CNtBu) (43) could be generated by the treatment of 40 with Gomberg's dimer, a source of the trityl radical.135,136 However, the trityl radical alone is not sufficiently reducing to generate the Co0 center present in 43, so its formation was hypothesized to be driven by the affinity of the Zr oxo for the trityl cation. Treatment of 40 with Ph3CCl in the presence of NaBPh4 supported this hypothesis, as the cationic complex [(Ph3CO)Zr(MesNPiPr2)3Co(CNtBu)][BPh4] (44) was generated.135 These reactions serve to highlight a unique example of bimetallic cooperativity in which one transition metal serves as a redox-active electron reservoir while new bonds are formed with a nucleophilic ligand bound to a second redox-inert metal.
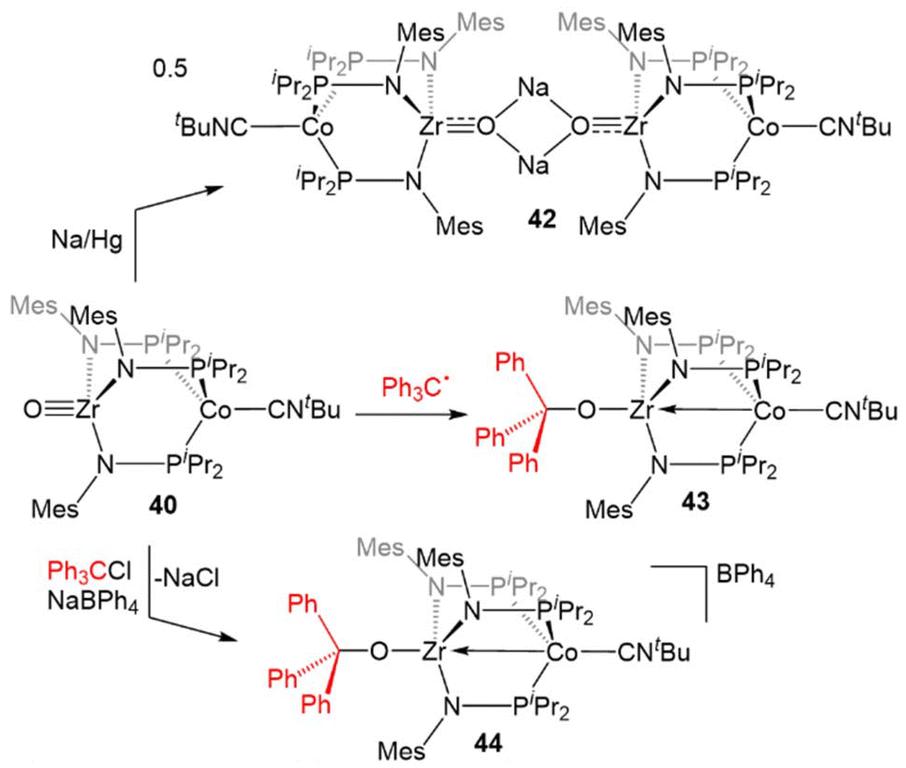 | ||
| Scheme 11 Reactivity of the Zr-oxo complex 40. | ||
Ti/Co and Hf/Co tris(phosphinoamide) complexes
Ti/Co tris(phosphinoamide) complexes
Naturally, there are multiple similarities and subtle differences between the Zr-containing heterobimetallic compounds and their Ti/Co congeners. Most notably, due to the 0.13 Å decrease in atomic radius while moving up group IV to Ti,28 three mesityl-substituted phosphinoamide ligands cannot be coordinated to the metal center. As such, all comparative studies with Ti/Co tris(phosphinoamide) complexes were carried out using the smaller [XylNPiPr2]− ligand (Xyl = 3,5-dimethylphenyl), necessitating the synthesis of a direct Zr/Co analogue, (THF)Zr(XylNPiPr2)3CoN2 (45) (Fig. 8).137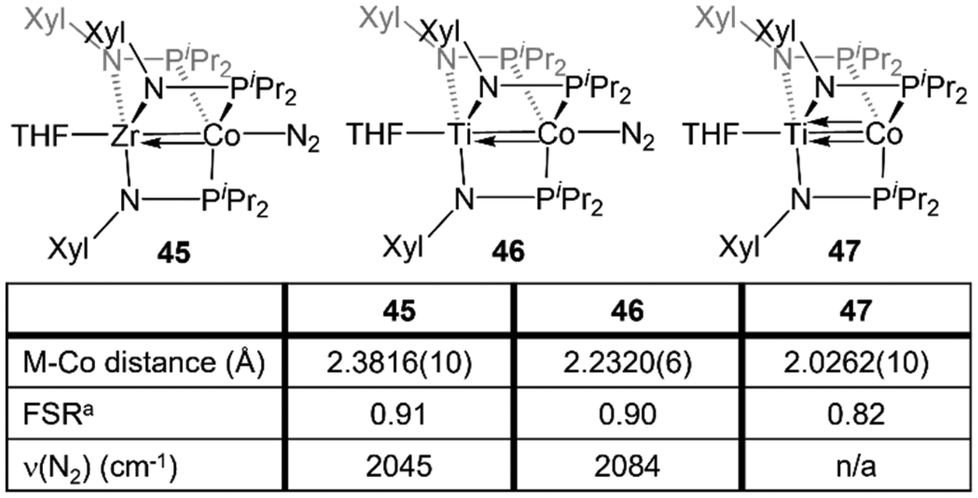 | ||
| Fig. 8 A comparison of the metal–metal bond metrics and ν(N2) stretching frequencies of ZrIV/Co−I and TiIV/Co−I complexes. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. | ||
The TiIV/Co−I complex (THF)Ti(XylNPiPr2)3Co(N2) (46) can be synthesized via a stepwise metalation/reduction strategy similar to that previously described for the Zr/Co analogue 5.138 Exposure to vacuum or preparation in the absence of N2 affords (THF)Ti(XylNPiPr2)3Co (47), a direct analogue of 7. Similar to its heavier congener,111 the Ti–Co bond in 47 consists of one σ and two π bonding orbitals with two Co-centered non-bonding orbitals of δ symmetry. N2 binding in 46 results in elongation of the metal–metal bond by ∼0.21 Å, indicative of a decrease in metal–metal bond order.138 The metal–metal distances in 46 and 47 are shorter than the corresponding distances in 45 and 7 but, owing to the smaller size of Ti, the FSRs are nearly identical (Fig. 8). Through computational studies, the Ti–Co multiple bonds in 46 and 47 were found to be more covalent than the Zr–Co multiple bonds in 5 and 7, as expected based on better overlap between the 3d orbitals of Ti and Co compared to the 4d/3d overlap in the Zr/Co congeners. This subtle difference in metal–metal bonding manifests in a large difference in the degree of backbonding to N2 in 45 and 46. By analyzing the N2 stretching frequencies by IR spectroscopy, the Ti/Co complex 46 was found to have a markedly less activated N2 moiety in comparison to its Zr analogue 45 (2084 vs. 2045 cm−1, respectively), indicating that Ti–Co bonding withdraws significantly more electron density from the Co center.
