
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined solid state, solution and DFT study of a dimethyl-cyclen-Pd(II) complex†
Daniele
Paderni‡
 a,
Maria
Voccia‡§
b,
Eleonora
Macedi
*a,
Mauro
Formica
a,
Luca
Giorgi
a,
Lucia
Caporaso
b and
Vieri
Fusi
*a
a,
Maria
Voccia‡§
b,
Eleonora
Macedi
*a,
Mauro
Formica
a,
Luca
Giorgi
a,
Lucia
Caporaso
b and
Vieri
Fusi
*a
aDepartment of Pure and Applied Sciences, University of Urbino, via Ca’ le Suore 2-4, 61029 Urbino, Italy. E-mail: eleonora.macedi@uniurb.it; vieri.fusi@uniurb.it
bDepartment of Chemistry and Biology, University of Salerno, via Giovanni Paolo II 132, 84084 Fisciano, Salerno, Italy
First published on 30th July 2024
Abstract
A new palladium(II) complex containing the previously synthesized 4,10-bis[(3-hydroxy-4-pyron-2-yl)methyl]-1,7-dimethyl-1,4,7,10-tetraazacyclododecane ligand maltonis was prepared and characterized both in solution and in the solid state. Hirshfeld surface and energy framework analyses were also performed. Because maltonis already showed antineoplastic activity, the complexation of Pd(II), chosen as an alternative to Pt(II), was investigated to study its possible biological activity. UV-vis and NMR studies confirmed the formation and stability of the complex in aqueous solution at physiological pH. X-ray diffraction data revealed a structure where the Pd(II) ion is lodged in the dimethyl-cyclen cavity, with maltol rings facing each other (closed shape) even if they are not involved in the coordination. DFT analysis was performed in order to understand the most stable shape of the complex. In view of evaluating its possible bioactive conformation, the DFT study suggested a slight energetic preference for the closed one. The resulting closed complex was stabilized in the X-ray structure by intermolecular interactions that replace the intramolecular interactions present in the optimized complex. According to the DFT calculated formation energies, notwithstanding its rarity, the Pd(II) complex of maltonis is the thermodynamically preferred one among analogous complexes containing different metal ions (Pt(II), Co(II), and Cu(II)). Finally, to study its possible biological activity, the interaction between the Pd(II) complex of maltonis and nucleosides was evaluated through NMR and DFT calculations, revealing a possible interaction with purines via the maltol moieties.
Introduction
The role of metal complexes in medicinal biochemistry, and in particular as anticancer agents, experienced a pronounced advancement after the discovery of the potent drug cisplatin, [PtCl2(NH3)2], whose FDA approval in 1978 for the treatment of testicular cancer gave birth to medicinal inorganic chemistry. The impact of metal compounds in modern medicine has been growing ever since; however, very few complexes have been successfully translated into clinical use. Nevertheless, transition metal complexes possess several properties that can be tuned via ligand and metal selection, paving the way for the rational design of new drugs and diagnostic or theranostic agents.1,2,3–10,11–15Besides their considerable therapeutic effect, platinum-based drugs feature significant disadvantages, such as non-negligible side effects on several organs due to their non-specificity, dose-limiting toxicity and increased resistance in some types of tumors.16,17 Because of these limitations, new anticancer agents based on metals different from platinum are being developed.6,18,19 Among the possible alternatives, the use of palladium-based compounds as potential anticancer drugs20–23 has aroused particular interest due to the similar coordination chemistry of Pd(II) and Pt(II) (both are soft metals with a rich chemistry that is not limited to soft bases (P and S) but extended to hard ligands (N and O);24,25 same coordination number (four) and geometry (square planar); similar bond lengths and sizes), their activity towards cisplatin-resistant tumor cell lines and their different modes of action compared to cisplatin.26,27
Compared to Pt(II) analogues, Pd(II) complexes show faster exchange kinetics (104–105 times), making them more reactive in solution with possibly higher toxicity and different biological behaviors. This aspect could affect the study of palladium complexes as potential anticancer agents; the choice of the ligand thus turns out to be crucial. In particular, the use of macrocyclic ligands that could strongly bind to the metal center can stabilize the Pd(II) complex and lower the risk of hydrolysis of palladium–ligand bonds.6,15,21,28,29
The tetraazacyclododecane macrocycle, cyclen, is an easily functionalizable N-donor analogue of crown ether that has been widely used in molecular sensing, catalysis and biomedicine for a long time. Many metal complexes of cyclen-based ligands have been reported in the literature, showing different applications.30–48 Functionalization of dimethyl-cyclen with two 3-hydroxy-2-methyl-4H-pyran-4-one (maltol) units has been previously reported by our group (4,10-bis[(3-hydroxy-4-pyron-2-yl)methyl]-1,7-dimethyl-1,4,7,10-tetraazacyclododecane, maltonis, Chart 1),49 with the ligand showing interesting and unique coordination properties towards transition, alkali, alkaline-earth and rare earth metal ions.50–54 Maltonis features multiple donor atoms of different hard/soft nature; therefore, it can bind different transition metal ions (Cu(II), Co(II)) that are able to preorganize the ligand in distinct ways depending on the coordination requirements of the metal center. By using a suitable preorganizing transition metal ion, very stable complexes that are generally difficult to form in aqueous solutions can be obtained, such as those involving alkali, alkaline-earth and rare earth metal ions.
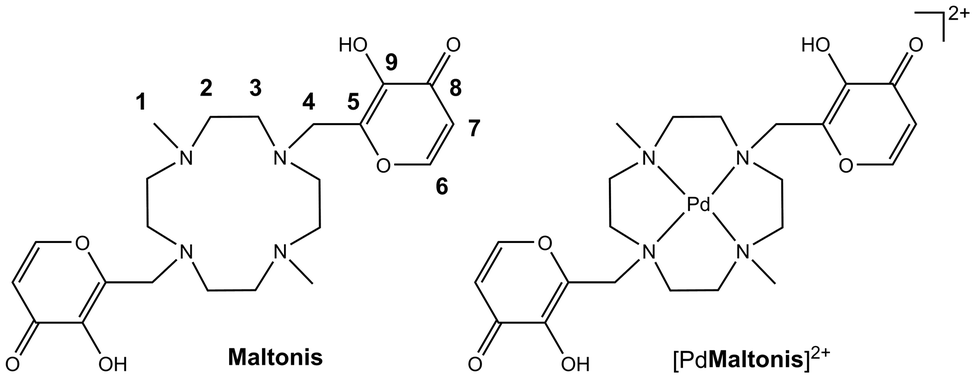 | ||
| Chart 1 The ligand maltonis and its palladium(II) complex studied in this work, along with the NMR labelling scheme. | ||
Similarly to other bis-maltol polyamine ligands,49,55–58 maltonis revealed an interesting antineoplastic activity.49,59,60 Preliminary mechanistic observations revealed the ability of maltonis to interfere with the chromatin structure,49 inducing a DNA damage-like response characterized by γ-H2AX recruitment.59 More recently, maltonis was shown to induce a profound remodulation of the histone code and the associated remodulation of the gene expression in the NB4 cell line, which is considered the most representative in vitro cellular model of human acute promyelocytic leukemia (APL). Maltonis can epigenetically reprogram the gene expression profile of APL cells, inducing an intriguing antiviral-like response, concomitantly with the downregulation of c-MYC-related pathways, thus making it an attractive candidate for antileukemic therapy.60
For these reasons, the binding behavior of maltonis towards the Pd(II) ion was herein studied, exploiting the versatile coordination abilities of the ligand and combining its antineoplastic activity with the promising antitumor potential coming from Pd(II) complexation.
In this paper, the synthesis of the Pd(II) complex of maltonis is reported: to the best of our knowledge, only three Pd(II)-cyclen-based metal complexes61–63 have been deposited in the Cambridge Structural Database (CSD)64 so far, suggesting a non-straightforward complexation of such metal ion in that specific cavity. The complex was studied from both solution and solid state points of view by means of UV-vis and NMR spectroscopies for the former, and single crystal X-ray diffraction for the latter.
The molecular structure of the Pd(II) complex was investigated by DFT calculations, comparing the possible conformations, the formation energies and the stabilities of the Pd(II) complex and analogous complexes with different metal ions. The solid state assembly of the Pd(II) complex was also investigated. The intermolecular interactions and their quantitative contributions towards the crystal packing were assessed via Hirshfeld and fingerprint plot analysis, and by using the energy framework module of CrystalExplorer.65–67
All studies were performed to gain insight into the possible shape assumed by the complex. The bioactive conformation of a compound is indeed crucial to produce a possible biological response. This is due to the necessary recognition of the compound by the target receptor.68,69 In this regard, metal chelation could also lead to an activity enhancement.70
Finally, in view of testing the Pd(II) complex of L as an anticancer agent, a possible interaction with nucleosides was explored through NMR and DFT studies.
Experimental
General methods
All chemicals were purchased in the highest quality that was commercially available. Solvents were RP grade, unless otherwise indicated, and used without further purification. All aqueous solutions were prepared with deionized water.Synthesis of [Pdmaltonis]·(ClO4)2 (1)
Maltonis·3HClO4 (5.6 mg, 8.7 μmol) was dissolved in H2O (2.5 mL) at acidic pH values (HClO4). Then, 1 equiv. of Pd(II) (as K2PdCl4, 2.8 mg) was added and the pH was adjusted to around 5–6 (litmus paper) by using a NaOH 0.1 mol dm−3 solution. The solution was stirred for 30 minutes at 50 °C, and then it was left to cool down to room temperature. Crystals of 1 suitable for X-ray diffraction were obtained by slow evaporation of this solution (5 mg, 76%). 1H NMR (400 MHz, D2O pH 7.4, 25 °C): δ = 2.31 (s, 6H), 3.09 (m, 4H), 3.27 (m, 4H), 3.83 (m, 8H), 3.99 (s, 4H), 6.56 (d, 2H, J = 5.4), 8.13 (d, 2H, J = 5.5) ppm (Fig. S15†). 13C NMR (100 MHz, D2O pH 7.4, 25 °C): δ = 46.18, 53.73, 57.83, 60.06, 113.19, 133.74, 144.30, 154.69, 181.84 ppm (Fig. S16†).UV-vis absorption measurements
The spectrophotometric measurements were carried out at 298.1 K using a Varian Cary-100 spectrophotometer equipped with a temperature control unit. UV-vis absorption pH-titration of the maltonis/Pd(II) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 system was performed in aqueous solution starting from acidic to basic pH by using a NaOH 0.1 mol dm−3 solution and a ligand concentration of 2.0 × 10−5 mol dm−3. Due to the slow Pd(II) complexation kinetics, the UV-vis absorption titration of maltonis with Pd(II) at pH 7.4 (HEPES buffer solution, 2.0 × 10−3 mol dm−3) was performed by preparing distinct solutions of maltonis (2.0 × 10−5 mol dm−3, 3 mL) containing increasing amounts of Pd(II) (as K2PdCl4) from 0.5 to 3 equivalents, and analysing them after two hours to be sure of Pd(II) complex formation.
:1 system was performed in aqueous solution starting from acidic to basic pH by using a NaOH 0.1 mol dm−3 solution and a ligand concentration of 2.0 × 10−5 mol dm−3. Due to the slow Pd(II) complexation kinetics, the UV-vis absorption titration of maltonis with Pd(II) at pH 7.4 (HEPES buffer solution, 2.0 × 10−3 mol dm−3) was performed by preparing distinct solutions of maltonis (2.0 × 10−5 mol dm−3, 3 mL) containing increasing amounts of Pd(II) (as K2PdCl4) from 0.5 to 3 equivalents, and analysing them after two hours to be sure of Pd(II) complex formation.
NMR spectroscopy
1H and 13C NMR spectra were recorded on a Bruker Avance 400 instrument, operating at 400.13 and 100.61 MHz, respectively, and equipped with a variable temperature controller. The temperature of the NMR probe was calibrated using 1,2-ethanediol as a calibration sample. Chemical shifts (δ scale) are presented in ppm and referenced by residual solvent peak. Coupling constants (J values) are given in hertz (Hz). 1H–1H and 1H–13C correlation experiments were performed by using standard Bruker pulse sequences to assign the signals. All measurements were performed in D2O.The 1H NMR pH titration of the maltonis/Pd(II) 1:1 system was performed by starting from acidic to basic pH using a KOD solution in D2O and a ligand concentration of 4.0 × 10−3 mol dm−3. 1H NMR titrations of 1 with nucleosides were performed by dissolving L and 1 equiv. of Pd(II) in D2O, and adjusting the pH to 7.4 with a KOD solution in D2O ([L] = 5.8 × 10−3 mol dm−3). After stirring this solution at 50 °C for 30 minutes to allow for complex formation, increasing amounts of a solution of the nucleosides at pH 7.4 were added ([A] = [G] = [T] = [U] = 0.1 mol dm−3). The stability of the complex was evaluated by recording 1H NMR spectra of 1 at pH 7.4 and 310 K over a weekend.
X-ray crystallography
Intensity data for compound 1 were collected at 100 K using a Bruker Apex-II CCD diffractometer and Cu-Kα radiation (λ = 1.54184 Å). Data were collected with the Bruker APEX2 program,71 integrated and reduced with the Bruker SAINT software,72 and the absorption correction was performed with SADABS-2016/2.73The crystal structure was solved using the SIR-2004 package,74 and refined by full-matrix least squares against F2 using all data (SHELXL-2018/375).
All non-hydrogen atoms were refined with anisotropic displacement parameters, while all of the hydrogen atoms, with the exception of the one bonded to O1, were introduced in idealized positions. Their coordinates and thermal parameters were refined in agreement to those of the atom to which they are bonded. Hydrogen atom H1O (bonded to O1) was found in the Fourier difference map. Its coordinates were freely refined, while its thermal parameter was set in accordance to that of O1.
Geometrical calculations were performed by PARST97,76 and molecular plots were produced by the program Mercury (2024.1.0).77
Crystallographic data and refinement parameters are reported in Table 1. In Fig. 3, an ORTEP view of the [PdL]2+ cation in 1 is reported.
| 1 | |
|---|---|
| Empirical formula | C22H32Cl2N4O14Pd |
| Formula weight | 753.81 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.54178 |
| Crystal system, space group | Monoclinic, C2/c |
| Unit cell dimensions (Å, °) | a = 19.6006(9) |
| b = 9.2491(4), β = 119.8500(10) | |
| c = 17.4562(12) | |
| Volume (Å3) | 2744.8(3) |
| Z, Dc (mg cm−3) | 4, 1.824 |
| μ(mm−1) | 7.961 |
| F(000) | 1536 |
| Crystal size (mm) | 0.17 × 0.18 × 0.20 |
| θ range (°) | 5.203 to 72.482 |
| Reflections collected/unique (Rint) | 21120/2719 (0.0672) |
| Data/parameters | 2719/198 |
| Final R indices [I > 2σ] | R1 = 0.0397, wR2 = 0.1109 |
| R indices (all data) | R1 = 0.0399, wR2 = 0.1111 |
| Gof | 1.063 |
Computational analysis
Details about the optimizations and Cartesian coordinates of the optimized species are reported in the ESI.†Results and discussion
Synthetic procedures
The synthesis of ligand maltonis was previously reported.49The [Pdmaltonis]·(ClO4)2 (1) complex was synthesized by dissolving maltonis·3HClO4 in an aqueous solution, followed by the addition of 1 equiv. of Pd(II) and adjustment to pH 6 (NaOH 0.1 mol dm−3). After 30 minutes of stirring at 50 °C, the solution was cooled down to room temperature. After a few days, crystals of 1 suitable for X-ray diffraction were obtained by slow evaporation.
Solution studies
The UV-vis absorption behavior of ligand maltonis in aqueous solution as a function of pH has been previously investigated.49 Briefly, maltonis shows an absorption band with λmax at 275 nm (ε = 19600 cm−1 mol−1 dm3) at acidic pH values, relative to the maltol unit in its neutral form, and a band with λmax at 317 nm (ε = 13800 cm−1 mol−1 dm3) at alkaline pH values, relative to the maltol unit in its deprotonated form. This latter form is visible starting from pH 5, where the HL+ species is present in solution, and reaches its maximum at pH 10, where the neutral L species is fully formed. HL+ and L bear one and two maltolate functions, respectively (Fig. S1†).49
In the present paper, the formation and the UV-vis absorption behavior of the Pd(II) complex of maltonis in aqueous solution as a function of pH has been investigated. The formation of the metal complex occurs immediately after the addition of Pd(II) ion (1 equiv.) in solution, even at acidic pH values. This is suggested by the growth of the band with λmax at 320 nm, attributed to the absorption of the maltolate function, that started right off the metal addition at pH 2 (see black line and inset in Fig. 1). Contrarily, the same band in the free ligand started to grow only at pH > 5.49 It is probable that this is truly due to the complex formation since the coordination of the Pd(II) ion slightly increases the acidity of the OH group of maltols, even if they do not seem to be directly involved in the coordination (vide infra). Based on a comparison between the UV-vis absorption measurements on the complex and the free ligand, it can be hypothesized that the two maltolate species in the Pd(II) complex feature similar acid–base properties to those of the free ligand, exhibiting protonation constants of 8.11 and 6.67.49
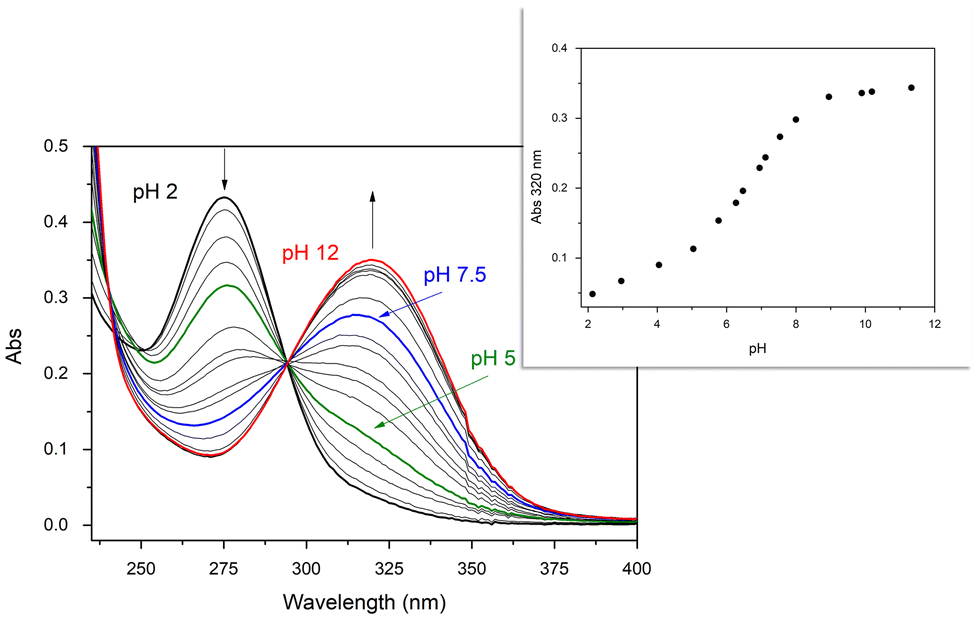 | ||
| Fig. 1 UV-vis spectra of the maltonis/Pd(II) 1:1 system in aqueous solution at different pH values. [maltonis] = 2.0 × 10−5 M. Inset: trend of absorbance for maltonis/Pd(II) 1:1 at λ = 320 nm in aqueous solution at different pH values. | ||
During the pH titration, the band of neutral maltol (λmax at 275 nm) continuously decreased starting from pH 2, exchanging with the band of the deprotonated maltol (λmax at 320 nm). The results are clearly visible at pH > 5 (green line in Fig. 1), and reached the maximum absorption at alkaline pH values (ε = 17500 cm−1 mol−1 dm3). Interestingly, at neutral pH (blue line in Fig. 1), the maltolate form is mainly visible in the spectrum, in contrast to what is observed in the case of free maltonis, where the band of the neutral maltol was still visible.49
A UV-vis titration of maltonis with Pd(II) in aqueous HEPES buffer solution (Fig. S2†) highlighted the formation of the Pd(II) complex at fixed physiological pH. The addition of increasing amounts of metal ion at pH 7.4 indeed induced the deprotonation of the maltol moieties (vide supra). Due to the slow kinetics of Pd(II), the experiment was performed by preparing different solutions of maltonis containing increasing concentrations of the metal ion up to three equivalents, which were then left at room temperature for two hours before the analysis (see Experimental section).
The formation of the complex in solution was also studied via1H NMR measurements by performing a pH titration similar to the above cited procedure. The spectrum of the mixture maltonis/Pd(II) 1:1 at pH < 2 is basically the same as that of the free ligand (Fig. S3†); namely, the complex does not form at pH < 2.
Beginning at pH 2.1, all resonances (and especially the aliphatic ones) underwent significant shifts, thus suggesting the complexation of the metal ion by the ligand starting from pH ≥ 2 (Fig. S3† and 2). It should be noted that at pH = 2.1, the spectrum shows two distinct series of resonances attributable to two species slowly exchanging on the NMR time scale, whose molar ratio changes up to pH < 5.3. The main species is ascribed to the Pd(II)-maltonis complex, while the other one is ascribed to the free maltonis. This suggests an existing equilibrium between the free and complexed L on the NMR time-scale, although it strongly shifted towards the complexed form. By looking at the main resonances attributed to the Pd(II)-complex, the aromatic (H6 and H7) and benzylic (H4) resonances shifted downfield compared to those of free maltonis, while the signal of the methyl groups (H1) showed a marked upfield shift (see Chart 1 for NMR atom labelling). The aliphatic signals belonging to the macrocycle also showed a pronounced variation, suggesting both the stiffening of the system upon coordination of the metal ion and the involvement of the macrocycle amine functions in the coordination. In particular, the multiplet assigned to H2 shifted upfield, appearing as two distinct signals, while the triplet assigned to H3 markedly shifted downfield and became broader. The spectrum shows a number of 1H and 13C signals in the whole investigated pH range that are in agreement with a C2v symmetry of both maltonis and the complex on the NMR time scale.
 | ||
| Fig. 2 1H NMR chemical shifts of maltonis (pH < 2) and the Pd(II) complex (pH ≥ 2) in aqueous solution as a function of pH. See Chart 1 for NMR atom labelling. | ||
Starting from pH ≥ 5.3, only one pattern of resonances ascribed to the Pd(II)-maltonis complex can be detected, suggesting the full formation of the species.
Moving from pH 5.3 to pH 7, all resonances shifted upfield (Fig. 2 and S2†). By moving towards more alkaline pH values, the aliphatic signals remained substantially unchanged. Thus, it can be suggested that the Pd(II) ion is fully coordinated by the macrocyclic skeleton of maltonis and is not released in the alkaline field. On the contrary, in the same basic pH environment, the two aromatic protons showed variations, shifting towards different directions.
Considering the whole NMR measurement, the resonances of maltonis in the Pd(II) complex showed a trend as a function of pH that is similar (concerning the maltol resonances) and different (concerning the aliphatic signals) to that of the free ligand.49 This suggests that Pd(II) is stabilized by the tetraamine macrocyclic framework, while the maltol rings are not involved in the coordination.
The complexation of Pt(II) was also investigated in solution, but the UV-vis measurement revealed a behaviour that was quite similar to that of the free ligand. Upon the addition of the metal ion, the 1H NMR spectrum did not change compared to that of the free ligand (Fig. S4†). These findings suggest that the ligand maltonis is not able to coordinate Pt(II) under the present experimental conditions.
Single crystal X-ray diffraction
The formation of a 1:1 Pd(II) complex was also confirmed in the solid state by X-ray diffraction. Fig. 3 shows an ORTEP view of the [Pdmaltonis]2+ cation in [Pdmaltonis]·(ClO4)2 (1), along with the atom-labelling scheme. The X-ray crystallographic analysis revealed that 1 crystallizes in the C2/c space group (full crystallographic data and refinement parameters in Table 1).
 | ||
| Fig. 3 ORTEP view of the [PdL]2+ cation in 1 along with the X-ray structure atom-labelling scheme. Hydrogen atoms are omitted for the sake of clarity, except for those of the maltol hydroxyl groups. Thermal ellipsoids are shown at 30% probability. | ||
In the asymmetric unit of 1, one half of the Pd(II) complex and a perchlorate anion are present, with the metal ion lying on a two-fold rotation axis. The perchlorate anion interacts via hydrogen bond with the –OH maltol group (vide infra, Table S1†). The cyclen macrocycle adopts the usual [3333]C-corners conformation.78 Meanwhile, the Pd(II) ion is tetracoordinated by the four N atoms of the macrocycle, being out of the plane defined by such atoms (distance Pd(II)-plane 0.3927(3) Å, see Table S2†). This is in line with the square planar coordination of the metal ion.
On the basis of a CSD survey (Fig. S5a†), only 3 out of 1422 deposited 1,4,7,10-tetraazacyclododecane-based metal complexes contain Pd(II).61–63 Among them, only one is a trans-dimethyl-cyclen similarly to 1 (refcode: JODSUU61).
JODSUU is the only Pd(II) compound among 50 trans-dimethyl-cyclen metal complexes found in the CSD (Fig. S5b†) containing other transition metal ions (Mn(II), Ni(II), Cu(II), Co(II), Zn(II), Y(III)), post-transition (Pb(II), Bi(III)), rare earth (Eu(III), Lu(III)) and alkali (Li(I), Na(I)).
Due to the lack of numerous similar structures and to obtain more significant data, the N–Pd bond lengths found in 1 (see Table S3†) were compared to those found in the Pd-complexes of the (i) three cited cyclen compounds, (ii) linear fragment in Fig. S5c,† and (iii) tetraazamacrocycles (similar, but bigger than cyclen; Fig. S6†), revealing comparable values in all cases.
In the [PdMaltonis]2+ cation, the two maltol rings are not involved in the coordination of Pd(II). This is in agreement with its geometrical requirements being fulfilled by the four N atoms of the macrocycle. Nevertheless, even if the O atoms of the maltol units do not coordinate to the metal centre, the two rings are arranged to face each other above the macrocycle cavity and are almost orthogonal to the mean plane formed by the four N donor atoms (Table S2†). In other words, the [Pdmaltonis]2+ cation features a closed shape, rather than an open or partially open one (Fig. 4), with both maltol fragments located on the same side with respect to the macrocycle plane. Moreover, from DFT calculations, the closed shape appears to be slightly energetically favoured compared to the others (vide infra, Table 3).
 | ||
| Fig. 4 Possible ligand conformations within the complexes: (a) open; (b) closed; (c) partially open. | ||
In the crystal packing of 1, complexes stack upon each other, interacting via weak HC–H⋯OC hydrogen bonds between the methylene hydrogen of the macrocycle of a complex and the carbonyl group of another complex, forming columns that develop along the b axis (Fig. 5 and 6a, see interaction 1 in Table 2). Columns having the same direction align side by side along the a axis (Fig. 5a, view along the b axis), while columns developing the opposite way alternate along the c axis (Fig. 5b, view along the a axis).
 | ||
| Fig. 5 Crystal packing of 1. View along the b (a) and a (b) axes, showing columns with opposite directions (magenta and blue). (c) Perspective view along the b axis. Hydrogen atoms are omitted for clarity. Palladium ions in dark cyan (ball-and-stick), oxygen atoms in red, carbon atoms in light grey, nitrogen atoms in light blue, chlorine atoms in green. Perchlorate anions are depicted in ball-and-stick style. | ||
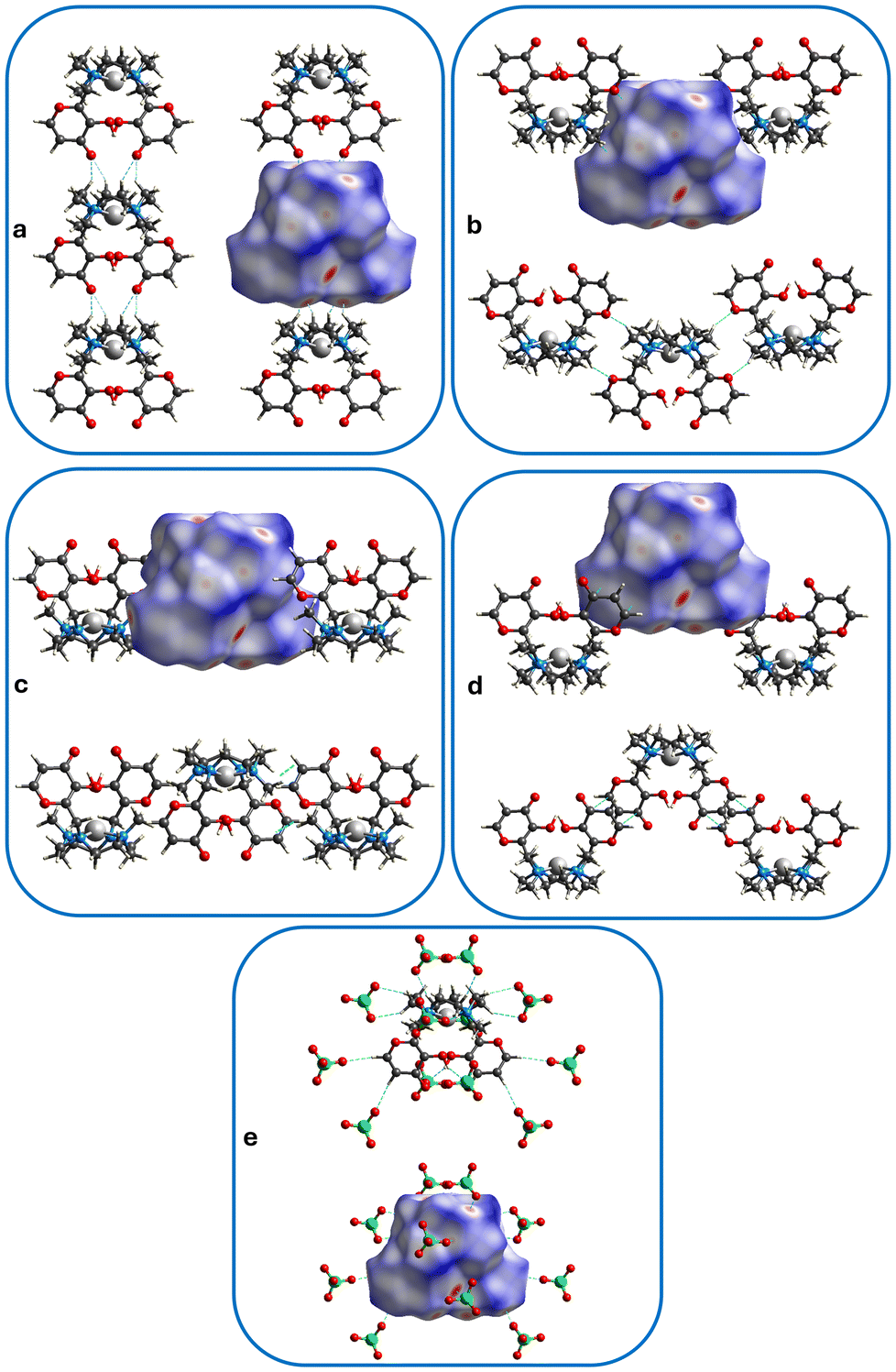 | ||
Fig. 6 Hirshfeld surface showing contacts (see Table 2) belonging to: (a) interaction 1 (HC–H⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, columns), (b) interaction 2 (HC–H⋯O), (c) interaction 3 (H2C–H⋯CC, CH⋯π), (d) interaction 4 (OC⋯CC, π⋯π); (e) hydrogen bonds between complexes and perchlorate anions (see Table S1†). Contacts are depicted as light-blue dotted lines. Palladium ions in light grey, oxygen atoms in red, carbon atoms in grey, nitrogen atoms in blue, chlorine atoms in green, hydrogen atoms in white. O, columns), (b) interaction 2 (HC–H⋯O), (c) interaction 3 (H2C–H⋯CC, CH⋯π), (d) interaction 4 (OC⋯CC, π⋯π); (e) hydrogen bonds between complexes and perchlorate anions (see Table S1†). Contacts are depicted as light-blue dotted lines. Palladium ions in light grey, oxygen atoms in red, carbon atoms in grey, nitrogen atoms in blue, chlorine atoms in green, hydrogen atoms in white. | ||
| Interaction | D–H⋯A | D⋯Aa | H⋯Ab | D–H⋯Ab | Symmetry operation |
|---|---|---|---|---|---|
| a Crystallographic data from the CIF file. b data from CrystalExplorer: the lengths of the X—H bonds are normalized using standard X–H distances from Allen.79 Thus, the reported X–H distances and X⋯H contacts are not equal to those calculated from the original CIF files. | |||||
| 1 | C8–H8B⋯O2 | 3.080(5) | 2.28 | 129 | x, 1 + y, z |
| C9–H9B⋯O2 | 3.230(5) | 2.31 | 142 | ||
| 2 | C7–H7A⋯O3 | 3.687(5) | 2.72 | 148 | 0.5 − x, 1.5 − y, −z |
| 3 | C11–H11C⋯C2 | 4.012(7) | 3.09 | 143 | x, 1 − y, z − 1/2 |
| 4 | C1⋯C3 | 3.491(6) | 1.5 − x, ½ −y, 1 − z | ||
Columns are connected in the three dimensions through weak HC–H⋯O, CH⋯π and π⋯π contacts between the Pd(II)-complexes (respectively between macrocyclic methylene hydrogen atoms and maltol heterocyclic oxygen atom) (interaction 2 in Table 2), macrocyclic methyl hydrogen atoms and maltol double bonds (interactions 3 and 4 in Table 2), as well as hydrogen bonds between maltol –OH or –CH groups, or macrocyclic –CH2 groups of the complexes and perchlorate anions (Table S1†). The latter arrange in the crystal packing in a sandwich-like fashion, creating anion-complex-anion patterns, favouring the formation of a three-dimensional net throughout the whole crystal (vide infra).
Hirshfeld surface and fingerprint plot analysis
The main intermolecular interactions in the crystal were assessed by means of Hirshfeld surface (HS) and fingerprint plots analysis (Fig. 6, 7 and S6†). In addition, intermolecular interaction energies were calculated using CrystalExplorer1765,66,80 (Fig. 8 and S7†).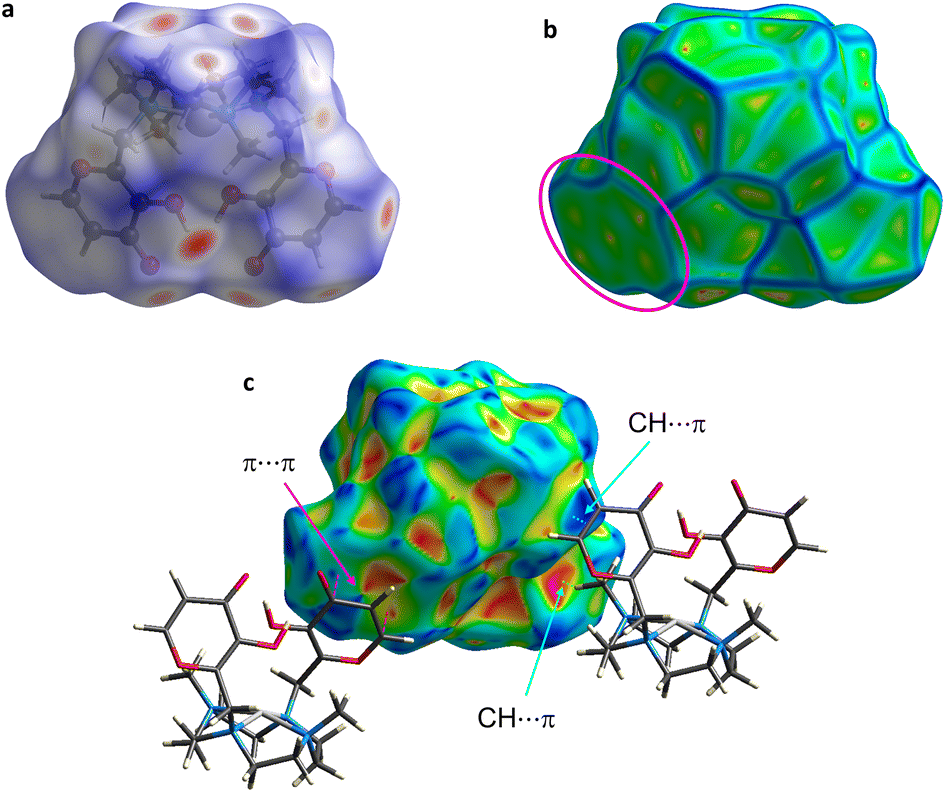 | ||
| Fig. 7 The Hirshfeld surface (volume: 535.9 Å3, area: 387.9 Å2, globularity 0.822, asphericity 0.029) of the palladium complex in 1 mapped over (a) dnorm, (b) curvedness and (c) shape index. The π–π stacking is indicated by the magenta oval (b) and arrow (c); the CH⋯π contact is represented by the light blue arrows (c). | ||
 | ||
| Fig. 8 Energy frameworks for the crystal structure of 1. View of total energies (blue cylinders), Coulomb energies (red cylinders) and dispersion energies (green cylinders) along the a (a and b), b (c) and c (d) axes. Cylinders relative to motifs 1–4 (see Fig. S8†) are highlighted. The cluster displays the contents of two unit cells along b. Hydrogen atoms have been omitted for clarity. | ||
Fig. 6a–e shows the normalized contact distance (dnorm) values mapped onto the Hirshfeld surface (see ESI† for more details). The intracolumnar HC–H⋯OC hydrogen bonds appear on the Hirshfeld surface as red spots, and they represent the shortest contacts that develop between complexes. The O2 oxygen atom behaves as a bidentate acceptor towards two CH2 groups of the macrocycle (Fig. 6a, interaction 1 in Table 2). All other red spots refer to contacts between complexes and perchlorate anions (C–H⋯O and O–H⋯O bond types, Fig. 6e and Table S1†). The HC–H⋯O (Fig. 6b, interaction 2 in Table 2), H2C–H⋯CC (CH⋯π,81 interaction 3 in Table 2 and Fig. 6c) or OC⋯CC (π⋯π, interaction 4 in Table 2 and Fig. 6d) contacts between complexes equal to the sum of vdW radii82 appear as white spots on the surface.
HS mapped over dnorm, shape index and curvedness are presented in Fig. 7. The measures of curvature (shape index and curvedness) provide further information about the packing.83 The shape index mapped onto HS highlights the presence of the above described CH⋯π contact as a red π-hole between the H atom of the methyl group and the aromatic maltol ring (red hollow – and mutual blue bump – indicated by light blue arrows in Fig. 7c). The above cited π–π stacking between the maltol rings appear instead on the same surface as adjacent red and blue triangles (“blue-red bow-tie” pattern indicated by the magenta arrow in Fig. 7c).84,85 In addition, the π–π stacking is highlighted onto the curvedness surface as the indicated flat region (Fig. 7b).80
Fingerprint plots are reported in Fig. S7 (see ESI† for more details). On the HS of the Pd(II) complex, the O⋯H interactions appear as the largest (57.2%) and more asymmetrical region of the fingerprint plot. The O(inside)⋯H(outside) interactions (bottom right) represent 9.9% of the plot, with the spike being due to intracolumnar contacts (de + di ≈ 2.3 Å). Meanwhile, the H(inside)⋯O(outside) interactions (top left) account for 47.4% of the plot, with the sharp long spike embodying contacts with perchlorate anions (de + di ≈ 2 Å). The frequency of occurrence of H(inside)⋯O(outside) interactions is increased in the regions of de ≈ 1.2–1.9 Å and di ≈ 0.8–1.7 Å on the plot diagonal, as suggested by the presence of light blue to green to red areas.
H⋯H interactions contribute to 30.3% of the Hirshfeld surface, with the shortest contacts being ≈ 2.4 Å.
The combined C⋯H/H⋯C contacts contribute to 5.8% of the surface. They correspond to CH⋯π contacts (in agreement with the shape index surface, Fig. 7), and are depicted as characteristic wings at the top and bottom sections of the related plot.
The C⋯C contacts contribute to 2.7% of the surface, and feature the typical stacking kite motif on the middle plot. This is due to the formation of π⋯π interactions between aromatic rings, which is in agreement with the shape index and curvedness surfaces (Fig. 7).
All other contacts contribute to 4% of the HS (Fig. S7†).
Energy frameworks analysis
CrystalExplorer intermolecular energy calculations (TONTO B3LYP method with 3-21G basis set level) were performed to quantify the intermolecular interactions within the crystal packing of 1 and visualize them as cylinders, whose width is proportional to the strength of the interaction (see ESI† for more details).The computed interaction energies in kcal mol−1, the symmetry operations, and the distances between molecular centroids (R) relative to selected motifs corresponding to the strongest contacts in the crystal packing of 1 are reported in Fig. S8.†
Cylinders relative to motifs 1, 2 and 3 (see Fig. S8†) are reported in Fig. 8, and are all comparable in size. This means that the corresponding interaction energies among the complexes are very similar to each other. Thus, a three-dimensional net forms, with intracolumnar interactions along b (interaction 1 in Table 2) and intercolumnar interactions along c (interaction 3 in Table 2), forming planes parallel to the bc plane and connected to each other by intercolumnar interactions along the ac plane bisector (interaction 2 in Table 2). Fig. 8 and S7† also show the presence of destabilizing energies (motif 4, yellow cylinders), forming zig-zag chains developing along the ac plane bisector. Being that 1 is an ionic crystal, it necessarily incorporates large positive destabilizing energies (cation–cation or anion-anion) due to a large positive electrostatic contribution (Ecoul). However, such energies are counterbalanced by a larger number of stabilizing energies.
Geometry optimizations
The analysis of the molecular structure of [Pdmaltonis]2+ showed that, notwithstanding the non-involvement of the –OH maltol groups in the coordination of Pd(II), the two rings are arranged to form a closed structure, rather than an open or a partially open one (Fig. 4).The three mentioned shapes were previously observed in the solid state for the same ligand, depending both on the presence of a metal ion and on the type of the metal ion coordinated to maltonis (Fig. 9).
 | ||
| Fig. 9 Structures of the diprotonated ligand L (a, refcode: SELGAT)49 and metal complexes of maltonis with Cu(II) (b1: tetranuclear dimer, b: mononuclear monomer within the dimer; refcode: WELGUR)50 and Co(II) (c, refcode: WELGEB).50 Hydrogen atoms are omitted for clarity, except for those of the maltol hydroxyl groups and the protonated nitrogen atoms in a. Copper ion in orange, cobalt ion in blue, oxygen atoms in red, carbon atoms in light grey, nitrogen atoms in light blue, hydrogen atoms in white. | ||
In the absence of a metal ion, the structure of the diprotonated H2L2+ species featured an open conformation (Fig. 4a and 9a), where the two maltol groups do not interact and are located far away from each other.49 In this case, the N–CH3 groups are protonated, with the protons pointing in toward the cavity of the macrocycle.
In the presence of metal ions, different ligand conformations within the complexes have been observed, depending on the geometrical requirement of the metal centre. To match the geometrical requirement of Cu(II), for instance, the ligand participates with an oxygen atom of a single maltol group, together with the four nitrogen atoms of the macrocyclic base, to give a penta-coordination. This maltol unit is forced indeed by the coordination of the metal to move from a position almost parallel to the macrocyclic scaffold (as in the open structure) to a position that is almost perpendicular to the polyamine mean plane. The second maltol unit instead maintains a parallel position, and is involved in the coordination of a second Cu(II) ion, whose coordination sphere is saturated by the maltol unit of another copper complex. The structure thus emerges as a tetranuclear dimer (Fig. 9b1), in which a single copper complex assumes a partially open shape (Fig. 4c and 9b).50
To match the octahedral geometrical requirement of the Co(II) ion, the ligand is involved in the coordination with both maltol units and the four nitrogen atoms of the macrocycle. In this case, the convergence of the two maltol groups above the macrocycle is achieved, leading to a closed shape (Fig. 4c and 9c).50
It is important to underline that, differently from 1, all crystal structures of maltonis complexes reported so far showed both deprotonated maltols involved in metal cation coordination. This can be attributed to the different coordination requirements of the bound metal ions that forced the deprotonation of the maltol-OH functions, contrarily to Pd(II).
In view of testing the biological activity of the Pd(II) complex, the knowledge of the most stable overall shape of the complex would be of help. This is due to the crucial role of the bioactive conformation of a drug. To this purpose, a DFT geometry optimization of the palladium complex with an implicit model of water as the solvent was performed, so as to release the molecular conformation from the crystal packing potential and avoid the influence of the intermolecular contacts (for the computational details see the ESI†). The X-ray structures of the diprotonated ligand49 and of the Cu(II) and Co(II) complexes50 (a, b and c in Fig. 9) were used in the DFT calculations as starting models for the open, partially open and closed structures respectively. A Pd(II) cation was then added to each structure. In addition, such analysis was undertaken to try to understand the reasons for the closed shape observed for the [Pdmaltonis]2+ complex, even if maltol rings are not directly involved in the metal coordination.
The DFT calculations revealed that the closed shape observed in the solid state for the palladium complex is slightly energetically preferred with respect to all those calculated (Table 3). Nevertheless, the optimized molecule differs from that obtained by X-ray diffraction analysis (1a and 1, respectively, in Tables S4 and S5†). There is an intramolecular hydrogen bond between the two maltol rings, involving the carbonyl and hydroxyl functions (O–H⋯CO: 1.47 (O⋯O: 2.50) Å, 162.6° in 1a (Table S5†) vs. 3.31(6) (O⋯O: 3.823(4)) Å, 142.9(1)° in 1) (Fig. 10) by virtue of a greater bending of the maltol rings towards each other, with the Pd(II) ion being located farther away from the mean plane formed by the four N donor atoms than in the crystal structure of 1 (Table S5†).
 | ||
| Fig. 10 Top: top view of the [Pdmaltonis]2+ cation in 1 (colored by element) and 1a (magenta). Bottom: superimposition of the two complexes (RMSD: 0.4446 Å). Hydrogen bonds in 1a are depicted as dashed blue lines. Hydrogen atoms are omitted for the sake of clarity, except for those of the maltol hydroxyl groups. For the [Pdmaltonis]2+ cation in 1: palladium ions are given in dark cyan (ball-and-stick), oxygen atoms in red, carbon atoms in light grey, nitrogen atoms in light blue, and hydrogen atoms in white. | ||
| Metal ion | Optimized geometry | ΔG (kcal mol−1) | |
|---|---|---|---|
| Ligand conformer | Metal coordination number | ||
| a Formation of an intramolecular hydrogen bond between maltol groups; bold values: complex geometries found in the crystal structures. | |||
| Pd(II) | Open | 4 | 1.6 |
| Partially open | 4 | 3.4 | |
| Closed (1a) | 4 | 0.0 | |
| Pt(II) | Open | 4 | 1.6 |
| Partially open | 4 | 2.6 | |
| Closeda (2a) | 4 | 0.0 | |
| Co(II) | Open | 4 | 10.3 |
| Partially open | 5 | 0.9 | |
| Closed (3a) | 6 | 0.0 | |
| Cu(II) | Open | 4 | 5.5 |
| Partially open (4a) | 5 | 0.0 | |
| Closed | 6 | 4.1 | |
The intramolecular hydrogen bond between the two maltol rings observed in 1a most likely stabilizes the closed shape compared to the others. It can be thus hypothesized that the X-ray crystal structure of the palladium complex does not represent the absolute minimum, and that the observed structure in the solid state can be possibly ascribed to the crystal packing interactions.
By looking at the crystal packing analysis of 1 (vide supra), it can be thus hypothesized that the intramolecular hydrogen bonds observed in 1a are replaced by the intracolumnar interactions along with some H-bonds with perchlorate ions. These interactions and H-bonds stabilize the closed shape also in the X-ray structure, and possibly explain why this structure is the observed one in the solid state.
Clearly, the nature of the coordinated metal ion affects the topology of the complex. For this reason, the geometry optimization was extended to three more metal ions (Co(II), Cu(II), and Pt(II)) in addition to Pd(II), considering all three open, partially open and closed possible geometries.
As discussed above, complexes of maltonis with Co(II), Cu(II) and Pd(II) were obtained in the solid state from aqueous solutions. Quite the opposite, the Pt(II) complex could not be obtained by using the same experimental conditions. Nevertheless, Pt(II) was included in the computational analysis to possibly gain hints about the failure in obtaining the corresponding complex with maltonis.
To compare the energy values of analogous species, the same calculations were run on complexes where the hydroxyl functions of the maltol units were either protonated (as in the Pd(II) complex, 1a–4a in Table 3) or deprotonated (as in the Cu(II) and Co(II) complexes, 1b–4b in Table S6†). Fig. 11 and S8† show the most stable complex shapes obtained with each different metal cation (Pd(II), Pt(II), Cu(II), Co(II)), starting from maltonis having either protonated –OH groups (Fig. 11) or deprotonated –O− groups (Fig. S9†).
 | ||
| Fig. 11 Most stable shape of complexes of the ligand maltonis (with protonated –OH maltol groups) with Pd(II) (1a), Pt(II), Cu(II) and Co(II). Hydrogen atoms are omitted for clarity, except for those on the maltol hydroxyl groups that were included in the calculations. Metal ions are depicted in ball-and-stick style. The palladium ion is depicted in dark cyan, platinum ion in silver, copper ion in orange, cobalt ion in blue, oxygen atoms in red, carbon atoms in light grey, nitrogen atoms in light blue, hydrogen atoms in white. | ||
By looking at the complex geometries obtained with the different metal ions, the most stable ones in all cases correspond to those found in the crystal structures (bold values in Tables 3 and S6†). In other words, the closed shapes are energetically preferred in the case of Pd(II) and Co(II), whereas the partially open one is the most stable in the case of Cu(II).
In the case of Pd(II), the closed shape is slightly favoured compared to the others, with the open and partially open structures being just 1.6 and 3.4 kcal mol−1 higher, respectively. As for Pt(II), the DFT calculations showed a behaviour similar to Pd(II) (Fig. 11, S8† and Tables 3, S6†).
As expected, the presence of –OH or –O− maltol groups changes the stability of the complexes obtained with Pd(II) and Pt(II): for both metal ions, the closed shape was found to be the most stable with the OH groups (Fig. 11 and Table 3), while the open one is the preferred one with the deprotonated groups (Fig. S9 and Table S6†). This is probably due to the formation of the intramolecular hydrogen bond, which can occur only if the –OH groups are present and can drive the shape towards the closed one. The protonation degree of the maltol groups does not affect the complex stability in the case of Cu(II) and Co(II) (partially open and closed structures, respectively). Indeed, despite the degree of protonation, oxygen atoms are always engaged in coordination due to the metal geometry requirement.
Finally, it must be noted that during the optimization of the Cu(II) and Co(II)-complexes with maltonis featuring OH-maltol groups, a proton transfer from the OH to the adjacent carbonyl group is observed (Fig. 11, bottom). On the one hand, the loss of a proton is in line with the deprotonation of the OH-maltol functions observed following the coordination of a transition metal cation. On the other hand, the transfer of the proton to the neighbour carbonyl moiety has been recently observed in the Pd(II) complex of a similar maltol-based ligand obtained in the solid state from aqueous solution (unpublished results).
Computed bond dissociation energies (BDEs)
In addition to the possible shapes, the bond dissociation energies (BDEs), known to be a fundamental thermodynamic property that measures the strength of a chemical bond,86,87 have been investigated by DFT calculations for the [M(L)]2+ → M]2+ + (L) process (L = maltonis).In analogy to the work reported by Dyke88 on tetraaza-macrocyclic [M(Me4cyclen)]+ complexes with M = Li+, K+, Rb+, and Cs+, the relative stability of the complexes with different metal ions in water as solvent were compared (for the computational details, see the ESI†). Also, calculations were performed to possibly determine the reason for the successful experimental formation of complexes only with some of the tested metal ions. The considered reactions and the relative calculated energies are reported in Fig. 12.
 | ||
| Fig. 12 Left: dissociation reactions of complexes of maltonis with different metal ions. Right: calculated energies (kcal mol−1) for the [M(L)]2+ → [M]2+ + (L) dissociation process. | ||
By looking at the calculated energies in Fig. 12, it can be inferred that the formation of all [M(L)]2+ complexes is possible, being that all BDEs ≫0 kcal mol−1. If not considering the copper complex, which is part of a tetranuclear dimer structure (see Fig. 9), the palladium complex (1a in Fig. 12) shows the highest BDE value, representative of the highest stability within the series of [M(L)]2+.
According to the values reported in Fig. 12, the platinum complex (2a) could be obtained in principle. Unfortunately, the Pt(II) complex was not obtained under the present experimental conditions, neither in solution nor in the solid state. It can be suggested that factors, such as the complementary host–guest size, the requested optimal pH value, the kinetic inertness of Pt(II) and many other experimental factors not taken into account (ions solvation, reactants concentrations, etc.), counterbalance the thermodynamic possibility to obtain the complex.
Interaction with nucleosides
In view of a future biological investigation of the Pd(II) complex of maltonis that can possibly be used as an antineoplastic agent, the interaction of the complex with nucleosides was evaluated. To this purpose, 1H NMR experiments were performed in D2O solution at neutral pH.First, the stability of the complex was evaluated in aqueous solution at pH 7.4 at 37 °C, revealing an invariance of the spectrum up to 48 hours, which is compatible with possible biological studies (Fig. S10†).
1H NMR titrations of the Pd(II) complex at pH 7.4 were then performed with adenosine (A), guanosine (G), thymidine (T) and uridine (U) for up to 3 equiv. of nucleoside (only 2 equiv. for G, due to low solubility; 10 equiv. for A, vide infra). Fig. S11† shows that no variations in the spectrum of the complex occurred upon the addition of G, T and U, suggesting the absence of interaction between maltonis and those species under these experimental conditions. It must be noted that a possible interaction with G is not observed maybe due to its scarce solubility.
On the contrary, the addition of adenosine induced a shift on both complex and nucleoside resonances, suggesting the formation of an interaction between the Pd(II) complex and A (Fig. 13). Looking at Fig. 13, the more perturbed resonances resulted from those of the maltol and purine functions (green and red dots, respectively), while the aliphatic ones remained unvaried. The maltol signals showed an overall upfield shift by increasing the amount of A. On the contrary, the A resonances shifted downfield by increasing the presence of the Pd(II) complex in solution. This suggests the host–guest complex formation between the two species, mainly involving interactions between maltol and purine functions.
 | ||
| Fig. 13
1H NMR titration at pH 7.4 of the [Pd maltonis]2+ complex with adenosine (A). Bottom: spectrum of [PdL]2+; top: spectrum of A. Complex and A resonances are highlighted by green and red dots, respectively. The 1:1 complex to A molar ratio is highlighted. | ||
The down- and up-field shift is highlighted in Table S7† as Δδ chemical shifts of the main moving resonances for A and the Pd(II) complex when their ratio was 1 to 1.
In any case, all resulting shifts were quite small. From the analysis of their trend, it is difficult to safely affirm the stoichiometry of the host–guest adduct formed, although the formation of the 1:1 Pd(II) complex-A adduct could be hypothesized.
DFT calculations were performed to investigate the possible interaction between the Pd(II) complex of maltonis and the nucleosides. Since the NMR measurements revealed a preference for the purines compared to the pyrimidines (vide supra), only A and G were included in the DFT study.
The optimized interaction between the Pd(II) complex and A (1-A, Fig. 14) suggests the formation of a hydrogen bond between the –NH2 of A and the CO of maltol (Table S5†). Moreover, a π–π stacking interaction between the two moieties could also be suggested (Table S5†). This would be in agreement with the observed tendency of the complex to form π–π interactions in the solid state (vide supra).
 | ||
| Fig. 14 Front (left) and side (right) views of the optimized interaction between the Pd(II) cation in 1 (stick style) and Adenosine (ball and stick style) (1-A). Hydrogen bonds are depicted as dashed lines (intermolecular: light blue; intramolecular: blue). Centroids are visible as green and magenta spheres. The distance between them is depicted as a green dashed line. Hydrogen atoms are omitted for the sake of clarity, except for those linked to heteroatoms. Palladium ion in dark cyan, oxygen atoms in red, carbon atoms in light grey, nitrogen atoms in light blue, hydrogen atoms in white. | ||
The formation of a H-bond is also computed between A (–NH2 group) and the Pd(II) complex (maltolate –O− group) featuring deprotonated maltol moieties (1b-A, N–H⋯O 1.62 Å, 171.4°, see Fig. S12†).
The computed interaction with A did not seem to induce a marked structural variation in the palladium complex compared to 1a (Tables S4 and S5†): the Pd–N distances and angles remained very similar to those retrieved in 1a, while the Pd–O distances were very different from each other due to the interaction with A (Table S4†). Moreover, the intramolecular hydrogen bonds between the two maltol rings of the complex were maintained (Table S5† and Fig. 14).
Thus, the DFT analysis seems to support both the above mentioned tendency of the complex to form π–π stackings in the solid state, as well as the interaction observed during the NMR titration of the Pd(II) complex of maltonis with A, which mainly involved the aromatic moieties of both compounds.
The optimized interaction between the Pd(II) complex and G (1-G, Fig. S13†) suggested the formation of two hydrogen bonds involving the –NH and CO groups of G and, respectively, and the CO and –OH groups of a maltol ring (Table S5 and Fig. S13†). Also in this case, a π–π stacking is suggested between a maltol ring and the pyrimidine moiety of G, confirming this already mentioned ability of the complex (Table S5 and Fig. S13†). The computed possible interaction with G seemed to produce more structural variations in the palladium complex compared to A; indeed, in this case the intramolecular H-bond between the two maltol rings was not observed (Tables S4 and S5†).
It must be noted that the optimized structure shows a proton transfer from the –OH group of maltol to the carbonyl group of G: this could be favored by the formation of the adjacent H-bond between COmaltol and –NHguanosine, which enriches the electron density of the Nguanosine atom. At the same time, it changes the aromaticity of the purine ring, shifting the equilibrium towards the enol form. To possibly support this finding, an additional optimization of the interaction between the Pd(II) complex and G in the presence of an explicit water molecule (namely 1-G-H2O) was performed to better understand the nature of this interaction. The proton transfer was observed also in this case (Fig. S14†). Unfortunately, it was not possible to confirm this process in solution by NMR measurements, due to the scarce solubility of G in aqueous solution at pH 7.4, that prevented to evaluate a possible interaction between the two species.
Finally, it should be noted that both 1-A and 1-G (as well as 1-G-H2O), featuring non-coordinated maltol moieties, show the ability of such groups to interact with the nucleosides. A possible maltol-dependent biological mechanism, already observed for the free L, could be thus maintained.
Conclusions
A dimethyl-cyclen-based Pd(II) complex, containing the 4,10-bis[(3-hydroxy-4-pyron-2-yl)methyl]-1,7-dimethyl-1,4,7,10-tetraazacyclododecane ligand (maltonis), has been synthesized and characterized both in solution and in the solid state. UV-vis and NMR spectroscopies suggested that the complex formation in solution started from acidic pH and the presence of a single species at neutral pH. Such a cyclen-based Pd(II) complex is not so represented within the CSD, pointing at a non-straightforward complexation of this metal ion in that specific cavity. The structure shows that the –OH groups of the maltol rings are not involved in the coordination of the metal ion. Nevertheless, a closed shape, with maltol rings facing each other, is observed. The same closed shape is slightly preferred upon DFT optimization, with the maltol moieties interacting through an intramolecular hydrogen bond. More likely, this intramolecular hydrogen bond is replaced in the X-ray structure of 1 by the intermolecular interactions found in the crystal packing, forming columns and some interactions with perchlorate counterions. Furthermore, a more extensive study of the solid state assembly by Hirshfeld surfaces, 2D-fingerprint plots and energy frameworks revealed that complexes in 1 arrange to form a three-dimensional net. A DFT geometry optimization including different metal ions (Pd(II), Pt(II), Co(II), Cu(II)) revealed that the most stable complex shape with each metal ion corresponds to that observed in the X-ray structure previously obtained with the same metal. The BDEs calculated by DFT showed that the palladium complex found in 1 is the preferred one within the series in water. DFT calculations also suggested that the formation of a closed Pt(II) complex is theoretically possible, but such species was not obtained in these experimental conditions.Due to the antineoplastic activity of the ligand maltonis, which may be increased by the presence of Pd(II), a possible interaction of the palladium complex with nucleosides was evaluated by both NMR and DFT, revealing a preference for purines through π–π stacking and hydrogen bonds involving the maltol moieties, that are indeed not involved in the coordination of Pd(II). These observed interactions pave the way towards future biological investigations.
Author contributions
Daniele Paderni: investigation, writing – reviewing and editing; Maria Voccia: investigation, formal analysis, writing – reviewing and editing; Eleonora Macedi: conceptualization, investigation, formal analysis, writing – original draft, project administration; Mauro Formica: writing – reviewing and editing, funding acquisition; Luca Giorgi: writing – reviewing and editing, funding acquisition; Lucia Caporaso: supervision; Vieri Fusi: supervision, writing – reviewing and editing, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1 have been deposited at the CCDC under 2360175,† and can be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CRIST (Centro di Cristallografia Strutturale, University of Florence), where the X-ray diffraction data were collected, is greatly acknowledged. Ms A. Pierleoni is greatly acknowledged for help with NMR measurements. This work was supported by the European Union – NextGenerationEU – under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY – CUP [H33C22000430006]; University of Urbino (Department of Pure and Applied Sciences – Grant DISPEA_Giorgi_Prog22 and Dispea Assegnazione Ateneo Sicurezza Alimentare).References
- Y. Zhang, Q. Zhou, N. Tian, C. Li and X. Wang, Inorg. Chem., 2017, 56, 1865–1873 CrossRef CAS PubMed.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2018, 119, 1519–1624 CrossRef PubMed.
- J. Karges and S. M. Cohen, ChemBioChem, 2021, 22, 2600–2607 CrossRef CAS PubMed.
- Y. Lin, H. Betts, S. Keller, K. Cariou and G. Gasser, Chem. Soc. Rev., 2021, 50, 10346–10402 RSC.
- M. J. S. A. Silva, P. M. P. Gois and G. Gasser, ChemBioChem, 2021, 22, 1740–1742 CrossRef CAS PubMed.
- I. Yousuf, M. Bashir, F. Arjmand and S. Tabassum, Coord. Chem. Rev., 2021, 445, 214104 CrossRef CAS.
- L. Conti, E. Macedi, C. Giorgi, B. Valtancoli and V. Fusi, Coord. Chem. Rev., 2022, 469, 214656 CrossRef CAS.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F. X. Wang, W. Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- P. Chellan and P. J. Sadler, Chem. – Eur. J., 2020, 26, 8676–8688 CrossRef CAS PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed.
- B. S. Murray and P. J. Dyson, Curr. Opin. Chem. Biol., 2020, 56, 28–34 CrossRef CAS PubMed.
- D. Cirri, F. Bartoli, A. Pratesi, E. Baglini, E. Barresi and T. Marzo, Biomedicines, 2021, 9, 504 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed.
- S. Y. Lee, C. Y. Kim and T. G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044–7100 RSC.
- S. Amatori, G. Ambrosi, A. Errico Provenzano, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, J. Inorg. Biochem., 2016, 162, 154–161 CrossRef CAS PubMed.
- T. Lazarević, A. Rilak and Ž. D. Bugarčić, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef PubMed.
- M. Vojtek, M. P. M. Marques, I. M. P. L. V. O. Ferreira, H. Mota-Filipe and C. Diniz, Drug Discovery Today, 2019, 24, 1044–1058 CrossRef CAS PubMed.
- T. J. Carneiro, A. S. Martins, M. P. M. Marques and A. M. Gil, Front. Oncol., 2020, 10, 1–8 CrossRef PubMed.
- M. Voccia, L. Falivene, L. Cavallo, C. Tubaro, A. Biffis and L. Caporaso, Organometallics, 2019, 38, 3730–3739 CrossRef CAS.
- M. Voccia, L. Odenwald, M. Baur, F. Lin, L. Falivene, S. Mecking and L. Caporaso, J. Am. Chem. Soc., 2022, 144, 15111–15117 CrossRef CAS PubMed.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC.
- T. Scattolin, V. A. Voloshkin, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS.
- E. Z. Jahromi, A. Divsalar, A. A. Saboury, S. Khaleghizadeh, H. Mansouri-Torshizi and I. Kostova, J. Iran. Chem. Soc., 2016, 13, 967–989 CrossRef CAS.
- R. Czarnomysy, D. Radomska, O. K. Szewczyk, P. Roszczenko and K. Bielawski, Int. J. Mol. Sci., 2021, 22, 8271 CrossRef CAS PubMed.
- Z. Zhao, J. Ru, P. Zhou, Y. Wang, C. Shan, X. Yang, J. Cao, W. Liu, H. Guo and Y. Tang, Dalton Trans., 2019, 48, 16952–16960 RSC.
- Q. Guo, Y. Liu, G. Mu, L. Yang, W. Wang, J. Liu and J. Liu, Biomater. Sci., 2020, 8, 5638–5646 RSC.
- X. Liu, X. Dong, C. He, X. Zhang, G. Xiang and X. Ma, Bioorg. Chem., 2020, 96, 103574 CrossRef CAS PubMed.
- F. Xiang, B. Li, P. Zhao, J. Tan, Y. Yu and S. Zhang, Adv. Synth. Catal., 2019, 361, 5057–5062 CrossRef CAS.
- J. Dai, Z. Liu, L. Wang, G. Huang, S. Song, C. Chen, T. Wu, X. Xu, C. Hao, Y. Bian, E. A. Rozhkova, Z. Chen and H. Yang, J. Am. Chem. Soc., 2023, 145, 1108–1117 CrossRef CAS PubMed.
- Z. F. Wang, X. F. Zhou, Q. C. Wei, Q. P. Qin, J. X. Li, M. X. Tan and S. H. Zhang, Eur. J. Med. Chem., 2022, 238, 114418 CrossRef CAS PubMed.
- Y. Ichimaru, K. Kato, R. Nakatani, K. Sugiura, H. Mizutani, E. Kinoshita-Kikuta, T. Koike, W. Jin, M. Imai and H. Kurosaki, Inorg. Chem. Commun., 2023, 147, 110221 CrossRef CAS.
- L. Yang, X. He, Z. Zeng, J. Tang, D. Qi, H. Ma, H. Chen, X. Ning and X. Feng, Chem. Sci., 2021, 12, 8394–8400 RSC.
- L. Yang, Y. Zhu, L. Liang, C. Wang, X. Ning and X. Feng, Nano Lett., 2022, 22, 4207–4214 CrossRef CAS PubMed.
- J. Gao, Z. Wang, Q. Guo, H. Tang, Z. Wang, C. Yang, H. Fan, W. Zhang, C. Ren and J. Liu, Theranostics, 2022, 12, 1286–1302 CrossRef CAS PubMed.
- X. Ma, J. Hua, M. Wang, D. Zhang, X. Pei, X. Zhao, Y. Niu and Y. Wang, Catalysts, 2022, 12, 695 CrossRef CAS.
- H. F. Chau, Y. Wu, W. Y. Fok, W. Thor, W. C. S. Cho, P. Ma, J. Lin, N. K. Mak, J. C. G. Bünzli, L. Jiang, N. J. Long, H. L. Lung and K. L. Wong, J. Am. Chem. Soc., 2021, 1, 1034–1043 CAS.
- M. Kahnt, S. Hoenke, L. Fischer, A. Al-Harrasi and R. Csuk, Molecules, 2019, 24, 2254 CrossRef PubMed.
- N. G. Chabloz, H. L. Perry, I. C. Yoon, A. J. Coulson, A. J. P. White, G. J. Stasiuk, R. M. Botnar and J. D. E. T. Wilton-Ely, Chem. – Eur. J., 2020, 26, 4552–4566 CrossRef CAS PubMed.
- S. Hafner, M. Raabe, Y. Wu, T. Wang, Z. Zuo, V. Rasche, T. Syrovets, T. Weil and T. Simmet, Adv. Thermoelectr., 2019, 2, 1900084 CAS.
- J. Feng, Q. Luo, Y. Chen, B. Li, K. Luo, J. Lan, Y. Yu and S. Zhang, Bioconjugate Chem., 2018, 29, 3402–3410 CrossRef CAS PubMed.
- J. Hormann, J. Malina, O. Lemke, M. J. Hülsey, S. Wedepohl, J. Potthoff, C. Schmidt, I. Ott, B. G. Keller, V. Brabec and N. Kulak, Inorg. Chem., 2018, 57, 5004–5012 CrossRef CAS PubMed.
- Q. Guo, Y. Liu, Z. Wang, J. Zhang, G. Mu, W. Wang and J. Liu, Acta Biomater., 2021, 122, 343–353 CrossRef CAS PubMed.
- R. Zhao, K. Ploessl, Z. Zha, S. Choi, D. Alexoff, L. Zhu and H. F. Kung, Mol. Pharm., 2020, 17, 4589–4602 CrossRef CAS PubMed.
- S. Amatori, G. Ambrosi, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, J. Org. Chem., 2012, 77, 2207–2218 CrossRef CAS PubMed.
- E. Borgogelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, Dalton Trans., 2013, 42, 2902–2912 RSC.
- C. Benelli, E. Borgogelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, Dalton Trans., 2013, 42, 5848–5859 RSC.
- P. Paoli, E. Macedi, P. Rossi, L. Giorgi, M. Formica and V. Fusi, Acta Crystallogr., 2017, E73, 1806–1811 CrossRef PubMed.
- P. Rossi, E. Macedi, P. Paoli, L. Giorgi, M. Formica and V. Fusi, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2017, 73, 1959–1965 CrossRef CAS PubMed.
- P. Rossi, S. Ciattini, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni and P. Paoli, Inorg. Chim. Acta, 2018, 470, 254–262 CrossRef CAS.
- S. Amatori, I. Bagaloni, E. Macedi, M. Formica, L. Giorgi, V. Fusi and M. Fanelli, Br. J. Cancer, 2010, 103, 239–248 CrossRef CAS PubMed.
- S. Amatori, G. Ambrosi, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli and P. Rossi, Chem. – Eur. J., 2014, 20, 11048–11057 CrossRef CAS PubMed.
- E. Macedi, D. Paderni, M. Formica, L. Conti, M. Fanelli, L. Giorgi, S. Amatori, G. Ambrosi, B. Valtancoli and V. Fusi, Molecules, 2020, 25, 943 CrossRef CAS PubMed.
- L. Giorgi, G. Ambrosi, D. Paderni, L. Conti, S. Amatori, F. Romagnoli, P. Rossi, M. Formica, E. Macedi, C. Giorgi, P. Paoli, M. Fanelli and V. Fusi, New J. Chem., 2021, 45, 2659–2669 RSC.
- C. Guerzoni, S. Amatori, L. Giorgi, M. C. Manara, L. Landuzzi, P. L. Lollini, A. Tassoni, M. Balducci, M. Manfrini, L. Pratelli, M. Serra, P. Picci, M. Magnani, V. Fusi, M. Fanelli and K. Scotlandi, BMC Cancer, 2014, 14, 137 CrossRef PubMed.
- S. Amatori, G. Persico, F. Cantatore, M. Rusin, M. Formica, L. Giorgi, E. Macedi, F. Casciaro, A. Errico Provenzano, S. Gambardella, R. Noberini, T. Bonaldi, V. Fusi, M. Giorgio and M. Fanelli, Cancer Gene Ther., 2022, 1–12 Search PubMed.
- V. Monini, M. Bonechi, C. Bazzicalupi, A. Bianchi, P. Gentilesca, W. Giurlani, M. Innocenti, A. Meoli, G. M. Romano and M. Savastano, Dalton Trans., 2024, 53, 2487–2500 RSC.
- W. Gong, Y. Xie, X. Wang, K. O. Kirlikovali, K. B. Idrees, F. Sha, H. Xie, Y. Liu, B. Chen, Y. Cui and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 2679–2689 CrossRef CAS PubMed.
- S. Li, C. Liu, Q. Chen, F. Jiang, D. Yuan, Q. F. Sun and M. Hong, Chem. Sci., 2022, 13, 9016–9022 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, Univ. West. Aust., 2017 Search PubMed.
- J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488–499 CrossRef CAS PubMed.
- S. R. Laplante, H. Nar, C. T. Lemke, A. Jakalian, N. Aubry and S. H. Kawai, J. Med. Chem., 2013, 57, 1777–1789 CrossRef PubMed.
- F. Bosica, S. A. Andrei, J. F. Neves, P. Brandt, A. Gunnarsson, I. Landrieu, C. Ottmann and G. O'Mahony, Chem. – Eur. J., 2020, 26, 7131–7139 CrossRef CAS PubMed.
- Bruker APEX2, Bruker AXS Inc., 2012 Search PubMed.
- Bruker SAINT, Bruker AXS Inc., 2012 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick and IUCr, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. Nardelli and IUCr, J. Appl. Crystallogr., 1995, 28, 659–659 CrossRef CAS.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- I. Bernal, Stereochemical and Stereophysiscal behaviour of macrocycles, Elsevier, Amsterdam, 1987 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, International Tables for Crystallography, Dordrecht: Kluwer, 1995, vol C Search PubMed.
- S. L. Tan, M. M. Jotani and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 308 CrossRef CAS PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- I. Dance, New J. Chem., 2003, 27, 22–27 RSC.
- J. J. Koenderink and A. J. van Doorn, Image Vis. Comput., 1992, 10, 557–564 CrossRef.
- C. Valverde, W. F. Vaz, J. M. F. Custodio, V. S. Duarte, P. S. Carvalho-Jr, A. S. Figueredo, G. L. B. de Aquino, B. Baseia and H. B. Napolitano, New J. Chem., 2017, 41, 11361 RSC.
- M. Keikha, M. Pourayoubi and A. van der Lee, J. Chem. Crystallogr., 2020, 50, 88–98 CrossRef CAS.
- X. Qu, D. A. R. S. Latino and J. Aires-de-Sousa, J. Cheminf., 2013, 5, 1–13 Search PubMed.
- P. C. St John, Y. Guan, Y. Kim, S. Kim and R. S. Paton, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- H. Bhakhoa, L. Rhyman, E. P. Lee, D. K. W. Mok, P. Ramasami and J. M. Dyke, Dalton Trans., 2017, 46, 15301–15310 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: More UV–vis and NMR spectroscopic details, CSD searches, fingerprint plots, interaction energies, DFT calculations details, selected distances and angles. CCDC 2360175. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01791a |
| ‡ These authors contributed equally to this work. |
| § Present Address: Department of Materials Science, University of Milano-Bicocca, 20125 Milano, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |