
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring C–H amination activity via modification of the triazole-derived carbene ligand†
Luke A.
Hudson
 ,
Wowa
Stroek
and
Martin
Albrecht
*
,
Wowa
Stroek
and
Martin
Albrecht
*
Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012, Bern, Switzerland. E-mail: martin.albrecht@unibe.ch
First published on 15th August 2024
Abstract
Two new C,O-bidentate chelating triazolylidene-phenolate ligands were synthesized that feature a diisopropylphenyl (dipp) and an adamantyl (Ad) substituent respectively on the triazole scaffold. Subsequent metalation afforded iron(II) complexes [Fe(C^O)2] that are active catalysts for the intramolecular C–H amination of organic azides. When compared to the parent complex containing a triazolylidene with a mesityl substituent (Mes) the increased steric bulk led to slightly lower activity (TOFmax = 23 h−1vs. 30 h−1), however selectivity towards pyrrolidine formation increases from 92% up to >99%. Kinetic studies indicate that the mechanism is similar in all three complexes and includes a half-order dependence in [Fe(C^O)2], congruent with the involvement of a dimetallic catalyst resting state within this catalyst class. Structural analysis suggests that enhanced bulkiness disfavors N2 loss and nitrene formation, yet shields the nitrene from intermolecular processes and thus favors intramolecular nitrene insertion into the C–H bond. This model rationalizes the high selectivity and the lower reaction rate observed with dipp and with Ad substituents on the ligand.
The direct amination of C–H bonds is an attractive method for constructing C–N bonds,1 as unlike classical methods,2–4 no carbon functionalization is required. Direct C–H amination relies on the availability of a nitrene as transient and active species, which is typically generated from azides upon N2 loss at a transition metal center.1,5–11 Iron complexes have been established as particularly active catalysts for this reaction, especially when using organic azides as nitrene precursors.8,12–18 Within this context, we recently demonstrated that complex 1 containing a triazole-derived carbene ligand is one of the best performing catalysts for C–H amination (Scheme 1), reaching record-high turnover numbers and state-of-the-art turnover frequencies.19
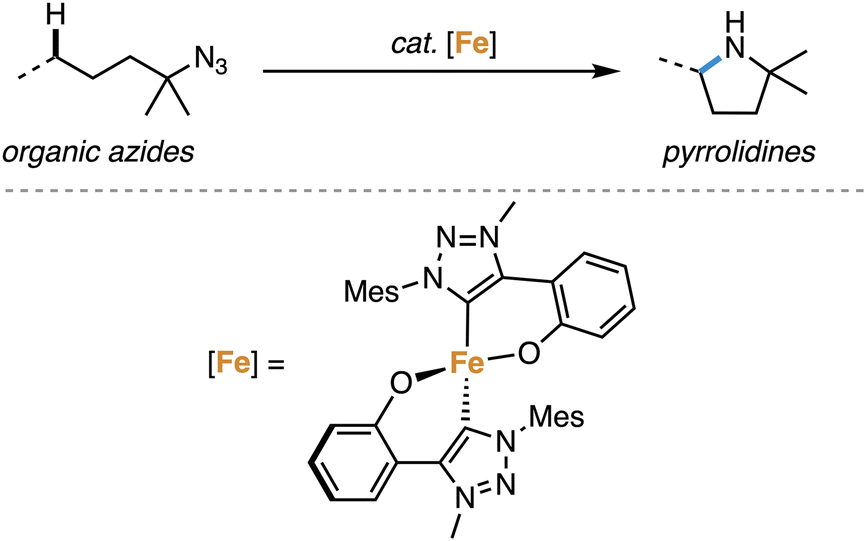 | ||
| Scheme 1 Application of mesoionic carbene iron complex 1 in intramolecular C–H amination catalysis. | ||
Kinetic studies of this system revealed an unusual half-order rate dependence on iron catalyst concentration. Therefore, a mechanism was postulated that involves an off-cycle dimeric species as catalyst resting state that needs to be cleaved en-route to coordination of the azide and rate-limiting release of N2 to generate the reactive iron nitrene intermediate. Considering the synthetic flexibility of the triazole core of the ligand in complex 1,20–23 ligand tailoring provides a plausible strategy for modulating catalytic activity and selectivity.24–28 Specifically, we hypothesized that the use of bulkier wingtip groups on the carbene will destabilize the dimeric resting state and thus accelerate catalytic turnover.
Here, we demonstrate that substitution of the mesityl wingtip group of the carbenes in complex 1 for diisopropylphenyl (dipp) and adamantyl (Ad) substituents indeed affects the catalytic rates, whilst preserving the overall mechanism. Moreover, steric tailoring leads to enhanced chemoselectivity.
Results and discussion
Synthesis of the complexes
The new ligand precursors L2 and L3 were synthesised according to a procedure adapted from the preparation of L1 (Scheme 2).19 Thus, “click”-type copper-catalysed azide–alkyne cycloaddition (CuAAC)21,22,29 of a silyl-protected alkynylphenol and the corresponding organic azide followed by methylation with methyl triflate (MeOTf) yielded L2 and L3 in good yields. Subsequent metalation with Fe(HMDS)2 in the presence of KHMDS afforded complexes 2 and 3 as highly air-sensitive, bright orange solids in 26% and 53% yield, respectively (HMDS = hexamethyldisilazide, N(SiMe3)2−). | ||
| Scheme 2 Synthesis of complexes 2 and 3. | ||
Analytically pure complexes 2 and 3 were isolated from the reaction mixture after precipitation with n-hexane, subsequent washing of the solid residue with Et2O, and finally extraction into benzene followed by filtration. The 1H NMR spectra of both complexes 2 and 3 showed characteristic multiplicity-free signals in the +55 to −25 ppm range (Fig. S10 and S11†), indicative of an open-shell electronic structure. Magnetic susceptibility measurements in C6D6 solution using Evans’ method30 yielded magnetic moments μeff = 4.87 and 4.88μB for complexes 2 and 3, respectively. These values are consistent with the spin-only value (4.90μB) for a quintet (S = 2) spin system and high-spin iron(II) complexes. These data are very similar to those of complex 1 and suggest a similar coordination environment around the Fe center. Furthermore, bulk purity of both compounds 2 and 3 was deduced from the corresponding CHN combustion elemental analysis.
Furthermore, single crystals of complexes 2 and 3 were grown from concentrated THF solutions upon slow diffusion of hexane. X-ray diffraction analysis confirmed the molecular structures deduced from solution analysis (Fig. 1). Both complexes feature homoleptic geometries, each possessing two C,O-bidentate chelating phenolate-carbene ligands coordinated to the metal centre. The complexes exhibit highly distorted tetrahedral structures with τ4 = 0.79–0.80, (τ′4 = 0.77; Table 1).31,32 The bite angle of the C,O-bidentate ligands is 90 ± 1° (Table 1), with the Fe–O bond lengths consistently at 1.945(5) Å, while the Fe–C bonds oscillate around 2.06(2) Å and vary slightly more across complexes 1–3. Notably, complex 2 and 3 feature the longest Fe(II)–Ctrz bonds known thus far for a monometallic complex with 2.0607(14) and 2.0827(14) Å, respectively.33–37 This bond lengthening may be a direct consequence of the steric bulk introduced at the wingtip substituents. These ligand modifications also induce a considerable widening of the O–Fe–O angle in complex 2 (128.69(4)°) similar to 1 (126.92(5)°) while in complex 3, this angle is 114.01(4)° and thus more commensurate with the 109.5° in an ideal tetrahedral coordination geometry. The larger angles for complexes 1 and 2 may be rationalized by electrostatic O⋯O repulsion together with interligand π-stacking of the aryl N-substituents with the phenolate of the other C,O-ligand (dipp⋯phenolate 3.603(1) Ň).38 In contrast, the adamantyl substituents in complex 3 lack the potential for such interligand interactions. Instead, the enhanced 3-dimensional bulk increases the repulsion with the phenolate, which outweighs the electrostatic O⋯O repulsion of the two phenolate ligands.
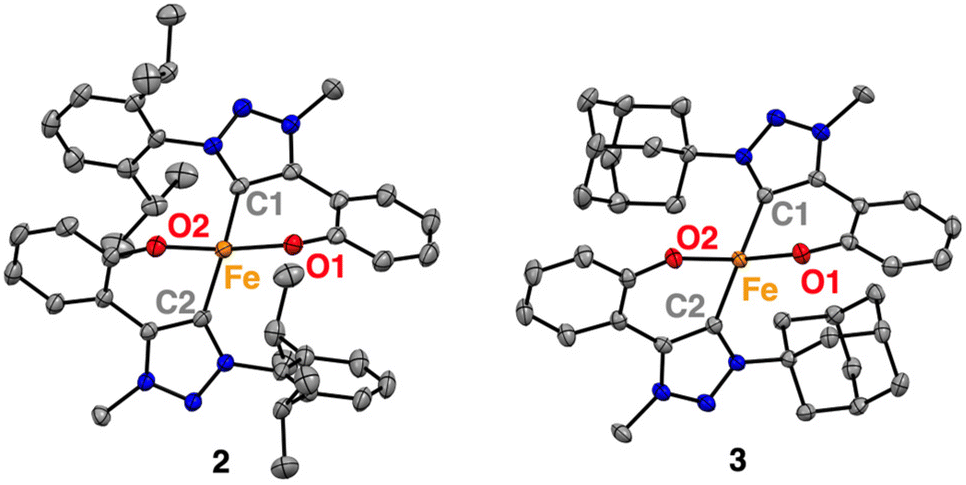 | ||
| Fig. 1 Molecular structures for complexes 2, and 3 determined by single-crystal X-ray diffraction analysis (50% probability ellipsoids, hydrogen atoms and any co-crystallised solvent molecules omitted for clarity). | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| a Data for 1 from ref. 19, with C2 = C1′ and O2 = O1′. b Calculated according to ref. 31 (τ4) and ref. 32 (τ′4). | |||
| Fe–C1 | 2.0407(12) | 2.0554(14) | 2.0827(14) |
| Fe–C2 | 2.0407(12) | 2.0607(14) | 2.0778(14) |
| Fe–O1 | 1.9474(9) | 1.9402(10) | 1.9518(10) |
| Fe–O2 | 1.9474(9) | 1.9451(10) | 1.9442(9) |
| O1–Fe–O2 | 126.91(5) | 128.69(4) | 114.01(4) |
| C1–Fe–O1 | 90.29(4) | 89.96(5) | 89.38(5) |
| C2–Fe–O2 | 90.29(4) | 90.20(5) | 89.50(5) |
| C1–Fe–O2 | 115.66(4) | 114.66(5) | 121.66(5) |
| C2–Fe–O1 | 115.66(4) | 118.09(5) | 126.92(5) |
| τ 4 (τ′4)b | 0.79 (0.78) | 0.80 (0.77) | 0.79 (0.77) |
Catalytic C–H bond amination
Complexes 2 and 3 were evaluated as catalyst precursors for the intramolecular C–H amination utilizing 4-azido-4-methyl-pentyl-benzene 4 as an organic azide substrate to afford pyrrolidine product 5 (Table 2). Complexes 2 and 3 display activity very similar to that of complex 1 and reach almost quantitative conversion after 7 ± 1 h at 1 mol% catalyst loading and 120 °C. Comparison of the pertinent time–conversion profiles reveal subtle differences (Fig. 2). Complexes 2 and 3 reach their maximum turnover frequencies (TOFmax) after ca. 3 h and thus later than complex 1 (1.5 h). Their TOFmax are slightly lower, 22 h−1 and 23 h−1 respectively, vs. 30 h−1 with complex 1, yet they all remain in the same order of magnitude (Table 2). With all complexes 1–3, the reaction proceeds to full conversion of the azide 4 according to 1H NMR spectroscopy, however the yield of pyrrolidine 5 is slightly higher for complex 2 than for 1 (95% vs. 92%) and almost quantitative for complex 3 (97%), indicating better suppression of side products such as amines and cyclic imines. | ||
| Fig. 2 Time–yield profiles for the formation of C–H aminated product 5 with complexes 1–3. Conditions: [5]0 = 447.3 mM, and [Fe] = 4.47 mM (1 mol%), toluene-d8 (0.5 mL). Product quantification by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. | ||
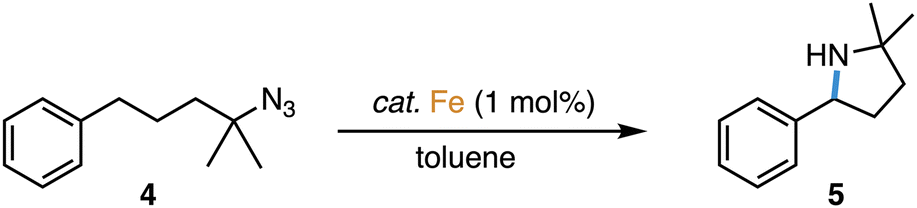
The slightly decreased activity of complexes 2 and 3 suggests an energetically elevated transition state for the rate-limiting step. Mechanistic work with complex 1 has shown this step to be N2 loss from the coordinated azide to form the metal-coordinated nitrene intermediate.19 According to this model, N2 loss should be hampered by the increased steric requirements of the dipp and Ad groups in complexes 2 and 3 compared to 1. Speculatively, this difference may induce less strong azide coordination in these complexes and hence lower the catalytic activity, though obviously, also the longer Fe–Ctrz bond may play a considerable role in increasing the Lewis acidity of the iron center. The same reasoning may rationalize the enhanced selectivity with complexes 2 and 3, as the increased steric protection limits the reactivity of the formed nitrene towards exogenous hydrogen atom sources, which would lead to terminal amine side products. Instead, the intramolecular insertion into the benzylic C–H bond is favored. According to this model, bonding of the pyrrolidine N-heterocyclic product, once formed, is sterically disfavored, thus preventing either hydrogen abstraction or dehydrogenation to form cyclic imine side products with these bulkier complexes 2 and especially 3.
The kinetics of the C–H amination reaction catalyzed by complexes 2 and 3 were evaluated by varying the catalyst and initial substrate concentrations. Modulation of the loading of the iron complex 2 from 0.5, 0.75, 1, and 2 mol% provided quantitative conversions and product yields that remained at 95%. The time to reach full conversion increased to almost 14 h for the 0.5 mol% catalyst loading (Fig. S20†). For complex 3, the same variation had a moderate impact on the selectivity, with 90% yield at 0.5 mol% catalyst loading, 97% at 1 mol%, and >99% of pyrrolidine 5 at 2 mol% in just 6 h (Fig. 3). This essentially quantitative selectivity toward pyrrolidine formation indicates an efficient suppression of any side reactions such as undesired intermolecular hydrogen atom abstraction (Table 3).
 | ||
| Fig. 3 Kinetic profile of the C–H amination with complex 3 at varying concentrations of [3] = 2.24–8.94 mM, [4]0 = 447 mM, in toluene-d8. Product quantities determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as internal standard. Inset shows rate dependence on catalyst concentration and reveals 0.5 order rate dependence in complex (Ratemax in mM min−1; R2 = 0.993). | ||
| Entry | Fe | Conc./mM | Yieldb | Time/h | TOFmax/h−1 | |
|---|---|---|---|---|---|---|
| [Fe] | [4]0 | |||||
| a For general conditions, see Table 2. b Yields determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as internal standard. | ||||||
| 1 | 2 | 4.47 | 447 | 95% | 8 | 22 |
| 2 | 3 | 4.47 | 447 | 97% | 7 | 23 |
| 3 | 2 | 2.24 | 447 | 96% | 13.5 | 26 |
| 4 | 3 | 2.24 | 447 | 90% | 8.5 | 38 |
| 5 | 2 | 8.94 | 447 | 95% | 8 | 12 |
| 6 | 3 | 8.94 | 447 | >99% | 6 | 17 |
| 7 | 2 | 4.47 | 224 | 93% | 12.5 | 7 |
| 8 | 3 | 4.47 | 224 | 92% | 9.5 | 6 |
| 9 | 2 | 4.47 | 894 | 95% | 7.5 | 45 |
| 10 | 3 | 4.47 | 894 | >99% | 7 | 51 |
The time–yield profiles reveal for both complexes consistently a significant induction period of around 30–60 min (cf.Fig. 2). Moreover, the maximum rate of the reaction increases with increasing catalyst concentration, though the correlation is not linear and instead points to a half-order rate dependence in catalyst concentration for both complexes (inset Fig. 3 and S16†). This conclusion was also supported by the strong correlation of a variable time normalization analysis (VTNA)39 when assuming a 0.5 order in catalyst rather than a first-order dependence (Fig. S24–S28†). This dependence corroborates the mode of operation established for complex 1,19 and it therefore reinforces the significance of a dimeric species as catalyst resting state.
Similar variation of the substrate concentration gave full conversion at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 1:100, 1:150, and 1:200 complex/substrate ratios, albeit at longer reaction time for lower substrate concentrations (Table 3 and Fig. 4, S22, S23†). For example, at 1:50 ratio, >12 h were required to reach 93% yield with complex 2, while at a 1:200 catalyst/substrate ratio, 95% yield were accomplished in less than 8 h. Again, the reaction stoichiometry impacted the yield considerably more with complex 3, than the other two complexes. At low substrate concentration, the yield of pyrrolidine 5 leveled at only 92%, while at highest measured 0.9 M substrate concentration, full selectivity to the desired product was observed with >99% yield of 5. Notably, the changes of maximum rates correlate linearly with initial substrate concentrations, indicative of a first-order rate dependence with respect to substrate (Fig. 4 and Fig. S22, S23†).
:50, 1:100, 1:150, and 1:200 complex/substrate ratios, albeit at longer reaction time for lower substrate concentrations (Table 3 and Fig. 4, S22, S23†). For example, at 1:50 ratio, >12 h were required to reach 93% yield with complex 2, while at a 1:200 catalyst/substrate ratio, 95% yield were accomplished in less than 8 h. Again, the reaction stoichiometry impacted the yield considerably more with complex 3, than the other two complexes. At low substrate concentration, the yield of pyrrolidine 5 leveled at only 92%, while at highest measured 0.9 M substrate concentration, full selectivity to the desired product was observed with >99% yield of 5. Notably, the changes of maximum rates correlate linearly with initial substrate concentrations, indicative of a first-order rate dependence with respect to substrate (Fig. 4 and Fig. S22, S23†).
 | ||
| Fig. 4 Kinetic profile of the C–H amination catalyzed by complex 3 at varying substrate concentrations. Conditions: [3] = 4.47 mM, [4]0 = 224–894 mM, toluene-d8. Product quantities determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as internal standard. Inset shows rate dependence on substrate concentration and reveals first order rate dependence in substrate (Ratemax in mM min−1, R2 = 0.997). | ||
The coherent appearance of an induction period, and the kinetic profile, especially the first-order dependence in substrate and the half-order in catalyst indicate that complexes 1–3 operate according to a common mechanism that is general for these types of iron carbene complexes. These data therefore reinforce the formation of a bimetallic catalyst resting state and a rate limiting step that involves this dimer and the substrate. While originally we proposed an azide- or nitrene-bridged dimer, one might also speculate about other dimeric species. For example, carbene–iron bond dissociation may be plausible when considering the high substitutional lability of iron(II) paired with the scattered observations of carbene dissociation as catalyst activation pathways.40–42 Such a scenario would also be consistent with the observed induction time as the strongly bound chelating ligand is expected to require significant rearrangement for being cleaved from the iron center.43
Conclusions
This work demonstrates that the catalytic C–H bond amination reactivity of iron triazolylidene complexes is directly affected by ligand modulations. Specifically, increasing bulk on the triazole N-substituent enhances the selectivity of the catalyst, accomplishing up to 99% yield of the pyrrolidine and thus suppressing efficiently side reactions that are typically observed in C–H amination such as the formation of terminal amines through nitrene quenching. Structural analysis of the complexes suggests that the smaller pocket available for substrate coordination reduces the propensity for intermolecular H atom abstraction, and thus maximizes the intramolecular reaction. At the same time, this smaller pocket also rationalizes the slightly lower activity when using bulkier ligands, as azide bonding is surmised to be weaker. These structure–activity trends indicate that the iron center remains ligated under catalytic conditions. In addition, the half-order rate dependence in iron complex is a common feature for this class of catalysts, suggesting a prominent role of dimers as catalyst resting states. These insights offer opportunities for further ligand modifications to rationally enhance catalytic activity.Experimental section
General
Complex 1,19 2-((trimethylsilyl)ethynyl)phenol,44 substrate 4,1 and organic azides dipp-N3,45 and Mes-N346 were synthesised according to previously reported procedures. The synthesis of ligands L2 and L3 is described in the ESI.† All other reagents were commercially available and used as received unless stated otherwise. All manipulations involving the handling of transition metal complexes were performed in a MBraun glovebox with <0.1 ppm O2 and H2O levels using dry and degassed solvents. Benzene, hexane, pentane and diethyl ether were taken from an MBraun SPS system, degassed by three freeze–pump–thaw cycles and dried with 4 Å molecular sieves before use. THF was distilled from sodium benzophenone ketyl radical, degassed by three freeze–pump–thaw cycles and dried over 4 Å molecular sieves. THF-d8, C6D6 and toluene-d8 were distilled over NaK, degassed over three freeze–pump–thaw cycles and dried over 4 Å. Molecular sieves were pre-dried in a 1000 W microwave for 10 minutes, in 30 seconds intervals and subsequently dried under vacuum for 3 days. All organic syntheses were performed under aerobic conditions with commercially available solvents unless stated otherwise. All other chemicals were used as received without further purification from commercial sources. All 1H, 13C and 19F NMR spectra were recorded on a Bruker AVANCE III HD 300 or a Bruker AVANCE III HD 400 at room temperature. The chemical shifts are reported relative to SiMe4 using the chemical shift of residual solvent signals as reference. Mass spectrometric analyses were performed on a LTQ Orbitrap XL (Thermo Scientific) high resolution mass spectrometer, equipped with a static nano electrospray ion source using Econo12 platinated quartz emitters (New Objective Inc.). Determination of contents of carbon, hydrogen and nitrogen was performed on a Thermo Scientific Organic Elemental Analyzer. Air and moisture sensitive samples were sealed in Santis tin capsules for liquids (2.9 × 6 mm) inside an argon filled glovebox, taken outside the glovebox and measured directly.
General synthetic procedure of complexes
In an argon-filled glovebox the solid ligand (2.0 eq.) was suspended in THF (2 mL) and cooled to −30 °C. A pre-cooled solution (−30 °C) of KHMDS (2.0 eq.) in THF (2 mL) was added dropwise to the suspension of ligand and stirred for 1 h at room temperature and subsequently cooled to −30 °C. A pre-cooled solution (−30 °C) of Fe(HMDS)2 (1.0 eq.) in THF (2 mL) was added dropwise to the ligand/KHMDS mixture and stirred for 18 h at room temperature. The reaction mixture was concentrated to approximately 1 mL and hexane (10 mL) was added. The resulting orange precipitate was collected by filtration, washed with Et2O (3 × 5 mL) and extracted into benzene. The resulting solution was freeze-dried and washed with pentane (5 × 5 mL) to obtain the title complexes.Complex 2
According to the general method, L2 (400 mg, 0.82 mmol), KHMDS (164 mg, 0.82 mmol) and Fe(HMDS)2 (155 mg, 0.41 mmol) afforded complex 2 as a bright orange powder (78 mg, 26%). Single crystals suitable for XRD analysis were obtained by laying a concentrated THF solution of the complex with hexane. 1H NMR (300 MHz, C6D6): δ = 47.7 (brs), 47.0 (brs), 18.0 (brs), 5.1 (brs), −1.9 (brs), −7.9 (brs), −22.6 (brs); μeff (C6D6, 299 K): 4.87μB; elemental analysis calcd for C42H48FeN6O2 (724.73 g mol−1): C 69.61, H 6.68, N 11.60%. Found: C 69.97, H 6.81, N 11.21%.Complex 3
According to the general method, L3 (200 mg, 0.44 mmol), KHMDS (87 mg, 0.22 mmol) and Fe(HMDS)2 (82 mg, 0.22 mmol) yielded complex 3 as a bright orange powder (77 mg, 53%). Single crystals suitable for XRD analysis were obtained by laying a concentrated THF solution of 3 with hexane. 1H NMR (300 MHz, THF-d8): δ = 54.2 (brs), 51.0 (brs), 18.4 (brs), −0.1 to −1.8 (m), −14.7 (brs); μeff (C6D6, 299 K): 4.88μB; elemental analysis calcd for C38H44FeN6O2 (672.66 g mol−1): C 67.85, H 6.59, N 12.44%. Found: C 67.59, H 6.77, N 12.53%.General catalytic procedure
Inside an argon filled glovebox, a stock solution of complex was prepared by dissolving a known amount of iron complex in toluene-d8 (2 mL). A stock solution of standard was prepared by dissolving 1,3,5-trimethoxybenzene (45.5 mg. 0.0271 mmol) in toluene-d8 (1.0 mL). The azide 4 (25, 50, 75, or 100 mg) was weighed into a vial, portions of the iron complex stock solution (0.2 mL), internal standard solution (0.1 mL), and toluene-d8 (0.2 mL) were added. This mixture was transferred into an oven-dried J Young NMR tube, transported out of the glove box, and heated in an oil bath at 120 °C. Conversion was measured at given time intervals after removing the NMR tube from the oil bath and cooling it in an ice bath before measuring a 1H NMR spectrum. Times listed are the cumulative amount of time in the oil bath at the specified temperature. Yields were determined by 1H NMR analysis relative to the internal standard following the proton signal of the benzylic hydrogens of 4 at 2.28 ppm (toluene-d8) for substrate and the appearance of a quartet at 4.03 ppm (toluene-d8) corresponding to the cyclized product 5 (Fig. S18 and S19†). Lower catalyst loadings were achieved by appropriate dilution of the complex stock solution to maintain a constant reaction volume.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for complexes 2 and 3 have been deposited at the CCDC under 2361237 and 2361238.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the X-ray crystal structure determination service unit of the University of Bern for their work and the Swiss National Science Foundation for generous financial support (grants 200020_212863, 206021_177033).Notes and references
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed.
- N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703–2802 CrossRef CAS PubMed.
- J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- Y. Baek and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed.
- Y. Dong, R. M. Clarke, G. J. Porter and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 10996–11005 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed.
- P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed.
- J. Qin, Z. Zhou, T. Cui, M. Hemming and E. Meggers, Chem. Sci., 2019, 10, 3202–3207 RSC.
- M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed.
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., 2017, 129, 15805–15808 CrossRef.
- B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117–5124 CrossRef CAS PubMed.
- Y.-D. Du, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Org. Lett., 2019, 21, 895–899 CrossRef CAS PubMed.
- Z.-Y. Qin, S. Gao, Y. Zou, Z. Liu, J. B. Wang, K. N. Houk and F. H. Arnold, ACS Cent. Sci., 2023, 9, 2333–2338 CrossRef CAS PubMed.
- D. Possenti and G. Olivo, ChemCatChem, 2024, 16, e202400353 CrossRef.
- S. Roy, D. A. Vargas, P. Ma, A. Sengupta, L. Zhu, K. N. Houk and R. Fasan, Nat. Catal., 2024, 7, 65–76 CrossRef CAS PubMed.
- T. You, S.-H. Zeng, J. Fan, L. Wu, F. Kang, Y. Liu and C.-M. Che, Chem. Commun., 2021, 57, 10711–10714 RSC.
- W. Stroek, M. Keilwerth, D. M. Pividori, K. Meyer and M. Albrecht, J. Am. Chem. Soc., 2021, 143, 20157–20165 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- G. Rigoni, P. V. S. Nylund and M. Albrecht, Dalton Trans., 2023, 52, 7992–8002 RSC.
- W. Stroek and M. Albrecht, Chem. Soc. Rev., 2024, 53, 6322–6344 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- C. Johnson and M. Albrecht, Organometallics, 2017, 36, 2902–2913 CrossRef CAS.
- Y. Liu, K. S. Kjær, L. A. Fredin, P. Chábera, T. Harlang, S. E. Canton, S. Lidin, J. Zhang, R. Lomoth, K.-E. Bergquist, P. Persson, K. Wärnmark and V. Sundström, Chem. – Eur. J., 2015, 21, 3628–3639 CrossRef CAS PubMed.
- P. V. S. Nylund, N. C. Ségaud and M. Albrecht, Organometallics, 2021, 40, 1538–1550 CrossRef CAS.
- P. Dierks, A. Kruse, O. S. Bokareva, M. J. Al-Marri, J. Kalmbach, M. Baltrun, A. Neuba, R. Schoch, S. Hohloch, K. Heinze, M. Seitz, O. Kühn, S. Lochbrunner and M. Bauer, Chem. Commun., 2021, 57, 6640–6643 RSC.
- W. Stroek, N. A. V. Rowlinson, M. Keilwerth, D. M. Pividori, K. Meyer and M. Albrecht, Organometallics, 2024, 43, 1386–1392 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, A. Yu. Chernenko, A. V. Astakhov, D. B. Eremin, D. V. Pasyukov, A. S. Kashin and V. P. Ananikov, Chem. Sci., 2018, 9, 5564–5577 RSC.
- V. M. Chernyshev, E. A. Denisova, D. B. Eremin and V. P. Ananikov, Chem. Sci., 2020, 11, 6957–6977 RSC.
- N. Ségaud, C. Johnson, A. Farre and M. Albrecht, Chem. Commun., 2021, 57, 10600–10603 RSC.
- The use of azides that cannot undergo intramolecular amination such as adamantyl-N3 led to a mixture of products that was intractable in our hands and therefore did not add to any mechanistic understanding with these compounds.
- A. Arcadi, S. Cacchi, M. Del Rosario, G. Fabrizi and F. Marinelli, J. Org. Chem., 1996, 61, 9280–9288 CrossRef CAS.
- S.-K. Chen, W.-Q. Ma, Z.-B. Yan, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, X.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2018, 140, 10099–10103 CrossRef CAS PubMed.
- D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 135, 12354–12357 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of the ligand precursors, analytical data of the complexes, catalytic and crystallographic details. CCDC 2361237 (2) and 2361238 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01715c |
| ‡ Stacking distance was calculated using Olex2-1.5 software by creating a plane between the phenolate ring connected to O2 and the dipp ring connected to C1 and using the ‘esd’ tool to calculate the distance between the centroids. |
| This journal is © The Royal Society of Chemistry 2024 |