The reactivity of 46 was investigated, with the goal of comparing its reactivity patterns with those of 5 (Scheme 12). In this pursuit, 46 and 47 were found to readily react with diaryl ketones to form adducts at either Ti or Co.126 Addition of one equivalent of benzophenone to 46 affords the Ti-bound ketone adduct (Ph2CO)Ti(XylNPiPr2)3Co(N2) (48). In solution, 48 was found to be in equilibrium with (η2-XylNPiPr2)Zr(μ-XylNPiPr2)2Co(η2-OCPh2) (49), in which N2 and one of the phosphinoamide ligands has dissociated from Co allowing benzophenone to bind to Co in an η2 fashion. Product 49 was formed exclusively when the benzophenone addition reaction was performed under an argon atmosphere. Unlike the reaction of 5 with benzophenone, no evidence for ketyl radical formation was observed and thermolysis did not afford an oxo/carbene complex.128
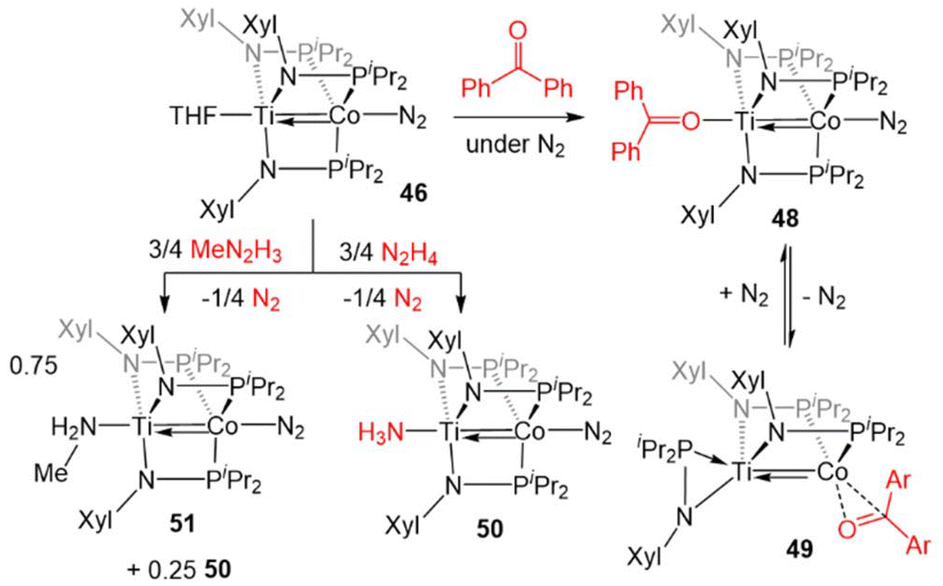 | ||
| Scheme 12 Examples of the divergent reactivity of TiIV/Co−I compound 46 compared to ZrIV/Co−I compound 5. | ||
In another example of reactivity that diverges between Zr/Co and Ti/Co combinations, 46 was treated with both hydrazine and methyl hydrazine. While hydrazine was found to undergo one-electron reduction and spontaneous liberation of H2 upon reaction with ZrIV/Co−I complex 5 (vide supra, Scheme 3),120 the reaction between 46 and hydrazine generates a diamagnetic TiIV/Co−I ammonia complex (H3N)Ti(XylNPiPr2)3Co(N2) (50).138 With methyl hydrazine, 50 and the analogous methylamine complex (51) are generated in a 1:3 ratio (Scheme 12). In both cases, the reaction is balanced via the formation of N2. Exposure of 46 to a 50-fold excess of hydrazine resulted in catalytic disproportionation of hydrazine into ammonia and dinitrogen with a maximum of 18 turnovers in 30 minutes.138 In an attempt to generate a model complex that may give information about the catalytically relevant intermediates in N–N bond cleavage, azobenzene was added to 46, resulting in the cleavage of the NN double bond, imido insertion into the Co–P bond, formation of a bridging imido ligand, and dimerization of the compound into an N2-bridged, tetrametallic complex (52) (Scheme 13). Product 52 is also generated in the reaction of 46 with 1,2-diphenylhydrazine.138 Although 52 likely represents a model of an off-cycle product, its isolation indicates that both Ti and Co play an essential role in the disproportionation of hydrazine. The differences in reactivity between the Ti/Co compounds and their Zr/Co analogues are likely due to the increased covalency of the Ti–Co bond and the resulting more positive reduction potential of the Ti/Co compounds.126
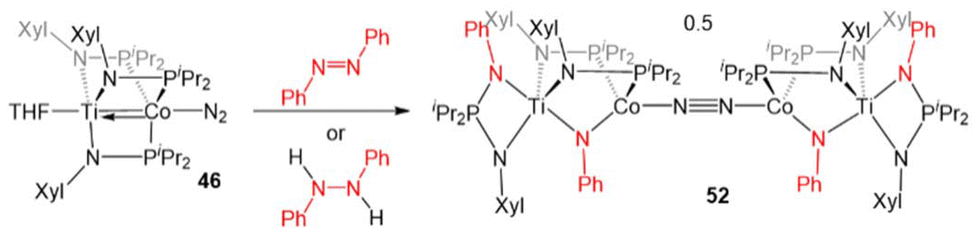 | ||
| Scheme 13 N–N bond cleavage reaction of TiIV/Co−I compound 46. | ||
Hf/Co tris(phosphinoamide) complexes
Despite the nearly identical atomic radii of Zr and Hf and their similar coordination chemistry, tris(phosphinoamide) Hf/Co complexes displayed distinct reactivity from their Zr/Co analogues. A series of HfIV/CoI precursors, ClHf(R′NPR2)3CoI (R′ = iPr, R = Ph (53); R′ = Mes, R = iPr (54); R′ = R = iPr (55)) were synthesized in an identical manner to their Zr analogues (Fig. 9).112 In comparison to the Zr compounds (Fig. 5) the HfIV/CoI complexes 53–55 exhibited slightly elongated M–Co bonds and slightly more negative reduction potentials, with the exception of 55, which exhibits more positive CoI/0 and Co0/−I potentials (Fig. 9). Both the longer M–Co distances and cathodically shifted reduction potentials were ascribed to weaker Co→M dative bonds in the Hf/Co compounds, which is expected based on the larger disparity in orbital energies between a 3d and 5d metal compared to a 3d/4d combination.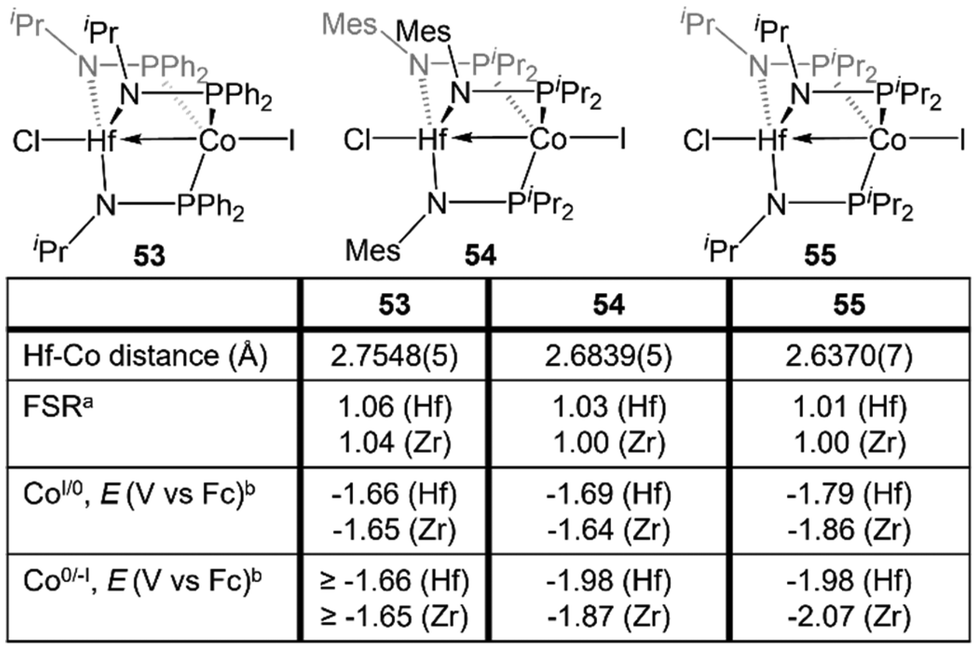 | ||
| Fig. 9 A comparison of the metal–metal bond metrics and redox potentials of tris(phosphinoamide) HfIV/CoI and ZrIV/CoI complexes. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. bThe listed potentials represent either E1/2 or Epc values, depending on the reversibility of the redox events, but reversibility was consistent between Hf and Zr derivatives. | ||
While the differences in the structural and redox properties of otherwise analogous Zr/Co and Hf/Co complexes were subtle, more tangible differences were observed in the reduced MIV/Co−I species (Fig. 10). Reduction of 53 and 55 results in the formation of HfIV/Co−I dinitrogen complexes [{ClHf(iPrNPPh2)3CoN2}2Na(THF)4][Na(THF)6] (56) and Hf(iPrNPiPr2)3Co(N2) (57),112 which adopt structures analogous to their ZrIV/Co−I congeners.111 Upon reduction of 54, a dinitrogen complex is generated, but the halide ligand remains bound at the Hf site to afford (THF)5Na–X–Hf(MesNPiPr2)3Co(N2) (58).112 Unlike a similar intermediate generated in the synthesis of 5,110 dissolution in benzene does not liberate the sodium halide. The short Hf–Co distances in 56 (2.5470(3) Å, FSR = 0.98) and 58 (2.455(5) Å, FSR = 0.94) are indicative of metal–metal bonds, and their FSRs are greater than that in the Zr complex (THF)5Na–X–Zr(MesNPiPr2)3Co(N2) (59, FSR = 0.92).110,112 A way to indirectly measure the electron density at the Co center in these compounds is by examination of the IR stretching frequencies of the apically coordinated dinitrogen ligand. The N–N stretches in 56 (2010 cm−1), 58 (1992 cm−1), and 57 (2046 cm−1) are consistently lower than those in the closely related Zr complexes 4 (2015 cm−1), 59 (2023 cm−1), and 6 (2056 cm−1).110–112 The relatively long intermetallic distances in 56 and 58 suggests that the metal–metal interactions in the Hf/Co complexes are weaker than those in their Zr/Co analogues, likely resulting from the decreased bond covalency owing to the energetic mismatch between the Hf and Co d orbitals. This results in a more electron-rich cobalt center in the Hf/Co complexes, which is exemplified by the increased backbonding to the apical dinitrogen ligands in 56–58 compared to their Zr/Co analogues. Decreased Co→Hf donation renders the early metal center more Lewis acidic, accounting for the decreased halide lability in 58.
