
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of air stable Ni(II) complexes with isocyanides and dialkyldithiophosphate ligands: acyclic diaminocarbene formation†
Lucía
Álvarez-Miguel‡
 *a,
Clara
del Carmen-Rodríguez‡
b,
María
Valle
b,
Celedonio M.
Álvarez
b,
José M.
Martín-Álvarez
b,
Raúl
García-Rodríguez
b and
Daniel
Miguel
b
*a,
Clara
del Carmen-Rodríguez‡
b,
María
Valle
b,
Celedonio M.
Álvarez
b,
José M.
Martín-Álvarez
b,
Raúl
García-Rodríguez
b and
Daniel
Miguel
b
aSOSCATCOM Group, Departamento de Química Orgánica y Química Inorgánica, Facultad de Farmacia and Instituto de Investigación Química “Andrés M. del Río” (IQAR) Universidad de Alcalá Campus Universitario, 28871 Alcalá de Henares, Madrid, Spain. E-mail: lucia.alvarezm@uah.es
bGIR MIOMET/IU CINQUIMA/Química Inorgánica, Facultad de Ciencias Universidad de Valladolid, 47011, Valladolid, Spain
First published on 27th August 2024
Abstract
A library of new neutral and cationic Ni(II) complexes containing isocyanide ligands and mono- or dialkyl-dithiophosphate have been easily prepared and fully characterized. The synthesis of the neutral complexes unfolds through the alkyl transfer from one alkyldithiophosphate leaving group coordinated to the Ni(II) complex. The alkyl transfer is controlled by steric factors and is highly solvent-dependent. These complexes shown to constitute excellent precursors to obtain new families of air stable Ni(II)-based acyclic diaminocarbene complexes (Ni(II)-ADCs) by nucleophilic attack with various alkyl-substituted amines. Remarkably, the ADC is only produced at one of the isocyanide ligands, keeping the other isocyanide unreacted. This was subsequently exploited to prepare the unprecedented neutral and cationic dinuclear Ni(II) complexes containing a bridging bis-carbene ligand using piperazine.
Introduction
Ni(II)-based carbene complexes (see Fig. 1) are the object of continual interest due to their well-known catalytic potential,1–3 as well as the lower cost and greater abundance of Ni compared to Pd and Pt. Thus, many reports of N-heterocyclic carbene Ni(II) complexes have appeared in the wake of a tremendous outburst of studies of NHC complexes of d metals with the goal of obtaining easy-to-prepare, user-friendly, low-cost Ni pre-catalysts.4 Additionally, metal acyclic amino carbenes (AACs) are attracting growing attention focussed on both their fundamental chemistry and catalytic activity.5,6 They are expected to have a wider range of steric and electronic properties than their cyclic NHC congeners due to their structural flexibility/versatility, the stronger donor character along with the electrophilicity and the Brønsted-acidity of NH hydrogens.7,8 Although free AAC carbenes have been known from the pioneering work of Alder,9–11 the slower development of the chemistry of AACs has been attributed to their lower stability as free carbene species compared to their cyclic counterparts.12 However, AACs are easy to build within the coordination sphere of the metal, as shown in the seminal work of Fischer, in which alkoxy or amino carbenes were obtained via nucleophilic attack on a coordinated CO ligand. In this context, the use of metal-coordinated isocyanide complexes as precursors for carbene ligands has become a very well-known method to obtain acyclic diaminocarbene (ADC) complexes of transition metals through the nucleophilic attack of amines in recent years.13,14 In the context of the formation of square planar compounds, this procedure has frequently been applied to obtain Pd(II), Pt(II) and Au(III) complexes with synthetic, catalytic or bioactive applications;15 however, reports on Ni(II) species are comparatively scarce, probably due to the limited number of isocyanide complexes available as suitable starting compounds due to their poor stability. Several Ni(II)-ADCs have been prepared using different approaches such as protonation of Ni(0) isocyanide complexes followed by hydrocarbation of alkenes16 and the oxidative insertion of [Ni(cod)]/PPh3 into the C–Cl bond of 2-chloroamidinium salts.17 As far as we know, there is only one report about the synthesis of Ni(II)-ADCs complexes as a result of nucleophilic attack of amines to metal coordinated isocyanides surrounded by fluorocarbon ligands18 Additionally, non-isolated intermediate Ni(II)-ADCs complexes have been proposed as catalytic carbene species that propagate the chain-growth in the poly(iminomethylenes) polymerization processes.19–22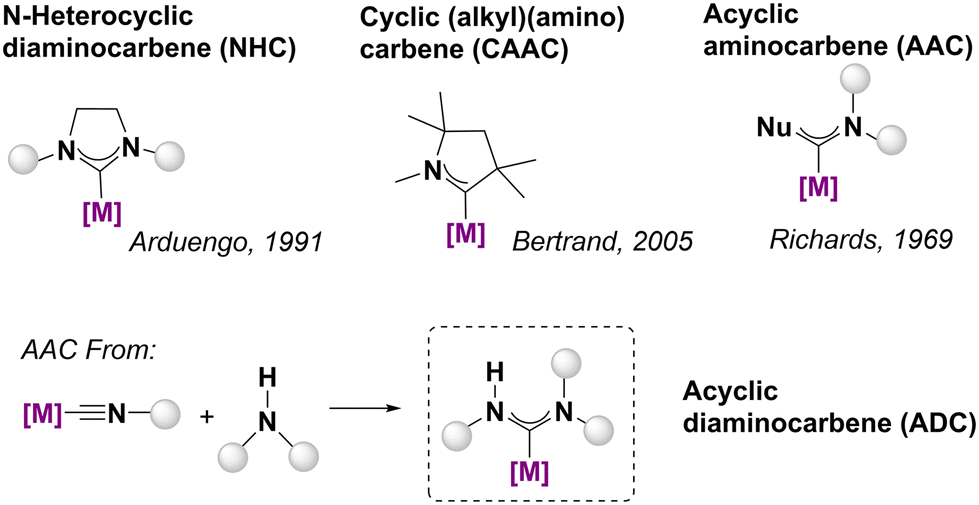 | ||
| Fig. 1 General overview of different metal carbene families.23 | ||
Therefore, we thought it would be interesting to develop a novel, air stable and soft methodology for the preparation of new families of Ni(II) complexes with isocyanide ligands and to test their potential for the preparation of new acyclic amino carbene derivatives. We had previously described the preparation of square-planar [Ni{S2P(O)(OEt)}(CNXyl)2] (3a) (see Scheme 1) containing ethyldithiophosphate and 2,6-dimethyl xylyl isocyanide ligands.24 The formation of this complex was remarkable, as it proceeded through the octahedral diisocyanide complex [Ni{S2P(OEt)2}2(CNXyl)2] followed by ethyl transfer to the leaving dithiophosphate group, producing O,O′,S-triethyl dithiophosphate byproduct. However, the reaction required the assistance of PCy3, leading to an ill-defined mixture of compounds that necessitated chromatographic purification.
 | ||
| Scheme 1 General synthetic route for the formation of diisocyanide mono- or dialkyildithiophosphate Ni(II) complexes 3a–3f/4a–4g and their related acyclic amino carbenes 5a–5g/6a–6h. The colour of the central atoms represents the colour of the isolated products. | ||
In the present work, we have re-examined this reactivity to develop a generalizable air tolerant protocol for the preparation of a variety of square-planar Ni(II) diisocyanide dithiophosphate complexes (see Scheme 1). This synthetic route provides two series of neutral and cationic complexes with ancillary alkyl-/dialkyldithiophosphate and isocyanide ligands. Through further reaction with alkylamines, both the neutral and cationic complexes can be converted into Ni(II)-ADCs via nucleophilic attack of the amines at one of the isocyanides. Notably, in these complexes, both dithiophosphate and isocyanide behave as soft base ligands, which is necessary to stabilize the square planar geometry around Ni(II).25
Results and discussion
Neutral and cationic isocyanide Ni(II) complexes
We first synthesized a family of neutral [Ni{S2P(O)(OR1)}(CNR2)2] (R1 = Me, Et, Bz; R2 = Xyl, Dipp) 3a–3f complexes following the previously reported updated protocol (see ESI† for details),24 by mixing the square-planar precursors with the formulae [Ni(S2P(OR1)2)2] with 2 equivalents of the appropriate isocyanide (2,6-dimethylphenyl isocyanide, CNXyl, or 2,6-diisopropylphenyl isocyanide, CNDipp). The optimization of this reaction showed that the process is highly solvent-dependent, (see Table S1 in ESI†) proceeding smoothly in a virtually quantitative manner in chlorinated solvents at both reflux and at high temperatures, and that it occurs cleanly without the assistance of PCy3. Significantly, the reaction rate is strongly dependent on the nature of the alkyl group to be transferred. R1 groups with a greater number of carbons in the alkyl chain are worse leaving groups and, therefore, will not promote the formation of the O,O,S-trialkyl species. The sharp dependence of the reaction rate on steric factors (with no trace of alkyl transfer detected for longer secondary (iPr, Cy) or tertiary (tBu) alkyls) strongly suggests that formation of complexes 3a–3f proceeds via nucleophilic attack of one dialkyldithiophosphate rather than by a radical pathway. This was supported by the fact that the addition of TEMPO did not affect the evolution of the reaction. We propose that the observed alkyl-transfer is the result of the nucleophilic attack of one dialkyldithiophosphate on the other, leading to C–O cleavage and C–S bond formation, in a SN2 reaction at carbon with the trialkyl dithiophosphate (S = P(SR1)(OR1)2) as the leaving group. However, the precise details of this reaction are unclear at this stage, and more work will be needed to ascertain the SN2 mechanism.The new complexes 3b–3f were isolated and fully characterized using analytical and spectroscopic methods (see ESI† and experimental part), and X-ray crystal structure determinations were carried out for 3c (Fig. 2, above) and 3d–3e (see ESI†). The Ni atom of compound 3c maintains a square-planar geometry and is coordinated by two carbons from the isocyanide ligands and two sulfur atoms from one alkyldithiophosphate.
