
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of stereochemistry in the anticancer activity of Re(I) tricarbonyl complexes†
Atena B.
Solea
 *a,
Gozde
Demirci
b,
Freya M.
Harvey
a,
Aurelien
Crochet
b,
Fabio
Zobi
*b and
Olimpia
Mamula Steiner
*a
*a,
Gozde
Demirci
b,
Freya M.
Harvey
a,
Aurelien
Crochet
b,
Fabio
Zobi
*b and
Olimpia
Mamula Steiner
*a
aUniversity of Applied Sciences of Western Switzerland, HES-SO, HEIA-FR, Pérolles 80, CH-1705 Fribourg, Switzerland. E-mail: olimpia.mamulasteiner@hefr.ch
bUniversity of Fribourg, Department of Chemistry, Chemin du Musée 9, CH-1700 Fribourg, Switzerland
First published on 12th July 2024
Abstract
Cancer is a leading cause of death worldwide, accounting for about one among six deaths, so the quest for new and improved therapies is of crucial importance. The discovery of cisplatin as an anticancer agent has paved the way for the development of other metal-based therapeutic agents and Re(I)-based candidates have been recently found to show promising results. It is known as well that chirality plays a central role in the interactions of metal-based drugs with intrinsically chiral biomolecules such as membrane transport proteins or DNA. To further exploit this property, we have developed a series of diastereomeric dinuclear Re(I) complexes with chiral ligands containing pinene-bipyridine units. These complexes offer unique insights into the relation between stereochemistry and biological activity. Single-crystal X-ray diffraction studies, spectroscopic analysis, including UV-Vis and circular dichroism (CD), confirmed the chiral structures of these complexes. Biological activity assessments were carried out against various cancer cell lines, with a particular focus on breast and colon cancer. The diastereomers exhibited distinct anticancer activities, with some displaying promising results. Notably, one diastereomer showed exceptional cytotoxicity against HCT116 and MCF-7 cancer cells. This research underscores the significance of chirality in the design of novel anticancer agents, providing insights into the potential of dinuclear Re(I) complexes as effective candidates for cancer treatment.
Introduction
The serendipitous discovery of cisplatin as a chemotherapy agent back in 19651 was a major breakthrough in the development of anticancer treatments. To date, this compound and its Pt(II) analogues are still used as a first line treatment drug for several cancers, but they come with the drawback of serious side effects and the risk of acquired resistance.2 Nonetheless, metal complexes are important anticancer candidates due to their ability to interact with biomolecules involved in cancer. Their intrinsic flexibility in terms of number and coordination geometry and preferred interaction with certain types of atoms (hard or soft Lewis bases) can lead to targeted delivery. Due to the plethora of interactions they can share, (i.e. electrostatic, coordinative binding, supramolecular), in recent years, noteworthy candidates based on copper, gallium, ruthenium or gold have been obtained.3 More recently, Re(I)-based complexes with a fac-tricarbonyl core have been investigated for the same purpose, and have shown highly promising activities.4–12 Since then, the field has been rapidly expanding.13–20 An important feature to note is that if the three remaining coordinative positions bound to the fac-Re(I) tricarbonyl core are non-equivalent, the Re(I) complexes are intrinsically chiral. This intrinsic chirality was mostly ignored in the biological studies of fac-Re(CO)3+ complexes. Considering the central role played by chirality in the living systems (quoting Albert Eschenmnoser: “Chirality is the signature of life”) we believe it deserves a closer inspection. Only a handful of studies so far have focused on the synthesis and diastereomeric separation of Re(I) tricarbonyl complexes with chiral ligands for biological applications,19,21–23 and even fewer have focused on a systematic study of the anticancer activity of the stereoisomers.24 We have recently started investigating in depth the influence of chirality on the biological activity of chiral fac-Re(CO)3+ with bipyridine, and the very promising results25 motivated us to further explore this field on chiral dinuclear complexes. This approach doubles the number of active sites compared to mononuclear complexes, which could improve their potency. Moreover, the biological activity, and by default, the cellular uptake of Re(I) complexes is correlated with their lipophilicity.26–28 We have previously shown how enantiopure pinene-annealed compounds can have useful reactivity,29 as well as interesting self-assembly30 and sensing31 applications. Most importantly, they can induce chirality in a variety of metal complexes for tuning the properties of the final compounds.32,33 For our purpose, annealing a chiral pinene unit onto diimine ligands has a dual role: providing a chirality element and increasing the lipophilicity of the final complex. We synthesised both enantiomers of the chiral ligands 5 and 6 (Fig. 1), which possess a bis-bipyridine skeleton and two pinene units, and we prepared the corresponding dinuclear complexes with the formula [Re2(CO)6(L)Cl2] in which the bis-bidentate ligand L bridges the two metal centres. For each enantiopure ligand, two sets of major diastereomers were isolated, which differ through the relative orientations of the Cl atoms and the methylene bridge of the pinene units. Yeung and co-workers also employed the ligand 5a for the formation of dinuclear Re(I) complexes, but only isolated one of the diastereomers.34 For each enantiopure ligand, two diastereomers were isolated and fully characterised by spectroscopic means, as well as single crystal X-ray diffraction. Further, different cytotoxicity values of each stereoisomer were identified, emphasizing the impact of chirality on the biological activity of the molecules. Two candidates displayed promising activities against breast and colon cancer. | ||
| Fig. 1 Structures of the four ligands used in this study with numbering scheme for the non-equivalent atoms. | ||
Results and discussion
Synthesis of chiral ligands and dinuclear Re(I) complexes
The synthetic route for the formation of ligands 5a–b, 6a–b involves a classic procedure developed in our group, starting from the previously reported Kröhnke salts 1 and 2 and reacting them with the two enantiomers of pinocarvone. This leads to the formation of enantiopure, brominated precursors 3a–b and 4a–b, which then undergo Ni(0)/ZnCl2-assisted coupling,26 to give the final enantiopure ligands 5a–b and 6a–b (Scheme 1).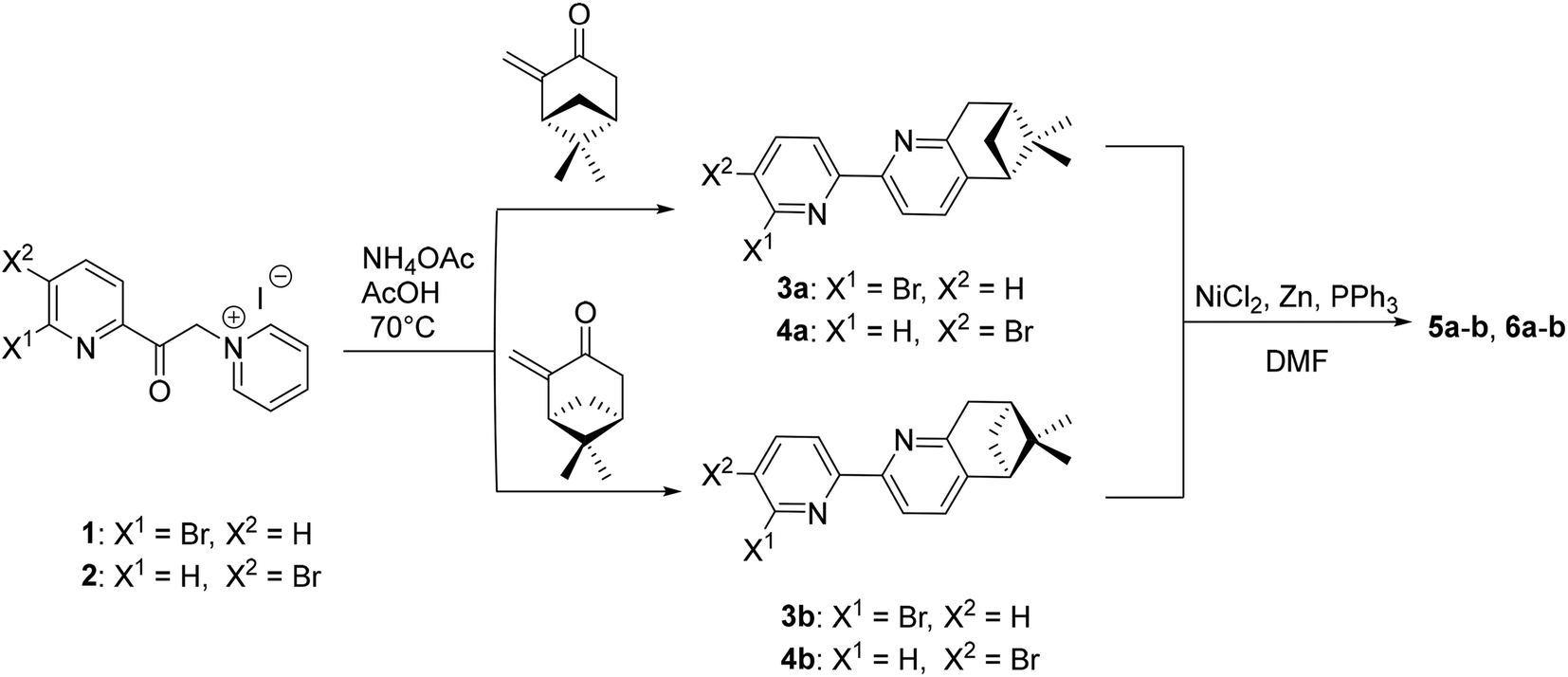 | ||
| Scheme 1 Synthetic route for the formation of the ligands. | ||
All ligands were fully characterised by NMR, UV-Vis, CD and HRMS. Single crystals of 6a and 6b were obtained through slow evaporation from acetone solutions, and their structures were determined by single-crystal X-ray diffraction (Fig. 2). The ligand 6a crystallises in the P21 (4) space group with one molecule in the asymmetric unit. Each bipyridine unit is, as expected, in relative trans conformation, giving a zig-zag arrangement of the N atoms in the ligand. The pyridine units adopt almost coplanar arrangements, with angles of 14.39°, 24.21°, respectively 4.86° between adjacent pyridine rings. Similarly, its enantiomer, 6b crystallises in the same space group with one molecule in the asymmetric unit and with the same zig-zag arrangement of the pyridine units. Similar angles are also observed between the adjacent pyridine rings, with angles of 13.70°, 24.26° and 4.77°.