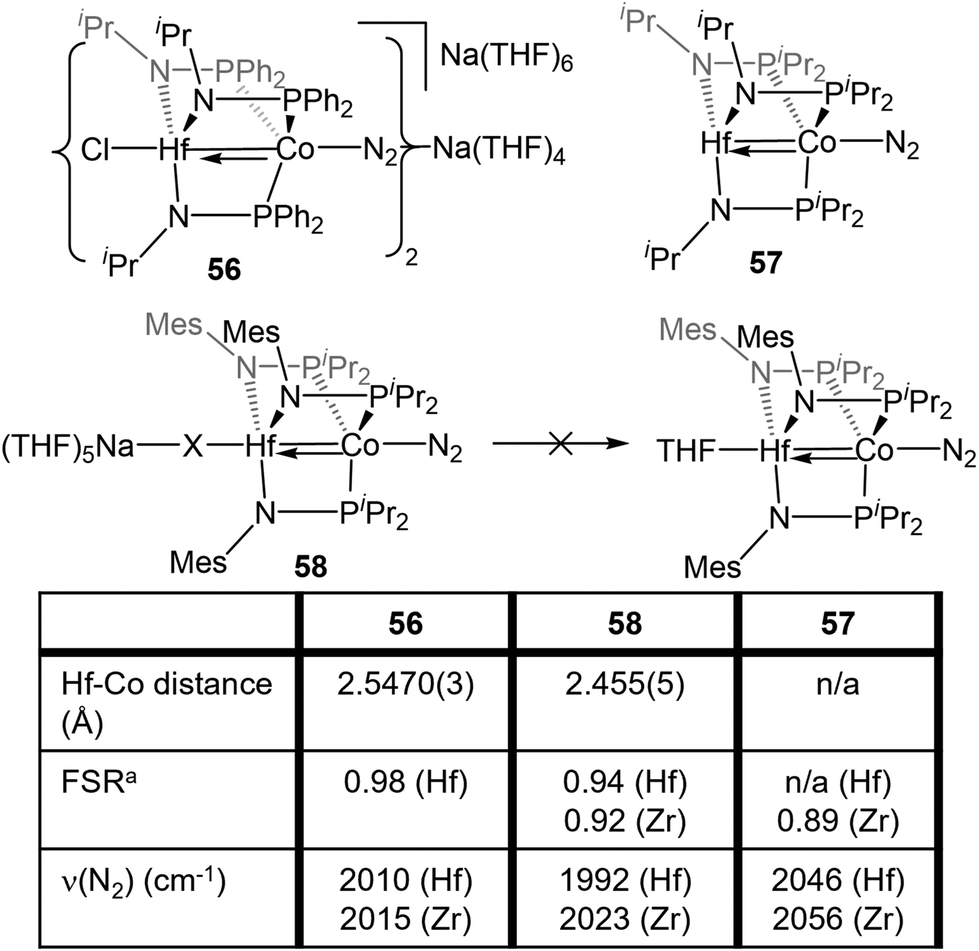 | ||
| Fig. 10 A comparison of the metal–metal bond distances and infrared N2 stretching frequencies of tris(phosphinoamide) HfIV/Co−I and ZrIV/Co−I complexes. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. | ||
Studies of the further reactivity of the Hf/Co compounds revealed stark differences from the Zr/Co analogues but was limited to the mesityl-substituted amide complexes 54 and 58. As shown in Scheme 14, the exposure of 58 to stoichiometric methyl iodide results in the one-electron oxidation of the complex to form XHf(MesNPiPr2)3Co(N2) (60),112 in contrast to the two-electron oxidation observed after addition of MeI to ZrIV/Co−I complex 5 (Scheme 1, vide supra).115 In a catalytic study, 54 was found to be much less active than its Zr analogue in Kumada coupling reactions.112,116
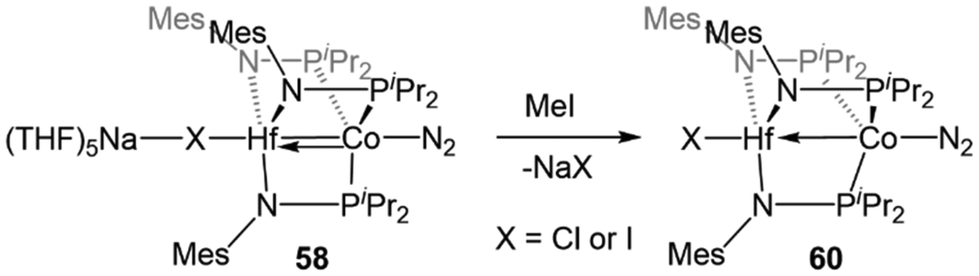 | ||
| Scheme 14 One-electron oxidation of HfIV/Co−I complex 58 with MeI, illustrating divergent reactivity compared to the ZrIV/Co−I analogue (cf.Scheme 1). | ||
M/Co bis(phosphinoamide) complexes (M = Ti, Zr)
In studying the reactivity of the group IV/cobalt tris(phosphinoamide) complexes, a common structural pattern was observed: in many two-electron oxidative addition reactions and reactions where substrates are bound to Co in an η2 fashion, phosphine dissociation from the Co center is required. This results not only in an η2-phosphinoamide ligand bound at Zr, congesting its coordination sphere, but may contribute to undesirable phosphinoamide degradation reactions that hamper the potential uses of these heterobimetallic complexes in catalysis. Therefore, an alternative bis(phosphinoamide) ligand scaffold was developed to target more coordinatively unsaturated heterobimetallic complexes. These endeavors were successful and both Ti/Co and Zr/Co bis(phosphinoamide) complexes were synthesized. A similar degree of metal–metal multiple bonding was observed with just two buttressing ligands, but the reactivity of the bis(phosphinoamide) compounds was found to be significantly enhanced.Ti/Co bis(phosphinoamide) complexes
The combination of the TiIV metalloligand Cl2Ti(XylNPiPr2)2 with CoI2 in the presence of Zn powder yields the tetrametallic dimer [ClTi(XylNPiPr2)2CoI]2 (61, Scheme 15).139 Unlike the MIV/CoI precursors observed in the tris(phosphinoamide) system, this is a TiIV/Co0 complex and its 2.2051(4) Å intermetallic distance is indicative of a Ti–Co double bond (FSR = 0.89).139 Furthermore, an examination of the frontier molecular orbitals shows that there is a σ-bonding interaction and two weak π bonding interactions, heavily polarized toward cobalt, between the two metal centers, consistent with a bond order of ∼2. Further reduction of 61 with excess KC8 in the presence of PMe3 yields the TiIV/Co−I complex ClTi(XylNPiPr2)2Co(PMe3) (62). The metal–metal bond in 62 is further contracted to 2.0234(9) Å (FSR = 0.82, Fig. 11) and a computational examination of the frontier molecular orbitals reveals a (σ)2(π)4(Conb)4 electronic configuration consistent with a metal–metal triple bond.139 The metal–metal distance in 62 is similar to that seen in 47 (2.0262(5) Å; FSR = 0.81), further supporting its assignment as a Ti–Co triple bond.138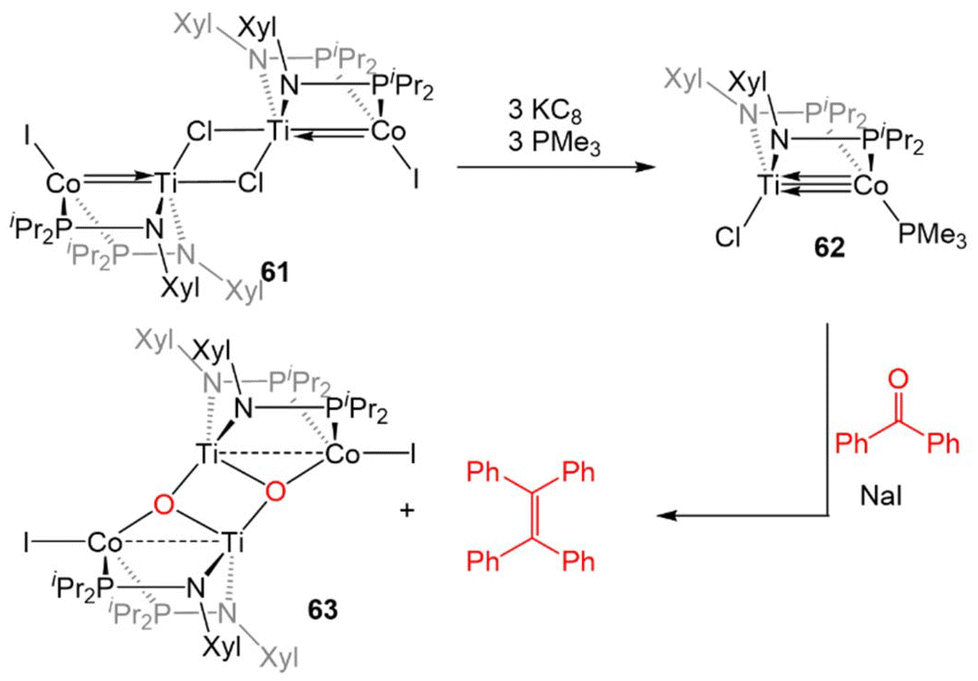 | ||
| Scheme 15 Synthesis of a bis(phosphinoamide) TiIV/Co−I complex and its reactivity in a stoichiometric McMurray coupling reaction with benzophenone. | ||
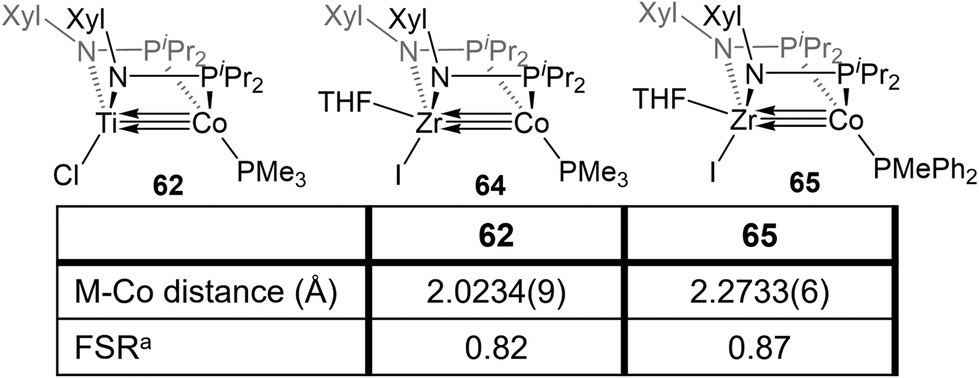 | ||
| Fig. 11 A comparison of the metal–metal bond metrics of bis(phosphinoamide) TiIV/Co−I and ZrIV/Co−I complexes. aFSR is defined as the ratio of the metal–metal distance to the sum of the single bond atomic radii of the two metals. | ||
The highly reduced bimetallic core of 62 reacts readily with aryl ketones in the presence of NaI to give the μ3-oxo dimer [Ti(μ-O)(XylNPiPr2)2CoI]2 (63) and tetraphenylethylene in a stoichiometric McMurray reaction (Scheme 15).139 This stoichiometric coupling reaction is not observed in the tris(phosphinoamide) Ti/Co complexes, wherein only ketone adducts are observed (Scheme 12, vide supra).126 Due to this vast difference in reactivity, it was concluded that bis(phosphinoamide) complexes are more reactive than their tris(phosphinoamide) analogues.
Zr/Co bis(phosphinoamide) complexes
Bis(phosphinoamide)-linked ZrIV/Co−I compounds, (THF)IZr(XylNPiPr2)2Co(PR3) (PR3 = PMe3 (64), PMePh2 (65)), were successfully synthesized and proven even more reactive than bis(phosphinoamide) compound 62 or the aforementioned tris(phosphinoamide) Zr/Co compounds.140 As shown in Fig. 11, the Zr–Co distance in 65 is 2.2733(6) Å (FSR = 0.87), which is elongated compared to that observed in the N2-free ZrIV/Co−I tris(phosphinoamide) complex 7 (FSR = 0.82) and the TiIV/Co−I bis(phosphinoamide) analogue 62 (FSR = 0.82).111,139 As expected, the electronic configuration of 65 was found to be (σ)2(π)4(Conb)4, corresponding to a metal–metal triple bond. However, despite the similar electron configuration, the metal–metal bonding orbitals in 65 were found to be significantly more polarized with far lower Zr contributions, leading to weaker bonding.The increased polarization and steric accessibility of the metal–metal bond in 64 and 65 renders these complexes significantly more reactive than either their Ti/Co or tris(phosphinoamide) analogues. For example, 64 and 65 react instantaneously and irreversibly with H2 (1 atm) to form the ZrIV/CoI dihydride complex (THF)IZr(μ-H)(XylNPiPr2)2Co(H)(PR3) (PR3 = PMe3 (66), PMePh2 (67)) (Scheme 16).140 This reactivity is in stark contrast to the tris(phosphinoamide) Zr/Co complex 5, which reacted sluggishly and destructively with H2 over the course of 12 hours to form 11 (Scheme 1; vide supra).115 The Zr–Co distance elongates to 2.4695(3) Å (FSR = 0.94) upon oxidative addition of H2, but remains shorter than observed for previous ZrIV/CoI complexes owing to a three-center–two-electron bond between Zr, Co, and the bridging hydride ligand.140 Upon exploring the utility of the H2 activation reaction, it was discovered that 64 and 65 are effective catalysts for the semi-hydrogenation of alkynes.140 At 5 mol% loading of 65 under 1 atm H2 at 60 °C, diphenylacetylene was hydrogenated to a mixture of stilbene isomers with 98% conversion in 6 h (Scheme 16). The PMe3 derivative 64 was also an effective catalyst but was more sluggish, suggesting that phosphine dissociation is a requisite step in the catalytic mechanism. Indeed, phenylacetylene was shown to displace the PMePh2 ligand in 65 to generate (THF)IZr(XylNPiPr2)2Co(PhCCPh) (68) and both 67 and 68 were observed by 31P{1H} NMR spectroscopy during catalysis. Additionally, independently synthesized 68 was shown to be catalytically competent, with comparable activity to 65 and enhanced activity compared to 64. In all cases, very little over-hydrogenation occurred (∼2% 1,2-diphenylethane), but the stilbene product was generated as a statistical mixture of isomers (Z:E = 57:43).140 Computational investigations of this catalytic cycle suggest that the cleavage of dihydrogen takes place primarily at the cobalt center, with the cleavage of the metal–metal triple bond playing an important role in the formation of the stable bridging hydrides in 66/67.141 Alkyne insertion then primarily takes place at the terminal cobalt hydride and then reductive elimination occurs, with the Z : E selectivity depending primarily on the steric environment at cobalt.141
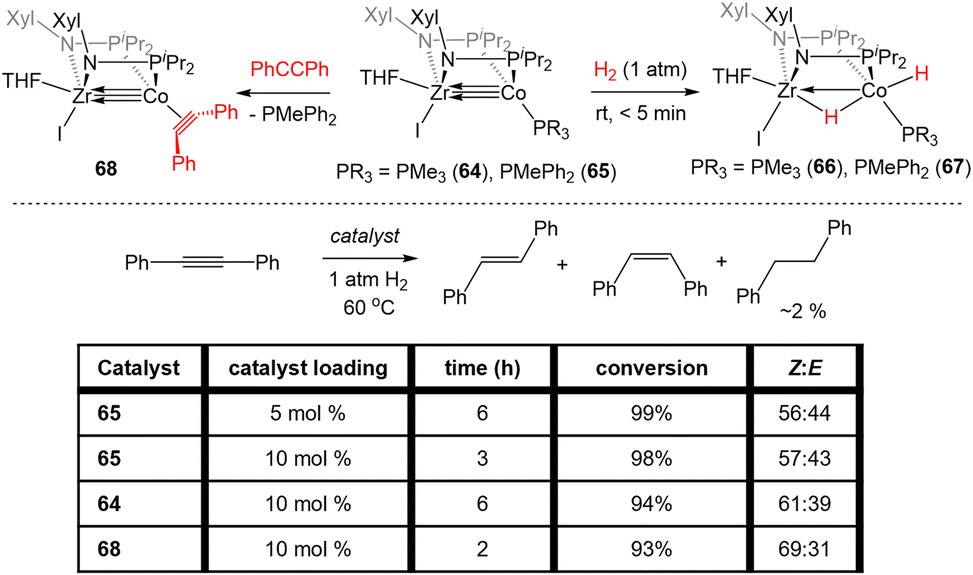 | ||
| Scheme 16 Reactivity of bis(phosphinoamide) ZrIV/Co−I complexes towards H2 and applications of these compounds as catalysts for the semi-hydrogenation of internal alkynes. | ||