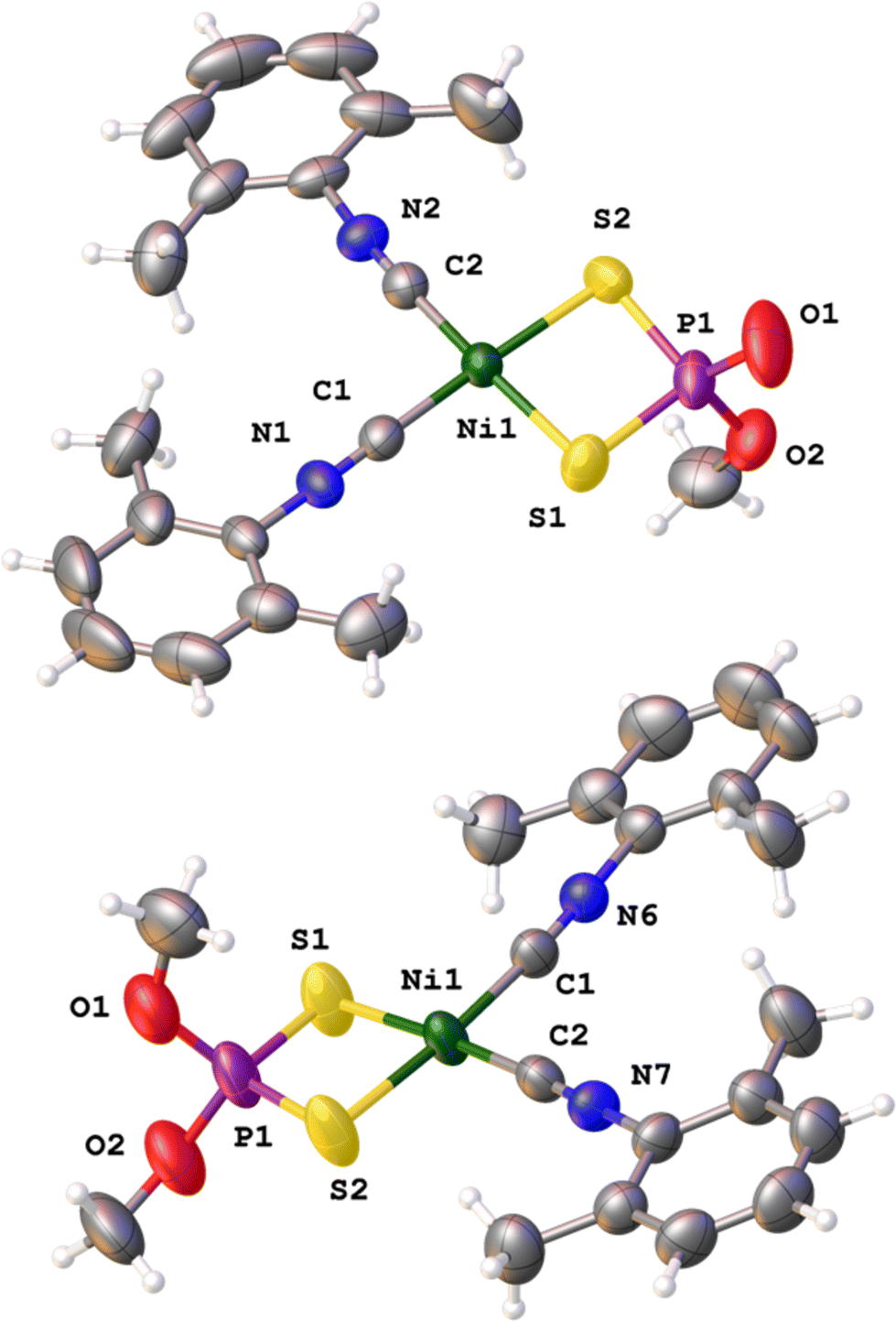 | ||
| Fig. 2 X-ray crystal structures obtained for neutral complex 3c (above) and cationic moiety of complex 4c (below). Complete structural information can be found in the ESI.† | ||
We were also interested in exploring the formation of a family of cationic complexes, as we expected that the isocyanides in such cationic complexes would display enhanced electrophilicity. It has been proposed that the electrophilic character of an isocyanide carbon, and therefore its ability to undergo nucleophilic attack, can be estimated from the shift in the frequency of its FTIR peak upon coordination, i.e., Δν = ν(CN)coord − ν(CN)free, with positive values of Δν ≥ 40 cm−1 indicating that the CNR ligand is susceptible to nucleophilic attack.14 We obtained cationic complexes [Ni{S2P(OR1)2}(CNR2)2]X (X = ClO4 or BF4) (4a–g) containing dialkyldithiophosphate and isocyanide ligands through a redistribution reaction by simply mixing Ni complexes 1-R1 with an equimolar amount of Ni(X)2·6H2O and the appropriate isocyanide (i.e., a twofold excess with respect to the total amount of Ni), as summarized in Scheme 2.
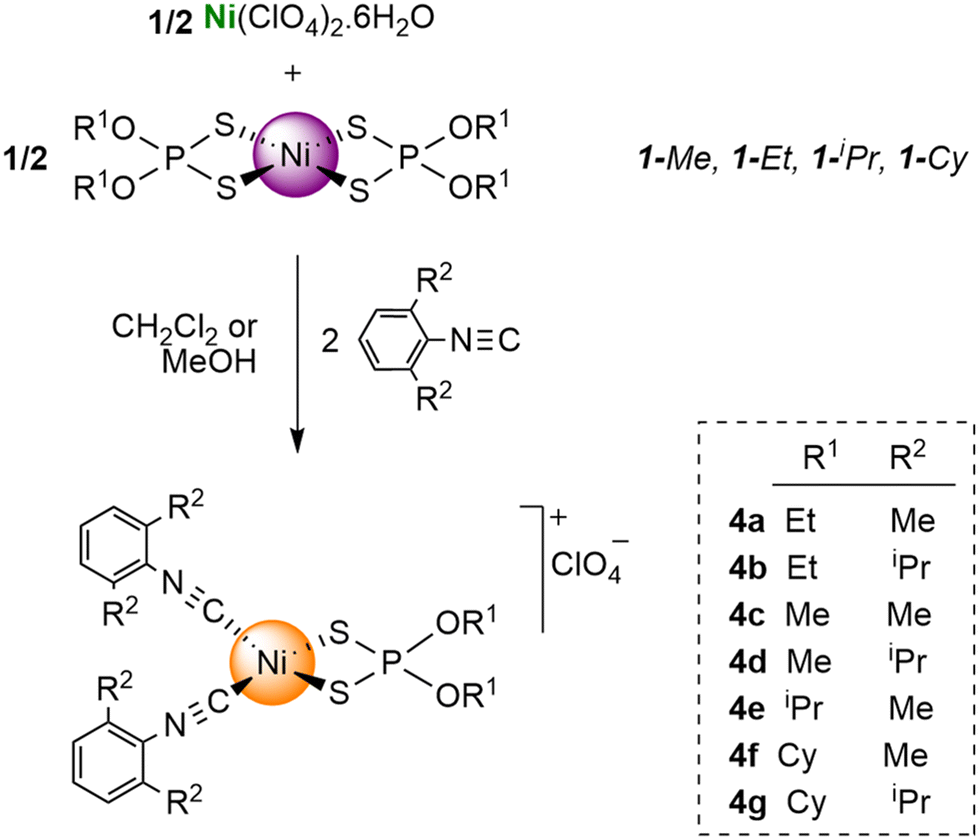 | ||
| Scheme 2 Formation of cationic diisocyanide dialkyldithiophosphate nickel(II) complexes from the Ni(ClO4)2·6H2O precursor. | ||
X-ray quality crystals could be grown for complexes 4a, 4c, 4e and 4f (see Fig. 2 below for 4c and ESI† for the rest), which allowed confirmation of their structure. As expected, these complexes show square-planar geometry, as also observed in the neutral 3 complexes. The Ni–S bond distances are significantly longer in cationic 4c than in its neutral analogue 3c (2.225(1) and 2.219(1), cf. 2.199(1) and 2.197 (1) Å respectively) while the Ni–C bond distances in both complexes are similar (1.843(4) and 1.844(4), cf. 1.842(2) and 1.838(2) Å respectively). The S–Ni–S bite angle in the cationic complex is increased (89.01(5) cf. 87.72(3)°) but the C–Ni–C angle is decreased (94.24(16), cf. 96.0(3)°) with respect to the neutral complex. These same trends are also observed in the aforementioned bond distances and bite angles in the X-ray structures of complexes 4a and 3a.24
The FTIR spectra also show that the displacement Δν of the isocyanide is about 50 cm−1 for the neutral and 70 cm−1 for the cationic complexes compared to that of the free ligand (2123 cm−1 for CNXyl), indicating that although both families are susceptible to nucleophilic attack, the cationic complexes should be more reactive. This is consistent with the lower electron density at the Ni atom, which forces stronger σ-donation from the isocyanide to Ni and weaker π-back-donation from Ni to the isocyanide. Both effects should increase the electrophilicity at the isocyanide carbon and, subsequently, make it more reactive towards nucleophilic attack.
Neutral and cationic isocyanide Ni(II) carbene complexes
With the two families of neutral and cationic complexes 3 and 4 in hand, we focused on converting these diisocyanide dialkyldithiophosphate Ni(II) complexes into Ni(II)-ADCs. Compounds 3a–3f react with diethylamine (NHEt2) or piperazine (ppze) to afford yellow carbene complexes 5a–5f after 30 min at room temperature, as shown in Scheme 3. The spectroscopic data for the complexes and X-ray structural determination carried out for 5e (see Fig. 3) show the formation of the carbene ligand by the addition of NHEt2 to one isocyanide, while the other isocyanide remains unreacted. However, IR monitoring shows that even when a greater than a twofold excess of amine is used, attack on the second isocyanide is not observed, as was observed for Pt and Pd complexes.26 This is consistent with the guideline regarding the frequency shift of the CN band. The band of the remaining isocyanide in 5e appears at 2155 cm−1, with a shift of only 22 cm−1 relative to that of the free ligand. This is smaller than the proposed threshold of Δν ≥ 40 cm−1, indicating that the isocyanide is not sufficiently activated towards nucleophilic attack of the amine. | ||
| Scheme 3 (Above) Formation of neutral 5a–5g and cationic 6a–6h isocyanide dithiophosphate carbene Ni(II) complexes by reaction with a secondary amine. (Below) Carbene isomerism Z/E of Ni(II)-ADC complexes. | ||
 | ||
| Fig. 3 Structure of Z-carbene complex 5e. Selected bond lengths (Å) and angles (°): Ni(1)–C(1) 1.812(4), Ni(1)–C(2) 1.918(4), Ni(1)–S(1) 2.209(2), Ni(1)–S(2) 2.235(1), S(1)–Ni(1)–S(2) 88.17(4), C(1)–Ni(1)–C(2) 90.15(16). | ||
The isolated neutral aminocarbene products show Z/E ratios between 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 and 80:20 (see 31P{1H} NMR spectra in the Experimental part), with the Z product being the major compound, as was reported in DFT studies for an associative reaction mechanism in analogous cis-Pt compounds.27 We carried out high-temperature NMR experiments to determine whether interconversion between the Z/E isomers was accessible at higher temperatures.28 However, upon heating of 5b above 60 °C in DMSO-d6, decomposition was observed from the appearance of additional signals in the 31P{1H} spectrum before any interconversion was evident, indicating a relatively high activation energy for the interconversion process.
:25 and 80:20 (see 31P{1H} NMR spectra in the Experimental part), with the Z product being the major compound, as was reported in DFT studies for an associative reaction mechanism in analogous cis-Pt compounds.27 We carried out high-temperature NMR experiments to determine whether interconversion between the Z/E isomers was accessible at higher temperatures.28 However, upon heating of 5b above 60 °C in DMSO-d6, decomposition was observed from the appearance of additional signals in the 31P{1H} spectrum before any interconversion was evident, indicating a relatively high activation energy for the interconversion process.
From the IR data and based on their cationic nature, it was expected that the complexes of the cationic series 4a–g would be more activated than those of the neutral family. Thus, we anticipated that they would be more electrophilic and more prone to undergo nucleophilic attack by the amines. Indeed, complex 4c readily produced cationic 6c in moderate yield (60%, see Scheme 3) in just 10 min (as monitored by IR) after the addition of the NHEt2 in dichloromethane. However, the outcome of the reaction was not as clean as for the neutral congeners, as indicated by 1H NMR studies on the reaction crude. This was attributed to the Ni center being highly electrophilic and therefore prone to incorporate additional ligands or promote side reactions. Nonetheless, complexes 6a–6h could be easily purified by addition of ether and hexane to the reaction mixtures, leading to their isolation as yellow microcrystalline powders in moderate yields (45–60%). Complexes 6a–6h were characterized using analytical and spectroscopic methods, and the structures of 6c (Fig. 4), 6e, 6g and 6h (see ESI†) were confirmed unambiguously by X-ray diffraction analysis, which showed the formation of one diaminocarbene ligand. In contrast with the neutral ADC complexes, the cationic final products present the Z isomer almost in quantitative yield (>95%).
 | ||
| Fig. 4 Structure of the cationic carbene compound 6c, showing the atom numbering. Selected bond lengths (Å) and angles (°): Ni(1)–C(1) 1.799(4), Ni(1)–C(2) 1.899(3), H(2)⋯N(4) 2.152(3), Ni(1)–S(1) 2.243(2), Ni(1)–S(2) 2.213(2), S(1)–Ni(1)–S(2) 88.38(3), C(1)–Ni(1)–C(2) 88.59(14). | ||
Importantly, even for these cationic complexes, the attack of only one of the isocyanides was again observed. Even using an excess of the amine (>2 equivalents), nucleophilic attack on the remaining isocyanide of complexes 6 did not proceed, as evidenced by IR monitoring of the reaction. It is worth pointing out that the ν(CN) frequency value of this isocyanide is around 48 cm−1 higher than that of the free isocyanide; this shift is thus at the borderline of the threshold discussed above.5 The non-formation of mononuclear bis-carbene complexes seems to be related to the reduced electrophilicity of the remaining isocyanide carbon, which is directly linked to lower values of ν(CN) in the unchanged isocyanides. Apparently, the steric hindrance does not prevent this second amine reaction, since the surroundings of the unreacted isocyanide ligand retain their planarity, with enough space for the attack.
Inspection of particular bond distances in the X-ray structures of the comparable complexes 5e and 6c show different trends than the neutral and cationic 3c and 4c precursors. The Ni–C and Ni–S distances are almost identical in the neutral carbene than in the cationic carbene complex (i.e.: Ni–C bond distances 1.812(4) and 1.918(4), cf. 1.799(4) and 1.899(3) Å, respectively) whereas the S–Ni–S bite angles are similar (88.17(4), cf. 88.38(3)°) and the C–Ni–C angle is significantly decreased (90.15(16), cf. 88.59(14)°).
Formation of dinuclear Ni(II) complexes with a bis-carbene bridge
The unreactivity of the second isocyanide ligand can be exploited to build dinuclear complexes containing a bis-carbene bridging two metal atoms in the reaction with a diamine. Some examples of dinuclear Au(I) complexes have been reported but as far as we know, no evidence of Ni(II) bis-carbene bridge structures have been described in the literature.29,30 When neutral complex 3c was reacted with 1/2 equivalent of ppze in CH2Cl2, the dinuclear complex 7 was obtained, whose structure was confirmed unambiguously by X-ray analysis. As shown in Fig. 5, compound 7 consists of two {Ni(S2P(O)(OMe))(CNXyl)} fragments held together by a bis-carbene ligand that results from the controlled intermolecular attack of ppze on one isocyanide each of two 3c complexes, rather than the intramolecular attack on the two isocyanide ligands of the same molecule (see Scheme 4, above). Further proof of this intermolecular selectivity came from the reaction of complex 3c with 1 equivalent of piperazine (see ESI†), which allowed the formation of 5f (clearly indicating that intramolecular attack is prevented). Reaction of 5f with one equivalent of 3c leads to dinuclear complex 7 through a controlled intermolecular nucleophilic attack of the free amine group in ppze on the isocyanide from the other molecule of 3c. Interestingly, both pathways (direct, route a, and stepwise, route b) yielded the same mixture of Z/E isomers (77:23), as determined by 31P{1H} NMR experiments. Four signals appear in the 31P{1H} NMR spectra, one pair for each isomer (δ 57.61 and 57.54 for the Z isomer and 57.24 and 57.11 for the E). The signal splitting was tentatively attributed to the presence of the second Ni(II) fragment, which would result in additional stereoisomers.
 | ||
| Fig. 5 Structure of the neutral bis-carbene 7, showing the atom numbering. Selected bond lengths (Å) and angles (°): Ni(1)–C(1) 1.839(9), Ni(1)–C(2) 1.922(7), Ni(1)–S(1) 2.218(2), Ni(1)–S(2) 2.203(2), S(1)–Ni(1)–S(2) 90.6(3), C(1)–Ni(1)–C(2) 88.33(8). | ||
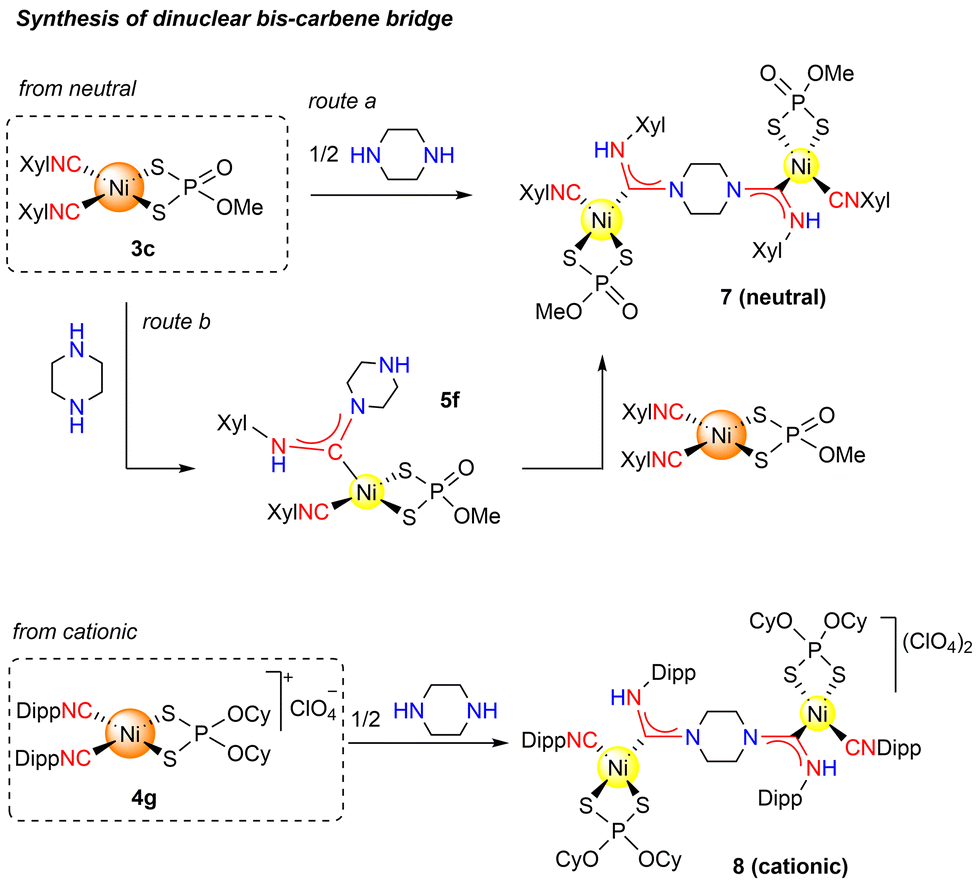 | ||
| Scheme 4 Formation of dinuclear complex 7 by different synthetic routes: (a) direct reaction of neutral 3c with the ppze under stoichiometric conditions; (b) starting from neutral carbene 5f and an equimolar amount of neutral complex 3c. Formation of dinuclear cationic complex 8 by reaction of cationic 4g with the ppze under stoichiometric conditions. | ||
In order to extend the synthetic scope, the use of cationic complex 4g to prepare the corresponding dicationic dinuclear bis-carbene bridge by reaction with 1/2 equivalent of ppze was tested (Scheme 4, below). Complex 8 was isolated and despite the low solubility of this complex which prevented characterization by NMR, the structure was established through single-crystal, X-ray studies (see Fig. 6). We are currently exploring the possibility of preparing non-symmetrical metal bis-carbenes bridge, using both neutral and cationic precursors.
 | ||
| Fig. 6 Structure of the cationic moiety bis-carbene 8, showing the atom numbering. Selected bond lengths (Å) and angles (°): Ni(1)–C(1) 1.908(4), Ni(1)–C(2) 1.826(5), Ni(1)–S(1) 2.235(1), Ni(1)–S(2) 2.197(2), S(1)–Ni(1)–S(2) 88.95(6), C(1)–Ni(1)–C(2) 89.9(2). | ||
Conclusions
In conclusion, we have developed a simple and open-air methodology to prepare new neutral and cationic isocyanide complexes of Ni(II), which are otherwise scarce. The synthesis was applicable to a wide variety of Ni(II) dithiophosphate precursors by the simple reaction with CNXyl or CNDipp. The neutral and cationic complexes exhibit Δν(CN) frequencies approximately 50 cm−1 and 70 cm−1 higher than those of the respective free isocyanides, respectively, which reflects the electrophilic character of the isocyanide carbon. Notably, ADCs are easily formed at one of the coordinated isocyanides by the simple addition of different amines, affording the new families of neutral and cationic air stable Ni(II)-ADCs complexes. In addition, we demonstrated the synthetic opportunities of these structures towards the formation of the unprecedented dinuclear neutral and cationic bis-carbene bridge Ni(II) complexes. We are currently working to extend and control this reactivity to access mixed bis-carbene bridge species. Additionally, further research is now in progress to explore the use of other fragments on the Ni(II) precursors described here in order to enhance the electrophilicity of the isocyanide carbon with the goal of achieving the bis-carbene mononuclear complexes and exploring their catalytic properties.Experimental section
Materials and general methods
All reagents were purchased from commercial suppliers and used without further purification. Reactions can be carried out at air atmosphere. Solvents were used as received. Kieselguhr (diatomaceous earth, Merck) was used for filtration. NMR spectra (see Fig. 7) were recorded on Agilent DD2 500 instruments. 1H and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) and are referenced to TMS, using solvents as internal references. Coupling constants (J) are reported in Hertz (Hz). Standard abbreviations are used to indicate multiplicity: s = singlet, d = doublet, t = triplet, m = multiplet. 1H and 13C assignments were performed using 2D NMR methods (COSY, gradient CRISIS-HSQC, and gradient CRISIS-HMBC). Some quaternary carbon atoms were not detected in the 13C spectra but were observed via a 1H–13C HMBC experiment. IR spectra of solid samples were recorded with a Frontier PerkinElmer Spectrum RX I FT-IR instrument. Elemental analyses were performed using a PerkinElmer 2400B microanalyzer. High-resolution mass spectra were recorded at the mass spectrometry service of the Laboratory of Instrumental Techniques of the University of Valladolid (L.T.I., https://www.laboratoriotecnicasinstrumentales.es). A MALDI-TOF system Bruker Autoflex Speed (N2 laser (337 nm, pulse energy 100 μJ, 1 ns), acceleration voltage 19 kV, reflector and linear positive mode) was used. A UPLC-MS system (UPLC: Waters ACQUITY H-class UPLC; MS: Bruker Maxis Impact) using electrospray ionization (ESI positive and negative) was utilized as well. HRMS spectra were analyzed using Bruker Data Analysis 4.1© (https://www.bruker.com). | ||
| Fig. 7 Abbreviations of the different RX groups for NMR elucidation. | ||
General synthesis of cationic diisocyanide dialkyldithiophosphate Ni(II) complexes (4a–4h)
A mixture of [Ni{S2P(OR)2}2] 1-R (1 mmol) and Ni(ClO4)2·6H2O (0.366 g, 1 mmol) in methanol (15 mL) was stirred at room temperature for 10 minutes. CNXyl (0.266 g, 2 mmol) or CNDipp (0.374 g, 2 mmol) were then added, and after stirring for 20 minutes the solvent was evaporated in vacuo. The solid residue was dissolved in CH2Cl2 and filtered. Slow evaporation gave 4a–4h as orange microcrystals.General synthesis of neutral (5a–5g) and cationic Ni(II)-ADC complexes (6a–6h)
A mixture of 3a–d or 4a–g (1 mmol) and the appropiate secondary amine (2 mmol) in CH2Cl2 (15 mL) was stirred at room temperature. Slow evaporation of the solvent and further precipitation with hexane gave 5a–5g and 6a–6h as orange-yellow microcrystalline solids.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O)(OEt))(CNXyl){C(NHXyl)(NEt2)}] (5a).
From 3a and NHEt2 in a CH2Cl2 solution. Yield 0.485 g, 88%. Anal. calcd for C24H34N3NiO2PS2 (550.34 g mol−1): C, 52.37; H, 6.24; N, 7.64 Found: C, 52.31 H, 6.15; N, 7.60. IR (CH2Cl2, cm−1): 2155 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.85 (s, 1H, NHcarbene), 7.29–7.10 (m, 6H, ArH), 4.72 (dq, J = 14.1, 7.0 Hz, 1H, NCH2aCH3), 4.53 (dt, J = 13.9, 7.0 Hz, 1H, NCH2a′CH3), 3.57 (m, 2H, NCH2cCH3), 3.16–2.97 (m, 2H, OCH2CH3), 2.26–2.29 (m, 12H, ArCH3), 1.41 (t, J = 7.1 Hz, 3H, NCH2CH3b), 1.17 (t, J = 6.5 Hz, 3H, NCH2CH3d), 0.95 (t, J = 7.1 Hz, 3H, OCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.9 (NCcarbene), 130.46 (ArC4), 128.88(ArC3,5), 52.22 and 52.15 (NCH2a,a′CH3), 41.92 (NCH2cCH3), 62.18 (OCH2CH3), 18.96 (ArCH3) 14.36 (NCH2CH3b), 13.20 (NCH2CH3d), 6.06 (OCH2CH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 55.54, 55.34 (Z/E, 85:15).
O)(OEt))(CNXyl){C(NHXyl)(ppze)}] (5b).
From 3a and ppze in a CH2Cl2 solution, 30 min. Yield: 0.450 g, 84%. Anal. calcd for C24H33N4NiO2PS2 (563.34 g mol−1): C, 51.17; H, 5.90; N, 9.95 Found: C, 50.96 H, 5.73; N, 9.72. HR-MS (ESI-TOF, m/z); calcd for C24H34N4NiO2PS2 = 563.1209; obtained = 563.1215 [M + H]+. IR (DMSO, cm−1): 2154 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 9.01 (s, 1H, NHcarbene), 7.39–7.13 (m, 6H, ArH), 4.98–4.86 (m, 1H, NCH2a,ppze), 4.48–4.37 (m, 1H, NCH2a,ppze), 3.93–3.76 (m, 2H, NCH2ppze), 3.38–3.24 (m, 2H, NCH2ppze), 3.15–2.90 (m, 4H, OCH2CH3 and NCH2ppze), 2.32–2.23 (m, 12H, ArCH3), 0.94 (t, J = 7.1 Hz, 3H, OCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.90 (NCcarbene), 134.79 (ArC), 129.83 (ArC4), 128.42 (ArC3,5), 128.22 and 128.15 (ArC), 61.72 (OCH2CH3), 61.66 (NCH2ppze), 46.21 (NCH2ppze), 19.62 and 18.55 (ArCH3), 17.98 (OCH2CH3) ppm. 31P{1H} NMR (162 MHz): δ 55.65, 55.32 (Z/E 78:22) ppm.
O)(OMe))(CNDipp){C(NHDipp)(ppze)}] (5c).
From 3d and ppze in a CH2Cl2 solution, 30 min. Yield 0.512 g, 79%. Anal. calcd for C31H47N4NiO2PS2 (661.53 g mol−1): C, 56.28; H, 7.18; N, 8.47 Found: C, 56.14 H, 7.28; N, 8.23. IR (CH2Cl2, cm−1): 2157 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.77 (s, 1H, NHcarbene), 7.67–7.12 (m, 6H, ArH), 5.43–5.25 (m, 2H, NCH2ppze), 4.29–4.10 (m, 3H, NCH2ppze), 3.54–3.45 (m, 3H, NCH2ppze), 3.11 (m, 2H, ArCHiPr), 3.06 (m, 2H, ArCHiPr), 2.84–2.74 (m, 3H, OCH3), 1.36–0.98 (m, 24H, ArCH3iPr) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 145.12 (ArC), 135.65 (ArC), 130.76 (ArC), 128.51 (ArC4), 124.19 (ArC3,5), 64.90 (NCH2ppze), 52.51 (OCH3), 30.68 (ArCHiPr), 29.33 (ArCHiPr), 28.92 (ArCH3iPr), 28.26 (ArCH3iPr), 25.31 (ArCH3iPr), 24.56 (ArCH3iPr), 22.54 (ArCH3iPr), 15.15 (ArCH3iPr) ppm. 31P NMR (162 MHz, CDCl3): δ 58.53, 58.11 (Z/E 85:15) ppm.
O)(OMe))(CNDipp){C(NHDipp)(NEt2)}] (5d).
From 3d and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.512 g, 79%. Anal. calcd for C31H48N3NiO2PS2 (648.53 g mol−1): C, 57.41; H, 7.46; N, 6.48 Found: C, 57.33 H, 7.28; N, 6.53. IR (CH2Cl2, cm−1): 2152 (ν CN). 1H NMR (500 MHz, CDCl3) Z-isomer: δ 7.36 (t, J = 7.5 Hz, 2H, ArH4), 7.19 (d, J = 8.7 Hz, 4H, ArH3,5), 6.75 (s, 1H, NHcarbene), 5.08 (dq, J = 14.2, 7.1 Hz, 1H, NCH2CH3), 4.51 (dq, J = 13.9, 7.0 Hz, 1H, NCH2CH3), 3.62–3.52 (m, 4H, ArCHiPr), 3.15 (sept, J = 6.9 Hz, 2H, NCH2CH3), 2.82 (d, J = 14.9 Hz, 3H, OCH3), 1.52 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.41–1.33 (t, J = 7.1 Hz, 6H, NCH2CH3 and ArCH3iPr), 1.24 (d, J = 6.9 Hz, 12H, ArCH3iPr), 1.08 (t, d, J = 6.9 Hz, 9H, ArCH3iPr). ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 129.25 (ArC4), 124.00 (ArC3,5), 53.68 (OCH3), 53.62 and 53.49 (NCH2CH3), 29.73 (ArCHiPr), 23.38 (ArCH3iPr), 13.84 and 12.74 (NCH2CH3) ppm. 31P NMR (162 MHz, CDCl3): δ 61.06, 60.67 (Z/E 85:15) ppm.
O)(OMe))(CNXyl){C(NHXyl)(NEt2)}] (5e).
From 3c and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.450 g, 84%. Anal. calcd for C23H32N3NiO2PS2 (536.32 g mol−1): C, 51.51; H, 6.01; N, 7.84. Found: C, 51.76; H, 6.13; N, 7.64. IR (CH2Cl2, cm−1): 2156 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.86 (s, 1H, NHcarbene), 7.26 (m, 6H, ArH), 4.73 (m, 1H, NCH2aCH3), 4.55 (dt, J = 14, 7.0 Hz, 1H, NCH2a′CH3), 3.55 (m, 2H, NCH2cCH3), 2.84 (d, J = 14.9 Hz, 3H, OCH3), 2.26 (s, 12H, ArCH3), 1.41 (t, J = 7.1 Hz, 3H, NCH2CH3b), 1.17 (t, J = 7.1 Hz, 3H, NCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.88 (NCcarbene), 129.90 (ArC4), 128.61(ArC3,5), 52.08 (OCH3), 51.15 (NCH2a,a′CH3), 41.22 (NCH2cCH3), 19.83 and 18.02 (ArCH3) 13.67 (NCH2CH3b), 12.52 (NCH2CH3d) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 58.16, 57.85 (Z/E 72:28) ppm.
O)(OMe))(CNXyl){C(NHXyl)(ppze)}] (5f).
From 3c and ppze in a CH2Cl2 solution, 30 min. Yield 0.495 g, 90%. Anal. calcd for C23H31N4NiO2PS2 (549.31 g mol−1): C, 50.29; H, 5.70; N, 10.20 Found: C, 50.33; H, 5.70; N, 10.13. HR-MS (ESI-TOF, m/z); calcd for C23H32N4NiO2PS2 = 549.1052; obtained = 549.1072 [M + H]+. IR (CH2Cl2, cm−1): 2157 (ν CN). 1H NMR (500 MHz, DMSO-d6) Z-isomer: δ 9.03 (s, 1H, NHcarbene), 7.42–7.05 (m, 6H, ArH), 5.01–4.83 (m, 1H, NCH2ppze), 4.56–4.37 (m, 1H, NCH2ppze), 3.99–3.72 (m, 1H, NCH2ppze), 3.38–3.19 (m, 3H, NCH2ppze) 3.12–2.93 (m, 2H, NCH2ppze), 2.89–2.75 (m, 3H, OCH3), 2.31 (s, 6H, ArCH3), 2.27 (s, 6H, ArCH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): 197.13 (NCcarbene), 134.78, 134.61, 129.90 (ArC4), 128.41 (ArC3,5), 128.30, 128.14, 125.93, 54.90 (NCH2ppze), 52.77 (OCH3), 45.60 (NCH2ppze), 19.59 (ArCH3), 18.54 (ArCH3), 17.97 (ArCH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 58.15, 57.72 (Z/E 85:15) ppm.
O)(OBz)}(CNXyl){C(NHXyl)(NEt2)}] (5g).
From 3e and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.575 g, 94%. Anal. calcd for C29H36N3NiO2PS2 (612.41 g mol−1): C, 56.88; H, 5.93; N, 6.86. Found: C, 56.71; H, 5.81; N, 6.79. IR (CH2Cl2, cm−1): 2156 (ν CN). 1H NMR (500 MHz, CDCl3) Z-isomer: 8.15 (s, 1H, NHcarbene), 7.36–7.16 (m, 6H, ArH3,4,5) 7.16–6.97 (m, 5H, OArHBz), 4.76–4.63 (m, 2H, NCH2CH3), 4.24 (qd, J = 7.6, 3.7, 2.5 Hz, 2H, OCH2Bz), 3.61 (m, 2H, NCH2CH3), 2.28 (m, 12H, ArCH3), 1.43 (t, J = 7.1 Hz, 3H, NCH2CH3) 1.27 (t, J = 7.1 Hz, 3H, NCH2CH3) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 199.11 (NCcarbene), 138.35, 138.26, 137.70, 135.35 (ArC*), 128.39 (OArBz), 127.63 (ArC2,6), 127.21 (ArC4), 68.34 (OCH2Bz), 52.58 (NCH2CH3), 42.05 (NCH2CH3), 18.93 (ArCH3), 14.21 and 12.99 (NCH2CH3) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ 61.57, 61.32 (Z/E 76:14) ppm.
O)(OMe))(CNXyl)2C(NHXyl)}2(N2C4H8)] (7).
A red-orange mixture of 3c (0.463 g, 1 mmol) and ppze (0.048 g, 0.5 mmol) in CH2Cl2 (15 mL) was stirred at room temperature for 60 min. At this time, a yellow precipitate appeared, which was collected in a fritted funnel. Slow evaporation gave 7 as yellow microcrystals. Yield 0.920 g, 91%. Anal. calcd for C42H52N6Ni2O4P2S4 (1012.49 g mol−1): C, 49.82; H, 5.19; N, 8.30. Found: C, 49.66; H, 4.98; N, 8.23. HR-MS (ESI-TOF, m/z); calcd for C42H52N6Ni2O4P2S4 = 1011.1188; obtained = 1011.1181 [M + H]+. IR (CH2Cl2, cm−1): 2161 (ν CN). 1H NMR (400 MHz, DMSO): δ 9.36 (s, 1H, NHcarbene), 7.34 (m, 2H, ArH), 7.19 (m, 4H, ArH), 5.53 (m, 1H, OCH2), 4.86 (m, 1H, OCH2), 4.13 (m, 1H, OCH2), 3.93 (m, 1H, OCH2), 2.86 (m, H OCH3), 2.30 (s, 12H, ArCH3) ppm. 13C{1H} NMR (101 MHz, DMSO): δ 135.75(ArC2,6), 134.87 (ArC4), 130.04 (ArC4), 128.43 (ArC3,5), 128.01 (ArC3,5), 52.83 (OCH3), 50.41 (NCH2ppze), 48.00 (NCH2ppze), 19.59 (ArCH3), 18.56 (ArCH3), 18.27 (ArCH3), 18.02 (ArCH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 57.61 and 57.54 (Z), 57.24 and 57.11 (E) ppm.
O)(OEt))(CNXyl){C(NHXyl)(NEt2)}] (5a).
From 3a and NHEt2 in a CH2Cl2 solution. Yield 0.485 g, 88%. Anal. calcd for C24H34N3NiO2PS2 (550.34 g mol−1): C, 52.37; H, 6.24; N, 7.64 Found: C, 52.31 H, 6.15; N, 7.60. IR (CH2Cl2, cm−1): 2155 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.85 (s, 1H, NHcarbene), 7.29–7.10 (m, 6H, ArH), 4.72 (dq, J = 14.1, 7.0 Hz, 1H, NCH2aCH3), 4.53 (dt, J = 13.9, 7.0 Hz, 1H, NCH2a′CH3), 3.57 (m, 2H, NCH2cCH3), 3.16–2.97 (m, 2H, OCH2CH3), 2.26–2.29 (m, 12H, ArCH3), 1.41 (t, J = 7.1 Hz, 3H, NCH2CH3b), 1.17 (t, J = 6.5 Hz, 3H, NCH2CH3d), 0.95 (t, J = 7.1 Hz, 3H, OCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.9 (NCcarbene), 130.46 (ArC4), 128.88(ArC3,5), 52.22 and 52.15 (NCH2a,a′CH3), 41.92 (NCH2cCH3), 62.18 (OCH2CH3), 18.96 (ArCH3) 14.36 (NCH2CH3b), 13.20 (NCH2CH3d), 6.06 (OCH2CH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 55.54, 55.34 (Z/E, 85:15).
O)(OEt))(CNXyl){C(NHXyl)(ppze)}] (5b).
From 3a and ppze in a CH2Cl2 solution, 30 min. Yield: 0.450 g, 84%. Anal. calcd for C24H33N4NiO2PS2 (563.34 g mol−1): C, 51.17; H, 5.90; N, 9.95 Found: C, 50.96 H, 5.73; N, 9.72. HR-MS (ESI-TOF, m/z); calcd for C24H34N4NiO2PS2 = 563.1209; obtained = 563.1215 [M + H]+. IR (DMSO, cm−1): 2154 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 9.01 (s, 1H, NHcarbene), 7.39–7.13 (m, 6H, ArH), 4.98–4.86 (m, 1H, NCH2a,ppze), 4.48–4.37 (m, 1H, NCH2a,ppze), 3.93–3.76 (m, 2H, NCH2ppze), 3.38–3.24 (m, 2H, NCH2ppze), 3.15–2.90 (m, 4H, OCH2CH3 and NCH2ppze), 2.32–2.23 (m, 12H, ArCH3), 0.94 (t, J = 7.1 Hz, 3H, OCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.90 (NCcarbene), 134.79 (ArC), 129.83 (ArC4), 128.42 (ArC3,5), 128.22 and 128.15 (ArC), 61.72 (OCH2CH3), 61.66 (NCH2ppze), 46.21 (NCH2ppze), 19.62 and 18.55 (ArCH3), 17.98 (OCH2CH3) ppm. 31P{1H} NMR (162 MHz): δ 55.65, 55.32 (Z/E 78:22) ppm.
O)(OMe))(CNDipp){C(NHDipp)(ppze)}] (5c).
From 3d and ppze in a CH2Cl2 solution, 30 min. Yield 0.512 g, 79%. Anal. calcd for C31H47N4NiO2PS2 (661.53 g mol−1): C, 56.28; H, 7.18; N, 8.47 Found: C, 56.14 H, 7.28; N, 8.23. IR (CH2Cl2, cm−1): 2157 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.77 (s, 1H, NHcarbene), 7.67–7.12 (m, 6H, ArH), 5.43–5.25 (m, 2H, NCH2ppze), 4.29–4.10 (m, 3H, NCH2ppze), 3.54–3.45 (m, 3H, NCH2ppze), 3.11 (m, 2H, ArCHiPr), 3.06 (m, 2H, ArCHiPr), 2.84–2.74 (m, 3H, OCH3), 1.36–0.98 (m, 24H, ArCH3iPr) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 145.12 (ArC), 135.65 (ArC), 130.76 (ArC), 128.51 (ArC4), 124.19 (ArC3,5), 64.90 (NCH2ppze), 52.51 (OCH3), 30.68 (ArCHiPr), 29.33 (ArCHiPr), 28.92 (ArCH3iPr), 28.26 (ArCH3iPr), 25.31 (ArCH3iPr), 24.56 (ArCH3iPr), 22.54 (ArCH3iPr), 15.15 (ArCH3iPr) ppm. 31P NMR (162 MHz, CDCl3): δ 58.53, 58.11 (Z/E 85:15) ppm.