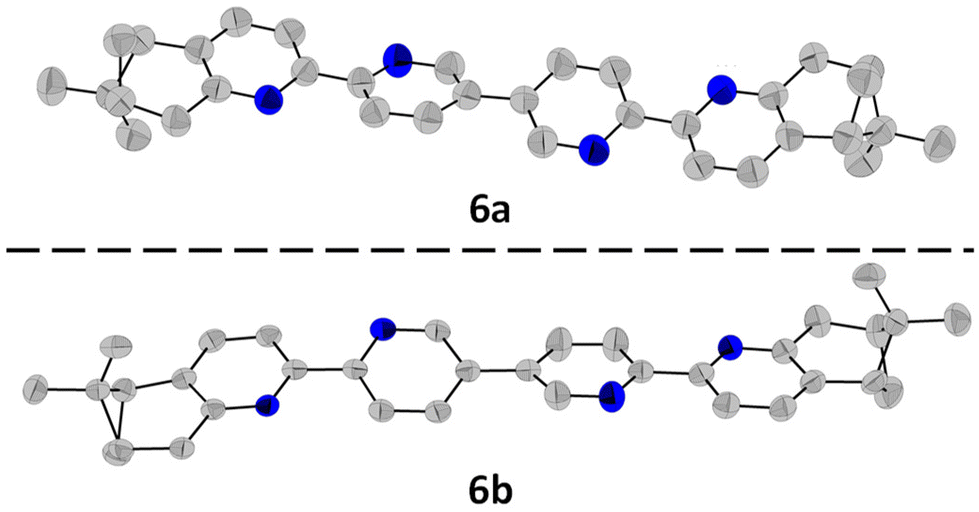 | ||
| Fig. 2 Molecular structures of 6a and 6b determined by single-crystal X-ray diffraction, with the ellipsoids at 50% probability. The H atoms were omitted for clarity. | ||
Further, the dinuclear Re(I) complexes with the general formula [Re2(CO)6(L)Cl2] (L = 5a–b, 6a–b) were synthesised following a modified literature procedure,34 by reacting 1 eq. L with 2.1 eq. Re(CO)5Cl under inert atmosphere, in a mixture of anhydrous toluene and THF (4/1 v/v), at 85 °C, for 16 h (Scheme 2).
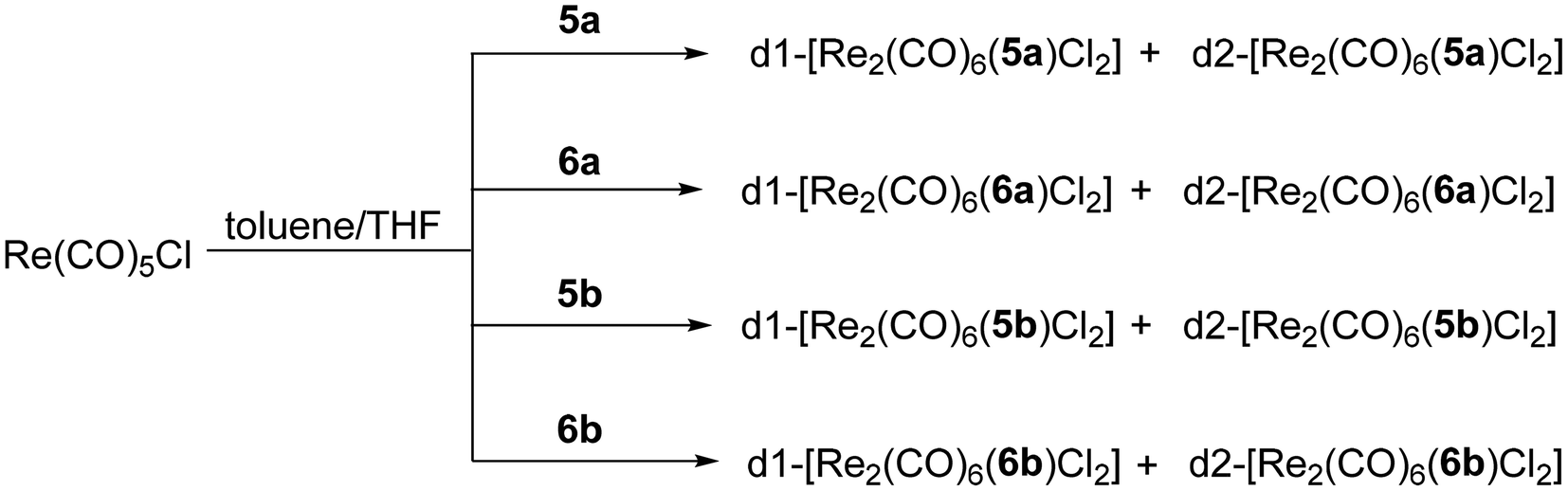 | ||
| Scheme 2 Synthesis of the chiral complexes. | ||
The reported diastereomers could be isolated and fully characterised following column chromatography purification. This separation step however led to significant losses due to decomposition on silica, explaining the modest yields obtained. Stacked 1H NMR spectra of the complexes obtained with ligands 5a and 6a can be observed in Fig. 3. Considering the relative orientation of the Cl ligand and the methyl groups of the pinene moiety belonging to the bipy unit coordinated to the same metal centre, there are several possible diastereomers, but in all cases the presence of two major diastereomers was observed. We assume that this is due to the extra stabilization provided through both intra and intermolecular Cl⋯H–C interactions.34 Nonetheless, these interactions are not producing significant shifts in the 1H NMR spectra of any of the diastereomers. For example, d1-[Re2(CO)6(5a)Cl2] and d2-[Re2(CO)6(5a)Cl2] show similar shifts for the Me units in the pinene unit (1.45/0.74 ppm, respectively 1.47/0.74 ppm vs. 1.44/0.71 ppm for 5a), despite the fact that d1-[Re2(CO)5(5a)Cl2] presents several intramolecular interactions between the Me and the Cl atom and d2-[Re2(CO)5(5a)Cl2] does not (see ESI, Fig. S9–S12†). In solid state, the complexes present two strong bands in IR (Fig. S45–S52†), at ∼2015 and ∼1880 cm−1, which are characteristic to the symmetric and asymmetric stretching of the coordinated CO units.
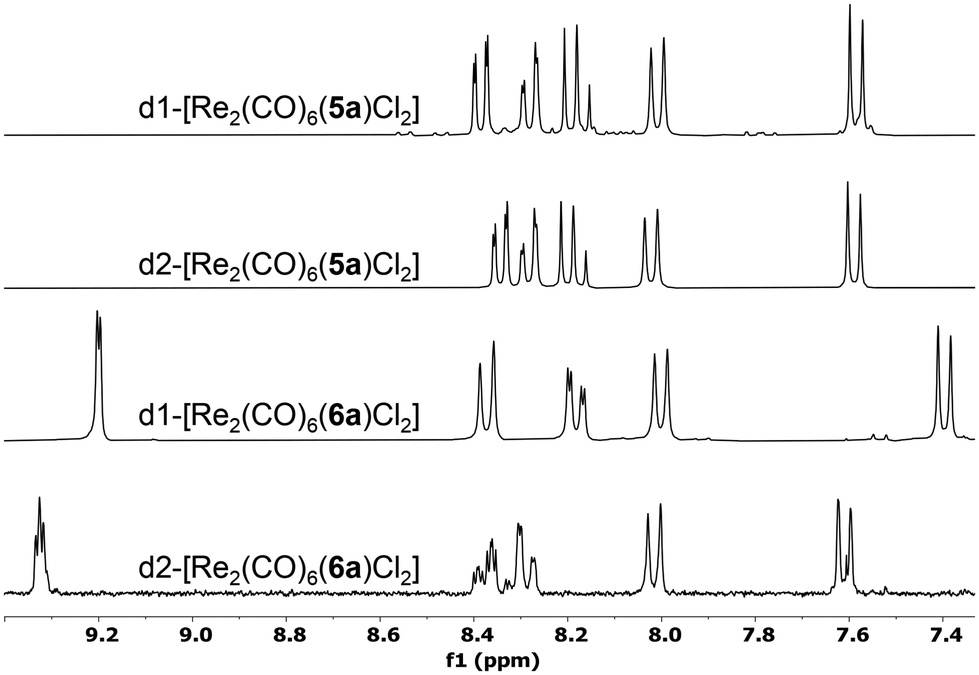 | ||
| Fig. 3 Stacked 1H NMR spectra of the diastereomeric pairs obtained with 5a, respectively 6a, in CDCl3. | ||
Single-crystal X-ray diffraction
All diastereomers were successfully characterised by single-crystal X-ray diffraction, revealing the intricate structures that these chiral dinuclear complexes adopt in the solid state, as well as several noteworthy intra- and intermolecular interactions. The X-ray structures of d1-[Re2(CO)6(5)Cl2] and d2-[Re2(CO)6(5)Cl2] with each enantiomer of the ligand are shown in Fig. 4. All complexes adopt a single-stranded helical arrangement in a diastereomer/enantiomer relationship to each other, as proven by their structures and CD spectra. All the Re(I) centres adopt a fac configuration, as expected. Each enantiopure ligand forms a mixture of two diastereomers which can be distinguished through the relative position of the Cl atoms, as well as the relative position of the Cl atoms and the methyl groups of the pinene unit leading to helicates with P or M helicity. The P/M chiral descriptors were assigned following the revisited principles made in the review about helicates by Piguet et al.35 common feature of all isolated complexes is the presence of intermolecular C–H⋯Cl electrostatic interactions which play an important role in stabilizing the helical structure of the complexes. | ||
| Fig. 4 Single-crystal X-ray diffraction structures of the four stereoisomers obtained with the ligands 5a and 5b, depicting the enantiomeric pairs. The H atoms were removed for clarity. | ||
For instance, d1-[Re2(CO)6(5a)Cl2] crystallizes in the P212121 (19) space group, with one molecule in the asymmetric unit. The complex adopts a helical shape with a M configuration, with a torsion angle of −91.1° between the two bipy units, while the two chlorine atoms are found on opposite (anti) axial positions. It is important to note the relatively small distance between the Cl atoms and a methyl H atom from the pinene unit, similar to what Yeung and co-workers have identified for the brominated analogue.34 In our case, these intramolecular distances are 3.502, respectively 3.474 Å and this is because both Cl atoms are on the same side to the methyl groups. Intermolecular electrostatic interactions between the Cl atoms and aromatic Hs are also observed, with Cl⋯H distances of 2.625, 2.684, respectively 2.609, 2.726 Å (Fig. S65†). Similarly, its enantiomer, d1-[Re2(CO)6(5b)Cl2] crystallizes in the P212121 (19) space group adopting a P helical configuration, with a torsion angle of +82.8° between the two bipy units and the Cl atoms again on opposite axial positions. The Cl atoms display short contacts of 3.806, respectively 3.838 Å with H atoms from the Me groups of the neighbouring pinene unit. Intermolecular electrostatic interactions between the Cl atoms and aromatic H are also observed, with Cl⋯H distances of 2.671, 2.775, 3.808 and 3.9882. Å (Fig. S66†).