In addition to the activation of H2, 64 and 65 were also found to activate the C–H bonds of terminal alkynes and pyridine derivatives. In a reaction similar to that described for the tris(phosphinoamide) complex 5 (Scheme 2, vide supra), treatment of 65 with TMSCCH or PhCCH resulted in Csp–H oxidative addition to afford a product with a terminal Co-hydride and a bridging acetylide fragment that binds κ1 to Co and η2 to Zr (69 and 70, Scheme 17).142 While the tris(phosphinoamide) ZrIV/Co−I complex 5 simply binds pyridine derivatives at the available Zr coordination site,143 the bis(phosphinoamide) complex 64 undergoes Csp2–H oxidative addition when exposed to pyridine derivatives. For example, the addition of excess 4-methylpyridine to 64 affords the ZrIV/CoI C–H activation product 71 (Scheme 17).142 The less electron-rich PMePh2-substituted analogue 65, on the other hand, proved unable to activate the C–H bond of pyridine derivatives, illustrating the delicate steric and electronic influences that dictate the favorability of this reaction. In fact, compound 71 was shown to take part in a dynamic equilibrium in solution through reversible C–H activation. The reversibility of the pyridine C–H activation process hampered efforts to promote further C–H functionalization reactions but, nonetheless, this stoichiometric reaction demonstrated the enhanced reactivity of the more coordinatively unsaturated bis(phosphinoamide)supported Zr/Co systems.
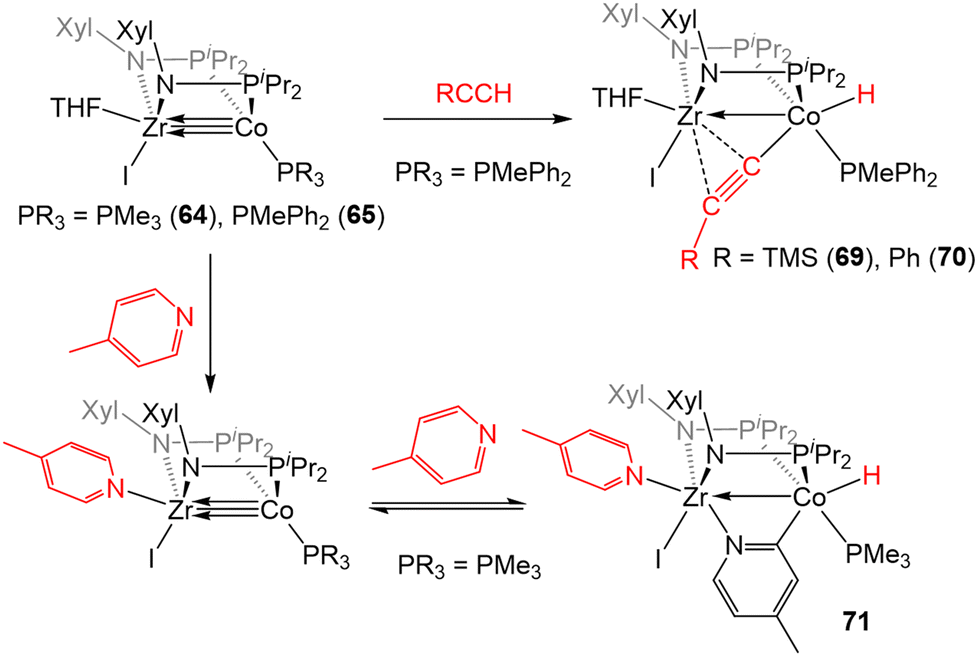 | ||
| Scheme 17 C–H bond activation promoted by bis(phosphinoamide) ZrIV/Co−I complexes. | ||
Zr/Co mono(phosphinoamide) complexes
The metal–metal bonding in both tris(phosphinoamide)- and bis(phosphinoamide)-supported ZrIV/Co−I complexes is similar, with both ligand frameworks supporting metal–metal triple bonds with remarkably short intermetallic distances (vide supra).111,140 However, the question remained whether the short metal–metal distances are primarily due to the constraints applied on these systems by multiple bridging ligands, or whether metal–metal orbital overlap is the key driving force for the formation of metal–metal multiple bonds. While Casey et al. and Bergman et al. synthesized ZrIV/group 9 complexes bearing only a single bridging ligand, the group 9 metals in these systems were either in a higher oxidation state or encumbered by multiple π-acidic ligands, hampering the formation of metal–metal multiple bonds.144,145 Therefore, to understand the unusual stability of phosphinoamide-supported Zr/Co multiple bonds and potentially generate even more reactive species, a mono(phosphinoamide)-supported ZrIV/Co−I complex was targetted.The tetrametallic mono(phosphinoamide)-bridged ZrIV/CoI precursor [I2Zr(μ-I)(μ-XylNPiPr2)CoI]2 (72) was synthesized and subsequently reduced with KC8 in the presence of PMe3 and 18-crown-6 to yield [(18-crown-6)K][I3Zr(XylNPiPr2)Co(PMe3)2] (73) (Scheme 18).113 In 73, the metal–metal distance was found to be 2.170(1) Å (FSR = 0.83), suggesting a high degree of metal–metal multiple bonding with just one bridging phosphinoamide ligand. A DFT analysis of the frontier molecular orbitals of the anionic, chloride analogue of 73 (73Cl) reveals a (σ)2(π)4(Conb)4 electronic configuration (Fig. 12).113 The Wiberg Bond Index (WBI) and Mayer Bond Order (MBO) of 73Cl were found to be 1.52 and 1.92, respectively. These values are consistent with those of 7 (WBI = 1.49, MBO = 1.73) and 64 (WBI = 1.50, MBO = 1.89), both of which were also found to contain metal–metal triple bonds.113 This led to the conclusion that the short intermetallic distances are the result of metal–metal multiple bonding rather than constraints imparted by multiple phosphinoamide ligands. Unfortunately, 73 was found to be too reactive to effectively perform stoichiometric or catalytic bond activation reactions.
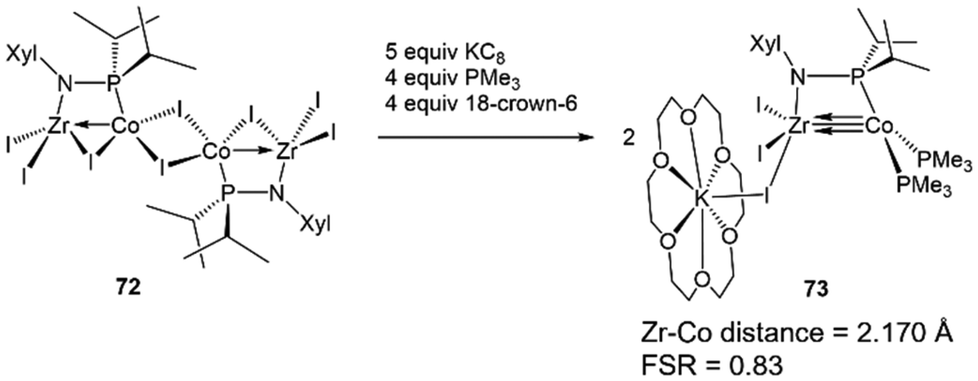 | ||
| Scheme 18 Synthesis of a mono(phosphinoamide)-supported ZrIV/Co−I complex. | ||
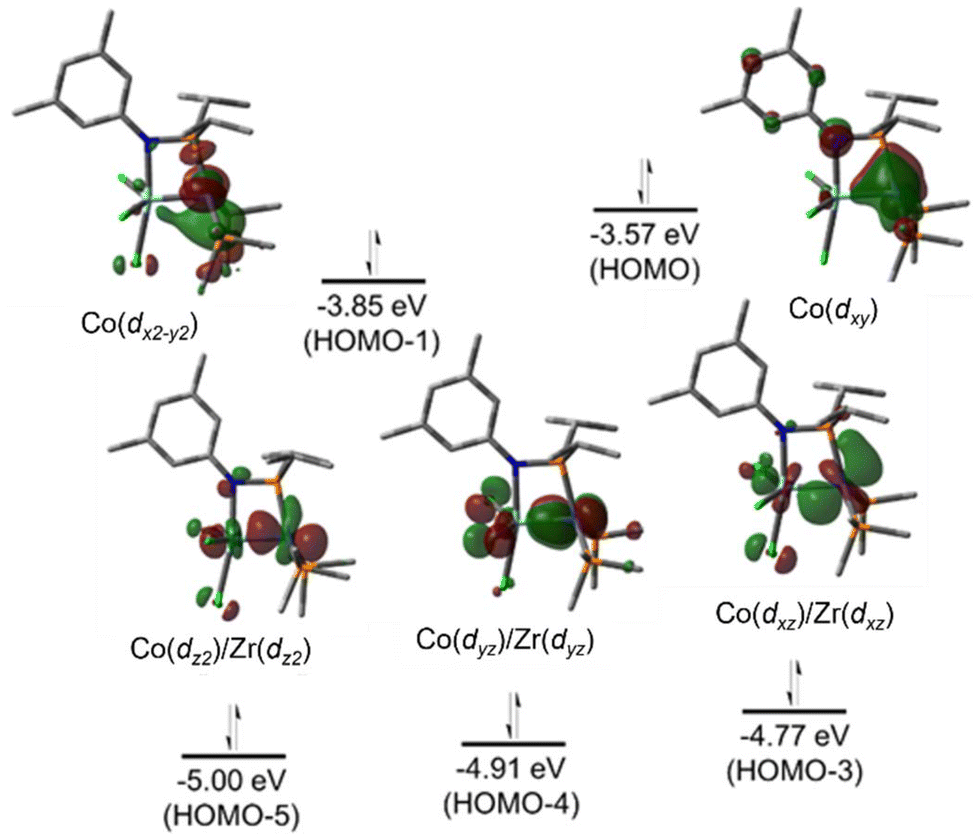 | ||
| Fig. 12 Pictorial representations of the frontier molecular orbitals of 73Cl (ωB97XD/def2SVP). | ||
Summary and outlook
An array of phosphinoamide-supported ZrIV/Co−I early/late heterobimetallic complexes have been synthesized and shown to display enhanced redox properties compared to monometallic analogues, polarized metal–metal multiple bonds, and reactivity towards small molecule activation and the cleavage of relatively inert σ and π bonds. The tris(phosphinoamide) ligand scaffold serves as a robust support to study metal–metal multiple bonding and fundamental aspects of the multielectron redox transformations facilitated by Zr/Co complexes. However, oxidative addition reactions across the Zr–Co bond require dissociation of a phosphinoamide ligand from cobalt to allow substrates to access the metal–metal bond. The more coordinatively unsaturated bis(phosphinoamide) complexes can access the same metal–metal multiple bonding motifs as their tris(phosphinoamide) analogues while displaying enhanced reactivity towards inert H–H and C–H bonds. Although a mono(phosphinoamide) ZrIV/Co−I complex was isolated and shown to have comparable metal–metal bonding properties to the bis- and tris(phosphinoamide) analogues, the absence of multiple buttressing ligands rendered this species too unstable for further well-defined reactions. Comparative studies with Ti/Co and Hf/Co complexes indicate that similar metal–metal bonding can be accessed, but that the increased covalency of the Ti–Co bonds and weaker Hf–Co Lewis acid/base interactions leads to diminished reactivity.The compilation of results described herein suggest that the most promising group IV M/Co (M = Ti, Zr, Hf) complexes for future reactivity studies are bis(phosphinoamide) Zr/Co compounds. Moreover, the reactivity of these molecules towards inert bonds is likely to be even further enhanced in the absence of strongly binding ancillary ligands (e.g. PMe3) coordinated to the reactive Co−I site. Efforts to realize more reactive ZrIV/Co−I synthons are currently ongoing and will be reported in due course.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research described in this Perspective has been consistently supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, most recently under Award DE-SC0019179. N. H. H. is grateful for the Dean's Distinguished University Fellowship from the Graduate School at The Ohio State University.Notes and references
- M. H. Chisholm, Anything One Can Do, Two Can Do, Too – And It's More Interesting, 2009.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef.
- A. Chaudhary, A. Singh and R. C. Kamboj, Chem. Sci. Rev. Lett., 2016, 5, 170–192 Search PubMed.
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef PubMed.
- N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef.
- M. K. Karunananda and N. P. Mankad, ACS Catal., 2017, 7, 6110–6119 CrossRef.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC.
- C. M. Farley and C. Uyeda, Trends Chem., 2019, 1, 497–509 CrossRef.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev.: Sci. Eng., 2012, 54, 1–40 CrossRef CAS.
- P. A. Lindahl, J. Inorg. Biochem., 2012, 106, 172–178 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef.
- M. H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385–2419 CrossRef.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–350 CrossRef PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- W.-F. Liaw, C.-Y. Chiang, G.-H. Lee, S.-M Peng, C.-H. Lai and M. Y. Darensbourg, Inorg. Chem., 2000, 39, 480–484 CrossRef CAS PubMed.
- T. Zhao, P. Ghosh, Z. Martinez, X. Liu, X. Meng and M. Y. Darensbourg, Organometallics, 2017, 36, 1822–1827 CrossRef CAS.
- S. Ding, P. Ghosh, M. Y. Darensbourg and M. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9775–E9782 CAS.
- M. Y. Darensbourg, E. L. Oduaran, S. Ding, A. M. Lunsford, K. D. K. Patherana, P. Ghosh and X. Yang, in Enzymes for Solving Humankind's Problems, ed. J. J. G. Moura, I. Moura and L. B. Maia, 2021, pp. 275–302 Search PubMed.
- A. McSkimming and D. L. M. Suess, Nat. Chem., 2021, 13, 666–670 CrossRef CAS.
- B. E. Petel and E. M. Matson, Chem. Commun., 2020, 56, 13477–13490 RSC.
- W. Zhang, C. E. Moore and S. Zhang, J. Am. Chem. Soc., 2022, 144, 1709–1717 CrossRef CAS.
- J. A. Kephart, B. S. Mitchell, W. Kaminsky and A. Velian, J. Am. Chem. Soc., 2022, 144, 9206–9211 CrossRef CAS PubMed.
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer Science and Business Media, Inc., New York, 2005 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- F. A. Cotton, Acc. Chem. Res., 1978, 11, 225–232 CrossRef CAS.
- J. A. Bertrand, F. A. Cotton and W. A. Dollase, J. Am. Chem. Soc., 1963, 85, 1349–1350 CrossRef CAS.
- M. J. Bennett, F. A. Cotton and R. A. Walton, J. Am. Chem. Soc., 1966, 88, 3866–3867 CrossRef.
- F. A. Cotton, Inorg. Chem., 1965, 4, 334–336 CrossRef.
- J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115–5127 RSC.
- J. E. McGrady, in Molecular Metal-Metal Bonds, ed. S. T. Liddle, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 1–22 Search PubMed.
- Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC.
- F. G. A. Stone, Angew. Chem., Int. Ed. Engl., 1984, 23, 89–99 CrossRef.
- K. M. Baines and W. G. Stibbs, in Multiply Bonded Main Group Metals and Metalloids, ed. F. G. A. Stone and R. West, Elsevier Masson SAS, 1st edn, 1996, vol. 39, pp. 275–324 Search PubMed.
- J. P. Collman and R. Boulatov, Angew. Chem., Int. Ed., 2002, 41, 3948–3961 CrossRef.
- M. H. Chisholm, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2563–2570 CrossRef.
- C. Ni and P. P. Power, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin, Heidelberg, 1st edn, 2010, vol. 136, pp. 59–111 Search PubMed.
- G. Parkin, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 113–145 Search PubMed.
- J. F. Berry, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin, Heidelberg, 1st edn, 2010, vol. 136, pp. 1–28 Search PubMed.
- M. H. Chisholm, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin, Heidelberg, 1st edn, 2010, vol. 136, pp. 29–57 Search PubMed.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- F. A. Cotton and M. Millar, J. Am. Chem. Soc., 1977, 99, 7886–7891 CrossRef.
- F. A. Cotton, L. M. Daniels and C. A. Murillo, Angew. Chem., Int. Ed. Engl., 1992, 31, 737–738 CrossRef.
- F. A. Cotton, L. M. Daniels and C. A. Murillo, Inorg. Chem., 1993, 32, 2881–2885 CrossRef.
- P. Berno, S. Hao, R. Minhas and S. Gambarotta, J. Am. Chem. Soc., 1994, 116, 7417–7418 CrossRef.
- F. A. Cotton and D. J. Timmons, Polyhedron, 1998, 17, 179–184 CrossRef.
- F. A. Cotton, E. A. Hillard, C. A. Murillo and X. Wang, Inorg. Chem., 2003, 42, 6063–6070 CrossRef PubMed.
- F. A. Cotton, E. A. Hillard and C. A. Murillo, J. Am. Chem. Soc., 2003, 125, 2026–2027 CrossRef PubMed.
- F. A. Cotton, Z. Li and C. A. Murillo, Eur. J. Inorg. Chem., 2007, 3509–3513 CrossRef.
- J. Haywood, F. A. Stokes, R. J. Less, M. McPartlin, A. E. H. Wheatley and D. S. Wright, Chem. Commun., 2011, 47, 4120 RSC.
- F. A. Cotton, L. M. Daniels and C. A. Murillo, Inorg. Chim. Acta, 1994, 224, 5–9 CrossRef.
- G. H. Timmer and J. F. Berry, C. R. Chim., 2012, 15, 192–201 CrossRef.
- F. A. Cotton and R. Poli, Inorg. Chem., 1987, 26, 3652–3653 CrossRef.
- L. P. He, C. L. Yao, M. Naris, J. C. Lee, J. D. Korp and J. L. Bear, Inorg. Chem., 1992, 31, 620–625 CrossRef.
- F. A. Cotton, L. M. Daniels, X. Feng, D. J. Maloney, J. H. Matonic and C. A. Murilio, Inorg. Chim. Acta, 1997, 256, 291–301 CrossRef.
- F. A. Cotton, Z. Li, C. Murillo, P. Poplaukhin and J. Reibenspies, J. Cluster Sci., 2008, 19, 89–97 CrossRef.
- M. Corbett, B. F. Hoskins, N. J. McLeod and B. P. O'Day, Aust. J. Chem., 1975, 28, 2377–2392 CrossRef.
- D. I. Arnold, F. A. Cotton, D. J. Maloney, J. H. Matonic and C. A. Murillo, Polyhedron, 1997, 16, 133–141 CrossRef.
- J. F. Berry, E. Bothe, F. A. Cotton, S. A. Ibragimov, C. A. Murillo, D. Villagrán and X. Wang, Inorg. Chem., 2006, 45, 4396–4406 CrossRef PubMed.
- K. Pang, J. S. Figueroa, I. A. Tonks, W. Sattler and G. Parkin, Inorg. Chim. Acta, 2009, 362, 4609–4615 CrossRef.
- T. Hamaguchi, R. Shimazaki and I. Ando, J. Mol. Struct., 2018, 1173, 345–348 CrossRef.
- S. O. Ojwach, T. T. Okemwa, N. W. Attandoh and B. Omondi, Dalton Trans., 2013, 42, 10735 RSC.
- M. Iqbal, I. Ahmad, S. Ali, N. Muhammad, S. Ahmed and M. Sohail, Polyhedron, 2013, 50, 524–531 CrossRef.
- P. L. Dunn, R. K. Carlson and I. A. Tonks, Inorg. Chim. Acta, 2017, 460, 43–48 CrossRef.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577–7581 CrossRef PubMed.
- L. M. Slaughter and P. T. Wolczanski, Chem. Commun., 1997, 2109–2110 RSC.
- C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14–38 CrossRef.
- R. J. Eisenhart, L. J. Clouston and C. C. Lu, Acc. Chem. Res., 2015, 48, 2885–2894 CrossRef.
- N. Wheatley and P. Kalck, Chem. Rev., 1999, 99, 3379–3419 CrossRef.
- D. W. Stephan, Coord. Chem. Rev., 1989, 95, 41–107 CrossRef.
- R. M. Bullock and C. P. Casey, Acc. Chem. Res., 1987, 20, 167–173 CrossRef CAS.
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS PubMed.
- K. Koessler and B. Butschke, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, pp. 1–31 Search PubMed.
- R. T. K. Baker, S. J. Tauster and J. A. Dumesic, Strong Metal-Support Interactions, American Chemical Society, Washington, DC, USA, 1986, vol. 298 Search PubMed.
- S. J. Tauster, ACS Symp. Ser., 1986, 298, 1–9 CrossRef CAS.
- S. Friedrich, H. Memmler, L. H. Gade, W. S. Li, I. J. Scowen, M. McPartlin and C. E. Housecroft, Inorg. Chem., 1996, 35, 2433–2441 CrossRef CAS.
- G. S. Ferguson and P. T. Wolczanski, J. Am. Chem. Soc., 1986, 108, 8293–8295 CrossRef CAS.
- G. S. Ferguson, P. T. Wolczanski, L. Parkanyi and M. C. Zonnevylle, Organometallics, 1988, 7, 1967–1979 CrossRef CAS.
- G. S. Ferguson and P. T. Wolczanski, Organometallics, 1985, 4, 1601–1605 CrossRef CAS.
- S. M. Baxter, G. S. Ferguson and P. T. Wolczanski, J. Am. Chem. Soc., 1988, 110, 4231–4241 CrossRef CAS.
- P. A. Rudd, S. Liu, L. Gagliardi, V. G. Young and C. C. Lu, J. Am. Chem. Soc., 2011, 133, 20724–20727 CrossRef CAS PubMed.
- J. T. Moore, S. Chatterjee, M. Tarrago, L. J. Clouston, S. Sproules, E. Bill, V. Bernales, L. Gagliardi, S. Ye, K. M. Lancaster and C. C. Lu, Inorg. Chem., 2019, 58, 6199–6214 CrossRef.
- L. J. Clouston, V. Bernales, R. C. Cammarota, R. K. Carlson, E. Bill, L. Gagliardi and C. C. Lu, Inorg. Chem., 2015, 54, 11669–11679 CrossRef.
- L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142–13148 CrossRef PubMed.
- R. J. Eisenhart, P. A. Rudd, N. Planas, D. W. Boyce, R. K. Carlson, W. B. Tolman, E. Bill, L. Gagliardi and C. C. Lu, Inorg. Chem., 2015, 54, 7579–7592 CrossRef PubMed.
- P. A. Rudd, S. Liu, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2013, 52, 4449–4452 CrossRef PubMed.
- R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef PubMed.
- J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef.
- M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS.
- B. L. Ramirez, P. Sharma, R. J. Eisenhart, L. Gagliardi and C. C. Lu, Chem. Sci., 2019, 10, 3375–3384 RSC.
- B. L. Ramirez and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 5396–5407 CrossRef CAS PubMed.
- J. T. Moore and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef CAS.
- H. Nagashima, T. Sue, T. Oda, A. Kanemitsu, T. Matsumoto, Y. Motoyama and Y. Sunada, Organometallics, 2006, 25, 1987–1994 CrossRef CAS.
- H. Tsutsumi, Y. Sunada, Y. Shiota, K. Yoshizawa and H. Nagashima, Organometallics, 2009, 28, 1988–1991 CrossRef CAS.
- T. Sue, Y. Sunada and H. Nagashima, Eur. J. Inorg. Chem., 2007, 2897–2908 CrossRef CAS.
- Y. Sunada, T. Sue, T. Matsumoto and H. Nagashima, J. Organomet. Chem., 2006, 691, 3176–3182 CrossRef CAS.
- S. Kuppuswamy, M. W. Bezpalko, T. M. Powers, M. J. T. Wilding, C. K. Brozek, M. Foxman and C. M. Thomas, Chem. Sci., 2014, 5, 1617–1626 RSC.
- S. Kuppuswamy, T. M. Powers, J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Sci., 2013, 4, 3557–3565 RSC.
- S. M. Greer, J. McKay, K. M. Gramigna, C. M. Thomas, S. A. Stoian and S. Hill, Inorg. Chem., 2018, 57, 5870–5878 CrossRef CAS PubMed.
- B. Wu, M. J. T. Wilding, S. Kuppuswamy, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2016, 55, 12137–12148 CrossRef CAS.
- G. Culcu, D. A. Iovan, J. P. Krogman, M. J. T. Wilding, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 9627–9636 CrossRef CAS PubMed.
- B. A. Barden, G. Culcu, J. P. Krogman, M. W. Bezpalko, G. P. Hatzis, D. A. Dickie, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2019, 58, 821–833 CrossRef CAS PubMed.
- D. A. Evers, A. H. Bluestein, B. M. Foxman and C. M. Thomas, Dalton Trans., 2012, 41, 8111–8115 RSC.
- J. W. Napoline, S. J. Kraft, E. M. Matson, P. E. Fanwick, S. C. Bart and C. M. Thomas, Inorg. Chem., 2013, 52, 12170–12177 CrossRef CAS PubMed.
- J. P. Krogman, J. R. Gallagher, G. Zhang, A. S. Hock, J. T. Miller and C. M. Thomas, Dalton Trans., 2014, 43, 13852–13857 RSC.
- B. P. Greenwood, S. I. Forman, G. T. Rowe, C.-H. Chen, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2009, 48, 6251–6260 CrossRef CAS.
- B. P. Greenwood, G. T. Rowe, C.-H. Chen, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2010, 132, 44–45 CrossRef CAS PubMed.
- V. N. Setty, W. Zhou, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2011, 50, 4647–4655 CrossRef CAS PubMed.
- N. H. Hunter, J. E. Stevens, C. E. Moore and C. M. Thomas, Inorg. Chem., 2023, 62, 659–663 CrossRef CAS.
- X. Yang, L. Zhao, T. Fox, Z.-X. Wang and H. Berke, Angew. Chem., Int. Ed., 2010, 49, 2058–2062 CrossRef CAS.
- C. M. Thomas, J. W. Napoline, G. T. Rowe and B. M. Foxman, Chem. Commun., 2010, 46, 5790–5792 RSC.
- W. Zhou, J. W. Napoline and C. M. Thomas, Eur. J. Inorg. Chem., 2011, 2029–2033 CrossRef CAS.
- J. Coombs, D. Perry, D.-H. Kwon, C. M. Thomas and D. H. Ess, Organometallics, 2018, 37, 4195–4203 CrossRef CAS.
- J. W. Beattie, C. Wang, H. Zhang, J. P. Krogman, B. M. Foxman and C. M. Thomas, Dalton Trans., 2020, 49, 2407–2411 RSC.
- Y. Sun, J. Zhang, Y. Zeng, L. Meng and X. Li, J. Org. Chem., 2024, 89, 605–616 CrossRef CAS PubMed.
- J. W. Napoline, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Commun., 2013, 49, 4388–4390 RSC.
- J. W. Napoline, J. P. Krogman, R. Shi, S. Kuppuswamy, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Eur. J. Inorg. Chem., 2013, 3874–3882 CrossRef CAS.
- J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS.
- J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2013, 52, 3022–3031 CrossRef CAS.
- W. Zhou, S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2013, 32, 1766–1772 CrossRef CAS.
- S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2014, 33, 2071–2079 CrossRef CAS.
- H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498–3507 CrossRef CAS.
- M. P. Crockett, H. Zhang, C. M. Thomas and J. A. Byers, Chem. Commun., 2019, 55, 14426–14429 RSC.
- S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2013, 135, 6018–6021 CrossRef CAS.
- B. Wu, R. Hernández Sánchez, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2014, 53, 10021–10023 CrossRef CAS PubMed.
- N. P. Mankad, P. Mueller and J. C. Peters, J. Am. Chem. Soc., 2010, 4083–4085 CrossRef CAS.
- J. P. Krogman, B. M. Foxman and C. M. Thomas, Organometallics, 2015, 34, 3159–3166 CrossRef CAS.
- J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Dalton Trans., 2016, 45, 11182–11190 RSC.
- H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed.
- W. A. Howard, M. Waters and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4917–4918 CrossRef CAS.
- H. Zhang, G. P. Hatzis, D. A. Dickie, C. M. Thomas and C. E. Moore, Chem. Sci., 2020, 11, 10729–10736 RSC.
- M. Gomberg, J. Am. Chem. Soc., 1903, 25, 1274–1277 CrossRef CAS.
- W. Zhou, N. I. Saper, J. P. Krogman, B. M. Foxman and C. M. Thomas, Dalton Trans., 2014, 43, 1984–1989 RSC.
- B. Wu, K. M. Gramigna, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2015, 54, 10909–10917 CrossRef CAS PubMed.
- B. Wu, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Sci., 2015, 6, 2044–2049 RSC.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS.
- Y. Li, P. Su, J. Jiang and Z. Ke, ACS Catal., 2021, 11, 13452–13462 CrossRef CAS.
- N. H. Hunter, E. M. Lane, K. M. Gramigna, C. E. Moore and C. M. Thomas, Organometallics, 2021, 40, 3689–3696 CrossRef CAS.
- B. Crown thesis, Brandeis University, 2013 Search PubMed.
- C. P. Casey and F. Nief, Organometallics, 1985, 4, 1218–1220 CrossRef CAS.
- A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 3822–3835 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |