
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective sensing of picric acid using a Zn(II)-metallacycle: experimental and theoretical validation of the sensing mechanism and quantitative analysis of sensitivity in contact mode detection†
Vishakha
Jaswal
a,
Sanya
Pachisia
 bc,
Jagrity
Chaudhary
d,
Krishnan
Rangan
e and
Madhushree
Sarkar
*a
bc,
Jagrity
Chaudhary
d,
Krishnan
Rangan
e and
Madhushree
Sarkar
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Pilani Campus, Rajasthan 333031, India. E-mail: msarkar@pilani.bits-pilani.ac.in
bDepartment of Chemistry, University of Delhi, India
cDepartment of Chemistry, University of California, Irvine, California, USA
dDepartment of Chemistry, Purdue University, West Lafayette, Indiana, USA
eDepartment of Chemistry, BITS Pilani, Hyderabad Campus, Jawahar Nagar Shameerpet Mandal, Ranga Reddy District, Hyderabad 500078, India
First published on 7th August 2024
Abstract
A combination of N,N′,N′′-tris(3-pyridyl)-1,3,5-benzenetricarboxamide (L1) and p-chlorobenzoic acid (HL2) with Zn(NO3)2·6H2O resulted in the formation of a dinuclear metallacycle [ZnL1(L2)2(DMF)2]2 (1(DMF)4). In 1(DMF)4, the Zn(II) centre adopts a square pyramidal geometry, while one of the pyridyl N out of the three pyridyl groups in L1 remained uncoordinated. Solvated DMF molecules are present in 1(DMF)4. The structural and chemical nature of 1(DMF)4 is effective for it to act as a potential fluorescent probe for the detection of nitroaromatic compounds. It is observed that the probe, 1(DMF)4, could selectively detect picric acid (PA) among various aromatic compounds in solution (DMSO), while the solid state (contact mode) detection showed a positive sensing response for the nitrophenols (PA: 87% quenching efficiency, 2,4-dinitrophenol (2,4-DNP): 57% quenching efficiency and 4-nitrophenol (4-NP): 40% quenching efficiency). The limit of detection (LOD) of PA by the probe in DMSO was found to be 6.8 × 10−11 M while the LOD in contact mode detection was estimated to be 0.49 ng cm−2. The mechanism of selective detection of PA by 1(DMF)4 in DMSO was analyzed through photophysical studies, 1H-NMR experiments and also by density functional theory (DFT) calculations. The effective overlap of the absorption spectrum of 1(DMF)4 and emission spectrum of PA in DMSO suggests that the Förster resonance energy transfer (FRET) is responsible for quenching phenomena in DMSO. The DFT calculations and molecular docking studies showed the adduct formation due to the favorable interactions between 1(DMF)4 and PA in DMSO, while negligible interactions were observed between 1(DMF)4 with other aromatic compounds. The experimental and DFT studies showed that the efficient sensing ability of PA by 1(DMF)4 in the solid-state was due to photoelectron transfer (PET) and FRET phenomena described herein.
Introduction
Organic–inorganic hybrid materials have gained a lot of interest due to their complex structures and applications in various fields. Based on the connectivity of metals and organic ligands as linkers, the hybrid materials can be categorized as extended networks like metal–organic frameworks (MOFs) and discrete coordination self-assemblies like metallacyclic polygons and polyhedra,1,2 which have shown applications in various fields.3–5 Several research groups have reported dinuclear metallacycles, some of which have demonstrated desirable properties such as host–guest interactions.6 Fujita et al. have synthesized dinuclear metallomacrocycles containing a Pd(II) center using a series of bis(pyridyl) ligands and analyzed the dynamic behavior of those macrocycles.7–10 Stang and co-workers have reported a series of dinuclear macrocyclic rhomboids using bis(triethylphosphine)platinum(II) and bis(triethylphosphine)palladium(II) bistriflates with the corresponding bis(pyridine) silanes.11 Mirkin and co-workers have synthesized a series of homodimetallic macrocyclic structures from the ligands derived from phosphinyl alkyl ether, thioether, and amine groups, which bind to many late transition metal centers such as rhodium(I) and palladium(II). Some of the macrocycles have incorporated fluorescent aromatic units, where the fluorescence emissions of the macrocycles are sensitive to guest molecules bound inside the macrocyclic cavity; hence, they show potential applications as sensors.12–14 Binuclear metallacycles are reported by the groups of Pandey and co-workers who synthesized a Zn(II) metallacycle, which shows fast trans–cis swift photoisomerization;15 Ozarowski16 and Long17 worked on first transition series metal salts to prepare metallacycles and have explored their magnetic properties; and Rosenthal18 and co-workers have performed the oligomerization of ethylene using a Cr-based metallocycle. Some of the luminescent binuclear metallacycles have been reported by Lu,19 Mukherjee,20 Shanmugaraju21 and Pandey.22N,N′,N′′-Tris(3-pyridyl)-1,3,5-benzenetricarboxamide (L1), being a flexible tridentate ligand and possessing amide functionalities, can serve as a potential candidate to generate coordination complexes and polymers of varied geometries. In 1997, Palmans et al. reported the crystal structure of L1,23 whereas Biradha et al. reported the three different crystalline phases of L1 in methanol in different concentrations.24L1 was used by Mukherjee et al. to synthesize the first nano-sized Pd(II) based 3D cage.25 Jia et al. reported a cyclic dimeric oligoamide zinc complex of L1, where Zn(II) in the dimeric complex adopted a tetrahedral coordination environment where two Zn(II) and two L1 units are linked alternately to form a 28-membered ring in which all the amide hydrogens of L1 are involved in hydrogen bond interactions, while one of the pyridyl nitrogens is free.26 Bai et al. worked on the mixed ligand strategy to prepare a coordination polymer with L1 and 1,4-benzenedicarboxylic acid with Cd(II), Co(II), and Zn(II). The presence of auxiliary ligands in these coordination polymers has extended the networks into higher dimensions.27
The surge in global environmental pollution has caused severe effects on human health, ecosystems, wildlife, and livestock.28 Among the various pollutants, nitroaromatic compounds (NACs) are significant energetic pollutants identified with their explosive nature.29 The presence of picric acid (PA) among NACs has versatile usage in different industries like medicines, explosives, pesticides, germicides, tanning, and dyes.30 Due to the exclusive usage of PA it is a need of the hour to detect PA. The two major categories of NACs based on chemical functionalities are (i) phenolic NACs and (ii) non-phenolic NACs. NACs are electron deficient, and a more significant number of nitro groups further increases the electron deficiency. This feature has been exploited to design probes for their detection. The development of various fluorescence-based detectors, such as organic models,31,32 metal–organic frameworks (MOFs),33–37 polymers,38,39 covalent organic frameworks (COFs),40,41 nanomaterials,42–44 and quantum dots,45–47 has proven effective for detecting NACs. Additionally, the advancement of discrete metal–organic cages has enabled the detection of NACs in both solid and liquid states.
In phenolic NACs, the hydroxyl group acts as an added chemical feature that helps in their selective detection through the rational design of probes. The ability of phenolic NACs to protonate basic sites such as amines or heterocyclic nitrogen present in a probe can induce electrostatic interactions, which may result in visual photophysical changes. Peng, Wang, and co-workers have reported N-acyl hydrazone-based fluorescent probes for selective detection of PA, which was achieved through the hydrogen bond interactions of pyridyl groups with PA. They confirmed the proton transfer from PA to pyridyl nitrogen by 1H-NMR studies and single crystal XRD.48–50 Various other groups have reported pyridyl-based probes for selective detection of PA where hydrogen bonding/transfer of a proton from PA to pyridyl nitrogen is responsible for the selective detection.51–53
Electron-rich amidic oxygen may also result in facile protonation, leading to electrostatic interactions between the PA and the probe. Many research groups have used this phenomenon where the amide-based probes showed fluorescence quenching upon protonation by PA.54–58 Gupta and co-workers have reported two Pd(II) based fluorescent macrocycles which have hydrogen bonding cavities for the selective detection of PA in ethanol. They have also performed docking studies to validate the effectiveness of the hydrogen bonding functionalities of the macrocycle for the selective binding of PA and, hence, its selective recognition.59 Tang and co-workers have synthesized the two macrocycles and performed the selective sensing of PA in DMSO.60 Nurnabi and co-workers have worked on the selective sensing of PA based on aggregation-induced emission (AIEE) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, THF:H2O) with rhenium macrocycles.61 In a recent report, Gupta and co-workers have synthesized three mononuclear nickel complexes of an amide-based pincer ligand, where all of them possessed hydrogen bonded cavities which were responsible for selective detection of PA.62 Pandey and co-workers have synthesized two binuclear Zn(II) complexes of bis-chelating salen type ligands possessing N,N-diethylamine moieties on the periphery of the molecules which were utilized in the selective detection of PA in different organic and aqueous media. They further performed DFT calculations, which showed a static mechanism through ground state charge transfer between the complexes and PA. The interactions between the complex and PA was also supported by 1H-NMR spectral studies.63
:50, THF:H2O) with rhenium macrocycles.61 In a recent report, Gupta and co-workers have synthesized three mononuclear nickel complexes of an amide-based pincer ligand, where all of them possessed hydrogen bonded cavities which were responsible for selective detection of PA.62 Pandey and co-workers have synthesized two binuclear Zn(II) complexes of bis-chelating salen type ligands possessing N,N-diethylamine moieties on the periphery of the molecules which were utilized in the selective detection of PA in different organic and aqueous media. They further performed DFT calculations, which showed a static mechanism through ground state charge transfer between the complexes and PA. The interactions between the complex and PA was also supported by 1H-NMR spectral studies.63
In the current work, a combination of two ligating molecules, an amide-based tripodal ligand having pyridyl groups, N,N′,N′′-tris(3-pyridyl)-1,3,5-benzenetricarboxamide, L1, and p-chlorobenzoic acid, HL2, are used in generating a dinuclear Zn(II)-macrocyclic complex, [ZnL1(L2)2(DMF)2]2 (1(DMF)4) (Scheme 1). In 1(DMF)4, L1 is equipped with chemical functionalities, which may result in hydrogen bonding interactions with organic guest molecules, while the presence of L2, equipped with a chloro group, may result in regions of varied electron distributions in the macrocycles. Features present in 1(DMF)4 are suitable for detecting NACs. The –NO2 groups of NACs can interact with the electron deficient regions created in 1(DMF)4, while the electron deficient ring in NAC may interact with the electron-rich sites of L1. In this work, 1(DMF)4 is studied for its potential application as a suitable fluorescent probe for sensing NACs. The interactions of –NO2 present in the compounds with the pyridyl and amidic functionalities of 1(DMF)4 may result in the close assembling of the NACs with 1(DMF)4 and lead to some distinctive changes in photophysical properties.
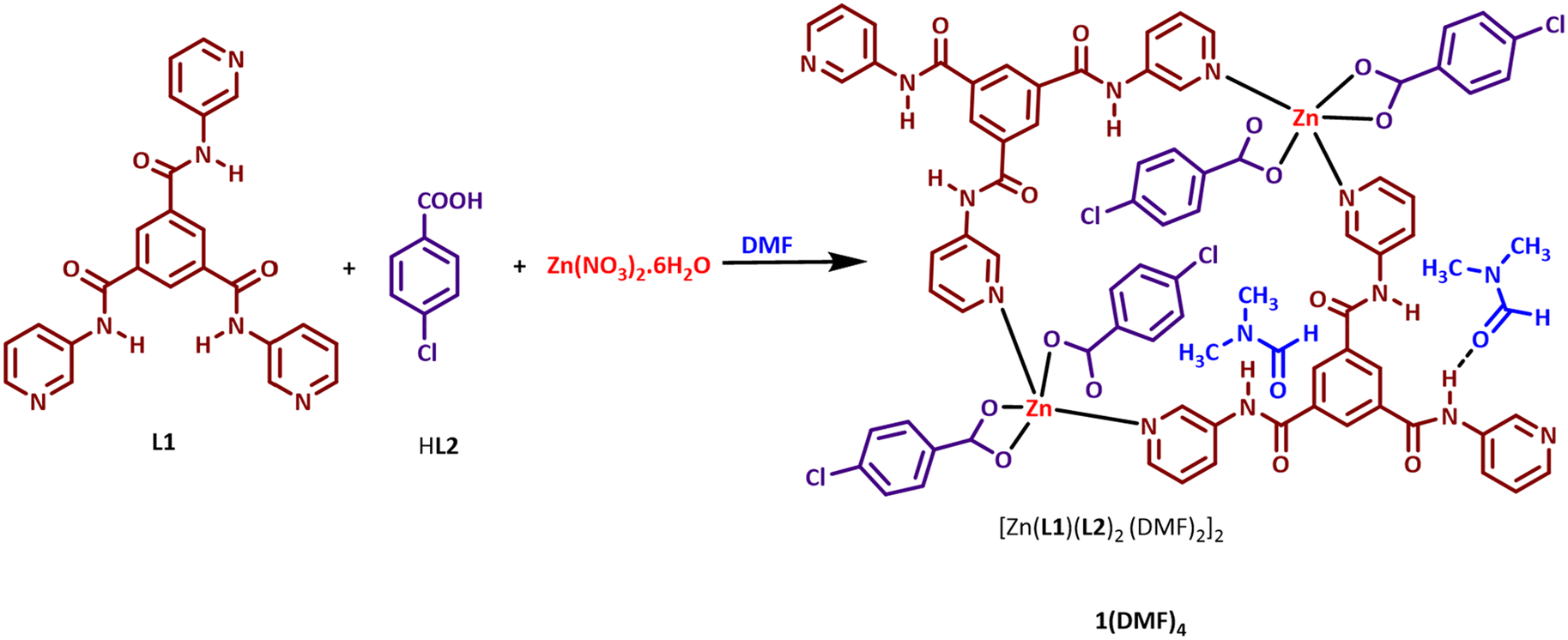 | ||
| Scheme 1 Synthesis of 1(DMF)4. | ||
Results and discussion
Structural description of 1(DMF)4
Crystal structure analysis of 1(DMF)4 shows that it crystallized in a monoclinic P21/n space group, and the asymmetric unit contains one Zn(II) center, one L1, two L2, and two DMF molecules. The Zn(II) center adopts a distorted square pyramidal geometry with two pyridyl nitrogen atoms (N4 and N5) from different L1 units and three carboxylate oxygen atoms (O4, O5, and O6) from two different L2 moieties satisfying the coordination environment (Fig. 1a). Bond lengths and angles in the metal coordination sphere in 1(DMF)4 are given in Table S2.† The linking of two asymmetric units resulted in the formation of a 28-membered ring containing a center of inversion. The distance between the two Zn(II) centers within a ring is 11.9548(7) Å. It is interesting to note that although L1 has three pyridyl nitrogen atoms (N4, N5 and N6) which are capable of forming coordinate bond interactions with the metal center, only two pyridyl nitrogen atoms (N4 and N5) are involved in coordination with the Zn(II) center, while the third pyridyl nitrogen (N6) is uncoordinated (Fig. 1a).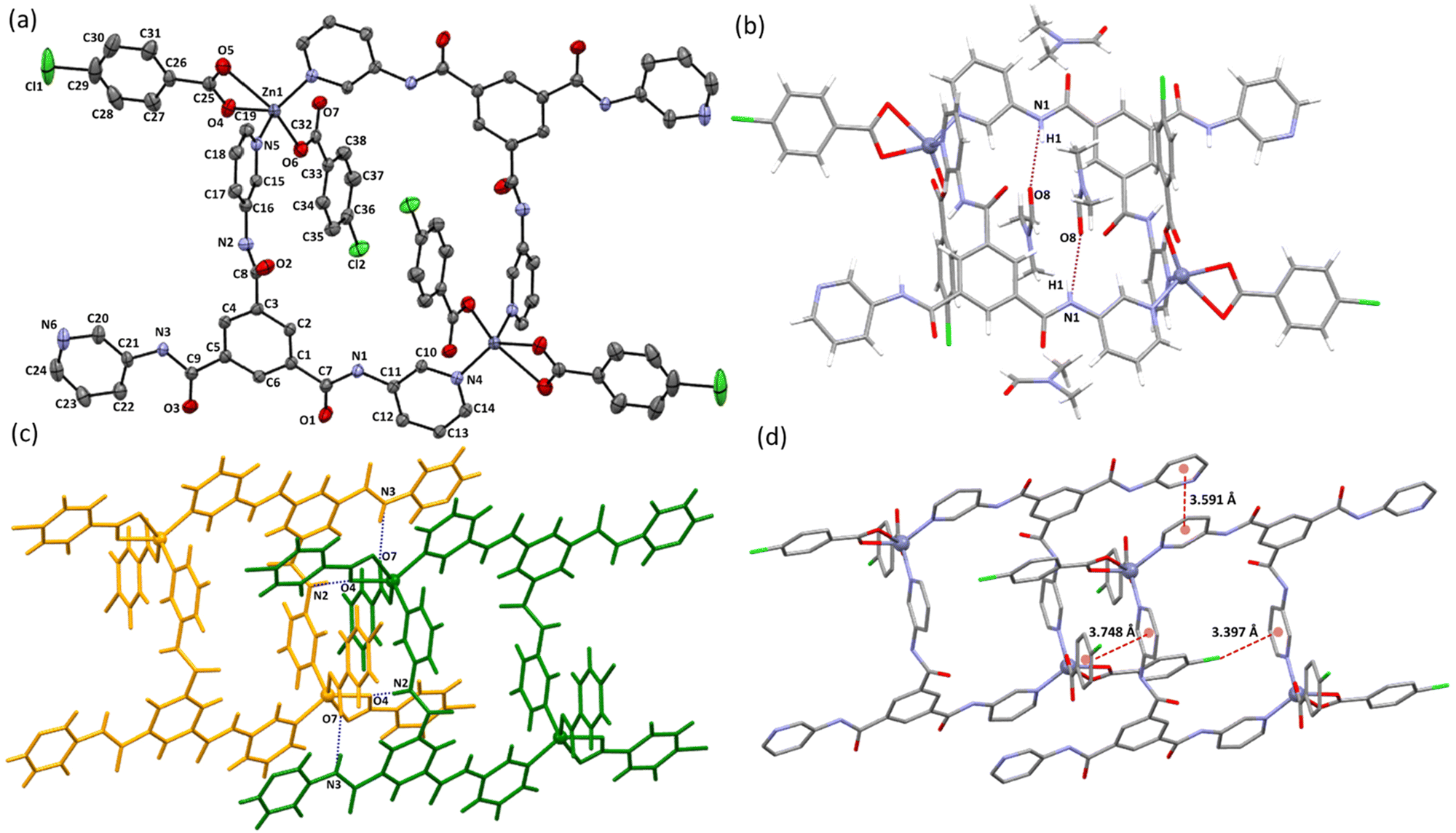 | ||
| Fig. 1 Illustrations of the crystal structure of 1(DMF)4: (a) ORTEP of 1(DMF)4; thermal ellipsoids shown at 50% probability (H atoms and solvated DMF are removed for clarity); notice the presence of the center of inversion in the macrocyclic ring; (b) one unit of the macrocyclic ring showing the coordination environment of the Zn(II) center and hydrogen bond interactions between amidic N–H and DMF molecules (N1–H1⋯O8a) (symmetry codes: a+x, +y, +z); (c) hydrogen bond interactions present between neighboring rings (N2–H2⋯O4b and N3–H3⋯O7b); (symmetry codes: b+x, 1 + y, +z); (d) aromatic π⋯π and aromatic Cl⋯π interactions between the adjacent rings (H-atoms are removed for clarity). | ||
Three amidic groups (involving the nitrogen atoms N1, N2, and N3) in a unit of L1 in 1(DMF)4 play an essential role in forming hydrogen bond interactions. The amide group involving the N1 atom forms hydrogen bond interactions with a DMF molecule (O8 of DMF), while N2 and N3 are involved in hydrogen bond interactions with carboxylate oxygen atoms (O4 and O7) of two different L2 units of the neighboring macrocyclic ring (Table S3† and Fig. 1b, c). In 1(DMF)4, the uncoordinated pyridyl nitrogen atom (N6) is not involved in hydrogen bond interactions.
Apart from the hydrogen bond interactions between the adjacent macrocyclic rings, which resulted in the stacking of the rings in 1D, the presence of an electron-rich heteroarene moiety in L1 and an electron-deficient aromatic ring in L2 resulted in very efficient π⋯π interactions, which in turn resulted in adequate packing of the macrocycles. Some of the important aromatic interactions include face-to-face π⋯π interactions between chlorobenzoate of L2 and pyridyl of L1 (centroid to centroid distance: 3.748 Å), face-to-face π⋯π interactions between two pyridyl of L1 of two different 1(DMF)4 rings (centroid to centroid distance: 3.591 Å), and Cl1⋯π interactions between L2 and pyridyl of L1 (centroid to Cl1 distance: 3.397 Å) (Fig. 1d). Assembling of the rings in 3D is shown in Fig. S9.†
The macrocyclic ring of L1 and Zn(II) reported by Jia et al. showed a tetrahedral coordination geometry of the Zn(II) center and a Zn⋯Zn distance of 11.162(5) Å within the ring.26 The tendency of one of the pyridyl nitrogen atoms out of the three in L1 to remain uncoordinated is also observed in the Zn(II) ring structure reported by Jia et al.26 and also in the coordination polymers reported by Bai et al.27 while the coordination complexes/polymers reported by using L1 with other metal centers such as Pd(II), Cu(II), and Ag(I) showed the utilization of all the three ligating nitrogen atoms for coordinating with metal centers.25,64–66
Photophysical studies on 1(DMF)4
The solid-state absorption spectrum of 1(DMF)4 showed the presence of two peaks at ∼245 nm and ∼290 nm, while the spectrum in DMSO showed a single peak at ∼278 nm (Fig. S10†). The solid-state emission spectrum of 1(DMF)4 showed peak maxima at 430 nm (Fig. S11a†). The emission spectrum of 1(DMF)4 in DMSO showed a peak maximum at 409 nm at a concentration of 10−2 M, and an increase in the concentration resulted in the bathochromic shift of the peak maxima (Fig. S11b†). Solid state emission spectra of L1, reported by Jia et al.,26 showed a broad emission with a maximum at ∼450 nm at an excitation wavelength of 389 nm, while the macrocycle reported by their group showed similar emission to that of L1, but split into two peaks at 450 and 471 nm.The emission peaks observed in 1(DMF)4 are due to intraligand transitions. The blue-shifted emission maxima in 1(DMF)4, as compared to that in L1 can be ascribed to the increased HOMO–LUMO gap on complexation due to non-planarity associated with L1 on coordinating with the Zn(II) center and the effect of the electron-withdrawing chloro group in the auxiliary ligand L2. The time-resolved fluorescence decay profile of 1(DMF)4 in DMSO is given in Fig. S20a.†
1(DMF)4 as a potential sensor for NACs
The chemical features present in 1(DMF)4 include (i) uncoordinated/free pyridyl nitrogen, (ii) the amidic group not involved in hydrogen bond interactions, and (iii) the electron withdrawing chloro group in the auxiliary ligand L2 along with a macrocyclic ring structure, making it a suitable candidate for the sensing of aromatic compounds. Sensing studies were carried out in DMSO solution and in the solid state. A control experiment for the sensing response of ligand L1 in DMSO was also conducted with PA and 2,4-dinitrophenol (2,4-DNP). Quenching of fluorescence was observed in both the cases (Fig. S12†).Study of 1(DMF)4 as a sensor in DMSO
The sensing ability of 1(DMF)4 in DMSO was studied for a range of aromatic compounds, which include 4-nitrophenol (4-NP), 2,4-dinitrophenol (2,4-DNP), 2,4,6-trinitrophenol (picric acid = PA), 2,4-dinitrotoluene (2,4-DNT), 4-nitrobenzoic acid (4-NB), 1-chloro-2,4-dinitrobenzene (ClDNB), 3-nitrotoluene (3-NT), 2,4-dichlorobenzene (2,4-DClB) and o-cresol (o-C). Fluorescence quenching titrations were carried out to analyze the sensing ability of 1(DMF)4 for aromatic compounds. For the sensing of an aromatic compound, 100 μL of DMSO solution of 10−5 M analyte (aromatic compound) was added to 1.99 mL of 10−2 M solution of 1(DMF)4 in DMSO to make a final volume of 2 mL (concentration of 1(DMF)4 is calculated by taking its asymmetric unit, which has a molecular weight of 961.11). Similarly, other solutions were prepared by varying the concentration of the analyte and 1(DMF)4 while maintaining the final volume at 2 mL. It was observed that a significant decrease in fluorescence intensity was observed only in the case of PA, while other aromatic compounds showed a negligible effect on the emission spectra of 1(DMF)4 (Fig. S13a–S15† and Fig. 2a). Here, it should be noted that as the experiment is carried out in DMSO, PA is present as picrate, which is also supported by the 1H-NMR spectrum of picric acid in DMSO (Fig. S16†).31 A Stern Volmer plot (S–V plot) in the presence of other nitroaromatics is shown in Fig. S13b.† The S–V plot of 1(DMF)4 in the presence of PA in DMSO showed a linear nature at lower concentrations (1–5 μM).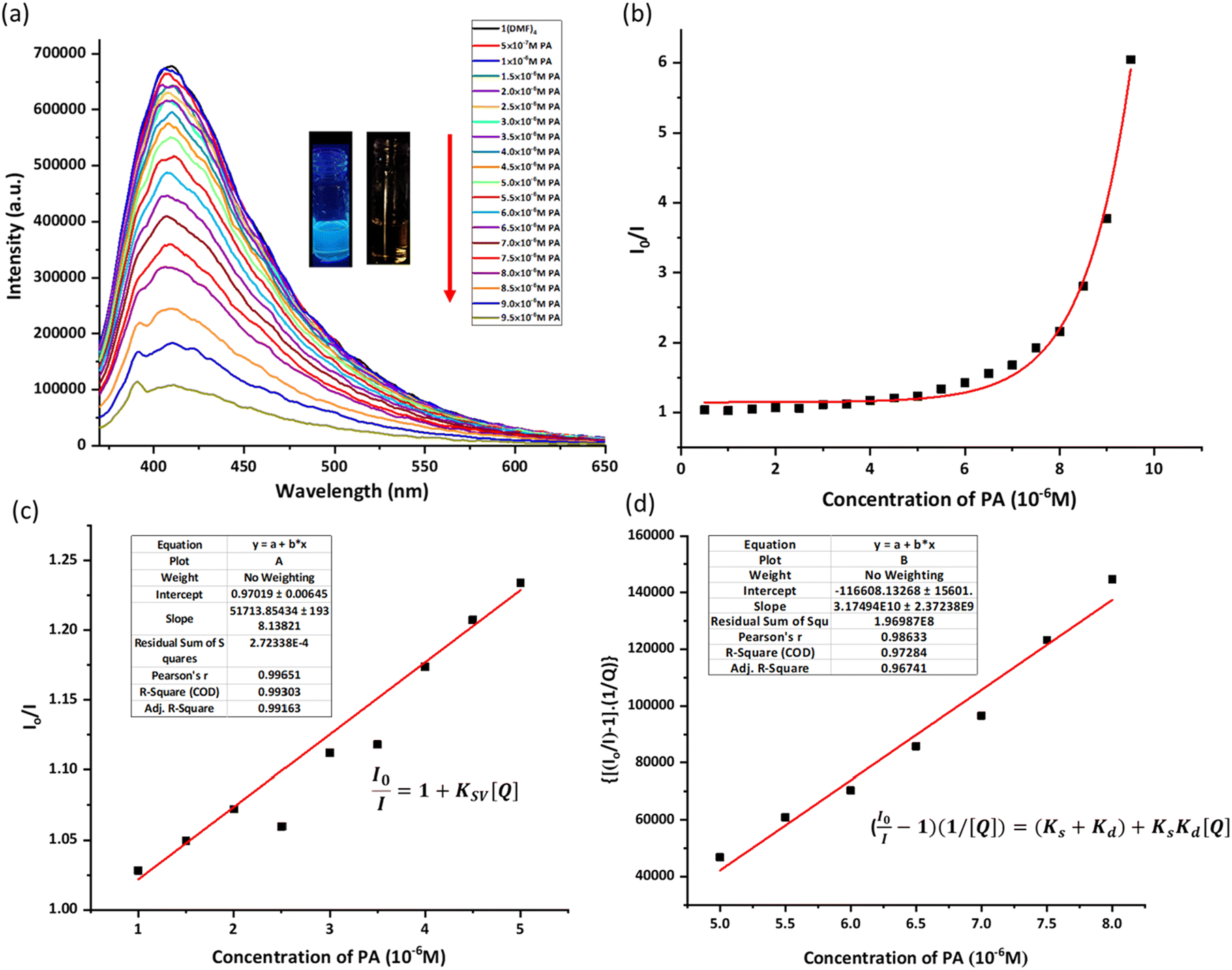 | ||
Fig. 2 (a) Fluorescence quenching titrations of 1(DMF)4 (10−2 M) with PA (10−5 M) in DMSO (excitation wavelength = 350 nm, slit width = 4 nm) (concentration of 1(DMF)4 is calculated by taking its asymmetric unit, with a molecular weight of 961.11); (b) S–V plot of 1(DMF)4 in the presence of PA in DMSO; (c) KSV for the lower concentration of PA (1–5 μM) shows the linear nature of the S–V plot; and (d) S–V plot  vs. [Q] to determine the Ks and Kd. vs. [Q] to determine the Ks and Kd. | ||
For the linear part, the Stern–Volmer constant (KSV) was determined to be 5.17 × 104 M−1 from the S–V plot in Fig. 2c using the linear equation:
 | (1) |
There is a deviation from linearity in the S–V plot (Fig. 2b), possibly due to static and dynamic quenching. So, to find out the values of KS and Kd for the nonlinear part, the following equation67–69 was used:
 | (2) |
Q is the concentration of the quencher i.e. PA concentration. The value of Kd of 1.7071 × 104 M−1 was found from the slope of the time resolved Stern–Volmer plot from Fig. S21.† Using eqn (2), a plot was drawn as shown in Fig. 2c, and the slope value is Ks × Kd using Kd from Fig. S21†; the Ks value was found to be 1.86 × 106. The higher magnitude of Ks signifies that the static quenching is dominant over dynamic quenching. The deviation from linearity at higher concentrations could be due to collision with the quencher in the solution.36,70
The binding ratio of PA with 1(DMF)4 in DMSO was estimated using Job's plot as 1:1 (PA: asymmetric unit of 1(DMF)4), while the binding constant was calculated to be 78.2 M−1 (Fig. S17 and S18†). The limit of detection of PA by 1(DMF)4 in DMSO was found to be 6.8 × 10−11 M (Fig. S19†). Time-resolved photoluminescence studies were performed, indicating the presence of static quenching at a low concentration of PA (Fig. S20 and S21†). The nonlinear nature of the Stern–Volmer plot at high concentrations can be related to energy transfer processes (Fig. 2b).
The spectral overlap of the absorption spectrum of PA with that of an emission spectrum of 1(DMF)4 in DMSO is shown in Fig. 3. The extent of energy transfer was determined by calculating the integral of overlap (Jλ) values using eqn (1), where FD is the corrected fluorescence intensity of the donor in the range λ to λ + dλ with the total intensity normalized to unity, and εA is the extinction coefficient of the acceptor at λ in M−1 cm−1. The overlap integral was calculated to be 9.49 × 1014 M−1 cm−1 nm4.
 | (3) |
 | ||
| Fig. 3 Spectral overlap of the absorption spectrum of PA in DMSO (picrate anions) and emission spectrum of 1(DMF)4 in DMSO (J(λ) = 9.49 × 1014 M−1 cm−1 nm4, R0 = 18.95 Å). | ||
It should be noted here that although there is a spectral overlap of the absorption spectra of some other aromatic compounds (2,4-DNP and 2,4-DNT) with the emission spectra of 1(DMF)4, still no/negligible quenching is observed in those cases (Fig. S22†). The role of the photoinduced electron transfer (PET) process in quenching was also analyzed using DFT calculations. Ground state geometry optimization of 1 in DMSO was performed using DFT calculations, and the respective frontier molecular orbital (FMO) energies were obtained (Fig. S23b†). It has been observed that the HOMO energy level of 1(DMF)4 obtained by cyclic voltammetry is in good agreement with the theoretically obtained one (Fig. S23a†). The DFT studies of an adduct/complex of 1 with picrate ions (in DMSO) indicated that there was no significant change of the energy gap (HOMO and LUMO) upon complex formation of 1 with picrate ions (Fig. S23b†). Furthermore, it was also observed that the HOMO and LUMO of the complex (1 with picrate ions) were primarily situated on the picrate ion, consequently precluding PET (Fig. S23b†) The spectral overlap of the absorption and emission spectra of picrate and 1(DMF)4 in DMSO and the energy ordering of their frontier orbitals suggest that the observed quenching phenomenon is due to the FRET. The selectivity of 1(DMF)4 towards PA sensing in DMSO is due to the ascribed chemical nature of 1(DMF)4, which interacts selectively with a picrate anion, resulting in a hydrogen bond interaction between 1(DMF)4 and the picrate ion with a feasible FRET process.
1H-NMR analysis to demonstrate the interaction of 1(DMF)4 with PA in DMSO
1H NMR analysis of 1(DMF)4 was performed in DMSO-d6 as shown in Fig. S2.† The peak at δ ppm 10.89 (s, 3H) corresponds to the –NH protons of L1 mentioned as Hc in Fig. S2,† peaks at δ ppm 9.00–8.24 correspond to the Hd, Hh, He and Hg protons of ligand L1 and peaks at 7.95 (dd, 8 Hz, 6H) and 7.48 (m, 7H) correspond to the Ha, Hb and Hf protons of L2 and L1. To validate the assignment of proton peaks, a 2D COSY NMR study was performed as shown in Fig. S3.† It can be observed that the protons of L2, Hb and Ha, correlate with each other. The protons in L1 are Hd, He, Hf, and Hg; proton Hf correlates with the protons He and Hg, whereas meta coupling is observed between Hg and Hd protons.1H NMR analysis was performed in DMSO-d6 to get more information related to the non-covalent interactions responsible for the interaction of 1(DMF)4 with PA in DMSO. For this purpose, a solution of 14.59 mM 1(DMF)4 and 14.59 mM PA was prepared in DMSO-d6. 1H-NMR spectra were recorded for eight solutions with different concentrations of 1(DMF)4 and PA, which were prepared separately using the stock solution (Fig. S24†). Fig. 4 presents the 1H-NMR spectra of four such solutions (solutions 1, 3, 5 and 8) with different concentrations of 1(DMF)4 and PA. It can be seen from the spectra that as the PA ratio increases in the solution, a shift in the peak position of some of the peaks can be observed. Some of the shifts are observed for the amidic N–H proton labelled as ‘c’ and the protons present in the chlorobenzoate unit of 1(DMF)4 labelled as ‘b’ in Fig. 4. Shifts are also observed for pyridyl protons of the L1 unit, while no change in the peak position is seen for the phenyl protons of L1. The shifts in some of the aromatic protons may be associated with the interaction of 1(DMF)4 with PA molecules. It should be noted that as the 1H-NMR spectra were recorded in DMSO-d6, PA was deprotonated and existed as picrate ions. The 1H-NMR spectra of 1(DMF)4 in the presence of 4-NP (Fig. S25†) and 2,4-DNT (Fig. S27†) do not show any changes in peak positions while some shifts in the peak positions of Hb and Hf are observed in the case of 2,4-DNP on increasing its concentration (Fig. S26†).
 | ||
| Fig. 4 1H-NMR spectra of different ratios of 1(DMF)4 (14.59 mM in DMSO-d6) and PA (14.59 mM in DMSO; solution 3: 0.1 mL of 1(DMF)4 + 0.05 mL of PA + 0.25 mL of DMSO-d6; solution 5: 0.1 mL of 1(DMF)4 + 0.15 mL of PA + 0.15 mL of DMSO-d6; and solution 8: 0.1 mL of 1(DMF)4 + 0.3 mL of PA). Note that PA in DMSO gets deprotonated to form picrate anions) (1H-NMR spectra of solutions 1, 2, 4, 6, 7 and 8 are shown in Fig. S24†). | ||
The electrostatic potential surface of optimized geometries of picrate ions and 1 in DMSO may explain the interactions between them which resulted in changes in chemical shifts of the protons of 1(DMF)4 in its 1H-NMR in response to concentration changes of PA in DMSO (picrate anions). It can be seen that a picrate ion has a more negative surface potential while the core of the 1 macrocycle has positive potentials (Fig. S28†). This might have resulted in the interaction of the picrate ion with the centre of the 1 ring. The effect of this interaction can be observed on the chemical shift of the protons of amidic N–H and the protons of the chlorobenzoate group in 1(DMF)4 as shown in Fig. 4. The amidic N–H protons in 1(DMF)4 appeared at 10.88 ppm, while on adding PA, it initially shifted upfield to 10.86 ppm and then downfield to 10.87 ppm. This may be due to the combined effect of hydrogen bonded DMF (solvated) and interaction with the electron rich environment of NO2 of picrate, while a downfield shift of protons of chlorobenzoate shows that an electron deficient region is created in L2 protons in 1(DMF)4 on interaction with picrate.
Molecular docking geometries and DFT optimized geometries to demonstrate the interaction of 1(DMF)4 with PA in DMSO
The geometry of the adduct (1 with picrate ions) obtained from DFT studies showed the interactions between the NH of 1 and NO2 of picrate ions with an interaction energy of ∼−178.49 kcal mol−1 (Fig. 5a). Molecular docking studies were also performed on the optimized geometry of 1 and picrate ions using Hex 8.0.0. Fig. 5b depicts one of the orientations which shows appreciable hydrogen bonding interactions between amidic N–H of 1 and NO2 of the picrate ion. The docking studies of 1 with 2,4-DNT did not show any possible intermolecular interactions (Fig. S29†), while few interactions were observed with 2,4-DNP and 4-NP (Fig. S30†).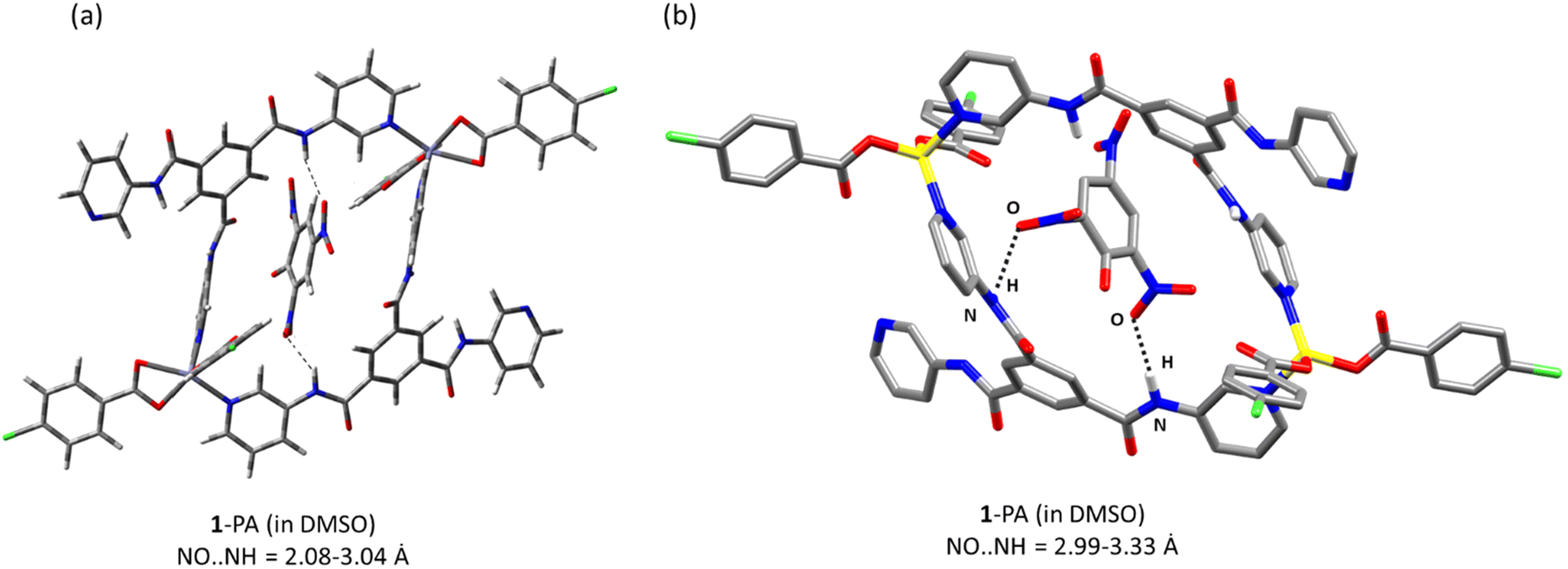 | ||
| Fig. 5 (a) Geometry of a 1-picrate ion along with possible interactions (dotted black line) obtained by DFT; interaction energy E = −178.49 kcal mol−1. (b) Representation of a possible orientation obtained by molecular docking studies on the optimized geometry of 1 and picrate ions (NO⋯NH = 2.986–3.327 Å) (distances between the heteroatoms are mentioned; estimated interaction energy E = −238.0 kcal mol−1). | ||
Study of 1(DMF)4 as a solid-state sensor for contact mode detection of aromatic compounds
Contact mode detection of aromatic compounds (4-NP, 2,4-DNP, PA, 2,4-DNT, 4-NB, and o-C) using 1(DMF)4 in the solid state was analysed. A qualitative representation of the response of 1(DMF)4 towards PA is shown in Fig. 6a, where filter paper strips coated with 1(DMF)4 were treated with different concentrations of PA and the resulting visual changes in the emission of 1(DMF)4 were observed under UV light. Fig. 6c presents a digital image of qualitative representation to show fluorescence quenching via a turn-off mechanism, where a TLC strip coated with 1(DMF)4 was used to observed the fluorescence on–off changes in the presence of PA. | ||
| Fig. 6 (a) Visual changes observed on the 1(DMF)4 coated filter paper test strips under UV-light after the addition of 10 μL of various concentrations of PA in CHCl3; (b) emission spectra of the thin film of 1(DMF)4 and PA; notice that on increasing the amount of PA, quenching along with a red shift of emission maxima is observed; and (c) digital image of the qualitative representation of fluorescence quenching on the TLC strip coated with 1(DMF)4. | ||
Quantitative estimation of the sensing response of 1(DMF)4 in the presence of aromatic compounds was performed by preparing thin films of 1(DMF)4 on glass slides (1 cm wide). On these films, a known volume of CHCl3 solution of aromatic compounds (analyte) of a particular concentration (10−3 M) was added and then dried. The emission spectra of the dried films were recorded. Fig. 6b presents the emission spectra of a thin film of 1(DMF)4 on adding different concentrations of PA. The emission spectrum of 1(DMF)4 shows quenching and a red shift of the peak maxima on gradually increasing the amount of PA. The estimated value of the detection limit of PA by the 1(DMF)4 solid was found to be 0.49 ng cm−2 (Fig. S31†).
The emission spectra of 1(DMF)4 in the presence of 4-NP and 2,4-DNP also showed quenching effects although no red shift was observed in the peak maxima for 4-NP while 2,4-DNP showed some red-shift in the peak maxima (Fig. S32†). No quenching was observed in the emission spectrum – of 1(DMF)4 was observed with 2,4-DNT, 4-NB and o-C (Fig. S33†). Furthermore, the extent of quenching observed in the case of 4-NP and 2,4-DNP is much less than that observed with PA. The percentage quenching observed in 1(DMF)4 due to PA, 2,4-DNP and 4-NP were 87%, 57% and 40%, respectively (Fig. 7).
 | ||
| Fig. 7 Bar graph showing the quenching percentages of 1(DMF)4 in the presence of PA, 2,4-DNP, 4-NP, 2,4-DNT, 4-NB, and o-C. | ||
Theoretical studies to analyze the quenching behavior of 1(DMF)4 in the solid state
In general, the mechanisms responsible for the quenching of fluorescence include ground state complex formation, electron transfer processes and energy transfer processes. To investigate the role of electron transfer in the quenching behavior observed for 1(DMF)4 (solid state) in the presence of nitrophenols, its electronic properties were investigated. The HOMO and LUMO of the aromatic compounds are shown in Fig. S34.† The lower the LUMO energy level of a NAC compound, the more feasible will be the electron transfer process from 1 to the aromatic compound. The maximum quenching is observed for PA, which is in good agreement with its LUMO energy level. Apart from the nitrophenols, other aromatic compounds did not show any quenching phenomena.To study the selective interactions of 1 with nitrophenols, the electrostatic potentials of the optimized geometries of 1 and the aromatic compounds were analyzed. It should also be noted that among all the studied aromatic compounds, nitrophenols are acidic and can undergo deprotonation/partial deprotonation. The pKa values of PA, 2,4-DNP and 4-NP are 0.42, 4.04 and 7.07, respectively. 1(DMF)4 has “free” pyridyl nitrogen and solvated DMF, which may assist in deprotonation in nitrophenols and result in the formation of partial anionic species for efficient interaction with the electron deficient centre of the 1 ring. The proximity between 1 and PA might have led to an efficient electron/energy transfer process, resulting in quenching. The electrostatic potential map of the optimized geometry of 1 (solvated DMF is removed from cif, followed by optimization) shows low electron density regions inside the cavity, while the potential surfaces of NACs show higher electron density along the nitro groups (Fig. S35†). The electrostatic potential surface of 2,4-DNT showed a very high negative potential along the nitro groups and a high positive potential along the aromatic and methyl regions. This might have resulted in lesser affinity of the 2,4-DNT towards the cavity of 1(DMF)4 and hence could not approach close enough to form an adduct/complex.
DFT calculations were also performed to analyze the adduct/complex formation between 1 and PA, 2,4-DNP and 4-NP complex in the gaseous state. The energy of interaction of 1 with PA, 2,4-DNP and 4-NP was found to be ∼−152.8 = 0, ∼−120.51 and ∼−80.11 kcal mol−1, respectively, which showed that the stable complex formation of 1 with PA occurred leading to a subsequent increase in the quenching percentage of 1 with PA. Fig. 8a shows the possible orientation of PA within 1 as obtained by optimization using DFT. The DFT study also supports the possible interactions (Fig. 8a) between amidic nitrogen of the host to the nitro oxygen of PA (N⋯O is in the range of 3.0 to 3.13 Å). DFT optimized geometries of 1-2,4-DNP and 1-4-NP are given in Fig. S39b and S40b.† As noted in Fig. 6b, the quenching of the emission intensity of 1(DMF)4 in the presence of PA also resulted in a significant red shift of peak maxima, while the emission spectra of 1(DMF)4 in the presence of 2,4-DNP and 4-NP showed quenching phenomena but without any shift of peak maxima (Fig. S32†). From the values of energy gaps between the HOMO and LUMO of the complex of 1 with the analyte (−2.35, −3.02 and −3.43 eV for 1-PA, 1-2,4-DNP and 1-4-NP, respectively), the red shift observed for the 1-PA complex can be explained (Fig. 8b and Fig. S36, S37†).37
 | ||
| Fig. 8 (a) Geometry of 1-PA along with possible interactions (dotted black line) obtained by DFT; interaction energy = −152.80 kcal mol−1. (b) HOMO–LUMO energies of 1 with PA (total energy of 1 is −2.7 × 10−5 eV and that of the complex is −3.0 × 10−5 eV). | ||
Molecular docking studies were performed on 1 with PA and with 2,4-DNT to get a better idea of possible interactions between 1 and the analyte (PA and 2,4-DNT) in the solid state. It was observed that the nitro groups of the PA interact very strongly with the amidic N–H of the cavity wall of 1. The best possible orientation to show the interactions is given in Fig. 9a. Hydrogen bonding interactions are clearly shown in Fig. S38.† The docking studies of 1 with 2,4-DNT showed no interactions of NO2 groups with any chemical functionalities of 1 (Fig. 9b), while the docking studies with 4-NP and 2,4-DNP showed some interactions with 1 (Fig. S39a and S40a†).
 | ||
| Fig. 9 Molecular docking structures for (a) 1 with PA (N2, N1⋯O 3.069–4.214 Å) (distances between the heteroatoms are mentioned) interaction energy E = −191.9 kcal mol−1 and (b) 1 with 2,4-DNT (notice the interactions between the NO2 and amidic N–H in the case of PA, while no interactions are observed for 2,4-DNT) interaction energy E = −165.3 kcal mol−1. | ||
The resonant energy transfer phenomena of 1(DMF)4 and PA were analyzed by examining the emission spectrum of 1(DMF)4 and absorption spectrum of PA in the solid state (Fig. S41a†). From Fig. S41,† it can be observed that the extent of spectral overlap of 1(DMF)4 and PA is larger than that in 4-NP and 2,4-DNP. All these studies indicated that the high sensing ability of 1(DMF)4 towards PA is due to the combination of all the factors which include effective interaction, energy ordering of frontier orbitals for efficient PET and good overlap integral for FRET.
Selectivity, recyclability and stability of 1(DMF)4 in contact mode detection
To check the selectivity of 1(DMF)4 towards PA in the presence of other aromatic compounds, fluorescence quenching titrations of 1(DMF)4 in the presence of PA along with other aromatic compounds (2,4-DNT, o-xylene, and o-C) were performed. These analytes were selected to compare the non-NAC (o-C), non-phenolic (2,4-DNT) and non-NAC, non-phenolic (o-xylene) groups with PA. For this purpose, the fluorescence spectrum of a thin film of 1(DMF)4 (in a glass slide) was recorded. On this film, a solution of 2,4-DNT (10 μL) in CHCl3 (10−3 M) was dispersed and allowed to dry, followed by recording of the PL spectra of the resultant film. Further 5 μL of PA in CHCl3 (10−3 M) was dispersed on the same film, dried and emission was recorded. 2,4-DNT was added initially, so that high affinity binding sites of 1(DMF)4 will be accessible to 2,4-DNT, but the quenching effect of 1(DMF)4 on adding 2,4-DNT was insignificant. On the other hand, the addition of PA solution to a 2,4-DNT containing 1(DMF)4 thin film showed significant fluorescence quenching (Fig. 10). Similar results were obtained by performing the fluorescence quenching titrations with other compounds in the presence of PA. This experiment clearly shows the selectivity of 1(DMF)4 towards PA in the presence of high concentrations of other compounds.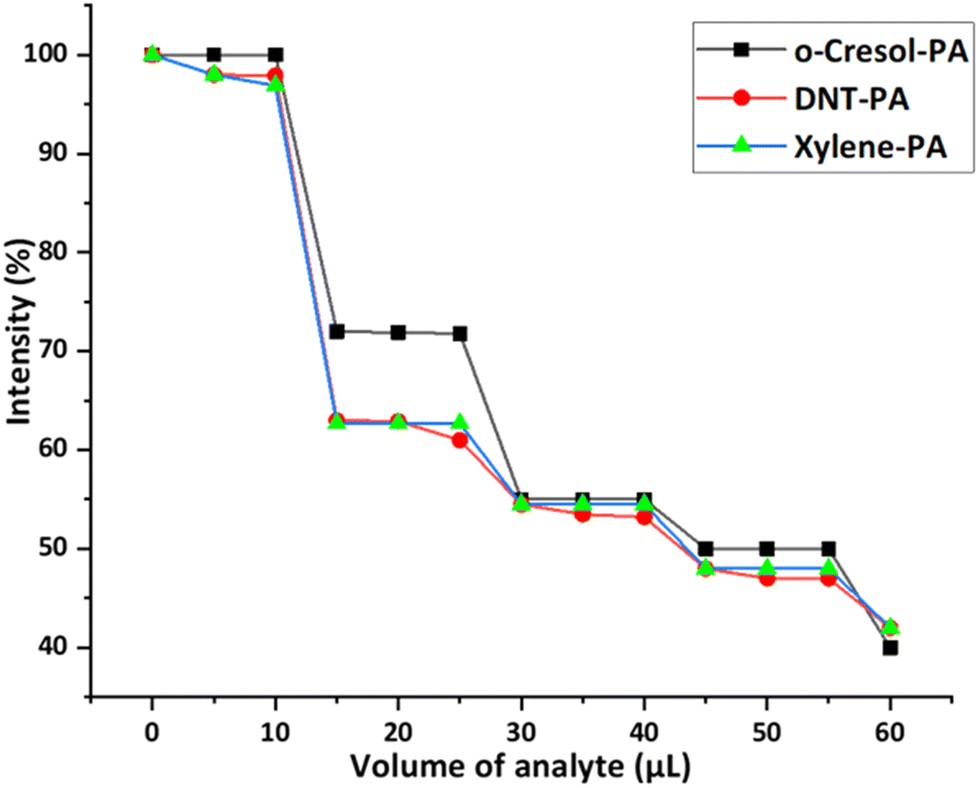 | ||
| Fig. 10 Decrease in the percentage of fluorescence intensity of λmax(emission) (430 nm) in 1(DMF)4 upon the addition of solutions of different aromatic compounds in chloroform followed by PA. | ||
Recyclability and stability are the important parameters in terms of the cost effectiveness and long-term real field selective detection of PA by the sensor 1(DMF)4. The stability of 1(DMF)4 in the presence of PA in the solid state was verified by IR and PXRD. The solid samples for recording IR spectra were prepared by taking an appropriate ratio of solid 1(DMF)4 (the asymmetric unit is considered for calculating the ratio) and PA in CHCl3, followed by uniform mixing and evaporating CHCl3 using a rotatory evaporator and vacuum drying. IR spectra of 1(DMF)4 in the presence of different ratios of PA showed the appearance of peaks corresponding to PA (some of the peaks are at 1627 cm−1 and 1426 cm−1) as the ratio of PA increases, while retaining the original peaks of 1(DMF)4 (some of the peaks are at 694 cm−1, 1435 cm−1, 1593 cm−1, and 1659 cm−1) (Fig. S42†). For recording the PXRD pattern of 1(DMF)4 in presence of PA, solid samples of 1(DMF)4 and PA from the thin film used in the contact mode detection experiments were taken. The PXRD pattern in Fig. S43† shows the stability of 1(DMF)4 in the presence of PA which matches well with the experimental PXRD pattern of 1(DMF)4.
For recyclability, the thin films of 1(DMF)4 were scratched off from the glass slides followed by washing with methanol to remove PA. 1(DMF)4 was recovered by centrifugation and was dried under vacuum. The recovered 1(DMF)4 was then used in the next cycle in the form of a thin film. Remarkably the fluorescence intensity after four cycles was nearly unchanged (Fig. S44 and S45†). Furthermore, to ensure the stability of the recovered 1(DMF)4 the PXRD analysis was performed after four cycles. In Fig. S46† the PXRD pattern after four cycles matches well with the experimental PXRD pattern.
The stability of solid 1(DMF)4 in the presence of PA was also verified by SEM-EDX. Sample preparation of SEM-EDX was performed by taking a 1:1 ratio of 1(DMF)4 and PA, and CHCl3 solution of PA was added to 1(DMF)4. Rotary evaporation of the suspension followed by vacuum drying resulted in the formation of a homogeneous solid sample. Fig. S47† shows the SEM-EDX of the 1:1 1(DMF)4 and PA sample, where the observed weight percentage of Zn is in good agreement with the calculated weight percentage. All these experiments revealed that 1(DMF)4 has good potential to act as a chemosensor for PA detection in terms of stability and recyclability.
Conclusion
A metallacycle, 1(DMF)4, is synthesized and it showed highly selective sensing towards PA in DMSO while the solid state (contact mode) detection showed a positive sensing response for the nitrophenols. The selectivity is due to the presence of favourable interactions between 1(DMF)4 and PA, which were supported by molecular docking studies and DFT optimized geometry of the complex of 1 and PA. The high selectivity of 1(DMF)4 in sensing PA in DMSO is due to a favourable energy transfer mechanism, while electron transfer and energy transfer mechanisms are actively involved in the quenching phenomena of solid state 1(DMF)4 in the presence of PA.Experimental
Materials
All reagents were obtained from commercial sources and used as received. Sigma Aldrich and TCI chemicals were used for the synthesis process. HPLC and UV grade solvents were used for the UV-visible and fluorescence spectral studies.Physical measurements
1H and 13C spectra were recorded on a 400 MHz spectrophotometer (Bruker). FTIR, UV-Visible absorption and fluorescence spectra were recorded using ABB Bomen MB-3000, Shimadzu UV-2450 and Fluorimax-4 0426C0809 spectrometers, respectively. Fluorescence quantum yields are determined by using a Quanta Phi instrument (Jobin Horiba, Fluorimax-4). A time-correlated single-photon counting (TCSPC) instrument Horiba-Jobin Yvon fluoro cube setup using a nano-LED (IBH UK) of wavelength 370 nm was used to estimate the excited-state decay curve and the lifetime of 1(DMF)4 in the presence of different quencher concentrations. Thermogravimetric analysis was performed using Shimadzu TGA-50 with a flow rate of N2 of 10 °C min−1. Powder X-Ray diffraction (XRD) was performed with a Rigaku miniflex ll, λ = 1.54 Å, Cu Kα. Cyclic voltammetry experiments were performed on a CHI electrochemical analyzer (Model 1120 A). SEM-EDX analysis was performed using an Apreo LoVac (Leica Ultra Microtome) EMUC7 model with 127 eV on Mn-Kα radiation. The concentration of 1(DMF)4, for UV-Visible, fluorescence and 1H-NMR studies, is calculated by taking its asymmetric unit, with a molecular weight of 961.11.Synthesis of 1(DMF)4
Ligand L1 (21.9 mg), which was prepared using a reported method,24p-chlorobenzoic acid (HL2) (51.0 mg), and Zn(NO3)2·6H2O (48 mg) were taken in a 1:6.5:4 ratio in a mortar and pestle and ground gently for 10 min until it showed a soggy white appearance. Then 8 mL of DMF was added and the contents were transferred to a glass vial and finally kept on a heating plate for 3 days. After 3 days, the temperature was decreased at a rate of 10 °C h−1 and then the vial was kept aside for 15–20 days. Needle-like white crystals were formed. Yield: 45.3% (21.7 mg), elemental analysis calculated for C44H40Cl2N8O9Zn (%): C = 54.98, H = 4.19, N = 11.66; found (%): C = 54.30, H = 3.62, N = 11.34; FTIR (cm−1, KBr pellet): 3309 (w), 3078 (w), 1658 (s), 1597 (s), 1535 (s), 1489 (s), 1396 (s), 694 (m) and 534 (s) (Fig. S1†); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.89 (s, 3H), 9.015 (d, 4 Hz, 3H), 8.80 (s, 3H), 8.38 (d, 4 Hz, 3H), 8.26 (tt, 8 Hz, 3H), 7.95 (dd, 8 Hz, 6H), 7.48 (m, 7H) (Fig. S2†); 2D COSY NMR (Fig. S4†); 13C NMR (100 MHz, DMSO-d6) δ ppm: 31.14, 31.24, 35.42, 36.25, 124.55, 128.43, 128.79, 130.75, 131.80, 134.39, 135.45, 136.16, 136.39, 142.35, 145.26, 162.79, 165.30, 167.34 and 170.56 (Fig. S4†).
Thermogravimetric analysis (TGA) of 1(DMF)4
Thermogravimetric analysis was performed using TGA-50 with a flow rate of N2 of 10 °C min−1 up to a temperature of 800 °C. The TGA of 1(DMF)4 shows three weight losses at 242.47 °C, 388.40 °C and 560.34 °C as shown in Fig. S6a.† The first derivative curve shows the %weight loss with respect to temperature as shown in Fig. S6b.† 3.5389 mg of 1(DMF)4 was taken for the analysis and a weight loss of 7.813% corresponds to the removal of one DMF molecule/asymmetric unit of 1(DMF)4, while weight losses of 50.537% and 6.879% correspond to the decomposition of 1(DMF)4. 1(DMF)4 is stable up to 284 °C.Single crystal X-ray crystallography
Single crystal XRD data were collected on an aXtaLAB Pro: Kappa dual offset/far diffractometer. The crystal was kept at 93(2) K during data collection. Using Olex2,71 the structure was solved with the SHELXT72 structure solution program using intrinsic phasing and refined with the SHELXL73 refinement package using least squares minimization. The crystal data and structure refinements of 1(DMF)4 are summarized in Table S1.† The crystal structure is twin modelled with component 1 (71%) and component 2 (29%). ORTEP of 1(DMF)4 is shown in Fig. S7.† Powder XRD (simulated and experimental PXRD) of 1(DMF)4 and experimental PXRD of 1 are provided in Fig. S8.†UV-Visible studies of 1(DMF)4
1(DMF)4 (4.795 mg, 5 mmol) was dissolved in 5 mL UV-grade DMSO in a 5 mL volumetric flask to make a 10−3 M stock solution. Furthermore, the stock solution was diluted to prepare 5 mL of a 10−5 M solution of 1(DMF)4 and the absorption spectra was recorded. For solid state UV-visible studies 2 mg of sample was taken and mixed with 10 mg of BaSO4 and a pellet was made and absorption spectra were recorded.Fluorescence quenching titrations in solution
Fluorescence emission spectra were recorded in UV-grade DMSO. For titration, a 1.99 mL stock solution (10−2 M) of 1(DMF)4 was taken in a quartz cell of 1 cm width and 10 μL solution of analyte (10−5 M) was added to make a total volume of 2 mL. The corresponding fluorescence emission spectra were recorded (Fig. S14 and S15†). A similar process was followed for all the respective measurements by varying the concentration of 1(DMF)4 and analyte, respectively, with a total volume of 2 mL. For all the measurements, the excitation wavelength was kept at 340 nm and the slit width was 3.5 nm.Fluorescence efficiency was calculated as

The binding stoichiometry of the 1(DMF)4 macrocycle with PA was calculated using Job's plot as shown in Fig. S17† and the binding constant (Kb) was calculated as
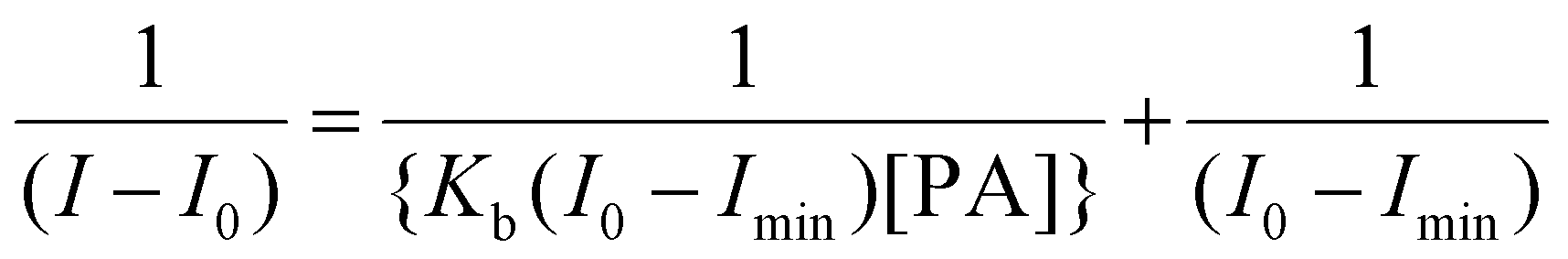
The limit of detection was calculated from 3σ/k, where σ is the standard deviation of ten blank measurements of the probe at the minimum slit width and k is the slope of a plot of fluorescence intensity vs. concentration of picric acid (Fig. S19 and S31†).
Lifetime measurements
Stock solutions of 10−2 M and 10−5 M 1(DMF)4 and PA, respectively, were prepared. For lifetime measurement, 1.9 mL of 1(DMF)4 and 100 μL of PA were taken in a cuvette by maintaining the total volume of 2 mL. Similarly, five different sets of solutions of 1(DMF)4 and PA were prepared as mentioned in Fig. S20 and S21† and the corresponding lifetime measurement was performed.Contact mode detection (visual qualitative detection)
For visual detection of PA, 0.1 mM solution of 1(DMF)4 was prepared and Whatman 42 filter paper strips (2 cm wide) were dipped and dried under vacuum. A drop of the solution of PA in CHCl3 of known concentration was put on the dried filter paper and was checked under a UV lamp (365 nm) for visual observation of the quenching effect.Contact mode detection (quantitative estimation)
Thin layers of 1(DMF)4 on a quartz slide are prepared by taking 50 μL of saturated solution of 1(DMF)4 in THF on a quartz slide. The quartz slide was dried under vacuum for 2 h. For each fluorescence quenching experiment, new slides with a thin layer were used.NMR titration studies of 1(DMF)4 with PA, 2,4-DNP, 4-NP and 2,4-DNT
NMR titration studies were performed to determine the interaction of analytes (PA, 2,4-DNP, 4-NP and 2,4-DNT) with 1(DMF)4. (The concentration of 1(DMF)4 is calculated by taking its asymmetric unit, with a molecular weight of 961.11.)Solution sets of 1(DMF)4 with PA
14.56 mM solution of 1(DMF)4 (16.8 mg of 1(DMF)4 in 1.2 mL of DMSO-d6) and 14.59 mM solution of PA (4 mg of PA in 1.2 mL of DMSO-d6) were used to prepare eight sets of solutions of 1(DMF)4 and PA: (i) NMR solution 1: 0.1 mL 1(DMF)4 solution + 0.3 mL DMSO-d6 solvent; (ii) NMR solution 2: 0.1 mL PA solution + 0.3 mL DMSO-d6 solvent; (iii) NMR solution 3: 0.1 mL 1(DMF)4 solution + 0.05 mL PA solution + 0.25 mL DMSO-d6 solvent; (iv) NMR solution 4: 0.1 mL 1(DMF)4 solution + 0.1 mL PA solution + 0.2 mL DMSO-d6 solvent; (v) NMR solution 5: 0.1 mL 1(DMF)4 solution + 0.15 mL PA solution + 0.15 mL DMSO-d6 solvent; (vi) NMR solution 6: 0.1 mL 1(DMF)4 solution + 0.2 mL PA solution + 0.1 mL DMSO-d6 solvent; (vii) NMR solution 7: 0.1 mL 1(DMF)4 solution + 0.25 mL PA solution + 0.05 mL DMSO-d6 solvent; and (viii) NMR solution 8: 0.1 mL 1(DMF)4 solution + 0.3 mL PA solution (Fig. S24†).Solution sets of 1(DMF)4 with 4-NP
14.59 mM solution of 1(DMF)4 (16.8 mg of 1(DMF)4 in 1.2 mL of DMSO-d6) and 14.59 mM solution of 4-NP (2.44 mg of 4-NP in 1.2 mL of DMSO-d6) were used to prepare the solutions of 1(DMF)4 and 4-NP. Solution 1: only 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 0.30 mL of DMSO-d6; solution 2: 4-NP (0.10 mL of 14.59 mM in DMSO-d6) + 0.30 mL of DMSO-d6; solution 3: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + NP (0.05 mL of 14.59 mM in DMSO-d6) + 0.25 mL of DMSO-d6; and solution 4: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + NP (0.25 mL of 14.59 mM in DMSO-d6) + 0.05 mL of DMSO-d6 (Fig. S25†).Solution sets of 1(DMF)4 with 2,4-DNP
14.59 mM solution of 1(DMF)4 (16.8 mg of 1(DMF)4 in 1.2 mL of DMSO-d6) and 14.59 mM solution of 2,4-DNP (3.2 mg of 2,4-DNP in 1.2 mL of DMSO-d6) were used to prepare the solutions of 1(DMF)4 and 2,4-DNP. Solution 1: only 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 0.30 mL of DMSO-d6; solution 2: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 2,4-DNP (0.05 mL of 14.59 mM in DMSO-d6) + 0.25 mL of DMSO-d6; solution 3: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 2,4-DNP (0.15 mL of 14.59 mM in DMSO-d6) + 0.15 mL of DMSO-d6; and solution 4: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 2,4-DNP (0.25 mL of 14.59 mM in DMSO-d6) + 0.05 mL of DMSO-d6 (Fig. S26†).Solution sets of 1(DMF)4 with 2,4-DNT
14.59 mM solution of 1(DMF)4 (16.8 mg of 1(DMF)4 in 1.2 mL of DMSO-d6) and 14.59 mM solution of 2,4-DNT (3.19 mg of 2,4-DNT in 1.2 mL of DMSO-d6) were used to prepare the solutions of 1(DMF)4 and 2,4-DNT. Solution 1: only 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 0.30 mL of DMSO-d6; solution 2: only DNT (0.10 mL of 14.59 mM in DMSO-d6) + 0.30 mL of DMSO-d6; solution 3: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 2,4-DNT (0.20 mL of 14.59 mM in DMSO-d6) + 0.10 mL of DMSO-d6; and solution 4: 1(DMF)4 (0.10 mL of 14.59 mM in DMSO-d6) + 2,4-DNT (0.30 mL of 14.59 mM in DMSO-d6) (Fig. S27†).Cyclic voltammetry
Cyclic voltammetry measurements were carried out at 25 °C in DMSO by using a three-electrode set-up comprising a glassy-carbon working electrode, a platinum wire auxiliary electrode, and a reference electrode. Stock solutions of 10−3 M 1(DMF)4 were prepared and potassium hexafluorophosphate (KPF6) was used as a supporting electrolyte.DFT calculations
DFT calculations were performed by taking the cif file coordinates of 1(DMF)4 and removing the coordinates of DMF molecules. Optimization of 1 in the gaseous phase was performed by using the B3LYP method and the 6-311G(d,p) basis set in the gaseous state using Gaussian 9.0 while the optimization of 1 in the solution state was performed using the Integral Equation Formalization Polarizable Continuum Model (IEFPCM) with the B3LYP method and 6-311G(d,p) basis set.The optimized geometries of nitroaromatic compounds (4-NP, 2,4-DNP, PA, 2,4-DNT, and 4-NB) were obtained using the B3LYP method and 6-311G(d,p) basis set. Picrate ion optimization was also performed in the solution phase using the B3LYP method and 6-311G(d,p) basis set using Gaussian 9.0. The frontier orbital values of the optimized nitroaromatic groups match well with those previously reported in the literature.74,75
DFT calculations on the adduct/complex were also performed on the cif optimized geometry of 1 by freezing the coordinates and optimization of only nitroaromatic compounds (PA, 2,4-DNP and 4-NP) was allowed within the host. For solution state studies, the integral equation formalization polarizable continuum model (IEFPCM) with the B3LYP method and 6-311G(d,p) basis set was used on the DFT results of the adduct/complex of 1 with PA in the gaseous phase after removing the proton of PA.
Molecular docking studies
Molecular docking studies for solid state analysis were performed by using the DFT optimized geometry of 1 in the gaseous phase while for solution phase studies, the DFT optimized geometry of 1 in the solution state was taken and energy minimized chemical drawings of various analytes (picrate ion, PA, 2,4-DNP, 4-NP and 2,4-DNT) were obtained using Hex 8.0.0.76Author contributions
V. J.: experimental investigation, analysis and writing – draft preparation; S. P.: molecular docking; J. C.: DFT calculations; K. R.: single crystal XRD; M. S.: conceptualization, formal analysis, funding acquisition, supervision, validation, and writing original draft, review and editing.Data availability
The data supporting this article have been included as part of the ESI available: Fig. S1–S47 and Tables S1–S6.†Crystallographic data for 1(DMF)4 have been deposited at the CCDC under the accession number 2195797.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the research grant from CSIR under the EMR scheme (01(3011)/21/EMR-II). We also acknowledge the Instrumentation facility from DST FIST & UGC-SAP to the Department of Chemistry, BITS-Pilani, Pilani Campus. We also acknowledge the use of the DST-FIST supported X-ray diffraction facility of the Physics Department, BITS-Pilani. VJ acknowledges BITS-Pilani for the fellowship.References
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- P. J. Stang, Chem. – Eur. J., 1998, 4, 19–27 CrossRef CAS.
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9–25 RSC.
- E.-S. M. El-Sayed and D. Yuan, Chem. Lett., 2020, 49, 28–53 CrossRef CAS.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- T. Habicher, J. F. Nierengarten, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed., 1998, 37, 1916–1919 CrossRef CAS.
- M. Fujita, F. Ibukuro, H. Hagihara and K. Ogura, Nature, 1994, 367, 720–723 CrossRef CAS.
- M. Fujita, F. Ibukuro, H. Seki, O. Kamo, M. Imanari and K. Ogura, J. Am. Chem. Soc., 1996, 118, 899–900 CrossRef CAS.
- M. Fujita, F. Ibukuro, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175–4176 CrossRef CAS.
- M. Fujita, S. Nagao, M. Iida, K. Ogata and K. Ogura, J. Am. Chem. Soc., 1993, 115, 1574–1576 CrossRef CAS.
- M. Schmitz, S. Leininger, J. Fan, A. M. Arif and P. J. Stang, Organometallics, 1999, 18, 4817–4824 CrossRef CAS.
- F. M. Dixon, A. H. Eisenberg, J. R. Farrell, C. A. Mirkin, L. M. Liable-Sands and A. L. Rheingold, Inorg. Chem., 2000, 39, 3432–3433 CrossRef CAS PubMed.
- J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 465–467 CrossRef CAS PubMed.
- B. J. Holliday, J. R. Farrell, C. A. Mirkin, K. C. Lam and A. L. Rheingold, J. Am. Chem. Soc., 1999, 121, 6316–6317 CrossRef CAS.
- A. Kumar, R. Pandey, R. K. Gupta, V. Mishra, S. M. Mobin and D. S. Pandey, Dalton Trans., 2014, 43, 6365–6376 RSC.
- D. L. Reger, A. E. Pascui, E. A. Foley, M. D. Smith, J. Jezierska, A. Wojciechowska, S. A. Stoian and A. Ozarowski, Inorg. Chem., 2017, 56, 2884–2901 CrossRef CAS PubMed.
- D. L. Reger, E. A. Foley, R. P. Watson, P. J. Pellechia, M. D. Smith, F. Grandjean and G. J. Long, Inorg. Chem., 2009, 48, 10658–10669 CrossRef CAS PubMed.
- S. Peitz, B. R. Aluri, N. Peulecke, B. H. Müller, A. Wöhl, W. Müller, M. H. Al-Hazmi, F. M. Mosa and U. Rosenthal, Chem. – Eur. J., 2010, 16, 7670–7676 CrossRef CAS PubMed.
- P. Thanasekaran, C.-C. Lee and K.-L. Lu, Acc. Chem. Res., 2012, 45, 1403–1418 CrossRef CAS PubMed.
- S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, Inorg. Chem., 2011, 50, 11736–11745 CrossRef CAS PubMed.
- B. Mohan and S. Shanmugaraju, Dalton Trans., 2023, 52, 2566–2570 RSC.
- N. Thakur, M. D. Pandey and R. Pandey, New J. Chem., 2018, 42, 3582–3592 RSC.
- A. A. Palmans, J. J. Vekemans and A. Spek, Chem. Commun., 1997, 2247–2248 RSC.
- L. Rajput and K. Biradha, J. Mol. Struct., 2008, 876, 339–343 CrossRef CAS.
- P. S. Mukherjee, N. Das and P. J. Stang, J. Org. Chem., 2004, 69, 3526–3529 CrossRef CAS PubMed.
- T. Jia, Y. Zhao, F. Xing, M. Shao, S. Zhu and M. Li, J. Mol. Struct., 2009, 920, 18–22 CrossRef CAS.
- H.-Y. Bai, S.-M. Wang, W.-Q. Fan, C.-B. Liu and G.-B. Che, Polyhedron, 2013, 50, 193–199 CrossRef CAS.
- N. Middleton, The global casino: An introduction to environmental issues, Routledge, 2018 Search PubMed.
- J. Pichtel, Appl. Environ. Soil Sci., 2012, 2012, 617236 Search PubMed.
- W. L. Goodfellow Jr, D. T. Burton, W. C. Graves, L. W. Hall Jr and K. R. Cooper, J. Am. Water Resour. Assoc., 1983, 19, 641–648 CrossRef.
- A. Chowdhury and P. S. Mukherjee, J. Org. Chem., 2015, 80, 4064–4075 CrossRef CAS PubMed.
- U. Mithra and S. Sarveswari, Inorg. Chim. Acta, 2024, 569, 122118 CrossRef.
- S. Chongdar, U. Mondal, T. Chakraborty, P. Banerjee and A. Bhaumik, ACS Appl. Mater. Interfaces, 2023, 15, 14575–14586 CAS.
- P. Ju, E. Zhang, L. Jiang, Z. Zhang, X. Hou, Y. Zhang, H. Yang and J. Wang, RSC Adv., 2018, 8, 21671–21678 RSC.
- W. Liu, X. Huang, C. Chen, C. Xu, J. Ma, L. Yang, W. Wang, W. Dou and W. Liu, Chem. – Eur. J., 2019, 25, 1090–1097 CrossRef CAS PubMed.
- X.-H. Zhou, L. Li, H.-H. Li, A. Li, T. Yang and W. Huang, Dalton Trans., 2013, 42, 12403–12409 RSC.
- X. Xin, Y. Jing, P. Li, X. Zhang, J. Li, L. Li and L. Zhang, Cryst. Growth Des., 2023, 23, 3988–3995 CrossRef CAS.
- B. Mondal and G. Das, React. Funct. Polym., 2024, 194, 105800 CrossRef CAS.
- N. F. Suhendra, R. Sharma and H.-I. Lee, Sens. Actuators, B, 2024, 418, 136345 CrossRef CAS.
- K. Wang, T.-M. Geng, H. Zhu and C. Guo, Microporous Mesoporous Mater., 2024, 363, 112794 CrossRef CAS.
- K. Wang, C. Guo and T.-M. Geng, React. Funct. Polym., 2024, 197, 105863 CrossRef CAS.
- Z. Rahman, N. Tohora, M. Mahato, S. Ahamed, T. Sultana, M. Shah, A. Borah and S. Das, J. Photochem. Photobiol., A, 2023, 444, 114906 CrossRef CAS.
- K. J. Goswami, N. Sultana and N. Sen Sarma, Sens. Actuators, B, 2023, 374, 132778 CrossRef CAS.
- N. Mate, D. Khandelwal, K. Nabeela and S. M. Mobin, J. Mater. Chem. C, 2023, 11, 16201–16213 RSC.
- Ö. K. Koç, A. Üzer and R. Apak, ACS Appl. Mater. Interfaces, 2023, 15, 42066–42079 CrossRef PubMed.
- Y. Wang, X. Chang, N. Jing and Y. Zhang, Anal. Methods, 2018, 10, 2775–2784 RSC.
- S. Kadian and G. Manik, Luminescence, 2020, 35, 763–772 CrossRef CAS PubMed.
- M. Dong, Y.-W. Wang, A.-J. Zhang and Y. Peng, Chem. – Asian J., 2013, 8, 1321–1330 CrossRef CAS PubMed.
- Z.-H. Fu, Y.-W. Wang and Y. Peng, Chem. Commun., 2017, 53, 10524–10527 RSC.
- Y. Peng, A.-J. Zhang, M. Dong and Y.-W. Wang, Chem. Commun., 2011, 47, 4505–4507 RSC.
- X. Cao, N. Zhao, H. Lv, Q. Ding, A. Gao, Q. Jing and T. Yi, Langmuir, 2017, 33, 7788–7798 CrossRef CAS PubMed.
- G. Chakraborty and S. K. Mandal, ACS Omega, 2018, 3, 3248–3256 CrossRef CAS PubMed.
- B. Pramanik, S. Das and D. Das, Chem. – Asian J., 2020, 15, 4291–4296 CrossRef CAS PubMed.
- R. Chopra, V. Bhalla, M. Kumar and S. Kaur, RSC Adv., 2015, 5, 24336–24341 RSC.
- P. Lasitha and E. Prasad, RSC Adv., 2015, 5, 41420–41427 RSC.
- Q. Lin, X.-W. Guan, Y.-Q. Fan, J. Wang, L. Liu, J. Liu, H. Yao, Y.-M. Zhang and T.-B. Wei, New J. Chem., 2019, 43, 2030–2036 RSC.
- H. Ma, C. He, X. Li, O. Ablikim, S. Zhang and M. Zhang, Sens. Actuators, B, 2016, 230, 746–752 CrossRef CAS.
- E. Zhang, P. Ju, P. Guo, X. Hou, X. Hou, H. Lv, J.-J. Wang and Y. Zhang, RSC Adv., 2018, 8, 31658–31665 RSC.
- S. Kumar, R. Kishan, P. Kumar, S. Pachisia and R. Gupta, Inorg. Chem., 2018, 57, 1693–1697 CrossRef CAS PubMed.
- Q. Tang, Y. Sun, H.-Y. Li, J.-Q. Wu, Y.-N. Liang and Z. Zhang, Appl. Organomet. Chem., 2019, 33, e4814 CrossRef.
- M. Nurnabi, S. Gurusamy, J.-Y. Wu, C.-C. Lee, M. Sathiyendiran, S.-M. Huang, C.-H. Chang, I. Chao, G.-H. Lee, S.-M. Peng, V. Sathish, P. Thanasekaran and K.-L. Lu, Dalton Trans., 2023, 52, 1939–1949 RSC.
- D. Prabha, D. Singh, P. Kumar and R. Gupta, Inorg. Chem., 2021, 60, 17889–17899 CrossRef CAS PubMed.
- A. Kumar, A. Kumar and D. S. Pandey, Dalton Trans., 2016, 45, 8475–8484 RSC.
- D. Moon, S. Kang, J. Park, K. Lee, R. P. John, H. Won, G. H. Seong, Y. S. Kim, G. H. Kim and H. Rhee, J. Am. Chem. Soc., 2006, 128, 3530–3531 CrossRef CAS PubMed.
- J. Park, S. Hong, D. Moon, M. Park, K. Lee, S. Kang, Y. Zou, R. P. John, G. H. Kim and M. S. Lah, Inorg. Chem., 2007, 46, 10208–10213 CrossRef CAS PubMed.
- B.-C. Tzeng, B.-S. Chen, H.-T. Yeh, G.-H. Lee and S.-M. Peng, New J. Chem., 2006, 30, 1087–1092 RSC.
- P. Sarkar, N. Tohora, M. Mahato, S. Ahamed, T. Sultana and S. K. Das, J. Fluoresc., 2023 DOI:10.1007/s10895-023-03555-y.
- A. S. Tanwar, R. Parui, R. Garai, M. A. Chanu and P. K. Iyer, ACS Meas. Sci. Au, 2022, 2, 23–30 CrossRef CAS PubMed.
- B. Gole, W. Song, M. Lackinger and P. S. Mukherjee, Chem. – Eur. J., 2014, 20, 13662–13680 CrossRef CAS PubMed.
- A. Chowdhury and P. S. Mukherjee, ChemPlusChem, 2016, 81, 1360–1370 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- N. Sebbar, M. Ellouz, E. Essassi, Y. Ouzidan and J. Mague, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o999 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. H. Malik, S. Hussain, A. Kalita and P. K. Iyer, ACS Appl. Mater. Interfaces, 2015, 7, 26968–26976 CrossRef CAS PubMed.
- S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed.
- D. Ritchie, Hex, Inria, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S38 and Table S1. CCDC 2195797 for 1(DMF)4. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01771d |
| This journal is © The Royal Society of Chemistry 2024 |