The diastereomer d2-[Re2(CO)6(5a)Cl2] crystallizes with one molecule in the asymmetric unit in the P21 (4) space group. The molecule adopts a P helical structure. In this case, the torsion angle between the central pyridine units is +73.7°, while the Cl atoms adopt a relative syn orientation. More importantly, no intramolecular interactions between the Cl atoms and the Me units of the pinene ring are possible, as in this case they are on opposite sides. One short contact is observed between Cl2 and H7 from a neighbouring pyridine ring, at 2.725 Å (Fig. S67†). Its enantiomer, d2-[Re2(CO)6(5b)Cl2] similarly crystallizes in the P21 (4) space group and adopts an M helical structure. The torsion angle is −72.6° and again one intermolecular interaction is observed between one Cl atom and a neighbouring aromatic H atom, at a distance of 3.592 Å (Fig. S68†).
Four diastereomers of [Re(CO)6(6)Cl2] were also characterized by single-crystal X-ray diffraction (Fig. 5). Similar to the complexes with the enantiomers of ligand 5, we also obtained two pairs of enantiomers for ligand 6. However, the enantiomers of ligand 6 are not helically wrapped around the two metal centres, but bridge them in a linear (ladder-like) fashion. The four donor nitrogen atoms are almost linearly arranged (vide infra). d1-[Re2(CO)6(6a)Cl2] crystallizes in the C2221 (20) space group and half of the molecule was generated by symmetry. The two bipyridine units are almost coplanar, with the angle between the two central pyridine rings of only 4.2° and with a torsion angle of only −1.8°. The two Cl atoms have a relative anti arrangement, and each is on the same side as the Me groups of the corresponding pinene units. The distance between the Cl atoms and the closest methyl H is 4.000 Å. The Cl atom again interacts with a neighbouring molecule through two electrostatic Cl⋯H–Carom interactions of 2.950, respectively 2.730 Å (Fig. S69†). Its enantiomer, d1-[Re2(CO)6(6b)Cl2] crystallizes in the same space group, C2221 (20), with half of the molecule forming the asymmetric unit and with a torsion angle of −13.8°. The Cl–H(Me) distance is 4.068 Å, and the intermolecular Cl⋯H–Carom interactions show distances of 3.019 and 2.878 Å (Fig. S70†).
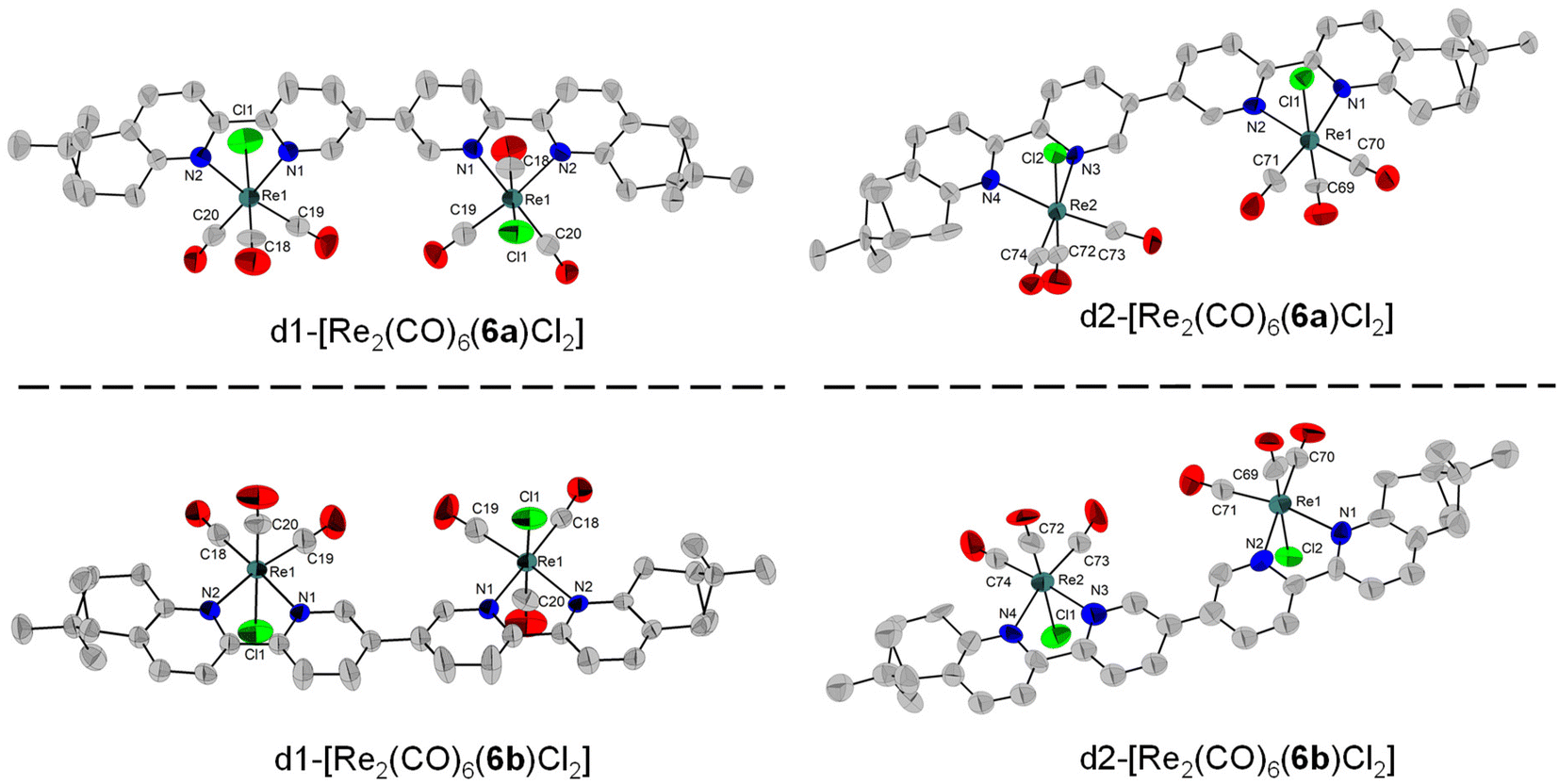 | ||
| Fig. 5 Single-crystal X-ray diffraction structures of the four stereoisomers obtained with the ligands 6a and 6b, depicting the enantiomeric pairs obtained. The H atoms were removed for clarity. | ||
d2-[Re2(CO)6(6a)Cl2] and d2-[Re2(CO)6(6b)Cl2] both crystallize with two molecules in the asymmetric unit, in the P1 (1) space group. In Fig. 5, one molecule from each enantiomer is shown. For d2-[Re2(CO)6(6a)Cl2], the torsion angles are 30.2° and 51.9°, respectively for each molecule in the asymmetric unit. The Cl atoms are in a relative syn orientation. One Cl atom is on the same side as the methylene bridge of the pinene unit, whereas the second one is on the same side as the two Me groups of the pinene unit, with intramolecular Cl⋯H–C distances of 4.318, respectively 4.910 Å. Short intermolecular Cl⋯H–C contacts are observed, ranging from 2.650 to 2.921 Å (Fig. S71†). As expected, the same arrangement is observed for d2-[Re2(CO)6(6b)Cl2], with a similar torsion angle of −28.6° and −77.7°, respectively for each molecule in the asymmetric unit. Intra- and intermolecular interactions of the type Cl⋯H–C are observed for both molecules in the asymmetric unit, with intermolecular Cl1⋯H–C, respectively Cl3⋯H–C distances of 4.242 Å and 4.202 Å between the Cl atoms and the methyl H from the pinene. Several short intermolecular Cl–H–C interactions are observed for both molecules in the asymmetric unit, with distances ranging from 2.653 to 2.931 Å (Fig. S72†).
Spectroscopical characterization
The solution behaviour of the complexes was further characterised spectroscopically, by UV-Vis, CD and fluorescence spectroscopy. As expected, the UV-Vis spectra in CH2Cl2 (Fig. 6, top) of all the diastereomers formed with ligands 5a/b are almost superposable, with a maximum of absorption at 345 nm and similar extinction coefficients. A similar trend is observed for the complexes formed with ligands 6a/b as well, which has a maximum of absorption at 357 nm. The emission and excitation spectra of the complexes formed with ligands 5a/b show maxima of emission around 600 nm (see ESI, Fig. S55–S62†), with an excitation maximum at 342 nm. The complexes formed with the ligands 6a/b emit at a slightly higher wavelength, ∼620 nm, with an excitation maximum at 350 nm for all the complexes. Differences are observed however in the quantum yields of the complexes. Despite the modest values obtained for all the complexes, the family of complexes with ligands 5a/b has substantially higher QY, between 3 and 5%, whereas the complexes obtained with ligands 6a/b all show QY values of 0.6% (Table S1†). | ||
| Fig. 6 UV-Vis (top) and CD (bottom) spectra of the complexes in CH2Cl2 (C = 0.005 mM). | ||
CD spectroscopy further confirmed the enantiomeric nature of the obtained complexes. As can be seen in Fig. 6, the CD spectra of each enantiomeric pair are mirror images of each other. Each ligand led to two diastereomers. The d1 has the two Cl in anti-conformation and each Cl is situated on the side of the methyl groups belonging to the pinene bipyridine coordinating the same metal centre. In the d2 diastereomer the two Cl atoms are in syn conformation, and each Cl is on the side with the methylene bridge belonging to the pinene bipyridine coordinating the same metal centre. For each diastereomer as defined before, the use of enantiomeric ligands (5a/5b or 6a/6b) produces mirror imaged CD spectra. Moreover, the CD activity of the monostranded helical complexes obtained with the ligands 5a/5b is superior to the one showed by the non-helical, “ladder-like” ones obtained with the ligands 6a/6b. This can be readily observed in the CD spectra where for example, a negative Cotton effects is observed for d1-[Re2(CO)6(5a)Cl2] (M helicate) and a positive Cotton effect for the P enantiomer d1-[Re2(CO)6(5b)Cl2]. As expected, for the other enantiomeric forms d2-[Re2(CO)6(5a)Cl2] and d2-[Re2(CO)6(5b)Cl2], complementary CD spectra with a mirror symmetry are obtained, confirming their enantiomeric relationship. It is noteworthy that the same enantiomeric form of ligand 5 led to opposite helicities in d1 and d2, i.e., M for d1 containing the ligand 5a and P for the d2. The situation is reversed for the enantiomer 5b, the M helical orientation is obtained for the diastereomer d2 and P for the diastereomer d1.
Further, in order to explain the biological activity of the complexes, we performed CD studies on water mixtures of calf thymus DNA (100 μM) and the complexes (5 μM) after 1 hour of incubation (Fig. S54†), following adapted reported procedures.24,36 No increase of any of the two bands was observed, suggesting that the complexes are not binding in the grooves of DNA. The slight decreases in band intensity that we observed could however be due to an intercalation mechanism. To verify this interaction, we performed docking calculations using a double stranded d(CpGpCpGpApApTpTpCpGpCpG) dodecamer (Fig. S63†). The calculations do not point to possible intercalation of the complexes, but confirm a weak interaction with the DNA grooves that might be responsible for the modest helix disruption.
Biological activity
Mononuclear fac-Re(CO)3+ complexes have been extensively evaluated13,16,17,37 for their anticancer properties, with several excellent examples also evaluated in vivo.6,7,11,12,38 In recent years, however, homonuclear multimetallic fac-Re(CO)3+ complexes have attracted increasing attention.13,26 The arrangement of two metallic fragments into one well-defined structure is intended to improve cellular uptake, by increased lipophilicity, and thus the cytotoxicity properties of the molecules.27,39 Prominent examples are the 1,2-di(pyridin-4-yl)ethane bridged phenanthroline species of Tan and Mao, which are ca. 3 times more cytotoxic than the mononuclear counterparts.40 However, it should be mentioned that this is not always the case, and often mononuclear fac-Re(CO)3+ complexes remain superior to their extended multimetallic derivatives.13,26 Herein, we have evaluated the cytotoxicity of our sets of diastereomers against three cancer and one healthy cell line to determine the selectivity index (SI) of the molecules. Selectivity index of the complexes was calculated by taking the ratio between the IC50 concentration of complexes in healthy cells (L929 cells) and IC50 concentration of complexes in cancer cells. Table 1 summarizes the data of our experiments.| Compounds | L929 | A549 | HCT116 | MCF-7 | |||
|---|---|---|---|---|---|---|---|
| IC50 (μM) | S.I. | IC50 (μM) | S.I. | IC50 (μM) | S.I. | ||
| 5a | >20 | >20 | — | >20 | — | >20 | — |
| 6a | 13.3 ± 1.1 | >20 | <0.7 | >20 | <0.7 | >20 | <0.7 |
| 5b | 13.7 ± 1 | >20 | <0.7 | >20 | <0.7 | >20 | <0.7 |
| 6b | 11.1 ± 1 | >20 | <0.6 | >20 | <0.6 | >20 | <0.6 |
| d1-[Re2(CO)6(5a)Cl2] | 12.3 ± 3.2 | 6.9 ± 0.6 | 1.8 | 2.6 ± 0.2 | 4.7 | 4.1 ± 0.6 | 3 |
| d2-[Re2(CO)6(5a)Cl2] | 11.1 ± 4.1 | 5.9 ± 0.5 | 1.9 | 5.6 ± 0.4 | 2 | 9.4 ± 0.9 | 1.2 |
| d1-[Re2(CO)6(6a)Cl2] | 12.8 ± 1.7 | 10.7 ± 1 | 1.2 | 10.4 ± 1 | 1.2 | 13.7 ± 1.3 | 0.9 |
| d2-[Re2(CO)6(6a)Cl2] | 2 ± 0.6 | 1 ± 0.2 | 1.9 | 0.6 ± 0.1 | 3.3 | 0.5 ± 0.1 | 3.8 |
| d1-[Re2(CO)6(5b)Cl2] | 5.2 ± 0.4 | 2.3 ± 0.2 | 2.3 | 1.5 ± 0.1 | 3.4 | 2.5 ± 0.2 | 2 |
| d2-[Re2(CO)6(5b)Cl2] | 9.8 ± 2.1 | 7.3 ± 0.9 | 1.3 | 3.7 ± 0.3 | 2.7 | 7.8 ± 0.7 | 1.3 |
| d1-[Re2(CO)6(6b)Cl2] | 12.9 ± 3 | 10.7 ± 1 | 1.2 | 8.7 ± 1.5 | 1.5 | 10.7 ± 1 | 1.2 |
| d2-[Re2(CO)6(6b)Cl2] | 3.3 ± 0.3 | 1.8 ± 0.3 | 1.9 | 1.2 ± 0.3 | 2.7 | 2.7 ± 0.5 | 1.2 |
| Cisplatin | 16.48 | 1.25 | 3.70 | ||||
The diastereomers display rather different anticancer activities (cytotoxicity), with perhaps the most striking results observed for the series of ligands 6a/b, in particular d2-[Re2(CO)6(6a)Cl2]. This compound showed an IC50 value of 0.6 and 0.5 μM against HCT116 and MCF-7 cells respectively. In comparison, d1-[Re2(CO)6(6a)Cl2] was ca. 18x and 25x less effective in the same lines. A similar, but less pronounced trend is observed if data of d2-[Re2(CO)6(6a)Cl2] are compared to those of d1-/d2-[Re2(CO)6(6b)Cl2]. In this series of compounds the increased cancer cells cytotoxicity was paralleled by an increased in toxicity versus healthy L929 cells (with d2-[Re2(CO)6(6a)Cl2] being the most toxic). While this trend may be expected, what is interesting is that the selectivity index (SI) of d2-[Re2(CO)6(6a)Cl2] (a value > 3) was significantly higher than all other isomers, which overall showed the same toxicity against cancer and healthy cells (i.e. no selectivity, with SI ca. 1). The series of complexes with ligands 5a/b further emphasizes the impact of chirality on the biological data. For these compounds, d1- isomers of 5a/b were the most active compounds and showed selectivity against HCT116 and MCF-7 cancers cells with SI values of 4.7 and 3.4 (HCT116) and 3 and 2 (MCF-7) for d1-[Re2(CO)6(5a)Cl2] and d1-[Re2(CO)6(5b)Cl2] respectively.
To rationalize the different activities of the diastereomers, we decided to study the cellular uptake and distribution of d1- and d2-[Re2(CO)6(6a)Cl2] in MCF7 and HCT116 cells by fluorescence light microscopy. These two complexes were selected as they showed the most significant difference in their cytotoxicity values against the two cell lines. Prior to the experiments we studied the emissive properties of d1- and d2-[Re2(CO)6(6a)Cl2]. Under aerobic conditions, the complexes are very weakly emissive (350 nm excitation, λmax of UV and excitation spectra). The emission range is very broad, from 500–700 nm, with the strongest band centred at around 620 nm (Fig. S57 and S58†). MCF7 and HCT116 cells were then incubated with the compounds for 4 h at 37 °C in separate imaging chambers, and the distribution of the complexes analysed. In MCF7 cells (Fig. 7), the result shows a clear difference in the uptake and distribution of the diastereomers. While d2-[Re2(CO)6(6a)Cl2] (IC50 of 0.5 μM) was able to enter the cells (within the 4 h incubation period) and was distributed in the cytoplasm, we found no evidence of d1-[Re2(CO)6(6a)Cl2] (IC50 of 13.7 μM) being taken up by MCF7 cells. The different uptakes were also confirmed by ICP-MS, where a clear difference in uptake was observed for the diastereomers obtained with 6a and 6b (see ESI, Table S2†). In HCT116 cells (Fig. 8) both compounds were found to accumulate in the nucleus. Thus, while the different intracellular uptake and distribution of the complexes may account for their different activity in MCF7 cells, the same argument cannot be valid for HCT116 cells. We do not know if DNA or nuclear proteins are the targets of the complexes in HCT116 cells, but docking calculations using a double stranded d(CpGpCpGpApApTpTpCpGpCpG) dodecamer as a model chiral nuclear molecule clearly show a different interaction of the two diastereomers with this possible biological target (Fig. 8). Indeed, while the lowest calculated energy pose of d2-[Re2(CO)6(6a)Cl2] (IC50 of 0.6 μM) suggests an association with the major groove of DNA, the same calculations point at a minor groove interaction of d1-[Re2(CO)6(6a)Cl2] (IC50 of 10.4 μM). Taken together, these results constitute evidence that the stereochemistry of the compounds may be playing a decisive role in determining the cytotoxicity of the complexes.
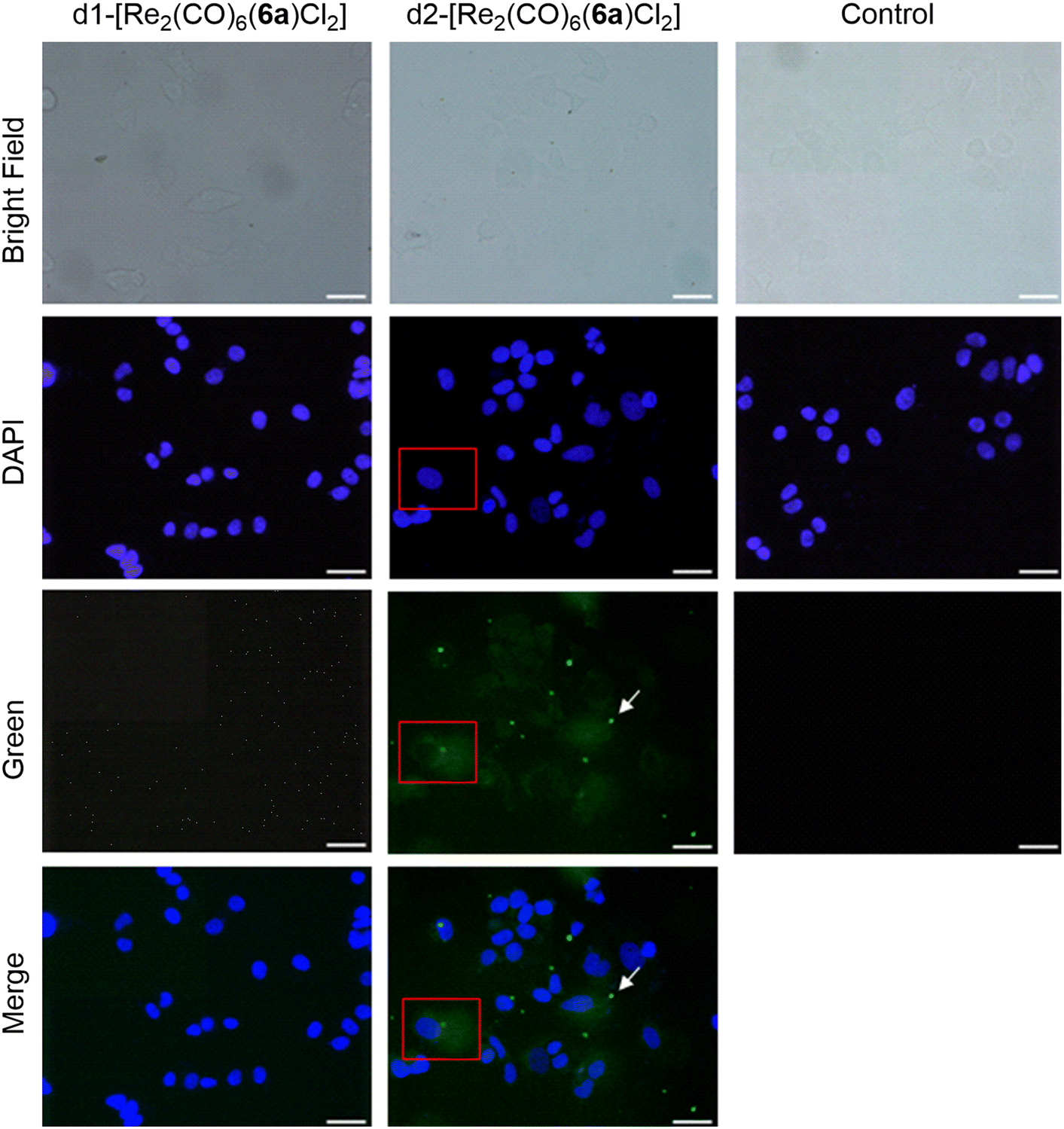 | ||
| Fig. 7 Fluorescence imaging of MCF7 cells incubated with d1-[Re2(CO)6(6a)Cl2] and d2-[Re2(CO)6(6a)Cl2]. Compound concentration: 10 μM, incubation time: 4 h, magnification: 40×, scale bar: 50 μm. White arrows indicate crystals of d2-[Re2(CO)6(6a)Cl2] (bright green spots) formed during the incubation period. | ||
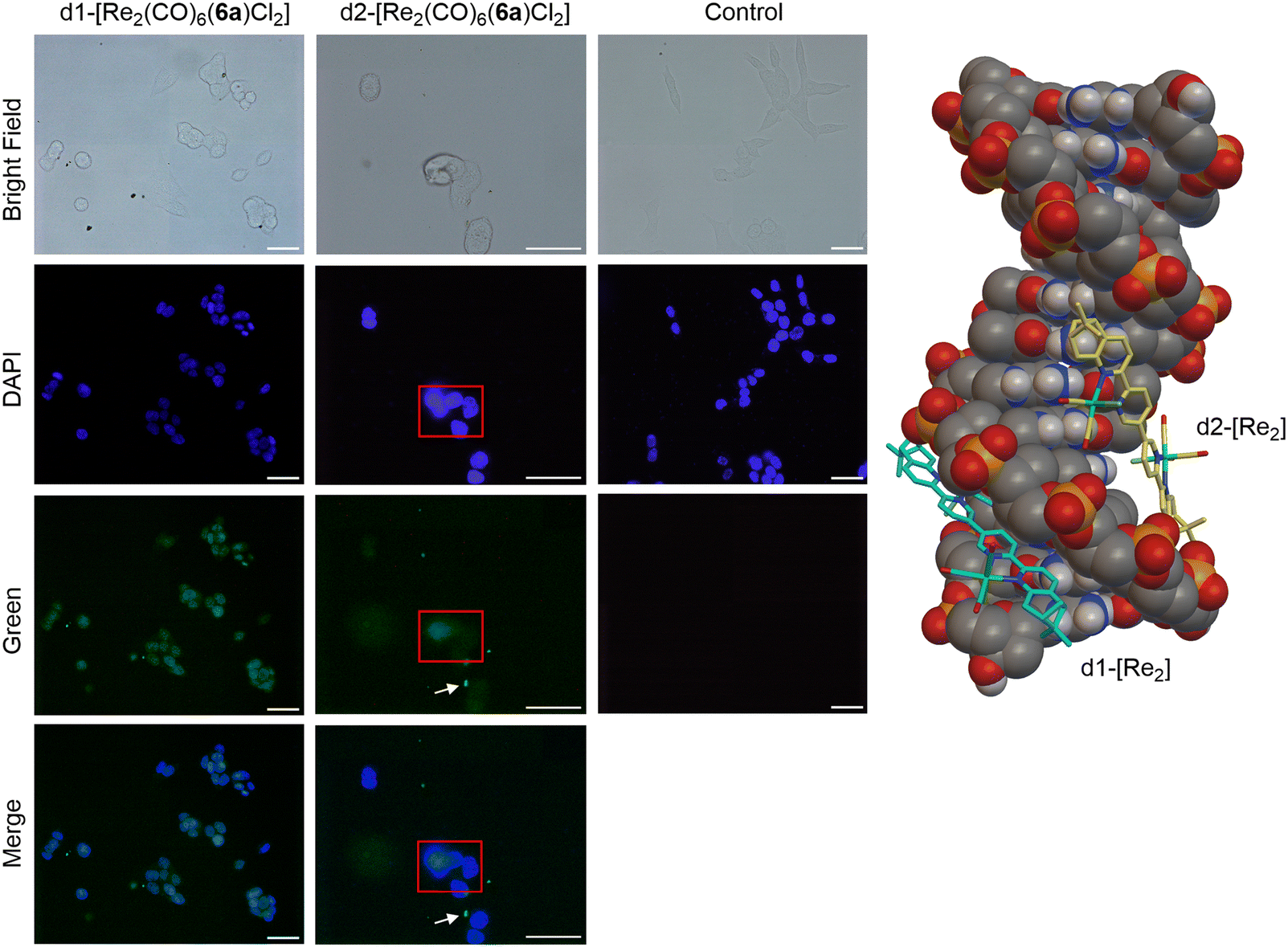 | ||
| Fig. 8 Left: fluorescence imaging of HCT116 cells incubated with d1-[Re2(CO)6(6a)Cl2] and d2-[Re2(CO)6(6a)Cl2]. Compound concentration: 10 μM, incubation time: 4 h, magnification: 40×, scale bar: 50 μm. White arrows indicate crystals of d2-[Re2(CO)6(6a)Cl2] (bright green spots) formed during the incubation period. Right: lowest calculated energy poses of d1- and d2-[Re2(CO)6(6a)Cl2] with double stranded d(CpGpCpGpApApTpTpCpGpCpG) dodecamer as a model chiral nuclear molecule. | ||
Conclusions
In conclusion, we have synthesised and fully characterised a library of chiral dinuclear Re(I) complexes with chiral bis-bidentate ligands containing pinene-bipyridine units and proved their potential as novel anticancer agents. The biological assessment of these dinuclear Re(I) complexes against various cancer cell lines, with a particular focus on breast and colon cancer, unveiled distinct anticancer activities among the diastereomers. Notably, one diastereomer, d2-[Re2(CO)6(6a)Cl2], displayed exceptional cytotoxicity against HCT116 and MCF-7 cancer cells with a clearly different uptake and distribution profile in the latter cell line. This finding underscores the importance of chirality in the design of novel anticancer agents and highlights the potential of dinuclear Re(I) complexes as effective candidates for cancer treatment. The study not only advances our understanding of the role of chirality in the biological activity of metal-based complexes, but also paves the way for further research in the development of innovative and targeted anticancer therapies.Experimental section
General
Reagent grade chemicals were purchased from Sigma Aldrich and Acros Organics and used without further purification. Yields reported are for isolated, spectroscopically pure compounds. When inert conditions were required, the reactions were performed under Ar atmosphere using glassware dried overnight at 120 °C. Analytical thin layer chromatography (TLC) was performed on SiO2 plates GF254 (0.25 mm layer thickness). Flash chromatography purifications were performed on a CombiFlash EZ Prep from Teledyne®. NMR spectra were recorded on a Bruker Avance DPX 300 spectrometer using TMS or the residual solvent proton as internal standard. HRMS spectra were recorded on FTMS 4.7T BioAPEX II and Waters SynaptG2-Si or at the Mass Services of Universities of Bern and Zurich. ICP-MS analyses were performed on a NexION 350D inductively coupled plasma mass spectrometer at the Mass Spectrometry service of EPFL. CD spectra were recorded on a Chirascan CD spectropolarimeter. Rotation angle values were recorded on an Anton Paar Modular Circular Polarimeter MCP 100. Fluorescence spectra were recorded on a Cary Eclipse fluorimeter. The determination of the quantum yields was performed using the comparative method, using Ru(bipy)Cl2 as reference. IR spectra were recorded on a Bruker ALPHA FTIR. Crystallographic data were collected on Stoe IPDS2T and Stoe STADIVARI diffractometers. For cytotoxicity experiments, Dulbecco's Modified Eagle Medium (DMEM) (with L-glutamine and glucose), L-glutamine penicillin/streptomycin (L-glu pen-strep) and trypsin-EDTA were purchased from Pan, Biotech. Thiazolyl blue tetrazolium bromide (MTT) was provided by Thermoscientific – Acros. Fetal bovine serum (FBS) and phosphate buffered saline (PBS, 1X) solution was obtained from Gibco. 96-Well plates were obtained from Nuncklon – Thermo. For cytotoxicity determination, Tecan – Infinity M Nano was used with the iControl program. MCF-7 – breast cancer cell line, HCT116 – human colon cancer cell line, A549 – lung carcinoma cell line and L929 – mouse fibroblast cells were kindly gifted from Prof. David Hoogewijs, Department of Medicine, University of Fribourg, Switzerland.Synthesis of the ligands: 5a, 5b, 6a, 6b
All the ligands were synthesised in a similar fashion, by following a reported literature procedure which was previously reported for 5a.34 For this, a solution of anhydrous NiCl2 (378 mg, 2.916 mmol, 1.2 eq.) in anhydrous and degassed DMF (15 mL) was heated under argon at 70 °C. Over this, PPh3 (3.059 g, 11.664 mmol, 4.8 eq.) was added, followed by Zn powder (411 mg, 6318 mmol, 2.6 eq.). The reaction mixture was stirred at 70 °C for 1 h. A DMF solution (3 mL) of the corresponding brominated precursor (3a–b/4a–b) (800 mg, 2.43 mmol, 1 eq.) was subsequently added over, and the reaction mixture was stirred for additional 3 h. The reaction mixture was left to reach room temperature and 35 mL NH3 25% was added. The mixture was extracted 3 times with 35 mL CH2Cl2 and washed twice with 35 mL H2O. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude was acidified with 10 mL HCl 6 M and extracted 3 times with 35 mL Et2O. The aqueous phase was neutralized with 4 M NaOH and afterwards extracted three times with 35 mL CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude product was recrystallized from EtOH to give the product as an off-white solid.5b: Yield: 73%. 1H NMR (500 MHz, CDCl3) δ 8.63 (d, J = 7.7 Hz, 2H, H2), 8.44 (d, J = 7.7 Hz, 2H, H4), 8.33 (d, J = 7.7 Hz, 2H, H7), 7.95 (t, J = 7.8 Hz, 2H, H3), 7.38 (d, J = 7.8 Hz, 2H, H8), 3.22 (d, J = 2.9 Hz, 4H, H11), 2.84 (t, J = 5.6 Hz, 2H, H14), 2.73 (dt, J = 9.7, 5.7 Hz, 2H, H15a), 2.42 (tt, J = 6.0, 2.9 Hz, 2H, H12), 1.44 (s, 6H, H16), 1.35 (d, J = 9.6 Hz, 2H, H15b), 0.71 (s, 6H, H17). 13C NMR (126 MHz, CDCl3) δ 156.5, 156.0, 155.6, 153.9, 142.4, 137.8, 133.9, 120.8, 120.6, 118.1, 46.7, 40.4, 39.7, 36. 9, 32.1, 26.2, 21.5. HRMS calcd. for C34H35N4+ [M + CH3CN + H]+ 562.2947, obtained 562.20626. [α]20D (1 mM in CHCl3) + 77°10−1 deg cm2 g−1.
6a: Yield: 65%. 1H NMR (500 MHz, CDCl3) δ 8.96 (d, J = 2.4 Hz, 2H, H1), 8.49 (d, J = 8.2 Hz, 2H, H4), 8.12 (d, J = 7.7 Hz, 2H, H7), 8.05 (dd, J = 8.2, 2.4 Hz, 2H, H3), 7.36 (d, J = 7.8 Hz, 2H, H8), 3.22 (d, J = 2.9 Hz, 4H, H11), 2.84 (t, J = 5.7 Hz, 2H, H14), 2.73 (dt, J = 9.6, 5.7 Hz, 2H, H15a), 2.42 (tt, J = 6.0, 2.9 Hz, 2H, H12), 1.44 (s, 6H, H16), 1.34 (d, J = 9.7 Hz, 2H, H15b), 0.71 (s, 6H, H17). 13C NMR (126 MHz, CDCl3) δ 156.8, 156.5, 153.2, 147.5, 142.7, 135.1, 134.0, 132.7, 121.0, 118.2, 46.7, 40.4, 39.7, 36.9, 32.1, 26.2, 21.5. HRMS calcd. for C34H35N4+ [M + H]+ 499.28562, obtained 499.28556. [α]20D (1 mM in CHCl3) −117°10−1 deg cm2 g−1.
6b: Yield: 71%. 1H NMR (500 MHz, CDCl3) δ 8.96 (d, J = 2.4 Hz, 2H, H1), 8.49 (d, J = 8.2 Hz, 2H, H4), 8.11 (d, J = 7.8 Hz, 2H, H7), 8.04 (dd, J = 8.2, 2.4 Hz, 2H, H3), 7.35 (d, J = 7.8 Hz, 2H, H8), 3.21 (d, J = 2.9 Hz, 4H, H11), 2.83 (t, J = 5.6 Hz, 2H, H14), 2.72 (dt, J = 9.7, 5.7 Hz, 2H, H15a), 2.41 (tt, J = 6.1, 2.9 Hz, 2H, H12), 1.43 (s, 6H, H16), 1.33 (d, J = 9.7 Hz, 2H, H15b), 0.70 (s, 6H, H17). 13C NMR (126 MHz, CDCl3) δ 156.8, 156.4, 153.1, 147.5, 142.7, 135.0, 134.0, 133.9, 132.7, 121.0, 118.1, 46.7, 40.4, 39.7, 36.9, 32.1, 26.2, 21.5. HRMS calcd. for C34H35N4+ [M + H]+ 499.28562, obtained 499.28559. [α]20D (1 mM in CHCl3) 113°10−1 deg cm2 g−1.
Synthesis of the complexes with the general formula [Re2(CO)6(L)Cl2] (L = 5a, 5b, 6a, 6b)
A mixture of enantiopure L (100 mg, 0.2 mmol, 1 eq.) and [Re(CO)5Cl] (152 mg, 0.42 mmol, 2.1 eq.) were dissolved under inert atmosphere in a mixture of anhydrous toluene (60 mL) and anhydrous THF (15 mL). The reaction mixture was stirred under inert atmosphere for 16 h at 85 °C. The reaction mixture was afterwards left to cool to RT and concentrated under reduced pressure. The crude compound was purified by column chromatography on SiO2 using a heptane/EtOAc = 5/95 mixture as eluent. Single crystals of the eight [Re2L(CO)6Cl2] complexes reported were obtained by vapour diffusion of Et2O or petroleum ether into a 4/1 = CH2Cl2/EtOH mixture.Cytotoxicity studies
Cytotoxicity of the complexes was evaluated using the standard MTT assay. For viability test, MCF-7, HCT116, A549 and L929 cells were seeded at a density of 5 × 104 cells per mL for cancer cells and 3.75 × 104 cells per mL for L929 cells in complete medium (supplemented with 10% (v/v) FBS and 1% (v/v) L-glu pen-strep) and incubated for 24 hours. Then, complexes, dissolved in DMSO were injected with fresh medium at a concentration between 0.63 and 10 μM. The volume ratio of the DMSO in the highest concentration was arranged to 1%. After 24 h incubation, MTT solution (5 mg ml−1 in PBS) was injected with fresh complete medium. After 4 hours incubation with MTT solution, the water insoluble formazan molecules, which is the result of the mitochondrial activity of the viable cells, were dissolved in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (1:1) mixture. Absorbance of the formazan molecules was read via microplate reader at 570 nm with reference wavelength 620 nm. Cells that were not exposed to the complexes and DMSO were used as controls. The viability of control cells was assumed as 100%. The relative cell viability was calculated by making the ratio with control cells. To check the effect of DMSO, 1% DMSO was added to cells and named "DMSO". Statistical analysis of the cytotoxicity of complexes were conducted by using ordinary one-way ANOVA analysis of variance followed by multiple Dunnett's comparison test of GraphPad Prism 8.4.2 software package. All measurements were expressed as mean values ± standard deviation (SD) based on 5 replicas. p < 0.05 was accepted as statistically significant difference. Statistical significance: (*) p < 0.0332, (**) p < 0.021, (***) p < 0.0002, (****) p < 0.0001. IC50 values were evaluated by using nonlinear regression analysis followed by variable slope.
:EtOH (1:1) mixture. Absorbance of the formazan molecules was read via microplate reader at 570 nm with reference wavelength 620 nm. Cells that were not exposed to the complexes and DMSO were used as controls. The viability of control cells was assumed as 100%. The relative cell viability was calculated by making the ratio with control cells. To check the effect of DMSO, 1% DMSO was added to cells and named "DMSO". Statistical analysis of the cytotoxicity of complexes were conducted by using ordinary one-way ANOVA analysis of variance followed by multiple Dunnett's comparison test of GraphPad Prism 8.4.2 software package. All measurements were expressed as mean values ± standard deviation (SD) based on 5 replicas. p < 0.05 was accepted as statistically significant difference. Statistical significance: (*) p < 0.0332, (**) p < 0.021, (***) p < 0.0002, (****) p < 0.0001. IC50 values were evaluated by using nonlinear regression analysis followed by variable slope.
Fluorescence light microscopy
Fluorescence light microscopy imaging was performed using an Olympus BX53 fluorescence microscope equipped with Olympus UC90 camera. The images were taken by staining the cells with DAPI (filter excitation: 360 nm, emission: 420–460 nm) for nucleus imaging, and a green filter (excitation: 470 nm, emission: 510–550 nm) for the complexes. The imaging glasses were sterilised and covered with a 0.1% gelatin solution in 12-well plate. After the gelatination, glasses were left in the laminar hood to dry. The cells were seeded in 60 × 103 cells per well in 0.75 mL per well. After 24 hours, the complexes were added in 10 μM final concentration and incubated for 4 hours. Then, cells were fixed with 10% formalin solution and treated with DAPI solution (1 μg mL−1). After the treatments, the cells on the glass were transferred to glass slide and sealed with transparent nail Polish.Cell uptake study
The quantification of internalised complexes was performed by the determination of Re content in the complexes treated cells. HCT116 and MCF-7 cells were seeded on a 6-well plate at a density of 1 × 105 cells per mL complete medium and grown for 24 h in an incubator. Next day, the growing cells were incubated with the complexes at 10 μM complex concentration for 4 h. Then, the cells were washed with PBS to remove uninternalised complexes and subsequently detached with trypsin to collect them into preweighted clean vials. The trypsin was evaporated and dried under vacuum. After acid treatment, the Re content was determined by ICP-MS.Docking study
Docking calculations were performed as previously described.41 The DNA target for investigated Re complexes was selected from Protein Data Bank (pdb code: 1BNA), as the double stranded d(CpGpCpGpApApTpTpCpGpCpG) dodecamer with two G![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C rich regions flanking one A
C rich regions flanking one A![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) T rich region.42 Molecular docking calculations were carried out with the AutoDock Vina (version 1.2.0)43 and the AutoDock4 (version 4.2.6)44 software. The Biovia Discovery Studio Visualizer software (version 21.1.0.20298) was employed to analyse possible interactions between the ligand and the DNA target.
T rich region.42 Molecular docking calculations were carried out with the AutoDock Vina (version 1.2.0)43 and the AutoDock4 (version 4.2.6)44 software. The Biovia Discovery Studio Visualizer software (version 21.1.0.20298) was employed to analyse possible interactions between the ligand and the DNA target.
Determination of the quantum yields
The quantum yields (QY) were determined through the comparison method, using [Ru(Bipy)3]Cl2 as a reference. Solutions of each complex were prepared in CH2Cl2 at a concentration of 2.5 μM, such as the absorption values at λmax were not exceeding 0.1. UV-Vis and emission spectra were measured for the reference and each complex and the areas under the emission spectra were integrated. Using the formula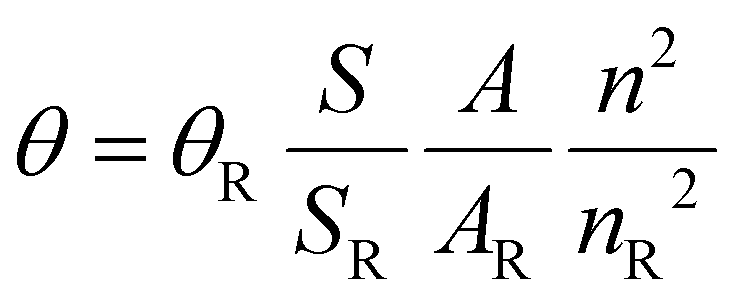 , where S is the area under the emission spectra of the complexes, A represents the absorption values at the excitation wavelength and n is the refraction index of the solvent, the QY values were determined (θR = 0.059, n = 1.4244 and nR = 1.3441).45
, where S is the area under the emission spectra of the complexes, A represents the absorption values at the excitation wavelength and n is the refraction index of the solvent, the QY values were determined (θR = 0.059, n = 1.4244 and nR = 1.3441).45
Single-crystal X-ray structure analysis
Suitable crystals were selected and mounted on loop with oil on a single-crystal diffractometer (Stoe IPDS2T or STADIVARI). The crystals were kept at 250(2) K during data collection. Using Olex2,46 the structures were solved with the SHELXT47 structure solution program using Intrinsic Phasing and refined with the SHELXL48 refinement package using Least Squares minimisation.Crystallographic data for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre. Copies of the data can be obtained by quoting the depository numbers CCDC 2310572–2310580 and 2366457.†
Data availability
The data supporting this article has been included in the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
ABS and OMS thank HES-SO for funding as well as our HEIA-FR students: Maxime Brunisholz, Rémy Schobinger and Guillaume Chapuis for their help in the ligands syntheses.References
- F. Arnesano and G. Natile, Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA, Coord. Chem. Rev., 2009, 253, 2070–2081 CrossRef CAS.
- R. Oun, Y. E. Moussa and N. J. Wheate, The side effects of platinum-based chemotherapy drugs: a review for chemists, Dalton Trans., 2018, 47, 6645–6653 RSC.
- K. B. Garbutcheon-Singh, M. P. Grant, B. W. Harper, A. M. Krause-Heuer, M. Manohar, N. Orkey and J. R. Aldrich-Wright, Transition metal based anticancer drugs, Curr. Top. Med. Chem., 2011, 11, 521–542 CrossRef CAS PubMed.
- A. P. King, S. C. Marker, R. V. Swanda, J. J. Woods, S. B. Qian and J. J. Wilson, A Rhenium Isonitrile Complex Induces Unfolded Protein Response-Mediated Apoptosis in Cancer Cells, Chemistry, 2019, 25, 9206–9210 CrossRef CAS PubMed.
- C. C. Konkankit, J. Lovett, H. H. Harris and J. J. Wilson, X-Ray, fluorescence microscopy reveals that rhenium(I) tricarbonyl isonitrile complexes remain intact in vitro, Chem. Commun., 2020, 56, 6515–6518 RSC.
- S. C. Marker, A. P. King, S. Granja, B. Vaughn, J. J. Woods, E. Boros and J. J. Wilson, Exploring the In Vivo and In Vitro Anticancer Activity of Rhenium Isonitrile Complexes, Inorg. Chem., 2020, 59, 10285–10303 CrossRef CAS PubMed.
- P. Collery, A. Mohsen, A. Kermagoret, S. Corre, G. Bastian, A. Tomas, M. Wei, F. Santoni, N. Guerra, D. Desmaële and J. d'Angelo, Antitumor activity of a rhenium(I)-diselenoether complex in experimental models of human breast cancer, Invest. New Drugs, 2015, 33, 848–860 CrossRef CAS PubMed.
- V. Veena, A. Harikrishnan, B. Lakshmi, S. Khanna, D. Desmaele and P. Collery, A New Model Applied for Evaluating a Rhenium-diselenium Drug: Breast Cancer Cells Stimulated by Cytokines Induced from Polynuclear Cells by LPS, Anticancer Res., 2020, 40, 1915–1920 CrossRef CAS PubMed.
- P. Collery, V. Veena, A. Harikrishnan and D. Desmaele, The rhenium(I)-diselenoether anticancer drug targets ROS, TGF-beta1, VEGF-A, and IGF-1 in an in vitro experimental model of triple-negative breast cancers, Invest. New Drugs, 2019, 37, 973–983 CrossRef CAS PubMed.
- P. V. Simpson, I. Casari, S. Paternoster, B. W. Skelton, M. Falasca and M. Massi, Defining the Anti-Cancer Activity of Tricarbonyl Rhenium Complexes: Induction of G2/M Cell Cycle Arrest and Blockade of Aurora-A Kinase Phosphorylation, Chemistry, 2017, 23, 6518–6521 CrossRef CAS PubMed.
- A. Domenichini, I. Casari, P. V. Simpson, N. M. Desai, L. Chen, C. Dustin, J. S. Edmands, A. van der Vliet, M. Mohammadi, M. Massi and M. Falasca, Rhenium N-heterocyclic carbene complexes block growth of aggressive cancers by inhibiting FGFR- and SRC-mediated signalling, J. Exp. Clin. Cancer Res., 2020, 39, 276 CrossRef CAS PubMed.
- J. Delasoie, A. Pavic, N. Voutier, S. Vojnovic, A. Crochet, J. Nikodinovic-Runic and F. Zobi, Identification of novel potent and non-toxic anticancer, anti-angiogenic and antimetastatic rhenium complexes against colorectal carcinoma, Eur. J. Med. Chem., 2020, 204, 112583 CrossRef CAS PubMed.
- K. Schindler and F. Zobi, Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives, Molecules, 2022, 27, 539 CrossRef CAS PubMed.
- Z. Huang and J. J. Wilson, Therapeutic and Diagnostic Applications of Multimetallic Rhenium(I) Tricarbonyl Complexes, Eur. J. Inorg. Chem., 2021, 2021, 1312–1324 CrossRef CAS.
- H. S. Liew, C.-W. Mai, M. Zulkefeli, T. Madheswaran, L. V. Kiew, N. Delsuc and M. L. Low, Recent Emergence of Rhenium(I) Tricarbonyl Complexes as Photosensitisers for Cancer Therapy, Molecules, 2020, 25, 4176 CrossRef CAS PubMed.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Organometallic and coordination rhenium compounds and their potential in cancer therapy, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium, Dalton Trans., 2018, 47, 9934–9974 RSC.
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Recent development of luminescent rhenium(I) tricarbonyl polypyridine complexes as cellular imaging reagents, anticancer drugs, and antibacterial agents, Dalton Trans., 2017, 46, 16357–16380 RSC.
- A. Leonidova and G. Gasser, Underestimated potential of organometallic rhenium complexes as anticancer agents, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- A. Sharma S., V. N., B. Kar, U. Das and P. Paira, Target-specific mononuclear and binuclear rhenium(I) tricarbonyl complexes as upcoming anticancer drugs, RSC Adv., 2022, 12, 20264–20295 RSC.
- R. S. Herrick, I. Wrona, N. McMicken, G. Jones, C. J. Ziegler and J. Shaw, Preparation and characterization of rhenium(I) compounds with amino ester derivatized diimine ligands. Investigations of luminescence. Crystal structures of Re(CO)3Cl(pyca-β-Ala-OEt) and Re(CO)3Cl(pyca-l-Asp(OMe)-OMe), J. Organomet. Chem., 2004, 689, 4848–4855 CrossRef CAS.
- F. Zobi, B. Spingler and R. Alberto, Guanine and plasmid DNA binding of mono- and trinuclear fac-[Re(CO)3]+ complexes with amino acid ligands, ChemBioBhem, 2005, 6, 1397–1405 CrossRef CAS PubMed.
- F. Zobi, B. Spingler and R. Alberto, Structure, reactivity and solution behaviour of [Re(ser)(7-MeG)(CO)3] and [Re(ser)(3-pic)(CO)3]: “nucleoside-mimicking” complexes based on the fac-[Re(CO)3]+ moiety, Dalton Trans., 2005, 2859–2865 RSC.
- G. A. Suarez-Ortiz, R. Hernandez-Correa, M. D. Morales-Moreno, R. A. Toscano, M. T. Ramirez-Apan, A. Hernandez-Garcia, M. Amezquita-Valencia and D. Araiza-Olivera, Diastereomeric Separation of Chiral fac-Tricarbonyl(iminopyridine) Rhenium(I) Complexes and Their Cytotoxicity Studies: Approach toward an Action Mechanism against Glioblastoma, J. Med. Chem., 2022, 65, 9281–9294 CrossRef CAS PubMed.
- K. Schindler, J. Horner, G. Demirci, Y. Cortat, A. Crochet, O. Mamula Steiner and F. Zobi, In Vitro Biological Activity of α-Diimine Rhenium Dicarbonyl Complexes and Their Reactivity with Different Functional Groups, Inorganics, 2023, 11, 139 CrossRef CAS.
- Z. Y. Huang and J. J. Wilson, Therapeutic and Diagnostic Applications of Multimetallic Rhenium(I) Tricarbonyl Complexes, Eur. J. Inorg. Chem., 2021, 2021, 1312–1324 CrossRef CAS.
- S. Clède, F. Lambert, R. Saint-Fort, M.-A. Plamont, H. Bertrand, A. Vessières and C. Policar, Influence of the Side-Chain Length on the Cellular Uptake and the Cytotoxicity of Rhenium Triscarbonyl Derivatives: A Bimodal Infrared and Luminescence Quantitative Study, Chem. – Eur. J., 2014, 20, 8714–8722 CrossRef PubMed.
- R. G. Balasingham, F. L. Thorp-Greenwood, C. F. Williams, M. P. Coogan and S. J. Pope, Biologically compatible, phosphorescent dimetallic rhenium complexes linked through functionalized alkyl chains: syntheses, spectroscopic properties, and applications in imaging microscopy, Inorg. Chem., 2012, 51, 1419–1426 CrossRef CAS PubMed.
- A. B. Solea, S. Wang, X. S. Xue, A. Crochet, K. M. Fromm, K. N. Houk, O. Mamula and C. Allemann, Efficient synthesis of isoindolones by intramolecular cyclisation of pyridinylbenzoic acids, Org. Biomol. Chem., 2021, 19, 8025–8029 RSC.
- A. B. Solea, I. Cornu, V. Deneva, A. Crochet, K. M. Fromm, L. Antonov, C. Allemann and O. Mamula, Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives, Molecules, 2020, 25, 298 CrossRef CAS PubMed.
- A. B. Solea, C. Curty, K. M. Fromm, C. Allemann and O. Mamula Steiner, A Rapid, Highly Sensitive and Selective Phosgene Sensor Based on 5,6-Pinenepyridine, Chemistry, 2022, 28, e202201772 CrossRef CAS PubMed.
- A. B. Solea, L. Yang, A. Crochet, K. M. Fromm, C. Allemann and O. Mamula, Complexation Behavior of Pinene–Bipyridine Ligands towards Lanthanides: The Influence of the Carboxylic Arm, Chemistry, 2021, 4, 18–30 CrossRef.
- A. B. Solea, T. Wohlhauser, P. Abbasi, Y. Mongbanziama, A. Crochet, K. M. Fromm, G. Novitchi, C. Train, M. Pilkington and O. Mamula, Versatile synthesis of chiral 6-oxoverdazyl radical ligands - new building blocks for multifunctional molecule-based magnets, Dalton Trans., 2018, 47, 4785–4789 RSC.
- H.-L. Yeung, W.-Y. Wong, C.-Y. Wong and H.-L. Kwong, Stereoselective Formation of Helical Binuclear Metal Complexes: Synthesis, Characterization, and Crystal Structures of Chiral Bis-Rhenium(I) Quaterpyridine Complexes, Inorg. Chem., 2009, 48, 4108–4117 CrossRef CAS PubMed.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Helicates as Versatile Supramolecular Complexes, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- V. Tsikaris, E. Panou-Pomonis, C. Sakarellos, M. Sakarellos-Daitsiotis and M. Marraud, Changes in the c.d. spectra of calf thymus DNA induced by sequential polypeptides in aqueous solutions. Part I, Int. J. Biol. Macromol., 1991, 13, 349–354 CrossRef CAS PubMed.
- P. Collery, D. Desmaele and V. Vijaykumar, Design of Rhenium Compounds in Targeted Anticancer Therapeutics, Curr. Pharm. Des., 2019, 25, 3306–3322 CrossRef CAS PubMed.
- S. Nasiri Sovari, I. Kolly, K. Schindler, A. Djuric, T. Srdic-Rajic, A. Crochet, A. Pavic and F. Zobi, Synthesis, characterization, and in vivo evaluation of the anticancer activity of a series of 5- and 6-(halomethyl)-2,2′-bipyridine rhenium tricarbonyl complexes, Dalton Trans., 2023, 52, 6934–6944 RSC.
- C. C. Konkankit, B. A. Vaughn, Z. Huang, E. Boros and J. J. Wilson, Systematically altering the lipophilicity of rhenium(I) tricarbonyl anticancer agents to tune the rate at which they induce cell death, Dalton Trans., 2020, 49, 16062–16066 RSC.
- R.-R. Ye, C.-P. Tan, M.-H. Chen, L. Hao, L.-N. Ji and Z.-W. Mao, Mono- and Dinuclear Phosphorescent Rhenium(I) Complexes: Impact of Subcellular Localization on Anticancer Mechanisms, Chem. – Eur. J., 2016, 22, 7800–7809 CrossRef CAS PubMed.
- K. Schindler, Y. Cortat, M. Nedyalkova, A. Crochet, M. Lattuada, A. Pavic and F. Zobi, Antimicrobial Activity of Rhenium Di- and Tricarbonyl Diimine Complexes: Insights on Membrane-Bound S. aureus Protein Binding, Pharmaceuticals, 2022, 15, 1107 CrossRef CAS PubMed.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Structure of a B-DNA dodecamer: conformation and dynamics, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179–2183 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- L. Prodi and A. Credi, in The Exploration of Supramolecular Systems and Nanostructures by Photochemical Techniques, 2012, ch. 5, pp. 97–129 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT - integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2310572–2310580, 2366457. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01643b |
| This journal is © The Royal Society of Chemistry 2024 |