
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSonohydrothermal synthesis of zeolite A and its phase transformation into sodalite†
William's
Nzodom Djozing
a,
Sabine
Valange
b,
Sergey I.
Nikitenko
 a and
Tony
Chave
*a
a and
Tony
Chave
*a
aUniv Montpellier, UMR 5257, ICSM, CEA, CNRS, ENSCM, Marcoule, F-30207 Bagnols Sur Cèze, France. E-mail: tony.chave@cea.fr
bUniversité de Poitiers, CNRS, Institut de Chimie des Milieux et Matériaux de Poitiers-IC2MP, ENSI Poitiers, B1, 1 rue Marcel Doré, F-86073 Poitiers Cedex 9, France
First published on 20th August 2024
Abstract
The sonohydrothermal (SHT) treatment is an innovative technique allowing the simultaneous coupling of low frequency ultrasound and hydrothermal conditions for the synthesis of materials. The aim of the present work was to investigate, for the first time, the synthesis of zeolite A and its formation mechanism under SHT conditions. The zeolite synthesis was carried out under sonohydrothermal conditions using a specially designed reactor that allows the application of ultrasonic irradiation at 20 kHz in an autoclave-type reactor heated up to 200 °C under autogenous pressure. The conversion kinetics of the amorphous hydrogel to zeolite A and its further conversion to sodalite were studied. Syntheses were performed in the SHT reactor at 80 and 100 °C, varying the synthesis time from 15 minutes to several hours. The required time to obtain fully crystalline zeolite A under sonohydrothermal conditions was only 25 minutes, highlighting a significantly improved crystallization rate compared to silent conditions (a 9.6-fold kinetic gain). In addition, the resulting zeolite A has smaller particles and a more homogeneous particle size distribution than the zeolite synthesized by hydrothermal treatment. These results can be explained by the sonofragmentation of the amorphous gel and the concomitant enhanced mass transfer of the building units at the interface between the crystallite surface and the solution resulting from the acoustic cavitation activity under SHT conditions. Compared to classical hydrothermal heating, a drastic kinetic increase of the transformation of zeolite A into the more stable sodalite phase was also observed under sonohydrothermal conditions.
1. Introduction
Zeolites are one of the most important classes of crystalline microporous materials. These hydrated microporous alumino-silicates feature a three-dimensional structure formed by the regular arrangement of tetrahedral TO4 units, where the T atoms (Si, Al) are connected by bridging oxygen atoms.1–3 The tetrahedra are bound together to form uniform narrow channels (and cages), typically less than 2 nm in size.4 The net negative charge of the crystalline framework, resulting from the presence of the alumina tetrahedra, is compensated by the positive charge of inorganic or organic cations located in the pores of the zeolite.4,5According to Breck,6 zeolites are metastable phases and they can be transformed into more stable and denser materials over the course of the synthesis process. For aluminosilicate zeolite materials, the synthesis consists of mixing the raw precursors (silicate and aluminate sources) in aqueous medium (typically highly alkaline), possibly in the presence of a suitable structure directing agent,7–12 at high temperature and pressure for different reaction times, from hours to days.10,13–16 This conventional hydrothermal (HT) method, however, is known to be a time- and energy-consuming process. This is because the crystallization of zeolites requires restructuring of the initial raw materials for nucleation of discrete particles of the new phase and subsequent growth of zeolite crystals. Among the parameters affecting zeolite crystallization, the synthesis temperature and crystallization time are the most important variables, due to the metastable nature of zeolites.16 Hence, strategies for accelerating the crystallization rate of zeolites have been developed, and have very recently been comprehensively reviewed by Yuan et al.17
In this respect, ultrasonic irradiation has emerged as an interesting alternative method for accelerating the crystallization kinetics of materials, including zeolites.17,18 Sonochemistry, or the use of power ultrasound for chemical applications, is based on the physicochemical effects generated by acoustic cavitation. Acoustic cavitation is characterized by the formation, growth and implosive collapse of bubbles in ultrasonically irradiated liquids.19 The chemical effects of acoustic cavitation occur preferentially when using high frequency ultrasound, specifically over 100 kHz. Free radicals are generated inside the cavitation bubble core where the formation of non-equilibrium plasma can be observed under extreme temperature and pressure conditions.19–26 At lower ultrasonic frequencies, 16–100 kHz, the physical effects of acoustic cavitation dominate, leading to microjets and shock waves in treated solutions. Indeed, due to the presence of extended surfaces or interactions between bubbles in the cavitation cloud, the cavitation bubble does not remain spherical during its collapse. This deformation leads to the formation of microjets which can be responsible for solid surface erosion. Coupled with violent shock waves and acoustic streaming, low ultrasonic frequency irradiation can thus be applied to enhance mass transfer, solid dispersion and media mixing, and to increase interparticle collisions which can lead to both physical and chemical solid surface modifications.22–24
Recently, ultrasonic pretreatment has received much attention for the preparation of zeolites due to significant acceleration of crystallization induced by the acoustic cavitation phenomenon. It is actually well known that sonocrystallization can influence particle morphology, reduce induction time, increase the nucleation rate and also improve particle size distribution as previously reported in the literature.27–31 In the case of zeolite Na-A, its ultrasound-assisted preparation was usually carried out in an ultrasonic bath at various synthesis temperatures.
For instance, the application of 47 kHz ultrasonic irradiation at 70 °C for 4–6 h to an amorphous hydrogel precursor made up of kaolin was shown to speed up the crystallization of zeolite Na-A (in 4 h) compared with the conventional synthesis under silent conditions (without ultrasound) that required 6 h.32 Erdem-Şenatalar and co-workers also studied the effect of ultrasound on zeolite A synthesis using an ultrasonic bath (35 kHz) at different temperatures and times (2–9 h). Upon application of ultrasound, the rates of nucleation and crystallization increased, thereby improving the yield of zeolite A crystals.33 However, whether or not ultrasound was used, the synthesis resulted in other crystalline phases for crystallization times over 10 h at 50 °C. In another study, it was shown that the use of ultrasound was decisive in the fast transformation of zeolite A into the more stable sodalite phase (SOD) under the experimental conditions studied (ultrasonic bath at 35 kHz, 240 W, 1–4 h, 60–75 °C). Complete transformation into the SOD phase took place after 4 h of sonication, whereas the progressive appearance of sodalite at the expense of Na-A zeolite began only after 4 h of treatment in the absence of ultrasound, demonstrating that zeolite A crystallization and phase transformation proceed via two different mechanisms under ultrasound and silent conditions.34 In addition to using ultrasonic baths for zeolite A synthesis, ultrasound can also be introduced into the system via a reactor fitted with an ultrasonic probe. In this way, Van Gerven and co-workers investigated the effect of ultrasound on the growth kinetics of zeolite A at 80 °C in a batch setup equipped with a transducer operating at 40 kHz, varying the synthesis time and sonication power (10–50 W). They concluded that ultrasound has a significant effect on the nucleation and growth steps. A reduction of up to 40% of the initial synthesis time could be achieved under the studied conditions. Compared with conventional conditions, zeolite A crystals synthesized under ultrasound have a narrower particle size distribution.35
Another ultrasound-based approach is the so-called sonohydrothermal (SHT) method. The application of ultrasound to the crystallization of materials has recently been investigated under SHT conditions, i.e. the simultaneous coupling of low frequency ultrasound (20 kHz) and hydrothermal conditions under autogenous pressure. The SHT treatment is an emerging synthesis route that allows the preparation of nanocrystalline materials with enhanced properties. This innovative approach has been successfully applied to the synthesis of porous (Ce,Zr)O2 mixed oxides with better physicochemical characteristics than those prepared by a two-step process based on ultrasound-assisted coprecipitation followed by hydrothermal treatment.36 Core–shell nanoparticles, such as Ti@TiO2 with enhanced characteristics and (photo)catalytic properties, have also been efficiently synthesized under SHT conditions.37,38
Therefore, in the current work, we have extended this concept, for the first time, to the preparation of zeolite materials, in particular zeolite A, in order to study how its synthesis under sonohydrothermal conditions is affected. For this purpose, the conversion of the amorphous aluminosilicate hydrogel into zeolite A and its subsequent transformation into higher-density sodalite are investigated under these coupling conditions. Synthesis experiments were conducted in a specially designed reactor for various sonication times at 100 °C. The effect of the applied power density on the crystallization of zeolite A and its phase transformation into SOD is also discussed. Finally, acoustic noise analyses were carried out during the zeolite synthesis under the simultaneous coupling of 20 kHz ultrasonic irradiation and hydrothermal conditions, in order to assess the evolution of the reaction medium during SHT heating.
2. Experimental
2.1. Materials
Fumed silica with particle sizes from 0.2 μm to 0.3 μm and sodium aluminate powder were purchased from Sigma Aldrich and used without any further purification. Sodium hydroxide pellets (ACS – ISO for analysis purity) were supplied by Carlo Herba. Milli-Q water with a resistivity higher than 18.2 MΩ cm at 25 °C was used for the preparation of all solutions described herein.2.2. Zeolite synthesis protocol
The zeolite A synthesis route considered herein is based on a classical two step procedure involving first the formation of an aluminosilicate hydrogel by mixing silica and sodium aluminate precursors in a sodium hydroxide solution.39 The precipitated amorphous aluminosilicate gel was then transferred to a hydrothermal or sonohydrothermal reactor for synthesis at a temperature of 100 or 150 °C with various treatment times ranging from 15 min to 48 h. Each experiment was repeated 2 to 3 times.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.1Al:4.1Na:2OH:102H2O. Precipitation of aluminosilicate gel was achieved by slow addition of the aluminate solution into the silica solution. This process was done either under vigorous mixing using a magnetic stirrer or under 20 kHz ultrasonic irradiation at 60 °C for 30 min. Ultrasonic irradiation during this precipitation step was carried out within a thermostated glass reactor through a 1 cm2 ultrasonic tip connected to a 750VCX Vibra-Cell device (Sonics Materials Inc.) as depicted in Fig. S1.† The acoustic power delivered to the system was measured by the thermal probe method at 30 W (70% amplitude), as already reported in the literature.40
:2.1Al:4.1Na:2OH:102H2O. Precipitation of aluminosilicate gel was achieved by slow addition of the aluminate solution into the silica solution. This process was done either under vigorous mixing using a magnetic stirrer or under 20 kHz ultrasonic irradiation at 60 °C for 30 min. Ultrasonic irradiation during this precipitation step was carried out within a thermostated glass reactor through a 1 cm2 ultrasonic tip connected to a 750VCX Vibra-Cell device (Sonics Materials Inc.) as depicted in Fig. S1.† The acoustic power delivered to the system was measured by the thermal probe method at 30 W (70% amplitude), as already reported in the literature.40
The sonochemical activity within the reactor under the studied hydrothermal conditions, i.e. within the range of 100 to 200 °C, has been demonstrated for the first time in our previous study.36 Precise measurement of the power dissipated in a heterogeneous system, particularly under hydrothermal conditions, is a difficult task.
However, based on the work of Mancier et al., it is interesting to note that in highly viscous media (for instance, in pure glycerol and in mixtures up to 1500 times more viscous than water), the dissipated acoustic power can be higher than in pure water.41 In our experiments, the displayed power supplied by the US generator was measured for water and the amorphous aluminosilicate hydrogel. A rough estimate of the dissipated power in the system could then be made on the basis of this displayed power. The initial power displayed is slightly higher in the viscous amorphous gel at room temperature than in water, with values around 35 and 32 W, respectively. As the temperature in the system increases, the displayed power decreases to 15 W for water at 100 °C and to around 15–20 W for the reaction medium at the same temperature. Although not identical, the power dissipated under both conditions remains in the same order of magnitude for both systems, with an estimated difference of approximately 5 W between pure water and the reaction medium, assuming that our heterogeneous system behaves in the same way as a pure liquid phase.
| # | Experiment | Reaction volume (mL) | Precipitation conditions | Heat treatment conditions | ||
|---|---|---|---|---|---|---|
| Ultrasonic irradiation | Time (hours) | Temperature (°C) | Ultrasonic irradiation | |||
| 1 | MS-HT-4 h-100 | 49 | No | 4 | 100 | No |
| 2 | US-HT-4 h-100 | 49 | Yes | 4 | 100 | No |
| 3 | MS-SHT-4 h-100 | 49 | No | 4 | 100 | Yes |
| 4 | US-SHT-4 h-100 | 49 | Yes | 4 | 100 | Yes |
| 5 | MS-HT-4 h-150 | 49 | No | 4 | 150 | No |
| 6 | MS-HT-16 h-100 | 49 | No | 16 | 100 | No |
| 7 | MS-SHT-25 min-100 | 49 | No | 0.4 | 100 | Yes |
| 8 | MS-SHT-30 min-100 | 49 | No | 0.5 | 100 | Yes |
| 9 | MS-SHT-1 h-100–71% | 60 | No | 1 | 100 | Yes |
| 10 | MS-SHT-1 h-100–86% | 70 | No | 1 | 100 | Yes |
| 11 | MS-SHT-1 h-100–100% | 80 | No | 1 | 100 | Yes |
| 12 | MS-HT-25 min-100 | 49 | No | 0.4 | 100 | No |
| 13 | MS-HT-1 h-100 | 49 | No | 1 | 100 | No |
| 14 | MS-HT-24 h-100 | 49 | No | 0.25 | 100 | No |
| 15 | MS-HT-48 h-100 | 49 | No | 48 | 100 | No |
| 16 | MS-SHT-15 min-100 | 49 | No | 0.25 | 100 | Yes |
| 17 | MS-SHT-1 h-100 | 49 | No | 1 | 100 | Yes |
| 18 | MS-SHT-25 min-80 | 49 | No | 0.4 | 80 | Yes |
| 19 | MS-HT-16 h-150 | 49 | No | 16 | 150 | No |
| 20 | MS-HT-25 min-100–1000 | 49 | No | 0.4 | 100 | No – 1000 rpm |
| 21 | MS-HT-30 min-100–1000 | 49 | No | 0.5 | 100 | No – 1000 rpm |
| 22 | MS-HT-1 h-100–1000 | 49 | No | 1 | 100 | No – 1000 rpm |
2.3. Specimen characterization
All obtained materials were characterized using the XRD technique on a Bruker D8-Advance device equipped with a Lynx eye detector. Analyses were conducted using Cu Kα radiation (λ = 1.5405 Å) over the 5–90° 2θ range with a step size of 0.01° and 1 s time per step. XRD patterns were corrected with Cu-Kα2 radiation stripping and background subtraction using a Diffrac.Eva V5.2 software integrated procedure. Crystallite size was estimated using the Scherrer equation with the FWHM determination method at the 30° 2θ value with a K constant of 0.89 as a first approximation.42 Sample morphology and particle sizes of the as-synthesized samples were assessed by SEM analyses using an FEI Quanta FEG 200 ESEM device operating at 20 kV without surface metallization. Crystal size distribution was determined using FIJI software with at least 25 to 60 particles analyzed on the basis of SEM image acquisitions by assuming that zeolite A crystals were perfect cubes.2.4. Acoustic noise measurement
Acoustic noise spectra were recorded using a Brüel & Kjær hydrophone (model 8103) calibrated in the frequency range of 0.1 Hz to 180 kHz with a receiving sensitivity of 211 dB per 1 V μPa−1. The hydrophone was positioned reproducibly at 9 cm from half the height of the SHT reactor (Fig. S4†). The microphone signal was recorded on a PicoScope 3424 oscilloscope with a resolution of 1 mV for the 2 V input signal after amplification by the Brüel & Kjær 2525 amplifier. The Fourier transform was calculated using the oscilloscope internal algorithm. Acoustic amplitude was measured in dBu with a reference voltage of 0.7746 V. Recordings were made every 10 minutes, enabling us to follow the evolution of noise in the reactor throughout the synthesis process as a function of the compound being formed.2.5. Gel viscosity measurement
Gel viscosity was determined using an RM 200 touch rheometer from Lamy Technology. Measurements were made using the Visco20 s−1 method with the CP4020 measuring system, allowing the sample to be heated up to 70 °C. The analysis lasted 60 s with a pre-shear time of 20 s and a shear time of 5 s, with data acquisition every second.3. Results and discussion
3.1. Influence of the ultrasonic irradiation during gel precipitation and heat treatment
Samples prepared at 100 °C for 4 hours, with or without the application of ultrasonic irradiation during gel formation or heat treatment (experiments #1 to #4), were first characterized by X-ray diffraction. XRD patterns shown in Fig. 1 reveal the presence of several intense diffraction peaks at 7.2°, 10.2°, 24.0° and 30.0° 2θ values, which are characteristic of the (200), (220), (622) and (644)/(820) zeolite A planes, respectively, according to the XRD database (COD 8104214). Whether or not ultrasound was applied, heating the aluminosilicate gel at 100 °C for 4 hours leads to the formation of the zeolite A phase in agreement with the literature.39 | ||
| Fig. 1 XRD patterns of the samples obtained after heat treatment at 100 °C during 4 hours under static hydrothermal treatment or SHT conditions with or without ultrasonic irradiation at 20 kHz during aluminosilicate gel formation (Experiments #1 to #4). Blue arrows indicate the main diffraction peaks of zeolite A (COD 8104214) while the dashed line refers to (110) and (211) sodalite plane reflections (COD 9003330). | ||
Further analyses of the XRD patterns shown in Fig. 1 reveal that a diffraction peak at the 13.9° 2θ value can only be observed when the sample is treated under the joint action of hydrothermal treatment and ultrasonic irradiation. According to the crystallography open database, this peak indicates the formation of the sodalite compound due to the reflection of the (110) plane in MS-SHT-4 h-100 and US-SHT-4 h-100 samples (COD 9003330). A closer examination of these XRD patterns also reveals a strong decrease of the main zeolite A diffraction peak intensity while the formation of sodalite seems to increase with the concomitant appearance of the sodalite reflection line (211) at 24.2° 2θ.
These observations could be confirmed by scanning electron microscopy analyses as shown in Fig. 2. SEM images of the two samples heated under hydrothermal conditions, i.e. MS-HT-4 h-100 and US-HT-4 h-100, prove the formation of cubic particles within the range of 1–2 μm whether or not ultrasonic irradiation was applied during the gel formation stage (Fig. 2A and C). This cube morphology is commonly observed for the zeolite A phase and could also be observed in the two SHT treated samples (Fig. 2B and D). Nevertheless, SEM images of the samples obtained after sonohydrothermal treatment (MS-SHT-4 h-100 and US-SHT-4 h-100) display a mixture of cubic crystals of zeolite A and desert rose-shaped particles of sodalite. In agreement with the XRD patterns (Fig. 1), the amount of sodalite formed within the sample prepared under ultrasonic irradiation during both the precipitation and heat treatment stages (US-SHT-4 h-100) seems to be higher than that for the sample obtained after mechanical formation of the amorphous aluminosilicate gel and SHT treatment (MS-SHT-4 h-100).
 | ||
| Fig. 2 SEM images of the samples obtained after heat treatment at 100 °C during 4 hours under static hydrothermal treatment or SHT conditions with (C and D) or without (A and B) ultrasonic irradiation at 20 kHz during aluminosilicate gel formation (Experiments #1 to #4). | ||
These results suggested that after 4 hours of treatment under the joint action of 20 kHz ultrasound and hydrothermal conditions at 100 °C, the sodalite phase could crystallize at the expense of the initially formed zeolite A phase. Phase transformation of the metastable zeolite A exhibiting a lower framework density into the more thermodynamically stable sodalite with a higher framework density is expected according to Ostwald's law and is actually well documented in the literature, but it usually requires much longer reaction treatment times (more than 22 h) or higher temperatures (above 100 °C).39,43
In order to better understand the crystallization of the amorphous hydrogel to zeolite A and its further phase transformation into sodalite, additional experiments were conducted at shorter SHT treatment times of 25 min (MS-SHT-25 min-100) and 30 min (MS-SHT-30 min-100). Supplementary conditions under silent hydrothermal treatment were assessed by increasing the crystallization temperature to 150 °C (MS-HT-4 h-150) and extending the duration of the heat treatment to 16 h (MS-HT-16 h-100). For this part of the study, the gel formation stage was only achieved under strong mechanical stirring since no dramatic effect of the ultrasonic irradiation, during this first stage, had been demonstrated previously (experiments MS-SHT-4 h-100 and US-SHT-4 h-100, Fig. 1, 2B and D).
3.2. Influence of the heating temperature and treatment time on the sodalite phase transformation
The XRD patterns of the specimens prepared under hydrothermal conditions with an extended treatment time (MS-HT-16 h-100) and a higher heating temperature (MS-HT-4 h-150) are shown in Fig. 3. XRD analyses reveal that even after 16 h of heat treatment at 100 °C, no sodalite phase could be detected within the sample #6 diffraction pattern composed solely of zeolite A reflection lines (Fig. 3). Cubic crystals of about 2 μm with sharp edges were observed from SEM analyses (Fig. 4A), confirming the sole presence of zeolite A in the MS-HT-16 h-100 sample in agreement with the XRD results. | ||
| Fig. 3 XRD patterns of the samples obtained at shorter SHT treatment times of 25 min (#7 MS-SHT-25 min-100) and 30 min (#8 MS-SHT-30 min-100) and under classical hydrothermal conditions at higher heating temperature (#5 MS-HT-4 h-150) and for extended reaction time (#6 MS-HT-16 h-100). Asterisk symbol indicates the presence of the sodalite diffraction peak according to COD 9003330. | ||
 | ||
| Fig. 4 SEM images of the samples obtained at shorter SHT treatment times of 25 min (#7 MS-SHT-25 min-100) (C) and 30 min (#8 MS-SHT-30 min-100) (D) and under classical hydrothermal conditions at higher heating temperature (#5 MS-HT-4 h-150) (B) and for extended reaction time (#6 MS-HT-16 h-100) (A). | ||
Increasing the heating temperature under hydrothermal conditions from 100 °C to 150 °C for 4 hours leads to similar results and conclusions. The as-synthesized sample MS-HT-4 h-150 exhibits an XRD pattern very similar to that obtained for samples MS-HT-16 h-100 and MS-HT-4 h-100, characteristic of the zeolite A phase. As a matter of fact, the sodalite reflection line, at the 14° 2θ value, was hardly visible on the raw XRD pattern of sample #5 and could not be pinpointed in the XRD background-subtracted and Cu-Kα2 stripped data shown in Fig. 3. However, further characterization by SEM revealed the presence of very few desert rose-like sodalite particles among the zeolite A cubes for the specimen MS-HT-4 h-150 (Fig. 4B). Increasing the reaction time from 4 h to 16 h at 150 °C under hydrothermal conditions leads to the partial transformation of zeolite A into sodalite, as ascertained by the very intense reflection line at 14° (the MS-HT-16 h-150 sample in Fig. S6†). As already stated, this is in line with previously reported observations that higher crystallization temperatures favor the zeolite A phase transformation into sodalite.43 Thus, even for extended synthesis times (16 h) at 150 °C under HT conditions, the transformation of zeolite A into sodalite is not complete, giving rise to a mixture of the two phases.
XRD analysis was next performed on samples synthesized under sonohydrothermal conditions at 100 °C with treatment times as short as 15 minutes (MS-SHT-15 min-100, Fig. S6†), 25 minutes (MS-SHT-25 min-100, Fig. 3) and 30 minutes (MS-SHT-30 min-100, Fig. 3).
At the early synthesis time (15 min), the obtained sample remains mainly amorphous with weak diffraction reflections characteristic of zeolite A as observed in the XRD pattern (MS-SHT-15 min-100 Fig. S6†). However, when the sonohydrothermal treatment is extended from 15 minutes to 25 and 30 minutes, all the reflection lines of zeolite A can be unequivocally identified by XRD (Fig. 3). Note here that the XRD patterns shown in Fig. 1 and 3 are background subtracted and kα2 stripped, meaning that the amorphous part on the pattern is no longer visible. Nevertheless, according to the original raw files available in the ESI (Fig. S6†), the crystallization of the amorphous gel into the zeolite A phase under our studied conditions is almost complete after a reaction time as short as 25 min under SHT conditions at 100 °C. This significant result was further proved by the SEM images shown in Fig. 4C. Even more interestingly, similar results could be obtained by reducing the synthesis temperature from 100 °C to 80 °C. Indeed, almost complete conversion of the amorphous hydrogel to zeolite A was also observed after a reaction time of just 25 minutes at 80 °C under sonohydrothermal conditions as shown by the XRD analysis in Fig. S6† (MS-SHT-25 min-80).
Surprisingly, when the synthesis time was extended to 30 minutes (MS-SHT-30 min-100 sample), the conversion of zeolite A to sodalite had already started, as illustrated by the appearance of the XRD reflection at 14° 2θ (Fig. 3) and the SEM image (Fig. 4D).
Therefore, it should be highlighted that only a 30 min treatment at 100 °C under SHT conditions can induce the phase transformation from zeolite A into sodalite, while the sodalite phase could not be observed in the sample synthesized by hydrothermal heating for 16 hours at the same temperature. Further experiments were thus carried out at 100 °C under hydrothermal treatment for extended reaction times from 24 to 48 hours (MS-HT-24 h-100 and MS-HT-48 h-100). XRD patterns obtained for these two specimens reveal the sole presence of the zeolite A phase with sharp diffraction peaks without any traces of the sodalite phase (Fig. S6†). The drastic kinetic increase of phase transformation from low framework density zeolite A into higher framework density sodalite under SHT conditions is not yet fully understood and further experiments at various acoustic power densities were therefore conducted.
3.3. Effect of applied acoustic power density during heat treatment on the phase transformation
Due to the high viscosity of the amorphous gel precipitated under our conditions, additional experiments were conducted at different values of acoustic power density (applied acoustic power per unit of volume) while maintaining the same ultrasonic amplitude of 70% as in the previous syntheses. The ultrasonic amplitude corresponds here to the maximum physical displacement of the ultrasonic probe and implies, under our conditions, a delivered acoustic power of 19 W measured at near room temperature. In order to study the influence of the acoustic power density on sodalite and zeolite A formation, the total reaction volume in the sonohydrothermal reactor was increased from 49 mL to 60, 70 and finally 80 mL, resulting in a decrease of the applied power density. Under such conditions, the reactor filling rates rise from 61% to respectively 71%, 86% and 100%. One may therefore expect a drastic increase of the autogenous pressure measured within the system. However, due to the design of our SHT reactor and the presence of a dead gas volume, even with a filling rate of 100%, only a slight increase in internal pressure from 0.5 to 1 bar was observed at 100 °C.The SEM image and XRD pattern of the sample treated for 1 h at 100 °C under SHT conditions with a 71% filling rate (MS-SHT-1 h-100–71%) unequivocally show the presence of both zeolite crystals and the sodalite phase (Fig. 5A and D). This is in line with the SHT treatment experiment previously carried out with a reactor filling rate of 61% for 30 min (MS-SHT-30 min-100) or 1 h (MS-SHT-1 h-100) as demonstrated in the ESI (Fig. S5 and S6†). Interestingly, increasing the reactor filling rate to 86% (MS-SHT-1 h-100–86%) leads to a decrease in the XRD sodalite signature while very intense reflection lines of zeolite A could be identified (Fig. 5D). In agreement with XRD analyses, both zeolite A and sodalite phases are easily visible in the SEM image of MS-S HT-1h-100–86% (Fig. 5B). Finally, when the reactor is completely filled, meaning that the power density is the lowest, a strong decrease of zeolite A diffraction peak intensities and the disappearance of the sodalite XRD signature can be noticed in sample MS-SHT-1 h-100–100% (Fig. 5D). SEM analyses, depicted in Fig. 5C, confirm this observation since no regularly shaped particles were found in this specimen obtained after SHT treatment in a completely filled reactor.
 | ||
| Fig. 5 SEM images (A–C) and XRD patterns (D) of the samples obtained after SHT treatment for 1 hour at 100 °C with a reactor filling rate of 71%, 86% and 100% (Experiments # 9 to #11). Arrows indicate the position of the sodalite diffraction peak according to COD 9003330. | ||
This set of experiments highlighted the crucial importance of the acoustic power density in the crystallization of aluminosilicate hydrogel into zeolite A and the subsequent phase transformation into sodalite. The main effect of ultrasonic irradiation under hydrothermal conditions may be related to the enhancement of mass transfer and mixing of the reaction medium induced by acoustic cavitation. Further experiments were therefore conducted to investigate the effect of stirring under hydrothermal conditions on zeolite A formation and phase transformation into sodalite.
3.4. Effect of stirring during hydrothermal synthesis
To estimate the influence of mixing conditions on the phase formation of zeolite A and sodalite, stirred experiments were carried out at 100 °C under magnetic stirring at 1000 rpm for 25 min (MS-HT-25 min-100–1000), 30 min (MS-HT-30 min-100–1000) and 1 hour (MS-HT-1 h-100–1000). XRD analysis of the sample obtained under stirred HT conditions after a reaction time of 25 min indicates that the specimen remains amorphous, with an XRD pattern very comparable to what we obtained under static hydrothermal conditions (Fig. 6). Increasing the reaction time to 30 min (MS-HT-30 min-100–1000) leads to the appearance of very weak reflection lines characteristic of zeolite A. It is important to remember that under sonohydrothermal conditions, the complete conversion of the amorphous hydrogel to zeolite A was observed after just 25 min (MS-SHT-25 min-100) and the start of the zeolite A phase transformation into sodalite was noticeable after 30 min of treatment (MS-SHT-30 min-100), as shown in Fig. 3. Interestingly, after 1 hour of hydrothermal treatment under static conditions, the sample remains totally amorphous, as observed in the XRD diagram (Fig. 6), whereas under stirring conditions, clear diffraction peaks attributed to the zeolite A phase are observed. It should be noted here that, unlike the sample sonohydrothermally synthesized for 1 hour (MS-SHT-1 h-100), no traces of sodalite could be detected in the sample synthesized under stirred hydrothermal conditions (MS-HT-1 h-100–1000). Finally, these results allow us to assert that the improvement of mass transfer under hydrothermal conditions plays a significant role in crystallization of the amorphous gel to zeolite A. Nevertheless, despite a rotation speed of 1000 rpm, the effects observed under stirring are much less than those under the action of 20 kHz ultrasonic irradiation.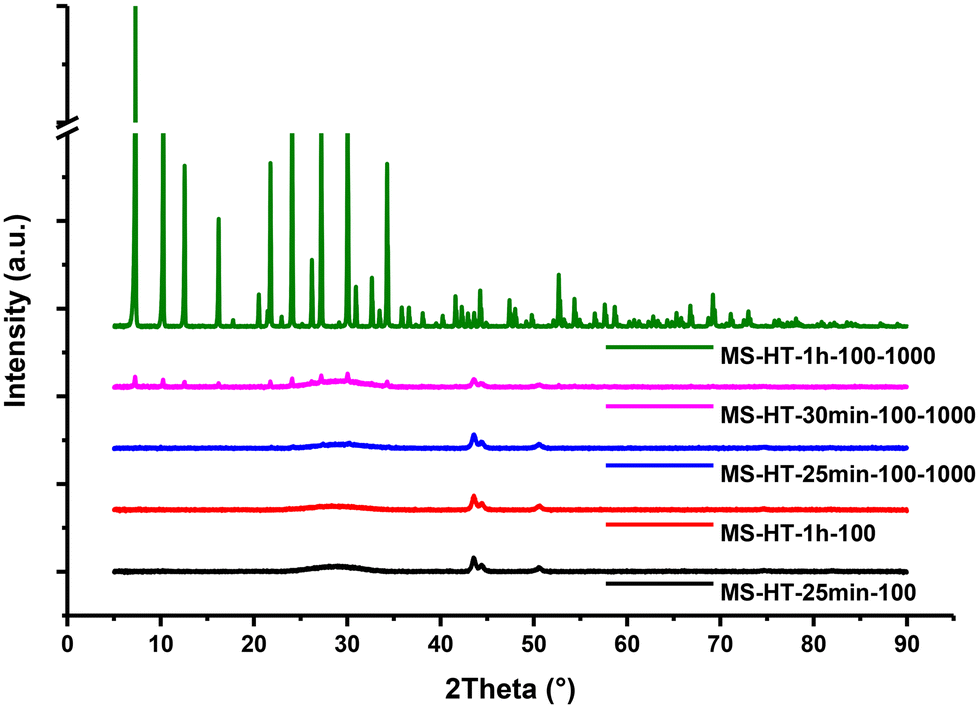 | ||
| Fig. 6 XRD patterns of the samples obtained under static hydrothermal conditions at 100 °C after a reaction time of 25 min (#12 MS-HT-25 min-100) and 1 hour(#13 MS-HT-1 h-100) compared to stirred hydrothermal conditions at 100 °C with a rotation speed of 1000 rpm after a reaction time of 25 min (#20 MS-HT-25 min-100–1000), 30 min (#21 MS-HT-30 min-100–1000) and 1 hour (#22 MS-HT-1 h-100–1000). | ||
3.5. Acoustic noise spectrum analysis
In order to assess the evolution of the reaction medium during the SHT treatment, acoustic noise spectrum analyses were conducted during zeolite synthesis at 100 °C under SHT conditions for 1 h (MS-SHT-1 h-100) and compared to pure water data. The acoustic noise spectrum recorded in pure water at 100 °C (Fig. 7A) exhibits a strong fundamental frequency band at 20 kHz (f0) and several harmonics nf0 (n = 2–6) associated with nonlinear bubble oscillations in agreement with recently published data.44 In contrast, Fig. 7B reveals a significant effect of the SHT treatment time on the acoustic noise spectra recorded during zeolite A synthesis. The spectrum recorded after 10 min of SHT treatment is strongly attenuated. The ratio of band intensities to background noise is lower than for pure water and some harmonics (namely n = 4–6) have completely disappeared. However, the acoustic spectra changed rapidly during SHT treatment. In fact, after 20 min of SHT treatment, the relative intensity of the harmonics is increased and the harmonic bands n = 4f0 and n = 5f0 reappear. Another harmonic band n = 6f0 reappears after 60 min of treatment and the acoustic spectrum of the reaction mixture becomes similar to that in pure water. We can assume that the acoustic spectrum evolution during SHT treatment is due to the change in solution viscosity during the synthesis process. At the first stage, the viscosity of the solution is expected to increase due to gel formation. The attenuation of acoustic modes as a function of liquid viscosity was first discussed by Rayleigh in 1945 and then studied by many research groups.45–47 It was highlighted that the viscosity of the medium significantly affects wave attenuation, in particular at high frequency. Nevertheless, even at low ultrasonic frequencies, the increase in medium viscosity hinders the acoustic propagation and leads to a reduction in the active cavitation zone size observed from 50 mPa s for 30 kHz ultrasonic irradiation.48 | ||
| Fig. 7 Acoustic noise spectrum of water at 100 °C under SHT treatment (A) and evolution in time of the acoustic noise spectra during the zeolite synthesis at 100 °C under SHT treatment (B). | ||
To verify this hypothesis, the viscosity of the reaction medium was measured at different temperatures. At 25 °C, the gel viscosity measured was 93.3 mPa s, which is almost one hundred times higher than that of pure water. When the aluminosilicate hydrogel is heated during SHT treatment, the viscosity increases during the first minutes of treatment. At 100 °C, the viscosity measured was 111 mPa s. This increase in viscosity during the first few minutes is due to the polycondensation of the silicate anions.49 The signal attenuation observed after 10 minutes of SHT processing (Fig. 7B) results from silica polymerization and inorganic polymer chain formation in the reaction medium. Using X-ray diffraction, we have shown that the system begins to crystallize within 20 minutes in the SHT reactor (gel conversion was complete after 25 min of treatment, as shown in Fig. 3). The conversion of the gel into zeolite A particles tends to reduce the viscosity of the medium. This is what we observe at 20 minutes of treatment time (Fig. 7B), when we see the reappearance of the harmonic bands, indicating better propagation of the ultrasonic waves. After 1 h of SHT treatment, the mixture obtained is composed of sodalite and zeolite A crystals (Fig. 3). At the end of SHT treatment, the measured viscosity of the suspension was 91.3 mPa s. It is important to note that the acoustic spectra clearly show the presence of cavitation in the whole range of studied SHT conditions.
3.6. General discussion
The results obtained in this study provide various insights into the effect of ultrasonic irradiation on both the aluminosilicate gel transformation into zeolite A and its further evolution into sodalite. First, a more detailed analysis of the SEM images allows us to determine the zeolite A crystal size distribution based on the experimental conditions assessed here, as reported in Fig. 8. Fig. 8C and D show that the application of 20 kHz ultrasonic irradiation during the precipitation step, prior to classical hydrothermal treatment, results in a slight decrease in particle size, with histograms showing a more homogenous and centered particle size distribution for the US-HT-4 h-100 sample compared to the MS-HT-4 h-100 compound. Pretreatment of the amorphous gel prior to sonohydrothermal treatment seems less decisive since both SHT treated samples for 4 h at 100 °C show advanced conversion into sodalite, making the size distribution determination of zeolite A crystals difficult (Fig. 8A and B) due to the abundant presence of desert rose crystals at the surface of the cubic particles (Fig. 2B and D). Fig. 8A and B data should therefore be treated with caution. | ||
| Fig. 8 Crystal size distribution of the zeolite A obtained under experimental conditions #1 to #8 as listed in Table 1 (A to H). Note that for samples #4 US-SHT-4 h-100 (A) and #3 MS-SHT-4 h-100 (B) (dashed columns) the abundant presence of sodalite hindered the determination of zeolite A particle size by masking the zeolite A particles. | ||
As expected, longer reaction times result in larger crystals, regardless of the experimental conditions. Thus, increasing the reaction time from 25 min (MS-SHT-25 min-100) to 30 min (MS-SHT-30 min-100) and finally to 4 hours (MS-SHT-4 h-100) under sonohydrothermal conditions results in an increase in zeolite A crystal size from 0.81 μm to 1.1 μm. A similar conclusion can be drawn from the particle size distribution of the samples obtained under conventional hydrothermal treatment. Indeed, a comparison of the size distribution of MS-HT-4 h-100 and MS-HT-16–100 crystals reveals an increase in size from 1.37 μm to 1.64 μm consistent with the Ostwald ripening process.
Zeolite A crystallite sizes determined from XRD data using Scherrer's equation were also achieved for experiments #1 to #8 and are presented in Fig. 9. These results suggest that ultrasonic irradiation performed during the formation of the amorphous hydrogel leads to the reduction of the zeolite A crystallite size from 132 nm (MS-HT-4 h-100) to 123 nm (US-HT-4 h-100) for hydrothermally treated samples and, to a lesser extent, from 114 nm (MS-SHT-4 h-100) to 109 nm (US-SHT-4 h-100) for materials obtained under sonohydrothermal conditions at 100 °C. Note here that zeolite A crystallite sizes obtained under hydrothermal conditions for an extended treatment time (MS-HT-16 h-100) or at a higher heating temperature (MS-HT-4 h-150) remain within the same range as for MS-HT-4 h-100, that is, around 130 nm (Fig. 9). However, in these cases, zeolite A exhibits particle sizes greater than 2 μm (Fig. 8E and F). On the other hand, a shorter SHT treatment leads to a decrease in both the zeolite A crystallite size down to 95 nm for the 25 min treatment and also the zeolite A particle size to around 1 μm (MS-SHT-25 min-100 and MS-SHT-30 min-100 Fig. 8G and H).
 | ||
| Fig. 9 Evolution of zeolite A crystallite sizes determined by Scherrer's equation in function of the hydrothermal and sonohydrothermal temperature and treatment time (experiments #1 to #8 as listed in Table 1). | ||
To our delight, the influence of ultrasonic irradiation on the phase evolution during the heat treatment of the amorphous gel is of a completely different order of magnitude compared to that under silent conditions. As a matter of fact, sonohydrothermal treatment appears to considerably enhance the crystallization of zeolite A. Indeed, the XRD patterns shown in Fig. S6† reveal the presence of zeolite A reflections after only a 15 min sonohydrothermal treatment of the aluminosilicate hydrogel. The complete conversion of the amorphous gel to fully crystalline zeolite A was observed in less than 25 min under SHT conditions, whereas after a 1-hour conventional hydrothermal treatment, the presence of zeolite A could not be revealed by XRD diffraction. This demonstrates a significant improvement in crystallization kinetics thanks to the combined action of 20 kHz ultrasound and hydrothermal conditions (a 9.6-fold kinetic gain). In the same way, the subsequent phase transformation of zeolite A into sodalite started after a reaction time two orders of magnitude shorter under sonohydrothermal conditions than during silent hydrothermal treatment at 100 °C. It is important to underline that applying ultrasonic irradiation in highly viscous heterogeneous systems, such as an amorphous gel with a viscosity of around 100 mPa S, can induce a drastic effect on the hydrogel crystallization rate and phase conversion despite the attenuation of the ultrasonic irradiation due to the medium. The reason for such a dramatic kinetic increase is not fully understood; however, some assumptions can be drawn. First, it can be stated that ultrasonic irradiation during the heat treatment leads to the fragmentation of the amorphous gel into smaller aggregates, resulting in a greater number of zeolite A nuclei. This statement, which is based on the sonofragmentation of crystalline material due to the strong local physical effects induced by cavitation bubbles, is commonly accepted by the scientific community.30 Here, it is suggested that the sonofragmentation occurred during the formation of amorphous aluminosilicate hydrogel. Ultrasonic irradiation treatment would thus lead to a larger amount of smaller zeolite A crystallites also exhibiting a faster crystallization rate, as observed in this study when the acoustic power density was varied (experiments #9 to #11). This second point can be explained by the enhanced mass transfer of the building units at the interface between the crystallite surface and the solution, resulting from the acoustic cavitation activity under SHT conditions. Indeed, when comparing static and stirred hydrothermal conditions, it was observed that materials synthesized under magnetic stirring at 1000 rpm showed better crystallinity than those synthesized under static conditions. Nevertheless, crystallization of the amorphous gel to zeolite A was very weak and not comparable to that obtained under sonohydrothermal conditions. The mass transfer and mixing effect induced by acoustic cavitation may be one of the main reasons for this dramatic increase in the rate of phase transformation. Furthermore, it has already been observed for other crystalline materials that local heating around cavitation bubbles could also enhance phase transformation and induce crystallization under ultrasonic irradiation.50 In the end, the zeolite crystals have smaller particles and a more homogenous particle size distribution than the hydrothermally treated samples, as observed here.
On the other hand, to explain the influence of ultrasonic irradiation on the transformation of zeolite A into sodalite, two different approaches can be considered, namely the classical growth theory and the reversed growth mechanism. Based on the classical growth approach (Fig. 10), the transformation of zeolite A into sodalite results from a dissolution–reprecipitation mechanism in solution rather than a direct solid state transformation of zeolite A into sodalite, which has been established as very unlikely.39 Therefore, the dissolution of zeolite A and the formation of the more thermodynamically stable sodalite would benefit from both the enhanced mass transfer rate induced by the acoustic cavitation phenomenon and the local heating effect. Recently, the second approach has been proposed in order to explain the formation of zeolite A and its further transformation into sodalite, based on a reversed crystal growth mechanism in the presence of organic or inorganic molecules rather than the classical Ostwald ripening (Fig. 10).51 This unusual mechanism was also considered by other authors in order to explain the effect of ultrasonic irradiation applied during amorphous gel formation as a pretreatment for the formation of zeolite A and its long time conversion into sodalite at room temperature.34 In brief, zeolite A would first crystallize on the surface of the amorphous gel aggregates, and after orientated aggregation of the crystallites, the formation of core–shell cubic particles with the amorphous gel remaining inside would be observed. Surface crystallization would then increase the pressure inside the particle core, leading to sodalite formation and expansion at the expense of the zeolite A phase. This reversed crystal growth approach can also be considered here, since SHT treatment has already been proved to induce oriented aggregation rather than the Ostwald ripening phenomenon.36 Thus, strong mixing of the reaction medium during heat treatment could lead to the rapid formation of small amorphous gel@zeolite A core–shell particles and their further conversion to sodalite. The formation of smaller cube-shaped particles would be beneficial for the conversion of the sodalite phase since the pressure gradient induced on the amorphous core would be more significant in smaller particles. This approach could therefore explain the fast appearance of sodalite under sonohydrothermal conditions, even at early stages.
 | ||
| Fig. 10 Suggested mechanism pathways for the amorphous aluminosilicate gel transformation into zeolite A and sodalite compared to the classical crystal growth route (adapted from Greer et al.51). | ||
To determine whether the conversion mechanism of zeolite A to sodalite could be driven by this reversed growth mechanism, TEM analyses were carried out on a specimen obtained after a SHT treatment of 3 h at 100 °C with a lower initial amount of aluminosilicate gel (dilution factor of 15). This protocol was considered to synthesize smaller zeolite crystals with a mixture of zeolite A and sodalite in order to determine whether the desert rose structures are located inside or next to the cubic zeolite A particles. As shown in Fig. 11, both typical sodalite and zeolite A morphologies can be observed in this sample, in agreement with the previous SEM and XRD analyses. Nevertheless, it is difficult to state whether the sodalite phase is growing outwards from the core of the zeolite A cubes or is just located nearby due to the potential particle superposition inherent in TEM sample analyses.
 | ||
| Fig. 11 TEM images (A and B) of the sample obtained after a 3 h SHT treatment at 100 °C from an aluminosilicate gel 15 times more diluted than our reference experimental protocol (see text for explanation). | ||
Based on the reversed crystal growth mechanism proposed by Greer et al. (Fig. 10), sodalite crystals are expected to nucleate within the amorphous gel inside the core of the zeolite A cubes. This means that the composition of the cubic particles should be somewhere between the composition of the initial aluminosilicate gel, with a Si/Al ratio of 0.5, and that of zeolite A, with a Si/Al ratio close to 1. Elemental profiles were therefore acquired during TEM analyses on zeolite A particles observed in the sample synthesized from a diluted aluminosilicate gel and are reported in Fig. 12. These results clearly show that the silicon to aluminum ratio remains constant and equal to 1 for both analyses made on zeolite A cubic particles (Fig. 12A and B). This observation indicates that the cubic crystals are homogeneous and not composed of an amorphous aluminosilicate gel core and a crystalline shell. Nevertheless, it is important to note that the Si/Al ratio for sodalite is also equal to 1, as shown in Fig. 12A. Therefore, it cannot be excluded that the particle core is already converted into sodalite and is no longer composed of amorphous aluminosilicate gel.
 | ||
| Fig. 12 Elemental profiles obtained on the 3 hours SHT treated sample at 100 °C exhibiting two different crystal morphologies characteristic of sodalite and zeolite A (A) and zeolite A alone (B). | ||
Moreover, probing the core of the material is all the more difficult as the samples analyzed here are thick and only partially penetrated by the TEM electron beam. It should also be noted that TEM analyses are highly localized and not necessarily representative of the entire sample.
Similarly, even though the silicon to aluminum ratio is initially close to 0.5 in solution, EDX analyses carried out on the freshly precipitated amorphous gel and after 15 min of heat treatment revealed that the Si/Al ratio could vary between 0.5 and 1 before heat treatment and can reach 1.5 after 15 minutes at 100 °C. These observations highlight that determining the nature of the solid phase on the basis of this elemental ratio is tricky. At this stage, there is no evidence that the formation of zeolite A and its subsequent conversion to sodalite follow the reversed growth mechanism, so the classical growth theory seems more plausible under SHT conditions.
According to our study, the main effects of the sonohydrothermal treatment on the synthesis of zeolite A and its phase transformation into sodalite are related to (i) the sonofragmentation of the amorphous aluminosilicate hydrogel, leading to smaller gel aggregates, (ii) the enhancement of acoustic cavitation induced mass transfer, leading to a faster crystallization rate of the hydrogel into zeolite A and (iii) the increased dissolution–reprecipitation rates during the phase transformation of zeolite A into sodalite. It should be noted here that local heating around the cavitation bubble might also contribute to the dramatic increase in reaction rate observed during the crystallization of the amorphous gel, explaining the almost complete conversion of the hydrogel in just 25 min at 80 °C under sonohydrothermal conditions.
The synthesis technique developed here, compared with the conventional hydrothermal treatment, offers major advantages such as a drastic reduction in the reaction time required to synthesize zeolite A by an order of magnitude. The simultaneous coupling of hydrothermal conditions with ultrasonic irradiation can therefore be described as a time- and energy-saving technique that produces smaller and more homogeneous zeolite crystals. However, the main limitation lies in the design of the reactor, which is still developed as a small-volume batch reactor.
As a final remark, this entire study was carried out using a viscous hydrogel as the starting material. Further experiments with systems of lower or higher viscosity could be envisaged to explore the extent to which the viscosity of the medium can have an effect on ultrasonic irradiation, and vice versa, and finally on the rate of crystallization of the hydrogel to zeolite A and its phase conversion to the more stable sodalite.
4. Conclusion
Our work highlighted that the simultaneous coupling of hydrothermal conditions with 20 kHz power ultrasound has proved to be an innovative alternative for zeolite synthesis. Conversion of the amorphous aluminosilicate gel into crystalline zeolite A under SHT treatment at 100 °C has been shown to be dramatically faster, exhibiting a 9.6-fold kinetic gain, under the joint action of the ultrasonic irradiation and hydrothermal conditions compared to a classical hydrothermal synthesis. The almost complete conversion of the aluminosilicate gel to zeolite A could also be achieved in just 25 min at 80 °C under sonohydrothermal conditions. Ultrasonic treatment during the amorphous gel crystallization also leads to smaller crystallites (30% reduction) and zeolite A particles (50% smaller), exhibiting a more homogeneous particle size distribution (mean deviation three times lower) than the zeolite synthesized by hydrothermal treatment. These observations can be explained by the sonofragmentation of the amorphous gel and a concomitant enhanced mass transfer of the building units at the crystallite surface due to the acoustic cavitation activity under SHT conditions. Finally, a drastic kinetic increase, of two orders of magnitude, in the transformation of zeolite A into the more stable sodalite was also observed during sonohydrothermal treatment, compared to silent hydrothermal conditions, highlighting the critical importance of the joint action of ultrasonic irradiation and hydrothermal conditions.Author contributions
William's Nzodom Djozing: Experiments, analyses, data treatment and writing. Sabine Valange: Design of experiments, discussion, supervision, review and editing. Sergueï Nikitenko: Discussion, supervision, review and editing. Tony Chave: Design of experiments, experiments, data collection and treatment, analyses, supervision, and writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
No conflicts of interest are declared by the authors.Acknowledgements
William's Nzodom Djozing is grateful to the University of Montpellier for PhD position and funding. PEPR-ECOCHEM and France 2030 are also acknowledged for their financial support (ANR-22-PESP-0006). Sabine Valange acknowledges financial support from the European Union (ERDF) and Région Nouvelle Aquitaine. This work pertains to the French government program “Investissements d'Avenir” (EUR INTREE, reference ANR-18-EURE-0010). All the authors would like to acknowledge Xavier Le Goff for TEM measurements.References
- K. Stocker, M. Ellersdorfer, M. Lehner and J. G. Raith, BHM, Berg- Huettenmaenn. Monatsh., Suppl., 2017, 162, 142–147 CrossRef CAS.
- Q. Wu, H. Luan and F.-S. Xiao, Natl. Sci. Rev., 2022, 9, nwac023 CrossRef CAS PubMed.
- S. Wang and Y. Peng, Chem. Eng. J., 2010, 156, 11–24 CrossRef CAS.
- C. Rios, C. Williams and M. Fullen, Appl. Clay Sci., 2009, 42, 446–454 CrossRef CAS.
- K. M. Leung, P. P. Edwards, E. Jones and A. Sartbaeva, RSC Adv., 2015, 5, 35580–35585 RSC.
- D. W. Breck, Zeolite Molecular Sieves, Wiley-Interscience, New York, 1974 Search PubMed.
- S. Alfaro, C. Rodríguez, M. A. Valenzuela and P. Bosch, Mater. Lett., 2007, 61, 4655–4658 CrossRef CAS.
- A. Soualah, M. Berkani and M. Chater, C. R. Chim., 2004, 7, 713–720 CrossRef CAS.
- P. Reinert, C. Schott-Darie and J. Patarin.
- Y. Pan, M. Ju, J. Yao, L. Zhang and N. Xu, Chem. Commun., 2009, 7233, 10.1039/b917949f.
- Y. Wang, T. Nishitoba, Y. Wang, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, H. Gies, D. De Vos, U. Kolb, M. Feyen, R. McGuire, A.-N. Parvulescu, U. Müller and T. Yokoi, Ind. Eng. Chem. Res., 2020, 59, 7375–7382 CrossRef CAS.
- B. Liu, H. Kita and K. Yogo, Sep. Purif. Technol., 2020, 239, 116533 CrossRef CAS.
- D. O. Shestakova, K. A. Sashkina and E. V. Parkhomchuk, Pet. Chem., 2019, 59, 838–844 CrossRef CAS.
- A. Nakhaei Pour and A. Mohammadi, J. Inorg. Organomet. Polym. Mater., 2021, 31, 2501–2510 CrossRef CAS.
- F. J. Machado, C. M. López, M. a. A. Centeno and C. Urbina, Appl. Catal., A, 1999, 181, 29–38 CrossRef CAS.
- J. Grand, H. Awala and S. Mintova, CrystEngComm, 2016, 18, 650–664 RSC.
- E.-H. Yuan, R. Han, J.-Y. Deng, W. Zhou and A. Zhou, ACS Appl. Mater. Interfaces, 2024, 16, 29521–29546 CrossRef CAS PubMed.
- S. Askari, S. Miar Alipour, R. Halladj and M. H. Davood Abadi Farahani, J. Porous Mater., 2013, 20, 285–302 CrossRef CAS.
- K. S. Suslick, D. A. Hammerton and R. E. Cline, J. Am. Chem. Soc., 1986, 108, 5641–5642 CrossRef CAS.
- N. M. Navarro, T. Chave, P. Pochon, I. Bisel and S. I. Nikitenko, J. Phys. Chem. B, 2011, 115, 2024–2029 CrossRef CAS PubMed.
- P. Riesz, D. Berdahl and C. L. Christman, Environ. Health Perspect., 1985, 64, 233–252 CrossRef CAS PubMed.
- T. G. Leighton, Ultrason. Sonochem., 1995, 2, S123–S136 CrossRef.
- R. E. Apfel, Ultrasonics, 1984, 22, 167–173 CrossRef.
- K. S. Suslick, Y. Didenko, M. M. Fang, T. Hyeon, K. J. Kolbeck, W. B. McNamara, M. M. Mdleleni and M. Wong, Philos. Trans. R. Soc., A, 1999, 357, 335–353 CrossRef CAS.
- D. J. Flannigan and K. S. Suslick, Nat. Phys., 2010, 6, 598–601 Search PubMed.
- S. I. Nikitenko and R. Pflieger, Ultrason. Sonochem., 2017, 35, 623–630 CrossRef CAS PubMed.
- A. Abbas, M. Srour, P. Tang, H. Chiou, H.-K. Chan and J. A. Romagnoli, Chem. Eng. Sci., 2007, 62, 2445–2453 CrossRef CAS.
- C. Virone, H. J. M. Kramer, G. M. van Rosmalen, A. H. Stoop and T. W. Bakker, J. Cryst. Growth, 2006, 294, 9–15 CrossRef CAS.
- H. Li, H. Li, Z. Guo and Y. Liu, Ultrason. Sonochem., 2006, 13, 359–363 CrossRef CAS PubMed.
- S. Nalesso, M. J. Bussemaker, R. P. Sear, M. Hodnett and J. Lee, Ultrason. Sonochem., 2019, 57, 125–138 CrossRef CAS PubMed.
- S. Sivalingam and S. Sen, Appl. Surf. Sci., 2019, 463, 190–196 CrossRef CAS.
- J. Park, B. C. Kim, S. S. Park and H. C. Park, J. Mater. Sci. Lett., 2001, 20, 531–533 CrossRef CAS.
- O. Andac, M. Tatlier, A. Sirkecioglu, I. Ece and A. Erdem-Senatalar, Microporous Mesoporous Mater., 2005, 79, 225–233 CrossRef CAS.
- C. Belviso, A. Lettino and F. Cavalcante, Molecules, 2018, 23, 2122 CrossRef PubMed.
- R. M. Dewes, H. R. Mendoza, M. V. L. Pereira, C. Lutz and T. V. Gerven, Ultrason. Sonochem., 2022, 82, 105909 CrossRef CAS PubMed.
- C. Cau, Y. Guari, T. Chave, J. Larionova, P. Pochon and S. I. Nikitenko, J. Phys. Chem. C, 2013, 117, 22827–22833 CrossRef CAS.
- S. I. Nikitenko, T. Chave, C. Cau, H.-P. Brau and V. Flaud, ACS Catal., 2015, 5, 4790–4795 CrossRef CAS.
- S. El Hakim, T. Chave, A. A. Nada, S. Roualdes and S. I. Nikitenko, Front. Catal., 2021, 1, 669260 CrossRef.
- H. Greer, P. S. Wheatley, S. E. Ashbrook, R. E. Morris and W. Zhou, J. Am. Chem. Soc., 2009, 131, 17986–17992 CrossRef CAS PubMed.
- S. I. Nikitenko, C. Le Naour and P. Moisy, Ultrason. Sonochem., 2007, 14, 330–336 CrossRef CAS PubMed.
- V. Mancier and D. Leclercq, Ultrason. Sonochem., 2008, 15, 973–980 CrossRef CAS PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- A. Gualtieri, P. Norby, G. Artioli and J. Hanson, Phys. Chem. Miner., 1997, 24, 191–199 CrossRef CAS.
- S. I. Nikitenko, M. Brau and R. Pflieger, Ultrason. Sonochem., 2020, 67, 105189 CrossRef CAS PubMed.
- R. F. Lambert, J. Acoust. Soc. Am., 1953, 25, 1068–1083 CrossRef.
- E. A. G. Shaw, Acustica, 1953, 3, 87–95 Search PubMed.
- J. W. S. Rayleigh, The theory of sound, Dover New York, New York, 2nd edn, vol. 2, 1945 Search PubMed.
- A. Bampouli, Q. Goris, J. Van Olmen, S. Solmaz, M. Noorul Hussain, G. D. Stefanidis and T. Van Gerven, Ultrason. Sonochem., 2023, 97, 106444 CrossRef CAS PubMed.
- A. Palčić, B. Subotić, V. Valtchev and J. Bronić, CrystEngComm, 2013, 15, 5784–5791 RSC.
- T. Chave, S. I. Nikitenko, D. Granier and T. Zemb, Ultrason. Sonochem., 2009, 16, 481–487 CrossRef CAS PubMed.
- H. F. Greer, Mater. Sci. Technol., 2014, 30, 611–626 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01943a |
| This journal is © The Royal Society of Chemistry 2024 |