O)(OMe))(CNDipp){C(NHDipp)(NEt2)}] (5d).
From 3d and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.512 g, 79%. Anal. calcd for C31H48N3NiO2PS2 (648.53 g mol−1): C, 57.41; H, 7.46; N, 6.48 Found: C, 57.33 H, 7.28; N, 6.53. IR (CH2Cl2, cm−1): 2152 (ν CN). 1H NMR (500 MHz, CDCl3) Z-isomer: δ 7.36 (t, J = 7.5 Hz, 2H, ArH4), 7.19 (d, J = 8.7 Hz, 4H, ArH3,5), 6.75 (s, 1H, NHcarbene), 5.08 (dq, J = 14.2, 7.1 Hz, 1H, NCH2CH3), 4.51 (dq, J = 13.9, 7.0 Hz, 1H, NCH2CH3), 3.62–3.52 (m, 4H, ArCHiPr), 3.15 (sept, J = 6.9 Hz, 2H, NCH2CH3), 2.82 (d, J = 14.9 Hz, 3H, OCH3), 1.52 (t, J = 7.1 Hz, 3H, NCH2CH3), 1.41–1.33 (t, J = 7.1 Hz, 6H, NCH2CH3 and ArCH3iPr), 1.24 (d, J = 6.9 Hz, 12H, ArCH3iPr), 1.08 (t, d, J = 6.9 Hz, 9H, ArCH3iPr). ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 129.25 (ArC4), 124.00 (ArC3,5), 53.68 (OCH3), 53.62 and 53.49 (NCH2CH3), 29.73 (ArCHiPr), 23.38 (ArCH3iPr), 13.84 and 12.74 (NCH2CH3) ppm. 31P NMR (162 MHz, CDCl3): δ 61.06, 60.67 (Z/E 85:15) ppm.
O)(OMe))(CNXyl){C(NHXyl)(NEt2)}] (5e).
From 3c and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.450 g, 84%. Anal. calcd for C23H32N3NiO2PS2 (536.32 g mol−1): C, 51.51; H, 6.01; N, 7.84. Found: C, 51.76; H, 6.13; N, 7.64. IR (CH2Cl2, cm−1): 2156 (ν CN). 1H NMR (400 MHz, DMSO-d6) Z-isomer: δ 8.86 (s, 1H, NHcarbene), 7.26 (m, 6H, ArH), 4.73 (m, 1H, NCH2aCH3), 4.55 (dt, J = 14, 7.0 Hz, 1H, NCH2a′CH3), 3.55 (m, 2H, NCH2cCH3), 2.84 (d, J = 14.9 Hz, 3H, OCH3), 2.26 (s, 12H, ArCH3), 1.41 (t, J = 7.1 Hz, 3H, NCH2CH3b), 1.17 (t, J = 7.1 Hz, 3H, NCH2CH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): δ 196.88 (NCcarbene), 129.90 (ArC4), 128.61(ArC3,5), 52.08 (OCH3), 51.15 (NCH2a,a′CH3), 41.22 (NCH2cCH3), 19.83 and 18.02 (ArCH3) 13.67 (NCH2CH3b), 12.52 (NCH2CH3d) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 58.16, 57.85 (Z/E 72:28) ppm.
O)(OMe))(CNXyl){C(NHXyl)(ppze)}] (5f).
From 3c and ppze in a CH2Cl2 solution, 30 min. Yield 0.495 g, 90%. Anal. calcd for C23H31N4NiO2PS2 (549.31 g mol−1): C, 50.29; H, 5.70; N, 10.20 Found: C, 50.33; H, 5.70; N, 10.13. HR-MS (ESI-TOF, m/z); calcd for C23H32N4NiO2PS2 = 549.1052; obtained = 549.1072 [M + H]+. IR (CH2Cl2, cm−1): 2157 (ν CN). 1H NMR (500 MHz, DMSO-d6) Z-isomer: δ 9.03 (s, 1H, NHcarbene), 7.42–7.05 (m, 6H, ArH), 5.01–4.83 (m, 1H, NCH2ppze), 4.56–4.37 (m, 1H, NCH2ppze), 3.99–3.72 (m, 1H, NCH2ppze), 3.38–3.19 (m, 3H, NCH2ppze) 3.12–2.93 (m, 2H, NCH2ppze), 2.89–2.75 (m, 3H, OCH3), 2.31 (s, 6H, ArCH3), 2.27 (s, 6H, ArCH3) ppm. 13C{1H} NMR (101 MHz, DMSO-d6): 197.13 (NCcarbene), 134.78, 134.61, 129.90 (ArC4), 128.41 (ArC3,5), 128.30, 128.14, 125.93, 54.90 (NCH2ppze), 52.77 (OCH3), 45.60 (NCH2ppze), 19.59 (ArCH3), 18.54 (ArCH3), 17.97 (ArCH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 58.15, 57.72 (Z/E 85:15) ppm.
O)(OBz)}(CNXyl){C(NHXyl)(NEt2)}] (5g).
From 3e and NHEt2 in a CH2Cl2 solution, 30 min. Yield: 0.575 g, 94%. Anal. calcd for C29H36N3NiO2PS2 (612.41 g mol−1): C, 56.88; H, 5.93; N, 6.86. Found: C, 56.71; H, 5.81; N, 6.79. IR (CH2Cl2, cm−1): 2156 (ν CN). 1H NMR (500 MHz, CDCl3) Z-isomer: 8.15 (s, 1H, NHcarbene), 7.36–7.16 (m, 6H, ArH3,4,5) 7.16–6.97 (m, 5H, OArHBz), 4.76–4.63 (m, 2H, NCH2CH3), 4.24 (qd, J = 7.6, 3.7, 2.5 Hz, 2H, OCH2Bz), 3.61 (m, 2H, NCH2CH3), 2.28 (m, 12H, ArCH3), 1.43 (t, J = 7.1 Hz, 3H, NCH2CH3) 1.27 (t, J = 7.1 Hz, 3H, NCH2CH3) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 199.11 (NCcarbene), 138.35, 138.26, 137.70, 135.35 (ArC*), 128.39 (OArBz), 127.63 (ArC2,6), 127.21 (ArC4), 68.34 (OCH2Bz), 52.58 (NCH2CH3), 42.05 (NCH2CH3), 18.93 (ArCH3), 14.21 and 12.99 (NCH2CH3) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ 61.57, 61.32 (Z/E 76:14) ppm.
O)(OMe))(CNXyl)2C(NHXyl)}2(N2C4H8)] (7).
A red-orange mixture of 3c (0.463 g, 1 mmol) and ppze (0.048 g, 0.5 mmol) in CH2Cl2 (15 mL) was stirred at room temperature for 60 min. At this time, a yellow precipitate appeared, which was collected in a fritted funnel. Slow evaporation gave 7 as yellow microcrystals. Yield 0.920 g, 91%. Anal. calcd for C42H52N6Ni2O4P2S4 (1012.49 g mol−1): C, 49.82; H, 5.19; N, 8.30. Found: C, 49.66; H, 4.98; N, 8.23. HR-MS (ESI-TOF, m/z); calcd for C42H52N6Ni2O4P2S4 = 1011.1188; obtained = 1011.1181 [M + H]+. IR (CH2Cl2, cm−1): 2161 (ν CN). 1H NMR (400 MHz, DMSO): δ 9.36 (s, 1H, NHcarbene), 7.34 (m, 2H, ArH), 7.19 (m, 4H, ArH), 5.53 (m, 1H, OCH2), 4.86 (m, 1H, OCH2), 4.13 (m, 1H, OCH2), 3.93 (m, 1H, OCH2), 2.86 (m, H OCH3), 2.30 (s, 12H, ArCH3) ppm. 13C{1H} NMR (101 MHz, DMSO): δ 135.75(ArC2,6), 134.87 (ArC4), 130.04 (ArC4), 128.43 (ArC3,5), 128.01 (ArC3,5), 52.83 (OCH3), 50.41 (NCH2ppze), 48.00 (NCH2ppze), 19.59 (ArCH3), 18.56 (ArCH3), 18.27 (ArCH3), 18.02 (ArCH3) ppm. 31P{1H} NMR (162 MHz, DMSO-d6): δ 57.61 and 57.54 (Z), 57.24 and 57.11 (E) ppm.
X-Ray diffraction studies
Diffraction data were collected using an Oxford Diffraction Supernova diffractometer, equipped with an Atlas CCD area detector and a four-circle kappa goniometer. For the data collection, Mo source with multilayer optics was used. Data integration, scaling, and empirical absorption correction were carried out using the CrysAlis Program package.31 The structures were solved using direct methods and refined by Full-matrix-least-squares against F2 with SHELX under OLEX2.32,33 The non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed at idealized positions and refined using the riding model. Full-matrix least-squares refinements were carried out by minimizing ∑w(Fo2 − Fc2)2 with the SHELXL weighting scheme and stopped at shift/err < 0.001. The final residual electron density maps showed no remarkable features. Graphics were made with OLEX2 and MERCURY.33–35 Crystal data, particular details, and CCDC reference numbers are given in ESI.† Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre: 2308594 (2e) 2298997 (3c), 2298999 (3d), 2308595 (3e), 2299006 (4a), 2299001 (4c), 2299268 (4e), 229969 (4f), 2299273 (5b), 2299274 (5c), 2299270 (5e), 2299288 (6c), 2299303 (6e), 2299286 (6g), 2299289 (6h), 2299483 (7) and 2300163 (8).Some structures are affected by an incipient disorder which could not be satisfactorily modelled. This leads to large ellipsoids and diminishes the formal quality of the structure. However, despite some remaining Alerts B (for 6e and 7) the quality is good enough to demonstrate without any doubt the connectivity of the molecule concerned, which was the purpose of the determination. In general, the geometric parameters which are more affected correspond to the side chains in the periphery of the molecule while the geometry of the atoms pertaining to the coordination sphere around Ni are less affected and can be taken with a better level of confidence.
Author contributions
Lucía Álvarez-Miguel and Clara del Carmen-Rodríguez contributed equally. The article was written through contributions of all authors. All authors have given approval to the final version of the article.Data availability
The data supporting this article have been included as part of the ESI.†Experimental details, including all synthesis, characterization data and CCDC. See https://doi.org/10.1039/x0xx00000x.
This work has been previously published as a preprint.36
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thanks to the Spanish Ministry of Science and Innovation (MCIN) (RG-R, PID2021-124691NB-I00, funded by MCIN/AEI/10.13039/501100011033/FEDER, UE) for financial support. C. C. R. acknowledges the Junta de Castilla y León (Spain) for a predoctoral contract (Orden EDU/875/2021).References
- B. C. Lee, C.-F. Liu, L. Q. H. Lin, K. Z. Yap, N. Song, C. H. M. Ko, P. H. Chan and M. J. Koh, Chem. Soc. Rev., 2023, 52, 2946–2991 RSC.
- K. Matsubara, Chem. Rec., 2021, 21, 3925–3942 CrossRef CAS PubMed.
- L. B. Junquera, F. E. Fernández, M. C. Puerta and P. Valerga, Eur. J. Inorg. Chem., 2017, 2547–2556 CrossRef CAS.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- L. M. Slaughter, ACS Catal., 2012, 2, 1802–1816 CrossRef CAS.
- V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS.
- M. A. Kinzhalov and K. V. Luzyanin, Coord. Chem. Rev., 2019, 399, 213014 CrossRef CAS.
- L. M. Slaughter, in N–Heterocyclic Carbenes, 2014, pp. 499–524 Search PubMed.
- R. W. Alder, M. E. Blake, C. Bortolotti, S. Bufali, C. P. Butts, E. Linehan, J. M. Oliva, A. G. Orpen and M. J. Quayle, Chem. Commun., 1999, 241–242 RSC.
- R. W. Alder, C. P. Butts and A. G. Orpen, J. Am. Chem. Soc., 1998, 120, 11526–11527 CrossRef CAS.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 CrossRef CAS.
- T. Schulz, M. Leibold, C. Färber, M. Maurer, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2012, 48, 9123–9125 RSC.
- R. A. Michelin, A. J. L. Pombeiro and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2001, 218, 75–112 CrossRef CAS.
- K. V. Luzyanin and A. J. L. Pombeiro, in Isocyanide Chemistry, 2012, pp. 531–550 Search PubMed.
- See selected examples in. (a) A. Maurya and R. Tyagi, Rev. Inorg. Chem., 2024, 44, 255–270 CrossRef CAS; (b) G. Minghetti, F. Bonati and G. Banditelli, Inorg. Chem., 1976, 15, 1718–1720 CrossRef CAS; (c) S. Montanel-Pérez, A. Izaga, A. Laguna, M. D. Villacampa and M. C. Gimeno, ACS Omega, 2018, 3, 13097–13103 CrossRef PubMed; (d) M. Williams, A. I. Green, J. Fernandez-Cestau, D. L. Hughes, M. A. O'Connell, M. Searcey, B. Bertrand and M. Bochmann, Dalton Trans., 2017, 46, 13397–13408 RSC; (e) S. Montanel-Pérez, R. Elizalde, A. Laguna, M. D. Villacampa and M. C. Gimeno, Eur. J. Inorg. Chem., 2019, 4273–4281 CrossRef; (f) A. Marchenko, G. Koidan, A. Hurieva, Y. Vlasenko, A. Kostyuk and A. Biffis, Dalton Trans., 2016, 45, 1967–1975 RSC; (g) S. A. Katkova, M. A. Kinzhalov, P. M. Tolstoy, A. S. Novikov, V. P. Boyarskiy, A. Y. Ananyan, P. V. Gushchin, M. Haukka, A. A. Zolotarev, A. Y. Ivanov, S. S. Zlotsky and V. Y. Kukushkin, Organometallics, 2017, 36, 4145–4159 CrossRef CAS; (h) S.-W. Lai, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Angew. Chem., Int. Ed., 1998, 37, 182–184 CrossRef CAS.
- H. Hou, P. K. Gantzel and C. P. Kubiak, Organometallics, 2003, 22, 2817–2819 CrossRef CAS.
- D. Kremzow, G. Seidel, C. W. Lehmann and A. Fürstner, Chem. – Eur. J., 2005, 11, 1833–1853 CrossRef CAS PubMed.
- C. H. Davies, C. H. Game, M. Green and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1974, 357–363 RSC.
- G. A. Metselaar, E. Schwartz, R. de Gelder, M. C. Feiters, S. Nikitenko, G. Smolentsev, G. E. Yalovega, A. V. Soldatov, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, ChemPhysChem, 2007, 8, 1850–1856 CrossRef CAS PubMed.
- P. C. J. Kamer, R. J. M. Nolte and W. Drenth, J. Am. Chem. Soc., 1988, 110, 6818–6825 CrossRef CAS.
- P. C. J. Kamer, R. J. M. Nolte and W. Drenth, J. Chem. Soc., Chem. Commun., 1986, 1789–1791 RSC.
- W. Drenth and R. J. M. Nolte, Acc. Chem. Res., 1979, 12, 30–35 CrossRef CAS.
- See original works: (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (b) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef; (c) E. M. Badley, J. Chatt, R. L. Richards and G. A. Sim, J. Chem. Soc. D, 1969, 1322–1323 RSC.
- E. Boíllos and D. Miguel, Organometallics, 2004, 23, 2568–2572 CrossRef.
- A. C. Dumke, T. Pape, J. Kösters, K.-O. Feldmann, C. Schulte to Brinke and F. E. Hahn, Organometallics, 2013, 32, 289–299 CrossRef CAS.
- K. V. Luzyanin, A. G. Tskhovrebov, M. C. Carias, M. F. C. Guedes da Silva, A. J. L. Pombeiro and V. Y. Kukushkin, Organometallics, 2009, 28, 6559–6566 CrossRef CAS.
- M. L. Kuznetsov and V. Y. Kukushkin, Molecules, 2017, 22, 1141–1156 CrossRef PubMed.
- G. M. D. M. Rúbio, T. T. Y. Tan, A. Prado-Roller, J. M. Chin and M. R. Reithofer, Inorg. Chem., 2022, 61, 7448–7458 CrossRef PubMed.
- Y.-M. Wang, C. N. Kuzniewski, V. Rauniyar, C. Hoong and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 12972–12975 CrossRef CAS PubMed.
- C. Bartolomé, D. García-Cuadrado, Z. Ramiro and P. Espinet, Inorg. Chem., 2010, 49, 9758–9764 CrossRef PubMed.
- V. CrysAlisPro-Data Collection and Integration Software, Agilent Technologies UK Ltd, Oxford, UK, 2011 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- L. Álvarez-Miguel, C. del Carmen-Rodríguez, M. Valle, C. M. Álvarez, J. M. Martin-Álvarez, R. García-Rodríguez and D. Miguel, This content is a preprint and has not been peer-reviewed, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-xv58c.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, including all synthesis and characterization data. CCDC 2308594, 2298997, 2298999, 2308595, 2299006, 2299001, 2299268, 229969, 2299273, 2299274, 2299270, 2299288, 2299303, 2299286, 2299289, 2299483 and 2300163. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01915f |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |